

Abstract
Objective:
Histone deacetylase inhibitors (HDACi) have proven to induce HIV-RNA and antigen expression in resting CD4+T cells of ART-treated HIV-infected individuals. However, to achieve viral eradication, immune clearance must follow latency reversal, and thus it is essential to understand the impact of latency reversal agents on immune function.
Design:
Here we evaluate the impact of in vivo administration of vorinostat (VOR) and panobinostat (PNB) during clinical trials on Natural Killer (NK) cell function and phenotype.
Methods:
Cryopreserved PBMC from HIV-positive participants receiving VOR (NCT01319383) or PNB (NCT01680094) were selected to assess the impact of the drugs on cell composition, activation, NK cell phenotype (CD16, NKG2D, NKp30, NKp46 and DNAM-1), cytotoxic activity (CD107a) and IFN-γ production.
Results:
No impairment of NK cell function was observed during treatment with either VOR or PNB. An increase in the frequency of CD3−CD56+ NK cells was consistently observed. Interestingly, after VOR administration, NK cells increased expression of NKp46 and CD16, and showed improved degranulation and IFN-γ production capacity. Moreover, taking together VOR and PNB samples, HIV DNA levels in CD4 cells were negatively correlated with NK cell frequency and NK cell expression of CD16.
Conclusions:
In vivo treatment with HDACi do not have measurable negative effects on NK cell function, with some evidence of improved function in vitro. These results have important implications for potential combinatorial approaches to target HIV reservoirs, suggesting that the use of HDACis as a latency reversal agent could be paired with interventions to enhance NK cell activity or recruitment.
Keywords: HIV, NK cells, Vorinostat, VOR, SAHA, Panobinostat, PNB, HDACi
INTRODUCTION
Research strategies geared towards curing persistent HIV infection have entered clinical testing[1]. The most advanced of these approaches involves the administration of compounds known as latency reversing agents (LRAs) to induce HIV gene transcription, thus allowing expression of latent virus within infected cells. Histone deacetylase inhibitors (HDACi) are drugs employed to treat certain cancers that have also demonstrated to reactivate HIV from latency [2–6]. Vorinostat (SAHA, VOR) was the first drug from this family to be tested in the clinic as an LRA[7–10]. Following VOR, two other HDACi, Panobinostat (PNB)[11] and Romidepsin (RMD)[12, 13] also entered clinical study. All three drugs increased cell-associated HIV RNA (ca-RNA) in CD4+T cells from ART suppressed individuals, but failed to reduce the frequency of latently infected cells. Therefore, approaches to enhance immune responses following latency reversal are required to eliminate infected cells.
Initial efforts towards enhancement of immune responses were focused on HIV-specific CD8+T cells [14–18], either through ex vivo expansion or vaccination strategies. In vitro studies have suggested that some LRAs might have a negative impact on the viability or function of CD8+T cells [19–21], although this has not been confirmed in vivo [14]. More recently, there has been a growing interest in the exploration of Natural Killer (NK) cells as cytotoxic effectors in HIV eradication strategies, as they have shown potential to be clinically exploited for HIV clearance [22]. Critical to those endeavors is the assessment of the effect of LRAs on NK cell function. To that end, we previously performed a study evaluating the effect of ex vivo LRA exposure on NK cells [23]. In that ex vivo study, we demonstrated that different compounds cause diverse effects on NK cells, with differences observed even within a given class of compounds. We observed that while VOR did not negatively affect NK cell function, PNB and RMD induced undesirable alterations [23]. However, given that the in vivo milieu cannot be fully replicated by ex vivo modeling, assessing the impact of in vivo exposure to LRAs on NK cell is extremely important. In the present study, we report a direct assessment of NK cell function in cells obtained from HIV+ participants receiving VOR or PNB in clinical studies.
METHODS
Study samples
Viably frozen peripheral blood mononuclear cells (PBMCs) were derived from participants included in two clinical trials involving two different HDAC inhibitors: vorinostat (VOR) and panobinostat (PNB). Basic characteristics of the clinical trials performed with HDACi in the context of HIV are summarized in Table 1.
Table 1.
Summary of the clinical trials conducted with HDACi in the context of HIV infection.
| Clinical Trial | # | Dose | Dosing | Reference | |
|---|---|---|---|---|---|
| VOR | NCT01319383 | 8 | 400 mg | 1 dose | Archin, Nature 2012 |
| NCT01319383 | 5 | 400 mg | 3 doses/week 8 weeks | Archin, JID 2014 | |
| NCT01365065 | 20 | 400 mg | 1 daily for 14 days | Elliott, PlosPath 2014 | |
| NCT01319383 | 16 | 400 mg | 1 dose | Archin, JCI 2017 | |
| 1+1 after 48h | |||||
| 1+1 after 72h | |||||
| 10 doses every 72 hrs | |||||
| PNB | NCT01680094 | 15 | 20 mg | 3 times/week every other week for 8 weeks | Rasmussen, Lancet HIV 2014 |
| RMD | NTC02092116 | 6 | 5 mg/m2 | 1 time/week for 3 weeks (intravenous) | Søgaard, PlosPath 2015 |
VOR samples were derived from 5 participants enrolled in the clinical trial NCT01319383[24], which evaluated the effect of 22 cyclical doses of VOR over a period of 12–16 weeks. Briefly, 400 mg of VOR was administered daily Monday to Wednesday for 4 weeks, for a total of 11 doses. After a rest period, participants received another 4 weeks of VOR doses Monday to Wednesday to reach a total of 22 doses. PBMCs from the participants were obtained by leukapheresis at baseline, after the first 4-week cycle (on the morning after the 11th dose, “11 VOR-doses”) and at the end of the study (on the morning after the 22nd dose, “22 VOR-doses”), and viably frozen until use.
PNB specimens were derived from the CLEAR study (NCT01680094)[11], in which 15 participants were administered 20 mg PNB three times per week (Monday, Wednesday and Friday) every other week for 8 weeks. PBMCs were obtained 8 hours after a PNB dose. In the present study we included samples from five of these participants, with one sample before treatment (“pre-PNB”), after 4 doses (“4 doses”), after 10 doses (“10 doses”) and 4 weeks after completing treatment (“follow-up”).
Participants included in both trials were virally suppressed (<50 HIV RNA copies/mL) on antiretroviral therapy for at least 2 years, and had CD4 counts above 470 cells/uL.
Cell processing
Viably frozen PBMCs from each participant at different time points were thawed simultaneously and rested on culture for 24 hours. An aliquot was used to analyze the frequency of each cell population (i.e., CD4, CD8 and NK cells), and assess proliferation and activation markers. A second aliquot was used to isolate NK cells (EasySep Human NK Cell negative selection Enrichment Kit, StemCell Technologies, Vancouver, BC) to study expression of activating receptors and immune function. After isolation, the purity of NK cells was consistently >85%.
Analysis of cell phenotype
PBMCs were stained with a panel of monoclonal antibodies to define cell populations. The antibody panel included CD3-PerCP (clone SK7, BD Biosciences), CD4-FITC (clone RPA-T4, BD Biosciences), CD8-AlexaFluor700 (clone SK1, Biolegend) and CD56-PE (clone NCAM16.2, BD Bioscience). Cell subsets were derived from the singlets gate within the lymphocyte population, and were classified as NK cells when they were CD3−CD56+, CD4+T cells when they co-expressed CD3 and CD4, and CD8+T cells when they were double positive for CD3 and CD8 (Fig. 1A). In addition, proliferation and activation markers were analyzed using Ki67-PE/Cy7 (Biolegend) and CD69-APC (clone FN50, Biolegend). Another antibody panel was designed to examine the expression of NK activating receptors on isolated NK cells. This panel included the following antibodies from Biolegend (San Diego, CA): CD16-AlexaFluor700 (clone 3G8), NKG2D-APC (clone 1D11), NKp30-PE (clone P30–15), NKp46-PE/Cy7 (clone 9E2) and DNAM1-FITC (clone 11A8). Briefly, cells were resuspended in staining buffer and incubated with an optimized antibody concentration for 20 minutes on ice in the dark. Cells were then washed twice, fixed and analyzed using the Attune NxT Acoustic Focusing Cytometer (Applied Biosystems, Forster City, CA). Fluorescence minus one controls (FMO) were used for each antibody combination to set the gates. Analysis was performed using the FlowJo software v10 (Ashland, OR).
Fig. 1. Frequency of cell populations, proliferation and activation during HDAC inhibitor treatment.

PBMC were stained with a panel of monoclonal antibodies to analyze cell populations and expression of proliferation and activation markers. A. Representative plot of cell gating strategy. B. VOR treatment: The proportion of CD4 and CD8+T cells did not change during VOR treatment, but frequency of NK cells increased after 11 doses of VOR, with increases maintained after 22 doses. Expression of the proliferation marker Ki67 and the activation marker CD69 decreased during VOR treatment. C. PNB treatment: CD4 and CD8+T cells remained stable during PNB treatment, but NK cells showed an increase, that was lost after the end of treatment. A decrease in CD69 was observed during PNB treatment. Each symbol represents cells from a different study participant. Wilcoxon matched-pairs signed rank test.
Functional assays
The function of NK cells was assessed by measuring degranulation, through the degranulation marker CD107a [25], and IFN-γ production by intracellular staining. Isolated NK cells were cocultured with the MHC-I lacking target cell line K562 at a 1:1 effector:target ratio, along with CD107a-PE/Cy7 (clone H4A3, Biolegend) for 2–4 hours, adding the protein transport inhibitor GolgiStop (BD Biosciences) after the first hour. Cells were then harvested and stained with CD3-APC/Fire750 (clone SK7, Biolegend) and CD56-FITC (clone HCD56, Biolegend). Cells were washed, and after fixation and permeabilization, intracellular staining with IFN-γ-PE (clone 4SB3, Biolegend) antibody was performed. Cells were acquired using the Attune NxT Acoustic Focusing Cytometer and analysis was performed using the Flowjo v.10. Cells were gated according to CD3 and CD56 expression to determine the proportion of NK cells expressing the degranulation marker CD107a and/or producing IFN-γ (Fig. 2A).
Fig. 2. NK cell cytotoxicity and cytokine production.
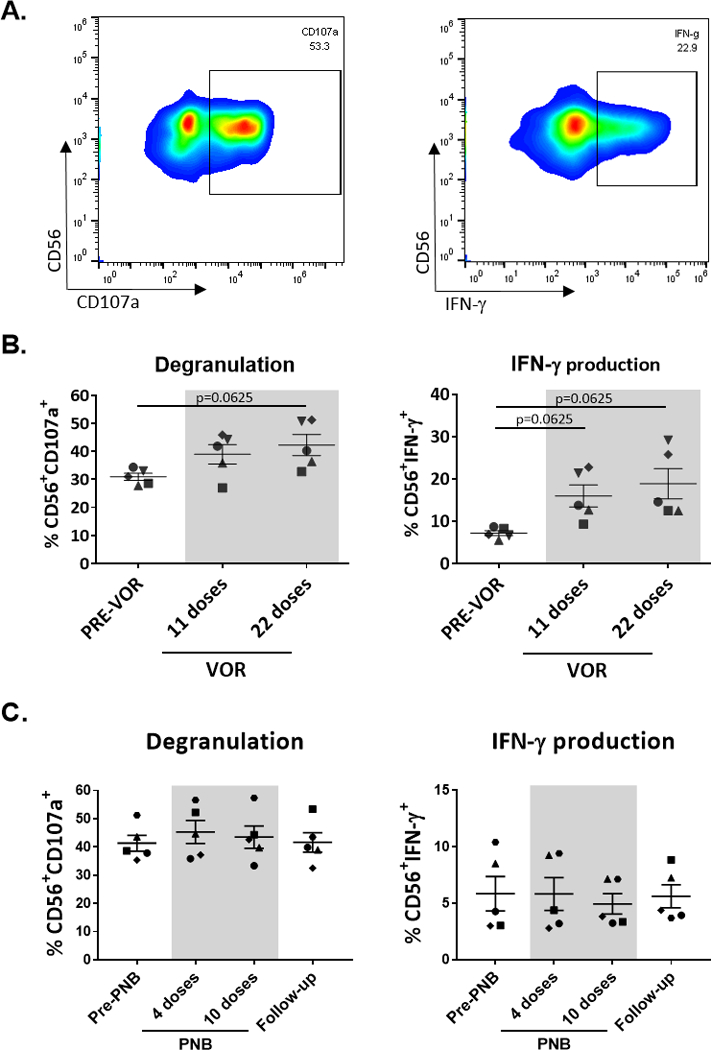
NK cells were negatively isolated from thawed PBMCs and cultured with the cell line K562 to measure degranulation through the marker CD107a and IFN-γ production by intracellular staining. A. Representative plots of cells from one participant. B. VOR treatment: An increase in both degranulation and IFN-γ production was observed after VOR exposure. C. PNB treatment: Cytotoxic and IFN-γ potential remained stable during PNB treatment. Each symbol represents cells from a different study participant. Wilcoxon matched-pairs signed rank test.
Frequency of latent HIV infection
Measurements of the frequency of latent HIV infection within each participant was determined at the different time points as described in detail by Archin et al. [24] for VOR, and Rasmussen et al.[11] for PNB. Briefly, measurement of the frequency of infection included quantification of HIV-1 pol DNA copies from resting or total CD4+T cells, analyzed by digital droplet PCR (ddPCR), and quantification of replication-competent virus using the quantitative viral outgrowth assays (QVOA), which provides a minimum estimation of the frequency of replication competent HIV reported as infectious units per million cells (IUPM).
Ethics statement
All donors provided written informed consent. Studies were approved by the University of North Carolina Institutional Review Board for the VOR samples and by the Danish Research Ethics Committee system for PNB samples. Specimens used in the study were anonymized.
Statistical analysis
Given the limited sample size, the statistical analysis was exploratory with no adjustment for multiple testing. Pre-treatment versus post-treatment measurements were compared with an exact two-sided Wilcoxon signed-rank test. For each study (VOR and PNB), measurements from 5 donors were available. Using a two-sided Wilcoxon signed-rank test, the smallest obtainable exact p-value with n=5 is p=0.0625. Analysis was performed with GraphPad Prism v7. The correlation between the different parameters and the size of the reservoir was assessed by pooling measurements from pre-treatment, mid-treatment, and post-treatment. A Spearman r was calculated, but we also performed a Kendall Tau correlation, since there were several measures from each participant.
RESULTS
NK cell frequency is increased after in vivo administration of HDACi
For VOR, phenotypic analysis on PBMCs was performed at three time points: before VOR administration (“pre-VOR”), after 11 and after 22 doses. Levels of CD4 and CD8 T cells remained constant over the treatment period. However, we observed an increase in the frequency of NK cells in all donors, who had an mean of 7.13% NK cells at baseline, 13.9% after 11 doses and 14.24% after 22 doses (Fig 1B).
For PNB, analysis was performed in PBMCs at four time points: pre-PNB, after 4 and 10 doses, and after completion of treatment (“follow-up”). As with VOR, there was no change over time in the proportion of CD4+ or CD8+ T cells, but consistent with VOR, we observed an increase in the frequency of NK cells during PNB treatment in all participants (3.6% pre-PNB, 5.6% at visit 6, 5.4% at visit 10 and back to 3.2% in the follow-up sample; Fig. 1C).
NK cells can be classified into bright and dim according to CD56 expression. Interestingly, we observed that CD56dim NK cells proportion increased in all participants (median of 93.72% before treatment and 96.1% after drug exposure), while CD56bright frequency decreased in most of them (median of 5.6% vs 3.47%).
Expression of the proliferation marker Ki67 and the early activation marker CD69 were also analyzed in total PBMCs and within the different cell subsets. Both VOR and PNB treatment resulted in a decrease of the expression of CD69, either analyzing the entire cell population (Fig. 1B, C) or the different cell subsets (data not shown). Conversely, Ki67 expression pattern differed after treatment with VOR or PNB. Ki67 expression significantly decreased after VOR treatment (Fig. 1B), while it remained unchanged after PNB treatment (Fig. 1C).
In vivo VOR treatment increases NK cell capacity to degranulate and produce IFN-γ
NK cell degranulation and IFN-γ production potential were evaluated after co-culture of isolated NK cells with the cell line K562. Degranulation was measured using surface CD107a expression, and IFN-γ production was assessed by intracellular staining. Due to limited sample availability, NK cell function could only be assessed in response to K562 cells, but we acknowledge that additional analysis of NK cell responses such as ADCC would provide further evidence of function alterations. Interestingly, VOR treatment improved both degranulation and IFN-γ production of NK cells after culture with K562 cells (Fig. 2B). Conversely, we did not find any function alteration after PNB treatment (Fig 2C). In addition, we found a positive correlation between the frequency of NK cells and both degranulation and IFN-γ production. Functional markers also correlated positively with the expression of NKp30, NKp46, NKG2D and CD16 (data not shown).
CD16 and NKp46 are upregulated after VOR treatment
NK cells express a wide variety of receptors that enable them to differentiate tumor or infected cells from healthy cells. These include inhibitory, activating, adhesion and cytokine receptors. The balance of these signals determines whether NK cells become activated or not [26]. Following NK cell isolation by negative selection, we analyzed the expression of a selection of activating receptors on the surface of NK cells. Expression of DNAM-1, NKp30 and NKG2D were not altered by VOR exposure, but interestingly NKp46 and CD16 expression were upregulated to some extent in most participants (Fig. 3A). Specifically, CD16 upregulation occurred exclusively within the CD56dim subset, while it decreased in the CD56bright population. In contrast, PNB treatment did not seem to impact in the expression of any receptor (Fig. 3B).
Fig. 3. Expression of activating receptors on NK cells.

NK cells were negatively isolated from thawed PBMC and stained with a panel of monoclonal antibodies to analyze the expression of activating receptors. A. VOR treatment: NKp30, DNAM-1 and NKG2D expression did not change during treatment, but expression of CD16 and NKp46 was increased to some extent after VOR administration. B. PNB treatment: No changes were observed during PNB treatment. Each symbol represents cells from a different study participant. Wilcoxon matched-pairs signed rank test.
Correlation with the HIV reservoir size
We analyzed whether the different parameters measured in the present study were correlated with the size of the reservoir, measured either as total DNA in CD4 cells or as IUPMs. Interestingly, after combining all timepoints from both VOR and PNB studies, some correlations were found. There was a negative correlation between the frequency of NK cells and the copies of DNA/million CD4 cells, [(Spearman r=−0.51, p=0.0017; Kendall’s Tau: −0.33 (95% CI:−0.57, −0.08)]. Similarly, there was a negative correlation between CD16 expression in NK cells and HIV-DNA [Spearman r=−0.43, p=0.01; Kendall’s Tau: −3.8 (95% CI:−0.72, 0.17), as well as a trend (p=0.09) towards a negative correlation between NK cell IFN-γ production and HIV-DNA content. Unexpectedly, a positive correlation was observed between NKG2D expression and HIV-DNA (Spearman r=0.35, p=0.041; Kendall’s Tau: 0.26 (95% CI:−0.08, 0.52) (Fig. 4). No significant correlation was found between HIV reservoir size measured as IUPM and any of the analyzed parameters (data not shown).
Fig. 4. Correlation with the HIV reservoir size.

Size of the HIV reservoir was measured as total DNA per million CD4 cells or as IUPMs after quantitative outgrowth assay. HIV DNA correlated negatively with NK cell frequency, CD16 expression and IFN-γ production, while it correlated positively with NKG2D expression. Circles correspond to samples from the VOR study and squares to the PNB study. Green represent samples before HDACi treatment, purple during treatment and red after treatment.
DISCUSSION
One of the current strategies aimed at eradicating persistent HIV infection consists of combining latency reversing agents with approaches to induce robust immune responses capable of identifying and clearing reactivated cells. Therefore, it is critical to discern if interventions used for latency reversal have any impact on the immune function needed for viral clearance. In the present study, we have evaluated the effect of in vivo administration of HDAC inhibitors on NK cell function. We have used samples from a clinical trial involving VOR (NCT01319383), in which 5 HIV-1 infected participants were administered 22 cyclical doses of 400 mg VOR over a period of 12 weeks[24], and a clinical trial involving PNB (NCT01680094)[11], in which 15 participants received 20 mg PNB three times a week every other week for 8 weeks. We analyzed the phenotype and the functionality of the immune cells in samples obtained before, during and after clinical exposure to the drugs, focusing on NK cells.
The main finding of the study is that NK cell immune function was not impaired in samples from participants treated with either of the two HDAC inhibitors. In addition, a moderate but consistent increase in the frequency of NK cells during HDACi treatment was observed, contrasting with stable frequencies of CD4+ and CD8+T cells. Unfortunately, whether the observed increase in NK cells in peripheral blood is due to proliferation or to cellular redistribution, cannot be concluded from this study. Interestingly, we observed an enhancement in the capacity of NK cells to degranulate and produce IFN-γ following in vivo administration of VOR. This improvement was accompanied by an increase in the expression of the activating receptors CD16 and NKp46, which in addition were positively correlated with degranulation and/or production of IFN-γ. PNB treatment did not cause any functional impairment, but no improvement was observed either. Finally, pooling together the data from both VOR and PNB studies, we observed an interesting negative correlation between the copies of HIV DNA/million CD4 and the frequency of NK cells, as well as with NK cell expression of CD16.
The question of whether HDACi suppress immune function is still under debate. Concern initially raised when Jones et al. reported that HDACis impaired CTL-mediated IFN-γ production and the elimination of HIV-infected cells in vitro [19]. However, later studies only partially confirmed these observations [15, 20], highlighting the importance of factors such as specific HDACi classes, drug concentrations and the length of drug exposure, which could have acted as confounding players in study outcomes.
This is the first evaluation of NK cell function performed after in vivo administration of VOR or PNB. Previous studies have evaluated the impact of VOR exposure on NK cell function in vitro or ex vivo. We performed a comprehensive analysis of the effect of ex vivo exposure of different HIV latency reversing agents on NK cell phenotype and function. In regards to VOR, we did not observe any major impact on NK cells [23], in agreement with the present in vivo results. However, other studies evaluating in vitro impact of VOR exposure on NK cell parameters reported contradictory results. Pace et al. [27] reported similar results as our group, detecting no impairment of NK cell function after exposure to VOR, while Ogbomo et al. [28] and Pfeiffer et al. [29] observed a decrease in NK cytotoxicity. One potential explanation for these discrepancies are the different concentration and time of exposure used. In our experiments, as well as in Pace’s, a concentration of 335 nM was used over 24 hours, to model peak VOR exposures in vivo, as VOR is cleared in less than 6 hours [30]. On the contrary, Ogbomo et al. used a continuous VOR concentration of 0.5–1 µM over 96 hours, and Pfeiffer et al. 1 µM over 48 hours. Regarding PNB, in our ex vivo study, we observed that a PNB exposure consisting of 20 ng/mL during 24 hours -which aimed to mimic physiologic conditions- increased NK cell death and reduced immune function [23], while in the present in vivo study we did not observe such impairment. Therefore, while in vitro studies are undoubtedly of interest, it is obvious that in vivo administration of the drug entails more complicated pharmacodynamics and cellular interactions that might provide different outcomes.
Studies of NK cell immune function after in vivo exposure to HDACi are limited. In the clinical trial using Panobinostat in HIV-1 infected individuals, Olesen et al.[31] found a correlation between HIV reservoir decline during Panobinostat treatment and NK cell frequency, reporting that the proportions of total and CD56dimCD16+ NK cells were inversely associated with HIV-1 DNA levels [31]. In agreement to these results, when we pooled the data obtained in the VOR and PNB study for sample size purposes, we observed a negative correlation between HIV DNA content and the proportion of NK cells (Fig. 4). However, we failed to detect such a correlation with the size of the replication competent reservoir (estimated as IUPMs by viral outgrowth assay), but this can be due to a reduced sample size, since IUPM data was available only in a subset of samples. In the present study, in addition to the correlation between HIV DNA and NK cell frequency, we observed an interesting negative correlation between HIV-DNA and CD16 expression, as well as a trend towards reduced HIV-DNA content and NK cell IFN-γ production. Interestingly, this observation has been reported previously [32], and thus the hypothesis that enhanced NK cell phenotype and function might have an impact in the HIV reservoir size deserves further investigation.
In summary, in vivo administration of HDAC inhibitors do not negatively impact NK cell function. Furthermore, we observed an increase in NK cell frequency during HDACi treatment, and interestingly VOR exposure improved cytotoxicity potential and capacity of NK cells to produce IFN-γ, as well as the expression of the activating receptor NKp46. Finally, there was a negative correlation between the size of the HIV reservoir measured as HIV DNA in CD4 T cells and the frequency of NK cells and expression of CD16. These results have important implications in light of HIV eradication strategies combining latency reversal with immune enhancement interventions.
ACKNOWLEDGEMENTS
We thank K. Sholtis, E. Stuelke and J. Kirchherr for technical assistance; J. Mathura and K. Mollan for statistical support; and J. Kuruc, C. Gay and C. Baker for clinical management. This work was supported by NIH U01-AI095052 and U19-AI096113 to DMM, and work in the UNC Flow Cytometry Core Facility is supported in part by the Center for AIDS Research award number P30-AI050410. We specially thank the participants in these clinical trials. Author contributions: CG conceived and designed the study; DMM and NSS provided input in study design and experiment planning; CG and BA performed the experiments; DMM, NMA, MT, TAR and OSS contributed reagents/materials; CG wrote the manuscript and NSS, DMM, NMA, MT and OSS reviewed it.
Footnotes
CONFLICT OF INTEREST
None declared.
REFERENCES
- 1.Archin NM, Sung JM, Garrido C, Soriano-Sarabia N, Margolis DM. Eradicating HIV-1 infection: seeking to clear a persistent pathogen. Nature reviews Microbiology 2014; 12(11):750–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van Lint C, Emiliani S, Ott M, Verdin E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. The EMBO journal 1996; 15(5):1112–1120. [PMC free article] [PubMed] [Google Scholar]
- 3.Coull JJ, Romerio F, Sun JM, Volker JL, Galvin KM, Davie JR, et al. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. Journal of virology 2000; 74(15):6790–6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. The EMBO journal 2006; 25(1):139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keedy KS, Archin NM, Gates AT, Espeseth A, Hazuda DJ, Margolis DM. A limited group of class I histone deacetylases acts to repress human immunodeficiency virus type 1 expression. Journal of virology 2009; 83(10):4749–4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Contreras X, Schweneker M, Chen CS, McCune JM, Deeks SG, Martin J, et al. Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. The Journal of biological chemistry 2009; 284(11):6782–6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012; 487(7408):482–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Archin NM, Espeseth A, Parker D, Cheema M, Hazuda D, Margolis DM. Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS research and human retroviruses 2009; 25(2):207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Archin NM, Kirchherr JL, Sung JA, Clutton G, Sholtis K, Xu Y, et al. Interval dosing with the HDAC inhibitor vorinostat effectively reverses HIV latency. The Journal of clinical investigation 2017; 127(8):3126–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elliott JH, Wightman F, Solomon A, Ghneim K, Ahlers J, Cameron MJ, et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS pathogens 2014; 10(10):e1004473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rasmussen TA, Tolstrup M, Brinkmann CR, Olesen R, Erikstrup C, Solomon A, et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. The lancet HIV 2014; 1(1):e13–21. [DOI] [PubMed] [Google Scholar]
- 12.Sogaard OS, Graversen ME, Leth S, Olesen R, Brinkmann CR, Nissen SK, et al. The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS pathogens 2015; 11(9):e1005142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tapia G, Hojen JF, Okvist M, Olesen R, Leth S, Nissen SK, et al. Sequential Vacc-4x and romidepsin during combination antiretroviral therapy (cART): Immune responses to Vacc-4x regions on p24 and changes in HIV reservoirs. The Journal of infection 2017; 75(6):555–571. [DOI] [PubMed] [Google Scholar]
- 14.Sung JA, Lam S, Garrido C, Archin N, Rooney CM, Bollard CM, et al. Expanded cytotoxic T-cell lymphocytes target the latent HIV reservoir. The Journal of infectious diseases 2015; 212(2):258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sung JA, Pickeral J, Liu L, Stanfield-Oakley SA, Lam CY, Garrido C, et al. Dual-Affinity Re-Targeting proteins direct T cell-mediated cytolysis of latently HIV-infected cells. The Journal of clinical investigation 2015; 125(11):4077–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patel S, Chorvinsky E, Albihani S, Cruz CR, Jones RB, Shpall EJ, et al. HIV-Specific T Cells Generated from Naive T Cells Suppress HIV In Vitro and Recognize Wide Epitope Breadths. Molecular therapy : the journal of the American Society of Gene Therapy 2018. [DOI] [PMC free article] [PubMed]
- 17.Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC, et al. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 2012; 36(3):491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lam S, Sung J, Cruz C, Castillo-Caro P, Ngo M, Garrido C, et al. Broadly-specific cytotoxic T cells targeting multiple HIV antigens are expanded from HIV+ patients: implications for immunotherapy. Molecular therapy : the journal of the American Society of Gene Therapy 2015; 23(2):387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones RB, O’Connor R, Mueller S, Foley M, Szeto GL, Karel D, et al. Histone deacetylase inhibitors impair the elimination of HIV-infected cells by cytotoxic T-lymphocytes. PLoS pathogens 2014; 10(8):e1004287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clutton G, Xu Y, Baldoni PL, Mollan KR, Kirchherr J, Newhard W, et al. The differential short- and long-term effects of HIV-1 latency-reversing agents on T cell function. Scientific reports 2016; 6:30749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walker-Sperling VE, Pohlmeyer CW, Tarwater PM, Blankson JN. The Effect of Latency Reversal Agents on Primary CD8+ T Cells: Implications for Shock and Kill Strategies for Human Immunodeficiency Virus Eradication. EBioMedicine 2016; 8:217–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garrido C, Abad-Fernandez M, Tuyishime M, Pollara JJ, Ferrari G, Soriano-Sarabia N, et al. Interleukin-15-Stimulated Natural Killer Cells Clear HIV-1-Infected Cells following Latency Reversal Ex Vivo. Journal of virology 2018; 92(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garrido C, Spivak AM, Soriano-Sarabia N, Checkley MA, Barker E, Karn J, et al. HIV Latency-Reversing Agents Have Diverse Effects on Natural Killer Cell Function. Frontiers in Immunology 2016; 7(356). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Archin NM, Bateson R, Tripathy MK, Crooks AM, Yang KH, Dahl NP, et al. HIV-1 expression within resting CD4+ T cells after multiple doses of vorinostat. The Journal of infectious diseases 2014; 210(5):728–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. Journal of immunological methods 2004; 294(1–2):15–22. [DOI] [PubMed] [Google Scholar]
- 26.Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nature reviews Immunology 2012; 12(4):239–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pace M, Williams J, Kurioka A, Gerry AB, Jakobsen B, Klenerman P, et al. Histone Deacetylase Inhibitors Enhance CD4 T Cell Susceptibility to NK Cell Killing but Reduce NK Cell Function. PLoS pathogens 2016; 12(8):e1005782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogbomo H, Michaelis M, Kreuter J, Doerr HW, Cinatl J Jr. Histone deacetylase inhibitors suppress natural killer cell cytolytic activity. FEBS Lett 2007; 581(7):1317–1322. [DOI] [PubMed] [Google Scholar]
- 29.Pfeiffer MM, Burow H, Schleicher S, Handgretinger R, Lang P. Influence of Histone Deacetylase Inhibitors and DNA-Methyltransferase Inhibitors on the NK Cell-Mediated Lysis of Pediatric B-Lineage Leukemia. Frontiers in oncology 2013; 3:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelly WK, O’Connor OA, Krug LM, Chiao JH, Heaney M, Curley T, et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2005; 23(17):3923–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olesen R, Vigano S, Rasmussen TA, Sogaard OS, Ouyang Z, Buzon M, et al. Innate Immune Activity Correlates with CD4 T Cell-Associated HIV-1 DNA Decline during Latency-Reversing Treatment with Panobinostat. Journal of virology 2015; 89(20):10176–10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marras F, Casabianca A, Bozzano F, Ascierto ML, Orlandi C, Di Biagio A, et al. Control of the HIV-1 DNA Reservoir Is Associated In Vivo and In Vitro with NKp46/NKp30 (CD335 CD337) Inducibility and Interferon Gamma Production by Transcriptionally Unique NK Cells. Journal of virology 2017; 91(23). [DOI] [PMC free article] [PubMed] [Google Scholar]