

Abstract
Osteogenesis imperfecta (OI) is a heritable connective tissue disorder primarily due to mutations in the type I collagen genes (COL1A1 and COL1A2), leading to compromised biomechanical integrity in type I collagen containing tissues such as bone. Bone is inherently mechanosensitive and thus responds and adapts to external stimuli, such as muscle mass and contractile strength, to alter its mass and shape. Myostatin, a member of the TGF-β superfamily, signals through activin receptor type IIB to negatively regulate muscle fiber growth. Due to the positive impact of myostatin deficiency on bone mass, we utilized a soluble activin receptor type IIB-mFc (sActRIIB-mFc) fusion protein in two molecularly distinct OI mouse models (G610C and oim) and evaluated their bone properties. Wildtype (WT), +/G610C, and oim/oim mice were treated from 2 to 4 months of age with either vehicle (Tris-Buffered Saline) or sActRIIB-mFc (10mg/kg). Femurs of sActRIIB-mFc treated mice exhibited increased trabecular bone volume regardless of genotype, while the cortical bone microarchitecture and biomechanical strength were only improved in WT and +/G610C mice. Dynamic histomorphometric analyses suggest the improved cortical bone geometry and biomechanical integrity reflect an anabolic effect due to increased mineral apposition and bone formation rates. Whereas, static histomorphometric analyses supported sActRIIB-mFc treatment also having an anti-catabolic impact with decreased osteoclast number per bone surface on trabecular bone regardless of sex and genotype. Together, our data suggest that sActRIIB-mFc may provide a new therapeutic direction to improve both bone and muscle properties in OI.
Keywords: Osteogenesis Imperfecta, Analysis/Quantification of Bone, Myostatin, Decoy Activin Receptor, Cell/Tissue Signaling
Introduction
Osteogenesis Imperfecta (OI) is a heritable connective tissue disorder with clinical manifestations of retarded growth and skeletal fragility, and often hearing loss, blue/grey sclera, dentinogenesis imperfecta and muscle weaknesses(1). The majority of individuals with OI possess mutations in their type I collagen genes, COL1A1 and COL1A2, which lead to compromised biomechanical integrity in type I collagen containing tissues, such as bone, skin, and blood vessels. The remaining individuals with OI have primarily recessive mutations in genes coding for proteins involved in posttranslational modification and folding of type I collagen, bone matrix mineralization and osteoblast function(1).
There is no cure for OI. The difficulties in developing effective OI treatments are associated with the heterogeneity of the disease causing OI mutations, which comprise over 1,500 dominant mutations in type I collagen genes as well as the additional recessive mutations (https://oi.gene.le.ac.uk, accessed November 27 2017)(2). Current treatment strategies are limited primarily to use of anti-resorptive drugs, bisphosphonates, and/or surgical intervention. Bisphosphonates have been widely used in OI patients with some success through increasing bone volume, and thus providing higher resistance to fracture(3). Despite the gain in bone mass, the bone matrix quality in OI is still poor due to disorganized collagen fibril assembly and abnormal matrix mineralization. The half-life of bisphosphonate is a decade long and therefore remains in the bone matrix for an extended time to inhibit the bone remodeling process, which allows accumulation of micro-damage in the OI bone matrix compromising bone quality even further(1,4). Surgical rodding to correct bone deformity or multiple fractures can present with several associated complications during and after surgery, including excess bleeding, delayed healing, rod migration, and device failure(1).
Myostatin, a member of the TGF-β superfamily, is a negative regulator of muscle growth, whose main mode of action is signaling through activin receptor type IIB (ActRIIB) to recruit nearby type I receptors, such as ALK4 and ALK5. This results in activation of the Smad-dependent pathway to regulate transcription of various downstream genes. Myostatin deficiency in mice leads to muscle hypertrophy and hyperplasia(5). Bone is mechanosensitive, which enables it to respond and adapt to external stimuli by altering its shape and mass. Muscle mass and contractile forces are some of the largest physiological loads bones may experience(6). In support of this, myostatin knock-out mice exhibit increased bone mass and biomechanical strength as compared to wildtype (WT) mice(7,8). Due to the potential benefit of myostatin inhibition on bone mass, postnatal myostatin inhibition has been tested in various mouse models with compromised musculoskeletal function, such as sex hormone deficiency(9), obesity(10), muscular dystrophy(11), and osteogenesis imperfecta(12). The positive influence of myostatin deficiency on bone mass has been recapitulated in our previous work with the oim mouse model of OI as well. We introduced congenic myostatin deficiency into heterozygous oim (+/oim) mice by breeding heterozygous myostatin (+/mstn) mice with +/oim mice to produce offspring that were double heterozygote mice (+/mstn+/oim) and demonstrated increased skeletal muscle mass with increased bone volume and strength in the +/mstn+/oim mice relative to +/oim mice(13).
As a preclinical strategy for postnatal myostatin inhibition in OI mouse models, we report herein the utilization of the soluble activin receptor type IIB-mFc (sActRIIB-mFc) fusion protein, which functions as a decoy receptor for myostatin and other ligands (GDF11, activins, and some BMPs) with high affinity for the ActRIIB(14). Treatment with sActRIIB-mFc prevents these ligands from binding to their endogenous cellular receptors, which inhibits activation of downstream signaling pathways. The primary aim of this study was to evaluate the effects of postnatal myostatin inhibition on the skeletal properties of two molecularly distinct OI mouse models, G610C and oim. Heterozygous G610C (+/G610C) mice have an autosomal dominant glycine to cysteine substitution at position 610 of proα2(I) collagen chain and model both the genotype and phenotype of a cohort of 64 individuals with OI type I/IV from an Old Order Amish kindred(15). Whereas, homozygous oim (oim/oim) mice have a functional null Col1α2 mutation resulting in abnormal proα2(I) collagen chains, which are unable to incorporate into normal heterotrimeric type I collagen [α1(I)2α2(I)](16). Instead, the oim/oim mice synthesize homotrimeric type I collagen [α1(I)3](16). We administered sActRIIB-mFc or vehicle (Tris-buffered saline) alone to WT, +/G610C, and oim/oim mice and evaluated its effect on the hindlimb skeletal microarchitecture and biomechanical integrity, as well as mechanistically at the cellular and molecular level.
Materials and Methods
Animals
Heterozygote G610C (+/G610C; Col1a2tm1.1Mcbr) mice on the C57BL/6J congenic background were a gift from Dr. Daniel McBride(15). Col1a2tm1.1Mcbr mice (stock #007248) and Col1a2oim (stock #001815) are publicly available from Jackson Laboratory (Bar Harbor, ME, USA). Both the +/G610C and +/oim mice were maintained on the C57BL/6J background and genotyped as previously described (15,17,18). Mice were housed in an AAALAC-accredited facility at the University of Missouri, and all experimental manipulations were performed under an approved University of Missouri Animal Care and Use protocol.
Drug Administration
The sActRIIB-mFc (RAP-031) was a kind gift from Acceleron Pharma Inc. (Cambridge, MA). Male and female WT, +/G610C, and oim/oim mice were each randomly distributed into two groups and treated bi-weekly with either vehicle [tris-buffered saline (TBS)] as control or sActRIIB-mFc in TBS (10mg/kg of bodyweight) by intraperitoneal injection beginning at 2 months of age as previously described (animal numbers were 10–12 per group and this was based on calculations of sample size using proc glmpower in SAS 9.1 (SAS Institute Inc., Cary, NC and Sigma Stat with a desired power of 0.80 and an alpha of 0.05) for bone microarchitectural [trabecular bone volume/total volume (BV/TV), number (Tb.N) and thickness (Tb.Th)] and biomechanical (torsional ultimate strength and energy to failure) parameters, and mineral apposition rate (MAR), bone formation rate/mineralizing surface (BFR/MS), and osteoblast and osteoclast numbers (19). At 4 months of age, mice were euthanized, and their serum, tibiae, and femurs harvested for analyses.
Femoral Microarchitecture
Trabecular microarchitecture was quantified from left femurs by vivaCT 40 (Scanco Medical AG, Bassersdorf, Switzerland) in accordance with recommended guidelines(20) using an X-ray energy of 55kV (145µA), a voxel resolution of 10.5µm, 200ms integration time with the number of projections set at 1000/180 degrees and using a 0.5mm aluminum low pass filter prior to dynamic and static histomorphometry analyses. The threshold was set to 316 (equivalent to 498.5mgHA/ccm) for trabecular bone to distinguish mineralized from non-mineralized tissue. 100 slices (1050µm) were contoured starting at the metaphyseal end of the distal growth plate, below the primary spongiosa, and progressing toward the midshaft. Trabecular bone properties were analyzed using Scanco bone evaluation software. Cortical bone microstructure from right femurs were evaluated by Siemens Inveon μCT equipped with Siemens Inveon Acquisition Workplace Software Version 1.5 (Siemens Preclinical Solutions, Knoxville, TN, USA) with an X-ray peak of 80kVp and an exposure time of 140ms prior to ex-vivo torsional loading to failure analyses. μCT image slices were reconstructed and analyzed using the Image J software (1.50e) with BoneJ plugin (NIH)(21) to give a cubic voxel dimension of 21μm3.
Torsional Loading to Failure
Following µCT analyses, right femurs were then potted into individualized cylindrical holders and evaluated by torsional loading to failure using the TA-HDi testing machine (Stable Micro Systems, Surrey, UK) as previously described(17). Applied torque T (Nmm) was calculated and plotted as a function of relative angular displacement θ (degrees). The whole-bone parameters of strength (torsional ultimate strength [Nmm], torsional stiffness [Nmm/rad] and strain energy to failure [Nmm]), which take into account bone geometric and material properties, were determined(17). The bone material properties, tensile strength (N/mm2), and stiffness (shear modulus of elasticity [N/mm2]) were also determined according to Roarck(22).
Serum Biochemical Markers
Blood collected by cardiac puncture at the time of sacrifice was centrifuged (14,000rpm, 15 minutes) and the collected serum stored at −80⁰C until analyses. Serum levels of the N-terminal propeptide of type I procollagen (PINP), a biomarker of collagen synthesis, and C-terminal cross-linked telopeptide of type I collagen (CTX), biomarker of collagen degradation, were quantified using the Rat/Mouse PINP EIA kit (AC-33F1) and the RatLaps™ EIA kit (AC-061F1) by Immunodiagnostics Systems (Scottsdale, Arizona), respectively. Samples and standards were measured in duplicate following the manufacturer’s instructions.
Dynamic and Static Histomorphometry
To evaluate the change in mineral apposition rate, a calcein label (5mg/kg i.p.; MP Biomedical, Santa Ana, CA) and an alizarin red label (20mg/kg i.p.; Acros Organics, Morris Plains, NJ) were administered 10 and 3 days prior to sacrifice, respectively. After sacrifice, left femurs were removed, fixed in 10% neutral buffered formalin (NBF) for 48 hours, transferred to 70% ethanol (4⁰C), subsequently dehydrated in graded ethanols (70–100%), and embedded in methyl methacrylate (MMA) without prior decalcification as described previously(23). 100μm sections at the femoral midshaft were cut using a Leica 1600 saw microtome. Unstained sections were viewed under epifluorescent illumination using a Leica SP8 spectral confocal microscope and a series of cross-sectional images of the femurs (20X magnification) obtained. Fluorescent labeling and the distance between the two fluorescent labels were quantified using image J software according to Egan et al.(24). Static histomorphometric analysis of trabecular bone was performed on left distal femurs. Undecalcified 5µm thin longitudinal serial sections were cut with a Thermo Scientific HM355S microtome. Three non-adjacent sections per animal were deplastified and stained with Von Kossa-Tetrachrome and tartrate resistant acid phosphatase (TRAP) to characterize osteoblasts and osteoclasts, respectively. A series of images were taken using a bright field microscope (Zeiss Axiovert 200M, Zeiss, Oberkochen, Germany) with 20X magnification, then compiled into a single image using MetaMorph (Molecular Devices, Sunnyvale, CA) software. Histomorphometric measurements of Von Kossa-Tetrachrome and TRAP stained sections were obtained using image J software (1.50e) according to Egan et al(24). Dynamic and static histomorphometric parameters were measured directly or derived from measured indices following the 2012 recommendations of the American Society for Bone and Mineral Research(25).
RNA isolation and analysis of gene expression by quantitative PCR
Snap frozen intact right tibiae (with marrow) were homogenized in TRIzol Reagent (Invitrogen, Carlsbad, CA) and total RNA was isolated using the RNeasy Kit (Qiagen, Germantown, MD) with in-column DNase treatment (Qiagen, Germantown, MD). Total RNA (2µg) was used to generate a cDNA library with a High Capacity Reverse Kit (Applied Biosystems, Foster City, CA). Gene expression levels for markers of differentiation and function of osteoblasts (Runx2, Sp7, Dlx5, Alpl, Bglap, and Serpinh1), osteoclasts (Tnfsf11, Csf1, Itgb3, and Ctsk), and osteocytes (Dmp1, Sost, Phex, and β-cat) were normalized to 18S, Pgk1, and Ubc as endogenous controls, using the ΔΔCT method (Quantstudio 3 Real Time PCR System, Applied Biosystems, Foster City, CA) (Supplemental Table S1). Results are plotted in bar graphs and the range of expression indicated by 2-(ΔΔCT+SD) and 2-(ΔΔCT-SD) (26).
Statistical Analyses
Statistical analyses were performed using SAS (SAS Institute Inc., Cary, NC). +/G610C and oim/oim data were analyzed independently as 2X2X2 factorials [2 genotypes (WT and +/G610C or WT and oim/oim), 2 sexes, and 2 treatments (vehicle and sActRIIB-mFc treated)] using Fisher’s Protected Least Significant Difference (27,28). If heterogeneous variations were present, a log transformation was used to stabilize the variation. If the log transformation failed to stabilize the variation, a nonparametric ranked analysis was performed according to Conover et al(29). Cohen’s effect size was calculated to evaluate the practical significance of sActRIIB-mFc treatment with d≥0.8 indicating high significance (Supplemental Tables S2-S6)(30). Bivariate Pearson correlation analysis was performed using SPSS (IBM Corp., Armonk, NY) to evaluate the relationships between skeletal muscle properties, biomechanical properties, and osteoblast/osteoclast function. Differences were considered to significant at p≤0.05.
Results
Skeletal maturity in C57BL/6J mice is reached at 4 months of age (the age of peak bone mineral density)(31) and we chose the 9–16 week treatment duration to encompass the pubertal growth period. Chiu et al. demonstrated no further increase in muscle mass with 30mg/kg of sActRIIB-mFc when compared to 10mg/kg dose(32). Thus, we chose the 10mg/kg dose and showed significant increases in body weight and skeletal muscle mass regardless of genotypes and sex without showing any adverse side-effects(19). Four of 94 mice (one +/G610C and three oim/oim) died during course of the study (Supplemental Table S7). Three of 35 +/G610C and 34 of 55 oim/oim mice had at least one fracture/callus of their hindlimb long bones (femurs and tibias) at the time of harvest (Supplemental Table S7) and the occurrence of fractures/calluses appeared independent of treatment.
Femoral Microarchitecture
Consistent with previously published works(17,18), femoral trabecular and cortical bone microarchitecture of 4 months old +/G610C and oim/oim mice were compromised relative to WT littermates (Figure 1). Importantly, distal femoral trabeculae of sActRIIB-mFc treated mice had at least 2-fold greater BV/TV regardless of genotype, with sActRIIB-mFc treated oim/oim femurs reaching 3 to 4-fold greater trabecular BV/TV values as compared to their vehicle treated counterparts (Figure 1A). Increased BV/TV with sActRIIB-mFc treatment was attributed to increased trabecular number (Tb.N) and decreased trabecular spacing (Tb.Sp), as trabecular thickness remained unchanged (Figures 1B-D). Similarly, femoral cortical bone microarchitecture of sActRIIB-mFc treated WT and +/G610C mice had increased femur length, cortical bone cross-sectional area (CSA) and polar moment of area as compared to their vehicle treated counterparts except for female +/G610C mice (Figures 1E-G). Oim/oim femurs did not show any changes in cortical bone geometry with sActRIIB-mFc treatment except for increased cortical bone CSA in female oim/oim mice (Figure 1F).
Figure 1.
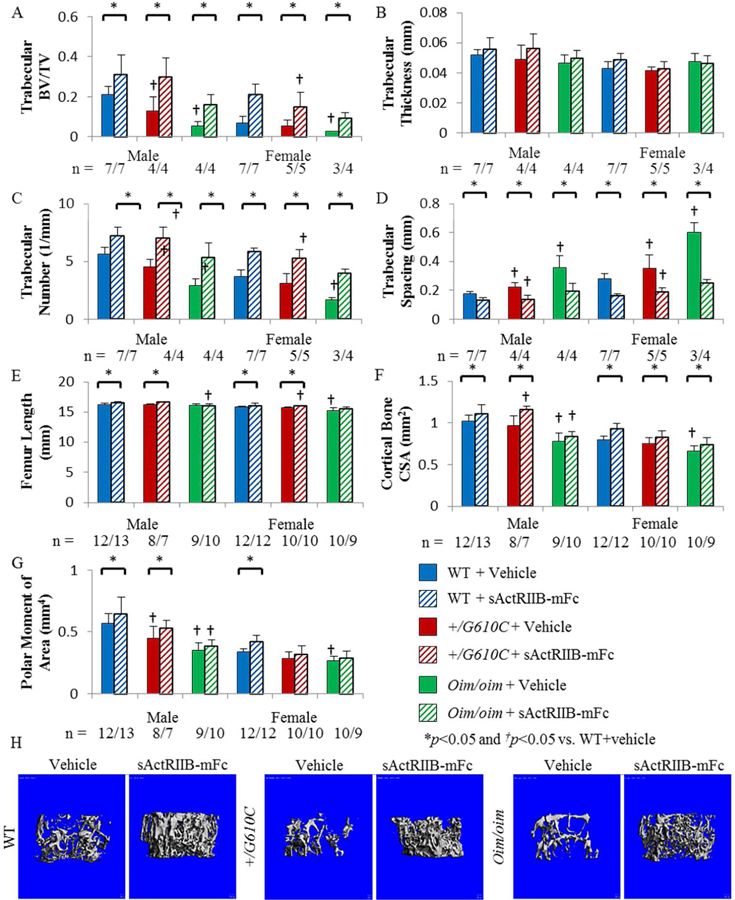
Femurs of sActRIIB-mFc treated mice exhibited improved trabecular bone microarchitecture regardless of sex and genotype. A) Trabecular bone volume/total volume (BV/TV), B) Trabecular thickness (Tb.Th), C) Trabecular number (Tb.N), D) Trabecular spacing (Tb.Sp), E) Femur Length, F) Cortical bone cross-sectional area (CSA), and G) Polar moment of area (K) of male and female WT, +/G610C, and oim/oim vehicle (solid color) and sActRIIB-mFc (diagonal) treated mice. (WT [blue], +/G610C [red], and oim/oim [green]). H) Representative µCT images of female femoral trabecular bone. Values are MEAN±SD. (n) is indicated for each genotype and treatment. *p<0.05; †p<0.05 vs. WT+vehicle treated mice.
Torsional Loading to Failure Biomechanical Testing
After μCT analyses, femoral torsional loading to failure testing demonstrated that +/G610C and oim/oim femurs had decreased torsional ultimate strength, tensile strength, and energy to failure as compared to their WT counterparts (Figure 2A, C, and E). The impact of sActRIIB-mFc treatment was evident in male WT and +/G610C femurs with increased torsional ultimate strength and energy to failure as compared to their vehicle treated counterparts (Figures 2A and E). Femurs of sActRIIB-mFc treated female WT mice had increased torsional ultimate strength as compared to their vehicle treated counterpart (Figure 2A). Oim/oim femurs did not exhibit changes in biomechanical properties with the sActRIIB-mFc treatment (Figure 2).
Figure 2.
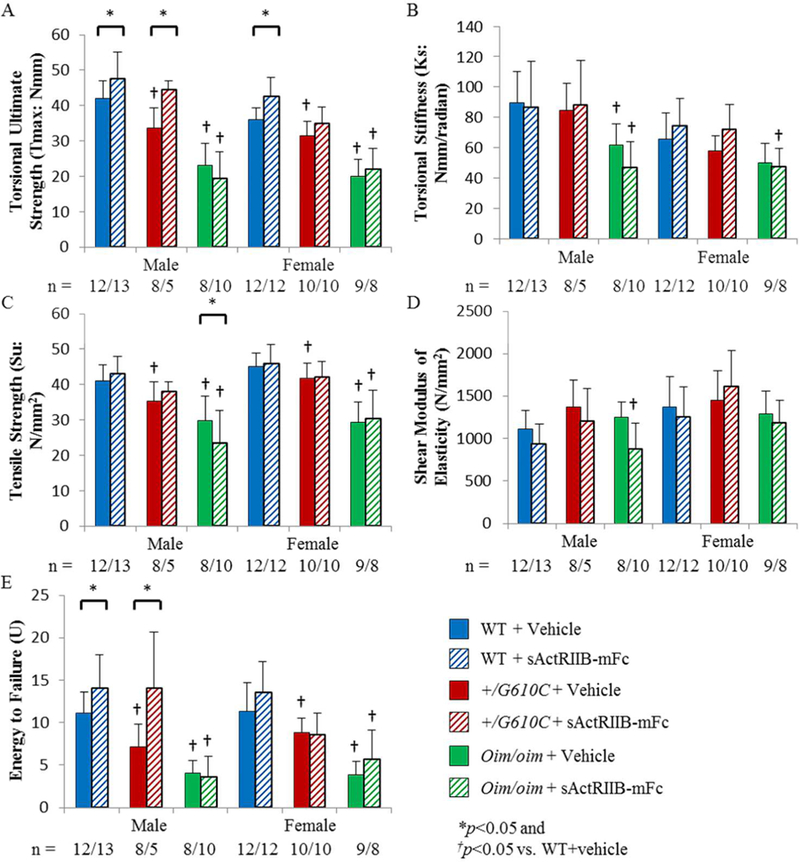
Femurs of sActRIIB-mFc treated male and female WT and male +/G610C mice demonstrated increased whole bone biomechanical function. A) Torsional ultimate strength (Tmax), B) Torsional stiffness (Ks), C) Tensile strength (Su), D) Shear modulus of elasticity (G), and E) Energy to failure (U) of male and female WT, +/G610C, and oim/oim vehicle (solid color) and sActRIIB-mFc (diagonal) treated mice. (WT [blue], +/G610C [red], and oim/oim [green]). (n) is indicated for each genotype and treatment. Male +/G610C + sActRIIB-mFc had n=5, as 3 samples could not be used due to malfunction of the biomechanical testing device. Values are MEAN±SD. *p<0.05; †p<0.05 vs. WT+vehicle treated mice.
Dynamic Histomorphometry
To evaluate the effects of sActRIIB-mFc on bone remodeling, we analyzed fluorochrome labeling at the mid-diaphysis of the left femur. Male +/G610C femurs displayed reduced endocortical bone formation as compared to their WT counterparts, while these differences were absent in female mice (Figures 3E and G). Oim/oim femurs had increased periosteal surface mineralization and bone formation relative to their WT counterparts regardless of sex (Figures 3B and F). On the endocortical surface, oim/oim femurs displayed increased mineralizing surface and decreased mineral apposition rate relative to their WT counterparts, although male oim/oim mice did not reach significance (Ec.MS: +32.4% [p=0.0613] & Ec.MAR: −21.1% [p=0.0783]) (Figures 3C and E). sActRIIB-mFc treatment effect was evident on the periosteal bone surface of WT and +/G610C mice regardless of sex. sActRIIB-mFc treated female WT mouse femurs had increased periosteal bone formation as compared to vehicle treated counterparts, while male WT mouse femurs also had non-significant increases in same parameters (Figures 3B, D and F). sActRIIB-mFc treated +/G610C mouse femurs had increases in periosteal bone formation relative to their vehicle treated counterparts regardless of sex, and male +/G610C femurs had additional increases in mineralizing surface and mineral apposition rate (Figure 3B, D, and F). The sActRIIB-mFc treatment effect on the endocortical bone surface was only present in female WT and +/G610C mouse femurs with increased endocortical surface mineralization and bone formation rate (Ec.BFR), although the +/G610C femurs did not reach significance in their Ec.BFR (+35.9%; p=0.134) (Figures 3C and G).
Figure 3.
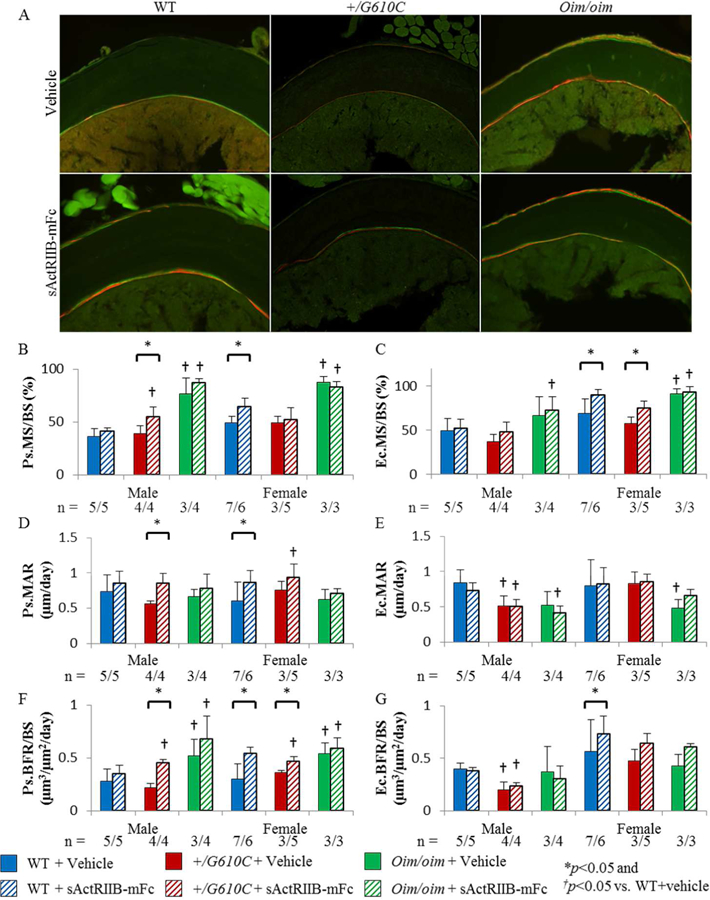
Femurs of sActRIIB-mFc treated female WT and male and female +/G610C mice exhibited increased periosteal bone formation rate. A) Representative fluorochrome labeled cortical cross-sectional images of female mice. B) periosteal mineralized surface per bone surface (Ps.MS/BS), C) endocortical mineralized surface per bone surface (Ec.MS/BS), D) periosteal mineral apposition rate (Ps.MAR), E) endocortical mineral apposition rate (Ec.MAR), F) periosteal bone formation rate per bone surface (Ps.BFR/BS), and G) endocortical bone formation rate per bone surface (Ec.BFR/BS) of male and female WT, +/G610C, and oim/oim vehicle (solid color) and sActRIIB-mFc (diagonal) treated mice. (WT [blue], +/G610C [red], and oim/oim [green]). (n) is indicated for each genotype and treatment, the number of samples are lower in +/G610C and oim/oim due to section fragility and limited number of callus free femurs. Values are MEAN±SD. *p<0.05; †p<0.05 vs. WT+vehicle treated mice.
Static Histomorphometry
To quantitatively elucidate the impact of sActRIIB-mFc treatment on +/G610C and oim/oim osteoblast and osteoclast, we performed static histomorphometry on the distal trabeculae of the left femur. +/G610C and oim/oim femurs had reduced total numbers of osteoblasts (N.Ob) relative to their WT counterparts regardless of sex except for male +/G610C mice (Figure 4A). Male oim/oim mice had reduced osteoblast number per bone surface (N.Ob/BPm) and osteoblast surface per bone surface (Ob.S/BPm), while female +/G610C mice had reduced Ob.S/BPm as compared to their WT counterparts (Figures 4C and E). Male +/G610C mice had increased osteoclast number per bone surface (N.Oc/BPm) as compared to WT littermates, and oim/oim mice exhibited increased N.Oc/BPm and osteoclast surface per bone surface (Oc.S/BPm) as compared to WT counterparts regardless of sex (Figures 4D and F). sActRIIB-mFc treated mice had increased N.Ob as compared to vehicle treated counterparts regardless of sex and genotype, with no changes in N.Ob/BPm and Ob.S/BPm (Figures 4A, C, and E). In contrast, N.Oc/BPm and Oc.S/BPm were significantly reduced in sActrRIIB-mFc treated mouse femurs as compared to vehicle treated counterparts regardless of genotype and sex, except for male WT mice (Figures 4D and F).
Figure 4.
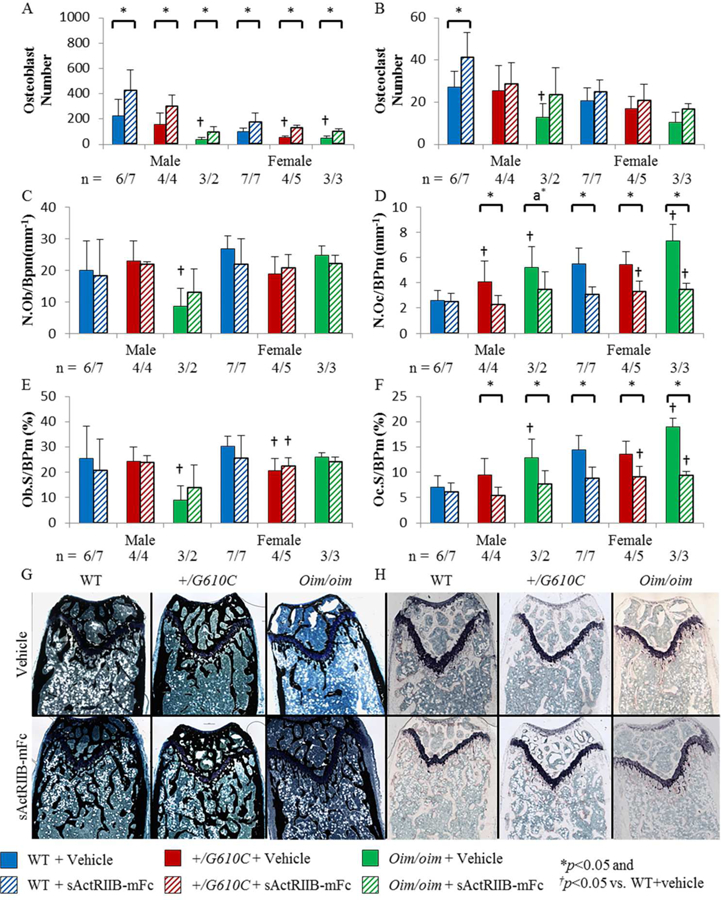
Femurs of sActRIIB-mFc treated mice demonstrated increased osteoblast number and decreased osteoclast number per bone surface and osteoclast surface per bone surface in the distal trabecular bone regardless of sex and genotype, except for male WT mice. A) Osteoblast number (N.Ob), B) Osteoclast number (N.Oc), C) Osteoblast number per bone surface (N.Ob/BPm), D) Osteoclast number per bone surface (N.Oc/BPm), E) Osteoblast surface per bone surface (Ob.S/BPm), and F) Osteoclast surface per bone surface (Oc.S/BPm) of male and female WT, +/G610C, and oim/oim vehicle (solid color) and sActRIIB-mFc (diagonal) treated mice. G) and H) Representative images of Von Kossa and TRAP stained longitudinal trabecular sections of female mice. (WT [blue], +/G610C [red], and oim/oim [green]). (n) is indicated for each genotype and treatment. The number of samples are lower in +/G610C and oim/oim due to fragility of the sections and limited number of callus free femurs. Values are MEAN±SD. *p<0.05; †p<0.05 vs. WT+vehicle treated mice; a*p=0.0673.
Serum Biochemical Markers
Serum PINP was decreased in female +/G610C and oim/oim mice as compared to their WT counterparts. Serum CTX was increased 10–15 fold in oim/oim mice relative to their WT counterparts regardless of sex; this dramatic increase in CTX likely reflects excess degradation of abnormal type I collagen, which has not been integrated into the bone matrix (Figure 5B). There was no major impact of sActRIIB-mFc treatment in either PINP or CTX levels, except increased PINP levels in female +/G610C mice, and increased CTX levels in male +/G610C and female oim/oim mice relative to their vehicle treated counterparts (Figure 5).
Figure 5.
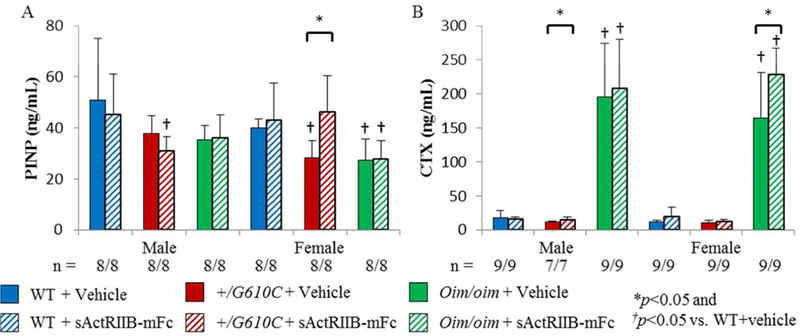
Oim/oim mice demonstrated a decrease in PINP and an increase in CTX serum levels as compared to WT counterparts. A) N-terminal propeptide of type I procollagen (PINP) and B) C-terminal cross-linked telopeptide of type I collagen (CTX) of male and female WT, +/G610C, and oim/oim vehicle (solid color) and sActRIIB-mFc (diagonal) treated mice. (WT [blue], +/G610C [red], and oim/oim [green]) Values are MEAN±SD. (n) is indicated for each genotype and treatment. *p<0.05; †p<0.05 vs. WT+vehicle treated mice.
RNA Analyses
To investigate the effects of sActRIIB-mFc on bone remodeling at the molecular level in oim and G610C OI mice, right tibial gene expression levels for specific transcripts critical to osteoblast, osteoclast, and osteocyte differentiation and function were evaluated. Vehicle treated male oim/oim mice demonstrated increases in gene expression levels of Alpl, Ctsk, and β-cat as compared to vehicle treated male WT mice (Figure 6). Tibiae of sActRIIB-mFc treated male +/G610C mice had increased Ctsk expression and female +/G610C mice had increased Sp7, Serpinh1, and Csf1expression relative to their vehicle treated counterparts (Figure 7B and Supplemental Figure S2). Lastly, tibiae of sActRIIB-mFc treated male oim/oim mice had enhanced gene expression levels of Alpl, Dmp1 and Phex as compared to vehicle treated counterparts (Figure 7C).
Figure 6.
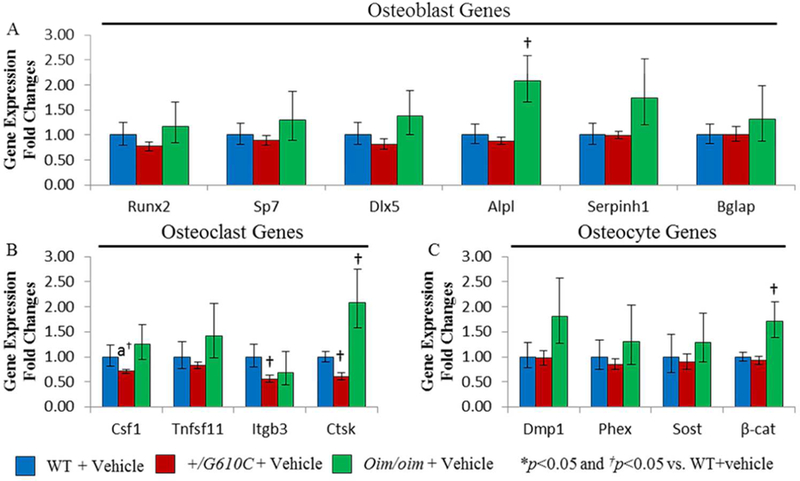
Male oim/oim mice exhibited upregulation of osteoblast, osteoclast, and osteocyte marker genes as compared to WT counterparts. A) Relative fold changes of vehicle treated +/G610C and oim/oim Runx2, Sp7 (Osterix), Dlx5, Alpl (Alkaline phosphatase), Serpinh1 (Hsp47), and Bglap (osteocalcin), B) Csf1 (M-CSF), Tnfsf11 (RANKL), Itgb3 (Integrinβ−3), and Ctsk (Cathepsin K), and C) Dmp1, Phex, Sost (sclerostin), and β-cat (β catenin) mRNA levels with respect to vehicle treated WT mice. (n=10 WT+vehicle [blue], n=8 +/G610C+vehicle [red], and n=11 oim/oim+vehicle [green]) (Alpl gene expression levels of WT and oim/oim sample size were 7 and 8, respectively). Values are MEAN±SD. *p<0.05; †p<0.05 vs. WT+vehicle treated mice; *ap=0.0676.
Figure 7.
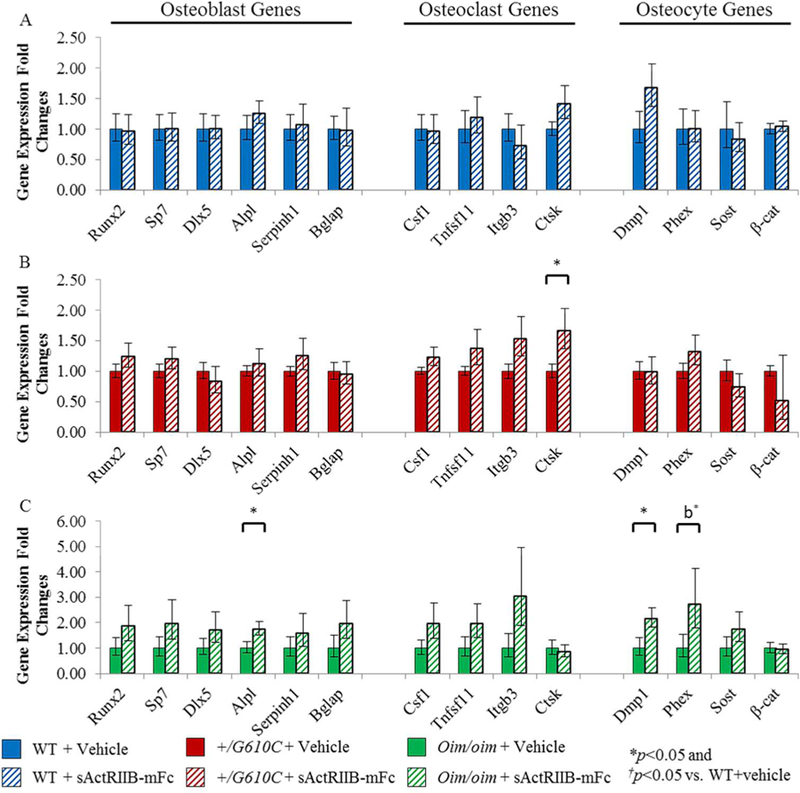
sActRIIB-mFc treated male oim/oim mice demonstrated upregulation of osteoblast and osteocyte marker genes as compared to vehicle treated counterparts. A) Relative fold changes of sActRIIB-mFc treated WT osteoblast (Runx2, Sp7, Dlx5, Alpl, Serpinh1, and Bglap), osteoclast (Csf1, Tnfsf11, Itgb3, and Ctsk), and osteocyte (Dmp1, Phex, Sost, and β-cat) mRNA levels with respect to vehicle treated WT mice, B) Relative fold changes of sActRIIB-mFc treated +/G610C osteoblast, osteoclast, and osteocyte mRNA levels with respect to vehicle treated +/G610C mice, and C) Relative fold changes of sActRIIB-mFc treated oim/oim osteoblast, osteoclast, and osteocyte mRNA levels with respect to vehicle treated oim/oim mice. (n=10 WT+vehicle [blue], n=10 WT+sActRIIB-mFc [blue diagonal], n=8 +/G610C+vehicle [red], n=8 +/G610C+sActRIIB-mFc [red diagonal], n=11 oim/oim+vehicle [green], and n=11 oim/oim+sActRIIB-mFc [green diagonal] [Alpl gene expression level of WT and oim/oim sample size were 7 and 8, respectively]). Values are MEAN±SD. *p<0.05; †p<0.05 vs. WT+vehicle treated mice; *bp=0.0545.
Discussion
ActRIIB ligand trapping for 8 weeks by sActRIIB-mFc treatment increased femoral trabecular bone volume regardless of sex and genotype. The impact was also evident by improved cortical bone geometry and biomechanical strength of WT and +/G610C femurs (Figures 1 and 2). The sActRIIB-mFc mediated increases in bone mass likely reflect anabolic effects with higher mineral apposition and bone formation rates at the femoral mid-diaphysis (Figure 3). Additionally, sActRIIB-mFc treatment was associated with an almost 2-fold increase in total osteoblast number concomitant with decreased osteoclast number per bone surface at the distal trabecular bone regardless of sex and genotype, thus synergistically causing a 2–4 fold increase in trabecular bone volume (Figure 1 and 4).
DiGirolamo et al. treated oim/oim mice with a soluble activin receptor type IIB fusion protein once a week for 4 weeks and demonstrated about 1.3 fold increase in trabecular BV/TV(12). Our study has taken this further to investigate the effect of extended sActRIIB-mFc treatment on biomechanical integrity and cellular and molecular mechanisms in two molecularly distinct OI mouse models. Torsional loading to failure testing showed increases in torsional ultimate strength and energy to failure without altering bone material properties (tensile strength and shear modulus of elasticity) with sActRIIB-mFc treatment, suggesting improvement in bone biomechanical strength appears mainly due to increased bone mass rather than altered bone quality (Figure 2). These increases were consistent with the increased cortical CSA and polar moment of area seen in the femoral mid-diaphysis of sActRIIB-mFc treated WT and +/G610C mice (Figure 1). In contrast, oim/oim mouse femurs did not show changes in cortical bone geometry or biomechanical parameters (Figures 1 and 2). It is difficult to discern if the absence of a difference in hindlimb fracture/callus numbers observed at sacrifice in the vehicle- versus sActRIIB-mFc treated oim/oim mice is due to the presence of fractures/calluses prior to the beginning of treatment (2 months) or to the absence of an impact of sActRIIB-mFc on fracture occurrence (Supplemental Table S7). However, the effect of sActRIIB-mFc treatment on trabecular bone microarchitecture was dramatic regardless of genotypes (Figure 1). Notably, sActRIIB-mFc treatment restored the femoral phenotype of +/G610C mice to normal levels, exhibiting greater trabecular bone volume and equivalent cortical bone geometric parameters and strength as vehicle treated WT mice. sActRIIB-mFc treated oim/oim femurs also acquired comparable trabecular bone volumes as vehicle treated WT femurs. Our findings suggest that oim/oim mice were still able to induce anabolic bone formation with sActRIIB-mFc treatment, but their ability to accumulate cortical bone appears reduced relative to WT and +/G610C mice. Similarly, in a clinical study, teriparatide (anabolic agent) treatment was more effective in mild type I OI patients with increases in vertebral BMD and bone formation biochemical markers, whereas there was no significant benefit in more severe type III/IV OI patients(33).
Increased bone formation in the periosteal regions was seen without major changes in the endocortical surface of sActRIIB-mFc treated WT and +/G610C mouse femurs. This suggests that the mechanism responsible for increased cortical bone quantity is likely an anabolic response of periosteal osteoblasts, which may reflect direct and/or indirect effects of myostatin inhibition. Bone marrow derived mesenchymal stem cells from myostatin knockout mice were shown to have increased bone mineralization capacity in osteogenic medium as compared to WT mice(34). In addition, direct regulation of osteocytes by myostatin to alter osteoblast function recently has been demonstrated by Qin et al.; specifically exosomes secreted by myostatin treated osteocyte cells can be engulfed by osteoblasts to decrease osteoblast differentiation via down-regulation of the Wnt signaling pathway, and these exosomes did not appear to have an impact on osteoclastogenesis(35).
Static histomorphometric analyses of trabecular bone further suggests that sActRIIB-mFc treatment suppresses bone resorption as indicated by reduced N.Oc/BPm and Oc.S/BPm in addition to increased total osteoblast numbers. Two recent studies demonstrated a potential direct role for myostatin in osteoclastogenesis, as they demonstrated stimulation of osteoclast differentiation from bone marrow derived macrophages with myostatin treatment in the presence of RANKL and/or M-CSF(36,37). These studies and our data suggest myostatin inhibition via sActRIIB-mFc treatment may have positive and negative regulatory effects on osteoblasts and osteoclasts, respectively. In addition to a potential direct effect on osteoblasts and osteoclasts by myostatin inhibition, significant increases in muscle mass occurred in our mice around 9–10 weeks of age(19), suggesting indirect regulation of osteoblasts and osteoclasts may occur as well. The positive effect of mechanical loading on bone formation is well established and increased muscle mass by myostatin inhibition increases load on bone, promoting bone formation. Negative correlations between Quad weight and osteoclast (N.Oc/BPm and Oc.S/BPm) were evident for all genotypes, however, positive correlations between biomechanical strength or osteoblast number and Quad weight were only present in WT and +/G610C, but not in oim/oim mice (Supplemental Table S8 and S9). The absence of a positive correlation in oim/oim mouse may reflect the compromised osteoblast function and/or the inherent muscle weakness associated with the oim gene defect(38,39).
Differences in the skeletal response of +/G610C and oim/oim may also be attributed to their specific collagen mutation; +/G610C mice are heterozygous for a glycine to cysteine substitution and oim/oim mice produce homotrimeric type I collagen [α1(I)3]. Different mechanisms promoting gains in trabecular bone mass have also been suggested with anti-sclerostin antibody treatment in dominant (+/G610C) and recessive (crtap−/−) OI mouse models; anti-sclerostin antibody treatment resulted in increased osteoblast surface and decreased osteoclast surface per bone surface in +/G610C and crtap−/− mice, respectively(40,41). Recently, anti-TGF-β antibody treatment has gained attention as another potential therapeutic option in OI, treatment with anti-TGF-β antibody inhibits the same downstream Smad2/3 signaling pathway as our study and demonstrated significant increases in vertebral trabecular bone volume in both +/G610C and crtap−/− mice. However, the mechanism of increasing bone volume in crtap−/− differed from our study by decreasing both osteoblast and osteoclast numbers per bone surface(42). sActRIIB-mFc treatment additionally targets myostatin to increase muscle mass, which may have prevented decreases in osteoblast number per bone surface via mechanotransduction in our study.
Gene expression levels of key bone metabolic markers were not altered in WT and +/G610C tibiae with sActRIIB-mFc treatment, suggesting increased bone mass was due to increased osteoblast numbers, which was consistent with our histomorphometric data. However, sActRIIB-mFc treated male oim/oim mouse tibiae had at least a 2 fold increase in expression of mature osteoblast and osteocyte biomarkers as well as osteoblast and osteoclast differentiation factors, although only a few of these biomarkers reached significance (Alpl, Dmp1, and Phex) (Figure 7). A limitation of this experiment is that we used intact bone (with marrow) to extract RNA; sActRIIB-mFc treated mouse tibial expression could reflect higher bone to marrow ratios which may impact relative expression of the biomarkers. Impairment of osteoblast lineage differentiation has also been implicated in oim/oim mice(43). We also cannot exclude the possibility that although sActRIIB-mFc treatment in oim/oim mice promoted bone formation, differentiation of oim/oim preosteoblasts to mature osteoblasts or osteocytes may remain compromised, and sActRIIB-mFc may drive constitutively higher expression of osteoblast, osteoclast, and osteocyte differentiation factors in response.
In addition to myostatin, activin A also has high affinity for the ActRIIB and regulates downstream signaling pathways, suggesting that gain in bone mass with sActRIIB-mFc treatment may not be due solely to the direct and/or indirect impact of myostatin inhibition. Activin A is known to be embedded in bone matrix(44,45) and also can be synthesized by osteoblasts and bone marrow cells to regulate osteoblast and osteoclast differentiation(46–48). Activin A promotes osteoclast differentiation and bone resorption in the presence of RANKL and/or M-CSF(46,47,49,50). Activin A’s effects on osteoblasts appears to be dependent on stage of differentiation; promoting differentiation during early phases of osteoblast maturation(47,51), while inhibiting the matrix mineralization process(52–54). Although there are conflicting reports concerning the influence of activin A on osteoblast and osteoclast cells, soluble ActRIIA and ActRIIB fusion protein-induced activin inhibition in-vivo were associated with improved bone geometry and strength in mice and primates (9,11,12,32,55–60). These studies suggest improvement in bone properties with sActRIIB-mFc treatment is potentially due in part to activin A and/or the synergistic effect of activin A and myostatin inhibition together. Another limitation of our study is the multiple ligand targets (myostatin, activin A, GDF-11, and some BMPs) of sActRIIB-mFc, which do not allow clear discernment of the impact of each ligand on bone remodeling, thus specific inhibition of myostatin, activin A, and GDF-11 independently is required in future studies.
In summary, we demonstrate that sActRIIB-mFc treatment improved cortical bone geometry and biomechanical strength in +/G610C but not in oim/oim femurs; however sActRIIB-mFc significantly improved trabecular microarchitecture in both +/G610C and oim/oim femurs with 2–4 fold increases in bone volume. Histomorphometric analyses demonstrated the improved bone microarchitecture was due to both an anabolic and anti-catabolic effects, overall, suggesting that the increased bone quantity with sActRIIB-mFc treatment could be a potential therapeutic target in OI.
Supplementary Material
Acknowledgements:
We would like to thank Dr. Mark R. Ellersieck (Department of Statistics, University of Missouri) for his assistance in the statistical analyses of this study. We would also like to thank the following funding sources: National Institutes of Health R01 AR055907, R21 AR062346 and P01 AG039355; NASA-EPSCoR Missouri Research Infrastructure Development; Leda J. Sears Trust Foundation; Kansas City Area Life Sciences Institute, Patton Trust Research Grant; March of Dimes Research Grant; Osteogenesis Imperfecta Foundation, Michael Geisman Fellowship; University of Missouri Research Board and Council; University of Missouri Interdisciplinary Intercampus Research Program.
Footnotes
Disclosures:
All authors state that they have no conflicts of interest. sActRIIB-mFc (RAP-031) was provided by Dr. R. Scott Pearsall, Acceleron Pharma Inc.
References:
- 1.Forlino A, Marini JC. Osteogenesis imperfecta. Lancet Review November 2 2015. Epub 2015/11/07. [DOI] [PMC free article] [PubMed]
- 2.Dalgleish R. The Human Collagen Mutation Database 1998. Nucleic Acids Res January 01 1998;26(1):253–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rauch F, Glorieux FH. Treatment of children with osteogenesis imperfecta. Current osteoporosis reports Research Support, Non-U.S. Gov’t Review December 2006;4(4):159–64. Epub 2006/11/23. [DOI] [PubMed] [Google Scholar]
- 4.Cundy T. Recent advances in osteogenesis imperfecta. Calcified tissue international Review June 2012;90(6):439–49. Epub 2012/03/28. [DOI] [PubMed] [Google Scholar]
- 5.McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. May 1 1997;387(6628):83–90. Epub 1997/05/01. [DOI] [PubMed] [Google Scholar]
- 6.Frost HM. Bone’s mechanostat: a 2003 update. The anatomical record Part A, Discoveries in molecular, cellular, and evolutionary biology Review December 2003;275(2):1081–101. Epub 2003/11/13. [DOI] [PubMed] [Google Scholar]
- 7.Hamrick MW. Increased bone mineral density in the femora of GDF8 knockout mice. The anatomical record Part A, Discoveries in molecular, cellular, and evolutionary biology Research Support, U.S. Gov’t, P.H.S. May 2003;272(1):388–91. Epub 2003/04/22. [DOI] [PubMed] [Google Scholar]
- 8.Hamrick MW, Samaddar T, Pennington C, McCormick J. Increased muscle mass with myostatin deficiency improves gains in bone strength with exercise. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research Research Support, N.I.H., Extramural March 2006;21(3):477–83. Epub 2006/02/24. [DOI] [PubMed] [Google Scholar]
- 9.Koncarevic A, Cornwall-Brady M, Pullen A, Davies M, Sako D, Liu J, et al. A soluble activin receptor type IIb prevents the effects of androgen deprivation on body composition and bone health. Endocrinology September 2010;151(9):4289–300. Epub 2010/06/25. [DOI] [PubMed] [Google Scholar]
- 10.Tang L, Yang X, Gao X, Du H, Han Y, Zhang D, et al. Inhibiting myostatin signaling prevents femoral trabecular bone loss and microarchitecture deterioration in diet-induced obese rats. Exp Biol Med (Maywood) Research Support, Non-U.S. Gov’t February 2016;241(3):308–16. Epub 2015/10/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Puolakkainen T, Ma H, Kainulainen H, Pasternack A, Rantalainen T, Ritvos O, et al. Treatment with soluble activin type IIB-receptor improves bone mass and strength in a mouse model of Duchenne muscular dystrophy. BMC musculoskeletal disorders January 19 2017;18(1):20 Epub 2017/01/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DiGirolamo DJ, Singhal V, Chang X, Lee SJ, Germain-Lee EL. Administration of soluble activin receptor 2B increases bone and muscle mass in a mouse model of osteogenesis imperfecta. Bone research 2015;3:14042 Epub 2015/07/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oestreich AK, Carleton SM, Yao X, Gentry BA, Raw CE, Brown M, et al. Myostatin deficiency partially rescues the bone phenotype of osteogenesis imperfecta model mice. Osteoporosis international : a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t January 2016;27(1):161–70. Epub 2015/07/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Souza TA, Chen X, Guo Y, Sava P, Zhang J, Hill JJ, et al. Proteomic identification and functional validation of activins and bone morphogenetic protein 11 as candidate novel muscle mass regulators. Mol Endocrinol December 2008;22(12):2689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daley E, Streeten EA, Sorkin JD, Kuznetsova N, Shapses SA, Carleton SM, et al. Variable bone fragility associated with an Amish COL1A2 variant and a knock-in mouse model. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research Research Support, N.I.H., Extramural Research Support, N.I.H., Intramural Research Support, Non-U.S. Gov’t February 2010;25(2):247–61. Epub 2009/07/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chipman SD, Sweet HO, McBride DJ Jr., Davisson MT, Marks SC Jr., Shuldiner AR, et al. Defective pro alpha 2(I) collagen synthesis in a recessive mutation in mice: a model of human osteogenesis imperfecta. Proceedings of the National Academy of Sciences of the United States of America March 1 1993;90(5):1701–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carleton SM, McBride DJ, Carson WL, Huntington CE, Twenter KL, Rolwes KM, et al. Role of genetic background in determining phenotypic severity throughout postnatal development and at peak bone mass in Col1a2 deficient mice (oim). Bone April 2008;42(4):681–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeong Y, Carleton SM, Gentry BA, Yao X, Ferreira JA, Salamango DJ, et al. Hindlimb Skeletal Muscle Function and Skeletal Quality and Strength in +/G610C Mice With and Without Weight-Bearing Exercise. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t October 2015;30(10):1874–86. Epub 2015/04/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeong Y, Daghlas SA, Kahveci AS, Salamango D, Gentry BA, Brown M, et al. Soluble activin receptor type IIB decoy receptor differentially impacts murine osteogenesis imperfecta muscle function. Muscle & nerve May 26 2017. [DOI] [PMC free article] [PubMed]
- 20.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research July 2010;25(7):1468–86. [DOI] [PubMed] [Google Scholar]
- 21.Doube M, Klosowski MM, Arganda-Carreras I, Cordelieres FP, Dougherty RP, Jackson JS, et al. BoneJ: Free and extensible bone image analysis in ImageJ. Bone Research Support, Non-U.S. Gov’t December 2010;47(6):1076–9. Epub 2010/09/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roarck RJ, Young WC. Formulas for Stress and Strain 5 ed: McGraw-Hill; 1975. [Google Scholar]
- 23.Tiede-Lewis LM, Xie Y, Hulbert MA, Campos R, Dallas MR, Dusevich V, et al. Degeneration of the osteocyte network in the C57BL/6 mouse model of aging. Aging (Albany NY) October 26 2017;9(10):2190–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Egan KP, Brennan TA, Pignolo RJ. Bone histomorphometry using free and commonly available software. Histopathology Comparative Study Research Support, N.I.H., Extramural December 2012;61(6):1168–73. Epub 2012/08/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, et al. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research January 2013;28(1):2–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greenhough J, Papadakis ES, Cutress RI, Townsend PA, Oreffo RO, Tare RS. Regulation of osteoblast development by Bcl-2-associated athanogene-1 (BAG-1). Sci Rep September 16 2016;6:33504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carmer SG SM. An Evaluation of Ten Pairwise Multiple Comparison Procedures by Monte Carlo Methods. Journal of the American Statistical Association 1973;68(341):66–74. [Google Scholar]
- 28.DJ S. Multiple Comparison Procedures: The Practical Solution. The American Statistician 1990;44(2):174–80. [Google Scholar]
- 29.Conover WJ, Iman RL. Analysis of covariance using the rank transformation. Biometrics September 1982;38(3):715–24. [PubMed] [Google Scholar]
- 30.Olejnik S, Algina J. Generalized eta and omega squared statistics: measures of effect size for some common research designs. Psychol Methods December 2003;8(4):434–47. [DOI] [PubMed] [Google Scholar]
- 31.Beamer WG, Shultz KL, Donahue LR, Churchill GA, Sen S, Wergedal JR, et al. Quantitative trait loci for femoral and lumbar vertebral bone mineral density in C57BL/6J and C3H/HeJ inbred strains of mice. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research July 2001;16(7):1195–206. [DOI] [PubMed] [Google Scholar]
- 32.Chiu CS, Peekhaus N, Weber H, Adamski S, Murray EM, Zhang HZ, et al. Increased muscle force production and bone mineral density in ActRIIB-Fc-treated mature rodents. The journals of gerontology Series A, Biological sciences and medical sciences Research Support, Non-U.S. Gov’t October 2013;68(10):1181–92. Epub 2013/03/26. [DOI] [PubMed] [Google Scholar]
- 33.Orwoll ES, Shapiro J, Veith S, Wang Y, Lapidus J, Vanek C, et al. Evaluation of teriparatide treatment in adults with osteogenesis imperfecta. The Journal of clinical investigation February 2014;124(2):491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamrick MW, Shi X, Zhang W, Pennington C, Thakore H, Haque M, et al. Loss of myostatin (GDF8) function increases osteogenic differentiation of bone marrow-derived mesenchymal stem cells but the osteogenic effect is ablated with unloading. Bone Research Support, N.I.H., Extramural Research Support, U.S. Gov’t, Non-P.H.S. June 2007;40(6):1544–53. Epub 2007/03/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qin Y, Peng Y, Zhao W, Pan J, Ksiezak-Reding H, Cardozo C, et al. Myostatin inhibits osteoblastic differentiation by suppressing osteocyte-derived exosomal microRNA-218: A novel mechanism in muscle-bone communication. The Journal of biological chemistry June 30 2017;292(26):11021–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dankbar B, Fennen M, Brunert D, Hayer S, Frank S, Wehmeyer C, et al. Myostatin is a direct regulator of osteoclast differentiation and its inhibition reduces inflammatory joint destruction in mice. Nature medicine Research Support, Non-U.S. Gov’t September 2015;21(9):1085–90. Epub 2015/08/04. [DOI] [PubMed] [Google Scholar]
- 37.Chen YS, Guo Q, Guo LJ, Liu T, Wu XP, Lin ZY, et al. GDF8 inhibits bone formation and promotes bone resorption in mice. Clinical and experimental pharmacology & physiology January 11 2017. Epub 2017/01/12. [DOI] [PubMed]
- 38.Kalajzic I, Terzic J, Rumboldt Z, Mack K, Naprta A, Ledgard F, et al. Osteoblastic response to the defective matrix in the osteogenesis imperfecta murine (oim) mouse. Endocrinology Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. May 2002;143(5):1594–601. Epub 2002/04/17. [DOI] [PubMed] [Google Scholar]
- 39.Gentry BA, Ferreira JA, McCambridge AJ, Brown M, Phillips CL. Skeletal muscle weakness in osteogenesis imperfecta mice. Matrix biology : journal of the International Society for Matrix Biology Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t September 2010;29(7):638–44. Epub 2010/07/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacobsen CM, Barber LA, Ayturk UM, Roberts HJ, Deal LE, Schwartz MA, et al. Targeting the LRP5 pathway improves bone properties in a mouse model of osteogenesis imperfecta. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research October 2014;29(10):2297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grafe I, Alexander S, Yang T, Lietman C, Homan EP, Munivez E, et al. Sclerostin Antibody Treatment Improves the Bone Phenotype of Crtap(−/−) Mice, a Model of Recessive Osteogenesis Imperfecta. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research May 2016;31(5):1030–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grafe I, Yang T, Alexander S, Homan EP, Lietman C, Jiang MM, et al. Excessive transforming growth factor-beta signaling is a common mechanism in osteogenesis imperfecta. Nature medicine June 2014;20(6):670–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H, Jiang X, Delaney J, Franceschetti T, Bilic-Curcic I, Kalinovsky J, et al. Immature osteoblast lineage cells increase osteoclastogenesis in osteogenesis imperfecta murine. The American journal of pathology May 2010;176(5):2405–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Funaba M, Ogawa K, Abe M. Expression and localization of activin receptors during endochondral bone development. European journal of endocrinology Research Support, Non-U.S. Gov’t January 2001;144(1):63–71. Epub 2001/02/15. [DOI] [PubMed] [Google Scholar]
- 45.Ogawa Y, Schmidt DK, Nathan RM, Armstrong RM, Miller KL, Sawamura SJ, et al. Bovine bone activin enhances bone morphogenetic protein-induced ectopic bone formation. The Journal of biological chemistry July 15 1992;267(20):14233–7. Epub 1992/07/15. [PubMed] [Google Scholar]
- 46.Fuller K, Bayley KE, Chambers TJ. Activin A is an essential cofactor for osteoclast induction. Biochemical and biophysical research communications February 05 2000;268(1):2–7. Epub 2000/02/01. [DOI] [PubMed] [Google Scholar]
- 47.Gaddy-Kurten D, Coker JK, Abe E, Jilka RL, Manolagas SC. Inhibin suppresses and activin stimulates osteoblastogenesis and osteoclastogenesis in murine bone marrow cultures. Endocrinology Research Support, U.S. Gov’t, Non-P.H.S. Research Support, U.S. Gov’t, P.H.S. January 2002;143(1):74–83. Epub 2001/12/26. [DOI] [PubMed] [Google Scholar]
- 48.Nicks KM, Perrien DS, Akel NS, Suva LJ, Gaddy D. Regulation of osteoblastogenesis and osteoclastogenesis by the other reproductive hormones, Activin and Inhibin. Molecular and cellular endocrinology Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S. Review October 30 2009;310(1–2):11–20. Epub 2009/07/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sugatani T, Alvarez UM, Hruska KA. Activin A stimulates IkappaB-alpha/NFkappaB and RANK expression for osteoclast differentiation, but not AKT survival pathway in osteoclast precursors. Journal of cellular biochemistry Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. September 01 2003;90(1):59–67. Epub 2003/08/26. [DOI] [PubMed] [Google Scholar]
- 50.Murase Y, Okahashi N, Koseki T, Itoh K, Udagawa N, Hashimoto O, et al. Possible involvement of protein kinases and Smad2 signaling pathways on osteoclast differentiation enhanced by activin A. Journal of cellular physiology Research Support, Non-U.S. Gov’t August 2001;188(2):236–42. Epub 2001/06/26. [DOI] [PubMed] [Google Scholar]
- 51.Centrella M, McCarthy TL, Canalis E. Activin-A binding and biochemical effects in osteoblast-enriched cultures from fetal-rat parietal bone. Molecular and cellular biology Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. January 1991;11(1):250–8. Epub 1991/01/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alves RD, Eijken M, Bezstarosti K, Demmers JA, van Leeuwen JP. Activin A suppresses osteoblast mineralization capacity by altering extracellular matrix (ECM) composition and impairing matrix vesicle (MV) production. Molecular & cellular proteomics : MCP Research Support, Non-U.S. Gov’t October 2013;12(10):2890–900. Epub 2013/06/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eijken M, Swagemakers S, Koedam M, Steenbergen C, Derkx P, Uitterlinden AG, et al. The activin A-follistatin system: potent regulator of human extracellular matrix mineralization. FASEB journal : official publication of the Federation of American Societies for Experimental Biology September 2007;21(11):2949–60. Epub 2007/04/24. [DOI] [PubMed] [Google Scholar]
- 54.Woeckel VJ, van der Eerden BC, Schreuders-Koedam M, Eijken M, Van Leeuwen JP. 1alpha,25-dihydroxyvitamin D3 stimulates activin A production to fine-tune osteoblast-induced mineralization. Journal of cellular physiology Research Support, Non-U.S. Gov’t November 2013;228(11):2167–74. Epub 2013/04/17. [DOI] [PubMed] [Google Scholar]
- 55.Pearsall RS, Canalis E, Cornwall-Brady M, Underwood KW, Haigis B, Ucran J, et al. A soluble activin type IIA receptor induces bone formation and improves skeletal integrity. Proceedings of the National Academy of Sciences of the United States of America May 13 2008;105(19):7082–7. Epub 2008/05/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fajardo RJ, Manoharan RK, Pearsall RS, Davies MV, Marvell T, Monnell TE, et al. Treatment with a soluble receptor for activin improves bone mass and structure in the axial and appendicular skeleton of female cynomolgus macaques (Macaca fascicularis). Bone January 2010;46(1):64–71. Epub 2009/09/29. [DOI] [PubMed] [Google Scholar]
- 57.Lotinun S, Pearsall RS, Davies MV, Marvell TH, Monnell TE, Ucran J, et al. A soluble activin receptor Type IIA fusion protein (ACE-011) increases bone mass via a dual anabolic-antiresorptive effect in Cynomolgus monkeys. Bone Research Support, Non-U.S. Gov’t April 2010;46(4):1082–8. Epub 2010/01/19. [DOI] [PubMed] [Google Scholar]
- 58.Chantry AD, Heath D, Mulivor AW, Pearsall S, Baud’huin M, Coulton L, et al. Inhibiting activin-A signaling stimulates bone formation and prevents cancer-induced bone destruction in vivo. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research Research Support, Non-U.S. Gov’t December 2010;25(12):2633–46. Epub 2010/06/10. [DOI] [PubMed] [Google Scholar]
- 59.Morse A, Cheng TL, Peacock L, Mikulec K, Little DG, Schindeler A. RAP-011 augments callus formation in closed fractures in rats. Journal of orthopaedic research : official publication of the Orthopaedic Research Society Research Support, Non-U.S. Gov’t February 2016;34(2):320–30. Epub 2015/07/18. [DOI] [PubMed] [Google Scholar]
- 60.Bialek P, Parkington J, Li X, Gavin D, Wallace C, Zhang J, et al. A myostatin and activin decoy receptor enhances bone formation in mice. Bone March 2014;60:162–71. Epub 2013/12/18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.