

Abstract
Importance:
Previous research indicates that cannabis use is associated with psychotic-like experiences (PLEs). Yet is unclear whether this relationship is due to predispositional (i.e., shared genetic) or individual-specific (e.g., causal processes, such as cannabis use leading to PLEs) factors.
Objective:
To estimate genetic and environmental correlations between cannabis use and PLEs, and to examine PLEs in twins/siblings discordant for exposure to cannabis use to disentangle predispositional from individual-specific effects.
Design:
Cross-sectional diagnostic interviews and self-report data were collected from two separate population-based samples.
Setting:
Data from the Human Connectome Project (HCP) were collected between 2012–2015 and data from the Australian Twin Registry Sample 3 (ATR3) were collected between 2005–2009.
Participants:
The study included data from 1,188 HCP participants and 3,486 ATR3 participants, totaling 4,674 participants.
Main Outcomes and Measures:
Three cannabis involvement variables were examined: frequent use (i.e., ≥100 times), a DSM-IV lifetime cannabis use disorder (CUD) diagnosis, and current cannabis use. Genetic and environmental correlations between cannabis involvement and PLEs were estimated. Generalized linear mixed models examined PLE differences in twin/sibling pairs discordant for cannabis use.
Results:
Analyses were performed in 4,674 participants (mean age=30.51 years, 62.5% female). PLEs were associated with frequent use (β=0.11; 95% CI, 0.08–0.14), CUD (β=0.13; 95% CI, 0.09–0.16), and current cannabis use (β=0.07; 95% CI, 0.04–0.10), even after adjustment for covariates. Correlated genetic factors explained between 69.2–84.1% of this observed association. Even so, within discordant pairs of twins/siblings (Npairs: 308–324), PLEs were more common in cannabis exposed individuals relative to their relative who used cannabis to a lesser degree (βs≥.23, ps<.05; e.g., mean PLE score difference between frequent and infrequent cannabis-using relatives=0.27, Z=−5.41, p<.001).
Conclusions:
Despite the strong contribution of shared genetic factors, frequent and problem cannabis use also relates to PLEs via person-specific pathways. Policy discussions surrounding legalization should consider the impact of escalations in cannabis use on trait-like indices of vulnerability, like PLEs, which could contribute to pervasive psychological and interpersonal burden.
Since first finding a 6.0 relative risk of schizophrenia in heavy cannabis users,1 researchers have debated the role of cannabis use in the etiology of psychotic disorders.2–7 Some posit that cannabis use causally impacts risk for psychosis, either via direct pharmacological pathways or by potentiating genetic susceptibility. For instance, a recent study found that first episode psychosis users were more likely to have used a high potency form of cannabis relative to controls, suggesting direct causation.8 In contrast, epidemiologists have demonstrated that while the prevalence of cannabis use has increased worldwide, the incidence of psychotic disorders remains largely stable,9 and further that attributable risk is rather small.10 Unmeasured confounders, including common genetic and environmental contributors, have also been inconsistently accounted for in causal calculations.11
Cannabis involvement is also linked to psychotic-like experiences (PLEs) that are more prevalent and easily assessed via self-report12–16. Lifetime PLEs are associated with mental health problems17 and independently increase risk for disability attributable to deficits in cognition, social interactions, and role functioning.18 Due to greater prevalence of PLEs (~7%)19 relative to psychotic disorders (~1% for schizophrenia), any causal effects of cannabis use on PLEs may have significant public health consequences.
Cannabis involvement (h2~51%)20, schizophrenia (h2~80%)21 and PLEs (h2~43–77%)14 are heritable. Until recently, the low prevalence of psychotic disorders precluded examination of the extent to which shared genetic factors were important (although see22). Using results from large genomewide association studies of schizophrenia, investigators have now found evidence for pleiotropic effects of schizophrenia loci on aspects of cannabis involvement.22–25 Others have interpreted this genetic commonality as evidence for causation,26,27 which is also consistent with the dopamine hypothesis of schizophrenia, since acute cannabis use releases dopamine,28 thus offering a biologically plausible pathway for cannabis use to lead to increased psychosis risk.
An alternative and frequently untested possibility is that both shared genetic influences and individual-specific factors of a causal nature might be implicated. One twin study reported a genetic correlation (rg=0.55) between cannabis use disorder (CUD) and PLEs and found that a model where CUD causally influenced PLEs fit better than the reverse14. However, the relative fits of the correlational and causal models were extremely close, precluding any definitive conclusion.
The current study examined the relationship between cannabis and PLEs in two large population-based twin/sibling samples, the Human Connectome Project (HCP) and a sample from the Australian Twin Registry (ATR3). First, we examined whether measures of cannabis use were associated with PLEs. Second, we estimated the extent to which additive genetic and individual-specific environmental factors contributed to their covariance. Next, we compared PLEs across twin/sibling pairs varying on cannabis exposure, including twins discordant for cannabis involvement. As twin and sibling pairs share at least 50% of their segregating loci identical-by-descent, any excess presence of PLEs in the cannabis-exposed twin relative to their unexposed co-twin may be viewed as evidence in favor of putatively causal individual-specific influences.
Methods
Participants
Participants were drawn from two sources: 1) HCP 1200 subjects release (N=1,206; collected between 2012–2015; mean age 29.33 [range 22–35]), and 2) ATR3 (N=3,856; collected between 2005–2009; mean age 30.91 [range 24–36], Supplemental text and eTable 1). Participants were excluded from the current analyses for missing relevant interview/questionnaire data (n=432; eTable 1 for details), resulting in a combined sample size of 4,674 individuals. For twin/sibling pair analyses, only individuals with a similarly-aged full sibling (≤2 years age difference) or twin with complete data were included, leaving 1733 pairs (758 MZ, 780 DZ, and 195 sibling pairs; note only same-sex sibling pairs were included in analyses of exposure effect; Supplemental text).
Measures
Psychotic-like Experiences (PLEs)
In the HCP sample, participants completed the Achenbach Adult Self Report (ASR).29 As in previous research,30 four questions were identified within the ASR as measuring PLEs. Although the ASR was not administered in the ATR3 sample, four questions mapping onto the ASR questions were assessed using items from a broad measure of personality (eTable 2 for items and prevalence). Endorsements for these four psychosis questions were summed to yield a PLE score (Supplemental text). 22.0% endorsed at least one PLE.
Cannabis Involvement
Cannabis involvement in both HCP and ATR3 was assessed using the Semi-Structured Assessment for the Genetics of Alcoholism (SSAGA).31 Specifically, we examined three variables: a) frequent cannabis use (1=cannabis use ≥100 times, 0=cannabis use <100 times in lifetime; 15.2% of sample); b) Cannabis Use Disorder, CUD (1=met criteria for DSM-IV abuse or dependence, 0=no CUD diagnosis; 14.3% of sample; see Supplemental Text for details), or c) current cannabis use [1=positive cannabis screen on either day of testing, 0=no positive cannabis screens (since the ATR3 sample did not conduct urine screens, current cannabis use was defined as past year cannabis use); 14.1% of sample].
Statistical Analysis
Analyses were performed using R version 3.4.3.32 Analyses were conducted individually for each of the three cannabis involvement variables using the combined HCP and ATR samples. Unless otherwise stated, the analyses used generalized linear mixed models (GLMMs; R package lme4)33, nesting individuals within families, and included the following covariates: sex (1=female, 0=male), age, MZ twin status (1=MZ twin, 0=not), DZ twin status (1=DZ twin, 0=not), sample (1=ATR, 0=HCP), total household income (Supplemental text), lifetime regular cigarette use (1=≥100 cigarettes, 0=<100 cigarette), lifetime regular alcohol use (1=average ≥2 drinks/day during heaviest period, 0=average <2 drinks/day), and lifetime non-cannabis illicit drug use (1=illicit drug use, 0=not); race/ethnicity was not included as a covariate due to lack of variability within the ATR3 sample; results in the HCP sample remained consistent when race/ethnicity was included (eTable 3)].
Estimation of Genetic and Environmental Correlation
The variance in and covariance between each cannabis involvement measure and PLEs was parsed into additive genetic (A), shared environmental (C) and individual-specific environmental (E) sources (Supplemental Text for details). Models were fitted to raw data using Full Information Maximum Likelihood estimation using the OpenMx34 and umx35 packages within R. These models also allowed for estimation of additive genetic (rg) and individual-specific environmental (re) correlations between PLEs and cannabis involvement.36 Akaike’s Information Criterion (AIC) was used to compare model fit.37
Twin/Sibling Pair Analyses of Exposure Effect
All possible same-sex twin/sibling pairs were drawn from the data (Npairs:2,022–2,041; Supplemental text). For each cannabis involvement measure, twin/sibling pairs were assigned to 4 groups: concordant unexposed pairs, concordant exposed pairs, exposed individuals from discordant pairs, and unexposed individuals from discordant pairs.38 Lifetime never users were included in the unexposed groups.
First, we used Helmert contrast coding to conduct sibling analyses by cannabis exposure, examining the relationship between cannabis involvement and PLEs using GLMMs, nesting individuals within twin/sibling pairs and nesting pairs within families.38 Three hypotheses were tested (eTable 4): 1) causal [i.e., cannabis involvement and PLEs are associated via person-specific, potentially causal factors; information regarding the onset of PLEs was not available for either dataset, precluding conclusions regarding the direction of causality, i.e., whether cannabis causes PLEs or vice versa], by testing whether cannabis exposed twins/siblings from discordant pairs differed in PLE scores from their unexposed co-twin/sibling; 2) predispositional (i.e., due to factors shared by members of twin/sibling pairs, including segregating loci), by testing whether the unexposed member of discordant pairs showed a similar liability to PLEs when compared to their exposed co-twin/sibling, and to individuals from concordantly exposed pairs; and 3) graded liability, a variation of the predispositional model (i.e., exposure does not lead to changes in PLEs within discordant pairs), testing whether unexposed individuals from discordant pairs exhibit increased liability to PLEs compared to unexposed members from concordant pairs. Importantly, these contrasts allowed examination of support for all three hypotheses as the likelihood of causation and correlated liabilities are not mutually exclusive. Post-hoc analyses examined cannabis exposure effects and whether each of the cannabis exposure groups showed significantly different PLEs, Bonferroni-corrected for multiple comparisons.
Second, we focused on the discordant pairs alone. We examined whether mean PLE scores were higher in the exposed twin/sibling relative to their genetically related co-twin/sibling, while accounting for covariates. Interaction terms between each cannabis exposure variable and zygosity (MZ vs. DZ; twin vs. non-twin sibling) were used to assess differences in the magnitude of the association between discordant MZ pairs and DZ/sibling pairs. An absence of a significant interaction term indicated equality of effect sizes in the MZ and DZ/sibling pairs. Significantly elevated PLE scores in MZ twins exposed to cannabis relative to their unexposed co-twin might be viewed as evidence in favor of putatively causal individual-specific environmental factors. MZ twins are fully matched for their segregating loci; therefore any excess association between cannabis and PLEs in these pairs cannot be related to segregating loci (or to those environmental factors that are shared by twins/siblings) and is therefore, attributed to person-specific influences, including causal processes.
Results
Associations between PLEs and Cannabis Use
Analyses were performed in 4,674 participants (mean age=30.51 years, 62.5% female). All three indices of cannabis involvement were associated with greater PLEs (Table 1). Those reporting frequent use, CUD and current use were 1.21–1.26 times more likely to report at least one PLE than their counterparts who used cannabis to a lesser extent or not at all. Associations persisted after including covariates, of which younger age, non-twin status, lower household income, lifetime regular smoking, and lifetime illicit drug use were also associated with greater PLEs. Interactions between cannabis exposure and sex were non-significant (βs≤.13, ps>.21; eTable 5). Interactions with sample were significant (frequent cannabis use: β=.03, p=.04; CUD: β=.04, p<.01; interaction with current cannabis use was non-significant, β=−.03, p=.06), with the HCP sample showing a weaker association with PLEs, but in the same direction (eTable 3). Age of onset of cannabis use was not related to PLEs (β=.013, p=.44).
Table 1.
Model Estimates for Associations Between Cannabis Involvement and PLEsa
| Frequent Cannabis Use | CUD | Current Cannabis Use | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| β | lower CI |
upper CI |
t | p | β | lower CI |
upper CI |
t | p | β | lower CI |
upper CI |
t | p | |
| Sexb | −0.02 | −0.05 | 0.01 | −1.08 | .28 | −0.01 | −0.04 | 0.01 | −0.91 | .36 | −0.02 | −0.05 | 0.01 | −1.32 | .19 |
| Age | −0.04 | −0.07 | −0.01 | −2.52 | .01 | −0.04 | −0.07 | −0.01 | −2.51 | .01 | −0.04 | −0.07 | −0.01 | −2.27 | .02 |
| MZ | −0.06 | −0.10 | −0.02 | −3.24 | .001 | −0.06 | −0.10 | −0.02 | −3.17 | .002 | −0.06 | −0.10 | −0.02 | −3.02 | .003 |
| DZ | −0.07 | −0.11 | −0.03 | −3.39 | .001 | −0.07 | −0.11 | −0.03 | −3.33 | .001 | −0.06 | −0.10 | −0.02 | −3.10 | .002 |
| Samplec | 0.03 | −0.01 | 0.07 | 1.66 | .10 | 0.02 | −0.02 | 0.06 | 1.11 | .27 | 0.02 | −0.01 | 0.06 | 1.23 | .22 |
| Household Income | −0.15 | −0.18 | −0.12 | −9.28 | <.001 | −0.15 | −0.18 | −0.12 | −9.56 | <.001 | −0.15 | −0.18 | −0.12 | −9.57 | <.001 |
| Lifetime Smoking | 0.03 | 0.00 | 0.06 | 1.88 | .06 | 0.03 | 0.00 | 0.06 | 1.97 | .049 | 0.05 | 0.02 | 0.08 | 3.06 | .002 |
| Lifetime Drinking | 0.01 | −0.02 | 0.04 | 0.89 | .37 | 0.01 | −0.02 | 0.04 | 0.79 | .43 | 0.02 | −0.01 | 0.05 | 1.17 | .24 |
| Lifetime Other Illicit Drug Use | 0.09 | 0.06 | 0.13 | 5.81 | <.001 | 0.09 | 0.06 | 0.12 | 5.60 | <.001 | 0.11 | 0.08 | 0.14 | 6.75 | <.001 |
| Cannabis Involvement Variable | 0.11 | 0.08 | 0.14 | 6.71 | <.001 | 0.13 | 0.09 | 0.16 | 7.89 | <.001 | 0.07 | 0.04 | 0.10 | 4.73 | <.001 |
Abbreviations: β=standardized regression coefficient; CI=95% bootstrapped (5000 iterations) Confidence Interval; t=t-test test statistic; p=p-value; CUD=cannabis abuse/dependence; MZ=monozygotic; DZ=dizygotic.
Significant model estimates are in bold.
Female as reference group for sex.
Australian Twin Registry as reference group for sample.
Estimation of Genetic and Environmental Correlation
Cannabis involvement (h2=0.69–0.77) and PLEs (h2=0.38) were heritable. The best fitting twin models did not include shared environmental influences (eTable 6). The observed association between frequent use, CUD, and current cannabis use measures and PLEs was generally attributable to genetic factors, and genetic correlations (rg) ranged from 0.41–0.56 (Table 2). These genetic factors accounted for 69.2–84.1% of the observed association with the remainder of covariance attributable to individual-specific environmental factors. Heritability remained significant although both heritability and the extent of rg were reduced (rg=0.26–0.46) when models were rerun adjusting for significant covariates (eTable 7). Genetic factors continued to contribute to much of the covariance, about 69.2–82.5%.
Table 2.
Estimation of Genetic and Environmental Correlation
| Phenotypic Correlation (rP) | Genetic Correlation (rg) | Environmental Correlation (re) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Trait | rP | %
rP Due to A |
%
rP Due to E |
rg |
lower CI |
upper CI |
re |
lower CI |
upper CI |
| Frequent Cannabis Use | 0.33 | 69.2% | 30.8% | 0.42 | 0.11 | 0.83 | 0.20 | 0.05 | 0.33 |
| CUD | 0.28 | 84.1% | 15.9% | 0.41 | 0.23 | 0.71 | 0.11 | −0.03 | 0.25 |
| Current Cannabis Use | 0.36 | 80.9% | 19.1% | 0.56 | 0.23 | 1.00 | 0.06 | −0.07 | 1.00 |
Abbreviations: % rP Due to A=proportion of the phenotypic correlation attributable to genetic factors; % rP Due to E= proportion of the phenotypic correlation attributable to environmental factors; CI=95% Confidence Interval; CUD=cannabis abuse/dependence.
Twin/Sibling Pair Analyses of Exposure Effect
PLEs were commonly reported by exposed members of discordant pairs (eTable 8 for Npairs) and by members of concordant exposed pairs, relative to unexposed members from discordant and concordant unexposed pairs, showing a robust main effect of exposure (βs=.08-.13, ps<.001; Figure 1 and Table 3). Using Helmert contrast coding, there was support for both the causal and graded liability contrasts (the predispositional contrast was also significant for the frequent cannabis and CUD variables, eTable 9).
Figure 1.
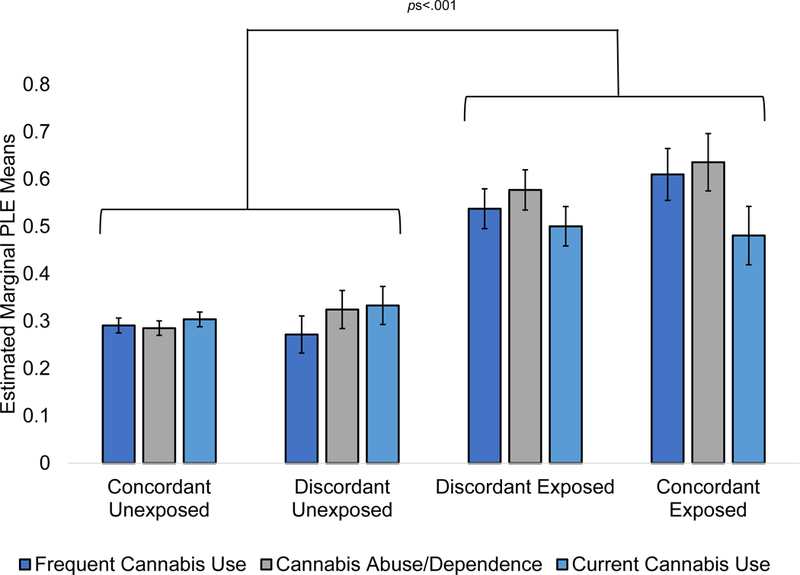
Estimated marginal means for psychotic-like experiences (PLEs) for cannabis exposure groups for each of the cannabis involvement measures (frequent cannabis use, cannabis abuse/dependence, and current cannabis use). Error bars reflect standard errors. Brackets indicate significant effects of cannabis exposure for each of the cannabis involvement measures.
Table 3.
Multiple Comparison Contrasts for Twin/Sibling Pair Analyses of Exposure Effecta
| Frequent Cannabis Use | CUD | Current Cannabis Use | ||||
|---|---|---|---|---|---|---|
| Z | p | Z | p | Z | p | |
| Exposed Discordant vs. Exposed Concordant | −1.15 | >.99 | −0.84 | >.99 | 0.28 | >.99 |
| Unexposed Concordant vs. Exposed Concordant | −5.52 | <.001 | −5.56 | <.001 | −2.78 | .03 |
| Unexposed Discordant vs. Exposed Concordant | −5.24 | <.001 | −4.43 | <.001 | −2.08 | .23 |
| Unexposed Concordant vs. Exposed Discordant | −5.44 | <.001 | −6.47 | <.001 | −4.47 | <.001 |
| Unexposed Discordant vs. Exposed Discordant | −5.41 | <.001 | −5.09 | <.001 | −3.41 | .004 |
| Unexposed Discordant vs. Unexposed Concordant | −0.46 | >.99 | 0.93 | >.99 | 0.70 | >.99 |
Abbreviations: Z=Z-test test statistic; p=p-value; CUD=cannabis abuse/dependence.
Significant model estimates are in bold. Multiple comparisons are Bonferroni-corrected.
Second, focusing on discordant pairs alone (eTable 10; with interactions: eTable 11), PLE endorsement was higher in exposed twin/siblings relative to their genetically related unexposed co-twin/sibling (βs=.23-.41, ps<.05) suggesting that even within twin/sibling pairs matched for 50 or 100% of their segregating loci and for familial environment, frequent or current use and CUD contributed to greater PLE endorsement. Interaction terms with zygosity were non-significant (MZ vs. DZ; twin vs. non-twin sibling; Zs≤−1.24, ps≥.22), indicating equality of effect sizes in MZ and DZ/sibling pairs. These significant associations in discordant MZ pairs provided evidence in favor of person-specific effects of a potentially causal nature.
Effect of Co-occurring Tobacco Smoking and Illicit Drug use.
All prior analyses accounted for lifetime history of regular tobacco smoking and illicit drug use. Use of other illicit drugs (22.3 – 43.5%) and regular tobacco smoking (27.3 – 41.5%) was not uncommon (eTable 12). Within discordant pairs (Supplemental Text), individuals who were cannabis exposed were more likely to report use of other illicit drugs and regular tobacco smoking than their cannabis unexposed relatives (eTable 13). The likelihood of PLEs was elevated in those reporting regular tobacco smoking (eTable 14) and use of other illicits > 11 times (eTable 15), over and above their cannabis exposure. However, there was also evidence that those reporting comorbid tobacco or illicit drugs were also likely to have significantly more CUD symptoms and somewhat more frequent use.
Discussion
Combining across U.S. and Australian datasets, we found cannabis involvement (i.e., frequent or current use and CUD) was associated with greater PLEs, even when including a variety of demographic variables and other substance use measures (e.g., lifetime tobacco, alcohol, other illicit drug use). While shared genetic influences were major contributors to their association, there was evidence for the role of person-specific influences (i.e., those over and above factors that twins and siblings are matched for) that might be of a causal nature on the relationship between cannabis involvement and PLEs.
Our study supports a growing body of literature outlining the extent of genetic overlap between cannabis involvement and psychotic disorders as well as PLEs.22–25,39 Our estimates of rg are also consistent with those from one prior twin study.14. It is too early to speculate the exact nature of the loci that might contribute to this genetic correlation as adequately powered GWAS of cannabis involvement are pending. Promising evidence arises from a recent GWAS of CUD in a Danish cohort that implicates a locus on chromosome 8,40 which is an expression quantitative trait locus for CHRNA2, and is also genomewide significant in the current largest schizophrenia GWAS.41 In contrast to one other study15, we did not find support for the role of shared environmental influences on either PLEs or cannabis involvement, or their covariance. Unlike the prior study which focused on cannabis use (ever trying cannabis) in adolescents, we focused on indices of more involved forms of use (e.g., CUD) in adults. Thus, our finding of no shared environment is consistent with the broader twin literature on the etiology of heavier cannabis use,20 including one prior study of CUD.14 Nonetheless, disentangling additive genetic from shared environmental influences requires very large sample sizes,42 and we cannot discount the role of shared environment, especially early life exposures (e.g., prenatal exposures, childhood adversity) as a contributor. Even so, as twin pairs are matched for these factors, our discordant pair analyses are likely unaffected by the extent of shared environmental overlap.
Importantly, even within pairs of individuals who share between 50% (DZ twins and non-twin siblings) and 100% (MZ twins) of their segregating loci and early environmental influences, cannabis involvement was related to greater PLE endorsement. While we cannot unequivocally ascribe this residual association to causal mechanisms (especially given the lack of data on PLE onset age), we can speculate that differences in PLE endorsement attributable to cannabis exposure, at least within related pairs (and as associations were of a similar magnitude in MZ pairs who share all their segregating loci, on average), can be viewed as evidence for causal processes. Potential causal pathways from cannabis involvement to PLEs may be related to dopaminergic dysfunction. Chronic drug use has been shown to modify the density and availability of dopamine D2/D3 receptors,43,44 although results for cannabis are mixed.45,46 Dopaminergic variants, including those in DRD2, have been implicated in schizophrenia,41 and dopamine receptor antagonists are generally effective in treating positive symptoms of schizophrenia, with the endocannabinoid system being involved in the modulation of dopamine neurotransmission.47,48 Therefore, increased sensitivity to the psychotomimetic effects of cannabis or further alteration of dopaminergic functioning upon significant cannabis use might lead to increased PLEs. On the other hand, purely environmental factors (e.g., early trauma) might also shape these potentially causal pathways.
Limitations
First, our data are cross-sectional and age at first PLE was not assessed. World Mental Health surveys indicate that PLEs have an average age of onset of 24–25 years.49 In our datasets, cannabis dependence age of onset was, on average, 19–21 years. Thus, cannabis use may precede PLEs, although we cannot rule out reverse causation (especially given other evidence for onset of PLEs in childhood and adolescence50,51). Unmeasured confounders (e.g., stressful life events) cannot be excluded. Second, while underpowered, our descriptive analyses suggest that there might be an independent effect of tobacco and other drug use on PLEs, over and above the association with cannabis severity. Third, we were unable to test for effects of variability in amount smoked or for varying strengths of delta-9-tetrahydrocannabinol (THC).11 While THC is associated with psychotic experiences,28 cannabidiol (CBD) may be associated with antipsychotic properties52. Fourth, our twin/sibling pair analyses treated never users of cannabis and less frequent/non-disordered users similarly, which does not adequately address whether associations extended beyond a simple effect of ever using cannabis. However, the association between using cannabis <11 times during the lifetime (versus never using) and PLEs was non-significant (p=0.75), suggesting that casual use may not be solely responsible for the observed association. Fifth, PLE measures were limited, although findings are generally consistent with extant literature.14,53,54 Sixth, even though we co-varied for sample, sample-specific differences cannot be discounted. For instance, HCP excluded participants for extended psychiatric hospitalization, which may have limited the severity of substance use and PLEs. Also, the definition of current cannabis use varied by sample (i.e., HCP=testing positive for THC on either day of testing; ATR3=past year cannabis use). However, results remained unchanged when current use was defined as past year use in both samples. Likewise, it is possible that we were underpowered to detect nuanced sex differences even though interactions with sex were not significant.
Conclusions
Psychosis is a major adverse health correlate of cannabis use.55,56 However, there is a lack of a consensus on the pathways underlying this robust association. While the association is primarily attributable to genetic overlap, the individual-specific component might serve as a target for intervention. If this person-specific pathway is causal, then policies that result in escalations in cannabis involvement should be further scrutinized. If they represent non-causal factors, such as severe early life stress, then such factors are critical to identify. Targeting cannabis use may be key in preventing exacerbation of PLEs amongst individuals at increased genetic liability to cannabis use and PLEs, should we be able to reliably identify those individuals in the future.
Supplementary Material
Key Points.
Question:
To what extent is the relationship between cannabis use and psychotic-like experiences (PLEs) attributable to shared genetic/predispositional and individual-specific factors?
Findings:
In a combined sample of 4,674 individuals, we found significant evidence for shared genetic factors between cannabis involvement and PLEs. Even after accounting for genetic overlap, individuals who were frequent users of cannabis were more likely to report PLEs when compared to their relatives who used cannabis less frequently.
Meaning:
Even though shared genetic influences are important, factors that are person-specific, and might be of a causal nature, impact the association between cannabis involvement and PLEs.
Acknowledgments
Funding/Support:
The Human Connectome Project research was funded by National Institute of Health Grant (1U54MH091657–01). The Australian Twin Registry research was funded by National Institute on Drug Abuse (NIDA) grant: DA18267 (ML) and facilitated through access to the Australian Twin Registry, a national resource supported by an Enabling Grant (ID 628911) from the National Health & Medical Research Council.
This work was also supported by National Institute on Health grants MH014677 (NRK), U01 DA041120 (DMB), K02DA032573 and R01DA023668 (AA), 2K05 AA017688 (ACH), T32-DA007313 and T32-GM081739 (CHD), T32-GM008151 (DAAB); National Science Foundation grant DGE-1745038 (DAAB; non-overlapping with NIH support).
Role of the Funder/Sponsor: The funding source had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Conflict of Interest Disclosures: Dr. Barch consults for Pfizer. No other authors report disclosures.
References
- 1.Andreasson S, Allebeck P, Engstrom A, Rydberg U. Cannabis and schizophrenia. A longitudinal study of Swedish conscripts. Lancet (London, England). 1987;2(8574):1483–1486. [DOI] [PubMed] [Google Scholar]
- 2.Casadio P, Fernandes C, Murray RM, Di Forti M. Cannabis use in young people: the risk for schizophrenia. Neuroscience and biobehavioral reviews. 2011;35(8):1779–1787. [DOI] [PubMed] [Google Scholar]
- 3.Henquet C, Di Forti M, Morrison P, Kuepper R, Murray RM. Gene-environment interplay between cannabis and psychosis. Schizophr Bull. 2008;34(6):1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moore TH, Zammit S, Lingford-Hughes A, et al. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet (London, England). 2007;370(9584):319–328. [DOI] [PubMed] [Google Scholar]
- 5.Murray RM, Di Forti M. Cannabis and Psychosis: What Degree of Proof Do We Require? Biol Psychiatry. 2016;79(7):514–515. [DOI] [PubMed] [Google Scholar]
- 6.Semple DM, McIntosh AM, Lawrie SM. Cannabis as a risk factor for psychosis: systematic review. Journal of psychopharmacology (Oxford, England). 2005;19(2):187–194. [DOI] [PubMed] [Google Scholar]
- 7.van Winkel R, Kuepper R. Epidemiological, neurobiological, and genetic clues to the mechanisms linking cannabis use to risk for nonaffective psychosis. Annual review of clinical psychology. 2014;10:767–791. [DOI] [PubMed] [Google Scholar]
- 8.Di Forti M, Marconi A, Carra E, et al. Proportion of patients in south London with first-episode psychosis attributable to use of high potency cannabis: a case-control study. The lancet Psychiatry. 2015;2(3):233–238. [DOI] [PubMed] [Google Scholar]
- 9.Hickman M, Vickerman P, Macleod J, Kirkbride J, Jones PB. Cannabis and schizophrenia: model projections of the impact of the rise in cannabis use on historical and future trends in schizophrenia in England and Wales. Addiction (Abingdon, England). 2007;102(4):597–606. [DOI] [PubMed] [Google Scholar]
- 10.Gage SH, Hickman M, Zammit S. Association Between Cannabis and Psychosis: Epidemiologic Evidence. Biol Psychiatry. 2016;79(7):549–556. [DOI] [PubMed] [Google Scholar]
- 11.Gage SH, Zammit S, Hickman M. Stronger evidence is needed before accepting that cannabis plays an important role in the aetiology of schizophrenia in the population. F1000 medicine reports. 2013;5:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duman B, Sedes N, Baskak B. Additive Effects of Former Methylenedioxymethamphetamine and Cannabis Use on Subclinical Psychotic Symptoms. Noro psikiyatri arsivi. 2017;54(1):38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griffith-Lendering MF, Wigman JT, Prince van Leeuwen A, et al. Cannabis use and vulnerability for psychosis in early adolescence--a TRAILS study. Addiction (Abingdon, England). 2013;108(4):733–740. [DOI] [PubMed] [Google Scholar]
- 14.Nesvag R, Reichborn-Kjennerud T, Gillespie NA, et al. Genetic and Environmental Contributions to the Association Between Cannabis Use and Psychotic-Like Experiences in Young Adult Twins. Schizophr Bull. 2017;43(3):644–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shakoor S, Zavos HM, McGuire P, Cardno AG, Freeman D, Ronald A. Psychotic experiences are linked to cannabis use in adolescents in the community because of common underlying environmental risk factors. Psychiatry research. 2015;227(2–3):144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Gastel WA, Wigman JT, Monshouwer K, et al. Cannabis use and subclinical positive psychotic experiences in early adolescence: findings from a Dutch survey. Addiction (Abingdon, England). 2012;107(2):381–387. [DOI] [PubMed] [Google Scholar]
- 17.McGrath JJ, Saha S, Al-Hamzawi A, et al. The Bidirectional Associations Between Psychotic Experiences and DSM-IV Mental Disorders. Am J Psychiatry. 2016;173(10):997–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Navarro-Mateu F, Alonso J, Lim CCW, et al. The association between psychotic experiences and disability: results from the WHO World Mental Health Surveys. Acta psychiatrica Scandinavica. 2017;136(1):74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Os J, Reininghaus U. Psychosis as a transdiagnostic and extended phenotype in the general population. World Psychiatry. 2016;15(2):118–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verweij KJ, Zietsch BP, Lynskey MT, et al. Genetic and environmental influences on cannabis use initiation and problematic use: a meta-analysis of twin studies. Addiction (Abingdon, England). 2010;105(3):417–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hilker R, Helenius D, Fagerlund B, et al. Heritability of Schizophrenia and Schizophrenia Spectrum Based on the Nationwide Danish Twin Register. Biol Psychiatry. 2018;83(6):492–498. [DOI] [PubMed] [Google Scholar]
- 22.Carey CE, Agrawal A, Bucholz KK, et al. Associations between Polygenic Risk for Psychiatric Disorders and Substance Involvement. Frontiers in genetics. 2016;7:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Power RA, Verweij KJ, Zuhair M, et al. Genetic predisposition to schizophrenia associated with increased use of cannabis. Mol Psychiatry. 2014;19(11):1201–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartz SM, Horton AC, Oehlert M, et al. Association Between Substance Use Disorder and Polygenic Liability to Schizophrenia. Biol Psychiatry. 2017;82(10):709–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reginsson GW, Ingason A, Euesden J, et al. Polygenic risk scores for schizophrenia and bipolar disorder associate with addiction. Addiction biology. 2018;23(1):485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gage SH, Jones HJ, Burgess S, et al. Assessing causality in associations between cannabis use and schizophrenia risk: a two-sample Mendelian randomization study. Psychol Med. 2017;47(5):971–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vaucher J, Keating BJ, Lasserre AM, et al. Cannabis use and risk of schizophrenia: a Mendelian randomization study. Mol Psychiatry. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murray RM, Englund A, Abi-Dargham A, et al. Cannabis-associated psychosis: Neural substrate and clinical impact. Neuropharmacology. 2017;124:89–104. [DOI] [PubMed] [Google Scholar]
- 29.Achenbach TM. The Achenbach System of Emprically Based Assessment (ASEBA): Development, Findings, Theory and Applications. Burlington, VT: University of Vermong Research Center for Children, Youth, and Families; 2009. [Google Scholar]
- 30.Sheffield JM, Kandala S, Burgess GC, Harms MP, Barch DM. Cingulo-opercular network efficiency mediates the association between psychotic-like experiences and cognitive ability in the general population. Biol Psychiatry Cogn Neurosci Neuroimaging. 2016;1(6):498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bucholz KK, Cadoret R, Cloninger CR, et al. A new, semi-structured psychiatric interview for use in genetic linkage studies: a report on the reliability of the SSAGA. Journal of studies on alcohol. 1994;55(2):149–158. [DOI] [PubMed] [Google Scholar]
- 32.Team RC. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: 2016. In:2017. [Google Scholar]
- 33.Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. arXiv preprint arXiv:14065823. 2014. [Google Scholar]
- 34.Neale MC, Hunter MD, Pritikin JN, et al. OpenMx 2.0: Extended structural equation and statistical modeling. Psychometrika. 2016;81(2):535–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bates TC, Maes HH, Neale MC. umx: Twin and Path-based Structural Equation Modeling in R. PeerJ PrePrints. 2017. [DOI] [PubMed] [Google Scholar]
- 36.Neale M, Cardon LR. Methodology for genetic studies of twins and families. Vol 67: Springer Science & Business Media; 2013. [Google Scholar]
- 37.Akaike H Factor analysis and AIC In: Selected Papers of Hirotugu Akaike. Springer; 1987:371–386. [Google Scholar]
- 38.Pagliaccio D, Barch DM, Bogdan R, et al. Shared Predisposition in the Association Between Cannabis Use and Subcortical Brain Structure. JAMA Psychiatry. 2015;72(10):994–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pasman JA, Verweij KJH, Gerring Z, et al. Genome-wide association analysis of lifetime cannabis use (N=184,765) identifies new risk loci, genetic overlap with mental health, and a causal influence of schizophrenia on cannabis use. bioRxiv. 2018. [Google Scholar]
- 40.Demontis D, Rajagopal VM, Thorgeirsson T, et al. Genome-wide association study implicates CHRNA2 in cannabis use disorder. bioRxiv. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Consortium SWGotPG. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511(7510):421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Visscher PM, Gordon S, Neale MC. Power of the Classical Twin Design Revisited: II Detection of Common Environmental Variance. Twin research and human genetics : the official journal of the International Society for Twin Studies. 2008;11(1):48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Volkow ND, Fowler JS, Wang GJ, Baler R, Telang F. Imaging dopamine’s role in drug abuse and addiction. Neuropharmacology. 2009;56 Suppl 1:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Volkow ND, Wang GJ, Fowler JS, Tomasi D, Telang F. Addiction: beyond dopamine reward circuitry. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(37):15037–15042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Albrecht DS, Skosnik PD, Vollmer JM, et al. Striatal D(2)/D(3) receptor availability is inversely correlated with cannabis consumption in chronic marijuana users. Drug and alcohol dependence. 2013;128(1–2):52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ginovart N, Tournier BB, Moulin-Sallanon M, Steimer T, Ibanez V, Millet P. Chronic Delta(9)-tetrahydrocannabinol exposure induces a sensitization of dopamine D(2)/(3) receptors in the mesoaccumbens and nigrostriatal systems. Neuropsychopharmacology. 2012;37(11):2355–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Covey DP, Mateo Y, Sulzer D, Cheer JF, Lovinger DM. Endocannabinoid modulation of dopamine neurotransmission. Neuropharmacology. 2017;124:52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mateo Y, Johnson KA, Covey DP, et al. Endocannabinoid Actions on Cortical Terminals Orchestrate Local Modulation of Dopamine Release in the Nucleus Accumbens. Neuron. 2017;96(5):1112–1126.e1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McGrath JJ, Saha S, Al-Hamzawi AO, et al. Age of Onset and Lifetime Projected Risk of Psychotic Experiences: Cross-National Data From the World Mental Health Survey. Schizophrenia Bulletin. 2016;42(4):933–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kelleher I, Connor D, Clarke MC, Devlin N, Harley M, Cannon M. Prevalence of psychotic symptoms in childhood and adolescence: a systematic review and meta-analysis of population-based studies. Psychol Med. 2012;42(9):1857–1863. [DOI] [PubMed] [Google Scholar]
- 51.Laurens KR, Hodgins S, Taylor E, Murray R. Is earlier intervention for schizophrenia possible? Identifying antecedents of schizophrenia in children aged 9–12 years. Schizophrenia: The final frontier. 2011:19–32. [Google Scholar]
- 52.Zuardi AW, Crippa JA, Hallak JE, et al. A critical review of the antipsychotic effects of cannabidiol: 30 years of a translational investigation. Curr Pharm Des. 2012;18(32):5131–5140. [DOI] [PubMed] [Google Scholar]
- 53.Bechtold J, Hipwell A, Lewis DA, Loeber R, Pardini D. Concurrent and Sustained Cumulative Effects of Adolescent Marijuana Use on Subclinical Psychotic Symptoms. Am J Psychiatry. 2016;173(8):781–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jones HJ, Gage SH, Heron J, et al. Association of Combined Patterns of Tobacco and Cannabis Use in Adolescence With Psychotic Experiences. JAMA Psychiatry. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Volkow ND, Compton WM, Weiss SR. Adverse health effects of marijuana use. The New England journal of medicine. 2014;371(9):879. [DOI] [PubMed] [Google Scholar]
- 56.Volkow ND, Swanson JM, Evins AE, et al. Effects of Cannabis Use on Human Behavior, Including Cognition, Motivation, and Psychosis: A Review. JAMA Psychiatry. 2016;73(3):292–297. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.