

Summary
Classical mechanisms through which brain-derived molecules influence behavior include neuronal synaptic communication and neuroendocrine signaling. Here we provide evidence for an alternative neural communication mechanism that is relevant for food intake control involving cerebroventricular volume transmission of the neuropeptide melanin-concentrating hormone (MCH). Results reveal that the cerebral ventricles receive input from approximately one third of MCH-producing neurons. Moreover, MCH cerebrospinal fluid (CSF) levels increase prior to nocturnal feeding and following chemogenetic activation of MCH-producing neurons. Utilizing a dual viral vector approach, additional results reveal that selective activation of putative CSF-projecting MCH neurons increases food intake. In contrast, food intake was reduced following immunosequestration of MCH endogenously present in CSF, indicating that neuropeptide transmission through the cerebral ventricles is a physiologically relevant signaling pathway for energy balance control. Collectively these results suggest that neural-CSF volume transmission signaling may be a common neurobiological mechanism for the control of fundamental behaviors.
Keywords: volume transmission; MCH; melanin-concentrating hormone; orexin,; cerebrospinal fluid; CSF; obesity; feeding
Introduction
The brain regulates physiology and behavior, in part, through neuronal release of neuropeptides, neurotransmitters, and other small molecules. These signals act primarily through “wired” synaptic communication between neurons, between non-neuronal target cells and neurons, and through neuroendocrine signaling via the vasculature. A complementary neural communication pathway examined in the present study involves release and dispersal of brain-derived molecules through the cerebral spinal fluid (CSF) to reach distal targets, a neuroendocrine-like humoral mechanism termed “volume transmission” or “bulk flow” communication (Agnati et al., 1986). While brain-derived biological systems have previously been postulated to utilize a CSF volume transmission pathway to regulate fundamental behaviors such as stress, energy balance, and reproduction (Caraty and Skinner, 2008; Hoistad et al., 2005; Hsu et al., 2015a; MacMillan et al., 1998; Skinner and Malpaux, 1999; Veening et al., 2010), the physiological relevance of neuronal communication through CSF transmission has not yet been systemically investigated.
The present study examines whether melanin-concentrating hormone (MCH), a neuropeptide produced primarily by neurons in the lateral hypothalamic area (LHA) and zona incerta (ZI) that increases appetite and food intake (Barson et al., 2013; Bittencourt et al., 1992; Qu et al., 1996; Shimada et al., 1998), exerts its stimulatory effects on feeding via release into CSF and volume transmission through the brain. Through the melanin-concentrating hormone receptor 1 receptor (MCH 1R), MCH both increases food intake and reduces energy expenditure, promoting overall elevated weight gain in rodents (Chambers et al., 1999; Kowalski et al., 2004; Lembo et al., 1999; Shearman et al., 2003). The importance of this neuropeptide system to overall energy balance is highlighted by findings showing that rodents lacking MCH (Shimada et al., 1998) or MCH 1R (Marsh et al., 2002) are lean, whereas transgenic MCH overexpression produces hyperphagia and obesity (Ludwig et al., 2001). While the central distribution of MCH 1R is extensive and spans throughout the brain neuraxis (Chee et al., 2013; Lembo et al., 1999), the sites of action and neurobiological mechanisms through which MCH stimulates feeding are poorly understood.
We hypothesized that MCH regulates feeding, in part, by neural-CSF volume transmission, and tested this hypothesis utilizing multiple levels of analysis in the rat.Collectively, our findings identify a population of MCH neurons that communicate with the CSF and we further reveal that MCH present in the CSF is functionally relevant to feeding behavior. These results implicate humoral volume transmission of brain-derived molecules through the cerebral ventricles as a physiological signaling pathway relevant to the control of fundamental behaviors.
Results
The cerebral ventricles receive direct input from MCH neurons.
We conducted a series of neuroanatomical investigations to establish a basis for possible CSF transmission of MCH. Tri-label immunofluorescence was performed for MCH, vimentin (a marker of cerebral ventricle-lining ependymal cells), and synaptophysin (a marker of synaptic vesicles). Confocal microscopic analysis revealed the presence of synaptophysin-immunoreactive (ir) elements colocalized with MCH-ir axon terminals in close apposition to vimentin-ir ependymal cells lining the cerebral aqueduct and the lateral, third, and fourth ventricles (Figure 1). These results support the possibility of release of MCH into the CSF from MCH-producing neurons.
Figure 1: MCH-producing neurons terminate in the ependymal layer lining the cerebral ventricles.
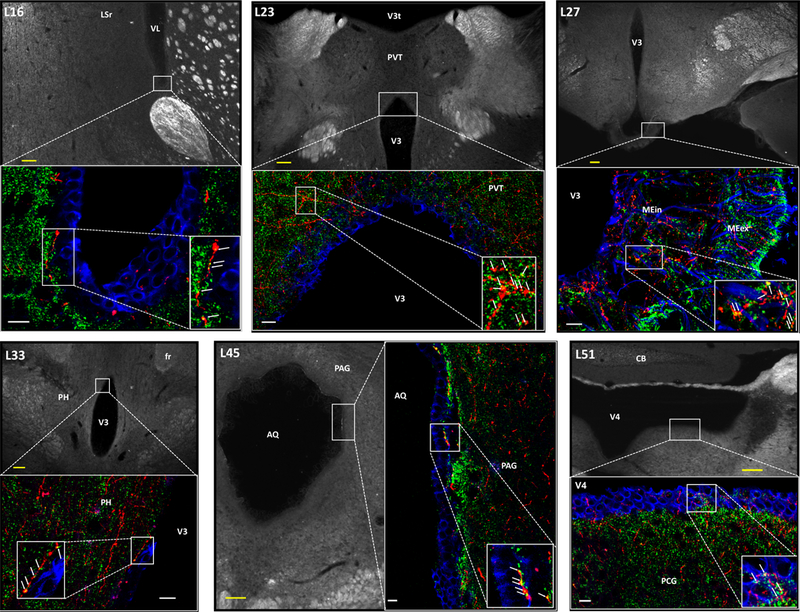
Immunofluorescence co-labeling of MCH and synaptophysin at the ventricular ependymal layer supports the possibility of cerebroventricular volume transmission. Red = MCH axons, green = synaptophysin (synaptic vesicle marker), blue = vimentin (ventricular ependymal cell marker). Arrows indicate MCH+ synaptophysin colabeling. Numbers in the upper left in each image preceded by “L” indicate correspondence to closest atlas levels of the rat brain atlas of Swanson (Swanson, 2004). aco = anterior commissure; AQ = cerebral aqueduct; CB = cerebellum; fr = fasciculus retroflexus; LSr = lateral septal nucleus, rostral part; MEex = median eminence; MEin = median eminence, internal lamina; PAG = periaqueductal gray; PCG = pontine central gray; PH = posterior hypothalamic nucleus; PVH, paraventricular hypothalamic nucleus; PVT = paraventricular thalamic nucleus; V3 = third ventricle; V3t = third ventricle, thalamic part; V4 = fourth ventricle; VL = lateral ventricle. Scale bars: white = 10 μm, yellow = 100 μm.
To determine the extent and distribution of MCH-expressing neurons involved in putative MCH-CSF-signaling, a complimentary set of experiments combined immunofluorescence detection of MCH with retrograde neural pathway tracing. Intracerebroventricular injections of cholera toxin B (CTB; a retrograde tracer that enters cells from axon terminals) combined with immunofluorescent labeling for MCH-expressing cell bodies revealed colocalization of CTB and MCH-ir labeling in approximately one third (32.7%) of MCH-ir somata within the lateral hypothalamic area (LHA) and zona incerta (ZI); this represented 43.8 % of CTB-labeled somata in the LHA and ZI. Furthermore, high spatial resolution analysis revealed a wide range of regional differences in the levels of colocalization (Figure 2A, B). These data, combined with the tri-label immunofluorescence data, establish a structural basis for putative neural-CSF MCH transmission, and suggest that it is highly organized and extensive.
Figure 2: Approximately one-third of all MCH neurons are CSF-projecting and these neurons are distinct from neuroendocrine neurons.
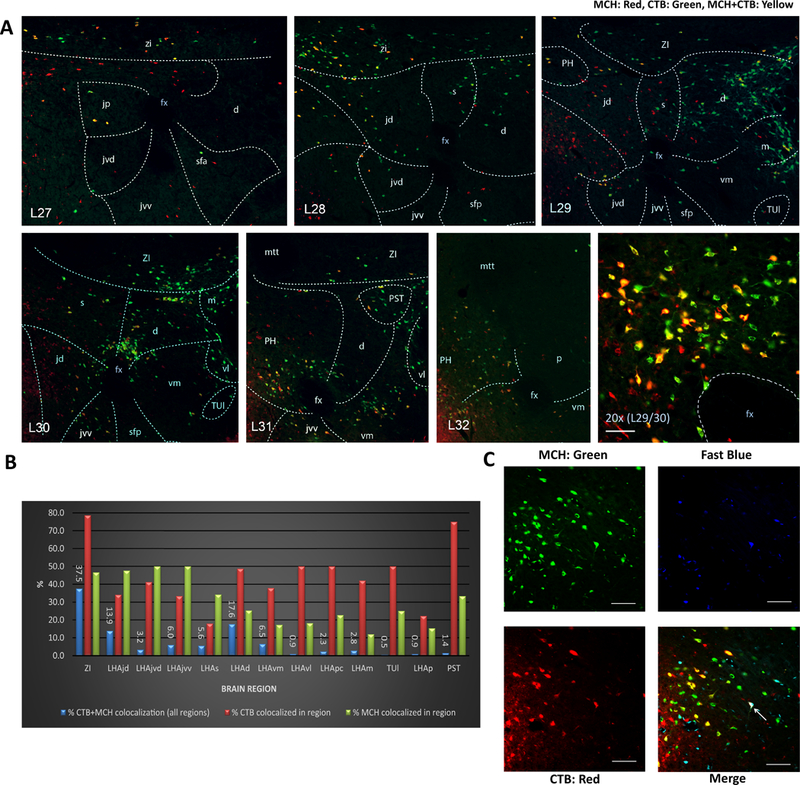
(A) Distribution of MCH immunolabeled and CTB retrogradely labeled neurons following lateral ventricular CTB injections; representative immunofluorescence for MCH (green), CTB (red), and colabeled cells (yellow). Numbers in the lower left in each image preceded by “L” indicate correspondence to closest atlas levels of the rat brain atlas of Swanson (Swanson, 2004). (B) Overall 32.7% of MCH-ir somata within the LHA and ZI were also CTB-ir, whereas 43.8 % of CTB-ir somata in the LHA and ZI were MCH-ir. Percentages of colabeling for the ZI and for specific LHA subregions are represented as follows: blue bars indicate the percentage of total (across all brain regions) colocalized MCH-ir + CTB-ir located in each region; red bars indicate the percentage CTB-ir neurons that are colocalized with MCH-ir in each region; green bars indicate the percentage MCH-ir neurons that are colocalized with CTB-ir in each region. (C) Representative images of immunofluorescence for MCH (green), CTB (red) and Fast Blue (FB, blue) in the lateral hypothalamic area. Intravenously injected FB retrogradely labelled neurons with access to the vasculature (putative neuroendocrine neurons). Overall 98.8% of MCH + CTB analyzed were not labeled with FB, with the arrow pointing to an extremely rare instance of a triple labeled cell (MCH + CTB + FB). Abbreviations: d = LHA dorsal region; fx = fornix; jd = LHA juxtadorsomedial region; jp = lateral hypothalamic area (LHA) juxtaparaventricular region; jvd = LHA juxtaventromedial region, dorsal zone; jvv = LHA juxtaventromedial region, ventral zone; m = LHA magnocellular nucleus; mtt = mammillothalamic tract; p = LHA posterior region; pc = LHA parvicellular region; PH = posterior hypothalamic nucleus; PST = preparasubthalamic nucleus; s = LHA suprafornical region; sfa = LHA subfornical region, anterior zone; sfp = LHA subfornical region, posterior zone; TUl = tuberal nucleus, lateral part; vl = LHA ventral region, lateral zone; vm = LHA medial zone, ventral region; ZI = zona incerta. Scale bar = 50 μm.
Based on the presence of MCH-ir terminals in the median eminence (Figure 1), a hypothalamic region where neuroendocrine cells interface with the vasculature, we sought to determine the extent to which CSF-signaling MCH neurons might also be neuroendocrine cells. To investigate this, we combined intravenous injections of fast blue (FB; a retrograde pathway tracer) with intracerebroventricular (ICV) injections of CTB and immunofluorescence detection of MCH. Established neuroendocrine regions contained FB, including the paraventricular nucleus of the hypothalamus (PVH) (Figure S1) and supraoptic nucleus (not shown). Analyses of the colocalization of CTB with FB in MCH neurons indicated that only very few MCH neurons are neuroendocrine (0.4%), and similarly very few CTB-containing MCH neurons (i.e., putative CSF-contacting MCH neurons) also project to the vasculature (~1.2%) (Figure 2C). Additionally, PVH neuroendocrine neurons do not express MCH and only rarely collateralize to CSF (indicated by rare occurrences of Fast Blue and CTB or MCH colabeling) (Figure S1). Taken together the data indicate that a substantial subset of MCH neurons project to CSF, almost all of which do not communicate via neuroendocrine pathways.
To determine whether other feeding-relevant neuropeptide systems are anatomically conducive to CSF-mediated ventricular transmission, comparable neuroanatomical analyses were conducted for the neuropeptide hypocretin/orexin (H/O). H/O was selected as a viable candidate because H/O-expressing neurons are intermingled with MCH neurons in the LHA, yet form a distinct population (Hahn, 2010; Swanson et al., 2005). Moreover, like MCH, H/O is implicated in control of feeding and energy balance and also (unlike MCH) promotes arousal, activity, and energy expenditure (for review see (Barson et al., 2013; Kotz et al., 2006)). Analyses revealed abundant H/O and CTB colabeling in the LHA (Figure 3A), accounting for 42% of LHA CTB-ir neurons retrogradely labeled following ICV CTB injections. Additional tri-label immunofluorescence and confocal microscopic analysis for H/O, synaptophysin and vimentin, indicated the presence of H/O terminals in close apposition to ependymal cells lining the lateral, third, and fourth cerebral ventricles (Figure 3B). These findings indicate that like MCH, H/O neurons contact CSF extensively and throughout the neuraxis.
Figure 3: Approximately 40% of hypocretin /orexin-producing neurons project to CSF.
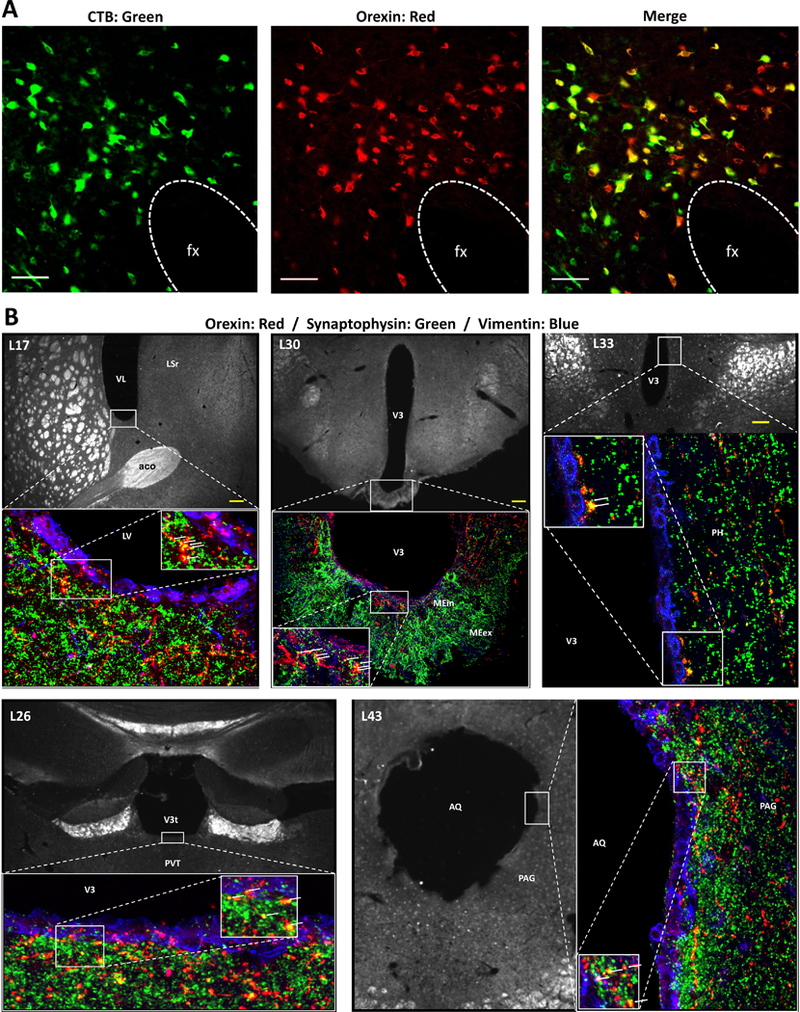
(A) Representative images of cholera toxin subunit B (CTB, red), hypocretin/orexin (H/O, green) and colabeled neurons (yellow) in the LHA. The presence of CTB retrograde labeling in H/O-ir neurons after CTB injection into the lateral ventricle indicates that some H/O neurons project to the CSF. Abbreviations: fx = fornix. Scale bars = 50 μm. (B) Immunofluorescence colabeling of H/O and synaptophysin at the cerebroventricular ependyma supports the possibility of volume transmission into the CSF. Synaptophysin (a synaptic vesicle marker) and H/O colabeling was present at the site of ependymal cells immunolabeled for vimentin (a marker of ependymal cells) at multiple locations within the cerebral ventricles. Red = H/O axons, green = synaptophysin, blue = vimentin. Arrows indicate H/O + synaptophysin colabeling. Numbers upper left in each image preceded by “L” indicate correspondence to closest atlas levels of the rat brain atlas of Swanson (Swanson, 2004). Abbreviations: aco =anterior commissure; AQ = cerebral aqueduct; LSr, lateral septal nucleus, rostral part; VL = lateral ventricle; ME = median eminence; PAG =periaqueductal gray; PH = posterior hypothalamic nucleus; PVH = hypothalamic paraventricular nucleus; PVT = paraventricular thalamic nucleus; V3, third ventricle. Scale bars = 100 μm.
Since a portion of MCH neurons are known to co-express the inhibitory neurotransmitter GABA (Harthoorn et al., 2005), we further characterized the CSF-contacting MCH neurons to reveal whether this population of cells is neurochemically distinct from other MCH neurons with regards to GAD 67-ir (a GABAergic marker). Analyses indicated that 45% of CTB labeled MCH-ir neurons were GAD-67-ir, compared to 23.5% of MCH-ir neurons that do not contact CSF (Figure 4A). These data suggest that CSF-signaling MCH neurons are more likely to be GABAergic compared to MCH neurons that do not communicate to the CSF. Furthermore, using a combined approach of fluorescence in situ hybridization with immunohistochemical staining we detected no measurable mRNA expression levels of the glutamatergic marker vGLUT2 in CTB positive MCH-ir neurons (Figure 4B), nor were CTB positive MCH-ir neurons colocalized with vGLUT1 or vGLUT3 mRNA expressing neurons (not shown). These findings suggest that CSF-signaling MCH neurons are likely not glutamatergic. Given that a large % of CSF-contacting MCH neurons are GABAergic, and that ventricular injections of both the GABA-A (Kokare et al., 2006) and GABA-B (Ebenezer, 1990) receptor agonists increase food intake, we reasoned it possible that MCH and GABA may act synergistically in the CSF to promote feeding. However, this is unlikely as we found that neither the GABA-A receptor agonist muscimol (Figure 4C,D) nor the GABA-B receptor agonist baclofen (Figure 4E) potentiated a feeding response when injected ICV in combination with a sub-effective dose of MCH. Thus, while none of the CSF-contacting MCH neurons are glutamatergic and almost half are GABAergic, GABA co-release does not appear to be critical for MCH feeding effects.
Figure 4: While 45% of CSF-projecting MCH neurons co-express GABAergic markers, GABA signaling is unlikely to interact with MCH-driven hyperphagia.
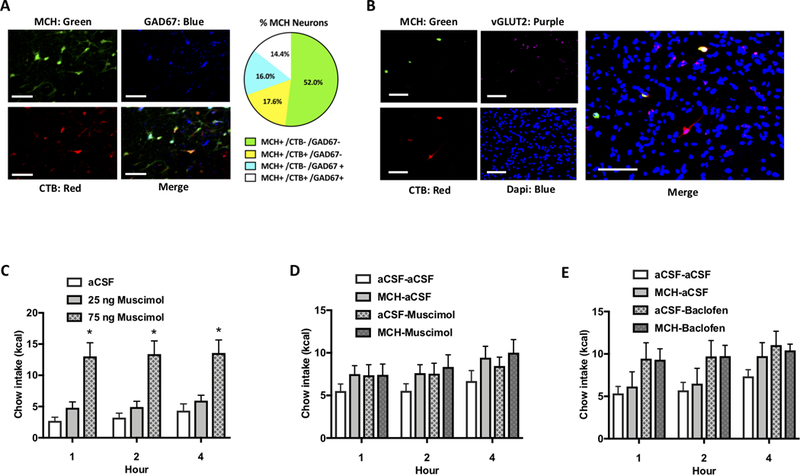
(A) CTB was injected into the lateral ventricle and immunofluorescent staining was performed for MCH (green), CTB (red) and the GABAergic marker GAD67 (blue). Quantification indicated that 14.4% of MCH neurons were both GABAergic (GAD67 immunolabeled) and CSF-signaling (CTB retrogradely labeled following CTB injection into the lateral ventricle), whereas 17.6 % of CTB positive MCH neurons did not contain the GABAergic marker. Thus 45% of putative CSF-signaling MCH neurons were GABAergic, compared to 23.5% of MCH neurons that are putative non-CSF-signaling. Scale bar: 50 μm. (B) CTB was injected into the lateral ventricle and immunofluorescent staining was performed for MCH (red) and CTB (green), followed by fluorescence in situ hybridization for the glutamatergic marker vGLUT2 (purple). Quantification indicated that there was no colocalization of vGLUT2 in CTB positive MCH-ir neurons (Scale bar: 100 μm), nor was there colocalization of vGLUT1 or vGLUT3 (data not shown). (C) A dose response for the effects of ICV injections of the GABA-A receptor agonist muscimol indicated a significant effect of drug where 75 ng significantly elevated food intake while 25 ng was ineffective (n=12). (D) ICV injection of a sub-effective dose of muscimol (20 ng) did not potentiate the feeding effects of a sub-effective dose of MCH (0.5 μg)(n=12). (E) ICV injection of a sub-effective dose of Baclofen (0.1 nmol) did not potentiate the feeding effects of a sub-effective dose of MCH (0.5 μg)(n=19) (*P<.05).
CSF MCH levels are increased following MCH neuron activation and prior to feeding
To determine if MCH levels in CSF are elevated following selective MCH neuron activation, rats were bilaterally injected in the LHA and ZI with an adeno-associated virus (AAV) containing an excitatory designer receptor exclusively activated by designer drugs (DREADDs) driven by an MCH-specific promoter, and containing the mCherry reporter (AAV2-rMCHp-hM3D(Gq)-mCherry). Immunofluorescence colocalization of MCH-ir and mCherry confirmed selective infection of MCH neurons with the DREADDs, as no mCherry-ir cell bodies were observed that were not also MCH-ir (Figure 5A). To confirm that hM3D(Gq) receptors are physiologically functional in MCH neurons, slice electrophysiological recordings from LHA mCherry positive DREADDs receptor-containing MCH neurons show that application of the DREADDs receptor ligand, Clozapine-N-Oxide (CNO), increases the firing rate of infected cells (Figure 5B-D). To confirm that hM3D(Gq) receptors are functional in MCH neurons at the behavioral level, ICV injections of CNO in rats that were bilaterally infected with the AAV2-rMCHp-hM3D(Gq)-mCherry significantly elevated food intake, an effect that was blocked by pretreatment with ICV injections of the MCH 1R antagonist H6408 (Figure 5E). While this experiment was performed during the dark cycle when animals normally eat, we also observed that animals infected with MCH DREADDs had elevated food intake when CNO injections were given prior to feeding during the light cycle (data not shown). Together these data show that hM3D(Gq) receptors are functional in MCH neurons and that chemogenetic activation of an indiscriminate population of MCH-producing neurons increases feeding via an MCH 1R-mediated mechanism. Moreover, these results support those from the previous experiment in suggesting that GABA co-release is not functionally relevant for MCH feeding effects, as the hyperphagic effects of chemogenetic MCH neuron activation were completely blocked with ICV co-delivery of an MCH 1R antagonist.
Figure 5: Viral transduction of Designer Receptors Exclusively Activated by Designer Drugs (DREADDs; hM3Gq) is a valid method of inducing selective and functionally-relevant activation of MCH neurons.
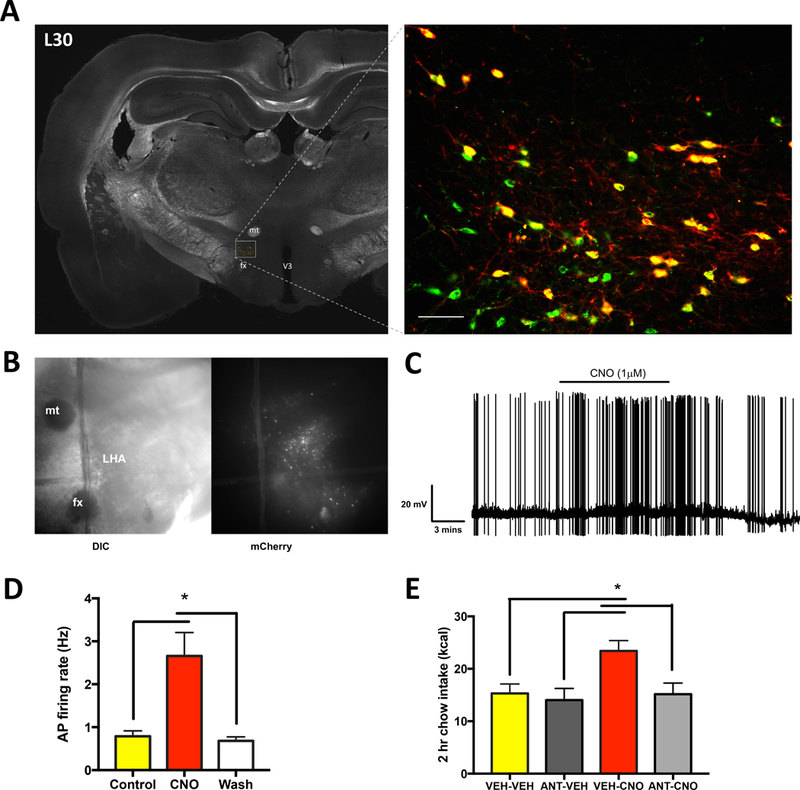
(A) Representative images of the mCherry reporter gene present in MCH neurons, indicating successful infection of the DREADDS AAV selective in MCH neurons. MCH: Green; mCherry: Red; Colocalization: Yellow. Scale bar: 50 μm. (B) Representative differential interference contrast (DIC) and fluorescence microscopy images of hypothalamic slice preparations. (C) Representative trace of electrophysiological recordings from mCherry positive DREADDs-expressing MCH neurons before, during, and after clozapine-N-oxide (CNO; the DREADDs ligand) application. (D) Rats were bilaterally injected in the lateral hypothalamic area and zona incerta with an AAV driving hM3Gq DREADDS expression under the control of a promoter for MCH (AAV2-rMCHp-hM3D(Gq)-mCherry). Data from electrophysiological recordings showing that CNO significantly increased the firing rate of mCherry positive DREADDs-expressing MCH neurons (n=8). (E) CNO (18 mmoles) was used to pharmacologically activate MCH neurons, which resulted in increased feeding that was blocked by pretreatment with the MCH 1R antagonist H6408 (ANT; 10μg)(n=7,8/group) (*P<.05). All data are represented as means ± SEM. mt, mammillothalamic tract; fx, fornix; V3, third ventricle.
We next reasoned that if MCH is transmitted through CSF to increase feeding behavior, then activation of MCH neurons should elevate MCH levels in CSF (Figure 6A). Following in vivo extraction of CSF from the cisterna magna of the rat (~200–250 μl/rat, approximating the entire CSF pool from an adult rat) (Figure 6B), MCH protein quantification detected via enzyme immunosorbent assay (EIA) revealed the presence of MCH in CSF under physiological conditions. Moreover, following chemogenetic activation of the MCH neurons (by ICV CNO injections first delivered at 120 minutes and a second injection 15 minutes prior to extracting CSF) and subsequent processing of the CSF as described above, animals had significantly higher CSF MCH levels compared to vehicle treatment. These results show that selective activation of MCH neurons increases MCH release into the CSF and thereby corroborate neuroanatomical data indicating that MCH neurons release MCH into the CSF (Figure 6C).
Figure 6: MCH levels in the CSF are increased by DREADDs-mediated activation of MCH neurons and prior to nocturnal feeding.
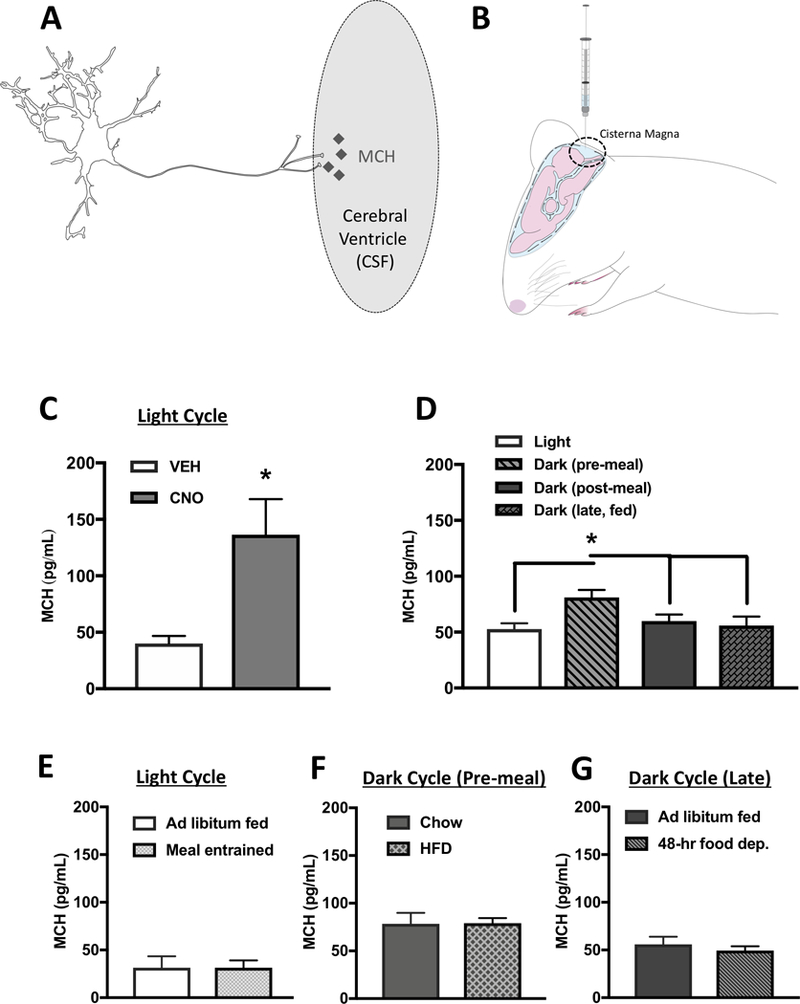
(A) A hypothetical model whereby MCH is transmitted into the CSF through axon terminals of ventricular-contacting terminals from MCH neurons. (B) Cartoon demonstrating the method of CSF extraction from the cisterna magna of an anesthetized rat. (C) MCH levels were elevated in CSF following MCH DREADDs activation (n=6,7). (D) Under physiological conditions, MCH levels in CSF are elevated during the early dark cycle prior to food consumption compared to during the light cycle and dark cycle postprandially (n=6–8). (E) There were no differences in CSF MCH levels prior to light cycle feeding in meal entrained animals compared to ad libitum fed controls (n=7/group). (F) Five days of exposure to a palatable high-fat diet had no effect on pre-prandial CSF MCH levels during the early dark cycle compared with chow-fed animals (n=6/group). (G) 48 hours of food deprivation had no effect on CSF MCH levels during the late dark cycle compared with ad libitum chow-fed controls. (n=5–6/group) (*P<.05). Data are means ± SEM.
To determine whether the concentrations of CSF MCH achieved after chemogenetic MCH neuron activation were comparable to those achieved following ICV pharmacological injection of a feeding-relevant dose of MCH, we first determined that 1μg is the lowest effective dose of ICV MCH to significantly increase food intake (Figure S2A). Next, we found that ICV injections of this dose of MCH raised MCH CSF levels to similar levels as MCH DREADDs activation (166.4 +/− 24.13 and 136.5 +/− 31.45 pg/mL, respectively) (Figure S2B). These findings indicate that compared to what would be predicted mathematically, pharmacologically effective doses of ICV MCH raise CSF MCH levels by relatively small incremental amounts. This outcome is not surprising, and is likely due to interactions with unidentified neurophysiological mechanisms that prolong MCH peptide integrity and/or enhance MCH ligand-receptor binding affinity under physiological conditions.
We next investigated whether MCH CSF levels are physiologically altered by feeding-relevant parameters. CSF MCH levels were measured during the inactive phase when rodents are not normally eating (light cycle), during the early dark cycle just prior to consumption of the 1st meal after light offset (typically one of the largest meals in a rodent’s feeding cycle (Johnson et al., 1986) (dark pre), during the dark cycle following ingestion of a standardized meal of 5 g of chow (dark post), and during the late dark cycle in ad libitum fed animals that were food restricted for only 1hr prior to CSF extraction (dark, late, fed). Results reveal that CSF MCH levels were elevated preprandially during the dark cycle compared to the postprandial state (dark post), late in the dark cycle (dark, late, fed), and compared to the inactive cycle (light cycle). These findings support the hypothesis that endogenous CSF MCH signaling plays a role in stimulating feeding at the beginning of the active dark cycle, when rodents normally consume their biggest meal (Figure 6D).
Several experiments were then conducted to determine whether MCH CSF levels are dynamically regulated by meal entrainment, maintenance diet, and/or acute food restriction. Relative to levels during the light cycle in ad libitum fed rats, MCH CSF levels were not different at the onset of a conditioned food access period in rats that were previously meal entrained to consume all of their daily chow within a 4-hr period during the light cycle (Figure 6E). This suggests that CSF MCH levels are not easily entrainable by scheduled light cycle feeding (food intake and body weight data depicted in Figure S3A-C). Moreover, consumption of a palatable high-fat diet (HFD) for two weeks did not elevate pre-prandial levels of CSF MCH during the dark cycle compared to animals fed ad-libitum chow (Figure 6F), despite the elevated caloric intake and weight gain of animals fed a HFD (Figure S3 D,E). Further, we did not observe elevations in MCH levels late during the dark cycle in ad-libitum fed animals compared with animals that were 48-hr food deprived (Figure 6G), despite significantly reduced body weights at the time of CSF extraction (Figure S3F). Taken together these data reveal that CSF MCH levels are not dynamically regulated by meal entrainment, maintenance diet, or acute food deprivation. Rather, the data suggest that MCH CSF release is under circadian control with a putative function of promoting the initial feeding bout during the nocturnal feeding cycle. This is consistent with previous literature revealing that MCH mRNA expression peaks at the beginning of the dark cycle (Bluet-Pajot et al., 1995).
Activation of CSF-contacting MCH neurons increases food intake
To directly examine the contribution of the CSF-signaling MCH neurons to feeding behavior, we utilized a dual virus approach to selectively activate the population of MCH neurons that project to CSF. The retrograde-transported canine adenovirus type 2 containing the genetic information for cre recombinase (CAV2-CRE) (Boender et al., 2014) was injected into the lateral ventricle and a cre-dependent MCH-DREADDs AAV type 2 (AAV2-DIO-MCH DREADDs-hM3D(Gq)-mCherry was injected into the LHA and ZI (Figure 7A). This dual virus approach targets the expression of the hM3D(Gq) DREADDs receptor only to the subset of MCH neurons that have axon terminals contacting the CSF. The selectivity of DREADDs expression in CSF-contacting MCH neurons was corroborated by colocalization of mCherry, MCH, and CTB following ICV injections of CTB (Figure 7B). Quantification of mCherry positive cells revealed that this dual virus approach infected ~8% of the number of cells infected using the non-cre-dependent dreads virus. Subsequent ICV CNO injections in these animals revealed that selective activation of CSF-projecting MCH neurons significantly elevated food intake (Figure 7C), indicating that chemogenetic-mediated activation of CSF-signaling MCH neurons is sufficient to produce a hyperphagic response.
Figure 7: Selective activation of ventricular-projecting MCH neurons increases feeding, whereas feeding is reduced by immunosequestration of MCH endogenously present in the CSF.
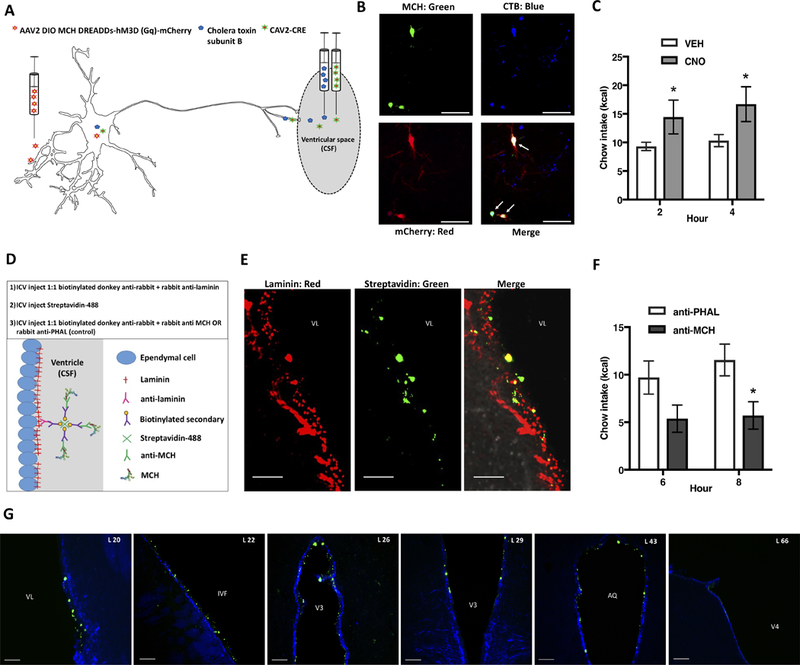
(A) To selectively activate ventricular projecting MCH neurons, we utilized a dual virus strategy 1) the retrograde canine adenovirus type 2 containing cre recombinase (CAV2-CRE) was injected into the lateral ventricle, and 2) a cre-dependent MCH-DREADDs AAV serotype 2 (AAV2-DIO-MCH DREADDs-hM3D(Gq)-mCherry was injected into the LHA and ZI. (B) Epifluorescence image showing mCherry labeling (red) colocalized with MCH immunolabeling (green), and cells retrogradely labeled by the retrograde tracer cholera toxin subunit B (CTB; blue), following lateral ventricle CTB injection. (C) Activation of ventricular projecting MCH neurons by lateral ventricular injections of CNO increases food intake (n=7). (D) A cartoon depicting the strategy and steps of an immunosequesteration approach designed to reduce the bioavailability of endogenous CSF MCH. (E) A representative image showing immunofluorescence labeling of laminin (red), streptavidin-Alexa Fluor 488 (green) and their colocalization (yellow) at the ventricular ependymal layer (indicating that anti-MCH immunoglobulins were localized with laminin-ir at the ventricular ependyma). (F) Results from the immuno-sequestration experiment indicated that under normal physiological conditions a reduction in the bioavailability of CSF MCH significantly reduces food intake (n=8,9). (G) Representative immunofluorescence images showing the rostral-caudal spread of streptavidin-Alexa Fluor 488 (green) localized at the ventricular ependyma (shown with vimentin immunofluorescence in blue), indicating extensive rostral caudal CSF MCH immunosequestration. The numbers in the upper left in each image preceded by “L” indicate correspondence to closest atlas levels of the rat brain atlas of Swanson (Swanson, 2004) (*P<.05). Data are means ± SEM. Scale bars in B, E = 50 μm, scale bars in F = 100μm. Abbreviations: AL = atlas level; AQ = cerebral aqueduct; IVF = Intraventricular foramen; V3 = third ventricle; V4 = fourth ventricle; VL = lateral ventricle.
From these data, we cannot exclude the possibility that the elevated food intake response was based, in part, on activation of collateral synaptic projections from MCH neurons that make contact with CSF. One region where parenchymal pharmacological injections of MCH have been shown to elevate feeding is the nucleus accumbens shell (ACBsh) (Georgescu et al., 2005). To determine whether MCH neurons that project to the CSF have collateral projections to the ACBsh, we first injected the retrograde tracer Fluoro-Gold into the ACBsh and the retrograde tracer CTB into the lateral ventricle and performed immunofluorescent labeling for MCH. Results indicated that an extremely small percentage (~0.08%) of MCH neurons that project to the CSF have collateral projections to the ACBsh (Figure S4 A,B).
Next, we examined whether DREADDs-mediated activation of ACBsh-projecting MCH neurons increases food intake. The retrograde-transported CAV2-CRE was injected into bilaterally into the ACBsh and a cre-dependent MCH-DREADDs AAV type 2 (AAV2-DIO-MCH DREADDs-hM3D(Gq)-mCherry was injected into the LHA and ZI. Quantification of 1 of 5 series from each injected animal revealed that an average of ~9% of cells infected with the non-cre-dependent DREADDs were infected using the dual virus approach with CAV2-CRE targeting the ACBsh, which is comparable to the percentage infected using intraventricular injections of CAV2-CRE (~8%). More detailed anatomical analyses revealed differences in the location of cell populations containing the mCherry reporter, with the highest expression of ACBsh-projecting MCH DREADDs transduced neurons in the zona incerta (atlas level 28) and the highest expression of ICV-projecting MCH DREADDs transduced neurons in the LHAd at atlas level 30 (Based on the Swanson Atlas (Swanson, 2004) [Figure S5]). Our behavioral results show that the same dose of ICV CNO that was effective for increasing food intake when DREADDs were targeted to CSF-contacting MCH neurons (Figure 7C; 18mmoles) had no effect on food intake when DREADDS were targeted to ACB-projecting MCH neurons (Figure S4C). Importantly, that ICV CNO injections did not affect food intake when DREADDs were targeted to ACBsh-projecting MCH neurons indicates that the hyperphagic effects of ICV CNO injections in rats with DREADDs targeted to CSF-projecting MCH neurons are not based on back-transformation of CNO into the non-inert molecule clozapine (and nonspecific clozapine binding), an outcome that has been demonstrated following peripheral injections of a high dose (5mg/kg) of CNO injections in rats (MacLaren et al., 2016). Collectively these results indicate that the elevated food intake following activation of CSF-contacting MCH neurons is not based on collateral projections to the ACBsh, nor is it based on nonspecific clozapine binding.
Blockade of MCH endogenously present in the CSF reduces food intake
In order to determine the endogenous relevance of MCH that is present CSF under physiological conditions to feeding behavior, we first injected animals with a neutralizing antibody against MCH or a non-endogenous molecule (control group; using primary antibodies directed against the lectin Phaseolus vulgaris leucoagglutinin (PHAL), and raised in the same species as the MCH primary) targeting the lateral ventricle. Injections were given just prior to the onset of the dark cycle, when animals normally consume their biggest meal. Using this approach, we found that food intake was significantly reduced in animals who received the MCH antibody injections compared with those in the control group (Figure S6A).
Post-mortem tissue analyses using fluorescent secondary antibody-based immunofluorescence staining revealed that some of the antibody likely diffused into the neuropil (Figure S6B, “neutralizing antibody approach”), which means that the reduction in food intake could potentially be based on neutralization of MCH present in the neuropil surrounding the ventricles in addition to the CSF. Thus, we developed a 3-step “immunosequestration” approach to selectively reduce the bioavailability of MCH present in the CSF while minimizing neuropil diffusion of antibody-protein complexes. The approach is as follows: [1] A primary antibody directed against the extracellular matrix protein laminin (expressed in ependymal cells lining the cerebral ventricles) was mixed with a biotinylated secondary antibody directed against the species of the primary antibody; this complex was then injected into the lateral ventricle. This first step was designed to anchor biotin molecules to laminin-expressing ependymal cells lining the cerebral ventricles. [2] Fluorescently-labeled streptavidin, which contains 4 binding sites for biotin, was injected into the lateral ventricle. This second step was designed to exploit the high binding affinity of biotin and avidin, resulting in fluorescently-labeled avidin molecules anchored to the cerebral ventricle walls. [3] Injections were given into the lateral ventricle of another primary/biotinylated-secondary antibody complex, with the primary antibody directed against either MCH (experimental group) or a non-endogenous molecule (PHAL; control group). The aim of this third step was to enable the binding of conjugated biotin in the complex to free streptavidin binding sites on the ependymal cells lining the ventricles (Figure 7D). This approach was designed to sequester endogenous MCH in the CSF to the ependymal ventricle/neuropil junction, reducing its bioavailability and preventing transport of MCH in the CSF into the neuropil. In contrast with the neutralizing antibody approach, we did not observe a haze surrounding the ventricular region at the injection site following our post-mortem immunofluorescence analyses, indicating it less likely that antibodies diffused into the neuropil using our immunosequesteration approach (Figure S6B). The fluorescent streptavidin was visualized along the ventricular lining, indicating successful anchoring of the complex to the walls of the ventricle and not into the neuropil (Figure 7E). Injections were given just prior to the beginning of the active phase, when animals normally eat and when endogenous CSF MCH levels are elevated (e.g., Figure 6D). Food intake was significantly reduced in animals subjected to CSF MCH immunosequestration compared with those who received the PHAL control complex (Figure 7F). Rosteral/caudal anatomical analyses through the ventricular system revealed that streptavidin was present throughout the forebrain and midbrain but was minimally present in the caudal hindbrain 30-min after the 3rd ICV injection (Figure 7G). Similar anatomical distribution was observed 8 hours post injection (data not shown). These data show that the ICV immunosequestration approach is effective for rapid and neuroanatomically extensive sequestration of antibody-protein complexes along the ventricular ependymal layer, and importantly, corroborate the hypothesis that CSF transmission of MCH is physiologically relevant for feeding behavior.
Discussion
Conceptually, “volume transmission” of brain-derived molecules (i.e., the transmission of signaling molecules through non-synaptic modalities) was explicitly proposed in 1986 by Agnati and Fuxe (Agnati et al., 1986) as a complement to “wiring transmission” (i.e., the synaptic transmission between neurons, and between neurons and non-neuronal target cells that are in direct contact). In contrast to synaptic transmission, where communication is discrete and dependent on neuroanatomical connections, CSF transmission of neuropeptides and small molecule neurotransmitters represents a neuroendocrine-like method of humoral transmission with the potential for widespread neuromodulation of distal regions of the brain (Veening and Barendregt, 2010). Indeed recent findings show that unidentified neuromodulators are present in human CSF in sufficient quantities to excite neurons, thereby supporting the plausibility of CSF transmission as a mechanism for system-wide modulation of neuronal activity (Bjorefeldt et al., 2015). However, the physiological contribution of CSF transmission to regulating behavior has remained hypothetical and largely understudied, possibly due in part to the technical challenge of separating the effects CSF-mediated signaling from concurrent synaptic transmission. Here we apply multiple levels of analysis to confirm the feasibility of endogenous CSF transmission of MCH, and further confirm that this pathway is a major contributor to the control of feeding behavior.
Prior findings revealed that MCH gene expression peaks during the early stages of the dark cycle, supporting a potential role for circadian fluctuations in driving MCH expression and subsequent peptide release (Bluet-Pajot et al., 1995). Consistent with this, we observed that endogenous CSF MCH levels are highest preprandially at the onset of the dark cycle compared to [1] postprandially following a fixed meal in the early nocturnal phase, [2] late in the dark cycle after ad libitum feeding, and [3] during the inactive light cycle. These findings suggest a framework in which a circadian mechanism stimulates an increase in CSF MCH release at the beginning of the feeding/nocturnal cycle with the putative goal of driving the first feeding bout of the active phase. Consistent with this model, our additional data suggest that endogenous CSF MCH signaling may not be driving food intake due to various other factors, including acute food restriction, palatable obesogenic diet maintenance, and light cycle meal entrainment.
Our data raise several important questions. Foremost, what are the sites of action through which CSF transmission of MCH (and other neuromodulators) elevates feeding? One consideration to take into account is the flow of CSF, which is rostral to caudal through the ventricular system and then proceeds through the cisternal subarachnoid space (Reiber, 2003). We observed immunoreactive terminals for MCH throughout the rostral-caudal extent of the ventricular system, suggesting that CSF MCH has widespread distribution throughout the neuraxis - a possibility that is consistent with the widespread central distribution of the MCH 1R (Lembo et al., 1999).
Our data also raise the question of in what form is MCH is released into the CSF. The CSF contains endopeptidases such as endopeptidase 24.11 and aminopeptidase M that are known to degrade MCH (Maulon-Feraille et al., 2002; Muraki et al., 1996). It is possible that MCH is released in CSF in a form that protects it against rapid proteolytic degradation. For example, the pro-hormone peptide form of MCH (NEI-MCH) has been shown to be resistant to proteolytic degradation (Maulon-Feraille et al., 2002). This is one possible mechanism to explain why a 1μg dose of exogenous MCH was required to elevate CSF MCH levels by only ~80 pg/mL (Figure S2). Alternatively, it is possible that MCH is released in the CSF in extracellular vesicles (exosomes). Indeed, exosomes that transport peptides have been identified in human CSF (Street et al., 2012). Future studies are necessary to identify the form in which MCH is transmitted in the CSF, as well as the downstream neural sites of action.
The extent to which MCH translocates from the CSF to the neuropil remains to be determined. Intercellular junctions between ependymal cells regulate the degree to which CSF contacts the extracellular fluid of the neuropil, and region-specific differences in these junctions exists (Vigh et al., 2004). One possibility is that MCH translocates into the neuropil through tanycytes, which are specialized ependymal cells lining the base of the lateral and third cerebral ventricles. For example, radiolabeled nerve growth factor injected into the CSF has been shown to translocate from the CSF through tanycytes and accumulate in the locus coeruleus (Feng et al., 2011). Moreover, the anorectic adipokine, leptin, is trafficked from the median eminence to mediobasal hypothalamic neurons via tanycyte-mediated transport into CSF (Balland et al., 2014). An additional possibility is that CSF MCH is able to alter feeding via activation of MCH 1R on cilia. Two types of cilia exist (primary and motile); interestingly, both types of cilia contain MCH 1R (Conductier et al., 2013; Hamamoto et al., 2016) and optogenetic-mediated activation of MCH neurons alters the tonic control of the ependymal motile cilia beat frequency and the CSF flow rate in mice (Conductier et al., 2013). It is unknown to what extent this function of MCH on motile cilia contributes to the feeding effects of CSF-transmitted MCH observed in the present study.
It is possible that CSF volume transmission through the cerebral ventricles is a commonly used means of biological communication for other brain-derived neuropeptides and small molecule neurotransmitters. We investigated this possibility at the neuroanatomical level for the neuropeptide hypocretin/orexin (H/O), that like MCH is produced in the lateral hypothalamic area in an intermingled yet distinct population of neurons (Hahn, 2010). As was observed for MCH, we identified H/O immunoreactive terminals at the site of ependymal cells lining multiple cerebral ventricular locations. Furthermore, ~40% of H/O neurons were labeled following injection of the retrograde pathway tracer, cholera toxin subunit B (CTB), into the lateral ventricle, suggesting that both H/O- and MCH-producing neurons are transmitted through the CSF. Common to MCH and H/O systems is the engagement of G protein coupled receptors to modulate core functions such as stress, energy balance, reproduction, glucose homeostasis, and sleep (Barson et al., 2013). It would be interesting for future studies to determine the relevance of CSF transmission of both H/O and MCH to each of these behaviors. A putative role for CSF H/O and MCH signaling in mediating sleep pathologies is suggested by findings that low levels of orexin A (hypocretin 1) were reported in the CSF of narcoleptic patients (Ripley et al., 2001) and CSF MCH levels were altered by paradoxical sleep deprivation in rodents (Dias Abdo Agamme et al., 2015). Future studies are needed to determine whether CSF MCH extends to the control of sleep and other non-feeding behaviors such as novelty detection, which was recently linked with MCH neuronal activity (Gonzalez et al., 2016). In addition to MCH and H/O, several different neuropeptide systems have been postulated to utilize a CSF volume transmission pathway, such as oxytocin (Veening et al., 2010), beta-endorphin (Hoistad et al., 2005; MacMillan et al., 1998), melatonin (Skinner and Malpaux, 1999), gonadotropin-releasing hormone (Caraty and Skinner, 2008), glucagon-like pepotide-1 (GLP-1) (Hsu et al., 2015a), and others. For example, intrastriatal injections of beta-endorphin can be detected in cerebrospinal fluid, suggesting that beta-endorphin may migrate out of the neuropil and be transmitted in CSF (Hoistad et al., 2005). Corroborating the likelihood of extrasynaptic beta-endorphin transmission (either via CSF or through interstitial volume transmission) is the presence of beta endorphin immunoreactivity in the cerebral cortex (a region lacking beta endorphin immunoreactive terminals) 60–90 minutes after stimulation of hypothalamic neurons (MacMillan et al., 1998). A similar receptor-terminal mismatch scenario was previously reported by our group for GLP-1 where we hypothesized that GLP-1 produced in hindbrain neurons reaches the hippocampus through CSF transmission (Hsu et al., 2015a). Future work is needed to determine whether these and other neuropeptide/ neurotransmitter systems utilize CSF transmission to mediate behavior and behavioral state changes. Such examination will fundamentally advance understanding of how various brain-derived molecules communicate to regulate physiology and behavior.
The possibility that behavioral and physiological state changes can be evoked by neuropeptidergic transmission in the CSF presents new avenues for clinical investigation. Lumbar spinal tap CSF sampling is commonly used to determine biomarkers for neurological disease such as Alzheimer’s disease and related pathologies (Sheline et al., 2014), a practice based on the predominant zeitgeist of thought that the CSF is primarily a medium for flushing metabolic waste and other solutes from the brain’s cellular tissue. However, the notion that the CSF is an active biological signaling medium opens up new possibilities for clinical insight derived from human CSF sampling. For example, CSF neuropeptide profiling may represent a biological thumbprint for susceptibility to future pathologies as opposed to simply indicating the presence of existing pathologies. Additionally, novel therapies may be developed that target neural-CSF signaling pathways for the prevention or treatment of conditions such as metabolic syndrome, narcolepsy, and disorders of reproduction.
Limitations of the study
While we observed a significant reduction in feeding using an antibody-based immunosequestration approach designed to reduce the bioavailability of MCH endogenously present in the CSF, a limitation of the study is that we were unable to accurately quantify the magnitude of the reduction of bioavailable MCH in CSF due to methodological limitations inherent in using an antibody based approach. Additionally, despite the use of immunohistochemistry amplification of fluorescent reporters, we cannot be certain regarding the percentage of cells that project to a given region (CSF or ACB) that were successfully targeted/transduced, and thus our quantification likely underestimated the total number of transduced cells that project to the respective regions. These limitations should be taken into account when interpreting our results.
Supplementary Material
STAR Methods
KEY RESOURCES TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Scott Kanoski (kanoski@usc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Male Sprague Dawley rats (Envigo, Indianapolis, IN, USA) weighing 300–400g and between ages of postnatal day 80–90 were individually housed in wire-hanging cages in a climate controlled (22–24°C) environment with a 12 hr: 12hr light/dark cycle. Animals were maintained on a diet of ad libitum access to standard rat chow (LabDiet 5001, LabDiet, St. Louis, MO), except where noted. Experiments were performed in accordance with NIH Guidelines for the Care and Use of Laboratory Animals, and all procedures were approved by the local committee of the Institute of Animal Care and Use (University of Southern California).
METHOD DETAILS
Immunofluorescence
Rats were anesthetized and sedated with a ketamine (90mg/kg)/xylazine (2.8 mg/kg)/acepromazine (0.72 mg/kg) cocktail, then transcardially perfused with 0.9% sterile saline (pH 7.4) followed by 4% paraformaldehyde in 0.1M borate buffer (pH 9.5). Brain dissection, tissue handling, and general immunofluorescence labeling procedures were performed as previously reported (Hsu et al., 2015b). The following antibody dilutions were used: rabbit anti-MCH (1:1000; Phoenix Pharmaceuticals, Burlingame, CA, USA), mouse anti-synaptophysin (1:1000; Progen, Heidelberg, Germany), rabbit anti-RFP (1:2000, Rockland Inc., Limerick, PA, USA), chicken anti-vimentin (1:10,000, Abcam, Cambridge, MA, USA), rabbit anti-orexin A (1:5ooo; Phoenix Pharmaceuticals, Burlingame, CA, USA), mouse anti-cholera toxin subunit B (1:500; Abcam, Cambridge, MA, USA), rabbit anti-laminin (1:10,000 Lifespan Biosciences, Seattle, WA, USA). Antibodies were prepared in 0.02 M KPBS solution containing 0.2% BSA and 0.3% Triton X-100 at 4°C overnight. After thorough washing with 0.02M KPBS, sections were incubated with secondary antibody. All secondary antibodies were obtained from Jackson Immunoresearch and used at 1:500 dilution at 4°C, with overnight incubations (Jackson Immunoresearch; West Grove, PA, USA). Sections were mounted and coverslipped using 50% glycerol in 0.02 M KPBS and the edges were sealed with clear nail polish. Photomicrographs were acquired using either a Nikon 80i (Nikon DS-QI1,1280X1024 resolution, 1.45 megapixel) under epifluorescence illumination, or as optical slices using a Zeiss LSM 700 UGRB Confocal System (controlled by Zeiss Zen software).
Fluorescence in situ hybridization
Rats were anesthetized and sedated with a ketamine (90mg/kg)/xylazine (2.8 mg/kg)/acepromazine (0.72 mg/kg) cocktail, then transcardially perfused with 0.9% sterile saline (pH 7.4) followed by 4% paraformaldehyde in 0.1M borate buffer (pH 9.5). Brains were coronally sectioned to 20 μm on a sliding microtome into 0.02 M KPBS solution. After immunofluorescence staining (see above), sections were mounted onto subbed slides. Slides were placed into a vacuum chamber overnight (~16 hours) to desiccate.
Sections were postfixed in 4% paraformaldehyde in 0.1M borate buffer (pH 7.4) for 1.75 hours followed by 5 washes in KPBS for 5 minutes each. Sections were then pretreated by incubating them at 37 °C in pretreatment buffer (100mM Tris buffer and 50mM EDTA in distilled deionized water, pH8) with 0.001% Proteinase K (Sigma P2308) for 30 minutes, followed by a 3-min wash in incubation buffer alone and a 3-min rinse in 100 mM Triethanolamine in water (pH 8). Sections were then incubated with 0.25% acetic anhydride in 100 mM triethanolamine for 10 minutes at room temperature followed by 2 × 2 min washes in saline-sodium citrate buffer (1% citric acid trisodium/2% sodium chloride in water (pH 7.0)). Slides were dehydrated in increasing concentrations of ethanol solution (50%, 70%, 95%, 100%, 100%) for 3 minutes each and air-dried prior to hybridization.
For hybridization, a hydrophobic barrier was drawn around each section and 3–4 drops of either vGlut1 (317001), vGlut2 (317011), and vGlut3 (476711) probe (Advanced Cell Diagnostics) was placed on each tissue section. Slides were incubated with the probe at 40 °C for 3 hours in a HybEz oven (Advanced Cell Diagnostics). Following a 2 -min wash with wash buffer (RNAscope®, Advanced Cell Diagnostics 320058) reagents from RNAscope® Fluorescent Multiplex Detection Reagent Kit (Advanced Cell Diagnostics, 320851) were applied in order to amplify the probe signal, with AMP1 applied for 45 minutes, AMP2 for 30 minutes, AMP 3 for 45 minutes, and AMP 4 for 30 minutes. Incubation steps occurred at 40 °C, and a 2-min wash followed each amplification step. Slides were coverslipped with ProLong® Gold Antifade mounting medium (Cell Signaling, 9071S). Photomicrographs were acquired using a Nikon 80i (Nikon DS-QI1, 1280×1024 resolution, 1.45 megapixel) under epifluorescent illumination, or as optical slices using a Zeiss LSM 700 UGRB Confocal System (controlled by Zeiss Zen software).
Cholera toxin subunit B (CTB) injections
Rats were anesthetized and sedated with a ketamine (90mg/kg)/xylazine (2.8 mg/kg)/acepromazine (0.72 mg/kg) cocktail, and 1 μl injections of 0.5% CTB-Alexa Fluor 594 (ThermoFisher Scientific, Waltham, MA, USA) dissolved in 0.1M phosphate buffered saline, pH 7.4 were delivered stereotaxically via a Hamilton syringe attached to a PE 20 catheter and injector. Injections were delivered over 30 seconds and injectors remained in the tissue for 2 min post-injection. Injection coordinates for the lateral ventricle were 0.9 mm caudal to bregma, 1.8 mm lateral to the midline (at bregma), and 4.6 mm ventral to the skull at the injection site. Post-operatively, the animals were sutured and analgesic was administered daily for 3 days. Ten days after the tracer injections, animals were transcardially perfused and immunofluorescent staining was performed for either orexin or MCH, as described above. Quantification of MCH, CTB, and CTB positive MCH neurons was performed by manually counting following previously described approaches (Hahn, 2010).
Fast blue injections
Rats were anesthetized and sedated with a ketamine (90mg/kg)/xylazine (2.8 mg/kg)/acepromazine (0.72 mg/kg) cocktail. Animals were shaved and an incision was made in the next to locate the left jugular vein. A 27.5 gauge syringe was inserted and jugular injections of 6 mg/kg body weight Fast Blue (FB) were delivered. Forty-eight hours later, under deep ketamine anesthesia, animals were sacrificed via transcardial perfusion (0.9% saline, followed by 4% paraformaldehyde pH 9.5). Ten days prior to sacrifice, and 8 days prior to FB injections, the animals were injected with CTB in the lateral ventricle, as described. Immunofluorescence labeling was performed for MCH, and quantification of CTB, MCH and FB labeled neurons in the lateral hypothalamic zone and zona incerta was performed based on one of five series from 3 animals.
Virus production
For AAVs [AAV2-MCHp-hM3D(Gq)-mCherry-WPRE, and AAV2-MCHp-DIO-hM3D(Gq)-mCherry-WPRE] transgenes were packaged into AAV2s (as previously described (Sterky et al., 2011), Vector Biolabs, Malvern, PA, USA) under the control of an MCH promoter (see below) and delivered at a titer of 8.8 × 10^12 GC/ml. The hM3D(Gq) plasmid was obtained from Addgene (#44361; Cambridge, MA, USA). CAV2 CRE was generated as described (Kremer et al., 2000) and had a titer of 3 × 10 12 viral genomes per ml.
MCH(462bp) promoter region(Konadhode et al., 2013):
(TCTAGAGATAACTTCTATTTAATAAGGATTAAAAGGGCATAAAGCTTTTATCCTTATTATCCATGAATTAAATAACACGTCTTAATATCTTAATCGGAAAGTACATAGAAGGTAACTGTAATATTGGGAGAGCCTGGTGTCCTATGGTCTTCCTTAATAACATCCCACTTTCAAGAGGCTATAATCTACTCACAGAAAGCCTTCTCTCAAAGACCTTAATCAGATTCTGGGAAAATTCACACACGAAATAAACAACAGGCCACTAAATCCAAAACAATGCAAGCATTAGCAGCTTTACAAAGGCGAGGCTCTACGGGGTGATTCATTTCTAAAAAGAAAAGATAAGGCCTTCAAGTGTTTTCTATTCAGGCACAAAGTATATAAAGGTAGGAATCATTCAGTCGCCAGCAGAGTGTGGCATTCTCCCCACATTCTCTTCGGCTTTACGGAGCAGCAAACAGG)
Virus injections
Rats were anesthetized and sedated with a ketamine (90mg/kg)/xylazine (2.8 mg/kg)/acepromazine (0.72 mg/kg) cocktail. Stereotaxic injections of MCH AAV2-rMCHp-hM3D(Gq)-mCherry were delivered using a microinfusion pump (Harvard Apparatus) connected to a 33-gauge microsyringe injector attached to a PE20 catheter and Hamilton syringe. Flow rate was calibrated and set to 5μl/min; injection volume was 300 nl/site. Injectors were left in place for 2 min post-injection. Bilateral injections of the AAV2 virus were given to the following coordinates (Paxinos and Watson, 2007) : injection 1) −2.6 mm AP, ± 1.8 mm ML, −8.0 DV; 2) −2.6 mm AP, ± 1.0 mm ML, −8.0 DV; 3) −2.9 mm AP, ± 1.1 mm ML, −8.8 DV; 4) −2.9 mm AP, ± 1.6 mm ML, −8.8 DV (from the skull surface at bregma). Animals were allowed to incubate with the virus for 21 days prior to experimentation. Successful virally mediated transduction of the DREADDs receptor was confirmed post-mortem in all animals (n=8) via immunohistochemical staining using an RFP primary antibody to enhance the mCherry signal followed by manual quantification under epifluorescence illumination using a Nikon 80i (Nikon DS-QI1,1280X1024 resolution, 1.45 megapixel).
The cre-dependent AAV2-DIO-rMCHp-hM3D(Gq)-mCherry was injected in four bilateral (8 total) injections as described in the methods above. Prior to injections with the MCH AAV2-DIO-rMCHp-hM3D(Gq)-mCherry, a unilateral injection of the canine adenovirus 2 Cre (CAV2 CRE; 1.5 μl) was stereotaxically injected into the lateral ventricle using the following coordinates (Paxinos and Watson, 2007): −0.9 mm AP, 1.8 mm ML, −4.6 mm DV (from the skull surface at the injection site). Animals were allowed to incubate with the virus for 21 days prior to experimentation. Successful virally mediated transduction of the DREADDs receptor was confirmed post-mortem in all animals (n=7) via immunohistochemical staining using an RFP primary antibody to enhance the mCherry signal followed by manual quantification under epifluorescence illumination using a Nikon 80i (Nikon DS-QI1,1280X1024 resolution, 1.45 megapixel).
In a separate cohort of animals, the cre-dependent AAV2-DIO-rMCHp-hM3D(Gq)-mCherry was injected in four bilateral (8 total) injections as described in the methods above. Prior to injections with the MCH AAV2-DIO-rMCHp-hM3D(Gq)-mCherry, bilateral injections of the canine adenovirus 2 Cre (CAV2 CRE; 200 nl/side) were stereotaxically injected into the nucleus accumbens shell using the following coordinates (Paxinos and Watson, 2007): 1.1 mm AP, ±0.8 mm ML, −7.5 mm DV (from the skull surface at bregma). Animals were allowed to incubate with the virus for 21 days prior to experimentation and successful transduction of the DREADDs was confirmed as described above.
Characterization of DREADDs expression
In all cases where DREADDs transduction occurred, the spread of DREADDs expression was quantified in 1 out of 5 series of brain tissue sections from perfused brains cut at 30 μm on a microtome with a frozen stage. Sections from Swanson Brain Atlas level 27–32 (Swanson, 2004), which encompasses all MCH containing neurons were selected using anatomical landmarks and immunofluorescence staining for red fluorescent protein (RFP) was conducted to amplify the mCherry signal (as described above). Cell counts were performed by 2 researchers under epifluorescence illumination using a Nikon 80i (Nikon DS-QI1,1280X1024 resolution, 1.45 megapixel) and the average of the 2 counts was taken. Researchers who performed the counting were kept consistent between cohorts and blind to experimental assignments. In cases where the anatomical location of the cells was identified, experimenters utilized anatomical landmarks from the Swanson Brain Atlas (Swanson, 2004) to characterize regions of cell localization.
ICV cannula implantation
Unilateral guide cannulae (26-gauge, Plastics One) were surgically implanted targeting the lateral ventricle as previously described (Hsu et al., 2015b) using the following coordinates (Paxinos and Watson, 2007): −0.9 mm AP, 1.8 mm ML, −2.6 mm DV. Placement for the lateral ventricle cannula was verified by elevation of cytoglucopenia resulting from an injection of 210μg (2μl) of 5-thio-D-glucose (5tg) (Ritter et al., 1981) using an injector that extended 2mm beyond the end of the guide cannula. A post-injection elevation of at least 100% of baseline glycemia as required for subject inclusion. Animals that did not pass the 5tg test were retested with an injector that extended 2.5 mm beyond the end of the guide cannula, and upon passing 5tg were subsequently injected using a 2.5 mm injector instead of a 2mm injector for the remainder of the study.
Effect of activation of MCH neurons on food intake
All injections were delivered through a 33-gauge microsyringe injector attached to a PE20 catheter and Hamilton syringe. The microsyringe injector extended either 2 or 2.5 mm beyond the end of the guide cannula targeting the lateral ventricle. For testing the effects of activating MCH neurons in rats with the MCH AAV2-rMCHp-hM3D(Gq)-mCherry on feeding, injections of artificial cerebrospinal fluid (Harvard Apparatus, Holliston, MA, USA; aCSF; 1μl), the MCH antagonist H6408 (Bachem, Torrance, CA, USA; 10μg in 1μl aCSF), Clozapine-N-Oxide (National Institute of Mental Health; 18 mmoles in 2μl), or 33% DMSO in aCSF (daCSF; 2 μl) were given in a mixed design, with aCSF and H6408 as a between subjects variable and CNO and daCSF as a within subjects variable (n=15). Food was removed one hour prior to injections, which were given 30 minutes prior to the onset of the dark cycle. Food hoppers were weighed and placed in cages post injections and food intake was measured by subtracting the hopper weight at 2 hours from the time the animal received the food from the initial hopper weight. For each animal, spillage was collected from papers that were placed beneath the wire hanging cages and subtracted from the differences of hopper weight to get a measure of total intake.
Activation of ventricular projecting MCH neurons
Food was removed at the beginning of the light cycle. Injections of Clozapine-N-Oxide (National Institute of Mental Health; 18 mmoles in 2μl), or 33% DMSO in aCSF (daCSF; 2 μl) were administered 1.5 hours into the light cycle, and food intake was monitored. Food hoppers were weighed and placed in cages post injections and food intake was measured by subtracting the hopper weight at 2 and 4 hours from the time the animal received the food from the initial hopper weight. For each animal, spillage was collected from papers that were placed beneath the wire hanging cages and subtracted from the differences of hopper weight to get a measure of total intake for each time point. A within subject’s design was used, where animals were injected with either CNO or vehicle in a counterbalanced fashion for a total of 2 treatments with 72 hours between treatments (n=7). This experiment was replicated once with similar results and was also replicated using a different retrograde virus (AAV6-CRE) with similar results.
Activation of ACB projecting MCH neurons
Food was removed at the beginning of the light cycle. Injections of Clozapine-N-Oxide (National Institute of Mental Health; 18 mmoles in 2μl), or 33% DMSO in aCSF (daCSF; 2 μl) were administered 1.5 hours into the light cycle, and food intake was monitored. Food hoppers were weighed and placed in cages post injections and food intake was measured by subtracting the hopper weight at 2 and 4 hours from the time the animal received the food from the initial hopper weight. For each animal, spillage was collected from papers that were placed beneath the wire hanging cages and subtracted from the differences of hopper weight to get a measure of total intake for each time point. A within subject design was used, where animals were injected with either CNO or vehicle in a counterbalanced fashion for a total of 2 treatments with 72 hours between treatments (n=11).
Brain slice preparation
Rats (n=7) were anesthetized by inhalation of isoflurane gas (2–4%) and decapitated by guillotine. The brain was removed and placed for 1 min in cold (0–4°C) artificial cerebrospinal fluid (aCSF) composed of the following (in mM): NaCl 125, KCl 3, KH2PO4 1.2, MgSO4 1.2, NaHCO3 25, dextrose 10, CaCl2 2, and bubbled with 95% O2/5% CO2 (pH 7.4, 306–310mOsM adjusted with sucrose). A tissue block was mounted in a vibrating microtome (Leica VT-1000S). Coronal slices (300μm thick) were cut with a sapphire knife (Delaware Diamond Knives, Wilmington, DE), submerged in a microscope chamber, and perfused with oxygenated aCSF at a rate of 2 ml/min.
Electrophysiology
Lateral hypothalamic area neurons were visually selected for electrophysiological patch clamp recordings using a fluorescence microscope (Olympus BX51WI). Patch electrodes were guided to neurons using differential interference contrast (DIC) optics illuminated with infrared light. Current-clamp recordings were made with a MultiClamp 700B amplifier, Digidata 1440A digitizer, and pClamp10.2 software (all from Molecular Devices, Sunnyvale, CA). Data were filtered at 10 kHz, and stored on a personal computer for offline analysis. For recordings, patch pipettes were pulled using a pipette puller (P-2000, Sutter Instruments, Novato, CA). The pipette resistance was 3.0–4.0 MΩ when filled with internal solution (in mM: NaCl 10, K-’gluconate 130, EGTA 11, CaCl2 1, MgCl2 2, HEPES 10, Na-ATP 2, Na-GTP 0.2, pH 7.3, 297–300 mOsM). After formation of a stable giga-ohm seal (>2 GΩ), the whole-cell configuration was established. Only neurons with holding currents not exceeding −100pA at VH = −60 mV for the 15 min control period (membrane resistance >250 MΩ) were studied further (n=8). Series resistance (Ra) was measured at the beginning and end of recordings and neurons were not included in additional analysis if it exceeded 20MΩ, or drifted >25%. Ra did not differ between control (aCSF) and drug treatment. aCSF was heated to 32 ± 0.5°C using a pre-heater, a temperature sensor positioned on the wall of the recording chamber next to the slice, and a temperature controller (Cell Micro Controls, Norfolk, VA). The extracellular solution was identical to oxygenated aCSF that was used for the brain tissue preparation.
CSF extraction and MCH Enzyme Immuno Assay
Rats were deeply anesthetized using a cocktail of ketamine 90mg/kg, xylazine, 2.8 mg/kg, and acepromazine 0.72 mg/kg by intramuscular injection. Shaved and prepped rats were then placed into stereotaxic ear bars, with the head angled downward. An incision was made along the back of the neck and the muscles were blunt dissected with a tissue retractor. A spatula was used to clear the spongy dura that is caudally adjacent to the base of the skull; this is the point of insertion into the medullary cisterna. A sterile 1ml syringe with 27-gauge needle was attached to the stereotaxic arm with the open side of the bevel facing the posterior end of the rat. After pulling 1 mL of air, the needle was lowered to below the caudal end of the occipital skull and the dura was punctured slowly, moving the stereotaxic arm up and down (less than 1mm each direction) to imitate a sawing motion. The needle was lowered another ~2mm and the syringe plunger pulled back, allowing the clear CSF to flow into the syringe. After extracting ~200 mL of CSF, the needle was raised quickly (to prevent suction of blood while coming out of the cisterna magna) and the CSF dispensed into a microfuge tube and immediately frozen in dry ice and stored at −80 degrees C until time of analysis. Quantification of MCH levels in CSF was performed according to manufacturer’s instructions using an EIA kit (Phoenix Pharmaceuticals, Burlingame, CA, USA).
CSF extraction following MCH DREADDS activation
For the effects of activation of MCH DREADDs on MCH levels in CSF, animals were injected with either 33% DMSO in aCSF (daCSF; 2 μl) or Clozapine-N-Oxide (National Institutes of Health; 18 mmoles in 2μl) 2 hours prior to perfusion and again 15 minutes prior to perfusion (n=6 and 7). CSF extractions and perfusions were performed in a counterbalanced manner beginning 2 hours into the light cycle.
Effect of physiological parameters on CSF MCH levels
To determine whether MCH levels in CSF fluctuate based on time of day, CSF was extracted either 1 hour into the dark cycle, or 3 hours into the light cycle (n=8/group). Food was removed 2 hours prior to CSF extraction. To determine whether MCH levels in CSF were altered as a function of prandial state (pre vs post) animals were meal entrained to the first 4 hours of the dark cycle. On the day of extraction, a standardized meal of 5 grams of chow was given at the start of the dark cycle. All animals consumed all of the chow in less than one hour, and CSF extractions occurred one hour into the dark cycle (n=8).
To determine whether MCH levels in CSF are elevated prior to feeding when animals are entrained to light cycle feeding, rats were first divided into two groups (light entrained or ad libitum fed) matched for body weight. On day zero, a baseline 4-hr cumulative food intake recording was measured during mid-light cycle period (ZT4-ZT8) when light entrained animals would subsequently receive all of their daily chow. The experimental group was then meal entrained for 18 days whereas the control group received ad libitum food access. Food intake and spillage were monitored during the 4-hr entrainment period for both the ad libitum fed control animals and the light entrained animals. CSF was extracted at ZT4 when food would normally be given to the light entrained animals (n=7/group) with food removed 1 hr prior to extraction from the ad libitum fed animals.
To determine whether consumption of a palatable high fat/sucrose diet increases pre-prandial levels of MCH, animals were randomized to receive either chow (controls) or a diet containing 45% kcal from fat (Research Diets, D12451). Animals were maintained on the diet for 5 days, and food intake, spillage and body weights were recorded. Food was pulled 1 hour prior to CSF extraction, and CSF was extracted at the time of the dark cycle onset (Z12) (n=6/group).
To determine whether MCH levels in CSF were affected by acute food restriction, animals were either food deprived for 48 hours (48-hr food dep., n=5) or fed ad libitum (n=6). Animals were divided based on body weight such that each group was of equal body weight at baseline. Food was removed 30 min prior to CSF extraction for the ad libitum fed animals. CSF was extracted during the late dark cycle (ZT18).
MCH dose response and CSF extraction
Male Sprague Dawley rats (n=12) were surgically implanted with ICV guide cannula and cannula placement was verified as described above. Animals were handled and habituated to injections prior to testing. On test day, food was removed 1 hour prior to injections, which occurred at ZT5. All injections were delivered through a 33-gauge micro-syringe injector attached to a PE20 catheter and Hamilton syringe. The micro-syringe injector extended either 2 or 2.5 mm beyond the end of the guide cannula targeting the lateral ventricle. Animals were randomized to receive either 0 (aCSF), 0.5μg, or 1 μg of MCH in 1 μl of aCSF vehicle in a counterbalanced within subject’s design. Injections were given during the light cycle such that food was replaced at ZT 6. On each treatment day, food was removed 1 hour prior to injections and replaced immediately following the last injection. Food intake and spillage under the cage was weighed and recorded.
For the CSF extraction following injections with the least effective dose of MCH for hyperphagia, rats (n=19) were injected with 1 μg of MCH or vehicle and anesthetized beginning either 45 min (n=4 and 3, respectively) or 1 hour and 45 minutes (n=6/group) later. CSF was extracted during the time window of either 60–90 minutes post MCH/vehicle injection or 120–150 minutes. Samples were treated with protease inhibitor and frozen and MCH levels were quantified in CSF using a commercial EIA kit as described above.
Co-injections of MCH and GABA-A or GABA-B agonists
Male Sprague Dawley rats were surgically implanted with ICV guide cannula and cannula placement was verified as described above. Animals were handled and habituated to injections prior to testing. On test day, food was removed 1 hour prior to injections, which occurred at ZT5. All injections were delivered through a 33-gauge microsyringe injector attached to a PE20 catheter and Hamilton syringe. The microsyringe injector extended either 2 or 2.5 mm beyond the end of the guide cannula targeting the lateral ventricle. For the dose response of muscimol, animals were randomized to receive either 0 (aCSF), 25 ng, or 75 ng of muscimol in 1 μl of aCSF vehicle in a counterbalanced within subject’s design (n=12; 3–4 days separating treatments). On each treatment day, food was removed 1 hour prior to injections and replaced immediately following the last injection. Food intake and spillage under the cage was weighed and recorded. As food intake readings were trending towards significance (P=.07 at 1 hr) at the 25 ng dose, we used a 20 ng dose of muscimol for our co-injection experiment to test whether muscimol (at a dose ineffective for food intake effects alone) would potentiate an orexigenic effect of a sub-effective dose of MCH (0.5 μg). Using a counterbalanced, within-subject’s design, animals (n=12) were randomized to receive either 0 (aCSF) or 0.5 μg of MCH and either 0 (aCSF) or 20 ng of muscimol (four treatments total: aCSF-aCSF, MCH-aCSF, aCSF-muscimol, MCH-muscimol). Injections of MCH were given one hour prior to injections of muscimol, beginning at ZT5. Food was replaced after the last muscimol injection and food intake and spillage under the cage were recorded. The same parameters and experimental design were used for MCH and Baclofen co-injection experiments, however for baclofen experiments a mixed design was used (n=17 animals total) with MCH vs aCSF (vehicle) as the within-subject’s variable and Baclofen vs aCSF (vehicle) as the between-subject’s variable. The dose of Baclofen used was 0.1 nmol in 1 μl of aCSF based on (Ebenezer, 1990).
Injections of neutralizing antibody in the CSF
Rats were surgically implanted with ICV cannula and cannula placement was verified as described above. On the day of testing, food was removed 1.5 hours prior to injections, which began 30 min prior to the onset of the dark cycle. All injections were delivered through a 33-gauge micro-syringe injector attached to a PE20 catheter and Hamilton syringe. The micro-syringe injector extended either 2 or 2.5 mm beyond the end of the guide cannula targeting the lateral ventricle. Injections of 2 μl of either rabbit anti-MCH (n=10) or rabbit anti-PHAL (control; n=9) in a 1:1 dilution of aCSF were delivered to the lateral ventricle. All injections were completed within 30 minutes and food (standard chow) was returned to the animals at the onset of the dark cycle. Food intake and spillage was recorded as described above. Animals were perfused following the 8-hr food intake reading and brains were dissected out and sectioned as described above.
Immunosequestration of MCH in CSF
Rats were surgically implanted with ICV cannula and cannula placement was verified as described above. On the day of testing, food was removed 30 minutes prior to injections, which began 2 hours prior to the onset of the dark cycle. All injections were delivered through a 33-gauge micro-syringe injector attached to a PE20 catheter and Hamilton syringe. The micro-syringe injector extended either 2 or 2.5 mm beyond the end of the guide cannula targeting the lateral ventricle. Three injections were given 3o minutes apart: injection 1) 1:1 mixture of biotinylated donkey anti-rabbit with rabbit anti-laminin; 2 μl injection volume; injection 2) Streptavidin conjugated to Alexa Fluor-488; 1 μl injection volume; and injection 3) 1:1 mixture of biotinylated donkey anti-rabbit premixed with either rabbit anti-MCH (n=9) or rabbit anti-PHAL (n=8). Antibody mixtures were prepared in 0.9% saline 24 hours prior to injections and stored at 4 °C. Food was replaced following the last injection and food intake was measured as described above.
Comparison of neuropil penetrance
Immunofluorescence visualization of the antibody in the tissue was performed using the immunofluorescence staining methods above but omitting the addition of primary antibody. The addition of secondary antibody (Donkey anti-rabbit-Cy3) enabled the detection of rabbit anti-MCH primary antibody in tissue sections from both groups. Tissue sections were stained in the same run to avoid variation in staining quality and allow for equal comparisons. Images were collected using identical microscope and imaging parameters using epifluorescence illumination using a Nikon 80i (Nikon DS-QI1,1280X1024 resolution, 1.45 megapixel).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses
ANOVA analyses were performed using GraphPad Prism 7.0 Software (GraphPad Software Inc.) and t tests were performed using either GraphPad Prism 7.0 Software or Microsoft Excel for Mac (v. 15.26; Microsoft Inc.). To investigate the effects of MCH DREADDs activation on CSF MCH levels, data were analyzed using a Student’s two-tailed unpaired t test with Welch’s correction for unequal variances. Comparisons of the effects of CNO on food intake in animals with or without prior injections of the MCH receptor 1 antagonist, which were analyzed using a mixed design with H6408 vs vehicle at the between subjects group and CNO vs vehicle as the within subjects group. These data were analyzed using Two-way repeated measures ANOVA with a mixed design and Newman-Keuls post hoc test to adjust for multiple comparisons. Statistica Software (StatSoft, Inc.). For the muscimol dose response experiment, data are analyzed at each time point using a repeated measures one-way ANOVA with Geisser-Greenhouse correction and Tukey’s multiple comparisons test. For the muscimol combined with a low dose of MCH experiment, data are analyzed using a repeated measures two-factor ANOVA (Drug 1 x Drug 2) at each time point. For the Baclofen experiment data are analyzed using a mixed design two-factor ANOVA (Drug 1 x Drug 2) at each time point with MCH vs vehicle as the within- subject variable and Baclofen vs vehicle as the between subject variable. For the electrophysiological recordings, data were analyzed using One-way ANOVA with Tukey’s post hoc test for multiple comparisons. For all statistical tests, the α level for significance was .05.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-MCH | Phoenix Pharmaceuticals | RRID: AB_10013632 |
| Mouse anti-synaptophysin | Progen | Cat # 61012 |
| Rabbit anti-RFP | Rockland Inc. | RRID: AB_2209751 |
| Chicken anti-vimentin | Abcam | RRID: AB_778824 |
| Rabbit anti-orexin | Phoenix Pharmaceuticals | RRID: AB_2315019 |
| Mouse anti-beta subunit cholera toxin | Abcam | RRID: AB_300499 |
| Rabbit anti-laminin | Lifespan Biosciences | RRID: AB_1808356 |
| Donkey anti-rabbit IgG-Cy3 | Jackson Immunoresearch | RRID: AB_2307443 |
| Donkey anti-rabbit IgG-AlexaFluor 488 | Jackson Immunoresearch | RRID: AB_2340619 |
| Donkey anti-mouse IgG-Cy3 | Jackson Immunoresearch | RRID: AB_2315777 |
| Donkey anti-mouse IgG-AlexaFluor 488 | Jackson Immunoresearch | RRID: AB_2341099 |
| Donkey anti-chicken IgG-AMCA | Jackson Immunoresearch | RRID: AB_2340361 |
| Donkey anti-rabbit IgG- biotin | Jackson Immunoresearch | RRID: AB_2340593 |
| Rabbit anti-PHAL | Vector Laboratories | RRID: AB_2313686 |
| Bacterial and Virus Strains | ||
| AAV2-MCHp-DIO-hM3D(Gq)-mCherry-WPRE | Vector Biolabs (this paper) | (custom) |
| AAV2-MCHp-hM3D(Gq)-mCherry-WPRE | Vector Biolabs (this paper) | (custom) |
| CAV2-CRE | Gift from Martin Darvas, PhD | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Clozapine N oxide | Generously provided by NIMH | |
| DMSO | MP Biomedicals, LLC | Cat #: 196055 |
| Protease inhibitor cocktail | Sigma | Cat #: P8340–5ML |
| aCSF | Harvard Apparatus | Cat #: 59–7316 |
| Melanin concentrating hormone | Bachem | Cat #: H-2218.1000 |
| Donkey serum | Jackson Immunoresearch | Cat #: 017–000-121 |
| Triton-X 100 | Sigma | Cat #: X100–500ML |
| Glycerol | EMD Millipore | Cat #: GX0185–5 |
| Ketaset (Ketamine), 100mg/mL | Henry Schein Animal Health | Cat #: 010177 |
| Anased (Xylazine) Injection, 100mg/mL | Henry Schein Animal Health | Cat #: 033198 |
| Aceproject (Acepromazine) Injection, 10mg/mL | Henry Schein Animal Health | Cat #: 003845 |
| Paraformaldehyde | Alfa Aesar | Cat # A11313 |
| Sodium hydroxide | EMD Millipore | Cat #: SX0590–1 |
| Sodium tetraborate | Alfa Aesar | Cat #: 40114 |
| EDTA Tetrasodium | EMD Millipore | Cat #: EX0550–5 |
| Trizma hydrochloride | Sigma Aldrich | Cat #: T3252–250G |
| Proteinase K | Sigma Aldrich | Cat #: P2308– 100MG |
| Triethanolamine | Sigma Aldrich | Cat #: T58300–1KG |
| Acetic anhydride | EMD Millipore | Cat #: AX0080–6 |
| Citric acid trisodium | VWR | Cat #: 0101–500G |
| ProLong Gold Antifade mounting medium | Cell Signaling | Cat #: 9071S |
| CTB AlexaFluor 594 | ThermoFisher Scientific | Cat #: C22842 |
| Streptavidin - Alexa Fluor 488 | Vector Laboratories | Cat #: SA-5488 |
| MCH Antagonist H6408 | Bachem | Cat #: H-6408 |
| Muscimol | Enzo Life Sciences | Cat #: ALX-550–127- M005 |
| (+/-) Baclofen | Sigma Aldrich | Cat #: B5399 |
| Critical Commercial Assays | ||
| MCH EIA Kit | Phoenix Pharmaceuticals | Cat #: FEK-070–47 |
| vGLUT1 probe | Advanced Cell Diagnostics | Cat #: 317001-C2 |
| vGLUT2 probe | Advanced Cell Diagnostics | Cat #: 317011-C2 |
| vGLUT3 probe | Advanced Cell Diagnostics | Cat #: 476711-C2 |
| RNAscope Fluorescent Multiplex Reagent Kit | Advanced Cell Diagnostics | Cat #: 320851 |
| RNAscope Wash Buffer | Advanced Cell Diagnostics | Cat #: 320058 |
Acknowledgements
The research was supported by DK104897 and institutional funds to S.E.K., institutional funds to M.D., DK083452 and institutional funds to S.M.A. (CVM, WSU), and DK107333 to T.M.H., and DK111158 to E.E.N. The authors thank Dr. Alan Watts, Dr. Ruth Wood, Brian Zingg, Sandhya Prathap, Kaitlin Sontag, Natalie Demirjian, Emily Nakamoto, April Banayan, Victor Lee, and Dr. Matthew Dean for their conceptual and experimental contributions. The authors have no conflicts of interest to disclose.
Footnotes
Declaration of Interests
The authors have no competing interests to declare.
DATA AND SOFTWARE AVAILABILITY
Data resources
All data generated and analyzed for this manuscript are available from the corresponding author (S.E.K.) upon reasonable request.
References
- Agnati LF, Fuxe K, Zoli M, Ozini I, Toffano G, and Ferraguti F (1986). A correlation analysis of the regional distribution of central enkephalin and beta-endorphin immunoreactive terminals and of opiate receptors in adult and old male rats. Evidence for the existence of two main types of communication in the central nervous system: the volume transmission and the wiring transmission. Acta physiologica Scandinavica 128, 201–207. [DOI] [PubMed] [Google Scholar]
- Balland E, Dam J, Langlet F, Caron E, Steculorum S, Messina A, Rasika S, Falluel-Morel A, Anouar Y, Dehouck B, et al. (2014). Hypothalamic tanycytes are an ERK-gated conduit for leptin into the brain. Cell metabolism 19, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barson JR, Morganstern I, and Leibowitz SF (2013). Complementary roles of orexin and melanin-concentrating hormone in feeding behavior. Int J Endocrinol 2013, 983964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittencourt JC, Presse F, Arias C, Peto C, Vaughan J, Nahon JL, Vale W, and Sawchenko PE (1992). The melanin-concentrating hormone system of the rat brain: an immuno- and hybridization histochemical characterization. J Comp Neurol 319, 218–245. [DOI] [PubMed] [Google Scholar]
- Bjorefeldt A, Andreasson U, Daborg J, Riebe I, Wasling P, Zetterberg H, and Hanse E (2015). Human cerebrospinal fluid increases the excitability of pyramidal neurons in the in vitro brain slice. The Journal of physiology 593, 231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluet-Pajot MT, Presse F, Voko Z, Hoeger C, Mounier F, Epelbaum J, and Nahon JL (1995). Neuropeptide-E-I antagonizes the action of melanin-concentrating hormone on stress-induced release of adrenocorticotropin in the rat. J Neuroendocrinol 7, 297–303. [DOI] [PubMed] [Google Scholar]
- Boender AJ, de Jong JW, Boekhoudt L, Luijendijk MC, van der Plasse G, and Adan RA (2014). Combined use of the canine adenovirus-2 and DREADD-technology to activate specific neural pathways in vivo. PLoS One 9, e95392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caraty A, and Skinner DC (2008). Gonadotropin-releasing hormone in third ventricular cerebrospinal fluid: endogenous distribution and exogenous uptake. Endocrinology 149, 5227–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers J, Ames RS, Bergsma D, Muir A, Fitzgerald LR, Hervieu G, Dytko GM, Foley JJ, Martin J, Liu WS, et al. (1999). Melanin-concentrating hormone is the cognate ligand for the orphan G-protein-coupled receptor SLC-1. Nature 400, 261–265. [DOI] [PubMed] [Google Scholar]
- Chee MJ, Pissios P, and Maratos-Flier E (2013). Neurochemical characterization of neurons expressing melanin-concentrating hormone receptor 1 in the mouse hypothalamus. J Comp Neurol 521, 2208–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conductier G, Martin AO, Risold PY, Jego S, Lavoie R, Lafont C, Mollard P, Adamantidis A, and Nahon JL (2013). Control of ventricular ciliary beating by the melanin concentrating hormone-expressing neurons of the lateral hypothalamus: a functional imaging survey. Front Endocrinol (Lausanne) 4, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias Abdo Agamme AL, Aguilar Calegare BF, Fernandes L, Costa A, Lagos P, Torterolo P, and D’Almeida V (2015). MCH levels in the CSF, brain preproMCH and MCHR1 gene expression during paradoxical sleep deprivation, sleep rebound and chronic sleep restriction. Peptides 74, 9–15. [DOI] [PubMed] [Google Scholar]
- Ebenezer IS (1990). The effect of intracerebroventricular administration of baclofen on food intake in rats. Neuroreport 1, 73–76. [DOI] [PubMed] [Google Scholar]
- Feng CY, Wiggins LM, and von Bartheld CS (2011). The locus ceruleus responds to signaling molecules obtained from the CSF by transfer through tanycytes. The Journal of neuroscience : the official journal of the Society for Neuroscience 31, 9147–9158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu D, Sears RM, Hommel JD, Barrot M, Bolanos CA, Marsh DJ, Bednarek MA, Bibb JA, Maratos-Flier E, Nestler EJ, et al. (2005). The hypothalamic neuropeptide melanin-concentrating hormone acts in the nucleus accumbens to modulate feeding behavior and forced-swim performance. J Neurosci 25, 2933–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez JA, Iordanidou P, Strom M, Adamantidis A, and Burdakov D (2016). Awake dynamics and brain-wide direct inputs of hypothalamic MCH and orexin networks. Nature communications 7, 11395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn JD (2010). Comparison of melanin-concentrating hormone and hypocretin/orexin peptide expression patterns in a current parceling scheme of the lateral hypothalamic zone. Neuroscience letters 468, 12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamoto A, Yamato S, Katoh Y, Nakayama K, Yoshimura K, Takeda S, Kobayashi Y, and Saito Y (2016). Modulation of primary cilia length by melanin-concentrating hormone receptor 1. Cellular signalling 28, 572–584. [DOI] [PubMed] [Google Scholar]
- Harthoorn LF, Sane A, Nethe M, and Van Heerikhuize JJ (2005). Multi-transcriptional profiling of melanin-concentrating hormone and orexin-containing neurons. Cell Mol Neurobiol 25, 1209–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoistad M, Samskog J, Jacobsen KX, Olsson A, Hansson HA, Brodin E, and Fuxe K (2005). Detection of beta-endorphin in the cerebrospinal fluid after intrastriatal microinjection into the rat brain. Brain research 1041, 167–180. [DOI] [PubMed] [Google Scholar]
- Hsu TM, Hahn JD, Konanur VR, Lam A, and Kanoski SE (2015a). Hippocampal GLP-1 receptors influence food intake, meal size, and effort-based responding for food through volume transmission. Neuropsychopharmacology 40, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu TM, Hahn JD, Konanur VR, Noble EE, Suarez AN, Thai J, Nakamoto EM, and Kanoski SE (2015b). Hippocampus ghrelin signaling mediates appetite through lateral hypothalamic orexin pathways. eLife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DF, Ackroff K, Peters J, and Collier GH (1986). Changes in rats’ meal patterns as a function of the caloric density of the diet. Physiology & behavior 36, 929–936. [DOI] [PubMed] [Google Scholar]
- Kokare DM, Patole AM, Carta A, Chopde CT, and Subhedar NK (2006). GABA(A) receptors mediate orexin-A induced stimulation of food intake. Neuropharmacology 50, 16–24. [DOI] [PubMed] [Google Scholar]
- Konadhode RR, Pelluru D, Blanco-Centurion C, Zayachkivsky A, Liu M, Uhde T, Glen WB Jr., van den Pol AN, Mulholland PJ, and Shiromani PJ (2013). Optogenetic stimulation of MCH neurons increases sleep. The Journal of neuroscience : the official journal of the Society for Neuroscience 33, 10257–10263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotz CM, Wang C, Teske JA, Thorpe AJ, Novak CM, Kiwaki K, and Levine JA (2006). Orexin A mediation of time spent moving in rats: neural mechanisms. Neuroscience 142, 29–36. [DOI] [PubMed] [Google Scholar]
- Kowalski TJ, Farley C, Cohen-Williams ME, Varty G, and Spar BD (2004). Melanin-concentrating hormone-1 receptor antagonism decreases feeding by reducing meal size. European journal of pharmacology 497, 41–47. [DOI] [PubMed] [Google Scholar]
- Kremer EJ, Boutin S, Chillon M, and Danos O (2000). Canine adenovirus vectors: an alternative for adenovirus-mediated gene transfer. Journal of virology 74, 505–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lembo PM, Grazzini E, Cao J, Hubatsch DA, Pelletier M, Hoffert C, St-Onge S, Pou C, Labrecque J, Groblewski T, et al. (1999). The receptor for the orexigenic peptide melanin-concentrating hormone is a G-protein-coupled receptor. Nature cell biology 1, 267–271. [DOI] [PubMed] [Google Scholar]
- Ludwig DS, Tritos NA, Mastaitis JW, Kulkarni R, Kokkotou E, Elmquist J, Lowell B, Flier JS, and Maratos-Flier E (2001). Melanin-concentrating hormone overexpression in transgenic mice leads to obesity and insulin resistance. J Clin Invest 107, 379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLaren DA, Browne RW, Shaw JK, Krishnan Radhakrishnan S, Khare P, Espana RA, and Clark SD (2016). Clozapine N-Oxide Administration Produces Behavioral Effects in Long-Evans Rats: Implications for Designing DREADD Experiments. eNeuro 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMillan SJ, Mark MA, and Duggan AW (1998). The release of beta-endorphin and the neuropeptide-receptor mismatch in the brain. Brain research 794, 127–136. [DOI] [PubMed] [Google Scholar]
- Marsh DJ, Weingarth DT, Novi DE, Chen HY, Trumbauer ME, Chen AS, Guan XM, Jiang MM, Feng Y, Camacho RE, et al. (2002). Melanin-concentrating hormone 1 receptor-deficient mice are lean, hyperactive, and hyperphagic and have altered metabolism. Proc Natl Acad Sci U S A 99, 3240–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maulon-Feraille L, Della Zuana O, Suply T, Rovere-Jovene C, Audinot V, Levens N, Boutin JA, Duhault J, and Nahon JL (2002). Appetite-boosting property of pro-melanin-concentrating hormone(131–165) (neuropeptide-glutamic acid-isoleucine) is associated with proteolytic resistance. J Pharmacol Exp Ther 302, 766–773. [DOI] [PubMed] [Google Scholar]
- Muraki K, Nakata Y, Simonaka H, Inoue J, Hirai Y, and Akiyama M (1996). Neutral endopeptidase activity in serum and cerebrospinal fluid. Hiroshima J Med Sci 45, 109–112. [PubMed] [Google Scholar]
- Paxinos G, and Watson C (2007). The rat brain in stereotaxic coordinates, 6th edn (Amsterdam; Boston;: Academic Press/Elsevier; ). [Google Scholar]
- Qu D, Ludwig DS, Gammeltoft S, Piper M, Pelleymounter MA, Cullen MJ, Mathes WF, Przypek R, Kanarek R, and Maratos-Flier E (1996). A role for melanin-concentrating hormone in the central regulation of feeding behaviour. Nature 380, 243–247. [DOI] [PubMed] [Google Scholar]
- Reiber H (2003). Proteins in cerebrospinal fluid and blood: barriers, CSF flow rate and source-related dynamics. Restor Neurol Neurosci 21, 79–96. [PubMed] [Google Scholar]
- Ripley B, Overeem S, Fujiki N, Nevsimalova S, Uchino M, Yesavage J, Di Monte D, Dohi K, Melberg A, Lammers GJ, et al. (2001). CSF hypocretin/orexin levels in narcolepsy and other neurological conditions. Neurology 57, 2253–2258. [DOI] [PubMed] [Google Scholar]
- Ritter RC, Slusser PG, and Stone S (1981). Glucoreceptors controlling feeding and blood glucose: location in the hindbrain. Science 213, 451–452. [DOI] [PubMed] [Google Scholar]
- Shearman LP, Camacho RE, Sloan Stribling D, Zhou D, Bednarek MA, Hreniuk DL, Feighner SD, Tan CP, Howard AD, Van der Ploeg LH, et al. (2003). Chronic MCH-1 receptor modulation alters appetite, body weight and adiposity in rats. European journal of pharmacology 475, 37–47. [DOI] [PubMed] [Google Scholar]
- Sheline YI, West T, Yarasheski K, Swarm R, Jasielec MS, Fisher JR, Ficker WD, Yan P, Xiong C, Frederiksen C, et al. (2014). An antidepressant decreases CSF Abeta production in healthy individuals and in transgenic AD mice. Sci Transl Med 6, 236re234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada M, Tritos NA, Lowell BB, Flier JS, and Maratos-Flier E (1998). Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature 396, 670–674. [DOI] [PubMed] [Google Scholar]
- Skinner DC, and Malpaux B (1999). High melatonin concentrations in third ventricular cerebrospinal fluid are not due to Galen vein blood recirculating through the choroid plexus. Endocrinology 140, 4399–4405. [DOI] [PubMed] [Google Scholar]
- Sterky FH, Lee S, Wibom R, Olson L, and Larsson NG (2011). Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proceedings of the National Academy of Sciences of the United States of America 108, 12937–12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street JM, Barran PE, Mackay CL, Weidt S, Balmforth C, Walsh TS, Chalmers RT, Webb DJ, and Dear JW (2012). Identification and proteomic profiling of exosomes in human cerebrospinal fluid. J Transl Med 10, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson LW (2004). Brain maps III : structure of the rat brain : an atlas with printed and electronic templates for data, models, and schematics, 3rd rev. edn (Amsterdam ; Boston: Elsevier, Academic Press; ). [Google Scholar]
- Swanson LW, Sanchez-Watts G, and Watts AG (2005). Comparison of melanin-concentrating hormone and hypocretin/orexin mRNA expression patterns in a new parceling scheme of the lateral hypothalamic zone. Neurosci Lett 387, 80–84. [DOI] [PubMed] [Google Scholar]
- Veening JG, and Barendregt HP (2010). The regulation of brain states by neuroactive substances distributed via the cerebrospinal fluid; a review. Cerebrospinal Fluid Res 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veening JG, de Jong T, and Barendregt HP (2010). Oxytocin-messages via the cerebrospinal fluid: behavioral effects; a review. Physiology & behavior 101, 193–210. [DOI] [PubMed] [Google Scholar]
- Vigh B, Manzano e Silva MJ, Frank CL, Vincze C, Czirok SJ, Szabo A, Lukats A, and Szel A (2004). The system of cerebrospinal fluid-contacting neurons. Its supposed role in the nonsynaptic signal transmission of the brain. Histology and histopathology 19, 607–628. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.