

Abstract
CPX-351 is a liposomally encapsulated 5:1 molar ratio of cytarabine and daunorubicin that recently received regulatory approval for the treatment of therapy-related acute myeloid leukemia (AML) or AML with myelodysplasia-related changes based on improved overall survival compared to standard cytarabine/daunorubicin therapy. Checkpoint kinase 1 (CHK1), which is activated by DNA damage and replication stress, diminishes sensitivity to cytarabine and anthracyclines as single agents, suggesting that CHK1 inhibitors might increase the effectiveness of CPX-351. The present studies show that CPX-351 activates CHK1 as well as the S and G2/M cell cycle checkpoints. Conversely, CHK1 inhibition diminishes the cell cycle effects of CPX-351. Moreover, CHK1 knockdown or addition of a CHK1 inhibitor such as MK-8776, rabusertib or prexasertib enhances CPX-351-induced apoptosis in multiple TP53-null and TP53-wildtype AML cell lines. Likewise, CHK1 inhibition increases the antiproliferative effect of CPX-351 on primary AML specimens ex vivo, offering the possibility that CPX-351 may be well suited to combine with CHK1-targeted agents.
Introduction
Despite advances in the molecular understanding of acute myeloid leukemia (AML), improvements in therapy are still needed. Most current regimens rely on treatment with the nucleoside analog cytarabine and an anthracycline for induction therapy. Depending on the subtype of AML, the driving oncogenic changes and the doses administered, these regimens have a 40–70% complete remission rate1,2. However, certain groups of patients, including those with antecedent hematological conditions or therapy-related AML, do particularly poorly with cytarabine/anthracycline-based induction therapy3–5.
A number of mechanisms of resistance to current AML induction therapy have recently been targeted, including increased expression of anthracycline exporters and increased activity of antiapoptotic BCL2 family members. However, resistance mechanisms involving the DNA damage and replication stress checkpoint pathways have received less attention. We and others previously reported that cytarabine activates the replication checkpoint kinases ATR and CHK1, which regulate a coordinated series of cellular responses that facilitate survival during replication stress6–10. Importantly, alterations that prevent activation of the replication checkpoint such as inhibition or downregulation of CHK1 sensitize AML cells to cytarabine7–10. CHK1 downregulation has also been reported to sensitize solid tumor cell lines to the anthracycline doxorubicin11,12, but this has not been tested using selective checkpoint kinase inhibitors in AML.
CPX-351 (Vyxeos®) is a liposomal formulation containing a synergistic 5:1 ratio of cytarabine and daunorubicin13. This formulation has a number of appealing properties, including the ability to kill AML cells harboring certain resistance mechanisms14 and a longer serum half-life than either cytarabine or daunorubicin15. CPX-351, which showed promising activity in AML clinical trials16,17, is now FDA approved for the treatment of therapy-related AML and AML with myelodysplasia-related changes18. Here we have assessed the impact of CHK1 inhibition on the cell cycle effects and cytotoxicity of CPX-351 in AML cell lines and clinical isolates.
Results
CPX-351 activates the replication checkpoint
In initial experiments, we examined the effects of CPX-351 on cell cycle distribution of the AML cell line U937. To assure that changes in propidium iodide staining were not a result of apoptosis-associated DNA fragmentation19,20, the caspase inhibitor Q-VD-OPh21 was included in these assays. Consistent with previous observations after cytarabine exposure6,7,22, U937 cells exposed to CPX-351 for 24 hours accumulated in S and G2 phases of the cell cycle, suggesting that the ATR/CHK1 replication checkpoint was activated (Fig. 1a,b).
Figure 1.

CPX-351 induces S phase arrest and activation of the ATR/CHK1 pathway. (a) DNA histograms of U937 cells treated for 24 h with diluent or CPX-351 (corresponding to cytarabine at 0.02, 0.04 and 0.08 µM in the fixed combination with doxorubicin) in the presence of the caspase inhibitor Q-VD-OPh21 (5 µM) to inhibit apoptosis-associated DNA degradation19,20. At the completion of the incubation, cells were stained with PI and analyzed by flow microfluorimetry as described in the Methods. (b) Percentage U937 cells in S phase after treatment with the concentrations of CPX-351 shown in panel a as well as additional concentrations. Results are mean ± sd of 3 independent experiments. (c) U937 cells were treated for 4.5 h with diluent (lanes 1–5) or CPX-351 (lanes 6–10) at a concentration equivalent to 2.5 µM cytarabine and 0.5 µM daunorubicin (abbreviated 2.5 µM cytarabine equivalents in subsequent figures) in combination with the indicated concentration of MK-8776 and immunoblotted for the indicated antigens. HSP90β served as a loading control. (d) Dot plots of U937 cells treated for 24 h with diluent or CPX-351 (corresponding to cytarabine at 0.31 µM in the fixed combination with doxorubicin) in the presence of 5 µM Q-VD-OPh and, where indicated, 500 nM MK-8776. At the completion of the incubation, cells were fixed, permeabilized, stained with PI and anti-phospho-Ser10-Histone H3 (Phospho-H3), and analyzed by flow microfluorimetry. R2 indicates the mitotic population. (e) Percentage U937 cells in M phase after treatment with diluent or CPX-351 along with MK-8776. Results are mean ± sd of 4 independent experiments. *Indicates p < 0.01 compared to diluent or treatment with CPX-351 in the presence of MK-8776.
Further experiments, performed in the absence of Q-VD-Ph, examined events at 4–8 hours after addition of CPX-351 to cells. Activation of the ATR/CHK1 pathway was confirmed by showing that CHK1 was phosphorylated on Ser345 (Fig. 1c, second panel), an ATR phosphorylation site required for CHK1 activation23, and on Ser296, an autophosphorylation site (Fig. 1c, top panel, lanes 1 and 6). Consistent with these findings, the CHK1 substrate KAP1 was phosphorylated on Ser473 and the ATR/ATM substrate H2AX was phosphorylated in Ser139 (Supplementary Fig. S1). In addition, the G2/M checkpoint24 was activated, as indicated by a decrease in the percentage of cells staining positive for the mitotic marker phospho-Ser10-Histone H3 after CPX-351 treatment (Fig. 1d,e).
In further experiments, the effects of the inhibitor MK-8776, which is highly selective for CHK125, were examined. When combined with CPX-351, MK-8876 diminished CPX-351-induced CHK1 Ser296 autophosphorylation (Fig. 1c, top panel, lanes 6–10) and KAP 1 Ser473 phosphorylation (Supplementary Fig. S1) as well as increased the levels of CDC25A (Fig. 1c, fourth panel), a protein that is degraded in a CHK1-dependent manner to initiate the replication checkpoint26. Moreover, MK-8776 partially restored the percentage of cells that stained for phospho-Ser10-Histone H3 (Fig. 1d,e), suggesting that the G2/M checkpoint has also been blunted24.
Effect of CHK1 inhibition or downregulation on CPX-351-induced apoptosis
In a parallel set of experiments, we omitted Q-VD-OPh and examined the ability of CPX-351 to kill AML cell lines by assessing DNA cleavage (Fig. 2a–c), phosphatidylserine externalization (Fig. 2d–f), PARP1 cleavage (Fig. 2g) and nuclear fragmentation (Supplementary Fig. S2), four separate hallmarks of apoptosis. For these studies, CPX-351 concentrations corresponding to those achieved in AML patients at the FDA-approved dose (equivalent to 5–10 µM cytarabine over the first 24 h)15 were applied. Each of these assays demonstrated that CPX-351 induced U937 cell apoptosis, which was increased by MK-8776 in a dose-dependent manner. In particular, MK-8776 increased the amount of CPX-351-induced DNA cleavage (Fig. 2a–c), phosphatidylserine externalization (Fig. 2d–f), nuclear fragmentation (Fig. S2) and caspase-mediated proteolysis of PARP1 (Fig. 2g). Additional analyses showed that MK-8776 enhanced the action of cytarabine and daunorubicin individually (Supplementary Fig. S3), demonstrating that MK-8776 impacts the cytotoxicity of both active agents in CPX-351.
Figure 2.

CHK1 inhibitors enhance CPX-351-induced apoptosis in U937 cells. (a–c) U937 cells were treated for 24 h with CPX-351 in the absence and presence of MK-8776, stained with PI and subjected to flow microfluorimetry. (a) DNA histograms from samples treated with diluent, 300 nM MK-8776, CPX-351 corresponding to 10 µM cytarabine, or the combination. (b) Summary of subdiploid DNA content in U937 cells shown in panel a and additional samples in the same experiment. (c) Summary of 10 independent experiments under conditions shown in panel a. Error bar, ±sd. *p < 0.0001 relative to CPX-351 plus diluent. (d–f) U937 cells were treated for 24 h with CPX-351 in the absence and presence of MK-8776, stained with Annexin V and analyzed by flow cytometry. (d) Histograms from samples treated with diluent, 300 nM MK-8776, CPX-351 corresponding to 10 µM cytarabine, or the combination. (e) Summary of annexin V binding in U937 cells shown in panel d and additional samples in the same experiment. (f) Summary of six independent experiments under conditions shown in panel d. Error bar, ± sd. **p = 0.011 relative to CPX-351 plus diluent. (g) After U937 cells were treated for 24 h with diluent, 300 nM MK-8776, CPX-351 at 5 µM cytarabine equivalents, or the combination, whole cell lysates were subjected to SDS-PAGE and blotted for procaspase-3, cleaved caspase 3, or PARP1. HSP90β served as a loading control.
In further experiments, siRNA-mediated CHK1 downregulation also enhanced CPX-351-induced apoptosis (Fig. 3a), confirming that loss of CHK1 signaling rather than an off-target effect of the kinase inhibitor was responsible for the sensitization. Moreover, additional inhibitors with selectivity for CHK1, including rabusertib27 and prexasertib28, also enhanced the cytotoxicity of CPX-351 (Fig. 3b–g), with similar effects demonstrated by analysis of DNA cleavage (Fig. 3b,e), annexin V binding (Fig. 3c,f), or caspase-mediated proteolysis (Fig. 3d,g). These observations argue that enhancement of CPX-351-induced apoptosis is not unique to one CHK1 inhibitor or one assay methodology.
Figure 3.

CHK1 siRNA and additional CHK1 inhibitors enhance CPX-351-induced apoptosis. (a) U937 cells were transfected with two different CHK1 siRNAs (siCHK1) or control luciferase siRNA (siLuc), incubated for 24 h, exposed to the indicated concentrations of CPX-351 for 24 h, stained with PI, and subjected to flow microfluorimetry. Left panel, results of one experiment. Middle panel, immunoblot of cell lysates prepared from cells 48 h after transfection with control or CHK1 siRNAs. Right panel, summarized results of 3 independent experiments. *p < 0.01 relative to control siRNA samples treated with CPX-351. (b–g) U937 cells were treated for 24 h with diluent or CPX-351 in the absence of presence of the indicated concentrations of rabusertib (b) or prexasertib (e) or in 300 nM rabusertib (c,d) or 3 nM prexasertib (f,g) in the absence or presence of CPX-351 at 10 µM cytarabine equivalents (b,c,e,f) or 5 µM cytarabine equivalents (d,g) and examined for subdiploid DNA by flow microfluorimetry (b,e), annexin V binding (c,f) or cleavage of procaspase-3 and PARP1 (d,g). In b and e, left hand panels show dose-response curves from individual experiments. Bar graphs show mean ± sd from 6 (b), 4 (c,f), or 3 (e) independent experiments. ** and ***p < 0.02 and p = 0.003 relative to CPX-351 plus diluent.
CHK1 inhibition also increased CPX-351-induced apoptosis in additional AML lines, including HL-60 (Fig. 4a), ML-2 (Fig. 4b and Supplementary Fig. S4a), THP.1 (Fig. 4c), and ML-1 cells (Supplementary Fig. S4b). In particular, MK-8776 sensitized all of these lines to CPX-351. While not every CHK1 inhibitor was tested in every cell line, sensitization was also observed when CPX-351 was combined with rabusertib (Fig. 4a) or prexasertib (Fig. 4a,b). We did not observe any instance where an AML cell line was sensitized to CPX-351 by MK-8776 but not by other inhibitors. Collectively, these observations suggest that CHK1-mediated resistance to apoptosis is not unique to U937 cells. In contrast, MV-4-11 cells were not appreciably sensitized by CHK1 inhibition (Fig. 4d), consistent with the possibility that these FLT3 mutation-positive cells are resistant to killing as a consequence of some other biochemical process such as alterations in apoptotic signaling29,30.
Figure 4.
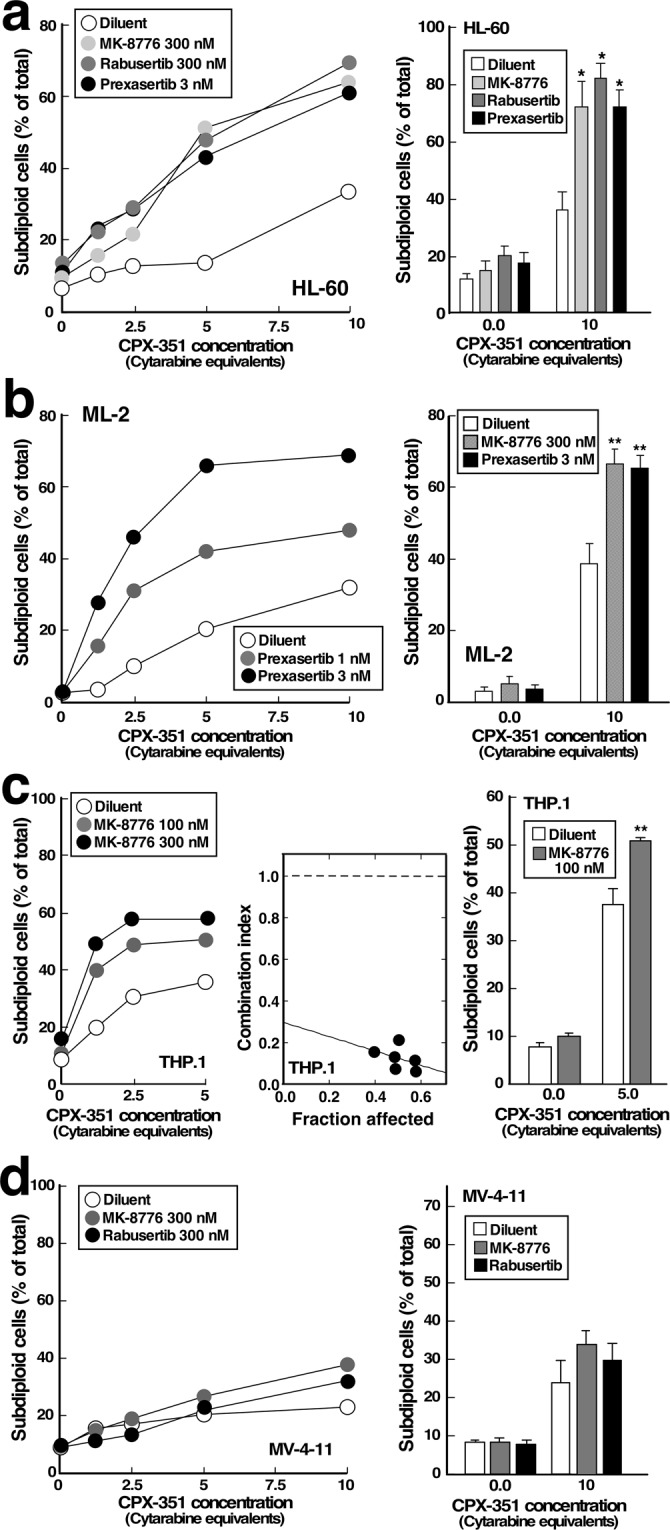
Effect of CHK1 inhibitors on CPX-351-induced apoptosis in additional AML cell lines. HL-60 (a), ML-2 (b), THP.1 (c) or MV-4-11 cells (d) were treated for 24 h with varying concentrations of CPX-351 in the absence of presence of MK-8776, rabusertib or prexasertib as indicated, stained with PI and subjected to flow microfluorimetry. Left panels show results from single experiment. Right panels show summary of 3–6 experiments. Results of single-agent MK-8776 in ML-2 cells are shown in Supplementary Figure S4a. Middle panel in c, plot showing combination index values for experiment shown in left panel. A combination index <1 indicates synergy47. * and **p < 0.002 (n = 4) and p < 0.02 (n = 3) relative to samples treated with CPX-351 plus diluent.
Effect of CHK1 inhibition on colony forming ability of leukemic cells
Additional assays examined the impact of MK-8776 on the antiproliferative effects of CPX-351 using colony-forming assays in soft agar. In these assays, MK-8776 sensitized U937 and HL-60 cell lines to CPX-351 (Fig. 5a,b). Moreover, in primary AML specimens (Supplementary Table S1), MK-8776 sensitized some AML specimens but not others to CPX-351 (Fig. 5c–e). In particular, sensitization occurred in samples that were relatively resistant to CPX-351 (e.g., Fig. 5e, IC90~0.0375 µM cytarabine equivalents) but not in cells that were highly sensitive to CPX-351 (e.g., Fig. 5c, IC90~0.01 µM cytarabine equivalents). Because CPX-351 suppresses normal hematopoiesis16,17, we examined the impact of the combination on normal marrow colony formation as well. As indicated in Supplementary Fig. S5, MK-8776 also sensitized committed normal progenitors to CPX-351, although their sensitivity did not approach that of sensitive AML samples treated with the combination.
Figure 5.

Effects of CPX-351 and MK-8776 on colony formation assays in human AML cell lines and primary AML specimens. (a,b) U937 (a) or HL-60 cells (b) were treated for 24 h with CPX-351 alone and in combination with 600 nM MK-8776, washed, plated in soft agar for 12 days and counted. (c–e) Marrow mononuclear cells from AML patients (Supplementary Table S1) were plated in cytokine-containing Methocult® methylcellulose containing the indicated concentration of CPX-351 in addition to diluent (0.1% DMSO) or 100 nM MK-8776. After a 14-day incubation, leukemic colonies were counted.
Discussion
Results of the present study demonstrate that (1) CPX-351 activates the ATR/CHK1-mediated replication checkpoint, (2) CHK1 signaling contributes to CPX-351 resistance, and (3) small molecule checkpoint kinase inhibitors sensitize AML cell lines and clinical samples to CPX-351 in vitro. These observations complement and extend our earlier studies indicating that CHK1 inhibition enhances the cytotoxicity of cytarabine as a single agent.
Several observations indicate that CPX-351 activates cell cycle checkpoints. First, cells accumulate in S phase after CPX-351 treatment (Fig. 1a,b). Second, increased phosphorylation of CHK1 on Ser345, which reflects ATR activation23, is observed within 4.5 hours (Fig. 1c). Third, increased phosphorylation of CHK1 substrates, including KAP1 Ser473 and CHK1 Ser296, is observed at the same time point, as are decreased CDC25A levels (Figs 1c and S1). All of these changes are consistent with activation of the replication checkpoint31,32. In addition, the number of cells traversing mitosis, as manifested by staining for phospho-Ser10-Histone H3, is also diminished by CPX-351 (Fig. 1d), suggesting activation of the G2/M checkpoint24. These cell cycle perturbations occurred at CPX-351 concentrations below those required to induce apoptosis in a substantial fraction of the cells (cf., Figs 1b and 2b), consistent with earlier results showing that cytarabine as a single agent induces replication checkpoint activation at 10–30 nM but requires 1000 nM to induce apoptosis in AML cell lines over the same 24-hour timeframe in vitro7.
The CPX-351-induced cell cycle perturbations shown in Fig. 1b,e were examined in the presence of the caspase inhibitor Q-VD-OPh to prevent potential apoptosis. While we are aware that Q-VD-OPh treatment has been associated with increased CDKN1A levels in doxorubicin-treated cells33, the work of Rebbaa utilized anthracycline levels 10- to 30-fold higher than those at which cell cycle perturbations were observed in the present study. Moreover, the changes in ATR/CHK1 signaling shown in Figs. 1c and S1 were observed at 4–8 hours in the absence of Q-VD-OPh, arguing that the cell cycle alterations reflect treatment with CPX-351 rather than Q-VD-OPh. Consistent with this interpretation, we were also able to reverse the G2/M checkpoint using a CHK1 inhibitor (Fig. 1d,e).
In further experiments, CPX-351 was used at concentrations achieved at the FDA-approved dose. While some apoptosis was observed with CPX-351 alone, CHK1 knockdown or inhibition increased the apoptosis observed with these higher CPX-351 concentrations in multiple AML cell lines (Figs 2–4). In these experiments, the spectrum of cell lines sensitized by the CHK1 inhibitors was particularly interesting. Among the genomic subsets of AML, those with TP53 mutations have historically exhibited particularly poor clinical outcomes with cytarabine/anthracycline-based induction therapy3–5. Additional studies have suggested that interruption of the replication checkpoint in conjunction with replication stress might be most toxic in cells lacking a G1 checkpoint as a consequence of TP53 loss or mutation34–37. In the present study, we have observed enhanced apoptosis when CHK1 inhibitors are combined with CPX-351 in TP53 mutant (THP.1) and TP53 null (U937, HL-60) AML lines in vitro, consistent with these earlier results. On the other hand, the sensitizing effects of CHK1 inhibitors are not limited to TP53-deficient AML cell lines. Instead, sensitization is also observed in TP53-wildtype lines such as ML-2 (Figs 4b and S4a) and ML-1 (Supplementary Fig. S4b), as would be expected based on current understanding of the role of the replication checkpoint in stabilization of stalled replication forks31,32. Importantly, the sensitizing effects of CHK1 knockdown (Fig. 3a) or CHK1 inhibition (Figs 2, 3 and 4a–c) were often observed at drug concentrations that did not appreciably kill AML cell lines on their own. Small but detectable MK-8776-induced apoptosis was observed in THP.1 cells, permitting analysis of synergy by the median effect method (Fig. 4c). While it was not possible to calculate synergy parameters such as the combination indices in other cell lines such as U937, where CHK1 knockdown or inhibition caused no toxicity, the ability of CHK1 knockdown or inhibition to enhance the effects of CPX-351 without inducing toxicity also meets the definition of synergy38.
As indicated above, the CPX-351 concentrations chosen for this study were based on the levels of CPX-351 achieved during treatment of AML patients with this agent15. In contrast, we chose CHK1 inhibitor concentrations based on the amount of drug required to inhibit CHK1 autophosphorylation and/or enhance CPX-351 activity. Table 1 compares the concentrations that sensitized to CPX-351 in the present study with drug levels sustained in the clinical setting. Our earlier studies, along with those of others, have demonstrated that MK-8776 enhances the cytotoxicity of cytarabine in AML cell lines and clinical samples ex vivo9,39. Importantly, the previously observed sensitization was most evident when cells were exposed to cytarabine for 24 h along with 300–600 nM MK-8776, with slightly higher concentrations required to sensitize in colony forming assays than in assays with apoptosis as an endpoint9. In the present study, sensitization to CPX-351 was also more evident at MK-8776 concentrations of 300 nM and above. In contrast, mean plasma concentrations at 24 h after administration of MK-8776 as a single agent at its maximum tolerated dose were 80 nM (Table 1), which is below the concentration that effectively sensitizes most AML lines. Dose-limiting off-target effects of MK-8776 on the cardiac conduction system precluded further dose escalation in the clinical setting. As a result, sustained exposure to free MK-8776 levels that would be required to sensitize cells to cytarabine or CPX-351 (Table 1)40 cannot readily be achieved clinically.
Table 1.
CHK1 inhibitor concentrations required to sensitize AML lines to CPX-351.
| Inhibitor | Concentration to sensitize to CPX-351 in vitro | Mean plasma concentration 24 h after administration at MTD* | Reference |
|---|---|---|---|
| MK-8776 | 300–600 nM (Figs 2 and 5a) | 30 ng/ml = 80 nM | 40, 48 |
| Rabusertib | 300 nM (Figs 3b and S4a) | 1000 ng/ml = 2.5 µM | 49 |
| Prexasertib | 3 nM (Figs 3c and 4b) | 15 ng/ml = 41 nM | 42 |
*Indicates the mean plasma concentration 24 h after the administration of the indicated inhibitor at the maximum tolerated dose (MTD) as a single agent except in the case of MK-8776, where the MTD with cytarabine is similar to that as a single agent as indicated in the cited references.
On the other hand, we also observed that the recently described CHK1 inhibitor prexasertib28 enhances the cytotoxicity of CPX-351 at low nanomolar concentrations (Figs 3e, 4a,b). Unlike MK-8776, prexasertib lacks detectable cardiac toxicity in the clinical setting at the maximum tolerated dose41. Moreover, prexasertib exposures in the clinical setting42 far exceed those required to sensitize to CPX-351 (Table 1), raising the possibility that further study of CPX-351 in conjunction with prexasertib might be particularly fruitful.
Methods
Materials
Reagents were purchased as follows: MK-8776 and rabusertib (LY2603618) from Chemietek (Indianapolis, IN); cytarabine, daunorubicin and propidium iodide (PI) from Sigma-Aldrich (St. Louis, MO); prexasertib (LY2606368) from Selleckchem (Houston, TX); Q-VD-OPh from SM Biochemicals (Anaheim, CA); murine monoclonal anti-CHK1 (catalog #sc-8408) from Santa Cruz Biotechnology (Santa Cruz, CA); murine monoclonal anti-phospho-Ser473-KAP1 from Biolegend (San Diego, CA); murine monoclonal anti-phospho-Ser139-histone H2AX (catalog #05-636) and rabbit polyclonal anti-phospho-Ser10-Histone H3 from Millipore (Billerica, MA); rabbit polyclonal antibodies to CDC25A (catalog #ab989) and KAP1 (catalog #ab10484) as well as rabbit monoclonal antibodies to CHK2 and phospho-Thr68-CHK2 (catalog #ab109413 and ab32148) from Abcam (Cambridge, MA); anti-caspase 3 (catalog #610323) from BD Biosciences (San Jose, CA); and rabbit polyclonal antibodies to phospho-Ser296-CHK1 (catalog #2349) and phospho-Ser345-CHK1 (catalog #2341) as well as rabbit monoclonal antibodies to glyceraldehyde phosphate dehydrogenase (catalog #2118), cleaved caspase 3 (#9664), PARP1 (catalog #9532), and total histone H2AX (catalog #7631) from Cell Signaling Technology (Beverly, MA). Murine monoclonal antibody H90-10 to heat shock protein 90β (HSP90β) was a kind gift from David Toft (Mayo Clinic, Rochester, MN). CPX-351 was provided by Lawrence Mayer (Celator Pharmaceuticals, Princeton, NJ) prior to the merger of Celator with Jazz Pharmaceuticals.
Cell culture
U937 and THP.1 cells (American Type Culture Collection, Manassas, VA) as well as HL-60 (Robert Abraham, Pfizer, La Jolla, CA), ML-1 (Michael Kastan, Duke University, Durham, NC), ML-2 (James Bogenberger, Mayo Clinic, Scottsdale, AZ) and MV-4-11 cells (Terra Lasho, Mayo Clinic, Rochester, MN) were authenticated by short tandem repeat profiling, reinitiated from frozen stocks at 3-month intervals, and maintained in RPMI 1640 containing 10% FBS, 100 U/ml penicillin G, 100 μg/ml streptomycin, and 2 mM glutamine (medium A) at concentrations below 1 × 106 cells/ml at all times. The TP53 mutation status of each line is as follows: TP53 null: HL-60, U937; TP53 mutant: THP.1 (pR174fs); TP53 wildtype: ML-1, ML-2 and MV-4-11. Aliquots were diluted to 2–4 × 105 cells/ml in medium A and treated with CPX-351 (added from 10X stocks freshly prepared in medium A) and CHK1 inhibitors (added from 1000X stocks in DMSO) for specific assays below.
Small interfering RNAs (siRNAs) were transiently transfected into U937 cells by electroporation (280 V, 10 ms) using a BTX 830 square wave electroporator (BTX, San Diego, CA) under conditions described previously43. siRNAs utilized included non-targeting control siRNA (ThermoFisher, Foster City, CA; catalog #AMB4635,), siCHK1 #1 (5′-GGAGAG-AAGGCAAUAUCCAtt-3′ (Thermo Fisher catalog #106), and siCHK1 #2 5′AAGCGU-GCCGUAGACUGUCCAtt3′(Dharmacon, Lafayette, CO, cat# HACJA-000033).
Analysis of cell cycle distribution and apoptosis
After incubation7 for 24 h with CPX-351 in the absence or presence of CHK1 inhibitors, cells were resuspended in ice cold 0.1% (wt/vol) sodium citrate containing 50 µg/mL PI and 0.1% (wt/vol) Triton X-100, incubated at 4 °C overnight, and analyzed by flow microfluorimetry in the FL2 channel on a Becton Dickinson (Franklin Lakes, NJ) FASCanto II flow cytometer. After collection of 20,000 events, files were analyzed using Modfit (Verity Software, Topsham, ME) for cell cycle distribution or Becton Dickinson CellQuest software for events with <2n DNA content. Alternatively, cells were fixed in 3:1 methanol:acetic acid, dropped onto coverslips and stained with 1 µg/ml Hoechst 33258 in 50% glycerol containing PBS, and examined for apoptotic morphological changes as previously described6,7.
To assess the G2/M checkpoint, cells were treated for 24 h as indicated in individual figures, fixed in 70% (v/v) ethanol, rehydrated in PBS, and simultaneously stained with antibody to phospho-Ser10-Histone H3 and propidium iodide as initially described by Xu et al.24.
Immunoblotting
U937 cells were exposed to the indicated CPX-351 concentrations without or with MK-8776 for 4–8 hours as indicated, washed with PBS, and lysed in 2X SDS-PAGE sample buffer (1 × 107 cells/mL). Lysates (2 × 105 cells/lane) were separated by SDS-PAGE, transferred to Immobilon P, and blotted for the indicated antigens. Alternatively, cells treated with siRNAs or with drugs for 24 hours were lysed in 6 M guanidine hydrochloride under reducing conditions and prepared for SDS-PAGE as described44.
Additional assays
Assays for the ability of AML cell lines to form colonies in 0.3% agar were performed as described7,9.
Leukemia and normal samples
After informed consent was obtained under the aegis of protocols approved by the Mayo Clinic Institutional Review Board and Johns Hopkins University Institutional Review Board, bone marrow aspirates were acquired from AML patients in accordance with relevant guidelines and regulations prior to therapy. Mononuclear cells were isolated on ficoll-Hypaque gradients, washed with ice cold serum-free RPMI 1640 medium, resuspended at 1.5 × 106 cells/ml in Iscove’s modified Dulbecco’s medium containing 20% (vol/vol) heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin G, 100 μg/ml streptomycin, and 2 mM glutamine, and plated at 300,000 cells/plate in MethoCult® methylcellulose (StemCell Technologies, Vancouver, British Columbia) containing the indicated concentrations of CPX-351 and MK-8776. Leukemic colonies were counted after a 14-day incubation45. WHO classification and karyotype were determined during the routine clinical care of the AML patients46.
Anonymized freshly isolated normal marrow samples, obtained from hip arthroplasty specimens, were provided by the Henry J. Predolin Biobank for Hematology Research (Mayo Clinic, Rochester, MN). After cells were cultured in Methocult® for 14 days as described above, normal myeloid colonies were scored45. Results are presented as the sum of BFU-E, CFU-GM and CFU-M, all of which showed similar sensitivities.
Statistical analysis
Experiments were performed 3–10 times independently as indicated in the figure legends. Statistical analyses were performed using StatView v.5 (SAS Institute). Results of various treatments were compared using 2-tailed t tests, with p < 0.05 considered to be statistically significant. Where multiple treatments were compared simultaneously, we conducted analysis of variance with post hoc t tests and applied a Bonferroni correction for multiple comparisons to all p values. Results of multiple experiments are summarized as bar graphs showing mean ± sd. To assess the presence of synergy, combination indices were calculated by the method of Chou and Talalay47 under the assumption that effects were mutually exclusive, which is equivalent to isobologram analysis38. With this method, synergy is indicated by a combination index <1, additivity by a combination index equal to 1, and antagonism by a combination index >147.
Supplementary information
Supplementary information for publication
Acknowledgements
Support for this project was provided, in part, by R01 CA190473 (L.M.K.), T32 GM0072474 (R.L.K.), T32 GM125633 (M.A.K.) and a fellowship from the Mayo Foundation for Education and Research (N.D.V.). We thank D. Toft for anti-HSP90β antibody; Lawrence Mayer for CPX-351, which was provided prior to acquisition of Celator by Jazz Pharmaceuticals; Kevin Peterson and Paula Schneider for assistance with immunoblotting; J. Karp and the members of the Kaufmann lab for advice; and the patients who provided consent for use of their AML samples.
Author Contributions
S.H.K. and L.M.K. conceived the project; N.D.V., H.D., A.M.H., R.L.K., K.S.F., M.A.K., S.H.K. and L.M.K. performed experiments; J.W., A.D.H., and K.W.P. procured patient samples; S.H.K. and L.M.K. wrote the manuscript. All authors reviewed the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Nicole D. Vincelette, Husheng Ding and Amelia M. Huehls contributed equally.
Larry M. Karnitz and Scott H. Kaufmann jointly supervised this work.
Contributor Information
Larry M. Karnitz, Email: Karnitz.Larry@Mayo.edu
Scott H. Kaufmann, Email: Kaufmann.Scott@Mayo.edu
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-40218-0.
References
- 1.Burnett A, Wetzler M, Lowenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol. 2011;29:487–494. doi: 10.1200/JCO.2010.30.1820. [DOI] [PubMed] [Google Scholar]
- 2.Dohner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med. 2015;373:1136–1152. doi: 10.1056/NEJMra1406184. [DOI] [PubMed] [Google Scholar]
- 3.Bowen D, et al. TP53 gene mutation is frequent in patients with acute myeloid leukemia and complex karyotype, and is associated with very poor prognosis. Leukemia. 2009;23:203–206. doi: 10.1038/leu.2008.173. [DOI] [PubMed] [Google Scholar]
- 4.Rucker FG, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119:2114–2121. doi: 10.1182/blood-2011-08-375758. [DOI] [PubMed] [Google Scholar]
- 5.Schoch C, et al. Acute myeloid leukemia with a complex aberrant karyotype is a distinct biological entity characterized by genomic imbalances and a specific gene expression profile. Genes Chromosomes Cancer. 2005;43:227–238. doi: 10.1002/gcc.20193. [DOI] [PubMed] [Google Scholar]
- 6.Loegering D, et al. Rad9 Protects Cells from Topoisomerase Poison-Induced Cell Death. J Biol Chem. 2004;279:18641–18647. doi: 10.1074/jbc.M313536200. [DOI] [PubMed] [Google Scholar]
- 7.Mesa RA, et al. Heat Shock Protein 90 Inhibition Sensitizes Acute Myelogenous Leukemia Cells to Cytarabine. Blood. 2005;106:318–327. doi: 10.1182/blood-2004-09-3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sampath D, et al. Pharmacodynamics of cytarabine alone and in combination with 7-hydroxystaurosporine (UCN-01) in AML blasts in vitro and during a clinical trial. Blood. 2006;107:2517–2524. doi: 10.1182/blood-2005-08-3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schenk EL, et al. Effects of selective checkpoint kinase 1 inhibition on cytarabine cytotoxicity in acute myelogenous leukemia cells in vitro. Clin Cancer Res. 2012;18:5364–5373. doi: 10.1158/1078-0432.CCR-12-0961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.David L, et al. CHK1 as a therapeutic target to bypass chemoresistance in AML. Sci Signal. 2016;9:ra90. doi: 10.1126/scisignal.aac9704. [DOI] [PubMed] [Google Scholar]
- 11.Luo Y, et al. Abrogation of G2 checkpoint specifically sensitize p53 defective cells to cancer chemotherapeutic agents. Anticancer Res. 2001;21:23–28. [PubMed] [Google Scholar]
- 12.Luo Y, et al. Blocking Chk1 expression induces apoptosis and abrogates the G2 checkpoint mechanism. Neoplasia. 2001;3:411–419. doi: 10.1038/sj.neo.7900175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tardi P, et al. In vivo maintenance of synergistic cytarabine:daunorubicin ratios greatly enhances therapeutic efficacy. Leuk Res. 2009;33:129–139. doi: 10.1016/j.leukres.2008.06.028. [DOI] [PubMed] [Google Scholar]
- 14.Gordon MJ, et al. CPX-351 exhibits potent and direct ex vivo cytotoxicity against AML blasts with enhanced efficacy for cells harboring the FLT3-ITD mutation. Leuk Res. 2017;53:39–49. doi: 10.1016/j.leukres.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 15.Feldman EJ, et al. Pharmacokinetics of CPX-351; a nano-scale liposomal fixed molar ratio formulation of cytarabine:daunorubicin, in patients with advanced leukemia. Leuk Res. 2012;36:1283–1289. doi: 10.1016/j.leukres.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 16.Feldman EJ, et al. First-in-man study of CPX-351: a liposomal carrier containing cytarabine and daunorubicin in a fixed 5:1 molar ratio for the treatment of relapsed and refractory acute myeloid leukemia. J Clin Oncol. 2011;29:979–985. doi: 10.1200/JCO.2010.30.5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lancet JE, et al. Phase 2 trial of CPX-351, a fixed 5:1 molar ratio of cytarabine/daunorubicin, vs cytarabine/daunorubicin in older adults with untreated AML. Blood. 2014;123:3239–3246. doi: 10.1182/blood-2013-12-540971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perl AE. The role of targeted therapy in the management of patients with AML. Hematology Am Soc Hematol Educ Program. 2017;2017:54–65. doi: 10.1182/asheducation-2017.1.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaufmann SH. Induction of Endonucleolytic DNA Cleavage in Human Acute Myelogenous Leukemia Cells by Etoposide, Camptothecin, and Other Cytotoxic Anticancer Drugs: A Cautionary Note. Cancer Res. 1989;49:5870–5878. [PubMed] [Google Scholar]
- 20.Enari M, et al. A Caspase-Activated DNase That Degrades DNA During Apoptosis, and Its Inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- 21.Caserta TM, Smith AN, Gultice AD, Reedy MA, Brown TL. Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis. 2003;8:345–352. doi: 10.1023/A:1024116916932. [DOI] [PubMed] [Google Scholar]
- 22.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Q, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. doi: 10.1101/gad.840500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu B, Kim ST, Lim DS, Kastan MB. Two molecularly distinct G(2)/M checkpoints are induced by ionizing irradiation. Mol Cell Biol. 2002;22:1049–1059. doi: 10.1128/MCB.22.4.1049-1059.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guzi TJ, et al. Targeting the Replication Checkpoint Using SCH 900776, a Potent and Functionally Selective CHK1 Inhibitor Identified Via High Content Screening. Mol Cancer Ther. 2011;10:591–602. doi: 10.1158/1535-7163.MCT-10-0928. [DOI] [PubMed] [Google Scholar]
- 26.Mailand N, et al. Rapid destruction of human Cdc25A in response to DNA damage. Science. 2000;288:1425–1429. doi: 10.1126/science.288.5470.1425. [DOI] [PubMed] [Google Scholar]
- 27.Klaeger, S. et al. The target landscape of clinical kinase drugs. Science358 (2017). [DOI] [PMC free article] [PubMed]
- 28.King C, et al. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol Cancer Ther. 2015;14:2004–2013. doi: 10.1158/1535-7163.MCT-14-1037. [DOI] [PubMed] [Google Scholar]
- 29.Lisovsky M, et al. Flt3 ligand stimulates proliferation and inhibits apoptosis of acute myeloid leukemia cells: regulation of Bcl-2 and Bax. Blood. 1996;88:3987–3997. [PubMed] [Google Scholar]
- 30.Levis M, et al. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood. 2002;99:3885–3891. doi: 10.1182/blood.V99.11.3885. [DOI] [PubMed] [Google Scholar]
- 31.Ashwell S, Zabludoff S. DNA damage detection and repair pathways–recent advances with inhibitors of checkpoint kinases in cancer therapy. Clin Cancer Res. 2008;14:4032–4037. doi: 10.1158/1078-0432.CCR-07-5138. [DOI] [PubMed] [Google Scholar]
- 32.Karnitz LM, Zou L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin Cancer Res. 2015;21:4780–4785. doi: 10.1158/1078-0432.CCR-15-0479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rebbaa A, Zheng X, Chou PM, Mirkin BL. Caspase inhibition switches doxorubicin-induced apoptosis to senescence. Oncogene. 2003;22:2805–2811. doi: 10.1038/sj.onc.1206366. [DOI] [PubMed] [Google Scholar]
- 34.Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell. 2007;11:175–189. doi: 10.1016/j.ccr.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reaper PM, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol. 2011;7:428–430. doi: 10.1038/nchembio.573. [DOI] [PubMed] [Google Scholar]
- 36.Morgan MA, Lawrence TS. Molecular Pathways: Overcoming Radiation Resistance by Targeting DNA Damage Response Pathways. Clin Cancer Res. 2015;21:2898–2904. doi: 10.1158/1078-0432.CCR-13-3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwok M, et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood. 2016;127:582–595. doi: 10.1182/blood-2015-05-644872. [DOI] [PubMed] [Google Scholar]
- 38.Berenbaum MC. What is Synergy? Pharmacol Rev. 1989;41:93–141. [PubMed] [Google Scholar]
- 39.Montano R, Chung I, Garner KM, Parry D, Eastman A. Preclinical development of the novel Chk1 inhibitor SCH900776 in combination with DNA-damaging agents and antimetabolites. Mol Cancer Ther. 2012;11:427–438. doi: 10.1158/1535-7163.MCT-11-0406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karp JE, et al. Phase I and pharmacologic trial of cytosine arabinoside with the selective checkpoint 1 inhibitor Sch 900776 in refractory acute leukemias. Clin Cancer Res. 2012;18:6723–6731. doi: 10.1158/1078-0432.CCR-12-2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee, J. M. et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: a first-in-class proof-of-concept phase 2 study. Lancet Oncol (2018). [DOI] [PMC free article] [PubMed]
- 42.Hong D, et al. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. J Clin Oncol. 2016;34:1764–1771. doi: 10.1200/JCO.2015.64.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ding H, et al. Histone deacetylase inhibitors interrupt HSP90*RASGRP1 and HSP90*CRAF interactions to upregulate BIM and circumvent drug resistance in lymphoma cells. Leukemia. 2017;31:1593–1602. doi: 10.1038/leu.2016.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaufmann SH, et al. Altered Formation of Topotecan-Stabilized Topoisomerase I-DNA Adducts in Human Leukemia Cells. Blood. 1997;89:2098–2104. [PubMed] [Google Scholar]
- 45.Eaves, C. & Lambie, K. Atlas of human hematopoietic colonies. (StemCell Technologies Inc., 1995).
- 46.Knorr KLB, et al. MLN4924 induces Noxa upregulation in acute myelogenous leukemia and synergizes with Bcl-2 inhibitors. Cell Death Differ. 2015;22:2133–2142. doi: 10.1038/cdd.2015.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chou T-C, Talalay P. Quantitative Analysis of Dose-Effect Relationships: The Combined Effects of Multiple Drugs or Enzyme Inhibitors. Adv Enzyme Regulation. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 48.Daud AI, et al. Phase I dose-escalation trial of checkpoint kinase 1 inhibitor MK-8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. J Clin Oncol. 2015;33:1060–1066. doi: 10.1200/JCO.2014.57.5027. [DOI] [PubMed] [Google Scholar]
- 49.Weiss GJ, et al. Phase I dose-escalation study to examine the safety and tolerability of LY2603618, a checkpoint 1 kinase inhibitor, administered 1 day after pemetrexed 500 mg/m(2) every 21 days in patients with cancer. Invest New Drugs. 2013;31:136–144. doi: 10.1007/s10637-012-9815-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information for publication