

Abstract
Nanoparticles (NPs) have been utilized to deliver drugs to the intestinal epithelium in vivo. Moreover, NPs derived from edible plants are less toxic than synthetic NPs. Here, we utilized ginger NP-derived lipid vectors (GDLVs) in a proof-of-concept investigation to test the hypothesis that inhibiting expression of divalent metal-ion transporter 1 (Dmt1) would attenuate iron loading in a mouse model of hereditary hemochromatosis (HH). Initial experiments using duodenal epithelial organ cultures from intestine-specific Dmt1 knockout (KO) (Dmt1int/int) mice in the Ussing chamber established that Dmt1 is the only active iron importer during iron-deficiency anemia. Further, when Dmt1int/int mice were crossed with mice lacking the iron-regulatory hormone, hepcidin (Hepc−/−), iron loading was abolished. Hence, intestinal Dmt1 is required for the excessive iron absorption that typifies HH. Additional experiments established a protocol to produce GDLVs carrying functional Dmt1 small interfering RNAs (siRNAs) and to target these gene delivery vehicles to the duodenal epithelium in vivo (by incorporating folic acid [FA]). When FA-GDLVs carrying Dmt1 siRNA were administered to weanling Hepc−/− mice for 16 days, intestinal Dmt1 mRNA expression was attenuated and tissue iron accumulation was blunted. Oral delivery of functional siRNAs by FA-GDLVs is a suitable therapeutic approach to mitigate iron loading in murine HH.
Keywords: hepcidin knockout mice, hepcidin, serum ferritin, Slc11a2, Ussing chamber, Dmt1 intestine-specific knockout mice, NTBI
Graphical Abstract
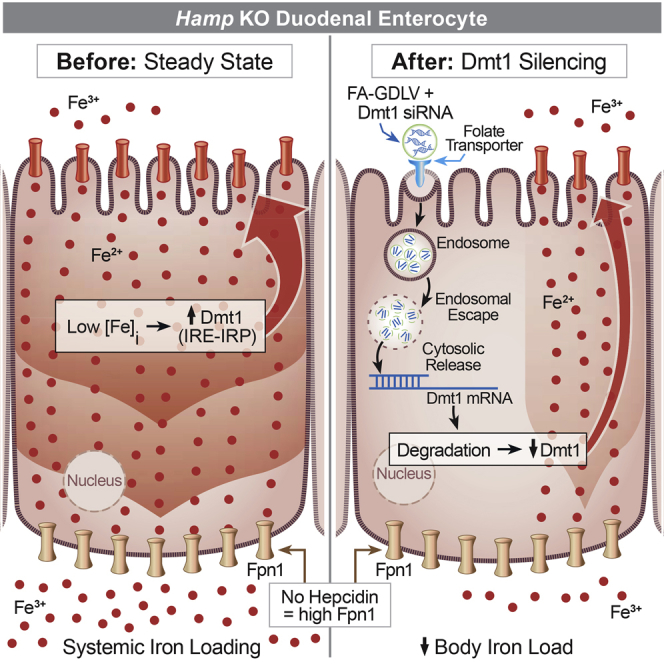
Collins and colleagues developed a novel experimental technique to mitigate excess iron accumulation in hepcidin knockout (KO) mice, which model human hereditary hemochromatosis. The approach involved oral intragastric gavage of lipid nanoparticles derived from edible ginger to deliver small interfering RNAs (siRNAs) targeting the main intestinal iron transporter Dmt1.
Introduction
Regulation of intestinal iron absorption is critical to maintain systemic iron homeostasis. Dietary iron exists in several molecular forms, with heme and non-heme (or inorganic) iron being predominant. Although heme iron makes up on average 10%–15% of iron intake in omnivorous humans, its higher absorption efficiency allows it to contribute up to 40% of total absorbed iron.1 Despite this, the processes and proteins that mediate heme iron absorption in humans and other mammals are not understood in detail. The mechanistic details of absorption of the most abundant dietary form of iron (i.e., non-heme) have, however, been clarified recently.2 First, ferric non-heme iron is reduced by duodenal cytochrome B (and endogenous and dietary factors); then, ferrous iron is transported into enterocytes by divalent metal-ion transporter 1 (DMT1). Ferrous iron may then be utilized in these cells or exported by ferroportin 1 (FPN1), followed by oxidation by hephaestin. Ferric iron then binds to transferrin in the interstitial fluids for delivery to the liver in the portal blood circulation. Hepcidin, the liver-derived, iron-regulatory hormone, modulates intestinal iron absorption by binding to and triggering internalization and degradation of FPN1 in duodenal enterocytes. Iron export from these cells is the rate-limiting step in iron absorption.
The iron import step mediated by Dmt1 is the focus of this investigation. Mice (microcytic anemia [mk])3, 4 and rats (Belgrade [b])5, 6 expressing a dysfunctional Dmt1 protein and global Slc11a2 (encoding Dmt1) knockout (KO) mice7 suffer from severe iron-deficiency anemia (IDA). Elucidating the causes of the iron deficiency in these animal models is complex, because Dmt1 also functions in iron acquisition from transferrin by developing erythrocytes (and other cells),8 and it may also facilitate iron reabsorption in the renal tubules.9 To clarify the specific contribution of Dmt1 to total iron flux, mice lacking Dmt1 only in the intestinal epithelium were generated (Dmt1int/int).7 These mice are severely anemic. When the anemia was corrected by iron supplementation, intestinal iron absorption was drastically reduced in these mice.10 Yet, paradoxically, most mutant animals live to ∼7 months of age without iron supplementation. Survival of these mice into mid-adulthood could be because of the existence of other, complementary intestinal iron transport systems. We therefore initially sought to test the hypothesis that additional, compensatory iron transport mechanisms exist when Dmt1 function is diminished. Accordingly, we optimized the ex vivo Ussing chamber technique to investigate intestinal iron transport in duodenal epithelial organ cultures isolated from intestine-specific Dmt1 KO (Dmt1int/int) mice and phenotypically normal littermates (Dmt1fl/fl). Because Dmt1int/int mice are anemic, and anemia stimulates intestinal iron absorption, controls were phlebotomized prior to experimentation to render the drive for intestinal iron transport equivalent between genotypes.
We next tested the hypothesis that intestinal Dmt1 mediates the excessive iron accumulation in murine hereditary hemochromatosis (HH). Although this possibility has been experimentally considered by previous investigators, definitive studies have not been reported to date. For example, HFE mice, which model the most common form of human HH,11 did not load iron when they carried an inactivating mutation in Slc11a2 (in mk mice) or when they were crossed with global Slc11a2 KO mice.7, 12 Based upon these observations, the authors suggested that impaired intestinal iron absorption explained the lack of an iron-overload phenotype; surprisingly, though, intestinal iron transport was not quantified. This omission is of concern because Dmt1 is expressed in several cell types and has multiple iron-related functions, making interpretation of the data in these studies challenging. Our experimental approach here was designed to eliminate the potential confounding influence of Dmt1 function in other organs by studying mice lacking Dmt1 only in the intestine. We also used a different model of HH, hepcidin KO mice, which represent early-onset (i.e., juvenile) iron loading in humans.13 Although mutations in Hamp (encoding hepcidin) represent a rare cause of HH in humans,14 observations in this most severe iron-loading condition should be applicable to other (less severe) forms of the disease. The iron-overload phenotype was thus assessed in hepcidin KO mice with and without intestinal Dmt1 function intact.
The final set of experiments was designed to test the hypothesis that blunting intestinal Dmt1 expression and activity will mitigate iron loading in murine HH. A therapeutic approach to inhibit intestinal Dmt1 function in vivo was thus developed and tested in hepcidin KO mice. The approach utilized a nanoparticle (NP)-based delivery system that has been used to target hormones, drugs, and small interfering RNAs (siRNAs) to the intestinal epithelium.15, 16, 17, 18 More recently, exosome-like NPs have been isolated from edible plants and utilized to produce NP lipid vectors,19, 20, 21, 22 circumventing some of the toxic properties of synthetic lipid NPs. Here, we utilized ginger NP-derived lipid vectors (GDLVs) to deliver functional Dmt1 (or negative control [NC]) siRNA to the duodenal epithelium of weanling hepcidin KO mice by oral intragastric gavage. After 16 daily treatments, iron loading was assessed using appropriate experimental procedures.
Results
The Ussing Chamber Is Suitable to Quantify Iron Transport in Duodenal Organ Cultures
To determine the optimal dose of 59Fe to be used for Ussing chamber analyses, different doses of 59Fe (15–40 μCi) were initially used in experiments with duodenal epithelial tissues isolated from wild-type (WT) mice. We found that adding 15 μCi of 59Fe to the mucosal side of the chamber was sufficient for detection of radioactivity in the buffer in the opposite chamber (data not shown). We then tested iron transport in duodenum and compared it with mid-jejunum. Results showed that iron flux in the duodenum was substantially higher, with most of the iron transport occurring within 1 h (data not shown). Also, to determine the appropriate concentration of cold Fe substrate (FeSO4) to use, we performed an iron substitution study. The addition of increasing concentrations of FeSO4 (100–2,000 μM) to Ringer’s buffer (pH 7.4) revealed the saturation kinetics of Fe Flux (Figure S1). The Michaelis constant (Km) and maximal rate (Vmax) were calculated by fitting data to the Hill equation using Origin software (v8.1). The Km was 540 μM and Vmax was 7.7 μEq/cm2/h. To ensure that iron transport was functioning near capacity, we used 1,000 μM FeSO4 for subsequent experiments. To define optimal conditions for Fe transport in the Ussing chamber system, we examined buffers with different pHs (Table S1). Buffers were first selected based on their pKa and then tested by acid-base titration. Results showed that Fe flux was significantly higher at pH 6.5 (Figure 1A); therefore, pH 6.5 was used for subsequent analyses. Redoing iron flux measurements at this pH while varying FeSO4 concentrations (100–2,000 μM) (Figure 1B) yielded Km and Vmax as 909 μM and 9.6 μEq/cm2/h, respectively. Again, to ensure that iron transport was functioning near capacity, 1,500 μM FeSO4 was used for the remaining experiments.
Figure 1.

Intestinal DMT1 Is Required for Vectorial Iron Flux across the Duodenal Epithelium during IDA
Duodenal epithelial sheets isolated from adult male WT mice were used initially to determine the optimal conditions for assessing iron flux in the Ussing chamber and then to define kinetic parameters of iron transport. Iron flux was then assessed in anemic adult Dmt1fl/fl and Dmt1int/int mice. Shown is iron (59Fe) flux at pH 5.0. 6.5, and 7.4 in WT mice in the presence of 1,000 μM FeSO4 (n = 6–9 mice/pH group) (A). Iron flux at different iron concentrations (at pH 6.5) in WT mice (n = 3–5 at each iron concentration) was subsequently measured (B), and kinetic parameters were calculated (see values in the upper left corner). Serum Hb concentrations in Dmt1fl/fl mice with facial vein bleeding and untreated Dmt1int/int mice are also shown (n = 5–6/group) (C). The dashed line indicates the approximate Hb concentration in untreated Dmt1fl/fl mice (for comparison). Iron flux was assessed in duodenal sheets isolated from these mice in the presence of 1,500 μM FeSO4 at pH 6.5 (n = 5–6 per group) (D). p = 0.345 for Fe flux being >0 in Dmt1int/int mice. Data were analyzed by one-way ANOVA (A) or t tests (C and D) and are presented as boxplots. Groups labeled with different letters are different from one another: (p = 0.0005) (A); ****p < 0.0001 (C); **p < 0.01 (D).
DMT1 Is the Main Intestinal Iron Importer in Mice
Having established the optimal conditions to quantify iron flux in the Ussing chamber, we next sought to test iron transport activity in Dmt1int/int mice. Daily, for 3 days prior to experimentation, Dmt1fl/fl mice were bled from the facial vein to induce anemia. After phlebotomy, serum hemoglobin (Hb) concentrations were reduced from the normal level (i.e., ∼15.0 g/dL) to ∼8.5 g/dL in control mice (Figure 1C). This manipulation ensured that the drive for intestinal iron absorption would be similar across genotypes, because Dmt1int/int mice are also anemic (and anemia stimulates iron transport2). Experimental results demonstrated that Fe flux was essentially nil for Dmt1int/int mice (Figure 1D). Thus, DMT1 is the predominant duodenal iron importer during anemia.
Hepc−/− Mice Lacking Intestinal DMT1 Grow Slower and Have Enlarged Spleens and Hearts
Hepc KO mice on the C57BL/6 genetic background were backcrossed to mice on the 129S6 genetic background for seven generations, thus producing mice that were >99% congenic for the 129S6 background. Hepc+/− mice were then bred with Dmt1int/+ mice (already 129S6 genotype). Littermate male and female mice of four genotypes were subsequently studied (Hepc+/+/Dmt1fl/fl, Hepc−/−/Dmt1fl/fl, Hepc−/−/Dmt1int/+, and Hepc−/−/Dmt1int/int). During breeding, we noticed that the Hepc−/−/Dmt1int/int mice were smaller and had pale extremities as adults. At 7 weeks of age, body and liver weights of Hepc−/−/Dmt1int/int mice were decreased with females also exhibiting decreased measures relative to males (Figures 2A and 2B). Also, spleens and hearts were enlarged (Figures 2C and 2D) in the double KOs, but no difference was noted between sexes for these parameters. Overall, these data exemplified impaired growth and alterations in organ weights in Hepc−/− mice lacking intestinal DMT1. These pathological perturbations are consistent with severe iron deficiency.
Figure 2.

Ablation of Intestinal DMT1 in Hepc−/− Mice Causes Reductions in Body and Liver Weights and Increases in Spleen and Heart Weights
Body and organ weights were assessed in Hepc+/+/Dmt1fl/fl, Hepc−/−/Dmt1fl/fl, Hepc−/−/Dmt1int/+, and Hepc−/−/Dmt1int/int mice (male and female littermates) at 7 weeks of age. Shown are body weight (A) and relative liver (B), spleen (C), and heart (D) weights. Data were analyzed by two-way ANOVA and are presented as boxplots for n = 6–8 mice. Data shown in (C) and (D) were log10 transformed because of unequal variance before performing statistical analyses.
Intestine-Specific Ablation of DMT1 Increases Erythropoietic Demand in Hepc KO Mice
Blood Hb and hematocrit (Hct) levels in adult Hepc−/−/Dmt1int/int mice were significantly lower than in the other genotypes, but there was no difference between sexes (Figures 3A and 3B). In plasma, total iron-binding capacity (TIBC) was higher in Hepc−/−/Dmt1int/int mice, whereas transferrin saturation (TSAT) was drastically reduced (Figures 3C and 3D). Again, no sex differences were noted for these parameters. Furthermore, renal erythropoietin (EPO) mRNA and protein levels increased, and bone marrow erythroferrone (Erfe) mRNA expression was elevated in Hepc−/−/Dmt1int/int mice (Figures 3E–3G), which is indicative of increased erythropoietic demand.23 Collectively, these observations demonstrate that intestinal DMT1 is required to prevent the development of anemia in Hepc−/− mice.
Figure 3.

Hepc−/− Mice Develop Severe Anemia When Intestinal Dmt1 Is Ablated
Hematological parameters and biomarkers of erythropoietic demand were assessed in Hepc+/+/Dmt1fl/fl, Hepc−/−/Dmt1fl/fl, Hepc−/−/Dmt1int/+, and Hepc−/−/Dmt1int/int mice (male and female littermates) at 7 weeks of age. Shown are Hb (A) and Hct (B) levels in whole blood. Plasma TIBC (C), TSAT (D), and plasma EPO protein levels (E) are also shown. Renal Epo (F) and bone marrow Erfe (G) mRNA expression was quantified by qRT-PCR. Data were analyzed by two-way ANOVA and are presented as boxplots for n = 6–8 (A and B) or 5–6 (C–G) mice/group. Groups labeled with different letters are different from one another (G). Data shown in (A), (B), and (E)–(G) were log10 transformed because of unequal variance before statistical analyses were performed.
Iron Loading Was Abolished in Hepc−/− Mice Lacking Intestinal DMT1
Tissue iron overload occurs in Hepc−/− mice because intestinal iron absorption is inappropriately elevated.24 While non-heme iron content was significantly increased in serum and all tissues tested in Hepc KO mice (as expected), lack of intestinal DMT1 prevented tissue iron accumulation in double-KO mice (Figures 4A–4E). Moreover, in the spleen, iron was depleted in all Hepc−/− mice, irrespective of whether they expressed functional intestinal DMT1 (Figure 4F). This was expected because it is well established that lack of Hepc leads to splenic iron depletion (from macrophages) because of high FPN1-mediated, iron-export activity.24 Iron loading in Hepc KO mice thus requires intestinal Dmt1.
Figure 4.

Lack of Intestinal DMT1 Prevents Iron Accumulation in Plasma and Tissues of Hepc−/− Mice
Non-heme iron levels were assessed in Hepc+/+/Dmt1fl/fl, Hepc−/−/Dmt1fl/fl, Hepc−/−/Dmt1int/+, and Hepc−/−/Dmt1int/int mice (male and female littermates) at 7 weeks of age. Shown are non-heme iron levels in plasma (A), liver (B), heart (C), kidney (D), pancreas (E), and spleen (F). Data were analyzed by two-way ANOVA and are presented as boxplots for n = 6–8 mice/group. Groups labeled with different letters are different from one another (B–D). Data shown in (B)–(F) were log10 transformed because of unequal variance before statistical analyses were performed.
Characterization of Ginger-Derived NPs and NP Lipid Vectors
Consistent with a previously published method of isolating and purifying ginger-derived NPs (GDNPs),25 NPs from the 30%–45% interface of the sucrose gradient were identified as GDNPs (Figure 5A). The average GDNP size was 202.3 nm, as determined by dynamic light scattering (DLS) (Figure 5B). The spherical shape and size of GDNPs were confirmed by transmission electron microscopy (TEM) and atomic force microscopy (AFM) (Figures 5C and 5D). The lipid profile of GDNPs was as follows: 45.1% phosphatidic acid (PA), 29.8% monogalactosyldiacylglycerol (MGDG), 15.6% digalactosyldiacylglycerol (DGDG), and other lipids in smaller amounts (Figure 5E). Lipids extracted from GDNPs spontaneously assemble into GDLVs, as previously described.16 The average size of GDLVs was 179.3 nm, as determined by DLS (Figure S2A). GDLVs were further confirmed to be nanosized and spheroid when visualized by TEM and AFM (Figures S2B and S2C).
Figure 5.

Characterization of Ginger-Derived Nanoparticles
Three bands were formed after step sucrose gradient ultracentrifugation (A); GDNPs from 30%/45% interface were measured by dynamic light scattering (DLS) (B) and visualized by transmission electron microscopy (TEM) (C) and atomic force microscopy (AFM) (D). Also shown is the lipid profile of GDNPs (n = 5) (E). DGDG, digalactosyldiacylglycerol; LPC, lysophosphatidylcholine; LPE, lysophosphatidylethanolamine; LPG, lysophosphatidylglycerol; MGDG, monogalactosyldiacylglycerol; PA, phosphatidic acid; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PG, phosphatidylglycerol; PI, phosphatidylinositol; PS, phosphatidylserine.
GDLVs Are Non-toxic to Cultured Intestinal Epithelial Cells
Synthetic liposomes or lipid NPs can be toxic when used in vivo or in vitro. To test the biocompatibility of GDLVs, the MTT assay was first used to measure viability of mouse Colon-26 cells after 24- and 48-h exposure to GDLVs or DC-Chol/DOPE (a synthetic liposome preparation used as a control). This experiment showed that GDLV exposure had minimal effects on the viability of Colon-26 cells, as compared with DC-Chol/DOPE, which showed notable toxicity at both time points (Figure S3A). Second, we monitored barrier function of Caco2-BBE human colorectal cancer cells by electrical cell-substrate impedance sensing (ECIS). After cells formed an intact monolayer, 100 μM GDLVs or DC-Chol/DOPE (or PBS as a control) was added to the culture media, and transepithelial electrical resistance (TEER) was measured. TEER was unaffected by GDLV exposure (i.e., similar to control), whereas it significantly decreased in cells exposed to DC-Chol/DOPE (Figure S3B). GDLVs are thus biocompatible with cultured mouse and human intestinal epithelial cells, providing an impetus for in vivo testing (as described below).
GDLVs Mediate Uptake of Functional Dmt1 siRNAs into Murine Intestinal Epithelial Cells
After exposing Colon-26 cells to GDLVs or GDLVs loaded with fluorescein isothiocyanate (FITC)-tagged siRNA (green) for 6 h, cells were stained with DAPI (blue) for visualization of nuclei and with phalloidin-tetramethylrhodamine (TRITC) (red) for actin. Confocal microscopic imaging revealed that green dots (siRNA labeled with FITC [siRNA-FITC]) were present inside the GDLVs-siRNA-treated cells (Figure 6A), but not in cells treated with empty GDLVs. Colon-26 cells were also treated with GDLVs-siRNA-FITC (10 and 20 nM) to assess cellular uptake of siRNAs by flow cytometry. The cell uptake efficiency increased from 2 to 6 h and reached more than 50% at both concentrations (Figure S4). Furthermore, to identify a functional Dmt1 siRNA, we used HEK293 cells that overexpressed mouse DMT1 when treated with Dox.26 Three different commercial Dmt1 siRNAs were tested. Dmt1 siRNA s70783 from Life Technologies showed the maximal knockdown percentage at ∼65% (Figure 6B) (data not shown for other siRNAs). To determine whether GDLVs can effectively deliver this functional Dmt1 siRNA into the cell cytoplasm, we incubated HEK293 and Colon-26 cells with Dmt1 siRNA-loaded GDLVs or Dmt1 siRNA-Lipofectamine for 48 h. In Colon-26 cells, both approaches decreased Dmt1 mRNA expression by ∼75%, as compared with NC siRNA-transfected cells (Figure 6B). In HEK293 cells, after treatment with Dox, Dmt1 mRNA expression increased about 17-fold in cells treated with NC-siRNA compared with control cells (i.e., not exposed to Dox). Dmt1 mRNA expression decreased ∼65% compared with NC-siRNA in the presence of Dox, after treatment with Dmt1 siRNA delivered by GDLVs or Lipofectamine (Figure 6C). These observations proved that GDLVs can deliver Dmt1 siRNA into cells in vitro and effectively knock down Dmt1 expression. This finding provided additional justification for in vivo experimentation.
Figure 6.

Ginger Nanoparticle-Derived Lipid Vectors Effectively Deliver siRNAs into Colon-26 and HEK293 Cells
Colon-26 cells were treated with GDLVs only or with GDLVs carrying FITC-labeled (control) siRNAs (green) for 6 h and then processed for confocal microscopy (A). Fixed cells were stained with DAPI (blue) for visualization of nuclei and with phalloidin-TRITC (red) for actin. Colon-26 (B) and Dmt1-overexpressing HEK293 cells (C) were untreated (control) or were transfected with NC- or Dmt1-siRNAs delivered by Lipofectamine (Dmt1-Lipo) or GDLVs (Dmt1-GDLVs). Dmt1 mRNA expression was subsequently assessed in the cells by qRT-PCR. Data were analyzed by one-way ANOVA and are presented as mean ± SD for n = 3 individual experiments. Groups labeled with different letters are different from one another (p < 0.0001). Dox, doxycycline.
FA-GDLVs Have Similar Biochemical Properties to GDLVs and Effectively Target the Duodenum In Vivo
Our aim was in vivo delivery of functional Dmt1 siRNA to epithelial cells of the duodenum and proximal jejunum, where intestinal iron absorption predominantly occurs (and where DMT1 is most highly expressed). Because GDLVs have been previously shown to mainly target the distal small intestine and colon,16, 25 we considered experimental approaches to more effectively target the proximal small intestine. The B vitamin folate is absorbed in the duodenum and jejunum by the proton-coupled folate transporter (PCFT),27 and the supplemental form of folate, folic acid (FA), has been used for enhancing NP absorption and bioavailability.28 We thus produced FA-GDLVs and then experimentally analyzed them to determine whether they had similar biophysical properties to GDLVs. We first demonstrated that, like GDLVs, FA-GDLVs formed a stable complex with siRNAs and protected them from degradation by RNase (Figure S5). Moreover, the size and zeta potential of FA-GDLVs and GDLVs, as assessed by DLS, was almost identical (Figure S6). Biocompatibility of FA-GDLVs and GDLVs was also very similar in that FA-GDLV exposure did not affect growth or proliferation of Colon-26 cells, as determined by MTT assay (Figure S7).
We then administered fluorescent-tagged FA-GDLVs, or GDLVs for comparison, to live mice. Oral gavage of DiIC18(7) (1,1’-dioctadecyl-3,3,3’,3’-tetramethylindotricarbocyanine iodide) (DiR)-labeled FA-GDLVs (or GDLVs) to adult mice and subsequent imaging of the duodenum revealed that FA-GDLVs had enhanced retention in the duodenal epithelium (as compared with GDLVs) (Figure 7A). This approach was thus successful at targeting the duodenal epithelium in live mice. Importantly, we also assessed fluorescence in other tissues to determine whether FA-GDLVs escaped the gut. No fluorescence was detected in heart, spleen, lung, liver, or kidney (Figure S8), demonstrating that orally delivered FA-GDLVs do not enter the portal blood circulation or lymph system.
Figure 7.

Oral Administration of FA-GDLVs Targeted the Upper GI Tract and Time Course of Iron Loading in Hepc−/− Mice
Six-week-old male mice were administered PBS, fluorescent-tagged GDLVs, or fluorescent-tagged FA-GDLVs by oral intragastric gavage. The proximal small bowel was imaged 4, 8, and 12 h after administration (the duodenum is at the top of the pictures) (A). Liver non-heme iron content was measured in both sexes of WT and Hepc−/− mice at different ages (2–9 weeks) (B). Data were analyzed by one-way ANOVA and are presented as boxplots for n = 4–5 mice/group. *p < 0.05; **p < 0.01; ****p < 0.0001, indicating differences as compared with WT mice at each age. FA, folic acid.
Both Sexes of Hepc KO Mice Begin Loading Hepatic Iron at 2–3 Weeks of Age
Next, because we sought to block iron loading preemptively in this mouse model of severe HH, the time course for iron loading in Hepc−/− mice had to be established to determine when the therapeutic intervention should begin. We therefore characterized the iron loading pattern in male and female Hepc−/− mice at different ages. Results showed that iron began accumulating in the liver between 2 and 3 weeks of age in both sexes (Figure 7B), consistent with considering this mouse mutant a model for juvenile HH. Given the similar, early chronology of iron loading in both sexes, we randomly chose females for further investigation.
FA-GDLVs-Dmt1 siRNA Treatment Does Not Alter Erythropoietic Biomarkers or Cause Inflammation
Prior to assessing the ability of FA-GDLVs to deliver functional Dmt1 siRNA in vivo, we sought to assess possible negative physiological outcomes associated with FA-GDLV administration. Serum Hb concentrations, Hct levels, and tissue weights were unaffected by FA-GDLV exposure (Figure S9). Bone marrow Erfe and Tfr1 mRNA expression was also unaltered, suggesting that erythropoietic demand and bone marrow iron levels were unaffected by the treatment (Figures S10C and S10D). Moreover, because serum ferritin is an acute-phase reactant known to be induced by proinflammatory cytokines,29 and serum ferritin levels did not increase in treated mice (Figure 8B), an inflammatory response likely did not occur. Supporting this inference, there was no change in TNF-α and IL-6 mRNA expression in the liver (Figures S10A and S10B), or myeloperoxidase (MPO) activity in the duodenal epithelium (Figure S11).
Figure 8.

Oral Administration of FA-GDLVs Loaded with DMT1 siRNA Mitigated Iron Loading in Hepc−/− Mice
19-day-old female Hepc−/− mice were orally gavaged with saline (control), negative control-siRNA FA-GDLVs (NC), or Dmt1-siRNA FA-GDLVs daily for 16 days. Mice were provided a standard chow diet on days 1–8 and then a semi-purified, low-iron diet on days 9–16. Shown is Dmt1 mRNA expression in isolated intestinal epithelial cells (A), serum ferritin concentrations (B), serum non-heme Fe concentrations (C), TSAT (D), and liver (E), kidney (F), pancreas (G), and heart (H) non-heme Fe concentrations. Data were analyzed by one-way ANOVA and are presented as boxplots for n = 6–11 mice/group. Groups labeled with different letters are different from one another: p < 0.0001 (A and F); p = 0.0138 (B); p = 0.0007 (C); p = 0.0001 (D); p = 0.0011 (E); p = 0.002 (G); p = 0.0003 (H).
FA-GDLVs-Dmt1 siRNA Administration Specifically Downregulated Dmt1 mRNA Expression in the Duodenal Epithelium and Mitigated Iron Loading in Hepc KO Mice
Because iron loading begins in neonates, we started the treatment at 19 days of age, just prior to weaning. Dmt1 mRNA expression decreased by ∼50% in the duodenal epithelium 24 h after oral administration of Dmt1 siRNA-loaded FA-GDLVs (as compared with NC siRNA-loaded FA-GDLVs) (Figure 8A). The treatment did not, however, affect hepatic, renal, or bone marrow Dmt1 mRNA expression (Figure S12), demonstrating specificity for the intestinal epithelium. Serum ferritin levels decreased ∼40% in mice treated with Dmt1 siRNA-loaded GDLVs compared with NC-siRNA, demonstrating that body iron load decreased (Figure 8B).30 In support of this contention, serum non-heme iron and TSAT were reduced, and non-heme iron content was significantly lower in the liver, kidney, pancreas, and heart of mice treated with Dmt1 siRNA-loaded FA-GDLVs (Figures 8C–8H). Collectively, these data provide proof-of-concept that: (1) Dmt1 is a suitable therapeutic target to blunt iron loading in HH; and (2) oral administration of an experimentally verified, functional Dmt1 siRNA by FA-GDLVs is a suitable treatment approach to mitigate iron loading in mouse models of genetic iron overload.
Discussion
Iron homeostasis in mammals depends critically on Dmt1, because it is required for optimal assimilation of dietary iron in the gut and acquisition of transferrin-bound iron by developing erythrocytes. Dmt1 may also function in iron reabsorption from the renal filtrate.9, 31, 32, 33 Perturbations in any of these physiological processes could disrupt systemic iron homeostasis. Humans harboring inactivating Dmt1 mutations are severely anemic,34, 35, 36, 37, 38, 39, 40 but the relative contributions of the different Dmt1 functions to the disease phenotype have not been established definitively. Mice and rats carrying an identical point mutation in the Slc11a2 gene,4, 6 and global Slc11a2 KO mice,7 all have very similar iron-deficient phenotypes; but similarly, how impairments in intestinal iron absorption, iron delivery to the erythron, and renal iron reabsorption contribute quantitatively to the noted IDA is unclear.
Dmt1int/int Mice Acquire Sufficient Iron to Support Growth and Development into Mid-adulthood
Mice lacking Dmt1 specifically in the intestinal epithelium develop severe IDA.7 Iron-supplemented Dmtint/int mice (with normal hematological parameters) assimilated <15% of the iron dose absorbed by control mice.10 The magnitude of iron absorption in untreated (anemic) Dmtint/int mice has, however, not been experimentally assessed. The need to understand the contribution of intestinal Dmt1 to total iron transport made it important to perform absorption studies in Dmtint/int mice with no other interventions because the drive for iron absorption increases in IDA (with concurrent hypoxia), and mechanisms of iron absorption could be quite different under these circumstances. This understanding is especially important given that these mice acquire enough iron to support growth and maturation into mid-adulthood (unpublished data)10 in the absence of intestinal Dmt1. So, the pertinent question here is: how do these mice acquire sufficient iron to survive to 6–7 months of age? Three logical possibilities come to mind. First, perhaps Dmtint/int mice are born with a sufficient iron endowment to live to this age in the absence of any significant absorption of dietary iron. Sole reliance on stores, however, seems unlikely because mice normally meet ∼50% of iron requirements from the diet,41, 42 with the other 50% presumably coming from iron recycling. Second, because the villin-Cre transgene may show a mosaic expression pattern in the intestinal epithelium, residual Dmt1 iron-transport activity could preserve life in these mice. Previous investigations, however, have not detected Dmt1 expression in the intestinal epithelium of these mice.7 And last, another transport process could provide adequate iron absorption capacity to prevent earlier mortality. This is a realistic possibility because, for example, some Zip transporters can transport iron43, 44 and some are expressed in the intestine.45 Here, we thus sought to test the hypothesis that another dietary iron transport process exists in the proximal small intestine of Dmtint/int mice.
Dmt1 Is the Principal Intestinal Iron Importer during IDA
We established the Ussing chamber technique to define mechanisms of intestinal iron transport and to determine the relative contribution of Dmt1 to intestinal iron flux. This ex vivo technique has provided novel insights into many intestinal transport processes.46 A principal strength of this approach is that it provides a short-term organ culture method that enables precise measurement of electrical and transport parameters of intact, polarized intestinal epithelium. After defining the optimal transport conditions and defining the kinetics of iron flux, we applied this technique to duodenal epithelial organ cultures isolated from control and Dmt1int/int mice. These experiments demonstrated that Dmt1 is required for the enhancement of intestinal iron transport associated with IDA. This finding complements and extends the previous observation that Dmt1 is the principal intestinal iron importer during physiological conditions in these mice.10 That Dmt1int/int mice live for several months may thus relate to compensatory mechanisms that decrease iron losses, given that iron excretion may play an important role in systemic iron homeostasis in mice.42 Alternatively, Dmt1-independent iron import processes could be active in more distal portions of the gastrointestinal (GI) tract in these mice.
Intestinal Dmt1 Mediates the Excessive Iron Absorption that Typifies Murine HH
Recent studies also suggested that Dmt1 was the main iron entry pathway in murine HH. This prediction was based upon the facts that when a mouse model of adult-onset HH, the HFE KO mouse, was bred to mk mice (expressing dysfunctional Dmt1)4 or global Slc11a2 Kos,7 tissue iron accumulation was prevented.7, 12 Interpretation of these data is, however, complicated by the fact that, as mentioned above, Dmt1 is expressed in multiple tissues and cell types, and has multiple notable physiologic functions. Importantly, intestinal iron transport was not measured in any of these previous investigations. Our goal here was thus to clarify this issue by experimentally defining the specific contribution of intestinal Dmt1 to the excessive iron absorption that typifies murine HH. By using mice lacking Dmt1 only in the intestinal epithelium, we avoided possible confounding influences of Dmt1 in other tissues and organs. Our experimental approach also utilized a model of early-onset (i.e., juvenile) HH, because we postulated that observations made in the most severe form of the disease would be applicable to all other types of (less severe) HH. This is a logical hypothesis because all forms of HH (except for “ferroportin” disease [i.e., type 4 HH]) share similar molecular underpinnings (due to inappropriate hepcidin expression and activity). Importantly, in the current investigation, experiments done with double-KO mice (i.e., hepcidin KO plus lack of intestinal Dmt1) demonstrated that intestinal Dmt1 is required for the iron loading that typifies murine HH. These observations are in agreement with earlier predictions and provide strong rationale for therapeutic targeting of intestinal Dmt1 to mitigate iron loading in HH.
Proof-of-Concept for a Novel Treatment Modality for HH
We next conceptualized, developed, and optimized a new experimental approach to blunt Dmt1 activity in vivo. First, a functional Dmt1 siRNA was developed and tested in vitro. Next, the GDLV-mediated siRNA delivery system was adapted to target the proximal small bowel (by incorporating FA). We subsequently demonstrated that orally administered FA-GDLVs carrying Dmt1 siRNAs successfully blunted tissue iron accumulation in Hepc−/− mice. Importantly, we found no evidence of inflammation or toxicity associated with FA-GDLVs administration, or off-target effects on Dmt1 expression in other tissues. The experimental approach utilized here thus represents a novel intervention to mitigate iron loading in murine HH. It also seems likely that this approach could be adapted to treat humans with HH, and possibly other iron-loading disorders including, for example, β-thalassemia. This is important because existing treatment modalities for iron overload in humans are either not specific for iron (repeat phlebotomies) or have significant side effects (use of oral iron chelators). Oral administration of FA-GDLVs carrying siRNAs targeting iron transporters (e.g., Dmt1 or Fpn1) could thus serve as an alternative therapeutic strategy to attenuate iron absorption, while avoiding some negative outcomes associated with currently used treatment regimens. Moreover, although here we demonstrated successful prevention of iron loading during the early stages of disease pathogenesis, we predict that this therapeutic approach could also reverse iron accumulation once established, especially if utilized in conjunction with iron removal by phlebotomy or chelation.
One limitation of this investigation is the use of laboratory mice, which cannot efficiently utilize heme iron, whereas omnivorous humans derive a significant portion of body iron from dietary heme (up to ≈40%). Heme is probably taken up into enterocytes via endocytosis, and the heme may then be degraded by heme oxygenase, releasing free iron into the lumen of the endosome. It is possible that Dmt1 then transports this iron out of endosomes into the cytosol for utilization by the cell or export via Fpn1. This would be analogous to the function of Dmt1 in the assimilation of transferrin-bound iron by developing erythrocytes. Because much of what we know about iron homeostasis in mammals comes from studies in mice, whether intestinal Dmt1 functions in the assimilation of diet-derived heme iron is, unfortunately, unknown (since mice cannot absorb heme iron). Moreover, it is not clear whether enhanced absorption of heme iron contributes to the development of iron overload in HH in humans; some studies support this possibility,47 and others do not.48 Nonetheless, because iron absorption is only marginally enhanced in humans with HH (from 1–2 mg/day up to ≈4 mg/day),49 silencing Dmt1, even if it only blunts non-heme iron absorption, should be effective at reducing body iron burden over time.
Materials and Methods
Mouse Models and Breeding Strategy
All animal studies were approved by the University of Florida Institutional Animal Care and Use Committee (IACUC). Intestine-specific Dmt1 KO (Dmt1int/int) mice and phenotypically normal (Dmt1fl/fl) control littermates7 and Hepc KO (Hepc−/−) mice24 were utilized for this investigation. The goal was to produce mice with intestine-specific Dmt1 KO plus (iron overload due to) Hepc KO. Mice with floxed Dmt1 and a villin-Cre transgene lose Dmt1 in an intestine-specific fashion (DMT1int/int), whereas those lacking the villin-Cre (DMT1flox/flox) serve as phenotypically normal controls. We obtained these mice from Dr. Bryan Mackenzie at the University of Cincinnati on the 129S6 inbred background (also note that these mice were originally derived from the laboratory of Dr. Nancy Andrews). Hepc KO mice on the C56BL/6 background were obtained from Dr. Ella Nemeth at the University of California Los Angeles (also note that these mice were originally derived from the laboratory of Dr. Sophie Vaulont). Hence, we first had to generate Hepc KO mice on the 129S6 background, by breeding Hepc KO mice with WT 129S6 mice for seven generations. Then the following crosses were done to generate control and experimental mice for this investigation:
F0: Hepc−/−|DMT1+/+|Cre−/− × Hepc+/+|DMT1flox/flox|Cre+/−
F1: Hepc+/−/DMT1+/fl/Cre+/− × Hepc+/−/DMT1+/fl/Cre−/−
F2: Hepc+/−/DMT1fl/fl/Cre−/− × Hepc+/−/DMT1+/fl/Cre+/−.
Keeping only these two genotypes from the F2 generation (representing just 1/16 of the progeny) for breeding allowed us to segregate parentals and produce the desired experimentals and controls: Hepc+/+/Dmt1fl/fl (normal controls), Hepc−/−/Dmt1fl/fl [Hepc KO, normal intestinal Dmt1 expression), Hepc−/−/Dmt1int/+ (Hepc KO with one copy of the intestinal Dmt1 gene ablated), and Hepc−/−/Dmt1int/int (Hepc KO lacking intestinal Dmt1, i.e., double KOs]. Littermate male and female mice of these four genotypes were studied. For some experiments, Dmt1fl/fl mice were bled from the facial vein to induce anemia (Dmt1int/int mice were similarly handled, but not bled, because they are already anemic).
Measurement of Transepithelial Fe Flux
Fresh duodenal mucosa was mounted between two halves of an Ussing chamber with 0.3 cm2 of exposed surface area (P2304; Physiologic Instruments, San Diego, CA, USA). For initial experiments, we used Ringer’s solution (in mM: 140 Na+, 119.8 Cl−, 5.2 K+, 2.4 HPO42−, 0.4 H2PO4−, 1.2 Mg2+, 1.2 Ca2+, and 25 HCO3− [pH 7.4]).50 Subsequently, transport buffers at pH 5.0, 6.5, and 7.4 were formulated specifically for Fe flux studies (Table S1). During experiments, buffers were maintained at 37°C and bilaterally infused with a 95% O2-5% CO2 gas mixture. FeSO4 was added to Ringer’s or transport buffers at 100, 200, 500, 750, 1,000, 1,250, 1,500, and 2,000 μM to define the kinetics of iron transport. Tissues were allowed to stabilize in buffers for 30 min after being placed in the chamber, and Fe flux across the duodenal epithelium was then analyzed by adding 59Fe (in 0.5 M HCl, neutralized with N-methyl-D-glucamine) to the mucosal or serosal chamber. Buffer samples were then collected from the opposite chamber every 15 min for 1 h. 59Fe activity in the collected buffers was measured using a gamma counter (Wizard 2, 2480 Automatic Gamma Counter; Perkin Elmer). During the transport assay, basal short-circuit current and conductance were recorded using a computer-controlled voltage and current clamp device (VCC MC-8; Physiologic Instruments), as previously described.51, 52 Unidirectional flux (J) was calculated as Jms (or Jsm) (where m is mucosal and s is serosal) = V × (S2 − S1 × dilution)/(specific activity × surface area × time), where V is the volume of sink superfusate (in mL); S1 and S2 are the cpm of samples taken at the beginning and end of the flux period, respectively; dilution is of the sink superfusate resulting from sample fluid replacement; specific activity is the isotopic activity in the source bath (cpm/μEq); surface area is the area of exposed mucosa; and time is the length of the flux period in hours. Fe flux was calculated as Jnet = Jms − Jsm.
Production of GDNP Lipid Vectors for siRNA Delivery
GDNPs were isolated and purified according to established methods.25 To produce GDLVs, 6 mL of methyl alcohol/chloroform (2:1) (v/v) was added to 1.6 mL of GDNPs (1 mg/mL) in PBS followed by thorough mixing. Next, chloroform (2 mL) and double-distilled H2O (ddH2O; 2 mL) were added sequentially and the solution was vortexed. The solution was then centrifuged at 2,000 × g for 10 min at room temperature to separate the aqueous and organic phases.16 To produce FA-GDLVs, 50 μL of FA (F7876; Sigma-Aldrich) (1.5 mg/mL in DMSO) was mixed with total lipids (organic phase), and the solution was dried in a round-bottomed flask to a thin lipid film in a rotary evaporator. The dried lipid layer was then suspended in 500 μL of 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (20 mM [pH 7.4]; Sigma-Aldrich). After 5 min of sonication, an equal volume of HEPES buffer was added and the solution was sonicated again for 5 min, finally producing FA-GDLVs.
For siRNA loading of GDLVs and FA-GDLVs, Dmt1- or negative control (NC)-siRNA (3.75 nmol) was dissolved in 300 μL of a sterile 5% glucose solution and vortexed gently. For in vitro GDLVs plus siRNA formulation, dried GDLVs (from 1.6 mg GDNPs) were immediately suspended in 500 μL of 20 mM HEPES buffer (pH 7.4), which contained 3.75 nmol DMT1 siRNA. After sonicating for 5 min, an equal volume of HEPES buffer was added and the mixture was sonicated for another 5 min. Finally, the solution was passed through liposomes extruded with a 200-nm polycarbonate membrane about 20 times. For in vitro studies, 15 pmol GDLVs/siRNA was added to test the transfection efficiency. TurboFect reagent (Life Technologies) (6 μL) was then added to the diluted siRNA solutions, and the sample was mixed immediately and incubated for 15 min at room temperature. For in vivo studies, high-performance liquid chromatography (HPLC)-purified siRNAs were used because they have better stability and less toxicity (than less pure preparations). For the in vivo FA-GDLVs/siRNA formulation, DMT1 siRNA (3.75 nmol) was first dissolved in sterile 300 μL 5% glucose solution, vortexed gently, and spun down. Then, TurboFect reagent was added (6 μL) to the diluted siRNA, which was mixed immediately and incubated for 15 min at room temperature. The packed siRNA plus TurboFect was then added to the GDLVs-FA lipid film with sonication to prepare FA-GDLVs/siRNA. For in vivo studies, 3.75 nmol GDLVs/siRNA per dose was used to knock down DMT1 expression in duodenum.
Cytotoxicity Assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to analyze cell viability, as previously described.18 Colon-26 cells were incubated with different concentrations of GDLVs, DC-Chol/DOPE (30/70 mol/mol; Avanti Polar Lipids) (0, 10, 20, 50, 100, 200 μM), or FA-GDLVs for 24 or 48 h. A cell attachment assay was used to observe real-time cytotoxicity by ECIS, as described in a previous study.18 Caco2-BBE cells were seeded on plates and grown to confluence. Control (PBS), GDLVs or FA-GDLVs (100 μmol/L), or DC-Chol/DOPE (30/70 mol/mol) (100 μmol/L) was added to the wells. Basal electrical resistance measurements were then performed using the ideal parameters for Caco2-BBE cells, 500 Hz and 1 V.
Characterization of GDNPs, GDLVs, and FA-GDLVs
NPs were characterized using TEM (for GDNPs and GDLVs only), AFM, and DLS. Particle size was measured using DLS (Malvern Instruments, Worcestershire, UK) at room temperature. For TEM imaging, one drop of sample was deposited onto the surface of Formvar-coated copper grids, and then 1% uranyl acetate was added for 15 s. Samples were allowed to dry at room temperature before imaging. AFM images were taken using a SPA 400 AFM (Seiko Instruments, Chiba, Japan). Lipid extract from GDNPs was analyzed by the Lipidomics Research Center at Kansas State University.
Cell Culture
HEK293 cells were engineered so that the mouse Dmt1 cDNA was expressed only when cells were treated with doxycycline (Dox; a tetracycline analog).26 Dmt1-overexpressing HEK293 cells or mouse Colon-26 cells were grown to 60% confluence and then transfected with Dmt1- (s70782, s70783, or s70784; Life Technologies, Gaithersburg, MD, USA) or NC-siRNA (4390843; Life Technologies) using Lipofectamine RNAiMAX (Thermo Fisher Scientific, Fair Lawn, NJ, USA) or GDLVs. Cells were analyzed 48 h later.
Cell Uptake of GDLVs Carrying siRNA Labeled with FITC
For fluorescence imaging of cellular uptake, Colon-26 cells were seeded at 1 × 105 cells/well on eight-chamber slides (Tissue-Tek; Sakura, USA) and cultured overnight at 37°C. Then, the medium was removed, cells were rinsed with PBS, and Opti-MEM containing GDLVs + siRNA-FITC (15 pmol/well) was added. After 8-h incubation at 37°C, cells were rinsed with PBS and fixed with 4% paraformaldehyde for 15 min and then dehydrated with acetone at −20°C for 5 min. After blocking with 1% BSA in PBS for 30 min, 100 μL of phalloidin-TRITC (1:40 dilution; Sigma-Aldrich) was added, and the cells were incubated for an additional 30 min. Finally, cells were coverslip-mounted with mounting medium containing DAPI (H-1500; Vector Laboratories). Cells were then imaged using a fluorescent microscope (BX63; Olympus, Japan). Cell uptake efficiency was quantified by flow cytometry by gating for FITC fluorescence (BD LSR Fortessa flow cytometer; BD Biosciences).
Animal Studies
Mice were anesthetized by CO2 narcosis and killed by cervical dislocation. Hb and Hct in whole blood were measured by standard methods.53, 54 Serum ferritin concentration was quantified with the Mouse Ferritin ELISA Kit (ab157713; Abcam). Plasma EPO concentration was measured by Mouse Erythropoietin Quantikine ELISA Kit (MEP00B; R&D Systems). Plasma non-heme iron concentrations were determined using a standard colorimetric method.54, 55 TIBC was assessed as previously described.53, 54 TSAT was calculated as serum iron/TIBC × 100. Non-heme iron content in tissues was determined using a standard acid digestion, chromogen-based colorimetric assay.53, 54
To evaluate the ability of NPs to target the mouse duodenum in vivo, 6-week-old male C57BL/6 mice were fasted for 24 h, and then PBS or DiR dye (PromoKine, Heidelberg, Germany)-labeled GDLVs or FA-GDLVs were administered to mice by oral intragastric gavage. The proximal small intestine was imaged 4, 8, and 12 h later by an IVIS series preclinical in vivo imaging system (Perkin Elmer, MA, USA). Subsequently, 19-day-old female WT and Hepc−/− mice were provided PBS (control), freshly made negative control-siRNA (NC-siRNA), or Dmt1-siRNA (3.75 nmol/dose)-loaded FA-GDLVs daily for 16 days by oral intragastric gavage. During the first 8 days, mice were fed a standard chow diet with 200 ppm Fe (#2918; Envigo), but were then switched to a low-iron diet (2–6 ppm Fe; TD.80396; Envigo) for the last 8 days. Because the standard chow diet contained excess iron (i.e., above the approximate 35 ppm requirement for adult mice56), we postulated that the low-iron diet would maximize the upregulation of Dmt1 expression, which occurs because of high Fpn expression (because Hepc is absent) and consequent intracellular iron depletion.57 This approach mimics HFE-related HH,58 the most common form of the disease in humans, in which Dmt1 expression in enterocytes is abnormally high with normal or low iron intakes. Moreover, given high Dmt1 and Fpn1 expression, and because in vivo delivery of anti-Dmt1 siRNA blunted Dmt1 expression by only ≈40%, we postulated that we would be more likely to see an effect when dietary iron intake was low. We also postulated that this experimental approach would nicely mimic humans with HH, because some published reports support the effectiveness of dietary (and supplemental) iron restriction in disease management.59
Real-Time qPCR
Total RNA was isolated using RNAzol RT reagent (Molecular Research Center, OH, USA). RNA concentration was measured using a NanoDrop spectrophotometer. SYBR-Green qRT-PCR was performed as described.60 Expression of experimental genes was normalized to expression of cyclophilin A. Primer sequences are listed in Table S2.
Statistical Analyses
Results are depicted as boxplots displaying the minimum, the lower (25th percentile), the median (50th percentile), the upper (75th percentile), and the maximum ranked sample. The mean values are indicated by a plus (+) sign. In Figures 6B and 6C, data are presented as mean ± SD. Statistical analysis was performed using GraphPad Prism (v 7.0), Stata (v 15.1), or JMP (v 12.2). Data were analyzed by Student’s t test or one- or two-way ANOVA followed by a Tukey’s multiple comparisons test. The Brown-Forsythe test or Levene’s test was used to check for equal variance when one-way or two-ANOVA was used, respectively. Some data were log10 transformed due to unequal distribution (as indicated in the figure legends). Note, however, that the non-transformed data are shown in the figures for ease of visual interpretation. p < 0.05 was considered statistically significant.
Author Contributions
S.V., M.D.G., D.M., and J.F.C. conceptualized this investigation. X.W., M.Z., S.R.L.F., R.R.W., C.Y., L.Y., P.X., and X.X. performed experiments. X.W., M.Z., M.D.G., S.V., D.M., and J.F.C. designed experiments and interpreted data. X.W. drafted the manuscript, performed statistical analyses, and made the figures. All authors participated in writing the manuscript and approved the final version of the paper.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This investigation was funded by grant R01 DK074867 from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and grant R01 DK109717 from NIDDK and the Office of Dietary Supplements (to J.F.C.). X.W. is a recipient of a doctoral scholarship from the China Scholarship Council. M.Z. was the recipient of a Research Fellowship Award from the Crohn's & Colitis Foundation of America. D.M. is a recipient of a Senior Research Career Scientist Award from the Department of Veteran Affairs. The authors wish to thank Dr. Bryan McKenzie for providing the Dmt1int/int mice, Drs. Sophie Vaulont and Ella Nemeth for providing the Hepc−/− mice, and Dr. Laura Garrick for critical reading of the manuscript.
Footnotes
Supplemental Information includes twelve figures and two tables and can be found with this article online at https://doi.org/10.1016/j.ymthe.2019.01.003.
Supplemental Information
References
- 1.Hurrell R., Egli I. Iron bioavailability and dietary reference values. Am. J. Clin. Nutr. 2010;91:1461S–1467S. doi: 10.3945/ajcn.2010.28674F. [DOI] [PubMed] [Google Scholar]
- 2.Gulec S., Anderson G.J., Collins J.F. Mechanistic and regulatory aspects of intestinal iron absorption. Am. J. Physiol. Gastrointest. Liver Physiol. 2014;307:G397–G409. doi: 10.1152/ajpgi.00348.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Russell E.S., Nash D.J., Bernstein S.E., Kent E.L., McFarland E.C., Matthews S.M., Norwood M.S. Characterization and genetic studies of microcytic anemia in house mouse. Blood. 1970;35:838–850. [PubMed] [Google Scholar]
- 4.Fleming M.D., Trenor C.C., 3rd, Su M.A., Foernzler D., Beier D.R., Dietrich W.F., Andrews N.C. Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat. Genet. 1997;16:383–386. doi: 10.1038/ng0897-383. [DOI] [PubMed] [Google Scholar]
- 5.Sladic-Simic D., Martinovitch P.N., Zivkovic N., Pavić D., Martinovic J., Kahn M., Ranney H.M. A thalassemia-like disorder in Belgrade laboratory rats. Ann. N Y Acad. Sci. 1969;165:93–99. doi: 10.1111/j.1749-6632.1969.tb27779.x. [DOI] [PubMed] [Google Scholar]
- 6.Fleming M.D., Romano M.A., Su M.A., Garrick L.M., Garrick M.D., Andrews N.C. Nramp2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport. Proc. Natl. Acad. Sci. USA. 1998;95:1148–1153. doi: 10.1073/pnas.95.3.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gunshin H., Fujiwara Y., Custodio A.O., Direnzo C., Robine S., Andrews N.C. Slc11a2 is required for intestinal iron absorption and erythropoiesis but dispensable in placenta and liver. J. Clin. Invest. 2005;115:1258–1266. doi: 10.1172/JCI24356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garrick L.M., Dolan K.G., Romano M.A., Garrick M.D. Non-transferrin-bound iron uptake in Belgrade and normal rat erythroid cells. J. Cell. Physiol. 1999;178:349–358. doi: 10.1002/(SICI)1097-4652(199903)178:3<349::AID-JCP9>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 9.Canonne-Hergaux F., Gros P. Expression of the iron transporter DMT1 in kidney from normal and anemic mk mice. Kidney Int. 2002;62:147–156. doi: 10.1046/j.1523-1755.2002.00405.x. [DOI] [PubMed] [Google Scholar]
- 10.Shawki A., Anthony S.R., Nose Y., Engevik M.A., Niespodzany E.J., Barrientos T., Öhrvik H., Worrell R.T., Thiele D.J., Mackenzie B. Intestinal DMT1 is critical for iron absorption in the mouse but is not required for the absorption of copper or manganese. Am. J. Physiol. Gastrointest. Liver Physiol. 2015;309:G635–G647. doi: 10.1152/ajpgi.00160.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou X.Y., Tomatsu S., Fleming R.E., Parkkila S., Waheed A., Jiang J., Fei Y., Brunt E.M., Ruddy D.A., Prass C.E. HFE gene knockout produces mouse model of hereditary hemochromatosis. Proc. Natl. Acad. Sci. USA. 1998;95:2492–2497. doi: 10.1073/pnas.95.5.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levy J.E., Montross L.K., Andrews N.C. Genes that modify the hemochromatosis phenotype in mice. J. Clin. Invest. 2000;105:1209–1216. doi: 10.1172/JCI9635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roetto A., Papanikolaou G., Politou M., Alberti F., Girelli D., Christakis J., Loukopoulos D., Camaschella C. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat. Genet. 2003;33:21–22. doi: 10.1038/ng1053. [DOI] [PubMed] [Google Scholar]
- 14.Sandhu K., Flintoff K., Chatfield M.D., Dixon J.L., Ramm L.E., Ramm G.A., Powell L.W., Subramaniam V.N., Wallace D.F. Phenotypic analysis of hemochromatosis subtypes reveals variations in severity of iron overload and clinical disease. Blood. 2018;132:101–110. doi: 10.1182/blood-2018-02-830562. [DOI] [PubMed] [Google Scholar]
- 15.Xiao B., Merlin D. Oral colon-specific therapeutic approaches toward treatment of inflammatory bowel disease. Expert Opin. Drug Deliv. 2012;9:1393–1407. doi: 10.1517/17425247.2012.730517. [DOI] [PubMed] [Google Scholar]
- 16.Zhang M., Wang X., Han M.K., Collins J.F., Merlin D. Oral administration of ginger-derived nanolipids loaded with siRNA as a novel approach for efficient siRNA drug delivery to treat ulcerative colitis. Nanomedicine (Lond.) 2017;12:1927–1943. doi: 10.2217/nnm-2017-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang M., Xu C., Liu D., Han M.K., Wang L., Merlin D. Oral delivery of nanoparticles loaded with ginger active compound, 6-shogaol, attenuates ulcerative colitis and promotes wound healing in a murine model of ulcerative colitis. J. Crohn’s Colitis. 2018;12:217–229. doi: 10.1093/ecco-jcc/jjx115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang M., Collins J.F., Merlin D. Do ginger-derived nanoparticles represent an attractive treatment strategy for inflammatory bowel diseases? Nanomedicine (Lond.) 2016;11:3035–3037. doi: 10.2217/nnm-2016-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mu J., Zhuang X., Wang Q., Jiang H., Deng Z.B., Wang B., Zhang L., Kakar S., Jun Y., Miller D., Zhang H.G. Interspecies communication between plant and mouse gut host cells through edible plant derived exosome-like nanoparticles. Mol. Nutr. Food Res. 2014;58:1561–1573. doi: 10.1002/mnfr.201300729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang M., Viennois E., Xu C., Merlin D. Plant derived edible nanoparticles as a new therapeutic approach against diseases. Tissue Barriers. 2016;4:e1134415. doi: 10.1080/21688370.2015.1134415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Q., Ren Y., Mu J., Egilmez N.K., Zhuang X., Deng Z., Zhang L., Yan J., Miller D., Zhang H.G. Grapefruit-derived nanovectors use an activated leukocyte trafficking pathway to deliver therapeutic agents to inflammatory tumor sites. Cancer Res. 2015;75:2520–2529. doi: 10.1158/0008-5472.CAN-14-3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Q., Zhuang X., Mu J., Deng Z.B., Jiang H., Zhang L., Xiang X., Wang B., Yan J., Miller D., Zhang H.G. Delivery of therapeutic agents by nanoparticles made of grapefruit-derived lipids. Nat. Commun. 2013;4:1867. doi: 10.1038/ncomms2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ganz T. Erythropoietic regulators of iron metabolism. Free Radic. Biol. Med. 2018 doi: 10.1016/j.freeradbiomed.2018.07.003. Published online July 5, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lesbordes-Brion J.C., Viatte L., Bennoun M., Lou D.Q., Ramey G., Houbron C., Hamard G., Kahn A., Vaulont S. Targeted disruption of the hepcidin 1 gene results in severe hemochromatosis. Blood. 2006;108:1402–1405. doi: 10.1182/blood-2006-02-003376. [DOI] [PubMed] [Google Scholar]
- 25.Zhang M., Viennois E., Prasad M., Zhang Y., Wang L., Zhang Z., Han M.K., Xiao B., Xu C., Srinivasan S., Merlin D. Edible ginger-derived nanoparticles: a novel therapeutic approach for the prevention and treatment of inflammatory bowel disease and colitis-associated cancer. Biomaterials. 2016;101:321–340. doi: 10.1016/j.biomaterials.2016.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garrick M.D., Kuo H.C., Vargas F., Singleton S., Zhao L., Smith J.J., Paradkar P., Roth J.A., Garrick L.M. Comparison of mammalian cell lines expressing distinct isoforms of divalent metal transporter 1 in a tetracycline-regulated fashion. Biochem. J. 2006;398:539–546. doi: 10.1042/BJ20051987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiu A., Jansen M., Sakaris A., Min S.H., Chattopadhyay S., Tsai E., Sandoval C., Zhao R., Akabas M.H., Goldman I.D. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell. 2006;127:917–928. doi: 10.1016/j.cell.2006.09.041. [DOI] [PubMed] [Google Scholar]
- 28.Roger E., Kalscheuer S., Kirtane A., Guru B.R., Grill A.E., Whittum-Hudson J., Panyam J. Folic acid functionalized nanoparticles for enhanced oral drug delivery. Mol. Pharm. 2012;9:2103–2110. doi: 10.1021/mp2005388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas C., Thomas L. Biochemical markers and hematologic indices in the diagnosis of functional iron deficiency. Clin. Chem. 2002;48:1066–1076. [PubMed] [Google Scholar]
- 30.Addison G.M., Beamish M.R., Hales C.N., Hodgkins M., Jacobs A., Llewellin P. An immunoradiometric assay for ferritin in the serum of normal subjects and patients with iron deficiency and iron overload. J. Clin. Pathol. 1972;25:326–329. doi: 10.1136/jcp.25.4.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferguson C.J., Wareing M., Ward D.T., Green R., Smith C.P., Riccardi D. Cellular localization of divalent metal transporter DMT-1 in rat kidney. Am. J. Physiol. Renal Physiol. 2001;280:F803–F814. doi: 10.1152/ajprenal.2001.280.5.F803. [DOI] [PubMed] [Google Scholar]
- 32.Wareing M., Ferguson C.J., Green R., Riccardi D., Smith C.P. In vivo characterization of renal iron transport in the anaesthetized rat. J. Physiol. 2000;524:581–586. doi: 10.1111/j.1469-7793.2000.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Veuthey T., Hoffmann D., Vaidya V.S., Wessling-Resnick M. Impaired renal function and development in Belgrade rats. Am. J. Physiol. Renal Physiol. 2014;306:F333–F343. doi: 10.1152/ajprenal.00285.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bardou-Jacquet E., Island M.L., Jouanolle A.M., Détivaud L., Fatih N., Ropert M., Brissot E., Mosser A., Maisonneuve H., Brissot P., Loréal O. A novel N491S mutation in the human SLC11A2 gene impairs protein trafficking and in association with the G212V mutation leads to microcytic anemia and liver iron overload. Blood Cells Mol. Dis. 2011;47:243–248. doi: 10.1016/j.bcmd.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 35.Beaumont C., Delaunay J., Hetet G., Grandchamp B., de Montalembert M., Tchernia G. Two new human DMT1 gene mutations in a patient with microcytic anemia, low ferritinemia, and liver iron overload. Blood. 2006;107:4168–4170. doi: 10.1182/blood-2005-10-4269. [DOI] [PubMed] [Google Scholar]
- 36.Lam-Yuk-Tseung S., Camaschella C., Iolascon A., Gros P. A novel R416C mutation in human DMT1 (SLC11A2) displays pleiotropic effects on function and causes microcytic anemia and hepatic iron overload. Blood Cells Mol. Dis. 2006;36:347–354. doi: 10.1016/j.bcmd.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 37.Lam-Yuk-Tseung S., Mathieu M., Gros P. Functional characterization of the E399D DMT1/NRAMP2/SLC11A2 protein produced by an exon 12 mutation in a patient with microcytic anemia and iron overload. Blood Cells Mol. Dis. 2005;35:212–216. doi: 10.1016/j.bcmd.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 38.Mims M.P., Guan Y., Pospisilova D., Priwitzerova M., Indrak K., Ponka P., Divoky V., Prchal J.T. Identification of a human mutation of DMT1 in a patient with microcytic anemia and iron overload. Blood. 2005;105:1337–1342. doi: 10.1182/blood-2004-07-2966. [DOI] [PubMed] [Google Scholar]
- 39.Priwitzerova M., Nie G., Sheftel A.D., Pospisilova D., Divoky V., Ponka P. Functional consequences of the human DMT1 (SLC11A2) mutation on protein expression and iron uptake. Blood. 2005;106:3985–3987. doi: 10.1182/blood-2005-04-1550. [DOI] [PubMed] [Google Scholar]
- 40.Iolascon A., d’Apolito M., Servedio V., Cimmino F., Piga A., Camaschella C. Microcytic anemia and hepatic iron overload in a child with compound heterozygous mutations in DMT1 (SCL11A2) Blood. 2006;107:349–354. doi: 10.1182/blood-2005-06-2477. [DOI] [PubMed] [Google Scholar]
- 41.Parmar J.H., Davis G., Shevchuk H., Mendes P. Modeling the dynamics of mouse iron body distribution: hepcidin is necessary but not sufficient. BMC Syst. Biol. 2017;11:57. doi: 10.1186/s12918-017-0431-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chappelle E., Gabrio B.W., Stevens A.R., Jr., Finch C.A. Regulation of body iron content through excretion in the mouse. Am. J. Physiol. 1955;182:390–392. doi: 10.1152/ajplegacy.1955.182.2.390. [DOI] [PubMed] [Google Scholar]
- 43.Liuzzi J.P., Aydemir F., Nam H., Knutson M.D., Cousins R.J. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc. Natl. Acad. Sci. USA. 2006;103:13612–13617. doi: 10.1073/pnas.0606424103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang C.Y., Jenkitkasemwong S., Duarte S., Sparkman B.K., Shawki A., Mackenzie B., Knutson M.D. ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading. J. Biol. Chem. 2012;287:34032–34043. doi: 10.1074/jbc.M112.367284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eide D.J. The SLC39 family of metal ion transporters. Pflugers Arch. 2004;447:796–800. doi: 10.1007/s00424-003-1074-3. [DOI] [PubMed] [Google Scholar]
- 46.Clarke L.L. A guide to Ussing chamber studies of mouse intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2009;296:G1151–G1166. doi: 10.1152/ajpgi.90649.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lynch S.R., Skikne B.S., Cook J.D. Food iron absorption in idiopathic hemochromatosis. Blood. 1989;74:2187–2193. [PubMed] [Google Scholar]
- 48.Hunt J.R., Zeng H. Iron absorption by heterozygous carriers of the HFE C282Y mutation associated with hemochromatosis. Am. J. Clin. Nutr. 2004;80:924–931. doi: 10.1093/ajcn/80.4.924. [DOI] [PubMed] [Google Scholar]
- 49.Brissot P., Troadec M.B., Loréal O. Intestinal absorption of iron in HFE-1 hemochromatosis: local or systemic process? J. Hepatol. 2004;40:702–709. doi: 10.1016/j.jhep.2004.01.020. [DOI] [PubMed] [Google Scholar]
- 50.Zhang H., Ameen N., Melvin J.E., Vidyasagar S. Acute inflammation alters bicarbonate transport in mouse ileum. J. Physiol. 2007;581:1221–1233. doi: 10.1113/jphysiol.2007.129262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yin L., Vijaygopal P., Menon R., Vaught L.A., Zhang M., Zhang L., Okunieff P., Vidyasagar S. An amino acid mixture mitigates radiation-induced gastrointestinal toxicity. Health Phys. 2014;106:734–744. doi: 10.1097/HP.0000000000000117. [DOI] [PubMed] [Google Scholar]
- 52.Zhang K., Yin L., Zhang M., Parker M.D., Binder H.J., Salzman P., Zhang L., Okunieff P., Vidyasagar S. Radiation decreases murine small intestinal HCO3- secretion. Int. J. Radiat. Biol. 2011;87:878–888. doi: 10.3109/09553002.2011.583314. [DOI] [PubMed] [Google Scholar]
- 53.Ha J.H., Doguer C., Flores S.R.L., Wang T., Collins J.F. Progressive increases in dietary iron are associated with the emergence of pathologic disturbances of copper homeostasis in growing rats. J. Nutr. 2018;148:373–378. doi: 10.1093/jn/nxx070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ha J.H., Doguer C., Wang X., Flores S.R., Collins J.F. High-iron consumption impairs growth and causes copper-deficiency anemia in weanling Sprague-Dawley rats. PLoS ONE. 2016;11:e0161033. doi: 10.1371/journal.pone.0161033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gulec S., Collins J.F. Investigation of iron metabolism in mice expressing a mutant Menke’s copper transporting ATPase (Atp7a) protein with diminished activity (Brindled; Mo (Br) (/y) ) PLoS ONE. 2013;8:e66010. doi: 10.1371/journal.pone.0066010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reeves P.G., Nielsen F.H., Fahey G.C., Jr. AIN-93 purified diets for laboratory rodents: final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J. Nutr. 1993;123:1939–1951. doi: 10.1093/jn/123.11.1939. [DOI] [PubMed] [Google Scholar]
- 57.Gunshin H., Allerson C.R., Polycarpou-Schwarz M., Rofts A., Rogers J.T., Kishi F., Hentze M.W., Rouault T.A., Andrews N.C., Hediger M.A. Iron-dependent regulation of the divalent metal ion transporter. FEBS Lett. 2001;509:309–316. doi: 10.1016/s0014-5793(01)03189-1. [DOI] [PubMed] [Google Scholar]
- 58.Fleming R.E., Migas M.C., Zhou X., Jiang J., Britton R.S., Brunt E.M., Tomatsu S., Waheed A., Bacon B.R., Sly W.S. Mechanism of increased iron absorption in murine model of hereditary hemochromatosis: increased duodenal expression of the iron transporter DMT1. Proc. Natl. Acad. Sci. USA. 1999;96:3143–3148. doi: 10.1073/pnas.96.6.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moretti D., van Doorn G.M., Swinkels D.W., Melse-Boonstra A. Relevance of dietary iron intake and bioavailability in the management of HFE hemochromatosis: a systematic review. Am. J. Clin. Nutr. 2013;98:468–479. doi: 10.3945/ajcn.112.048264. [DOI] [PubMed] [Google Scholar]
- 60.Gulec S., Collins J.F. Silencing the Menkes copper-transporting ATPase (Atp7a) gene in rat intestinal epithelial (IEC-6) cells increases iron flux via transcriptional induction of ferroportin 1 (Fpn1) J. Nutr. 2014;144:12–19. doi: 10.3945/jn.113.183160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.