

Abstract
LPS activated macrophages require metabolic reprogramming and glucose uptake mediated by hypoxia inducible factor (HIF-1) α and glucose transporter (Glut)1 expression for proinflammatory cytokine production, especially IL-1β. This process is tightly regulated through activation of mitogen-activated protein kinases (MAP) kinases including the MEK/ERK pathway as well as several transcription factors including HIF-1α. While Mitogen-Activated Protein Kinase Kinase (MEK) 2 deficiency had no significant effect on nitric oxide, TNF-α, and IL-12 production in response to LPS challenge, MEK2 deficient murine bone marrow derived macrophages (BMDMs) exhibited lower IL-10 production. Importantly, MEK2 deficient BMDMs exhibited a preserved ERK1/2 phosphorylation, higher HIF-1α, Glut1 levels and substantially increased IL-1β as well as IL-6 production in response to LPS stimulation. Knockdown of HIF-1α expression via small interfering RNA (siRNA) decreased the level of HIF-1α expression in MEK2 deficient BMDMs and decreased IL-1β production in response to LPS treatment. Furthermore, we performed gain of function experiments by overexpressing MEK2 protein in RAW264.7 cells. LPS stimulation of MEK2 overexpressed in RAW264.7 cells led to a marked decreased IL-1β production. Finally, we investigated the role of Mek1 and Mek2 double and triple mutation on ERK phosphorylation, HIF-1α expression and IL-1β production. We found that MEK2 is the major kinase, which inversely proportionally regulates HIF-1α and IL-1β expression independent of ERK activation. Our findings demonstrate a novel regulatory function for MEK2 in response to TLR4 activation in IL-1β production through modulating HIF-1α expression.
Keywords: Macrophages, BMDM, ERK, MEK1, MEK2, HIF-1alpha, IL-1 beta
Introduction
Mitogen-Activated Protein Kinase Kinase (MAP2K) 1 and 2, also known as MEK, is a member of the dual specificity protein kinase family. In mammals, there are two distinct gene isoforms of Mek present, Mek1 and Mek2. While the deletion of the Mek1 gene leads to embryonic lethality, interruption of Mek2 is compatible with life (1, 2). Both isoforms are considered to be directly upstream of extracellular signal-regulated kinases (ERK) (3). However, recent evidence suggests that each isoform has a unique biological role. For instance, MEK1 is capable of stimulating epidermal proliferation and in fibroblasts it has a regulatory function in cell migration (2, 4). Furthermore, MEK1 deficient mice exhibit a lupus-like syndrome through deregulation of phosphatase and tensin homolog (PTEN) and protein kinase B (AKT) activation (5). The physiological role of MEK2 versus MEK1 in the innate immune system, especially in macrophages is poorly understood (6, 7).
In contrast to the well-defined role of the MEK/ERK pathway in cell growth and cancer biology, the differential roles of MEK1 and MEK2 in response to Toll like receptor (TLR) activation is poorly understood. TLR receptors are type I transmembrane proteins that mediate the recognition of pathogen associated molecular patterns (PAMPs) (8). The TLR family of receptors is composed of up to 10 members in humans and 12 in mice (9). Docking of LPS to TLR4 recruits the adaptor protein MyD88, which activates mitogen-activated protein kinases (MAPKs), including extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 kinase. TLR4 activation leads to phosphorylation of MEK1/2 and subsequent ERK1/2 activation. ERK1/2 activation has been proposed to play a major role in NF-κB activation, ROS and cytokine production especially IL-1β (10, 11). IL-1β production is tightly regulated through activation of several transcription factors.
The hypoxia-inducible factor (HIF)-1α belongs to the oxygen-sensitive transcription factors and is known as a transcriptional regulator for several inflammatory cytokines including IL-1β and IL-6 (12–14). In normoxic conditions cytosolic HIF-1α is hydroxylated by prolyl-hydroxylases (PHDs) on the α-subunit regulating targeted polyubiquitination and degradation via the von Hippel-Lindau (VHL) dependent pathway (15). Mutations in pVHL and loss of its function may lead to HIF-1α accumulation and give rise to various cancers (16). In addition to pVHL loss of function, various conditions may lead to HIF-1α accumulation through VHL-independent pathways (17). Several mechanisms including ferritin-mediated iron sequestration or activation of pathways including PI3 kinase, mTOR, ERK1/2 and GSK3β have been proposed to regulate HIF-1α (18–22). It is well recognized that in response to TLR4 activation, HIF-1α protein escapes proteasomal degradation and dimerizes with HIF-1β, which facilitates its translocation to the nucleus (14, 23, 24). The exact LPS mediated signaling leading to accumulation of HIF-1α and IL-1β production has not been fully elucidated. It has been shown that endotoxins can induce HIF-1α at the transcriptional level and increase its stability (13, 18, 25, 26).
We investigated the role of MEK2 in macrophages in response to LPS mediated cytokine production applying a genetic approach. Using BMDMs derived from WT, Mek1d/d Sox2Cre/+ and Mek2−/− mice, we show that despite increased pVHL, MEK2 deficient BMDMs exhibit significantly higher HIF-1α levels at baseline and in response to LPS challenge. Higher HIF-1α levels in MEK2 deficient BMDMs was linked to a higher IL-1β production in response to LPS challenge. Furthermore, the abundance of HIF-1α and IL-β production was independent of ERK activation.
Material and Methods
Chemicals and antibodies.
LPS (055-B5 ultrapure) was purchased from InvivoGen (San Diego, CA). Phospho-specific antibodies against phospho-MEK1/2, ERK1/2, p38, JNK, as well as total ERK1/2, JNK, p38, MEK1, MEK2, VHL and β-actin were purchased from Cell Signaling Technology (Beverly, MA). Glut1 antibody was purchased from Thermo Fisher Scientfic (Waltham, MA). IL-1 β antibody was purchased (R&D Systems). The HIF-1α antibody was purchased from Bioss Inc (Woburn, Massachusetts, USA). NLRP3 antibody was obtained from Adipogen Inc (San Diego, CA). Horseradish peroxidase (HRP)-conjugated anti-mouse and anti-rabbit IgG secondary antibodies were purchased from Cell Signaling Technology, and horseradish peroxidase (HRP)-conjugated anti-goat antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Mice and Isolation of Bone Marrow Derived Macrophages (BMDMs).
Animal studies were approved by the University Committees on Use and Care of Animals. Wild-type (WT), Mek1d/d Sox2Cre/+ and Mek2−/− were generated onto the 129 cc background as previously described (1, 27). WT, Mek1d/d Sox2Cre/+ and Mek2−/− mice were maintained at animal facilities at the Centre de recherche sur le cancer de l’Université Laval (Québec, CA). Animal studies were approved by the corresponding Committee for Protection of Animals at Laval University (CPAUL) on Use and Care of Animals. All experiments were performed in accordance with relevant guidelines and regulations.
BMDMs from mice were prepared as described previously (28). Briefly, femurs and tibias from 6- to 12-week-old mice were dissected, and the bone marrow was flushed out. Macrophages were cultured with IMDM media supplemented with 30% L929 supernatant containing glutamine, sodium pyruvate, 10% heat-inactivated fetal FBS, and antibiotics for 5–7 days.
Cell Culture and transfections.
RAW 264.7 cells were obtained from ATCC and grown in Dulbecco’s modified Eagle’s medium with 10% FBS and 1% penicillin and streptomycin. DNA constructs: cDNA encoding murine MEK2, was sub-cloned into the p3XFLAG-myc-CMV-24 vector containing a C terminus Myc tag by PCR using specific primers. Transfection of RAW 264.7 cells was conducted using Lipofectamine LTX (Invitrogen) and a fixed amount of DNA (2.5 μg of DNA/per well of 6 well plate). 24 h after transfection, cells were treated with LPS and then harvested for immunoblotting.
Short interference RNA (siRNA) transfection.
BMDMs were seeded with a density of 2 × 105 per well in 6-well plates. Cells were transfected with either of HIF-1α siRNA (L-040794-00-0005, Thermo Scientific) or with a scrambled siRNA pool (D-001810-10-05, Thermo Scientific) using Lipofectamine 2000 (Invitrogen). After 24 h, transfected cells were treated with LPS and then harvested for immunoblotting.
Protein extraction and immunoblotting.
After the appropriate treatments, cells were washed with PBS, and harvested in RIPA buffer (Millipore, Massachusetts) containing protease inhibitor and anti-phosphatase cocktails, as previously described (29). Equal amounts of proteins (15μg) were mixed with the same volume of 2x sample buffer, separated on 10% SDS-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad, Hercules, CA) at 20V for 1h using a semi dry transfer cell (Bio-Rad) as previously described (29). The PVDF membrane was blocked with 5% dry milk in TBST (Tris-buffered saline with 0.1% Tween-20), rinsed, and incubated with primary antibody overnight. The blots were washed and incubated with HRP-conjugated secondary anti-IgG antibody. Membranes were washed and immunoreactive bands were visualized using a chemiluminescent substrate (ECL-Plus, GE Healthcare, Pittsburgh, PA). Images were captured on Hyblot CL film (Denville, Scientific Inc, Metuchen, NJ). Optical density analysis of signals was performed using ImageQuant software (version 5, Molecular Dynamics).
Enzyme linked immunosorbent assay (ELISA).
Murine TNF-α, IL-1β, IL-12, IL-10 and IL-6 cytokine levels in cell culture supernatants were measured using ELISA DuoKits (R&D Systems) as previously described (30).
Measurement of Nitrite.
The nitrite concentration in the culture media was used as a measure of NO production was determined by measuring nitrite accumulation in the medium using Griess reagent (30).
Separation of Cytoplasmic and Nuclear Fractions.
Cytoplasmic and nuclear fractions were separated as described previously (29). Briefly, after treatment the cells were resuspended in a hypotonic buffer (10 mm HEPES, pH 7.9, 0.5% Igepal, 2 mm MgCl2, 10 mm KCl, 0.1 mm EDTA, 0.5 mm phenylmethylsulfonyl fluoride, 1.0 μg/ml leupeptin, and 1.0 μg/ml aprotinin) and incubated on ice for 10 min. After centrifugation at 14,000 × g for 1 min at 4 °C, the supernatant (cytoplasmic) and the pellets (nuclear fraction) were collected.
Immunoprecipitation.
BMDMs were lysed in RIPA buffer (Millipore, Massachusetts) containing protease inhibitor and anti-phosphatase cocktails, and the lysate was centrifuged to pellet the cell debris. The resulting supernatant was used for immunoprecipitation. First the complex between immunoprecipitation matrix (IP-matrix, ImmunoCruz™ IP/WB Optima F from Santa Cruz Biotechnology, CA) was formed by incubating HIF-1α antibody with IP-matrix overnight at 4° C with gentle rotation. After the incubation, the IP-matrix pellet bound to HIF-1α antibody was washed three times with cold PBS. After the wash, the IP-matrix bound to HIF-1α antibody was incubated with the lysate overnight at 4° C with gentle rotation. Immunoprecipitation pellet was washed four times with cold PBS with a brief centrifugation. Samples from the IP-pellets were resuspended in the loading buffer and subjected to gel electrophoresis.
Statistical analyses.
Statistical analyses were performed using SPSS software, version 24.0 (SPSS Inc; Chicago, IL). One way analysis of variance (ANOVA) test and post hoc repeated measure comparisons (least significant difference (LSD) was performed to identify differences between groups. ELISA results were expressed as mean ± SEM. For all analyses, two-tailed p-values of less than 0.05 were considered significant.
RNA Extraction and Quantitative Reverse Transcriptase/Real Time-PCR.
Total RNA was extracted using Trizol reagent (Life Technology) and reverse-transcribed using the Reverse Transcription System (Promega, Madison, WI). The primers (targeting IL-1β, ARNT, VEGF, HIF-1α, and a reference gene, GAPDH) were used to amplify the corresponding cDNA by using iQ SYBR Green Supermix (Life Technology). Quantitative analysis of mRNA expression was performed using the MX3000p instrument (Stratagene, La Jolla, CA). PCR amplification was performed in a total volume of 20 μL containing 2 μL of each cDNA preparation and 20 pg of primers (Life Technology). The PCR amplification protocol was performed as described previously (29). Relative mRNA levels were calculated after normalizing to GAPDH. Data were analyzed using ANOVA or two-tailed Student’s t test, and the results were expressed as relative -fold change. The following primers were used in the PCR reactions: GAPDH, forward (5’- GTGAACGAGAAGGACTATAACCC-3’) and reverse (5’-GGCTGTGTACCAATGGACTG-3’); IL-1-β, forward (5’-CGCAGCAGCACATCAACAAGAGC-3’), and reverse (5’-TGTCCT CATCCTGGAAGGTCCACG-3’); HIF-1α forward (5’-TCAAGTCAGCAACGTGGAG-3’) and reverse(5’-TATCGAGGCTGTGTCGACG-3’);VEGF, forward (5′-GTAAGCTTGTACAAGATCCGCAGACG-3′) and reverse(5′-ATGGATCCGTATCAGTCTTTCCTGG-3′).
Results
MEK1 and MEK2 isoforms regulate LPS mediated cytokine production.
In macrophages it is not known whether MEK isoforms play a differential role in LPS mediated IL-1β production. Because there are no chemical inhibitors that specifically target MEK1 versus MEK2, we applied a genetic approach to further elucidate the differential role of MEK isoforms by using BMDMs derived from MEK1 and MEK2 deficient mice. Some studies suggested that MEK/ERK regulates TLR mediated nitric oxide (NO) production (31, 32). NO production by macrophages plays an important role in innate immunity. To investigate the relative role of MEK1 and MEK2 in LPS mediated NO production, murine BMDMs from WT and Mek1d/dSox2Cre and MEK2−/− mice were challenged with LPS for 24h followed by measurement of nitrite in conditioned media as a relative indicator of NO production. As shown in Figure 1A, the difference was short of statistical significance, but both MEK1 and MEK2 deficient BMDMs responded to LPS with lower production of NO compared to WT BMDMs. Next, we analyzed several other cytokines including, TNF-α, IL-10, IL-12, IL-6, and IL-1β. LPS challenge of all three strains of BMDMs led to the production of TNF-α (Fig. 1B). MEK2 deficiency had no significant effect on TNF-α production in response to LPS as compared to WT BMDMs. In contrast, MEK1 deficiency led to a significantly lower TNF-α production in response to LPS as compared to WT and MEK2−/− (Fig. 1B). Similarly, LPS treatment resulted in a significantly lower amount of IL-10 production in MEK1 and MEK2 deficient cells as compared to WT macrophages (Fig.1C). Consistent with our previous work, we observed that LPS challenge of MEK1 deficient BMDMs led to higher IL-12 production as compare to WT and MEK2 deficient cells (Fig. 1D). BMDMs from MEK2 deficient mice exhibited a higher IL-6 production in response to LPS (Fig. 1E). Interestingly, BMDMs lacking MEK2 responded to LPS challenge with a significant higher IL-1β production as compared to BMDMs from WT and Mek1d/dSox2Cre mice (Fig. 1F). It is known that MEK/ERK activation is a positive regulator of several cytokines (33, 34). Yet strikingly, we found that MEK2 and MEK1 differentially modulate the response to TLR ligands in macrophages. While MEK1 regulates IL-12, IL-10 and TNF-α, MEK2 regulates IL-1β and IL-6 production. MEK2 deficiency has minimal effect on TNF-α or NO production.
FIGURE 1. MEK1 and MEK2 isoform regulate LPS mediated IL-1β and IL-6 production in BMDMs.
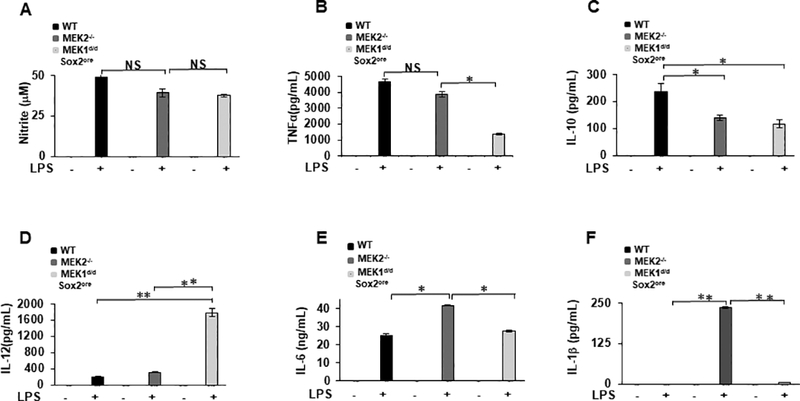
BMDMs derived from WT, Mek1d/dSox2Cre and Mek2−/− mice were cultured under similar conditions and treated with LPS (100 ng/mL) for 24 h. (A) Nitric Oxide production. After LPS challenge for 24h the generation of NO in the conditioned medium was determined by measuring nitrite accumulation using Griess reaction. Conditioned media were analyzed for (B) TNF-α, (C) IL-10, (D) IL-12, (E) IL-6 and (F) IL-1β using ELISA. Data presented as mean of three independent experiments. Using ANOVA Mann-Whitney U test, * signifies a p value <0.05, while ** signifies a p value <0.001, and error bars indicate SEM.
MEK2 deficient BMDMs respond to LPS with IL-1β production, and increased NLRP3 and HIF-1α.
IL-1β is one of the most potent inflammatory cytokines that regulates host responses to infections (14, 35, 36). IL-1β is regulated through complex mechanisms at the transcriptional and post-transcriptional level. HIF-1α is an important transcription factor regulating IL-1β production in response to TLR4 stimulation (14). To determine if the enhanced IL-1β expression in response to LPS is regulated through a transcriptional mechanism, we evaluated IL-1β mRNA expression levels using qRT-PCR. BMDMs from WT and MEK2 deficient mice were cultured side by side under equal conditions, stimulated with LPS for 1h and IL-1β mRNA expression was assessed. Figure 2A shows the mean of relative gene expression for IL-1β normalized to GAPDH. MEK2 deficient BMDMs responded to LPS with a marked increase in IL-1β mRNA expression (2-fold) as compared to WT BMDMs. Since our results showed an increase in mature IL-1β in MEK2 deficient BMDMs in response to LPS (Fig. 1F), we tested whether un-processed IL-1β (pro IL-1β) is similarly increased. BMDMs derived from WT, MEK2−/− and Mek1d/d Sox2Cre/+ mice were challenged with LPS for 3h. As shown in Figure 2B, LPS stimulation led to pro IL-1β expression in BMDMs derived from all three mice strains, however BMDMs deficient in MEK2 exhibited a significantly higher level of pro IL-1β as compared to WT. In contrast, we observed a lower pro IL-1β expression in response to LPS in MEK1 deficient BMDMs. The NLRP3 protein levels in resting macrophages are thought to be insufficient for inflammasome activation. Microbial components such as TLR ligands or endogenous molecules induce NLRP3 expression through the activation of NF-κB (37). Because the activation of NLRP3 is important for mature IL-1 β release and we have seen increased IL-1 β on multiple levels (mRNA, pro IL-1β and released IL-1β), we evaluated the effect of MEK1 and 2 deficiency on NLRP3 protein levels in response to LPS. WT BMDMs responded to LPS challenge after 60 min. with NLRP3 induction. Surprisingly, we observed a more rapid and robust increase in NLRP3 levels starting at 30 min post LPS challenge in MEK2 deficient macrophages. Interestingly, MEK1 deficient macrophages exhibited minimal NLRP3 induction in response to LPS (Figure 2D). Next, we investigated the effect of MEK2 deficiency on HIF-1α expression. We observed a significantly higher HIF-1α mRNA expression in MEK2 deficient BMDMs (6-fold) as compared to WT after LPS stimulation (Fig. 2F). HIF-1α heterodimerizes with the aryl hydrocarbon receptor nuclear translocator (ARNT-HIF-1β), which facilitates its translocation into the nucleus (38). In MEK2 deficient BMDMs, ARNT mRNA expression was significantly increased in response to LPS (Fig. 2G). Because HIF-1α transcriptional activity depends on HIF-1α protein stability, we assessed HIF-1α protein levels in BMDMs derived from WT and MEK2 deficient mice in response to LPS. LPS stimulation led to a time dependent higher HIF-1α protein level in MEK2 deficient cells as compared to WT (Figs. 2H and I). These results suggest that MEK2 deficiency leads to a higher HIF-1α and IL-1β expression both at the mRNA and at the protein levels in response to LPS challenge in murine BMDMs. Several studies have shown that HIF-1α accumulation largely depends on decreased degradation due to a decreased hydroxylation by prolyl-hydroxylases (PHDs) in response to LPS. There are three PHD isoforms with distinct specificity for different prolyl hydroxylation sites within HIF-1α (39). Furthermore, the all three PHD genes are under HIF-1α transcriptional control (40). We also assessed the mRNA expression for all three PHDs after LPS challenge. We observed that all three PHDs mRNA expressions were significantly higher in MEK2 deficient BMDMs (Data not shown). The augmentation of HIF-1α level in MEK2−/− BMDMs was not due to lack of PHDs expression.
FIGURE 2. MEK2 regulates IL-1β and HIF-1α expression at the transcript and protein level.
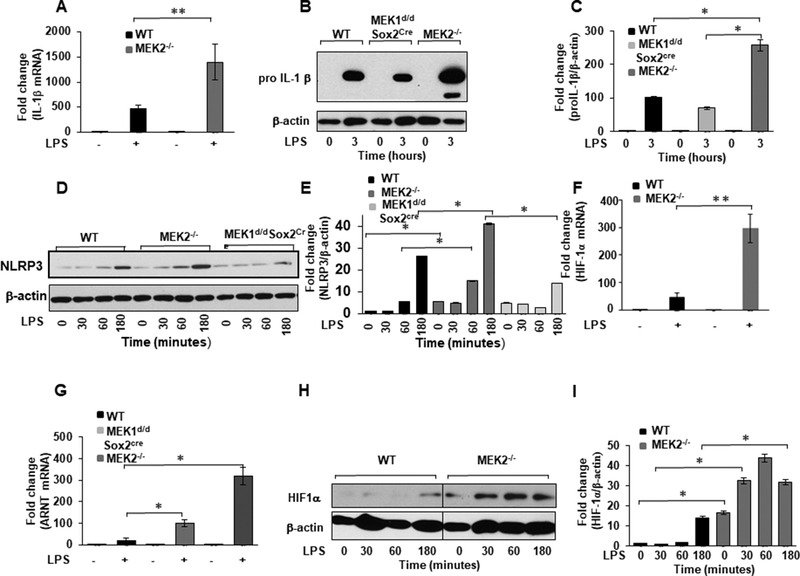
BMDMs derived from WT and MEK2−/− and Mek1d/d Sox2Cre mice were treated with LPS (100 ng/mL) for 1h. Total RNA was extracted and reverse-transcribed using the Reverse Transcription System. (A) The primers targeting IL-1β were used to amplify cDNA using iQSYBR Green Supermix. Relative mRNA levels were calculated by normalizing to GAPDH. Data were analyzed using the paired, two-tailed Student’s t test, and the results were expressed as fold change. (B) BMDMs derived from WT and MEK2−/− and Mek1d/d Sox2Cre mice were treated with LPS (100 ng/mL) for 3h. Whole cell extracts were prepared and subjected to SDS-PAGE and Western blot analysis using specific antibodies against pro IL-1β. (C) Equal loading was determined using antibodies against β-actin. (D) BMDMs derived from WT and MEK2−/− and Mek1d/d Sox2Cre were treated with LPS (100 ng/mL) for different time points as indicated. Whole cell extracts were prepared and subjected to SDS-PAGE and Western analysis using specific antibodies to NLRP3. (E) Densitometric values expressed as fold changes of the ratio NLRP3/ β-actin. (F) HIF-1α mRNA expression. RNA was isolated from cells and expression assessed using qRT-PCR. Values were normalized to GAPDH. Results represent mean values of 4 independent experiments. Using ANOVA Mann-Whitney U test a p value of <0.05 was considered significant and error bars indicate SEM. (G) ARNT (HIF-1 β) mRNA expression. Total RNA was extracted from WT and MEK2−/− and Mek1d/d Sox2Cre BMDMs treated with LPS (100 ng/mL) for 1h. The primers targeting ARNT were used to amplify cDNA. Relative mRNA levels were calculated by normalizing to GAPDH. Data were analyzed using the paired, two-tailed Student’s t test, and the results were expressed as fold change. (H) HIF-1α expression. Whole cell extracts were subjected to SDS-PAGE and Western blot analysis using specific antibodies against HIF-1α. Equal loading was determined using antibodies against β-actin. (I) Densitometric analysis of at least 3 independent experiments expressed as fold change of the ratio HIF-1α/ β-actin.
HIF-1α accumulates in nuclear extract of MEK2 deficient macrophages and interacts with p300/CBP.
Next, we assessed the nuclear accumulation of HIF-1α in BMDMs derived from WT, MEK1 and MEK2 deficient mice. BMDMs were cultured under normoxic conditions and nuclear and cytoplasmic protein fractions were assessed for HIF-1α expression at baseline and in response to LPS. The fractionated proteins were subjected to Western blot analysis assessing for HIF-1α and pVHL. HIF-1α levels were significantly increased in the nuclear fraction of MEK2 deficient BMDMs even in the absence of LPS challenge (Fig. 3A). We observed presence of pVHL in the cytoplasmic fractions, which increased in response to LPS in all three strains of mice. Interestingly, pVHL showed the highest expression in MEK2 deficient BMDMs (Fig. 3A). Members of the p300/CBP family interact(s) with HIF-1α in a DNA-bound complex to activate HIF-1α-dependent transcription of target genes (41, 42). Next, we assessed p300/CBP protein interaction with HIF-1α from MEK2 deficient BMDMs. BMDMs from WT and MEK2 deficient BMDMs were cultured and HIF-1α was immune-precipitated with anti-HIF-1α antibody. The extract was immunoblotted using HIF-1α (Fig. 3B) and p300/CBP antibodies (Fig. 3C). As shown in Fig. 3C, there is significantly higher p300/CBP in HIF-1α precipitated protein from MEK2 deficient BMDMs as compared to WT. These data indicate that in MEK2 deficient BMDMs transcriptionally active HIF-1α accumulates in the nuclear fraction and recruits the transcriptional coactivator p300/CBP. Glucose is the primary metabolic substrate of macrophages in response to endotoxin stimulation. In response to endotoxin, Glucose transporter (Glut1) expression rapidly increases and regulates metabolic reprogramming to drive pro-inflammatory cytokine production, including IL-1β (43, 44). Additionally, HIF-1 α and the RAS/MEK pathway are known to regulate Glut1 expression (45). We assessed Glut1 expression in response to LPS in BMDMs derived from WT, MEK2−/− and Mek1d/d Sox2Cre/+ mice. Cell lysates were subjected to immunoblotting using an antibody against Glut1 and equal loading was confirmed using β-actin antibody. As shown in Figs. 3D and E, BMDMs from all three mice strains responded to LPS with increased Glut1expression at 6h. Both MEK1 and MEK2 deficient BMDMs macrophages exhibit higher Glut1 at base line. In response to LPS MEK1 deficient BMDMs did not augment Glut1 expression, while MEK2 deficient BMDMs exhibit higher Glut1 at baseline, which was further augmented in response to LPS. These data indicate that MEK2−/− BMDMs exhibit significantly higher Glut1 and IL-1β, both of which are transcriptionally controlled by HIF-1α. In addition to Glut1 and IL-1β, HIF-1α regulates vascular endothelial growth factor (VEGF) expression (46). We determined the VEGF gene expression in BMDMs derived from WT, MEK1 and MEK2 deficient mice. Figure 3F shows a significantly higher expression for VEGF in MEK2 deficient BMDMs.
Figure 3. Despite higher pVHL HIF-1α accumulates in nuclear extract of MEK2 deficient BMDMs and co-immunoprecipitates with p300/CBP.
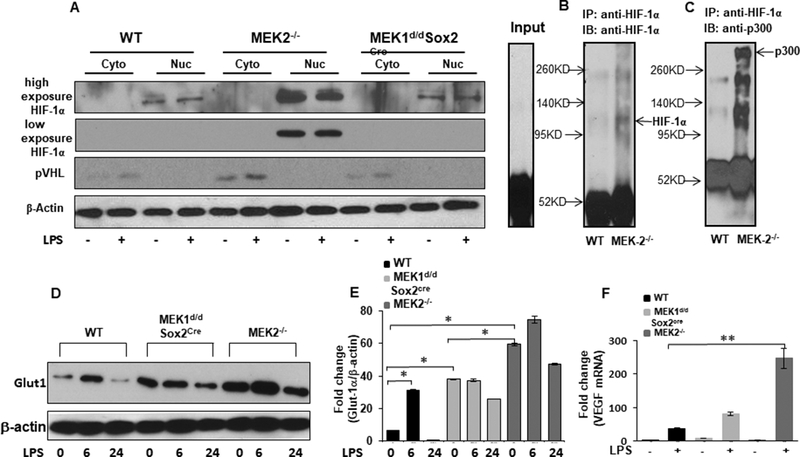
WT, MEK2−/−, and MEK1d/dSox2cre BMDMs were cultured and challenged with LPS (100 ng/mL) for 3h. Nuclear and cytosolic extracts were prepared and subjected to SDS-PAGE. (A) Western blot analysis was performed using specific antibodies against, VHL, HIF-1α and against β-actin. MEK2−/− BMDMs exhibit higher HIF-1α in nuclear extracts. Protein lysates prepared from untreated WT and MEK2−/− BMDMs were immunoprecipitated with HIF-1α specific antibody and equal amount of immunoprecipitates were subjected to SDS-PAGE. (Band C) Western blot analysis was performed using specific antibodies to HIF-1α and p300/CBP. MEK2−/− BMDMs exhibit higher p300/CBP protein in the lysates immunoprecipitated with HIF-1α specific antibody. (D) GLUT1 expression. BMDMs derived from WT and MEK2−/− and Mek1d/d Sox2Cre mice were treated with LPS (100 ng/mL) for 6 h and 24h. Whole cell extracts were subjected to SDS-PAGE and Western blot analysis using specific antibody against GLUT1. Equal loading was determined using antibody against β-actin. (E) Densitometric analysis of at least 3 independent experiments expressed as fold change of the ratio Glut1/ β-actin. MEK2 deficiency exhibited higher Glut1 levels in response to LPS. (F) VEGF mRNA expression. Total RNA was extracted from WT and MEK2−/− and Mek1d/d Sox2Cre BMDMs treated with LPS (100 ng/mL) for 1h and VEGF expression was assessed using qRT-PCR. Values were normalized to GAPDH. Results represent mean values of 4 independent experiments. Using ANOVA Mann-Whitney U test, * signifies a p value <0.05, while ** signifies a p value <0.001, and error bars indicate SEM. MEK2 deficient BMDMs showed highly significant expression of VEGF in response to LPS.
Targeted downregulation of HIF-1α in MEK2 deficient macrophages leads to a decreased IL-1β production.
Because HIF-1α is a transcription factor regulating both IL-1β and Glut1, we investigate whether targeted downregulation of HIF-1α via siRNA can modify IL-1β level in response to LPS in MEK2 deficient macrophages. MEK 2−/− BMDMs were transiently transfected with either nonsense vector (scrambled siRNA) or vector encoding HIF-1α siRNA. After 24 hours transfection, cells were challenged with LPS. We observed a significant decrease in HIF-1α expression with targeted HIF-1α siRNA and a 80% decrease in pro-IL-1β in response to LPS, as compared to non-targeted siRNA transfected cells (Fig. 4A–C). Similarly, when we measured released IL-1β in conditioned medium of transfected cells after 24 hours LPS challenge, we observed a significant reduction in IL-1β production (Fig. 4D). These data indicate that HIF-1α is an important transcription factor for IL-1β production in response to LPS in MEK2 deficient macrophages.
FIGURE 4. Targeted downregulation of HIF-1α in MEK2 deficient macrophages leads to a decreased IL-1β production.
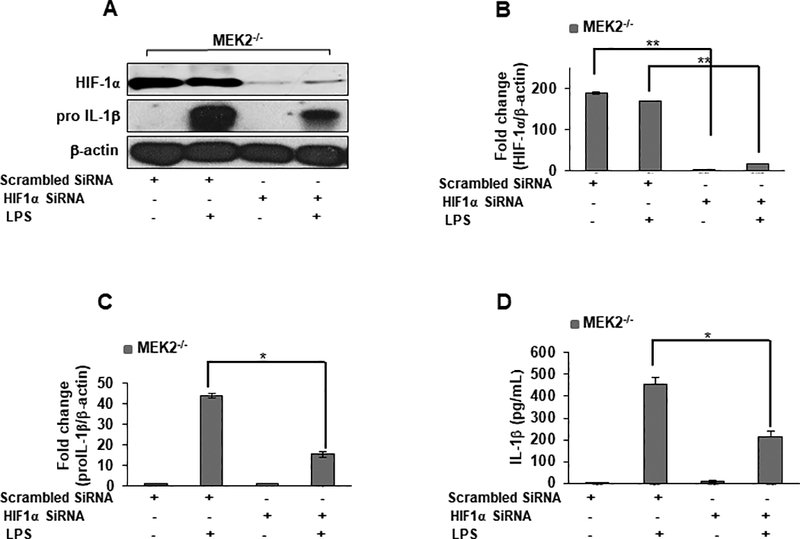
MEK2−/− BMDMs were transfected with HIF-1α or scrambled siRNA. 24 h post-transfection, cells were incubated with/without LPS for 6 h or 24. (A) Total cellular protein (of siRNA transfected cells) were subjected to Western blot analysis using antibodies against HIF-1α, pro IL-1β or β-actin. (B) Densitometric values expressed as fold increase of the ratio of HIF-1α/β-actin. (C) Densitometric values expressed as fold increase of the ratio of pro IL-1β/β-actin. (D) Conditioned media were collected for IL-1β analyses via ELISA. Targeted down regulation of HIF-1α resulted in a significant decrease of IL-1 β in response to LPS (p < 0.05). Data are presented as relative gene expression levels from three independent transfections each performed in triplicates. Data are representative results of three independent experiments each performed in triplicates. Using ANOVA Mann-Whitney U test, * signifies a p value <0.05, while ** signifies a p value <0.001, and error bars indicate SEM.
MEK2 is dispensable in LPS mediated ERK activation in BMDMs.
To address the relative contribution of MEK2 in LPS mediated MAPK activation, we first determined the level of MEK1 and MEK2 protein expression by Western blot analysis in BMDMs derived from MEK2−/− mice. As shown in Fig. 5A, MEK2 deficient BMDMs show no expression of MEK2 protein with comparable MEK1 protein levels in both WT and MEK2 deficient macrophages. To further investigate the contribution of MEK2 in LPS mediated MEK1/2 phosphorylation, BMDMs from WT and MEK2−/− mice were challenged with LPS for different time periods. LPS treatment of WT BMDMs led to phosphorylation of MEK1/2 (Ser217/221), which persisted up to 3h (Fig. 5B). MEK2−/− BMDMs showed a lower phosphorylation of MEK1/2 compared to WT BMDMs (Fig. 5B). The phosphorylation seen in MEK2 deficient BMDMs is mainly due to the presence of MEK1, as the antibody recognizes the phosphorylated forms of both MEK1 and MEK2. Furthermore, we assessed the role of MEK2 in the activation of other MAP kinases (ERK, p38 and JNK). Cell lysates were subjected to immunoblotting using antibodies against the phosphorylated forms of ERK (Thr202/Tyr204), p38 (Thr180/Tyr182) and SAPK/JNK (Thr183/Tyr185). As expected, LPS stimulation led to ERK1/2 phosphorylation in WT BMDMs and in MEK2 deficient cells. This phosphorylation peaked at 30 min and persisted for up to 3h (Fig. 5C). Densitometric analysis is shown in Figure 5D. LPS treatment led to a rapid and similar pattern of p38 phosphorylation in WT, in both MEK1 and MEK2 deficient BMDMs (Fig. 5G). Densitometric analysis is shown in Figure 5H. We also detected JNK phosphorylation. As shown in Figure 5E both WT, MEK1 and MEK2 deficient BMDMs responded to LPS challenge with similar JNK phosphorylation. Densitometric analysis is shown in Figure 5F. Taken together, our results suggest that MEK2 is dispensable in ERK, p38 and JNK phosphorylation in response to LPS stimulation of murine BMDMs.
FIGURE 5. MEK2 is dispensable for LPS mediated MAP kinase activation including ERK phosphorylation.
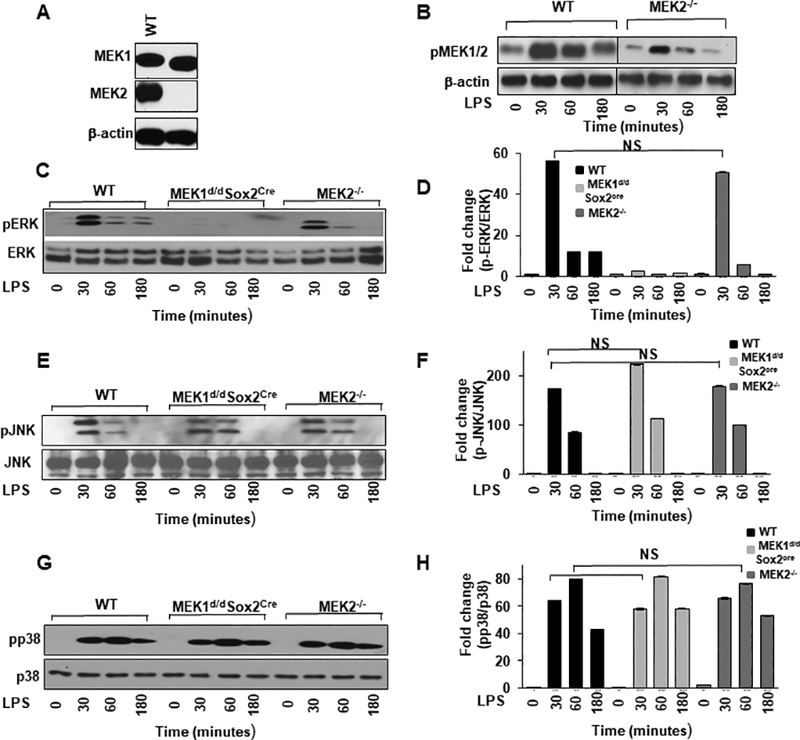
Murine BMDMs derived from WT, and Mek2−/− mice were treated with LPS (100 ng/mL) for different time points as indicated. (A) Detection of MEK1 and MEK2 isoforms. Whole cell extracts were prepared and subjected to SDS-gel electrophoresis and Western blot analysis using antibodies against MEK1 and MEK2. Equal loading was confirmed using β-actin antibodies. As shown, MEK2−/− BMDMs lack expression of MEK2. (B) LPS induced phosphorylation of MEK1/2 in WT and MEK2−/− BMDMs. Murine BMDMs were challenged with LPS for different time points as indicated. Whole cell extracts were prepared and 15μg of proteins were subjected to SDS-gel electrophoresis and Western blot analysis was performed using antibodies against the phosphorylated forms of MEK1/2 (Ser217/221). Equal loading was confirmed using β-actin antibodies. MEK2−/− BMDMs showed a lower phosphorylation of MEK1/2 which is mainly due to phosphorylation of MEK1. (C) LPS induced phosphorylation of ERK. Whole cell extracts were subjected to SDS-gel electrophoresis and Western blot analysis performed using antibodies against the phosphorylated forms of ERK (Thr202/Tyr204) and total ERK. (D) Densitometric analysis of at least 3 independent experiments expressed as fold change of the ratio phosphorylated/total ERK. (E) LPS induced phosphorylation of JNK. Western blot analysis was performed with antibodies against the phosphorylated form of SAPK/JNK (Thr183/Tyr185) and equal loading was determined measuring total JNK. (F) Densitometric analysis of at least 3 independent experiments expressed as fold change of the ratio phosphorylated/total JNK. (G) LPS induced phosphorylation of p38. Western blot analysis was performed with antibody against the phosphorylated form of p38 (Thr180/Tyr182) and equal loading was determined using antibody against total p38. (H) Densitometric analysis of at least 3 independent experiments expressed as fold change of the ratio phosphorylated/total p38. Using ANOVA Mann-Whitney U test, a p value <0.05 was considered significant and error bars indicate SEM.
MEK2 overexpression decreases IL-1β production with no significant effect on ERK phosphorylation.
Our data are consistent with the hypothesis that MEK2 negatively regulates IL-1β independent of ERK activation. Hence, we speculated that overexpression of MEK2 should prevent the production of IL-1β. To this end, we transfected RAW264.7 cells with CMV-MEK2 for 24h. As shown in Figure 6A, CMV-MEK2 transfected cells showed a significant increase in the expression of MEK2 protein compared to non-transfected (NT) cells. To further investigate the contribution of MEK2 in LPS mediated MEK1/2 phosphorylation, cells from NT and CMV-MEK2 were cultured in the presence of LPS for 30 min. LPS treatment led to a 2 fold increase in MEK1/2 phosphorylation in CMV-MEK2 transfected RAW264.7 cells as compared to NT cells (Fig. 6A). Densitometric analysis is shown in Figure 6B. To elucidate the role of MEK2 overexpression in regulating ERK activation in response to LPS, we challenged NT or CMV-MEK2 transfected RAW264.7 cells with LPS for 30 min and assessed for ERK phosphorylation. As shown in Figure 6c, LPS stimulation led to ERK phosphorylation in NT RAW264.7 cells. Interestingly, ERK phosphorylation was not significantly changed in MEK2 overexpressed cells (Fig. 6C). Densitometric analysis is shown in Figure 6D. Furthermore, we tested whether overexpression of MEK2 could decrease the level of pro IL-1β. Cell lysates were subjected to immunoblotting using antibodies against pro IL-1β. As shown in Figure 6e, as expected LPS stimulation led to a high pro IL-1β expression in NT RAW264.7 cells. While RAW264.7 cells overexpressing CMV-MEK2 responded to LPS with a significant lower pro IL-1β level as compared to NT cells (Fig. 6E). Densitometric analysis is shown in Figure 6F. These results suggest that MEK2 overexpression leads to a lower expression of IL-1β in response to LPS challenge with no significant effect on ERK phosphorylation.
FIGURE 6. MEK2 overexpression decreases IL-1β production with no significant effect on ERK activation.
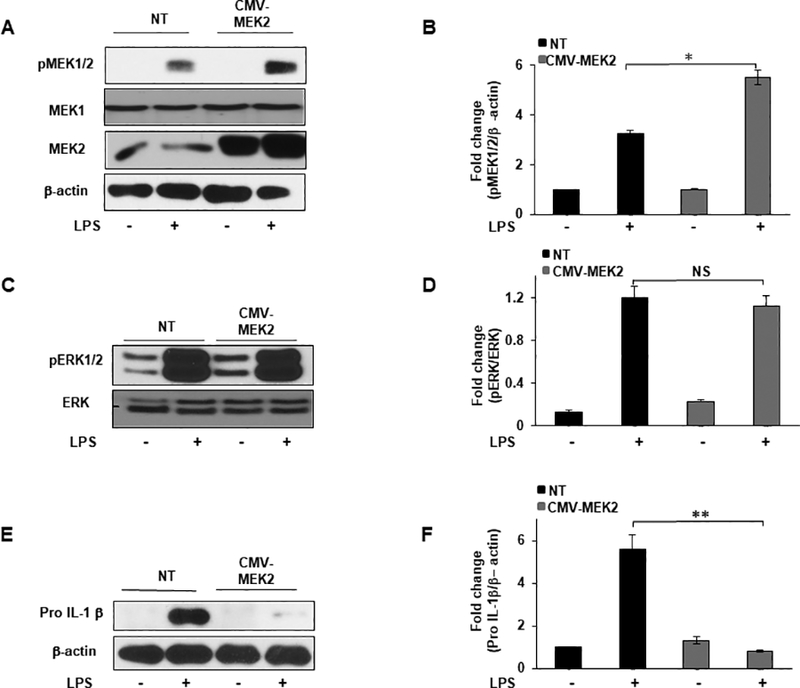
RAW264.7 cells were transfected with a Mek2-Myc construct (CMV-MEK2) for 24h or cultured without plasmid (NT). MEK2 overexpressed or NT cells were treated with LPS (100 ng/mL) for 30 minutes or 3h. (A) Whole cell lysates were subjected to SDS-PAGE followed by Western blot analysis using antibodies against MEK2 and phospho-specific antibody against MEK1/2. Equal loading was determined using antibody against β-actin. (B) Densitometric values expressed as fold change of the ratio pMEK1/2/β-actin. (C) Western blot analysis was performed using antibodies against phospho-specific ERK1/2 and total ERK. (D) Densitometric values expressed as fold change of the ratio pERK1/2/total ERK. (E) Western blot analysis was performed using antibody against pro IL-1β, equal loading was determined using antibody against β-actin. (F) Densitometric values expressed as fold increase of the ratio pro IL-1β/β-actin. Data presented for all experiments are representative of at least 3 independent experiments. Using ANOVA Mann-Whitney U test for all results, a p value <0.05 was considered significant and error bars indicate SEM.
MEK2 expression regulates the level of HIF-1α and IL-1β expression in BMDMs in response to LPS.
Because MEK1 and MEK2 structurally similar, they are thought to be functionally redundant. However, recently Catalanotti et. al. have shown that MEK1 and MEK2 build a heterodimer complex containing a negative feedback loop (47). This raises the question whether there is a role for a disrupted negative feedback loop through a lack of MEK1-MEK2 heterodimerization in MEK2 deficient BMDMs. To test this hypothesis, we generated mutants with three Mek allele deleted (triple mutants). Mice with only one Mek1 left (Mek1f/+ Mek2−/− Vav iCre+) are named 1M1L and triple mutant with only one Mek2 left (Mek1f/f Mek2+/− Vav iCre+) are named 1M2L. We then investigated the role of Mek1 and Mek2 double and triple mutation on ERK phosphorylation, HIF-1α expression and IL-1β production. BMDMs from WT, MEK2−/−, 1M1L and 1M2L were isolated and challenged with LPS. ERK1/2 phosphorylation in response to of LPS in WT BMDMs Mek2 double mutant mice were comparable, whereas or 1M1L mutant exhibited a decreased level of phosphorylated ERK1/2 (Figs 7A and B). BMDMs from triple mutant macrophages lacking two MEK1 (1M2L) responded with a significant diminished ERK phosphorylation to LPS challenge. Figure 7A lower panel shows the expression for MEK1 and MEK2 proteins. The mean densitometric analysis is shown in Figure 7b. Next, we determined the effect of these mutants on HIF-1α expression by Western blot analysis. WT BMDMs responded to LPS with a minimal expression of HIF-1α, while the absence of MEK2 led to increased HIF-1α expression in response to LPS challenge (Fig. 7C). BMDMs from the Mek2 double or 1M1L mutant mice exhibited a significantly increased level of HIF-1α protein expression in response to LPS challenge. BMDMs from triple mutant macrophages lacking two MEK1 (1M2L) exhibited higher baseline HIF-1α expression but no further increase in response to LPS (Fig. 7C). Mean densitometric analysis is shown in Figure 7D. Next, we assessed IL-1β production in response to LPS in Mek1 and Mek2 double and triple mutations. As shown in Figure 7E, the highest amount of IL-1β production was observed in macrophages derived from mice lacking MEK2 in response to LPS challenge. Importantly, introduction of one allele of MEK2 (1M2L) (Mek1f/f Mek2+/− Vav iCre+) led to a significant decrease of IL-1β production in response to LPS. These data suggest that rather a complete deletion of the MEK2 allele leads to higher IL-1β production and not the absence of MEK1, as the presence of MEK1 (1M1L) could not completely restore ERK phosphorylation and decreased IL-1β production. Figure 7F demonstrates the relationship of MEK2 protein expression of BMDMs and IL-1β production in response to LPS.
FIGURE 7. MEK2 expression determines IL-1β and HIF-1α expression in response to LPS.
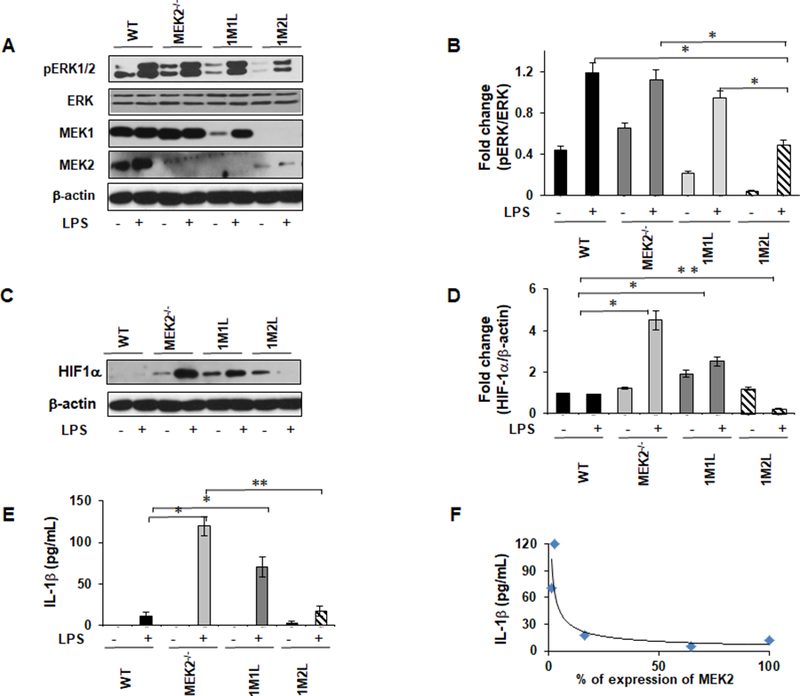
Murine BMDMs derived from WT, MEK2−/−, one MEK1 left (1M1L), and one MEK2 left (1M2L) were treated with LPS (100 ng/mL) for 30 minutes. (A) Whole cell lysates were subjected to SDS-PAGE followed by Western blot analysis using specific antibody against phospho ERK. Equal loading was determined using antibody against total ERK. (B) Densitometric analysis expressed as fold increase of the ratio pERK1/2/ERK. (C) Western blot analysis was performed using antibodies against HIF-1α and against β-actin. (D) Densitometric analysis expressed as fold increase of the ratio of HIF-1α/β-actin. (E) BMDMs were isolated from WT, MEK1-deficient mice, one MEK1 left (1M1L), and one MEK2 left (1M2L) and challenged with LPS (100 ng/mL) for 24h. Conditioned media were analyzed for IL-1β via ELISA. (F) Densitometric quantifications of three immunoblots of MEK2 protein expression (x-axis) plotted against pro-IL-1β (y-axis). Data presented for all experiments are representative of at least 4 independent experiments. Using ANOVA Mann-Whitney U test, a p value <0.05 was considered significant and error bars indicate SEM.
Discussion
Despite structural similarity between MEK1 and MEK2 evidence indicates that they are not functionally redundant. Several studies using genetic dissection of MEK1 and MEK2 in mouse models has suggested non-overlapping functions in disease development (5, 47). MEK1 and MEK2 show high homology in their kinase domains, while their N termini and their proline-rich domains show only 40% identity, which provides an opportunity for the two isoforms to interact and partner differently with scaffolding proteins and their activators and substrates (48, 49). Although the role of MEKs is well studied in developmental and cancer biology, the specific role of MEK2 versus MEK1 in innate immunity is not well understood. Most previous reports used chemical inhibitors (mostly U0126 or PD98059) to study the role of MEK/ERK in cancer biology or in TLR mediated activation (45, 50). However, these inhibitors are nonspecific for MEK1 versus MEK2 and have off-target effects on several other kinases (51). In most studies physiological consequences of MEK1/2 activation are judged by the effect on the activation of their best-known substrate ERK1/2. Our investigation is the first study to describe a unique role of MEK2 in HIF-1α expression and IL-1β production after LPS challenge that is independent of activation of ERK, p38 and JNK in murine macrophages. Interestingly, MEK2−/− macrophages exhibited early induction of NLRP3 in response to LPS associated with increased release of mature IL-1β. Furthermore, BMDMs derived from MEK2 deficient mice challenged with LPS exhibit a preserved ERK activation but significantly higher HIF-1α expression both at the mRNA and protein levels. In contrast, we observed a lower level of NLRP3 induction, reduced pro IL-1β and mature IL-1β production, and a lower HIF-1 α level in response to LPS challenge in MEK1 deficient BMDMs.
HIF-1α is recognized as the master regulator of the hypoxic response, which modulates transcription of numerous genes (52). HIF-1α expression is critical for the metabolic switch during inflammation and in cancer, as its induction regulates glucose uptake and the expression of glycolytic enzymes (53). In cancer and during inflammation aerobic glycolysis (Warburg effect) plays an important role in the maintenance of cellular energy supply, as the aerobic ATP production through the tricyclic acid cycle (TCA cycle) is suppressed (50, 54). This metabolic switch from oxidative phosphorylation to aerobic glycolysis is grossly coordinated by HIF-1α expression (53). Glucose is a major source of energy during inflammation and transported across the plasma membrane facilitated by glucose transporters. Among these glucose transporters, Glut1 regulates the enhanced glucose uptake in macrophages as well as in cancer cells. Its expression is rapidly induced in response to LPS (55, 56). Glut1 down regulation decreases inflammatory response, especially IL-1β production (57, 58). Various kinases including GSK3/TSC/mTOR, PI3K/AKT and RAS/MEK pathways have been proposed to regulate Glut1 expression (59). Surprisingly, in both MEK1 and MEK2 deficient macrophages we found increased Glut1 levels at baseline as compared to WT. MEK2 deficiency exhibited a higher Glut1 levels, which significantly increased in response to LPS.
Although HIF-1α accumulation was originally identified under hypoxia, as its name implies, now a large body of literature indicates that HIF-1α is strongly activated by TLRs, IFN-α, EGF and other cytokine receptors under well oxygenated conditions (13, 60, 61). There are several mechanisms underlying HIF-1α accumulation in response to LPS. TLR4 mediated induction of HIF-1α reflects a combination of increased HIF-1α transcription and decreased HIF-1α degradation (13). Here, we show that HIF-1α transcripts and protein expression were significantly increased in responses to LPS in MEK2 deficient macrophages as compared to WT. Additionally, the ARNT (HIF-1β), the binding partner of HIF-1α, was significantly increased in MEK2 deficient macrophages. Proline hydroxylation is important step in HIF-1α destabilization (39). In our study, augmented HIF-1α expression was not due to diminished expression of PHDs, as in MEK2 deficient BMDMs the expression for all three PHDs were higher in response to LPS. HIF-1α transcriptional activity is dependent on nuclear translocation(62). We observed that HIF-1α rapidly accumulates in the nuclear fraction in MEK2 deficient macrophages as compared to WT macrophages.
The molecular basis of how TLR4 mediates activation of RAS-GTPase upstream to MEK has not been precisely elucidated. Interaction of LPS or other lipid based mediators (e.g. lipid A) leads to association of TLR4 and MD2, which recruits several adaptor proteins (63). This is thought to lead to the subsequent activation of receptor tyrosine kinases (RTKs) and the classical RAS-RAF-MEK1/2 cascade (64–66). Additionally, TLR4 ligation can activate ERK1/2 through alternative pathways independent of RAS-RAF-MEK (67, 68). For instance, LPS can activate MEK/ERK through PKC ζ (69). Recently, we have shown that the MEK1 isoform plays a critical role in ERK1/2 activation in response to LPS (7). Despite intact MEK2, MEK1 deficient macrophages responded only minimally to LPS with ERK activation (7). While MEK1 deficient macrophages responded to LPS challenge with enhanced STAT4 phosphorylation and a heightened IL-12 production but decreased IL-10 production. The simultaneous challenge of TLR4 and retinoic acid (RA) or recombinant IL-10 restored ERK1/2 phosphorylation(7). This suggests that MEK2 mediated ERK1/2 activation requires additional signals (RA or IL-10) besides TLR receptor activation (7). Our current work shows that MEK2−/− BMDMs respond to LPS stimulation similar to MEK1 deficient macrophages with lower IL-10, in contrast IL-12 production in MEK2−/− macrophages were significantly lower as compared to MEK1 deficient macrophages. Furthermore, we observed in MEK2 deficient macrophages strikingly higher production of IL-1β production in response to LPS challenge. Furthermore, overexpression of MEK2 in RAW264.7 cells decreased IL-1β production in response to LPS, while MEK2 overexpression did not modulate ERK activation (Fig. 6). Most interestingly, the IL-1β producing phenotype of BMDMs from triple Mek knockout mice (lacking Mek1 but having one allele of Mek2) was rescued by introducing one allele of the Mek2 gene (Fig. 7). BMDMs of these mice challenged with LPS showed preserved ERK activation but a lower HIF-1α expression as compared to MEK2 deficient macrophages. This data indicates that the level MEK2 regulates IL-1β production in response to LPS independent of ERK activation. Our previous and current work suggests a differential role of MEK 1 and 2 in the regulation of cytokine networks in response to TLR4 activation. This observation are consistent with the differential role of the MEK isoforms in cancer cell biology (48), where HIF-1α is thought to be central in controlling neovascularization, survival and tumor spread in various cancers. Cancer associated enhanced HIF-1α expression is predominantly attributed to oxygen limitation in the tumor microenvironment (70). Because the deregulation of Ras/Raf/MEK/ERK signaling cascades and HIF signaling plays a crucial central role in tumor growth and spread (71, 72) and our data indicates that MEK2 plays an important role in HIF signaling, it would be interesting to further investigate the role of MEK2 in cancers.
Our data suggests an exquisite role of MEK2 as negative regulator of HIF-1α and Glut1 expression as well as IL-1β production. Yet, it remains unclear how the lack of MEK2 would influence the complex interplay of these pathways. (73, 74). Further studies need to determine whether MEK2 directly or indirectly regulates HIF-1α expression. In summary, our data indicates that there is a unique and un-dispensable role for MEK2 in regulating HIF-1α and thereby modulating IL-1β in response to LPS challenge that cannot be substituted by MEK1. Thus, modulation of MEK2 may represent a novel therapeutic target in HIF-1α mediated inflammation as well as cancer.
Acknowledgement:
Grant information: this work was supported by a grant (R01HL113508 (LS) as well as the Department of Medicine and the Center for Molecular Medicine and Genetics, Wayne State University School of Medicine (LS).
REFERENCES
- 1.Belanger LF, Roy S, Tremblay M, Brott B, Steff AM, Mourad W, Hugo P, Erikson R, and Charron J. 2003. Mek2 is dispensable for mouse growth and development. Molecular and cellular biology 23: 4778–4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giroux S, Tremblay M, Bernard D, Cardin-Girard JF, Aubry S, Larouche L, Rousseau S, Huot J, Landry J, Jeannotte L, and Charron J. 1999. Embryonic death of Mek1-deficient mice reveals a role for this kinase in angiogenesis in the labyrinthine region of the placenta. Curr Biol 9: 369–372. [DOI] [PubMed] [Google Scholar]
- 3.Seger R, and Krebs EG. 1995. The MAPK signaling cascade. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 9: 726–735. [PubMed] [Google Scholar]
- 4.Scholl FA, Dumesic PA, and Khavari PA. 2004. Mek1 alters epidermal growth and differentiation. Cancer research 64: 6035–6040. [DOI] [PubMed] [Google Scholar]
- 5.Zmajkovicova K, Jesenberger V, Catalanotti F, Baumgartner C, Reyes G, and Baccarini M. 2013. MEK1 Is Required for PTEN Membrane Recruitment, AKT Regulation, and the Maintenance of Peripheral Tolerance. Molecular cell 11:43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li X, Cao X, Zhang X, Kang Y, Zhang W, Yu M, Ma C, Han J, Duan Y, and Chen Y. 2016. MEK1/2 inhibitors induce interleukin-5 expression in mouse macrophages and lymphocytes. Biochem Biophys Res Commun. 13:939–946. [DOI] [PubMed] [Google Scholar]
- 7.Bouhamdan M, Bauerfeld C, Talreja J, Beuret L, Charron J, and Samavati L. 2015. MEK1 dependent and independent ERK activation regulates IL-10 and IL-12 production in bone marrow derived macrophages. Cell Signal 27: 2068–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akira S, and Takeda K. 2004. Toll-like receptor signalling. Nature reviews immunology 4: 499–511. [DOI] [PubMed] [Google Scholar]
- 9.Kawai T, and Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11: 373–384. [DOI] [PubMed] [Google Scholar]
- 10.Hsu H-Y, and Wen M-H. 2002. Lipopolysaccharide-mediated reactive oxygen species and signal transduction in the regulation of interleukin-1 gene expression. Journal of Biological Chemistry 277: 22131–22139. [DOI] [PubMed] [Google Scholar]
- 11.Gay NJ, Gangloff M, and Weber AN. 2006. Toll-like receptors as molecular switches. Nat Rev Immunol 6: 693–698. [DOI] [PubMed] [Google Scholar]
- 12.Frede S, Stockmann C, Winning S, Freitag P, and Fandrey J. 2009. Hypoxia-inducible factor (HIF) 1alpha accumulation and HIF target gene expression are impaired after induction of endotoxin tolerance. J Immunol 182: 6470–6476. [DOI] [PubMed] [Google Scholar]
- 13.Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, and Nizet V. 2007. Cutting edge: Essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J Immunol 178: 7516–7519. [DOI] [PubMed] [Google Scholar]
- 14.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, Jany SS, Corr SC, Haneklaus M, Caffrey BE, Pierce K, Walmsley S, Beasley FC, Cummins E, Nizet V, Whyte M, Taylor CT, Lin H, Masters SL, Gottlieb E, Kelly VP, Clish C, Auron PE, Xavier RJ, and O’Neill LA. 2013. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496: 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maxwell PH, Wiesener MS, Chang G-W, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, and Ratcliffe PJ. 1999. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399: 271–275. [DOI] [PubMed] [Google Scholar]
- 16.Haase VH 2009. The VHL tumor suppressor: master regulator of HIF. Current pharmaceutical design 15: 3895–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Isaacs JS, Jung Y-J, Mimnaugh EG, Martinez A, Cuttitta F, and Neckers LM. 2002. Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1α-degradative pathway. Journal of Biological Chemistry 277: 29936–29944. [DOI] [PubMed] [Google Scholar]
- 18.Frede S, Stockmann C, Freitag P, and Fandrey J. 2006. Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-kappaB. Biochem J 396: 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Treins C, Giorgetti-Peraldi S, Murdaca J, Semenza GL, and Van Obberghen E. 2002. Insulin stimulates hypoxia-inducible factor 1 through a phosphatidylinositol 3-kinase/target of rapamycin-dependent signaling pathway. J Biol Chem 277: 27975–27981. [DOI] [PubMed] [Google Scholar]
- 20.Flügel D, Görlach A, Michiels C, and Kietzmann T. 2007. Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1α and mediates its destabilization in a VHL-independent manner. Molecular and cellular biology 27: 3253–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kalucka J, Franke K, Ehrenschwender M, Schley G, Beneke A, rg Sutter J, Hentze MW, Koivunen P, Oefner PJ, and Bogdan C. Ferritin-Mediated Iron Sequestration Stabilizes Hypoxia-Inducible Factor-1a upon LPS Activation in the Presence of Ample Oxygen. [DOI] [PubMed]
- 22.Siegert I, Schödel J, Nairz M, Schatz V, Dettmer K, Dick C, Kalucka J, Franke K, Ehrenschwender M, and Schley G. 2015. Ferritin-Mediated Iron Sequestration Stabilizes Hypoxia-Inducible Factor-1α upon LPS Activation in the Presence of Ample Oxygen. Cell reports 13: 2048–2055. [DOI] [PubMed] [Google Scholar]
- 23.Frede S, Berchner-Pfannschmidt U, and Fandrey J. 2007. Regulation of hypoxia-inducible factors during inflammation. Methods in enzymology 435: 405–419. [DOI] [PubMed] [Google Scholar]
- 24.Chilov D, Camenisch G, Kvietikova I, Ziegler U, Gassmann M, and Wenger RH. 1999. Induction and nuclear translocation of hypoxia-inducible factor-1 (HIF-1): heterodimerization with ARNT is not necessary for nuclear accumulation of HIF-1alpha. Journal of cell science 112: 1203–1212. [DOI] [PubMed] [Google Scholar]
- 25.Hirota SA, Beck PL, and MacDonald JA. 2009. Targeting hypoxia-inducible factor-1 (HIF-1) signaling in therapeutics: implications for the treatment of inflammatory bowel disease. Recent patents on inflammation & allergy drug discovery 3: 1–16. [DOI] [PubMed] [Google Scholar]
- 26.Blouin CC, Pagé EL, Soucy GM, and Richard DE. 2004. Hypoxic gene activation by lipopolysaccharide in macrophages: implication of hypoxia-inducible factor 1α. Blood 103: 1124–1130. [DOI] [PubMed] [Google Scholar]
- 27.Bissonauth V, Roy S, Gravel M, Guillemette S, and Charron J. 2006. Requirement for Map2k1 (Mek1) in extra-embryonic ectoderm during placentogenesis. Development 133: 3429–3440. [DOI] [PubMed] [Google Scholar]
- 28.Bauerfeld CP, Rastogi R, Pirockinaite G, Lee I, Huttemann M, Monks B, Birnbaum MJ, Franchi L, Nunez G, and Samavati L. 2012. TLR4-mediated AKT activation is MyD88/TRIF dependent and critical for induction of oxidative phosphorylation and mitochondrial transcription factor A in murine macrophages. J Immunol 188: 2847–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rastogi R, Jiang Z, Ahmad N, Rosati R, Liu Y, Beuret L, Monks R, Charron J, Birnbaum MJ, and Samavati L. 2013. Rapamycin Induces Mitogen-activated Protein (MAP) Kinase Phosphatase-1 (MKP-1) Expression through Activation of Protein Kinase B and Mitogen-activated Protein Kinase Kinase Pathways. J Biol Chem 288: 33966–33977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Samavati L, Rastogi R, Du W, Huttemann M, Fite A, and Franchi L. 2009. STAT3 tyrosine phosphorylation is critical for interleukin 1 beta and interleukin-6 production in response to lipopolysaccharide and live bacteria. Mol Immunol 46: 1867–1877. [DOI] [PubMed] [Google Scholar]
- 31.Liu C, Zhang X, Zhou JX, Wei W, Liu DH, Ke P, Zhang GF, Cai GJ, and Su DF. 2013. The protective action of ketanserin against lipopolysaccharide-induced shock in mice is mediated by inhibiting inducible NO synthase expression via the MEK/ERK pathway. Free radical biology & medicine 65: 658–666. [DOI] [PubMed] [Google Scholar]
- 32.Moore TC, and Petro TM. 2013. IRF3 and ERK MAP-kinases control nitric oxide production from macrophages in response to poly-I:C. FEBS Lett 587: 3014–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu L, Ke Y, Jiang X, He F, Pan L, Xu L, Zeng X, and Ba X. 2012. Lipopolysaccharide activates ERK-PARP-1-RelA pathway and promotes nuclear factor-kappaB transcription in murine macrophages. Human immunology 73: 439–447. [DOI] [PubMed] [Google Scholar]
- 34.O’Neill LA 2008. When signaling pathways collide: positive and negative regulation of toll-like receptor signal transduction. Immunity 29: 12–20. [DOI] [PubMed] [Google Scholar]
- 35.Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, and Kanneganti TD. 2010. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32: 379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dinarello CA 2009. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 27: 519–550. [DOI] [PubMed] [Google Scholar]
- 37.He Y, Hara H, and Núñez G. 2016. Mechanism and regulation of NLRP3 inflammasome activation. Trends in biochemical sciences 41: 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang B-H, Rue E, Wang GL, Roe R, and Semenza GL. 1996. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. Journal of Biological Chemistry 271: 17771–17778. [DOI] [PubMed] [Google Scholar]
- 39.Appelhoff RJ, Tian Y-M, Raval RR, Turley H, Harris AL, Pugh CW, Ratcliffe PJ, and Gleadle JM. 2004. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. Journal of Biological Chemistry 279: 38458–38465. [DOI] [PubMed] [Google Scholar]
- 40.Marxsen JH, Stengel P, Doege K, Heikkinen P, Jokilehto T, Wagner T, Jelkmann W, Jaakkola P, and Metzen E. 2004. Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by induction of HIF-α-prolyl-4-hydroxylases. Biochemical Journal 381: 761–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ema M, Hirota K, Mimura J, Abe H, Yodoi J, Sogawa K, Poellinger L, and Fujii-Kuriyama Y. 1999. Molecular mechanisms of transcription activation by HLF and HIF1alpha in response to hypoxia: their stabilization and redox signal-induced interaction with CBP/p300. EMBO J 18: 1905–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA, Bunn HF, and Livingston DM. 1996. An essential role for p300/CBP in the cellular response to hypoxia. Proceedings of the National Academy of Sciences 93: 12969–12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gamelli RL, Liu H, He L-K, and Hofmann CA. 1996. Augmentations of glucose uptake and glucose transporter-1 in macrophages following thermal injury and sepsis in mice. Journal of leukocyte biology 59: 639–647. [DOI] [PubMed] [Google Scholar]
- 44.Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, Macintyre AN, Goraksha-Hicks P, Rathmell JC, and Makowski L. 2014. Metabolic reprogramming of macrophages glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. Journal of Biological Chemistry 289: 7884–7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lim J-H, Lee E-S, You H-J, Lee JW, Park J-W, and Chun Y-S. 2004. Ras-dependent induction of HIF-1α 785 via the Raf/MEK/ERK pathway: a novel mechanism of Ras-mediated tumor promotion. Oncogene 23: 9427. [DOI] [PubMed] [Google Scholar]
- 46.Palazon A, Goldrath AW, Nizet V, and Johnson RS. 2014. HIF transcription factors, inflammation, and immunity. Immunity 41: 518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Catalanotti F, Reyes G, Jesenberger V, Galabova-Kovacs G, de Matos Simoes R, Carugo O, and Baccarini M. 2009. A Mek1-Mek2 heterodimer determines the strength and duration of the Erk signal. Nature structural & molecular biology 16: 294–303. [DOI] [PubMed] [Google Scholar]
- 48.Ussar S, and Voss T. 2004. MEK1 and MEK2, different regulators of the G1/S transition. Journal of Biological Chemistry 279: 43861–43869. [DOI] [PubMed] [Google Scholar]
- 49.Kolch W 2000. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochemical Journal 351: 289–305. [PMC free article] [PubMed] [Google Scholar]
- 50.Koppenol WH, Bounds PL, and Dang CV. 2011. Otto Warburg’s contributions to current concepts of cancer metabolism. Nature Reviews Cancer 11: 325. [DOI] [PubMed] [Google Scholar]
- 51.Wauson EM, Guerra ML, Barylko B, Albanesi JP, and Cobb MH. 2013. Off-target effects of MEK inhibitors. Biochemistry 52: 5164–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu W, Shen S-M, Zhao X-Y, and Chen G-Q. 2012. Targeted genes and interacting proteins of hypoxia inducible factor-1. Int J Bichem Mol Biol 3: 165–178. [PMC free article] [PubMed] [Google Scholar]
- 53.Jose C, Bellance N, and Rossignol R. 2011. Choosing between glycolysis and oxidative phosphorylation: a tumor’s dilemma? Biochimica et Biophysica Acta (BBA)-Bioenergetics 1807: 552–561. [DOI] [PubMed] [Google Scholar]
- 54.Warburg O 1956. On the origin of cancer cells. Science 123: 309–314. [DOI] [PubMed] [Google Scholar]
- 55.Kelly B, and O’neill LA. 2015. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell research 25: 771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen C, Pore N, Behrooz A, Ismail-Beigi F, and Maity A. 2001. Regulation of glut1 mRNA by hypoxia-inducible factor-1 Interaction between H-ras and hypoxia. Journal of Biological Chemistry 276: 9519–9525. [DOI] [PubMed] [Google Scholar]
- 57.Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, Macintyre AN, Goraksha-Hicks P, Rathmell JC, and Makowski L. 2014. Metabolic reprogramming of macrophages: glucose transporter (GLUT1)-mediated glucose metabolism drives a pro-inflammatory phenotype. Journal of Biological Chemistry: jbc. M113. 522037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hotamisligil GS 2017. Inflammation, metaflammation and immunometabolic disorders. Nature 542: 177. [DOI] [PubMed] [Google Scholar]
- 59.Buller CL, Loberg RD, Fan M-H, Zhu Q, Park JL, Vesely E, Inoki K, Guan K-L, and Brosius FC III. 2008. A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. American Journal of Physiology-Cell Physiology 295: C836–C843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koh MY, Spivak-Kroizman TR, and Powis G. 2008. HIF-1 regulation: not so easy come, easy go. Trends in biochemical sciences 33: 526–534. [DOI] [PubMed] [Google Scholar]
- 61.Phillips RJ, Mestas J, Gharaee-Kermani M, Burdick MD, Sica A, Belperio JA, Keane MP, and Strieter RM. 2005. Epidermal growth factor and hypoxia-induced expression of CXC chemokine receptor 4 on non-small cell lung cancer cells is regulated by the phosphatidylinositol 3-kinase/PTEN/AKT/mammalian target of rapamycin signaling pathway and activation of hypoxia inducible factor-1α. Journal of Biological Chemistry 280: 22473–22481. [DOI] [PubMed] [Google Scholar]
- 62.Kallio PJ, Okamoto K, O’Brien S, Carrero P, Makino Y, Tanaka H, and Poellinger L. 1998. Signal transduction in hypoxic cells: inducible nuclear translocation and recruitment of theCBP/p300 coactivator by the hypoxia‐induciblefactor‐1α. The EMBO journal 17: 6573–6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Needham BD, and Trent MS. 2013. Fortifying the barrier: the impact of lipid A remodelling on bacterial pathogenesis. Nature reviews. Microbiology 11: 467–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Newton K, and Dixit VM. 2012. Signaling in innate immunity and inflammation. Cold Spring Harbor perspectives in biology 4: a006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sanghera JS, Weinstein SL, Aluwalia M, Girn J, and Pelech SL. 1996. Activation of multiple proline-directed kinases by bacterial lipopolysaccharide in murine macrophages. The Journal of Immunology 156: 4457–4465. [PubMed] [Google Scholar]
- 66.Büscher D, Hipskind RA, Krautwald S, Reimann T, and Baccarini M. 1995. Ras-dependent and-independent pathways target the mitogen-activated protein kinase network in macrophages. Molecular and Cellular Biology 15: 466–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schaeffer HJ, and Weber MJ. 1999. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Molecular and cellular biology 19: 2435–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Skarpen E, Flinder LI, Rosseland CM, Orstavik S, Wierod L, Oksvold MP, Skalhegg BS, and Huitfeldt HS. 2008. MEK1 and MEK2 regulate distinct functions by sorting ERK2 to different intracellular compartments. FASEB J 22: 466–476. [DOI] [PubMed] [Google Scholar]
- 69.Monick MM, Carter AB, Gudmundsson G, Mallampalli R, Powers LS, and Hunninghake GW. 1999. A phosphatidylcholine-specific phospholipase C regulates activation of p42/44 mitogen-activated protein kinases in lipopolysaccharide-stimulated human alveolar macrophages. The Journal of Immunology 162: 3005–3012. [PubMed] [Google Scholar]
- 70.Pouysségur J, Dayan F, and Mazure NM. 2006. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 441: 437–443. [DOI] [PubMed] [Google Scholar]
- 71.Roberts P, and Der CJ. 2007. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26: 3291–3310. [DOI] [PubMed] [Google Scholar]
- 72.Zassadowski F, Pokorna K, Ferre N, Guidez F, Llopis L, Chourbagi O, Chopin M, Poupon J, Fenaux P, Ann Padua R, Pla M, Chomienne C, and Cassinat B. 2015. Lithium chloride antileukemic activity in acute promyelocytic leukemia is GSK-3 and MEK/ERK dependent. Leukemia 29: 2277–2284. [DOI] [PubMed] [Google Scholar]
- 73.Rohde J, Heitman J, and Cardenas ME. 2001. The TOR kinases link nutrient sensing to cell growth. Journal of Biological Chemistry 276: 9583–9586. [DOI] [PubMed] [Google Scholar]
- 74.Wang C, Cigliano A, Delogu S, Armbruster J, Dombrowski F, Evert M, Chen X, and Calvisi D. 2013. Functional crosstalk between AKT/mTOR and Ras/MAPK pathways in hepatocarcinogenesis: implications for the treatment of human liver cancer. Cell cycle (Georgetown, Tex 12: 1999–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]