

HIV-1 maturation inhibitors are a class of small-molecule compounds that block a late step in the viral protease-mediated processing of the Gag polyprotein precursor, the viral protein responsible for the formation of virus particles. The first-in-class HIV-1 maturation inhibitor bevirimat was highly effective in blocking HIV-1 replication, but its activity was compromised by naturally occurring sequence polymorphisms within Gag. Recently developed bevirimat analogs, referred to as “second-generation” maturation inhibitors, overcome this issue. To understand more about how these second-generation compounds block HIV-1 maturation, here we selected for HIV-1 mutants that are resistant to these compounds. Selections were performed in the context of two different subtypes of HIV-1. We identified a small set of mutations at highly conserved positions within the capsid and spacer peptide 1 domains of Gag that confer resistance. Identification and analysis of these maturation inhibitor-resistant mutants provide insights into the mechanisms of resistance to these compounds.
KEYWORDS: antiretroviral, human immunodeficiency virus, retrovirus, virus assembly, virus maturation
ABSTRACT
A betulinic acid-based compound, bevirimat (BVM), inhibits HIV-1 maturation by blocking a late step in protease-mediated Gag processing: the cleavage of the capsid-spacer peptide 1 (CA-SP1) intermediate to mature CA. Previous studies showed that mutations conferring resistance to BVM cluster around the CA-SP1 cleavage site. Single amino acid polymorphisms in the SP1 region of Gag and the C terminus of CA reduced HIV-1 susceptibility to BVM, leading to the discontinuation of BVM’s clinical development. We recently reported a series of “second-generation” BVM analogs that display markedly improved potency and breadth of activity relative to the parent molecule. Here, we demonstrate that viral clones bearing BVM resistance mutations near the C terminus of CA are potently inhibited by second-generation BVM analogs. We performed de novo selection experiments to identify mutations that confer resistance to these novel compounds. Selection experiments with subtype B HIV-1 identified an Ala-to-Val mutation at SP1 residue 1 and a Pro-to-Ala mutation at CA residue 157 within the major homology region (MHR). In selection experiments with subtype C HIV-1, we identified mutations at CA residue 230 (CA-V230M) and SP1 residue 1 (SP1-A1V), residue 5 (SP1-S5N), and residue 10 (SP1-G10R). The positions at which resistance mutations arose are highly conserved across multiple subtypes of HIV-1. We demonstrate that the mutations confer modest to high-level maturation inhibitor resistance. In most cases, resistance was not associated with a detectable increase in the kinetics of CA-SP1 processing. These results identify mutations that confer resistance to second-generation maturation inhibitors and provide novel insights into the mechanism of resistance.
IMPORTANCE HIV-1 maturation inhibitors are a class of small-molecule compounds that block a late step in the viral protease-mediated processing of the Gag polyprotein precursor, the viral protein responsible for the formation of virus particles. The first-in-class HIV-1 maturation inhibitor bevirimat was highly effective in blocking HIV-1 replication, but its activity was compromised by naturally occurring sequence polymorphisms within Gag. Recently developed bevirimat analogs, referred to as “second-generation” maturation inhibitors, overcome this issue. To understand more about how these second-generation compounds block HIV-1 maturation, here we selected for HIV-1 mutants that are resistant to these compounds. Selections were performed in the context of two different subtypes of HIV-1. We identified a small set of mutations at highly conserved positions within the capsid and spacer peptide 1 domains of Gag that confer resistance. Identification and analysis of these maturation inhibitor-resistant mutants provide insights into the mechanisms of resistance to these compounds.
INTRODUCTION
More than two dozen drugs are currently approved for the treatment of HIV-1 infection (1). These drugs primarily target the viral enzymes reverse transcriptase (RT), protease (PR), and integrase (IN). Inhibitors targeting viral entry and fusion have also been developed (http://www.who.int/mediacentre/factsheets/fs360/en/). Although combination antiretroviral therapy (cART) can achieve sustained suppression of viral loads in infected individuals, toxicity can limit drug efficacy in some cases, and drug resistance may become an increasingly serious issue over time (2–6). Thus, novel inhibitors with targets that differ from those of current drugs are needed. Maturation inhibitors (MIs) represent a promising class of compounds that are currently at an early stage of clinical development.
The HIV-1 structural protein Gag is processed in a stepwise fashion by the viral PR during particle release from the infected cell (7–9). PR-mediated Gag processing generates matrix (MA), capsid (CA), spacer peptide 1 (SP1), nucleocapsid (NC), spacer peptide 2 (SP2), and p6. This PR-mediated Gag processing triggers virus maturation, during which the noninfectious, immature virus particle is converted to an infectious particle containing a conical capsid core inside the virion. The first-in-class MI bevirimat (BVM) acts by blocking CA-SP1 cleavage, the final step in the Gag processing cascade, resulting in the formation of poorly infectious viral particles lacking condensed conical cores (10, 11). Unlike PR inhibitors that bind the enzyme and globally block its proteolytic activity, BVM is thought to associate with a pocket in the assembled Gag complex and specifically disrupt processing at the CA-SP1 junction (12–14). A second MI, PF-46396 (PF96), although structurally distinct from BVM, appears to act via a similar mechanism (15–17). The CA-SP1 boundary region of Gag, in which the putative MI binding site is located, plays a central role in virus assembly and in the regulation of particle maturation (7, 18). Understanding how MIs act is thus of great interest not only for the development of this promising class of antiretroviral compounds but also for understanding basic aspects of HIV-1 assembly and maturation.
We previously conducted extensive resistance selection analyses with both BVM (19) and PF96 (17). BVM resistance mutations clustered around the CA-SP1 cleavage site at CA residues 226 and 231 and SP1 residues 1 and 3 (10, 19). Mutations that conferred resistance to PF96 were identified not only around the CA-SP1 cleavage site but also far upstream at CA residue 201 (15, 17) and at CA residues 156, 157, and 160 in the major homology region (MHR) (17). The MHR mutants were defective for assembly and replication in the absence of PF96 but in its presence were assembly competent and replication proficient. This compound-dependent behavior suggested that PF96 could correct an assembly defect conferred by the MHR mutations. The replication defect imposed by the compound-dependent mutations could also be corrected by second-site mutations downstream in SP1, most notably SP1-T8I (17, 20). On its own, the SP1-T8I mutation phenocopied MI binding by interfering with CA-SP1 processing and stabilizing the immature Gag lattice (17, 20). We also observed that mutations at SP1 residue 3 conferred a BVM-dependent phenotype (19). Based on current structural models for the immature Gag lattice (21, 22), it appears that the MI-dependent mutations destabilize an assembly domain spanning the base of the C-terminal domain of CA (CA-CTD) and the N-terminal six residues of SP1. MIs and the SP1-T8I mutation reverse the assembly defect conferred by the compound-dependent MHR and SP1 residue 3 mutations by stabilizing the CA-SP1 assembly domain. These findings illustrate the value of resistance selection studies in understanding the structural basis for MI activity.
Although the amino acid residues at which resistance mutations arise are very highly conserved across major subtypes of HIV-1, the central and C-terminal portions of SP1 are less conserved. In particular, SP1 residues 6 to 8 (Gln-Val-Thr, the so-called “QVT” motif) are highly polymorphic in non-subtype B isolates (https://www.hiv.lanl.gov/content/sequence/HIV). Clinical trials demonstrated that BVM was well tolerated, and in individuals infected with viruses containing the clade B consensus QVT motif in SP1, significant reductions in viral loads were achieved during a 10-day treatment period (23–25). However, polymorphisms in the QVT motif, in particular SP1-V7A, reduced the susceptibility of HIV-1 to BVM (26–28). To overcome this reduced potency elicited by SP1 polymorphisms, second-generation MIs are being developed by our group and others (29–36). These BVM analogs display marked improvements in potency against both consensus clade B isolates (e.g., NL4-3) and primary clinical isolates from multiple HIV-1 subtypes. Although second-generation BVM analogs clearly display increased potency and broad activity relative to BVM, the pathway(s) to resistance against these compounds in de novo selection experiments has not been determined.
In this study, we analyze the ability of second-generation MIs to inhibit BVM-resistant isolates and perform in vitro selection experiments with both subtype B and C clones to identify novel mutations that confer resistance to second-generation MIs. We identify a mutation in the CA MHR and a small set of mutations near the CA-SP1 junction that confer either partial or high-level resistance to these inhibitors.
RESULTS
Second-generation MIs retain significant activity against BVM-resistant CA mutants.
We recently identified a set of BVM analogs that demonstrate increased potency against the subtype B laboratory isolate NL4-3 and are active against an NL4-3 derivative bearing the SP1-V7A substitution (e.g., compounds 7m and 7r) (Fig. 1) (36, 37). These second-generation compounds are also active against a multisubtype panel of HIV-1 isolates (36, 37). As a first step toward defining pathways of resistance to these highly potent BVM analogs, we examined their activity against a set of previously selected BVM-resistant mutants: CA-H226Y, CA-L231M, CA-L231F, SP1-A1V, SP1-A3V, and SP1-A3T (19). Cells transfected with wild-type (WT) and mutant molecular clones were treated or not treated with 100 nM of the compounds 7m and 7r (36) and metabolically labeled with [35S]Met-Cys, and the released HIV-1 particles were collected by ultracentrifugation and analyzed directly. As previously reported (19), BVM impaired the processing of WT CA-SP1 to mature CA (leading to an increase in virion-associated CA-SP1) but was inactive against this panel of BVM-resistant mutants (Fig. 2A). In contrast, two representative second-generation BVM analogs, denoted 7m and 7r (36), markedly impaired CA-SP1 processing of the BVM-resistant CA mutants CA-H226Y, CA-L231M, and CA-L231F (Fig. 2A). To examine the effects of compounds 7m and 7r in spreading infections in a human T-cell line, Jurkat cells were infected with WT or mutant molecular clones. The infected cells were then propagated in the presence or absence of 7m or 7r, and virus replication was monitored by reverse transcriptase (RT) activity in the infected cell cultures. The results indicated that these second-generation MIs were able to block or markedly delay the replication of the BVM-resistant CA mutants CA-H226Y, CA-L231M, and CA-L231F in the Jurkat T-cell line (Fig. 2B).
FIG 1.
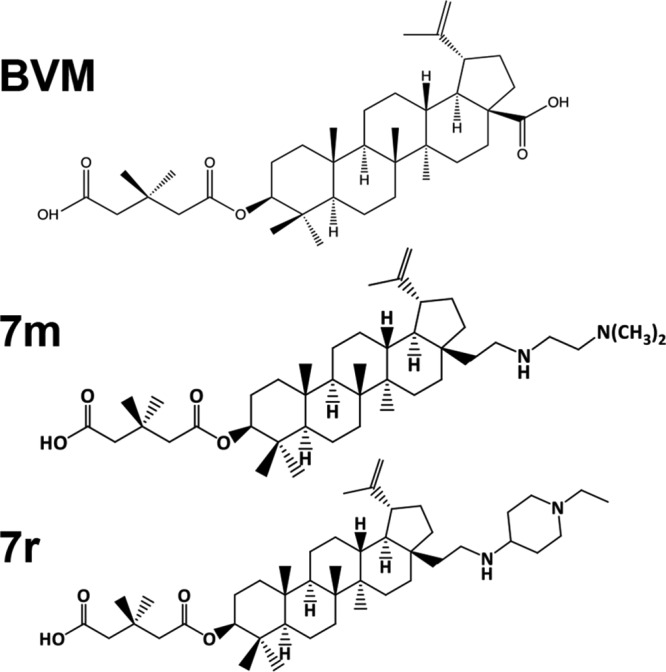
Structures of BVM and second-generation MIs 7m and 7r (36).
FIG 2.

Second-generation MIs can inhibit CA-SP1 processing of BVM-resistant C-terminal CA mutants. (A) CA-SP1 processing data for CA-H226Y, L231F, and L231M C-terminal CA mutants, with quantification of percent CA-SP1 accumulation indicated in the graph, using the radiolabeling-based CA-SP1 processing assay (see Materials and Methods) (n ≥ 3 independent assays; means ± standard deviations [SD]). (B) Replication kinetics of WT, CA-L231M, CA-H226Y, and CA-L231M in the absence (dimethyl sulfoxide [DMSO]) or presence of 7m or 7r. Virus stocks were generated in 293T cells, normalized for RT activity, and used to infect the Jurkat T-cell line. Cells were split every 2 days, and virus replication was monitored by RT activity.
In contrast to their activity against the BVM-resistant CA mutants CA-H226Y, CA-L231M, and CA-L231F, compounds 7m and 7r did not induce significant levels of CA-SP1 accumulation in the context of the BVM-resistant SP1 mutants SP1-A1V, SP1-A3V, and SP1-A3T (Fig. 3A). Likewise, the CA-V230I/SP1-V7A double mutant, which was selected as a BVM-resistant polymorph from mixed recombinant patient-derived HIV-1 isolates (38), was also resistant to the second-generation compounds (Fig. 3B). This result is consistent with our recent observation that the BVM analogs did not inhibit a subtype A isolate containing the CA-V230I/SP1-V7A double polymorph (36). The second-generation MIs were markedly more potent than BVM in their ability to block CA-SP1 processing of the SP1-V7A and CA-V230I single amino acid polymorphs (Fig. 3B).
FIG 3.

Quantification of CA-SP1 processing in the presence of 7m or 7r for SP1-A1V, A3T, and A3V (A) and SP1-V7A, CA-V230I, and CA-V230I/SP1-V7A (B). BVM and the DMSO vehicle were included as controls (n ≥ 3 independent assays; means ± SD).
Selection for resistance to second-generation HIV-1 MIs in subtype B HIV-1.
To define further the resistance pathways that enable HIV-1 to escape from second-generation MIs, we performed de novo selection experiments with compounds 7m and 7r. The Jurkat T-cell line was transfected with pNL4-3, and cells were cultured in the presence of a low concentration (2 nM) of the compound. Virus replication was monitored by RT activity. In untreated cultures, virus replication peaked at approximately 1 week posttransfection; in contrast, in cultures treated with 7m or 7r, no replication was observed for the first 3 weeks after transfection, but RT activity was detected thereafter (a representative selection experiment is shown in Fig. 4A). Virus was collected during peak replication and repassaged into fresh Jurkat cells in the presence of 2 or 10 nM compound 7m or 7r (data not shown). Sequencing of viral DNA from the peak of RT activity identified two mutations within the Gag-coding region: SP1-A1V (previously detected in our BVM selection experiments [10, 19]) and CA-P157A, located within the major homology region (MHR) of CA (Fig. 4B).
FIG 4.

Selection and analysis of mutants resistant to second-generation MIs in the subtype B context. (A) The Jurkat T-cell line was transfected with pNL4-3 and cultured in the presence or absence of 2 nM compound 7m or 7r. DMSO served as the vehicle control. Virus replication was monitored by an RT assay. (B) Location of the mutations identified in selection experiments: P157A in the CA major homology region (MHR) and SP1-A1V. The top shows the general organization of Pr55Gag; the sequences corresponding to the red boxes are indicated by arrows. The stippled arrow denotes the location of the CA-SP1 cleavage site. NTD, N-terminal domain. (C) Effect of the CA-P157A mutation on the ability of 7m and 7r to block CA-SP1 processing, using the radiolabeling-based CA-SP1 processing assay (see Materials and Methods). DMSO and BVM are included as controls. The 7m and 7r compounds caused a small but statistically significant (** denotes a P value of <0.02 by Student’s t test) increase in the accumulation of CA-P157A CA-SP1 relative to the DMSO control (n ≥ 3 independent experiments).
In Fig. 2, we present data indicating that SP1-A1V confers resistance to compounds 7m and 7r. To determine whether CA-P157A also confers resistance, we introduced this mutation into pNL4-3 and performed the CA-SP1 processing assay described above. The results indicated that treatment of virus-producing cells with 100 nM 7m or 7r resulted in a small but statistically significant increase in CA-SP1 levels in virions relative to levels observed in virions from untreated cells (Fig. 4C). These results demonstrate that the CA-P157A MHR mutation confers high-level, but not complete, resistance to the second-generation MIs 7m and 7r in the CA-SP1 processing assay.
To confirm these findings in the context of a spreading HIV-1 infection, we evaluated virus replication kinetics in the presence and absence of the inhibitor. The Jurkat T-cell line was transfected with WT pNL4-3 or derivatives containing the CA-P157A or SP1-A1V mutation. Transfected cells were cultured in several concentrations of the compounds (5, 10, 25, and 50 nM), and replication was monitored by RT activity. All concentrations of 7r significantly interfered with the replication of WT NL4-3; only at 5 nM 7r was any replication observed, and this occurred with a delay of approximately 1 month relative to that in the untreated culture (Fig. 5A). With SP1-A1V, little effect of the compounds was observed (Fig. 5B), consistent with this mutation conferring high-level MI resistance. The CA-P157A mutant showed an incompletely resistant phenotype, with the two highest concentrations of compound 7r (25 and 50 nM) causing a delay in virus replication of approximately 1 week but with the two lower concentrations (5 and 10 nM) not affecting virus replication kinetics (Fig. 5C). Similar results were obtained with compound 7m (data not shown). Together, the results demonstrate that SP1-A1V confers high-level resistance to the second-generation MIs tested here, whereas CA-P157A confers high-level, but not complete, resistance. The CA-P157A data highlight that (in the context of subtype B NL4-3) even small increases in CA-SP1 accumulation (Fig. 4C) are associated with significant delays in virus replication. These results also highlight the close correlation between increased CA-SP1 accumulation and delayed virus replication in the context of subtype B NL4-3.
FIG 5.

Resistance to second-generation MIs is conferred by SP1-A1V and CA-P157A mutations in spreading HIV-1 infections. The Jurkat T-cell line was transfected with WT pNL4-3 or the indicated mutant derivatives and cultured in the absence of MI or in the presence of the indicated concentrations of BVM or 7r. Virus replication was monitored by an RT assay.
To quantitatively measure the level of MI resistance conferred by the CA-P157A and SP1-A1V mutations, we performed 50% inhibitory concentration (IC50) analyses using single-cycle infectivity assays. 293T cells were transfected with WT or mutant molecular clones and treated with 22 concentrations of 7r from 0 to 8 μM (see Materials and Methods). Virus-containing supernatants were harvested, normalized for RT activity, and used to infect the TZM-bl indicator cell line (39). Luciferase activity was measured at 2 days postinfection. Infectivity data were analyzed with GraphPad Prism from four independent experiments (see Materials and Methods). Data from a representative experiment are shown in Fig. 6A. From these data, we also calculated the maximum percent inhibition (MPI) using the equation MPI = 1 − (signal from the average at the two highest drug concentrations)/(signal from the no-drug control) × 100 (40) (Fig. 6B). Against WT NL4-3, 7r demonstrated an IC50 of 9 nM and an MPI of 97%. In this analysis, the CA-P157A and SP1-A1V mutations demonstrated high-level resistance to 7r. A small amount of inhibition (MPI of ∼12%) was observed with CA-P157A, consistent with the results of the CA-SP1 processing and spreading replication assays.
FIG 6.

Quantitative assessment of MI resistance conferred by CA-P157A and SP1-A1V. 293T cell were transfected with WT or mutant molecular clones, and the transfected cells were treated with 22 concentrations of 7r from 0 to 8 μM (see Materials and Methods). Virus-containing supernatants were harvested, normalized for RT activity, and used to infect TZM-bl cells. Infectivity data were analyzed with GraphPad Prism 7 for Mac OS X from four independent experiments. Curves were fit using nonlinear regression as log(inhibitor) versus normalized response, with a variable slope using a least-squares (ordinary) fit. (A) Data from one representative experiment. (B) Maximum percent inhibition (MPI) calculated from the single-cycle (SC) assays described above for panel A, using the equation MPI = 1 − (signal from the average at the two highest drug concentrations)/(signal from the no-drug control) × 100. The SD is indicated (n = 4). NA, not applicable.
Selection for resistance to second-generation HIV-1 MIs in subtype C HIV-1.
BVM is ineffective against a subset of HIV-1 subtype B isolates with polymorphisms near the CA-SP1 cleavage site, including SP1-V7A within the “QVT” motif (SP1 residues 6 to 8) (26–28). These polymorphisms are more prevalent in subtype C isolates; as a result, many clade C viruses are naturally resistant to BVM (37). We recently reported that the BVM analog 7r showed a significantly lower IC50 against subtype C isolates than BVM (36, 37). To investigate pathways of resistance to second-generation BVM analogs in the context of subtype C HIV-1, we performed resistance selection experiments using the transmitted/founder subtype C isolate K3016 (41). This clone encodes QAN (rather than QVT) at SP1 residues 6 to 8 (Fig. 7A). K3016 replication was monitored in the HUTR5 T-cell line in the presence or absence of BVM analogs by a p24 enzyme-linked immunosorbent assay (ELISA). Virus replication was observed with a significant delay in compound-treated cultures relative to untreated controls (data not shown). Sequencing of the viral DNA isolated from infected cultures revealed the presence of several mutations, including CA-V230M, SP1-A1V, SP1-S5N, and SP1-G10R (Fig. 7A). SP1-S5N and G10R were detected together in the same culture, suggesting that they may have arisen as a double mutant.
FIG 7.

Selection and analysis of mutants resistant to second-generation MIs in the subtype C context. (A) Location of the mutations identified in selection experiments with the subtype C clone K3016: CA-V230M, SP1-A1V, S5N, and G10R. The top shows the general organization of Pr55Gag; the amino acid sequence at the CA-SP1 junction is indicated. The stippled arrow denotes the location of the CA-SP1 cleavage site. (B) Effect of the subtype C mutations on the ability of 7m and 7r to block CA-SP1 processing, using the radiolabeling-based CA-SP1 processing assay (see Materials and Methods). DMSO and BVM are included as controls. Statistical significance was calculated using Student’s t test. For SP1-S5N, G10R, and S5N/G10R, statistical significance for decreased CA-SP1 accumulation relative to the WT was calculated. ns, not significant (P > 0.05); *, P < 0.05; **, P < 0.02 (n ≥ 3 independent experiments).
To test the ability of the CA-V230M, SP1-A1V, SP1-S5N, and SP1-G10R mutations to confer resistance in the context of subtype C HIV-1, these substitutions were introduced individually into the K3016 clone; an SP1-S5N/G10R double mutant was also constructed. The effect of these mutations on CA-SP1 processing with and without second-generation MIs was evaluated in a radiolabeling assay (Fig. 7B). At concentrations of 100 nM, 7m and 7r had moderate and nearly equivalent effects (∼40% to 50% accumulation) on CA-SP1 processing of the SP1-S5N and SP1-G10R mutants. The SP1-S5N/G10R double mutant showed ∼35% to 40% CA-SP1 accumulation at 100 nM. At 500 nM concentrations, CA-SP1 accumulation of ∼40% to 60% was observed for these mutants. The SP1-A1V mutant showed full resistance, consistent with the effect of this mutation in the clade B context. Interestingly, the CA-V230M mutation showed high levels of CA-SP1 accumulation even in the absence of the inhibitor, and CA-SP1 levels were not significantly increased by MI treatment (Fig. 7B).
We also examined the effect of MI treatment on the replication of the subtype C-selected mutations in the context of a spreading infection. WT and mutant K3016 molecular clones were used to transfect the HUTR5 T-cell line, and virus replication was monitored by an RT assay (Fig. 8A to C). Replication of WT K3016 was completely blocked by 500 nM concentrations of compound 7m and 7r and was markedly delayed by 50, 100, or 200 nM of the compounds. Replication of SP1-S5N, G10R, and S5N/G10R was delayed by compounds 7m and 7r, consistent with the partially resistant phenotype observed in the CA-SP1 processing assay. In contrast, as observed in the clade B context, SP1-A1V conferred full resistance (Fig. 8A and B). The CA-V230M mutant displayed a significant delay (∼1 week) in replication relative to the WT (Fig. 8C), consistent with its inherent defect in CA-SP1 processing (Fig. 7B), and its replication was minimally affected by BVM, 7m, or 7r (Fig. 8C).
FIG 8.

Effect of MIs on the replication of subtype C mutants. (A) SP1-S5N, G10R, S5N/G10R, and A1V replication in the presence of BVM or 7m. (B) SP1-S5N, G10R, S5N/G10R, and A1V replication in the presence of BVM or 7r. (C) CA-V230M replication in the presence of BVM, 7m, or 7r. The HUTR5 T-cell line was transfected with the WT K3016 molecular clone or the indicated mutant derivatives. Virus replication was monitored by an RT assay. Replication curves for the WT without MIs and replication of the mutants with BVM are replotted in multiple panels for ease of comparison.
We next performed quantitative IC50 determinations for the subtype C resistant mutants with compound 7r, as we had done for the subtype B (NL4-3) clones (see above and Materials and Methods). The IC50 for the WT K3016 clone was approximately 250 nM, significantly higher than that observed with NL4-3. The SP1-S5N and SP1-G10R mutants displayed high but measurable IC50s, whereas SP1-A1V, SP1-S5N/G10R, and CA-V230M demonstrated high levels of resistance (Fig. 9A). Data from 3 to 6 independent experiments indicated that the MPIs were 88% for WT K3016, 62% for SP1-S5N, 69% for SP1-G10R, and 29% for CA-V230M but <5% for SP1-A1V and SP1-S5N/G10R.
FIG 9.

Quantitative assessment of MI resistance conferred by CA-V230M, SP1-A1V, S5N, G10R, and S5N/G10R. 293T cells were transfected with WT or mutant molecular clones, and the transfected cells were treated with 22 concentrations of compound 7r between 0 and 8 μM (see Materials and Methods). Virus-containing supernatants were harvested, normalized for RT activity, and used to infect TZM-bl cells. Infectivity data were analyzed with GraphPad Prism 7 for Mac OS X from 3 to 5 independent experiments, as described in the Fig. 6 legend and Materials and Methods. (A) Data from one representative experiment. (B) Maximum percent inhibition (MPI) calculated from the single-cycle (SC) assays as described in the Fig. 6 legend. The SD is indicated (n = 3 to 5).
To test whether resistance mutations obtained in the context of one subtype of HIV-1 would confer resistance when introduced into another subtype, the CA-P157A mutation that was selected during propagation of NL4-3 (subtype B) was introduced into K3016 (subtype C), and the SP1-S5N mutation that arose during K3016 propagation was introduced into pNL4-3. The SP1-G10R mutation could not be introduced into pNL4-3 because residue 10 of NL4-3 is a Pro and not a Gly. The effect of these mutations on the ability of compounds 7m and 7r to block CA-SP1 processing was evaluated (Fig. 10A and B). We observed that SP1-S5N displayed a modest but statistically significant reduction in CA-SP1 accumulation relative to WT NL4-3 in the presence of MIs (Fig. 10A). The P157A mutation conferred nearly complete resistance to 7m and 7r in the context of the K3016 (Fig. 10B), as shown above in the context of NL4-3 (Fig. 2C). These results demonstrate that the CA-P157A and SP1-S5N mutations confer similar levels of MI resistance in both the subtype B clone NL4-3 and the subtype C clone K3016.
FIG 10.

Effect of the SP1-S5N mutation in NL4-3 and the CA-P157A mutation in K3016 on inhibition by MIs. Cells were transfected with WT pNL4-3 or the SP1-S5N derivative (A) or WT K3016 or the CA-P157A derivative (B) and treated with 100 nM BVM, 7m, or 7r. CA-SP1 processing was quantified by using the radiolabeling-based CA-SP1 processing assay (see Materials and Methods). DMSO alone was included as a control. In panel A, statistical significance of decreased CA-SP1 accumulation relative to the WT controls was calculated using Student’s t test (*, P < 0.05; **, P < 0.02). In panel B, statistical significance of increased CA-SP1 accumulation relative to the DMSO control was calculated using Student’s t test. (*, P < 0.05; **, P < 0.02) (n ≥ 3 independent experiments).
Evaluation of the replicative fitness of MI-resistant mutants in primary T cells and their single-cycle infectivity.
To evaluate the ability of the MI-resistant mutants to replicate in primary human peripheral blood mononuclear cells (PBMCs), cells from two donors were infected with WT and mutant viruses, and replication was monitored by RT activity. The results of this analysis demonstrated that the mutants replicated with near-WT kinetics or showed modest replication defects relative to the WT (Fig. 11). The CA-P157A and CA-V230M mutants consistently showed lower RT peaks than the WT in the context of the K3016 clone. However, unlike the highly replication-deficient and compound-dependent CA MHR mutants selected during propagation in PF96 (including CA-P157S) (17), the CA-P157A mutant is replication competent and shows no indication of compound dependence (Fig. 5C and Fig. 11). To test the effect of the resistance mutations on specific particle infectivity, WT and mutant molecular clones were used to transfect 293T cells. Virus stocks were normalized for RT activity and used to infect the TZM-bl indicator line. The CA-P157A and SP1-A1V mutants showed single-round infectivities of approximately 40% relative to the WT. CA-V230M infectivity was reduced 3- to 4-fold. The other mutants displayed WT or near-WT infectivities (Fig. 12).
FIG 11.

Replicative fitness of the second-generation MI-resistant mutants in PBMCs. Virus stocks of WT K3016 and NL4-3 and the indicated mutant derivatives were prepared by transfection of 293T cells. RT-normalized virus stocks were used to infect PBMCs from two different donors. Virus replication was monitored by an RT assay.
FIG 12.

Specific infectivities of mutants resistant to second-generation MIs. 293T cells were transfected with the indicated WT and mutant molecular clones. Virus-containing supernatants were harvested, normalized for RT activity, and used to infect the TZB-bl indicator cell line. Luciferase activity was measured at 2 days postinfection. Infectivity of subtype B (NL4-3) clones is in blue, and that of subtype C (K3016) clones is in orange. The specific infectivities are presented relative to those of the WT (100%). Error bars indicate SDs (n = 4 independent assays performed in duplicate).
The CA-SP1 processing rate does not always correlate with MI resistance.
A possible model to explain MI resistance is that resistance mutations increase the kinetics of CA-SP1 processing, allowing the virus to overcome the effect of the compound on cleavage at this site (40, 42). We previously investigated the possibility that BVM resistance correlates with increased rates of CA-SP1 processing. While we (19) and others (42, 43) observed an increase in CA-SP1 processing kinetics for the SP1-A1V mutant, other BVM-resistant mutants displayed CA-SP1 processing kinetics indistinguishable from, or in some cases slower than, those of the WT (19). A recent study (40) used an in vitro CA-SP1 processing assay to confirm that the SP1-A1V mutation markedly increases the kinetics of CA-SP1 processing. This study also observed more-modest effects of other CA-SP1 region polymorphisms on CA-SP1 processing, leading to the suggestion that increased CA-SP1 cleavage kinetics were a major contributor to MI resistance. To investigate this issue using a subset of BVM-resistant Gag polymorphs and MI-resistant mutants, we measured the kinetics of CA-SP1 processing by using full-length HIV-1 molecular clones in a cell-based, pulse-chase radiolabeling assay. Cells transfected with WT pNL4-3 or the SP1-A1V, CA-P157A, SP1-V7A, and CA-V230I/SP1-V7A derivatives were metabolically radiolabeled for 20 min and then chased in cold medium for 30, 60, and 120 min. Cell lysates were radioimmunoprecipitated with HIV immunoglobulin (HIV-Ig). As previously reported (19), the SP1-A1V mutant displayed more-complete processing to mature CA (i.e., lower levels of CA-SP1) at each time point (Fig. 13). However, the other mutants tested displayed levels of CA-SP1 relative to CA that were indistinguishable from those of the WT. As indicated in Fig. 7B, CA-SP1 processing was highly impaired for the CA-V230M mutant. These data demonstrate that, for this set of mutants, MI resistance is not a consequence of significantly enhanced CA-SP1 processing kinetics.
FIG 13.

Effect of MI resistance mutations or CA-SP1 region polymorphisms on CA-SP1 processing kinetics. HeLa cells were transfected with WT pNL4-3 or the indicated mutants and cultured for 24 h. The cells were starved in Met-Cys-free medium for 30 min and then pulse-labeled with [35S]Met-Cys medium for 20 min. The cells were then washed, cultured, and lysed at 0, 30, 60, or 120 min. Lysates were immunoprecipitated with HIV-Ig (n = 3).
DISCUSSION
In this study, we investigated the development of HIV-1 resistance to second-generation MIs. Selection experiments were performed with both subtype B (NL4-3) and subtype C (K3016) clones. In the selections with NL4-3, we obtained CA-P157A and SP1-A1V mutations; in the selections with K3016, CA-V230M, SP1-A1V, SP1-S5N, and SP1-G10R changes arose. The SP1-A1V mutant was obtained in previous selections for BVM resistance (10, 19), but the other mutants reported here were not observed during selections with BVM. It is important to note that unlike the highly prevalent CA-SP1 region polymorphisms (e.g., SP1-V7A) that reduce the susceptibility of HIV-1 to BVM (26–28, 36), the amino acid positions at which second-generation MI resistance arose are highly conserved across multiple subtypes of HIV-1 (Table 1). For example, Pro-157 is conserved in 99.94% of the >30,000 sequences in the Los Alamos HIV sequence database (https://www.hiv.lanl.gov/content/sequence/HIV), whereas Ala is present in only 0.01% of these sequences. These results suggest that although these mutations might arise over time in the context of MI monotherapy, preexisting resistance is not likely to be a major concern. As with currently available antiretrovirals, if MIs are ultimately approved for clinical use, they would be administered in combination with other anti-HIV drugs.
TABLE 1.
Amino acid sequence conservation at maturation inhibitor-selected positionsa
| Gag amino acid | Amino acid (% conservation) |
|
|---|---|---|
| “WT” sequenceb | Resistant sequence | |
| CA residue 157 | Pro (99.94) | Ala (0.01) |
| CA residue 230 | Val (95.26) | Met (0.02) |
| SP1 residue 1 | Ala (99.63) | Val (0.017) |
| SP1 residue 5 | Ser (97.83) | Asn (0.56) |
More than 30,000 sequences analyzed across all subtypes.
Residue present in wild-type subtype B (NL4-3) and subtype C (K3016) molecular clones.
The selection experiments described here identified a series of mutants with wide-ranging phenotypes. The SP1-A1V mutant, identified previously in our BVM selections (10, 19), is highly MI resistant and, at least in culture, highly replication competent. The SP1-S5N and G10R mutants conferred low but statistically significant levels of resistance in terms of CA-SP1 processing, while in spreading infections and single-cycle infectivity assays, they demonstrated high levels of resistance to second-generation MI treatment. The CA-P157A mutant, located within the CA MHR, conferred high-level, but not complete, resistance in the context of both subtypes B and C. Unlike the highly PF96-dependent MHR mutant CA-P157S (17), CA-P157A replication was not enhanced by MI. Finally, the CA-V230M mutant displayed high levels of CA-SP1 accumulation even in the absence of MIs, and CA-SP1 levels were not significantly increased in the presence of MIs. As expected, based on its inherently high level of CA-SP1 accumulation, the CA-V230M mutant showed delayed replication in the HUTR5 T-cell line, somewhat lower RT peaks in PBMCs, and reduced single-cycle infectivity. While we cannot formally rule out the possibility that the cell line in which the selections were performed could have some impact on the mutations that arose, all of the IC50 analyses for both subtype B and C clones were performed using the same cell line (293T) as the virus producer cells, and results consistent with those obtained in the Jurkat and HUTR5 T-cell lines were obtained. These results indicate that the effects of the selected mutations on MI resistance are not specific to one or another T-cell line.
In the context of subtype B NL4-3, we observed a very close correlation between CA-SP1 accumulation and virus infectivity in both spreading infections and single-cycle assays. Indeed, in our screening efforts performed with hundreds of BVM analogs, we have consistently observed that CA-SP1 accumulation in our radiolabeling-based CA-SP1 processing assay is a strong predictor of antiviral potency (our unpublished results). The correlation between CA-SP1 processing and particle infectivity and replication appears to be less tight for the subtype C clone K3016. For example, we observe that 7m and 7r are able to significantly increase the levels of CA-SP1 in SP1-S5N, G10R, and S5N/G10R particles (Fig. 7B), yet these mutants display high-level resistance in both spreading infections (Fig. 8) and single-cycle assays (Fig. 9). Furthermore, the CA-V230M mutant displays high levels of virion CA-SP1 even in the absence of an inhibitor (Fig. 7) and yet is able to replicate, albeit with a delay relative to the WT, and shows only a 3- to 4-fold reduction in particle infectivity (Fig. 12). It will be of interest to determine whether the less-stringent correlation between CA-SP1 processing and replication/infectivity observed with K3016, relative to NL4-3, is a property of this particular clone or is a general feature of subtype C isolates.
It has long been suggested that the CA-SP1 boundary region adopts a helical conformation (44–47), and early cryo-electron tomography data led to the hypothesis that this region of Gag forms a six-helix bundle (48). Recent analyses performed by cryo-electron tomography and X-ray crystallography have confirmed the presence of a six-helix bundle at the CA-SP1 junction and have defined its structure to a resolution of ∼3 to 4 Å (21, 22). An intriguing feature of these recent structures is that the CA-SP1 processing site, which is cleaved slowly late in the Gag processing cascade, is buried within the six-helix bundle and is inaccessible to PR (21, 22). The buried nature of the CA-SP1 cleavage site leads to the suggestion that the six-helix bundle must undergo at least transient unfolding for PR to gain access to the cleavage site. MI binding may stabilize the six-helix bundle, thereby limiting PR access and blocking CA-SP1 processing. This is consistent with our previous data demonstrating that MIs stabilize the immature Gag lattice (49, 50). The SP1-T8I mutation, like MI binding, also blocks CA-SP1 processing and stabilizes the immature Gag lattice (17, 20). Recent data obtained by integrated nuclear magnetic resonance (NMR), cryo-electron microscopy (cryo-EM), and molecular dynamics simulations confirm that SP1-T8I stabilizes a helical conformation at the CA-SP1 junction (51). Wagner et al. (22) recently speculated that MIs may bind the immature Gag lattice by docking inside a central channel within the six-helix bundle, and density corresponding to BVM was observed at the center of this bundle (21, 52).
Multiple mechanisms appear to contribute to the ability of mutations and naturally occurring polymorphisms in CA and SP1 to confer MI resistance. (i) It has been suggested that increased rates of CA-SP1 processing can lead to MI resistance (40, 42). Because the putative inhibitor binding site spans the CA-SP1 junction in the assembled immature virus-like particle (VLP), and CA-SP1 processing prevents binding (10, 14, 21, 42), faster CA-SP1 processing would interfere with MI binding. This mechanism may contribute to MI resistance in the case of SP1-A1V, which exhibits a significantly enhanced rate of CA-SP1 processing (19, 40, 42, 43) (Fig. 13). However, other mutations/polymorphisms that induce MI resistance display only small increases, or no increases, in the kinetics of CA-SP1 processing (19, 40, 42, 43) (Fig. 13) or, in the case of CA-V230M, significantly impair CA-SP1 processing. (ii) Mutations in the CA-SP1 boundary region may directly prevent MI binding (53) without inducing dramatic effects on the rate of CA-SP1 processing (40). (iii) Resistance mutations may offset the ability of MI binding to stabilize the CA-SP1 helical bundle. The clearest examples of such mutations are those that confer MI dependence. For example, the SP1-A3V and A3T mutants are assembly defective in the absence of BVM but in its presence are assembly proficient and replication competent (19). Similarly, the G156E, P157S, and P160L mutations in the CA MHR and the G225D mutation near the C terminus of CA impose severe assembly defects that are rescued by the MI PF96 (17). These results are consistent with the hypothesis that the stability of the CA-SP1 region is fine-tuned to permit both assembly and CA-SP1 processing; mutations that destabilize this region, like the above-mentioned MHR mutations and SP1-A3V and A3V, impair assembly and are rescued by the MI with which they were selected. Conversely, substitutions that stabilize the CA-SP1 helical bundle (e.g., SP1-T8I) act, like MIs, to inhibit CA-SP1 processing (17, 20). Although the two chemical classes of MIs (BVM and its analogs and PF96) act in similar ways, they also exhibit interesting differences in their behaviors: unlike BVM, PF96 activity is not compromised by the SP1-V7A polymorphism, and PF96 but not BVM rescues the assembly defect imposed by the G156E, P157S, and P160L CA MHR mutations (17).
As mentioned above, recent structural studies have provided a 3- to 4-Å view of the six-helix bundle that spans the CA-SP1 boundary region (21, 22, 52). Figure 14 presents a structural model showing a cutaway of the six-helix bundle and the location of mutations selected in subtype B (Fig. 14A) and subtype C (Fig. 14B) clones. While these structures contribute significantly to our understanding of the basis for MI activity and the structural correlates of MI resistance, several key questions remain. In particular, it has been proposed (21, 22, 52) that the compounds dock inside the six-helix bundle, but at the current resolution, it is not clear how the compounds are oriented within the core of the bundle (with the extended C-28 side chain facing upwards into CA or downwards toward SP1). It is also not apparent what impact MI binding has on the structure of this region. These and other related questions will await higher-resolution structures for the six-helix bundle with second-generation MIs docked. Such structures will provide insights into why the second-generation MIs are more potent than the parental compound BVM and could be used to rationally design additional BVM analogs that are active against the MI-resistant mutants reported here. Ultimately, atomic-resolution structures of the six-helix bundle could be used as a launching point to develop novel MIs that are structurally distinct from either BVM or PF96.
FIG 14.

Crystal structural model for the CA-SP1 six-helix bundle and location of residues at which mutations arose during selection experiments with compounds 7m and 7r. Cutaway ribbon diagrams show the back three helices of the CA-SP1 six-helix bundle. The CA region is in gray, and SP1 residues 1 to 7 are in cyan. (A) Location of subtype B mutations CA-P157 and SP1-A1; (B) location of subtype C mutations CA-V230, SP1-A1, and SP1-S5. To-scale structures of compounds 7m and 7r are also included. The CA-SP1 structure is based on that reported under PDB accession number 5I4T (22).
MIs represent a promising class of anti-HIV compounds currently being pursued for clinical development. These inhibitors, and mutants resistant to them, have also provided valuable insights into the role of CA and SP1 in HIV-1 assembly and maturation. Higher-resolution structural information about the site of MI binding will further elucidate the molecular mechanism by which these inhibitors act and the function of the highly dynamic CA-SP1 boundary region in HIV replication.
MATERIALS AND METHODS
Cell culture and transfections.
The Jurkat and HUTR5 T-cell lines were maintained in RPMI 1640 medium with fetal bovine serum (FBS) (10%, vol/vol), glutamine (2 nM), penicillin (100 U/ml), and streptomycin (100 μg/ml). HeLa and 293T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with FBS (5% and 10%, vol/vol, respectively), glutamine (2 nM), penicillin (100 U/ml), and streptomycin (100 μg/ml). All cells were cultured at 37°C in a humidified incubator with 5% CO2. 293T and HeLa cells were transfected by using linear polyethylenimine (L-PEI) or Lipofectamine 2000 (Invitrogen) (54, 55). PBMCs (106) were infected for 2 h with RT-normalized virus stocks prepared by transfection of 293T cells using Lipofectamine 2000 (Invitrogen). Cells were washed and cultured in RPMI 1640 medium with 10% FBS in a 48-well plate. Supernatants from infected cells were collected every 2 to 3 days postinfection, and virus replication was monitored by RT activity.
Plasmids.
The subtype B molecular clone pNL4-3 (56) and the subtype C transmitted/founder viral clone CH185 (here denoted K3016 [41], a kind gift from Christina Ochsenbauer and John Kappes, University of Alabama) were used in this study. Point mutations were introduced into pNL4-3 and K3016 by site-direct mutagenesis using the QuikChange kit (Agilent Technologies).
CA-SP1 accumulation assay.
CA-SP1 accumulation assays were performed according to protocols previously outlined (17, 19, 36, 55), with minor changes. In brief, 293T or HeLa cells were transfected with WT pNL4-3 or K3016 molecular clones or their derivatives. MIs were added immediately following transfection. After 24 h, the transfected cells were starved in Met-Cys-free medium for 30 min, with MIs being added afterward to maintain levels during the labeling period. The cells were then metabolically labeled using [35S]Met-Cys express protein labeling mix (PerkinElmer) for 3 h. Culture supernatants were passed through 0.45-μm filters, and virus was pelleted by ultracentrifugation at 60,000 to 70,000 × g for 35 to 45 min. Virus pellets were then resuspended by boiling in a 1:1 mixture of Triton X-100 lysis buffer and 2× sample buffer. Proteins were separated by SDS-PAGE on 13.5% to 15% polyacrylamide gels. Gels were exposed to a phosphorimager plate (Fuji), scanned using a personal molecular imager (Bio-Rad), and quantified using Quantity One or Image Lab software (Bio-Rad).
Selection for MI-resistant mutants.
Selection experiments with NL4-3 were carried out using the Jurkat T-cell line according to previously reported methods (17), with minor modifications. Briefly, cells were transfected with the pNL4-3 WT and continually cultured in flasks with 2 nM compound 7m or 7r. Cells were split every 2 to 3 days, with viral replication being monitored by RT activity in the culture supernatants. At peak RT activity, virus-containing supernatants were harvested and used to reinfect fresh cells, which were cultured at 5-fold-higher concentrations of MI. At the new peak of RT activity, cells were collected, and genomic DNA was isolated by using a QIAamp DNA blood kit (Qiagen). The CA-SP1 region was then amplified using primers 1410F (5′-GGAAGCTGCAGAATGGGATA-3′) and 2897R (3′-AAAATATGCATCGCCCACAT-5′). PCR products were purified using ExoSAP-IT (Affymetrix) and sequenced using the primers 1410F (see above) and 1569R (3′-AAGAGGATGACCCTATCCACCTAATAC-5′). Selections with the subtype C clone K3016 were performed in the HUTR5 T-cell line. Briefly, HUTR5 cells were transfected with K3016 plasmid DNA and continually cultured in flasks with 100, 200, and 500 nM compound 7m or 7r. Cells were split every 3 days, and virus replication was monitored by quantifying p24 antigen using an HIV-1 p24 antigen capture kit (ABL). At peak HIV-1 p24 concentrations, cells were collected, and genomic DNA was isolated using a QIAamp DNA blood kit (Qiagen). The CA-SP1 region was then amplified using the K3016-752F (5′-CCGAATTTTATTTGACTAGCGGAG-3′) and K3016-2307R (5′-CTGGCCCCCTACTTTTATTGTG-3′) primers. PCR products were purified using ExoSAP-IT (Affymetrix) and sequenced using the primers K3016-752F (5′-CCGAATTTTATTTGACTAGCGGAG-3′), K3016-1306F (5′-ATCAGAAGGAGCCACTCCAC-3′), K3016-1795F (5′-ACCAGGGGCTACATTAGAAG-3′), K3016-1319R (5′-TGGCTCCTTCTGATAATGCTG-3′), K3016-1807R (5′-TGTAGCCCCTGGTCCTAATC-3′), and K3016-2307R (5′-CTGGCCCCCTACTTTTATTGTG-3′).
Measurement of antiviral IC50s.
To quantitatively measure the level of MI resistance conferred by selected mutations, we performed IC50 analyses using single-cycle infectivity assays. 293T cells were transfected with WT or mutant molecular clones, and cells were treated with 1, 2, 3, 4, 5, 6, 7, and 8 μM compound 7r and 1:2 serial dilutions starting from 1 μM for a total of 22 concentrations. Virus-containing supernatants were harvested, normalized for RT activity, and used to infect TZM-bl cells (39). Luciferase activity was measured at 2 days postinfection. Infectivity data were analyzed with GraphPad Prism 7 for Mac OS X. Curves were fit using nonlinear regression as log(inhibitor) versus normalized response, with a variable slope using a least-squares (ordinary) fit. From these data, we also calculated the MPI (40) using the equation MPI = 1 − (signal from the average at the two highest drug concentrations)/(signal from the no-drug control) × 100.
Pulse-chase analysis of CA-SP1 processing.
Pulse-chase analysis of CA-SP1 processing was performed as previously described (19). Briefly, HeLa cells were transfected with the pNL4-3 WT or mutants and cultured for 24 h. The cells were starved in Met-Cys-free medium for 30 min and then pulse-labeled with [35S]Met-Cys medium for 20 min at 37°C. The cells were washed, cultured at 37°C in DMEM with 10% FBS, and harvested at the 0-, 30-, 60-, and 120-min time points. The cells were lysed, and lysates were immunoprecipitated with HIV-Ig (obtained from the NIH AIDS Reagent Program). Proteins were separated by SDS-PAGE, and gels were exposed to a phosphorimager plate (Fuji), scanned using a personal molecular imager (Bio-Rad), and quantified using Quantity One or Image Lab software (Bio-Rad).
ACKNOWLEDGMENTS
We thank the members of the Freed laboratory for helpful discussions and critical reviews of the manuscript, Christina Ochsenbauer and John Kappes for the K3016 molecular clone, and Wei Shau for HIV-1 sequence analysis.
Work in the Freed laboratory is supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, NIH; the Intramural AIDS Targeted Antiviral Program; and funding from the NIH Intramural to India (I-I) Program. Work in the Gaur laboratory is supported by funding from the NIH Intramural to India (I-I) Program and the Department of Biotechnology, India.
REFERENCES
- 1.Kinch MS, Patridge E. 2014. An analysis of FDA-approved drugs for infectious disease: HIV/AIDS drugs. Drug Discov Today 19:1510–1513. doi: 10.1016/j.drudis.2014.05.012. [DOI] [PubMed] [Google Scholar]
- 2.Bertagnolio S, Perno CF, Vella S, Pillay D. 2013. The impact of HIV drug resistance on the selection of first- and second-line ART in resource-limited settings. J Infect Dis 207:S45–S48. doi: 10.1093/infdis/jit121. [DOI] [PubMed] [Google Scholar]
- 3.Guichet E, Aghokeng A, Serrano L, Bado G, Toure-Kane C, Eymard-Duvernay S, Villabona-Arenas CJ, Delaporte E, Ciaffi L, Peeters M. 2016. Short communication: high viral load and multidrug resistance due to late switch to second-line regimens could be a major obstacle to reach the 90-90-90 UNAIDS objectives in sub-Saharan Africa. AIDS Res Hum Retroviruses 32:1159–1162. doi: 10.1089/AID.2016.0010. [DOI] [PubMed] [Google Scholar]
- 4.Hosseinipour MC, Gupta RK, Van Zyl G, Eron JJ, Nachega JB. 2013. Emergence of HIV drug resistance during first- and second-line antiretroviral therapy in resource-limited settings. J Infect Dis 207:S49–S56. doi: 10.1093/infdis/jit107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iyidogan P, Anderson KS. 2014. Current perspectives on HIV-1 antiretroviral drug resistance. Viruses 6:4095–4139. doi: 10.3390/v6104095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Villabona-Arenas CJ, Vidal N, Guichet E, Serrano L, Delaporte E, Gascuel O, Peeters M. 2016. In-depth analysis of HIV-1 drug resistance mutations in HIV-infected individuals failing first-line regimens in West and Central Africa. AIDS 30:2577–2589. doi: 10.1097/QAD.0000000000001233. [DOI] [PubMed] [Google Scholar]
- 7.Freed EO. 2015. HIV-1 assembly, release and maturation. Nat Rev Microbiol 13:484–496. doi: 10.1038/nrmicro3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konvalinka J, Krausslich HG, Muller B. 2015. Retroviral proteases and their roles in virion maturation. Virology 479–480:403–417. doi: 10.1016/j.virol.2015.03.021. [DOI] [PubMed] [Google Scholar]
- 9.Lee SK, Potempa M, Swanstrom R. 2012. The choreography of HIV-1 proteolytic processing and virion assembly. J Biol Chem 287:40867–40874. doi: 10.1074/jbc.R112.399444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li F, Goila-Gaur R, Salzwedel K, Kilgore NR, Reddick M, Matallana C, Castillo A, Zoumplis D, Martin DE, Orenstein JM, Allaway GP, Freed EO, Wild CT. 2003. PA-457: a potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc Natl Acad Sci U S A 100:13555–13560. doi: 10.1073/pnas.2234683100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou J, Yuan X, Dismuke D, Forshey BM, Lundquist C, Lee KH, Aiken C, Chen CH. 2004. Small-molecule inhibition of human immunodeficiency virus type 1 replication by specific targeting of the final step of virion maturation. J Virol 78:922–929. doi: 10.1128/JVI.78.2.922-929.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adamson CS, Salzwedel K, Freed EO. 2009. Virus maturation as a new HIV-1 therapeutic target. Expert Opin Ther Targets 13:895–908. doi: 10.1517/14728220903039714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nguyen AT, Feasley CL, Jackson KW, Nitz TJ, Salzwedel K, Air GM, Sakalian M. 2011. The prototype HIV-1 maturation inhibitor, bevirimat, binds to the CA-SP1 cleavage site in immature Gag particles. Retrovirology 8:101. doi: 10.1186/1742-4690-8-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou J, Huang L, Hachey DL, Chen CH, Aiken C. 2005. Inhibition of HIV-1 maturation via drug association with the viral Gag protein in immature HIV-1 particles. J Biol Chem 280:42149–42155. doi: 10.1074/jbc.M508951200. [DOI] [PubMed] [Google Scholar]
- 15.Blair WS, Cao J, Fok-Seang J, Griffin P, Isaacson J, Jackson RL, Murray E, Patick AK, Peng Q, Perros M, Pickford C, Wu H, Butler SL. 2009. New small-molecule inhibitor class targeting human immunodeficiency virus type 1 virion maturation. Antimicrob Agents Chemother 53:5080–5087. doi: 10.1128/AAC.00759-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murgatroyd C, Pirrie L, Tran F, Smith TK, Westwood NJ, Adamson CS. 2016. Structure-activity relationships of the human immunodeficiency virus type 1 maturation inhibitor PF-46396. J Virol 90:8181–8197. doi: 10.1128/JVI.01075-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waki K, Durell SR, Soheilian F, Nagashima K, Butler SL, Freed EO. 2012. Structural and functional insights into the HIV-1 maturation inhibitor binding pocket. PLoS Pathog 8:e1002997. doi: 10.1371/journal.ppat.1002997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ganser-Pornillos BK, Yeager M, Pornillos O. 2012. Assembly and architecture of HIV. Adv Exp Med Biol 726:441–465. doi: 10.1007/978-1-4614-0980-9_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adamson CS, Ablan SD, Boeras I, Goila-Gaur R, Soheilian F, Nagashima K, Li F, Salzwedel K, Sakalian M, Wild CT, Freed EO. 2006. In vitro resistance to the human immunodeficiency virus type 1 maturation inhibitor PA-457 (bevirimat). J Virol 80:10957–10971. doi: 10.1128/JVI.01369-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fontana J, Keller PW, Urano E, Ablan SD, Steven AC, Freed EO. 2016. Identification of an HIV-1 mutation in spacer peptide 1 that stabilizes the immature CA-SP1 lattice. J Virol 90:972–978. doi: 10.1128/JVI.02204-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schur FK, Obr M, Hagen WJ, Wan W, Jakobi AJ, Kirkpatrick JM, Sachse C, Krausslich HG, Briggs JA. 2016. An atomic model of HIV-1 capsid-SP1 reveals structures regulating assembly and maturation. Science 353:506–508. doi: 10.1126/science.aaf9620. [DOI] [PubMed] [Google Scholar]
- 22.Wagner JM, Zadrozny KK, Chrustowicz J, Purdy MD, Yeager M, Ganser-Pornillos BK, Pornillos O. 2016. Crystal structure of an HIV assembly and maturation switch. Elife 5:e17063. doi: 10.7554/eLife.17063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin DE, Salzwedel K, Allaway GP. 2008. Bevirimat: a novel maturation inhibitor for the treatment of HIV-1 infection. Antivir Chem Chemother 19:107–113. doi: 10.1177/095632020801900301. [DOI] [PubMed] [Google Scholar]
- 24.Salzwedel K, Martin DE, Sakalian M. 2007. Maturation inhibitors: a new therapeutic class targets the virus structure. AIDS Rev 9:162–172. [PubMed] [Google Scholar]
- 25.Smith PF, Ogundele A, Forrest A, Wilton J, Salzwedel K, Doto J, Allaway GP, Martin DE. 2007. Phase I and II study of the safety, virologic effect, and pharmacokinetics/pharmacodynamics of single-dose 3-O-(3′,3′-dimethylsuccinyl)betulinic acid (bevirimat) against human immunodeficiency virus infection. Antimicrob Agents Chemother 51:3574–3581. doi: 10.1128/AAC.00152-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adamson CS, Sakalian M, Salzwedel K, Freed EO. 2010. Polymorphisms in Gag spacer peptide 1 confer varying levels of resistance to the HIV-1 maturation inhibitor bevirimat. Retrovirology 7:36. doi: 10.1186/1742-4690-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seclén E, González MDM, Corral A, de Mendoza C, Soriano V, Poveda E. 2010. High prevalence of natural polymorphisms in Gag (CA-SP1) associated with reduced response to bevirimat, an HIV-1 maturation inhibitor. AIDS 24:467–469. doi: 10.1097/QAD.0b013e328335ce07. [DOI] [PubMed] [Google Scholar]
- 28.Van Baelen K, Salzwedel K, Rondelez E, Van Eygen V, De Vos S, Verheyen A, Steegen K, Verlinden Y, Allaway GP, Stuyver LJ. 2009. HIV-1 susceptibility to the maturation inhibitor bevirimat is modulated by baseline polymorphisms in Gag SP1. Antimicrob Agents Chemother 53:2185–2188. doi: 10.1128/AAC.01650-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Z, Swidorski JJ, Nowicka-Sans B, Terry B, Protack T, Lin Z, Samanta H, Zhang S, Li Z, Parker DD, Rahematpura S, Jenkins S, Beno BR, Krystal M, Meanwell NA, Dicker IB, Regueiro-Ren A. 2016. C-3 benzoic acid derivatives of C-3 deoxybetulinic acid and deoxybetulin as HIV-1 maturation inhibitors. Bioorg Med Chem 24:1757–1770. doi: 10.1016/j.bmc.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 30.Nowicka-Sans B, Protack T, Lin Z, Li Z, Zhang S, Sun Y, Samanta H, Terry B, Liu Z, Chen Y, Sin N, Sit SY, Swidorski JJ, Chen J, Venables BL, Healy M, Meanwell NA, Cockett M, Hanumegowda U, Regueiro-Ren A, Krystal M, Dicker IB. 2016. Identification and characterization of BMS-955176, a second-generation HIV-1 maturation inhibitor with improved potency, antiviral spectrum, and Gag polymorphic coverage. Antimicrob Agents Chemother 60:3956–3969. doi: 10.1128/AAC.02560-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qian K, Bori ID, Chen CH, Huang L, Lee KH. 2012. Anti-AIDS agents 90. Novel C-28 modified bevirimat analogues as potent HIV maturation inhibitors. J Med Chem 55:8128–8136. doi: 10.1021/jm301040s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qian K, Kuo RY, Chen CH, Huang L, Morris-Natschke SL, Lee KH. 2010. Anti-AIDS agents 81. Design, synthesis, and structure-activity relationship study of betulinic acid and moronic acid derivatives as potent HIV maturation inhibitors. J Med Chem 53:3133–3141. doi: 10.1021/jm901782m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Regueiro-Ren A, Liu Z, Chen Y, Sin N, Sit SY, Swidorski JJ, Chen J, Venables BL, Zhu J, Nowicka-Sans B, Protack T, Lin Z, Terry B, Samanta H, Zhang S, Li Z, Beno BR, Huang XS, Rahematpura S, Parker DD, Haskell R, Jenkins S, Santone KS, Cockett MI, Krystal M, Meanwell NA, Hanumegowda U, Dicker IB. 2016. Discovery of BMS-955176, a second generation HIV-1 maturation inhibitor with broad spectrum antiviral activity. ACS Med Chem Lett 7:568–572. doi: 10.1021/acsmedchemlett.6b00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Swidorski JJ, Liu Z, Sit SY, Chen J, Chen Y, Sin N, Venables BL, Parker DD, Nowicka-Sans B, Terry BJ, Protack T, Rahematpura S, Hanumegowda U, Jenkins S, Krystal M, Dicker IB, Meanwell NA, Regueiro-Ren A. 2016. Inhibitors of HIV-1 maturation: development of structure-activity relationship for C-28 amides based on C-3 benzoic acid-modified triterpenoids. Bioorg Med Chem Lett 26:1925–1930. doi: 10.1016/j.bmcl.2016.03.019. [DOI] [PubMed] [Google Scholar]
- 35.Tang J, Jones SA, Jeffery JL, Miranda SR, Galardi CM, Irlbeck DM, Brown KW, McDanal CB, Han N, Gao D, Wu Y, Shen B, Liu C, Xi C, Yang H, Li R, Yu Y, Sun Y, Jin Z, Wang E, Johns BA. 2014. Synthesis and biological evaluation of macrocyclized betulin derivatives as a novel class of anti-HIV-1 maturation inhibitors. Open Med Chem J 8:23–27. doi: 10.2174/1874104501408010023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Urano E, Ablan SD, Mandt R, Pauly GT, Sigano DM, Schneider JP, Martin DE, Nitz TJ, Wild CT, Freed EO. 2016. Alkyl amine bevirimat derivatives are potent and broadly active HIV-1 maturation inhibitors. Antimicrob Agents Chemother 60:190–197. doi: 10.1128/AAC.02121-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Timilsina U, Ghimire D, Timalsina B, Nitz TJ, Wild CT, Freed EO, Gaur R. 2016. Identification of potent maturation inhibitors against HIV-1 clade C. Sci Rep 6:27403. doi: 10.1038/srep27403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Knapp DJ, Harrigan PR, Poon AF, Brumme ZL, Brockman M, Cheung PK. 2011. In vitro selection of clinically relevant bevirimat resistance mutations revealed by “deep” sequencing of serially passaged, quasispecies-containing recombinant HIV-1. J Clin Microbiol 49:201–208. doi: 10.1128/JCM.01868-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother 46:1896–1905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin Z, Cantone J, Lu H, Nowicka-Sans B, Protack T, Yuan T, Yang H, Liu Z, Drexler D, Regueiro-Ren A, Meanwell NA, Cockett M, Krystal M, Lataillade M, Dicker IB. 2016. Mechanistic studies and modeling reveal the origin of differential inhibition of Gag polymorphic viruses by HIV-1 maturation inhibitors. PLoS Pathog 12:e1005990. doi: 10.1371/journal.ppat.1005990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parrish NF, Gao F, Li H, Giorgi EE, Barbian HJ, Parrish EH, Zajic L, Iyer SS, Decker JM, Kumar A, Hora B, Berg A, Cai F, Hopper J, Denny TN, Ding H, Ochsenbauer C, Kappes JC, Galimidi RP, West AP Jr, Bjorkman PJ, Wilen CB, Doms RW, O’Brien M, Bhardwaj N, Borrow P, Haynes BF, Muldoon M, Theiler JP, Korber B, Shaw GM, Hahn BH. 2013. Phenotypic properties of transmitted founder HIV-1. Proc Natl Acad Sci U S A 110:6626–6633. doi: 10.1073/pnas.1304288110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sakalian M, McMurtrey CP, Deeg FJ, Maloy CW, Li F, Wild CT, Salzwedel K. 2006. 3-O-(3′,3′-Dimethysuccinyl) betulinic acid inhibits maturation of the human immunodeficiency virus type 1 Gag precursor assembled in vitro. J Virol 80:5716–5722. doi: 10.1128/JVI.02743-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fun A, van Maarseveen NM, Pokorna J, Maas RE, Schipper PJ, Konvalinka J, Nijhuis M. 2011. HIV-1 protease inhibitor mutations affect the development of HIV-1 resistance to the maturation inhibitor bevirimat. Retrovirology 8:70. doi: 10.1186/1742-4690-8-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Accola MA, Hoglund S, Gottlinger HG. 1998. A putative alpha-helical structure which overlaps the capsid-p2 boundary in the human immunodeficiency virus type 1 Gag precursor is crucial for viral particle assembly. J Virol 72:2072–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Datta SA, Temeselew LG, Crist RM, Soheilian F, Kamata A, Mirro J, Harvin D, Nagashima K, Cachau RE, Rein A. 2011. On the role of the SP1 domain in HIV-1 particle assembly: a molecular switch? J Virol 85:4111–4121. doi: 10.1128/JVI.00006-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liang C, Hu J, Russell RS, Roldan A, Kleiman L, Wainberg MA. 2002. Characterization of a putative alpha-helix across the capsid-SP1 boundary that is critical for the multimerization of human immunodeficiency virus type 1 gag. J Virol 76:11729–11737. doi: 10.1128/JVI.76.22.11729-11737.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morellet N, Druillennec S, Lenoir C, Bouaziz S, Roques BP. 2005. Helical structure determined by NMR of the HIV-1 (345-392)Gag sequence, surrounding p2: implications for particle assembly and RNA packaging. Protein Sci 14:375–386. doi: 10.1110/ps.041087605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wright ER, Schooler JB, Ding HJ, Kieffer C, Fillmore C, Sundquist WI, Jensen GJ. 2007. Electron cryotomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. EMBO J 26:2218–2226. doi: 10.1038/sj.emboj.7601664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keller PW, Adamson CS, Heymann JB, Freed EO, Steven AC. 2011. HIV-1 maturation inhibitor bevirimat stabilizes the immature Gag lattice. J Virol 85:1420–1428. doi: 10.1128/JVI.01926-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keller PW, Huang RK, England MR, Waki K, Cheng N, Heymann JB, Craven RC, Freed EO, Steven AC. 2013. A two-pronged structural analysis of retroviral maturation indicates that core formation proceeds by a disassembly-reassembly pathway rather than a displacive transition. J Virol 87:13655–13664. doi: 10.1128/JVI.01408-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang M, Quinn CM, Perilla JR, Zhang H, Shirra R Jr, Hou G, Byeon IJ, Suiter CL, Ablan S, Urano E, Nitz TJ, Aiken C, Freed EO, Zhang P, Schulten K, Gronenborn AM, Polenova T. 2017. Quenching protein dynamics interferes with HIV capsid maturation. Nat Commun 8:1779. doi: 10.1038/s41467-017-01856-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Purdy JG, Shi D, Chrustowicz J, Hattne J, Gonen T, Yeager M. 2018. MicroED structures of HIV-1 Gag CTD-SP1 reveal binding interactions with the maturation inhibitor bevirimat. Proc Natl Acad Sci U S A 115:13258–13263. doi: 10.1073/pnas.1806806115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou J, Chen CH, Aiken C. 2006. Human immunodeficiency virus type 1 resistance to the small molecule maturation inhibitor 3-O-(3′,3′-dimethylsuccinyl)-betulinic acid is conferred by a variety of single amino acid substitutions at the CA-SP1 cleavage site in Gag. J Virol 80:12095–12101. doi: 10.1128/JVI.01626-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brissault B, Kichler A, Guis C, Leborgne C, Danos O, Cheradame H. 2003. Synthesis of linear polyethylenimine derivatives for DNA transfection. Bioconjug Chem 14:581–587. doi: 10.1021/bc0200529. [DOI] [PubMed] [Google Scholar]
- 55.Waheed AA, Ono A, Freed EO. 2009. Methods for the study of HIV-1 assembly, p 163–184. In Prasad VR, Kalpana GV (ed), HIV protocols, 2nd ed Humana Press, New York, NY. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59:284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]