

While combination antiretroviral therapies (cART) have transformed HIV infection into a chronic manageable condition, they do not act upon the latent HIV reservoir and are therefore not curative. As HIV cure or remission should be more readily achievable in individuals with smaller HIV reservoirs, achieving a deeper understanding of the clinical, immunological, and virological determinants of reservoir size is critical to eradication efforts. We performed a post hoc analysis of 30 participants of a clinical trial of early cART who had previously been assessed in detail for their clinical, immunological, and reservoir size characteristics. We observed that the HIV subtype and autologous Nef-mediated HLA downregulation function correlated with the viral reservoir size measured approximately 1 year post-cART initiation. Our findings highlight virological characteristics—both genetic and functional—as possible novel determinants of HIV reservoir establishment and persistence.
KEYWORDS: CD4 downregulation, HIV reservoir, HIV-1, HLA downregulation, Nef, viral pathogenesis
ABSTRACT
The HIV accessory protein Nef modulates key immune evasion and pathogenic functions, and its encoding gene region exhibits high sequence diversity. Given the recent identification of early HIV-specific adaptive immune responses as novel correlates of HIV reservoir size, we hypothesized that viral factors that facilitate the evasion of such responses—namely, Nef genetic and functional diversity—might also influence reservoir establishment and/or persistence. We isolated baseline plasma HIV RNA-derived nef clones from 30 acute/early-infected individuals who participated in a clinical trial of early combination antiretroviral therapy (cART) (<6 months following infection) and assessed each Nef clone's ability to downregulate CD4 and human leukocyte antigen (HLA) class I in vitro. We then explored the relationships between baseline clinical, immunological, and virological characteristics and the HIV reservoir size measured 48 weeks following initiation of suppressive cART (where the reservoir size was quantified in terms of the proviral DNA loads as well as the levels of replication-competent HIV in CD4+ T cells). Maximal within-host Nef-mediated downregulation of HLA, but not CD4, correlated positively with post-cART proviral DNA levels (Spearman's R = 0.61, P = 0.0004) and replication-competent reservoir sizes (Spearman's R = 0.36, P = 0.056) in univariable analyses. Furthermore, the Nef-mediated HLA downregulation function was retained in final multivariable models adjusting for established clinical and immunological correlates of reservoir size. Finally, HIV subtype B-infected persons (n = 25) harbored significantly larger viral reservoirs than non-subtype B-infected persons (2 infected with subtype CRF01_AE and 3 infected with subtype G). Our results highlight a potentially important role of viral factors—in particular, HIV subtype and accessory protein function—in modulating viral reservoir establishment and persistence.
IMPORTANCE While combination antiretroviral therapies (cART) have transformed HIV infection into a chronic manageable condition, they do not act upon the latent HIV reservoir and are therefore not curative. As HIV cure or remission should be more readily achievable in individuals with smaller HIV reservoirs, achieving a deeper understanding of the clinical, immunological, and virological determinants of reservoir size is critical to eradication efforts. We performed a post hoc analysis of 30 participants of a clinical trial of early cART who had previously been assessed in detail for their clinical, immunological, and reservoir size characteristics. We observed that the HIV subtype and autologous Nef-mediated HLA downregulation function correlated with the viral reservoir size measured approximately 1 year post-cART initiation. Our findings highlight virological characteristics—both genetic and functional—as possible novel determinants of HIV reservoir establishment and persistence.
INTRODUCTION
The major barrier to achieving HIV cure or remission, where the latter is defined as a state where combination antiretroviral therapies (cART) could be discontinued without the risk of viremia recrudescence (1), is the long-term persistence of latently HIV-infected cells. Primarily comprising long-lived resting memory CD4+ T cells (2–4) that harbor integrated replication-competent HIV in a nearly transcriptionally quiescent state (5–7), these viral reservoirs can reactivate at any time to produce infectious virions (7–12). As individuals with smaller latent HIV reservoirs should be more amenable to cure, remission, and/or posttreatment virological control (13–15), the identification of clinical, immunological, and other determinants of reservoir size is a priority.
It is now well established that early cART initiation limits the latent HIV reservoir size (16–20) and that the set-point viral load and duration of viral suppression on cART represent additional positive and negative correlates of reservoir size, respectively (21). Immunological factors also play a role. The homeostatic proliferation (22) and clonal expansion (23–28) of latently HIV-infected CD4+ T cells directly influence the latent HIV reservoir size and dynamics, and evidence indicates that initial antiviral immune responses may also modulate reservoir establishment and persistence (29, 30). A recent longitudinal study of an acute infection cohort reported that the pretherapy levels of key cytokines correlated with HIV DNA levels after 96 weeks of cART (29). Moreover, a previous analysis of the cohort evaluated in the present study identified baseline HIV-specific granzyme B responses, contributed mainly by human leukocyte antigen class I (HLA-I)-restricted CD8+ T cells, to be significant negative correlates of the reservoir size at 48 weeks post-cART, as measured in terms of HIV proviral loads as well as the levels of replication-competent viral infectious units per million CD4+ T cells (IUPM) (30).
The observation that early HIV-specific adaptive immune responses correlate with reservoir size (30) prompts the hypothesis that viral factors that facilitate the evasion of such responses might also influence reservoir establishment and persistence. The HIV accessory protein Nef represents such a factor. Nef evades host adaptive immunity by downregulating cell surface HLA-A and -B (31) as well as CD4 (32, 33). The former function allows infected cells to evade HLA-restricted CD8+ T-cell responses (34), while the latter function allows infected cells to evade antibody-dependent cell-mediated cytotoxicity (ADCC) by reducing the capacity of cell surface Env to transit to its CD4-bound conformation, which is required for ADCC epitope exposure (35). As primary nef sequences differ in their ability to downregulate CD4 and, in particular, HLA (36–41), we hypothesized that individuals harboring nef sequences with a strong immune evasion function would display larger reservoirs due to Nef-mediated protection of infected cells from immune clearance. A role for Nef in maintaining the HIV reservoir is additionally supported by the recent observation that pharmacologic inhibition of Nef promotes CD8+ T-cell-mediated elimination of latently HIV-infected cells in vitro (42).
More broadly, given the vast genetic diversity of HIV globally (pandemic group M strains currently comprise 9 subtypes and 96 circulating recombinant forms [43]), modulatory effects of HIV genotype/phenotype variation on latent reservoir size are conceivable. In support of this, a recent study reported that HIV reservoir sizes among virally suppressed individuals in Uganda (where subtypes A and D predominate) were three times smaller than those among individuals in the United States (where subtype B predominates), where the differences were not clearly attributable to demographic or clinical characteristics (21). HIV subtype B nef sequences display, on average, the highest CD4 and HLA downregulation activities of all major group M subtypes (38), prompting the hypothesis that superior within-host Nef function may play a key role in establishing larger reservoirs in subtype B-infected persons.
Towards identifying novel virological correlates of HIV reservoir size, we performed HIV subtyping and assessed within-host Nef function among 30 individuals with acute/early (<6 months) infection who took part in a clinical trial comparing standard and intensive cART and who had previously been assessed in detail for their clinical, immunological, and reservoir size characteristics (20, 30). The original trial identified the timing of cART initiation, but not the initial treatment regimen, to be a significant correlate of the HIV reservoir size measured at 48 weeks post-cART (20). A subsequent analysis that pooled all participants regardless of the initial regimen identified HIV-specific CD8+ granzyme B responses directed against Tat/Rev, Env, Gag, and Vif, as well as proteome wide, to be additional negative correlates of reservoir size (30). With the present study, we extend these observations to identify the HIV subtype and Nef-mediated immune evasion function as additional novel correlates of reservoir size in early-treated individuals, supporting virological characteristics as critical modulators of the HIV reservoir.
RESULTS
Participant characteristics and Nef clonal isolation.
The participants included 30 initially cART-naive men (age range, 22 to 59 years) with acute/early (<6 months) HIV infection who took part in a clinical trial comparing standard and intensive cART (20). Blood was collected at baseline (cART initiation) as well as 48 weeks post-cART for virological, immunological, clinical, and reservoir size assessments, the last of which was measured in terms of proviral loads, as well as the levels of replication-competent infectious viral units per million CD4+ T cells (20). The original trial identified earlier cART initiation and baseline HIV-specific CD8+ granzyme B responses, but not the initial treatment regimen, to be significant correlates of a smaller HIV reservoir size at 48 weeks post-cART (20, 30). As such, for the present study we analyzed all participants together regardless of treatment regimen. At baseline, the pretreatment median plasma viral load (pVL) of the participants was 4.2 log10 copies HIV RNA/ml (interquartile range [IQR], 3.7 to 4.9 log10 copies HIV RNA/ml), the median CD4 count was 445 cells/mm3 (IQR, 358 to 690 cells/mm3), and 20 of 30 (67%) participants were estimated to be within 141 days of HIV seroconversion using a published multiassay algorithm (MAA+) (44).
For each participant, three nef sequences were isolated from baseline plasma HIV RNA, cloned into a green fluorescent protein (GFP) reporter plasmid using previously described methods (36, 38, 45), and confirmed to cluster with their respective bulk plasma HIV RNA nef sequence (Fig. 1). For 27 of 30 (90%) individuals, all three Nef clones were unique at the amino acid level, while for the remaining 3 individuals (participants 5286, 5305, and 5306), all isolated clones were identical at the amino acid level. Within-host nef diversity correlated significantly with the timing of cART initiation. Nef clones from participants who initiated cART very early (within 141 days of seroconversion; the MAA+ group) differed at a median of only 2 residues (IQR, 1 to 4 residues), while those who initiated cART slightly later, but still within 6 months of infection (the MAA− group), differed at a median of 6 residues (IQR, 3 to 8 residues) (Mann-Whitney test, P = 0.008). This is consistent with sexually acquired HIV infections being generally established by a single transmitted/founder virus (46, 47), followed by within-host diversification of this strain (48–52) until cART is initiated.
FIG 1.
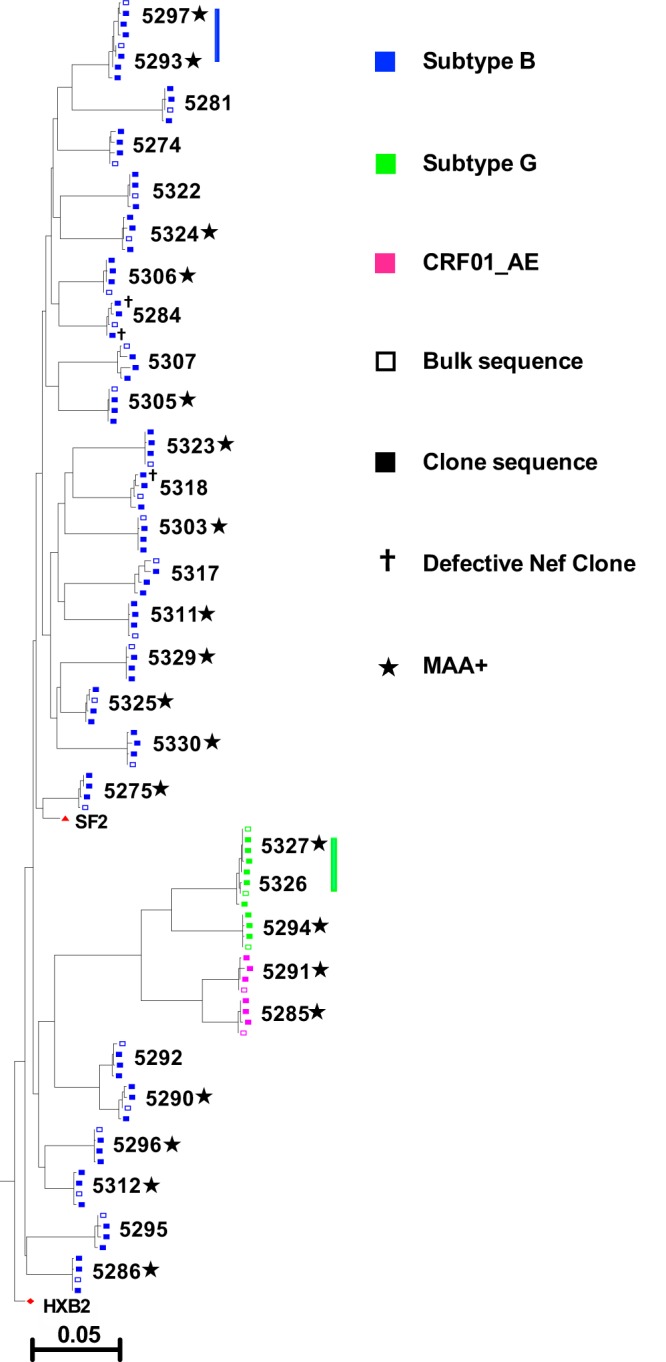
Maximum likelihood phylogeny relating participant bulk and clonal plasma HIV RNA Nef sequences. HIV nef isolates are colored by subtype, and bulk and clonal sequences are indicated. Defective Nef clones either encoded internal stop codons or lacked a stop codon. Participants who initiated cART very early (within an estimated 141 days of seroconversion), as determined by a multiassay algorithm (MAA+), are also indicated. Two participant pairs who shared highly genetically similar viruses, one pair infected with subtype B and the other pair infected with subtype G, are indicated by vertical lines. The phylogeny is rooted on the HIV subtype B reference strain HXB2. The scale is in the estimated number of nucleotide substitutions per site.
Of the 30 participants, 25 (83%) harbored HIV subtype B, while 5 (17%) harbored non-B subtypes (2 harbored subtype CRF01_AE and 3 harbored subtype G) (Fig. 1), a distribution that is consistent with recent HIV molecular epidemiological trends in Canada (53). Of note is the presence of two participant pairs, one each in the subtype B- and G-infected groups, who harbored highly similar viral strains, identifying them as putative transmission pairs (the HIV genetic similarity was confirmed via Gag sequencing; not shown). All but 3 of the 90 isolated Nef clones were genetically intact: 2 clones for participant 5284 harbored internal stop codons, while 1 clone for participant 5318 lacked the terminal stop codon (Fig. 1 and 2).
FIG 2.

CD4 and HLA-I downregulation functions of participant Nef clones. (A) Representative flow plots demonstrate surface CD4 and HLA-I expression following transfection of CEM cells with the empty vector (PSEL_GFP), positive-control Nef (PSEL_SF2), and example participant Nef clones. The median fluorescence intensity (MFI) of receptor staining in the GFP− (untransfected) and GFP+ (Nef-transfected) cells are displayed in the upper left and right corners of each gate, respectively. The normalized in vitro function of each Nef control and participant clone relative to that of NefSF2 is indicated as a percentage in the bottom right corner. (B) The normalized CD4 downregulation function of each participant Nef clone is shown and is expressed as the mean (box height) and standard deviation (error bars) from a minimum of triplicate independent experiments. Clones are colored by HIV subtype, as described in the key in Fig. 1. Defective Nef clones are indicated by a cross; these clones encoded either internal stop codons or lacked a stop codon. The dotted horizontal line indicates the population median. (C) The normalized HLA-I downregulation function of each participant Nef clone is shown, with all labeling being as described for panel B.
Functional assessment of participant-derived Nef clones.
The ability of each Nef clone to downregulate cell surface CD4 and HLA class I was assayed using an in vitro flow cytometry assay, as described previously (38, 39, 45, 54). The function of each clone was normalized to that of the HIV subtype B NefSF2 reference strain, which displays strong CD4 and HLA downregulation activities (36, 38, 39), such that a normalized value of 100% indicates a downregulation capacity equivalent to that of NefSF2, whereas values of <100% and >100% indicate downregulation capacities inferior or superior to those of NefSF2, respectively (for more details, see Materials and Methods). Representative raw data for negative-control (empty vector), positive-control (NefSF2), and participant-derived nef sequences, along with their corresponding NefSF2-normalized functions, are shown in Fig. 2A. Independent replicate measurements of the same Nef clone were highly consistent (Fig. 2B and C): median interreplicate (i.e., within-clone) standard deviations (SD) were 3.3% (IQR, 2.0% to 5.2%) for CD4 and 5.6% (IQR, 3.9% to 8.2%) for HLA downregulation. As expected, the within-host Nef function was highly consistent for the 3 participants for whom identical Nef clones were independently isolated but was more variable for the other 27 participants for whom all Nef clones were unique. Specifically, the median SD of within-host (i.e., between-clone) CD4 downregulation activities was 0.8% for the three individuals for whom identical Nef clones were independently isolated versus 3.3% for those for whom all isolated Nef groups were unique (Mann-Whitney test, P = 0.03), while the corresponding values for HLA downregulation were 3.6% versus 12.7%, respectively (P = 0.06). Moreover, among the Nef clones that displayed the poorest HLA downregulation function were those isolated from the subtype G putative transmission pair (participants 5326 and 5327), identifying their shared strain as partially attenuated for this function. Together, these observations demonstrate that our Nef functional assays are both sensitive and reliable. Nef clone amino acid sequences and their respective CD4 and HLA downregulation functions are provided in Table S1 in the supplemental material.
Overall, the range of Nef-mediated CD4 downregulation (Fig. 2B) was relatively narrow both within and between hosts. The median CD4 downregulation function of all participant-derived Nef clones was 96% (IQR, 89% to 99%); in fact, only 12 Nef clones (including the 3 with major genetic defects) exhibited normalized CD4 downregulation functions below 80% compared to those of NefSF2. Within-host variation in the Nef-mediated CD4 downregulation function was similarly limited (median standard deviation, 2.5% [IQR, 1.5% to 16.6%]); in fact, this distribution was not significantly different from that of the interreplicate standard deviations reported above (Mann-Whitney test, P = 0.8). Overall, these results are consistent with the CD4 downregulation function being highly conserved among primary Nef isolates (38, 39, 45, 54). In contrast, Nef-mediated HLA downregulation varied more widely both within and between hosts. The median HLA downregulation function of all participant-derived Nef clones was 72% (IQR, 48% to 89%), while the median standard deviation of within-host Nef clones was 9.5% (IQR, 3.5% to 25.4%) (Fig. 2C), where the latter distribution was significantly higher than the median interreplicate standard deviations reported above (Mann-Whitney test, P = 0.02).
Within-host Nef clone function, particularly for HLA downregulation, varied widely (e.g., whereas participant 5281's Nef clones ranged from 97.3% to 99.4% function, participant 5311's Nef clones ranged from 7% to 81% function, despite being genetically intact, where the latter participant's minority low-functioning clone differed solely by a rare P72L substitution; Table S1). Given that we cannot rule out the possibility of PCR or cloning errors as the cause of the minority of genetically and/or functionally defective clones, we elected to represent each participant's Nef CD4 and HLA-I downregulation function as the maximal value observed within each host. Sensitivity analyses were also performed using the median for all within-host clones tested. For 16 of 30 (53%) participants, the maximum within-host CD4 and HLA downregulation values were derived from different Nef clones, whereas for the remainder of the participants, the same clone displayed the greatest function for both activities. Overall, the maximal within-host CD4 and HLA downregulation function correlated positively (Spearman’s R = 0.56, P = 0.0013) (Fig. 3A). Steady-state Nef protein expression was also confirmed by Western blotting for all clones (Fig. 3B), where the variability in band intensity was likely primarily attributable to differential primary antibody binding to genetically diverse Nef isolates, since all clones displayed functional activities.
FIG 3.

Relationship between Nef-mediated CD4 and HLA-I downregulation functions and Nef steady-state protein expression. (A) Spearman's correlation between the maximal within-host Nef-mediated HLA-I and CD4 downregulation function. Nef isolates are colored according to HIV subtype. (B) Results of Western blot analyses of Nef and cellular β-actin are shown for assay controls and 30 participant clones that exhibited maximal intraindividual HLA downregulation activity. Sample identifiers are colored according to HIV subtype. Mw, molecular weight.
Validating established and identifying novel univariable correlates of reservoir size.
We next moved to our main objective of identifying correlates of reservoir size at 48 weeks post-cART, at which point plasma viremia had been suppressed to undetectable levels for a median of 40 weeks (IQR, 32 to 44.5 weeks) in the study participants. The reservoir size was measured in peripheral blood CD4+ T cells in terms of the total proviral burden (quantified by PCR and expressed as the number of log10 HIV DNA copies per million CD4+ T cells) and by the levels of replication-competent virus (measured by quantitative HIV coculture and expressed as the number of infectious units per million CD4+ T cells [IUPM]). As these two measurements correlated robustly (Spearman's R = 0.6, P = 0.001) and reservoir size is more routinely estimated by proviral DNA quantification rather than by laborious virus culture, the former was selected as the primary reservoir size measurement in our analyses.
We first verified that our study was sufficiently powered to identify established correlates of HIV reservoir size by confirming these relationships in our data (Fig. 4). As previously reported (30, 55), a very strong correlate of log10 proviral DNA levels at 48 weeks post-cART in peripheral blood CD4+ T cells was the log10 proviral DNA levels at baseline (Spearman's R = 0.84, P < 0.0001; Fig. 4A). In addition, log10 proviral DNA levels at 48 weeks correlated positively with baseline log10 pVL (Spearman's R = 0.45, P = 0.014), negatively with baseline CD4 counts (Spearman's R = −0.54, P = 0.0019), and negatively with baseline total HIV-specific granzyme B responses (Spearman's R = −0.57, P = 0.001; Fig. 4B to D) (30, 56–58). Also consistent with the findings of previous studies of the present (20, 30) and other (17, 18, 59) cohorts, early cART was also significantly associated with a smaller reservoir (median, 2.27 [IQR, 2.14 to 2.50] versus 2.91 [IQR, 2.42 to 3.22] log10 copies HIV DNA/million cells in MAA+ versus MAA− participants, respectively; P = 0.0045; Fig. 4E). Carriage of HLA-B*27 and/or HLA-B*57 was not associated with the reservoir size (P = 0.7), though only three participants expressed one of these protective alleles.
FIG 4.

Established and novel correlates of HIV reservoir size. (A to G) The correlations between log10 proviral DNA levels measured at 48 weeks post-cART and either baseline proviral DNA levels (A), baseline log10 plasma viral load (B), baseline CD4+ T cell counts (C), baseline granzyme B responses (D), timing of cART (E), baseline maximal Nef-mediated CD4 downregulation (F), or baseline maximal Nef-mediated HLA-I downregulation (G) are displayed. (H) The correlation between Nef-mediated HLA-I downregulation and replication HIV reservoir size measured at 48 weeks post-cART is shown. (I) The correlation between Nef-mediated HLA-I downregulation and baseline HIV-specific granzyme B responses is shown. (J) Week 48 proviral DNA levels are shown, stratified by HIV subtype. (K) Week 48 replication-competent reservoir sizes, assessed as the number of infectious units per million CD4+ T cells (IUPM), are shown, stratified by HIV subtype. Statistical significance was assessed using Spearman’s correlation or Mann-Whitney U tests. The Nef isolates in all panels are colored according to HIV subtype.
No significant relationship was observed between maximal (or median) Nef-mediated CD4 downregulation and log10 proviral DNA levels on cART (Spearman's R = 0.27, P = 0.15; Fig. 4F and data not shown). However, a significant positive association was observed between the maximal within-host Nef-mediated HLA downregulation function and HIV reservoir size (Spearman's R = 0.61, P = 0.0004; Fig. 4G). This relationship persisted when median within-host Nef-mediated HLA downregulation was evaluated (Spearman's R = 0.4, P = 0.027; data not shown). This observation prompted us to perform an exploratory codon function analysis to identify Nef amino acids significantly associated with the HLA downregulation function in the 30 maximally functioning Nef clones. We identified 39 residues at 28 of Nef's codons that were associated with the HLA downregulation function at a P value of <0.05 and a q value of <0.2 (Table S2). These included amino acid variants at Nef codon 20, a position that is indispensable for the HLA downregulation function (60), and variants at codons 8, 11, 14, 105, 108, 153, and 192, which have been previously reported as being associated with the HLA downregulation function in independent studies of natural Nef sequence variation (38, 39). Of note, with the exception of K204X near Nef's C terminus, all identified residues lie outside the motifs directly involved in Nef's interaction with HLA-I and the mu subunit of the adaptor protein complex 1 (these motifs have been identified to be the acidic cluster 62-EEEE-65, the PXXP motif at codons 69 to 78, and approximately codons 202 to 206 at the C terminus [61]).
Maximal within-host Nef-mediated HLA downregulation also correlated positively with the replication-competent reservoir size on cART (Spearman's R = 0.36, P = 0.056; Fig. 4H). Furthermore, the Nef-mediated HLA downregulation function correlated inversely with total HIV-specific granzyme B responses (Spearman's R = −0.37, P = 0.04; Fig. 4I), suggesting that the ability of an individual's autologous Nef to subvert antiviral cellular immune responses via HLA class I downregulation may influence the development of such responses in vivo.
We next stratified our data set based on HIV subtype and observed that subtype B-infected participants harbored significantly larger reservoirs than those harboring non-subtype B infections (median, 2.45 [IQR, 2.27 to 2.89] versus 1.72 [IQR, 0.96 to 2.10] log10 copies HIV DNA/million CD4+ T cells, respectively; P = 0.0008; Fig. 4J). This relationship persisted even after we excluded one member of each of the two putative transmission pairs (P = 0.002; data not shown), indicating that it is not simply driven by the relatively attenuated Nef-mediated HLA downregulation function observed in the subtype G-infected pair. Subtype B-infected participants also harbored larger replication-competent reservoirs in the setting of cART (P = 0.032; Fig. 4K).
Stratification by HIV subtype and multivariable analyses.
Having identified seven univariable correlates of reservoir size on cART (baseline log10 HIV DNA levels, pVL, CD4+ T cell counts, total HIV-specific granzyme B responses, and Nef-mediated HLA downregulation function, plus the timing of cART initiation and viral subtype), we next wished to tease apart their individual contributions to this outcome variable. In doing so, it became apparent that the subtype B- and non-subtype B-infected participants differed significantly with respect to some key parameters (Table 1). Specifically, subtype B-infected participants had significantly higher proviral DNA levels at baseline and 48 weeks post-cART (all P < 0.05) and displayed significantly better Nef-mediated HLA downregulation than non-subtype B-infected participants (median, 88% [IQR, 73 to 95%] versus 54% [IQR, 45 to 72%], P = 0.006), consistent with previous reports of the superior function of subtype B Nef isolates (38). Furthermore, a multivariable linear regression model, constructed using stepwise selection, that incorporated HIV subtype, baseline log10 pVL, the timing of cART, and Nef-mediated HLA downregulation identified HIV subtype to be the strongest independent correlate of reservoir size (after adjusting for other variables, subtype B infection was associated with an estimated 0.98 log10 higher proviral DNA level than non-subtype B infection; Table 2). The baseline log10 pVL and timing of cART explained an additional proportion of variation in reservoir size, and this was significant for the former variable.
TABLE 1.
Comparison of immunological and virological parameters between subtype B and non-subtype B infections
| Characteristic | Value(s) for participants infected with: |
P value | |
|---|---|---|---|
| Subtype B (n = 25) | Non-subtype B (n = 5) | ||
| Baseline | |||
| Median (IQR) plasma viral load (no. of log10 copies/ml) | 4.2 (3.7–4.8) | 4.8 (3.6–5.0) | 0.8 |
| Median (IQR) CD4+ T-cell count (no. of cells/mm3) | 440 (360–685) | 630 (345–750) | 0.7 |
| Median (IQR) proviral DNA load (no. of log10 copies/106 CD4+ T cells) | 3.4 (3.1–3.9) | 2.8 (2.1–3.2) | 0.01 |
| Median (IQR) no. of replication-competent HIV copies (no. of log10 IUPMa /106 CD4+ T cells) | 1.2 (0.9–2.0) | 1.2 (0.2–1.9) | 0.7 |
| % MAA+b for very early cARTc | 80 | 20 | 0.6 |
| Median (IQR) granzyme B response (no. of spot-forming cells/106 PBMCd ) | 2,710 (946–3,930) | 3,863 (2,380–8,860) | 0.095 |
| Median (IQR) maximal normalized Nef-mediated HLA downregulation (% function) | 88 (73–95) | 54 (45–72) | 0.006 |
| Maximal normalized Nef-mediated CD4 downregulation (% function) | 99 (96–101) | 97 (90–99) | 0.1 |
| Post-cART (wk 48) | |||
| Median (IQR) proviral DNA load (no. of log10 copies/106 CD4+ T cells) | 2.5 (2.3–2.9) | 1.7 (1.0–2.1) | 0.0008 |
| Median (IQR) no. of replication-competent HIV copies (log10 IUPM/106 CD4+ T cells) | −0.3 (−0.6 to 0.2) | −0.8 (−1.1 to −0.5) | 0.03 |
IUPM, infectious units per million CD4+ T cells.
MAA+, multiassay algorithm positive (participants who started cART within an estimated 141 days after infection).
cART, combination antiretroviral therapy.
PBMC, peripheral blood mononuclear cells.
TABLE 2.
Contribution of Nef-mediated HLA downregulation, baseline plasma viral load, cART timing, and HIV subtype on log10 proviral DNA levels at 48 weeks post-cART: final multivariable model
| Variable | Final model |
|
|---|---|---|
| Estimatea | P value | |
| Nef-mediated HLA downregulation | NDd | NDd |
| Very early cARTb (MAA+c) | −0.32 | 0.066 |
| Baseline log10 plasma viral load | 0.30 | 0.011 |
| HIV subtype B infection | 0.98 | 0.00001 |
β estimates are expressed as follows: Nef-mediated HLA downregulation (per percent function increment) and baseline plasma viral load (per log10 increment). For the timing of cART initiation, participants who started cART later than 141 days postinfection (MAA−) are the reference group. For HIV subtype, non-subtype B-infected participants were the reference group. The adjusted R2 value was 0.65, and P was 1.1 × 10−6.
cART, combination antiretroviral therapy.
MAA+, multiassay algorithm positive (participants who started cART within 141 days after infection).
NA, not applicable; variable was not selected in the final multivariable model.
Given the strong relationship between HIV subtype and reservoir size in our cohort (Table 1), we next confirmed that our previously identified correlates of reservoir size remained significant when the analysis was restricted to subtype B infections (n = 25). They did in all cases (Fig. 5A to F). From the strongest to the weakest, the correlates of proviral DNA levels in subtype B infections at 48 weeks post-cART were baseline proviral DNA levels (Spearman's R = 0.81, P < 0.0001), baseline CD4 count (R = −0.71, P < 0.0001), baseline pVL (R = 0.65, P = 0.0005), time of cART initiation (median, 2.4 versus 3.0 log10 copies in earlier- versus later-treated persons; P = 0.003), baseline HIV-specific granzyme B responses (R = −0.53, P = 0.0068), and maximal within-host Nef-mediated HLA downregulation function (R = 0.44, P = 0.026). In order to further tease apart the relationship between Nef function and reservoir size on cART, the remainder of our analyses therefore focused exclusively on the HIV subtype B infections.
FIG 5.

Confirmation of identified correlates in HIV subtype B infections only. (A to F) Correlations between log10 proviral DNA levels measured at 48 weeks post-cART and either baseline proviral DNA levels (A), baseline CD4+ T cell counts (B), baseline log10 plasma viral load levels (C), timing of cART (D), baseline granzyme B responses (E), or maximal baseline Nef-mediated HLA downregulation function (F) are shown, where all analyses are restricted to subtype B infections only. (G) A heat map summarizing Spearman's rho values (reported in boxes) and P values (depicted as colors) for the 15 possible pairwise comparisons between correlates of reservoir size in subtype B infections only is shown. NA, not applicable.
Determining the contributions of individual variables to a given outcome is challenging when these variables are highly intercorrelated, as in the present study. In the subtype B-infected subset, 10 of the 15 possible pairwise relationships between univariable correlates of reservoir size were statistically significant (Fig. 5G). Baseline proviral DNA levels, baseline pVL, and baseline HIV-specific granzyme B responses correlated most strongly with one another: Spearman's R = 0.86 (proviral DNA level/pVL), −0.71 (pVL/granzyme B response), and −0.62 (proviral DNA level/granzyme B response), respectively (all P < 0.001). Nef-mediated HLA downregulation, however, was the least interconnected variable. The only other factor that it significantly correlated with was baseline proviral DNA levels (R = 0.4, P = 0.045), consistent with a role of Nef in modulating infected cell levels in untreated HIV infection.
To identify which factors represented independent correlates of reservoir size in subtype B infection and, in particular, whether Nef-mediated HLA downregulation was among them, we constructed multivariable linear regression models using stepwise selection. To minimize the risk of overfitting, we followed the minimum criterion of 5 degrees of freedom per included variable (62, 63) (i.e., models restricted to the 25 participants with subtype B infections feature 20 degrees of freedom and could thus contain a maximum of 4 variables), and we ensured that the models would incorporate only one each of the three most highly intercorrelated variables (Fig. 5G). Given our specific interest in Nef, we also excluded the baseline proviral DNA level from the models due to its significant relationship with Nef-mediated HLA downregulation.
This left us with two models: the first (model 1) incorporated Nef-mediated HLA-I downregulation, baseline log10 pVL, baseline CD4 count, and early cART (Table 3), and the second (model 2) incorporated Nef-mediated HLA-I downregulation, baseline granzyme B response, baseline CD4 count, and early cART (Table 4). Notably, both final models retained Nef-mediated HLA downregulation as a variable and were highly statistically significant overall (adjusted R2 = 0.54 and P = 4.4 × 10−4 for model 1; adjusted R2 = 0.58 and P = 8.7 × 10−5 for model 2). Specifically, the final model 1 retained all four input variables, with none being individually significant, indicating that all four variables contribute to reservoir size to a comparable extent and together explain 54% of the variance in reservoir size (Table 3). Specifically, after adjustment for other parameters, model 1 estimated that each 10% increase in Nef-mediated HLA-I downregulation led to a 0.06 log10 increase in reservoir size on cART. The final model 2 retained three of the four input variables, Nef function, the granzyme B response, and early cART, where the last two remained significant, and these three variables together explained 58% of the variance in reservoir size. Consistent with the first model, model 2 estimated that each 10% increase in Nef-mediated downregulation function led to a 0.065 log10 increase in reservoir size after adjustment for other parameters. Taken together, these observations support Nef function as a novel contributor to reservoir size in multivariable models that explain >50% of the variance in reservoir size.
TABLE 3.
Model 1: contribution of Nef-mediated HLA downregulation, baseline plasma viral load, baseline CD4 T-cell count, and cART timing on log10 proviral DNA levels at 48 weeks post-cART
| Variable | Final model |
|
|---|---|---|
| Estimatea | P value | |
| Nef-mediated HLA-I downregulation | 0.006 | 0.11 |
| Baseline CD4+ T cell count | −0.0006 | 0.14 |
| Very early cARTb (MAA+c ) | −0.25 | 0.13 |
| Baseline log10 plasma viral load | 0.19 | 0.10 |
β estimates are expressed as follows: Nef-mediated HLA downregulation (per percent function increment), baseline plasma viral load (per log10 increment), and baseline CD4+ T cell count (per cell per cubic millimeter increment). For the timing of cART initiation, participants who started cART later than 141 days postinfection (MAA−) were the reference group. The adjusted R2 value was 0.54, and P was 4.4 × 10−4.
cART, combination antiretroviral therapy.
MAA+, multiassay algorithm positive (participants who started cART within 141 days after infection).
TABLE 4.
Model 2: contribution of Nef-mediated HLA downregulation, baseline CD4+ T-cell count, cART timing, and HIV-specific granzyme B responses on log10 proviral DNA levels at 48 weeks post-cART
| Variable | Final model |
|
|---|---|---|
| Estimatea | P value | |
| Nef-mediated HLA-I downregulation | 0.0065 | 0.06 |
| Baseline CD4+ T cell count | NAd | NAd |
| Very early cARTb (MAA+c) | −0.42 | 0.003 |
| Granzyme B response | −0.0001 | 0.009 |
β Estimates are expressed as follows: Nef-mediated HLA downregulation (per percent function increment), baseline CD4+ T cell count (per cell per cubic millimeter increment), and granzyme B production per spot-forming cell (per 106 PBMC increment). For the timing of cART initiation, participants who started cART later than 141 days postinfection (MAA−) were the reference group. The adjusted r2 value was 0.58, and P was 8.7 × 10−5.
cART, combination antiretroviral therapy.
MAA+, multiassay algorithm positive (participants who started cART within 141 days after infection).
NA, not applicable; variable was not selected in the final multivariable model.
DISCUSSION
Our results identify HIV subtype and Nef-mediated HLA downregulation function to be univariable and multivariable correlates of the viral reservoir size measured approximately 1 year following initiation of suppressive cART in persons who initiated treatment within 6 months of infection. These observations highlight virological characteristics—both genetic and functional—to be potentially novel determinants of HIV reservoir establishment and persistence. The identification of HIV subtype as a statistically significant multivariable correlate of reservoir size is particularly notable, given the modest number of non-subtype B infections in the cohort (5 of 30). Taken together with recent reports of smaller reservoir sizes among individuals infected with non-B subtypes (21), the relationship between infecting HIV subtype and reservoir size merits further study in larger cohorts. It will be particularly important to establish that viral subtype is not simply associated with a nonviral feature that influences HIV reservoir size (e.g., a host genetic factor that differs in frequency between populations in which the HIV subtype distribution also differs). Furthermore, it will be important to quantify what portion of the effect of HIV subtype on viral reservoir size is attributable to subtype-dependent differences in Nef-mediated HLA downregulation function (38), which were strongly apparent in the present cohort (Fig. 4J), versus what portions are attributable to other genetic and/or phenotypic viral features.
Similarly, our identification of Nef-mediated HLA downregulation as a correlate of reservoir size, even after stratification by HIV subtype and adjustment for established immunological and clinical correlates, also merits further investigation. In particular, it will be important to establish whether Nef-mediated HLA downregulation influences HIV reservoir size on cART primarily through Nef's ability to modulate pre-cART log10 proviral DNA levels (e.g., by allowing infected cells to evade cytotoxic T lymphocyte [CTL]-mediated clearance during active viremia, thereby increasing the absolute number of infected cells that subsequently enter and persist in a latent state) or whether the Nef-mediated HLA downregulation function additionally influences reservoir dynamics during suppressive cART (e.g., by shielding latently infected cells spontaneously reactivating from latency from detection by HLA-restricted HIV-specific CTL).
The results of our study suggest that Nef influences reservoir size, at least in part, by influencing HIV proviral DNA levels during untreated infection. Nef-mediated HLA downregulation correlated significantly with pre-cART proviral DNA levels (Fig. 5G); moreover, an exploratory multivariable model that incorporated proviral DNA levels, the baseline CD4 count, early cART, and Nef-mediated HLA downregulation (noting that the last three variables correlated significantly with the first one) identified baseline proviral DNA levels to be the single most significant predictor of reservoir size on cART (P = 1.2 × 10−6) (overall model-adjusted R2 = 0.74, P = 1.2 × 10−7), with early cART also surviving multivariable correction with P equal to 0.026. Though we did not directly investigate the mechanism in the present study, two lines of evidence support the notion that Nef influences pretreatment proviral DNA levels by allowing infected cells to evade HLA-mediated immune responses. First, Nef-mediated HLA downregulation correlates negatively with HIV-specific granzyme B responses (Fig. 4I). Furthermore, we have previously demonstrated in vitro that the extent of Nef-mediated HLA downregulation on target cells presenting the relevant HLA-bound peptide significantly inversely correlates with their ability to be recognized by effector T cells expressing a T-cell receptor specific to this peptide/HLA combination (45, 64). Whether Nef function additionally influences the subsequent persistence of latently infected cells during cART is less clear. In fact, our results do not directly support this (as evidenced by the above-mentioned observation that Nef function did not remain a significant correlate of reservoir size on cART after adjusting for baseline proviral DNA levels and the exploratory observation that Nef function did not correlate with the absolute level of decline of proviral DNA levels during cART; data not shown). Nevertheless, given the very strong relationship between baseline and post-cART proviral DNA levels (Fig. 4A) and the fact that reservoir size declines very slowly on cART (65–67), it would require larger cohorts with follow-up times substantially longer than those in the present study (48 weeks only) to adequately assess Nef's additional possible modulatory effects on reservoir decay dynamics during suppressive cART. The observation that pharmacologic inhibition of Nef promoted CD8+ T-cell-mediated elimination of latently HIV-infected cells in vitro (42) supports this notion, and given our study's modest size and follow-up time, we cannot conclusively rule this out. One caveat is that future studies, if undertaken using observational cohorts, would additionally need to control for the duration of cART suppression, a variable that we did not need to consider since ours was a post hoc analysis of a clinical trial where the cART duration was the same for all participants. Indeed, the latter point, as well as the fact that all participants were treated within 6 months of HIV infection, likely underlies our ability to identify correlates of reservoir size with a relatively modest number of participants.
In conclusion, the present study identifies Nef-mediated HLA downregulation function and, possibly, infecting HIV subtype, which are known regulators of HIV infectivity and pathogenesis (36, 39, 68–73), to be potential modulators of HIV reservoir size. Further research on the extent to which these and other virological characteristics—both genetic and functional—influence HIV reservoir establishment and persistence is merited in larger studies, particularly those featuring expanded HIV subtype distributions.
MATERIALS AND METHODS
Study participants.
Study participants, who were determined to have been infected with HIV for less than 6 months and who were initially naive to cART, were originally recruited from the Maple Leaf Clinic in Toronto, ON, Canada, as part of a treatment intensification clinical trial (ClinicalTrials.gov identifier NCT01154673). The original trial (20) recruited 32 males aged 22 to 59 years who self-identified as men who have sex with men; the present study included 30 of these participants for whom baseline plasma was available for analysis. All participants gave written informed consent, and the study was approved by the University of Toronto, St. Michael’s Hospital, and Simon Fraser University research ethics boards. As described previously (20), acute/early HIV infection was defined by one of the following criteria: (i) a positive HIV antibody test (Western blotting) with a documented negative test in the previous 6 months; (ii) a positive/weakly positive HIV enzyme-linked immunosorbent assay (ELISA) with indeterminate or evolving Western blotting result with demonstrated HIV antigenemia (p24) or viremia (HIV viral load ≥ 500 copies/ml); (iii) negativity for HIV antibodies in the setting of an illness compatible with acute seroconversion with demonstrated p24 antigenemia or plasma viremia; or (iv) a compatible clinical history of a recent seroconversion illness within the last 6 months with a documented high-risk exposure within 6 months and a negative HIV antibody test within the last year. Blood was collected at baseline, and participants were immediately initiated on cART; blood was additionally collected 48 weeks thereafter. The treatment regimen had no differential impact on HIV reservoir size (20); the present study therefore analyzed all participants regardless of regimen.
Determination of recent infection.
A published multiassay algorithm (MAA) which identifies infections that have occurred within a mean 141 days of seroconversion was used to refine HIV infection timing estimates (44). Briefly, baseline sera were tested with the BED capture enzyme immunoassay (BED-CEIA; Calypte Biomedical, Lake Oswego, OR), and the average normalized optical density (OD-n) was calculated. The antibody avidity of sera was measured using a modified Genetic Systems 1/2+O ELISA kit (Bio-Rad, Hercules, CA). Individuals were classified as having recent infection (MAA+) if their baseline sample exhibited a BED-CEIA normalized optical density of <1.0, an antibody avidity index of <80%, a positive viral load, and a CD4 count of >200 cells/μl; otherwise, individuals were classified as MAA−.
Reservoir size measurements.
The frequencies of CD4+ T cells carrying HIV proviral DNA and harboring replication-competent HIV were enumerated by real-time PCR and quantitative coculture assays, respectively, as previously described (20, 30). Briefly, for proviral DNA quantification, genomic DNA was isolated from 2 × 106 purified CD4+ T cells (Qiagen, Valencia, CA), and 1 μg of DNA was used as the template in real-time PCRs (7500 real-time PCR system; Applied Biosystems, Foster City, CA) utilizing both HIV-specific (long terminal repeat) and host-specific (RNase P) primers/probes in a 50-μl total reaction volume (TaqMan gene expression master mix; Applied Biosystems). All PCRs were performed in triplicate; the detection limit of the assay was 2.6 copies of HIV DNA. For replication-competent HIV quantification, highly enriched (>97% pure) CD4+ T cells were seeded to tissue culture plates, and irradiated peripheral blood mononuclear cells (PBMCs; 8 × 106) from HIV-negative donors were added to each well along with anti-CD3 antibody and incubated overnight in medium containing recombinant interleukin-2 (20 units/ml). CD8+-depleted and anti-CD3-stimulated PBMC blasts (1 × 106) from HIV-negative donors were added to each well on the following day and on day 7. The HIV p24 levels in the culture supernatants were quantified by ELISA between days 14 and 21, and the numbers of infectious units per million CD4+ T cells (IUPM) were determined as previously described (74).
Immunologic and immunogenetic assays.
Total HIV-specific granzyme B responses were determined by an enzyme-linked immunosorbent spot (ELISpot) assay at baseline as previously described (30). Briefly, cryopreserved peripheral blood mononuclear cells (PBMC) were plated at either 1 × 105 or 2 × 105 cells/well into 96-well plates that had been precoated with monoclonal granzyme B coating antibody (Mabtech, Cincinnati, OH) and stimulated with pools of 15-mer peptides with an 11-amino-acid overlap spanning the entire HIV proteome (NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH) for 48 h. Negative-control wells contained only cells in medium with 2, 4, or 12 μl of dimethyl sulfoxide; positive-control wells contained 1 μg/ml of staphylococcal enterotoxin B (Sigma, St. Louis, MO). After incubation, biotinylated antibody, streptavidin-alkaline phosphatase (both from Mabtech ELISpot assay kits), and alkaline phosphatase color development solution (Bio-Rad, Hercules, CA) were added sequentially, with thorough washes before and after each step. All samples were tested in duplicate. Image analysis was conducted using an ImmunoSpot series 3A analyzer (Cellular Technology, Ltd., Cleveland, OH). Human leukocyte antigen (HLA) class I typing was performed at allele-level resolution by PCR sequence-specific primer typing (AllSet+ Gold HLA ABC low-resolution kit; One Lambda) per the manufacturer's protocol. Ambiguous types for two participants were resolved by sequence-based typing as described elsewhere (75).
Nef isolation and cloning.
HIV-1 nef was amplified from plasma-derived HIV RNA in a one-step reverse transcription-PCR (RT-PCR) featuring a high-fidelity polymerase (Invitrogen Superscript III one-step RT-PCR Platinum Taq HiFi) using HIV-specific primers optimized to amplify all major HIV-1 subtypes: forward primer 5′-TAGCAGTAGCTGRGKGRACAGATAG-3′ (HXB2 nucleotides 8683 to 8707) and reverse primer 5′-TACAGGCAAAAAGCAGCTGCTTATATGYAG-3′ (HXB2 nucleotides 9536 to 9507). A nested PCR was then performed using a high-fidelity enzyme (Roche Expand HiFi) and primers that contained AscI (forward) and SacII (reverse) restriction enzyme sites for cloning: forward primer 5′-AGAGCACCGGCGCGCCTCCACATACCTASAAGAATMAGACARG-3′ (the AscI site is in boldface, HXB2 nucleotides 8746 to 8772 are italicized) and reverse primer 5′-GCCTCCGCGGATCGATCAGGCCACRCCTCCCTGGAAASKCCC-3′ (the SacII site is in boldface, HXB2 nucleotides 9474 to 9449 are italicized). Amplicons were then cloned into pSELECT (InvivoGen), an expression vector containing separate cytomegalovirus and composite human EF1/human T-cell leukemia virus promoters, allowing the simultaneous expression of GFP and nef, respectively. Briefly, a modified pSELECT-GFPzeo plasmid containing a linker bearing the AscI and SacII restriction sites was digested with the AscI and SacII enzymes and gel purified (GeneJet gel extraction kit; Thermo Scientific). Participant-derived nef amplicons were digested with AscI and SacII, ligated into cut pSELECT-GFPzeo (T4 ligase; Thermo Fisher), transformed into chemically competent Escherichia coli (E. cloni 10G DUOs; Lucigen), plated onto zeocin-containing Luria-Bertani (LB) agar plates, and incubated for 16 to 18 h. A minimum of three colonies per participant were subsequently propagated overnight in zeocin-containing LB broth, after which plasmid DNA was purified (Omega EZNA plasmid minikit; Thermo Fisher). The presence of an insert was verified by restriction enzyme digestion, followed by agarose gel electrophoresis.
Sequencing of bulk nef PCR products as well as clones containing correctly sized inserts was performed on an ABI 3130xl automated DNA analyzer using an ABI Prism BigDye Terminator (version 3.1) cycle sequencing kit (Applied Biosystems). Chromatograms were edited in Sequencher (version 5.0) software (GeneCodes). Nucleotide sequence alignments were performed using the HIVAlign tool (options, MAFFT program [76], codon alignment) hosted on the Los Alamos HIV sequence database (Los Alamos National Laboratory [LANL]) web server (77) and manually edited using the AliView program (78). Maximum likelihood phylogenies were constructed using the PhyML program (79) under a general time-reversible (GTR) model of codon substitution (80). HIV subtyping was performed using the recombinant identification program (RIP) hosted on the LANL web server (81).
Nef-mediated CD4 and HLA-I downregulation assays.
Three Nef clones per participant were assessed for their CD4 and HLA-I downregulation capacity using a published assay that involves transfection of nef plasmid DNA into a CEM-derived CD4+ T-cell line engineered to express HLA-A*02 (CEM-A*02) (38, 39, 45, 54). The use of HLA-A*02 as a representative HLA class I allele in this assay was previously validated by assessing the ability of a genetically diverse panel of 24 Nef clones to downregulate HLA-A*02 and HLA-B*07 in independent experiments, using CEM T-cell lines stably expressing each of these alleles individually, and observing that these two functions correlated robustly (Spearman’s R = 0.89 and P < 0.0001, as reported in the original published study [38]). The nef allele from HIV-1 subtype B reference strain SF2, cloned into pSELECT-GFPzeo (NefSF2), was used as the positive control, while empty pSELECT-GFPzeo served as a negative control. Briefly, 4 μg of participant-derived or control nef plasmid DNA was delivered into 500,000 CEM-A*02 cells by electroporation in 96-well plates (where positive and negative controls were included in each row), and the cells were incubated for 20 to 24 h. The cells were then stained with allophycocyanin-labeled anti-CD4 and phycoerythrin-labeled anti-HLA-A*02 antibodies (BD Biosciences), and the cell surface expression of these molecules was measured using flow cytometry. The receptor downregulation functions of participant-derived Nef clones were normalized to those of the positive control, NefSF2, using the following equation: {1 − [MFIparticipant (GFP+)/MFIparticipant (GFP−)]}/{1 − [MFISF2 (GFP+)/MFISF2 (GFP−)]}, where MFI (GFP+) and MFI (GFP−) refer to the median fluorescence intensity (MFI) of CD4 (or HLA-I) expression in the Nef-expressing (GFP positive [GFP+]) and Nef-nonexpressing (GFP negative [GFP−]) gates, respectively, and MFIparticipant and MFISF2 are the MFI for the participant Nef clone and NefSF2, respectively.
As such, normalized MFI values of 100% indicate a downregulation capacity equivalent to that of NefSF2, whereas values of <100% and >100% indicate downregulation capacities inferior or superior to those of NefSF2, respectively. All participant-derived Nef clones were assayed in a minimum of triplicate independent experiments, and the results are represented as the means of these measurements.
Western blotting.
Steady-state Nef protein levels were measured by Western blotting for selected clones exhibiting a maximal function for HLA downregulation. A total of 2.5 × 106 CEM cells were transfected with 10 μg of participant-derived or control plasmid DNA, and cell pellets were harvested 24 h later. Cells were lysed with Nonidet P-40 lysis buffer (1% Nonidet P-40, 50 mM Tris HCl, 150 mM NaCl) containing a protease inhibitor cocktail (catalog number P8340; Sigma). Following centrifugation, the resultant supernatants were subjected to SDS-PAGE, and proteins were electroblotted onto a polyvinylidene difluoride (PVDF) membrane. Nef was detected using sheep polyclonal anti-HIV-1 Nef serum (1:2,000 dilution; NIH AIDS Research and Reference Reagent Program, USA) primary antibody, followed by horseradish peroxidase (HRP)-conjugated donkey anti-sheep IgG (1:35,000; GE Healthcare). Blots were visualized using an ImageQuant LAS 4000 chemiluminescent imager (GE Healthcare).
Statistical analysis.
In univariable analyses, Spearman’s correlation was used to assess the relationship between continuous variables (e.g., Nef CD4 and HLA-I downregulation capacities, HIV clinical parameters, reservoir size measurements), while the Mann-Whitney U test was used for binary variables (e.g., the presence versus absence of protective HLA alleles, MAA+ versus MAA−, HIV subtype B versus not HIV subtype B). The Mann-Whitney U test was also used to identify Nef amino acids that were significantly associated with Nef-mediated HLA-I downregulation function, using the maximally functioning Nef clone per participant (see Table S2 in the supplemental material). This exploratory analysis was restricted to amino acids present in three or more Nef clones, and multiple comparisons were addressed using the q value, the P value analogue of the false discovery rate (FDR) (82). The FDR is the expected proportion of false-positive results among results deemed significant at a given P value threshold (e.g., at a q value of 0.2, we expect 20% of identified associations to be false positive). To identify correlates of reservoir size at 48 weeks post-cART, multivariable models were constructed by linear regression using a stepwise selection procedure. The variables investigated in the primary and sensitivity analyses were all those with P values of <0.05 in univariate analyses; these included Nef-mediated HLA downregulation function (per normalized percent functional increment), HIV-1 subtype (reference group, subtype B), baseline log10 plasma viral load (per log10 increment), baseline CD4 count (per cell per cubic millimeter increment), baseline proviral DNA levels (per log10 increment), total HIV-specific granzyme B responses (per spot-forming cell per 106 PBMC increment), and very early cART initiation (MAA+ versus MAA− [reference group]). Statistical tests were performed in Prism (version 5.0) software, and multivariable models were constructed in the R (version 3.5.0) program using the MASS package.
Accession number(s).
The GenBank accession numbers for the clonal nef sequences are MK076455 to MK076544.
Supplementary Material
ACKNOWLEDGMENTS
We thank Gursev Anmole and Natalie Kinloch for technical assistance and helpful discussions and Viviane Dias Lima for helpful statistical discussions. We gratefully thank the clinical trial participants, without whom this research would not be possible.
This research was funded in part by a Canadian HIV Cure Enterprise team grant from the Canadian Institutes for Health Research (CIHR), in partnership with the Canadian Foundation for AIDS Research (CANFAR) and the International AIDS Society (IAS) (HIG-133050 to M.A.B., M.O., and Z.L.B.); by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number UM1AI126617, with cofunding support from the National Institute on Drug Abuse, the National Institute of Mental Health, and the National Institute of Neurological Disorders and Stroke (to M.A.B., M.O., and Z.L.B.); by NIH award R21A127029 (to Z.L.B. and M.A.B.); and by project grants from the CIHR (PJT-148621 and PJT-159625 to Z.L.B. and M.A.B.). F.H.O. is supported by the Canadian Queen Elizabeth II Diamond Jubilee Scholarships program, a joint initiative of the Rideau Hall Foundation, Community Foundations of Canada, and the Association of Universities and Colleges of Canada, and also received a fellowship from the Sub-Saharan African Network for TB/HIV Research Excellence (SANTHE), a DELTAS Africa Initiative (grant number DEL-15-006). The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS) Alliance for Accelerating Excellence in Science in Africa (AESA) and is supported by the New Partnership for Africa’s Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust (grant number 107752/Z/15/Z) and the UK government. S.W.J. was supported by a Frederick Banting and Charles Best CIHR MSc award. M.A.B. holds a Canada Research Chair, Tier 2, in Viral Pathogenesis and Immunity. Z.L.B. is supported by a scholar award from the Michael Smith Foundation for Health Research.
The views expressed in this publication are those of the authors and not necessarily those of AAS, the NEPAD Agency, the Wellcome Trust, or the UK government.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01832-18.
REFERENCES
- 1.Pino M, Paiardini M, Marconi VC. 2018. Progress in achieving long-term HIV remission. Curr Opin HIV AIDS 13:435–455. doi: 10.1097/COH.0000000000000487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Soriano-Sarabia N, Bateson RE, Dahl NP, Crooks AM, Kuruc JD, Margolis DM, Archin NM. 2014. Quantitation of replication-competent HIV-1 in populations of resting CD4+ T cells. J Virol 88:14070–14077. doi: 10.1128/JVI.01900-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, Lloyd AL, Nowak MA, Fauci AS. 1997. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A 94:13193–13197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, Gallant J, Markowitz M, Ho DD, Richman DD, Siliciano RF. 1997. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278:1295–1300. [DOI] [PubMed] [Google Scholar]
- 5.Archin NM, Sung JM, Garrido C, Soriano-Sarabia N, Margolis DM. 2014. Eradicating HIV-1 infection: seeking to clear a persistent pathogen. Nat Rev Microbiol 12:750–764. doi: 10.1038/nrmicro3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pace MJ, Agosto L, Graf EH, O'Doherty U. 2011. HIV reservoirs and latency models. Virology 411:344–354. doi: 10.1016/j.virol.2010.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richman DD, Margolis DM, Delaney M, Greene WC, Hazuda D, Pomerantz RJ. 2009. The challenge of finding a cure for HIV infection. Science 323:1304–1307. doi: 10.1126/science.1165706. [DOI] [PubMed] [Google Scholar]
- 8.Shen L, Siliciano RF. 2008. Viral reservoirs, residual viremia, and the potential of highly active antiretroviral therapy to eradicate HIV infection. J Allergy Clin Immunol 122:22–28. doi: 10.1016/j.jaci.2008.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katlama C, Deeks SG, Autran B, Martinez-Picado J, van Lunzen J, Rouzioux C, Miller M, Vella S, Schmitz JE, Ahlers J, Richman DD, Sekaly RP. 2013. Barriers to a cure for HIV: new ways to target and eradicate HIV-1 reservoirs. Lancet 381:2109–2117. doi: 10.1016/S0140-6736(13)60104-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Durand CM, Blankson JN, Siliciano RF. 2012. Developing strategies for HIV-1 eradication. Trends Immunol 33:554–562. doi: 10.1016/j.it.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joos B, Fischer M, Kuster H, Pillai SK, Wong JK, Boni J, Hirschel B, Weber R, Trkola A, Gunthard HF, Swiss HIV Cohort Study. 2008. HIV rebounds from latently infected cells, rather than from continuing low-level replication. Proc Natl Acad Sci U S A 105:16725–16730. doi: 10.1073/pnas.0804192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pomerantz RJ. 2003. Reservoirs, sanctuaries, and residual disease: the hiding spots of HIV-1. HIV Clin Trials 4:137–143. doi: 10.1310/hct.2003.4.2.009. [DOI] [PubMed] [Google Scholar]
- 13.Rouzioux C, Richman D. 2013. How to best measure HIV reservoirs? Curr Opin HIV AIDS 8:170–175. doi: 10.1097/COH.0b013e32835fc619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saez-Cirion A, Bacchus C, Hocqueloux L, Avettand-Fenoel V, Girault I, Lecuroux C, Potard V, Versmisse P, Melard A, Prazuck T, Descours B, Guergnon J, Viard JP, Boufassa F, Lambotte O, Goujard C, Meyer L, Costagliola D, Venet A, Pancino G, Autran B, Rouzioux C, ANRS VISCONTI Study Group. 2013. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog 9:e1003211. doi: 10.1371/journal.ppat.1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharaf R, Lee GQ, Sun X, Etemad B, Aboukhater LM, Hu Z, Brumme ZL, Aga E, Bosch RJ, Wen Y, Namazi G, Gao C, Acosta EP, Gandhi RT, Jacobson JM, Skiest D, Margolis DM, Mitsuyasu R, Volberding P, Connick E, Kuritzkes DR, Lederman MM, Yu XG, Lichterfeld M, Li JZ. 2018. HIV-1 proviral landscapes distinguish posttreatment controllers from noncontrollers. J Clin Invest 128:4074–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ananworanich J, Chomont N, Eller LA, Kroon E, Tovanabutra S, Bose M, Nau M, Fletcher JLK, Tipsuk S, Vandergeeten C, O'Connell RJ, Pinyakorn S, Michael N, Phanuphak N, Robb ML, on behalf of the RV217 and RV254/SEARCH010 Study Groups. 2016. HIV DNA set point is rapidly established in acute HIV infection and dramatically reduced by early ART. EBioMedicine 11:68–72. doi: 10.1016/j.ebiom.2016.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Novelli S, Lecuroux C, Avettand-Fenoel V, Seng R, Essat A, Morlat P, Viard JP, Rouzioux C, Meyer L, Goujard C. 2018. Long-term therapeutic impact of the timing of antiretroviral therapy in patients diagnosed with primary HIV-1 infection. Clin Infect Dis 66:1519–1527. doi: 10.1093/cid/cix1068. [DOI] [PubMed] [Google Scholar]
- 18.Jain V, Hartogensis W, Bacchetti P, Hunt PW, Hatano H, Sinclair E, Epling L, Lee TH, Busch MP, McCune JM, Pilcher CD, Hecht FM, Deeks SG. 2013. Antiretroviral therapy initiated within 6 months of HIV infection is associated with lower T-cell activation and smaller HIV reservoir size. J Infect Dis 208:1202–1211. doi: 10.1093/infdis/jit311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buzon MJ, Martin-Gayo E, Pereyra F, Ouyang Z, Sun H, Li JZ, Piovoso M, Shaw A, Dalmau J, Zangger N, Martinez-Picado J, Zurakowski R, Yu XG, Telenti A, Walker BD, Rosenberg ES, Lichterfeld M. 2014. Long-term antiretroviral treatment initiated at primary HIV-1 infection affects the size, composition, and decay kinetics of the reservoir of HIV-1-infected CD4 T cells. J Virol 88:10056–10065. doi: 10.1128/JVI.01046-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ostrowski M, Benko E, Yue FY, Kim CJ, Huibner S, Lee T, Singer J, Pankovich J, Laeyendecker O, Kaul R, Kandel G, Kovacs C. 2015. Intensifying antiretroviral therapy with raltegravir and maraviroc during early human immunodeficiency virus (HIV) infection does not accelerate HIV reservoir reduction. Open Forum Infect Dis 2:ofv138. doi: 10.1093/ofid/ofv138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prodger JL, Lai J, Reynolds SJ, Keruly JC, Moore RD, Kasule J, Kityamuweesi T, Buule P, Serwadda D, Nason M, Capoferri AA, Porcella SF, Siliciano RF, Redd AD, Siliciano JD, Quinn TC. 2017. Reduced frequency of cells latently infected with replication-competent human immunodeficiency virus-1 in virally suppressed individuals living in Rakai, Uganda. Clin Infect Dis 65:1308–1315. doi: 10.1093/cid/cix478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, Boucher G, Boulassel M-R, Ghattas G, Brenchley JM, Schacker TW, Hill BJ, Douek DC, Routy J-P, Haddad EK, Sékaly R-P. 2009. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med 15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simonetti FR, Sobolewski MD, Fyne E, Shao W, Spindler J, Hattori J, Anderson EM, Watters SA, Hill S, Wu X, Wells D, Su L, Luke BT, Halvas EK, Besson G, Penrose KJ, Yang Z, Kwan RW, Van Waes C, Uldrick T, Citrin DE, Kovacs J, Polis MA, Rehm CA, Gorelick R, Piatak M, Keele BF, Kearney MF, Coffin JM, Hughes SH, Mellors JW, Maldarelli F. 2016. Clonally expanded CD4+ T cells can produce infectious HIV-1 in vivo. Proc Natl Acad Sci U S A 113:1883–1888. doi: 10.1073/pnas.1522675113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bui JK, Sobolewski MD, Keele BF, Spindler J, Musick A, Wiegand A, Luke BT, Shao W, Hughes SH, Coffin JM, Kearney MF, Mellors JW. 2017. Proviruses with identical sequences comprise a large fraction of the replication-competent HIV reservoir. PLoS Pathog 13:e1006283. doi: 10.1371/journal.ppat.1006283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hiener B, Horsburgh BA, Eden JS, Barton K, Schlub TE, Lee E, von Stockenstrom S, Odevall L, Milush JM, Liegler T, Sinclair E, Hoh R, Boritz EA, Douek D, Fromentin R, Chomont N, Deeks SG, Hecht FM, Palmer S. 2017. Identification of genetically intact HIV-1 proviruses in specific CD4+ T cells from effectively treated participants. Cell Rep 21:813–822. doi: 10.1016/j.celrep.2017.09.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee GQ, Orlova-Fink N, Einkauf K, Chowdhury FZ, Sun X, Harrington S, Kuo HH, Hua S, Chen HR, Ouyang Z, Reddy K, Dong K, Ndung'u T, Walker BD, Rosenberg ES, Yu XG, Lichterfeld M. 2017. Clonal expansion of genome-intact HIV-1 in functionally polarized Th1 CD4+ T cells. J Clin Invest 127:2689–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cohn LB, Silva IT, Oliveira TY, Rosales RA, Parrish EH, Learn GH, Hahn BH, Czartoski JL, McElrath MJ, Lehmann C, Klein F, Caskey M, Walker BD, Siliciano JD, Siliciano RF, Jankovic M, Nussenzweig MC. 2015. HIV-1 integration landscape during latent and active infection. Cell 160:420–432. doi: 10.1016/j.cell.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maldarelli F, Wu X, Su L, Simonetti FR, Shao W, Hill S, Spindler J, Ferris AL, Mellors JW, Kearney MF, Coffin JM, Hughes SH. 2014. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 345:179–183. doi: 10.1126/science.1254194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teigler JE, Leyre L, Chomont N, Slike B, Jian N, Eller MA, Phanuphak N, Kroon E, Pinyakorn S, Eller LA, Robb ML, Ananworanich J, Michael NL, Streeck H, Krebs SJ, RV254/RV217 Study Groups. 2018. Distinct biomarker signatures in HIV acute infection associate with viral dynamics and reservoir size. JCI Insight 3:98420. doi: 10.1172/jci.insight.98420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yue FY, Cohen JC, Ho M, Rahman AK, Liu J, Mujib S, Saiyed A, Hundal S, Khozin A, Bonner P, Liu D, Benko E, Kovacs C, Ostrowski M. 2017. HIV-specific granzyme B-secreting but not gamma interferon-secreting T cells are associated with reduced viral reservoirs in early HIV infection. J Virol 91:e02233-16. doi: 10.1128/JVI.02233-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Le Gall S, Erdtmann L, Benichou S, Berlioz-Torrent C, Liu L, Benarous R, Heard JM, Schwartz O. 1998. Nef interacts with the mu subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity 8:483–495. [DOI] [PubMed] [Google Scholar]
- 32.Anderson S, Shugars DC, Swanstrom R, Garcia JV. 1993. Nef from primary isolates of human immunodeficiency virus type 1 suppresses surface CD4 expression in human and mouse T cells. J Virol 67:4923–4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mariani R, Skowronski J. 1993. CD4 down-regulation by nef alleles isolated from human immunodeficiency virus type 1-infected individuals. Proc Natl Acad Sci U S A 90:5549–5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Collins KL, Chen BK, Kalams SA, Walker BD, Baltimore D. 1998. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 391:397–401. doi: 10.1038/34929. [DOI] [PubMed] [Google Scholar]
- 35.Veillette M, Desormeaux A, Medjahed H, Gharsallah NE, Coutu M, Baalwa J, Guan Y, Lewis G, Ferrari G, Hahn BH, Haynes BF, Robinson JE, Kaufmann DE, Bonsignori M, Sodroski J, Finzi A. 2014. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J Virol 88:2633–2644. doi: 10.1128/JVI.03230-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuang XT, Li X, Anmole G, Mwimanzi P, Shahid A, Le AQ, Chong L, Qian H, Miura T, Markle T, Baraki B, Connick E, Daar ES, Jessen H, Kelleher AD, Little S, Markowitz M, Pereyra F, Rosenberg ES, Walker BD, Ueno T, Brumme ZL, Brockman MA. 2014. Impaired Nef function is associated with early control of HIV-1 viremia. J Virol 88:10200–10213. doi: 10.1128/JVI.01334-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mahiti M, Toyoda M, Jia X, Kuang XT, Mwimanzi F, Mwimanzi P, Walker BD, Xiong Y, Brumme ZL, Brockman MA, Ueno T. 2016. Relative resistance of HLA-B to downregulation by naturally occurring HIV-1 Nef sequences. mBio 7:e01516. doi: 10.1128/mBio.01516-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mann JK, Byakwaga H, Kuang XT, Le AQ, Brumme CJ, Mwimanzi P, Omarjee S, Martin E, Lee GQ, Baraki B, Danroth R, McCloskey R, Muzoora C, Bangsberg DR, Hunt PW, Goulder PJ, Walker BD, Harrigan PR, Martin JN, Ndung UT, Brockman MA, Brumme ZL. 2013. Ability of HIV-1 Nef to downregulate CD4 and HLA class I differs among viral subtypes. Retrovirology 10:100. doi: 10.1186/1742-4690-10-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mwimanzi P, Markle TJ, Martin E, Ogata Y, Kuang XT, Tokunaga M, Mahiti M, Pereyra F, Miura T, Walker BD, Brumme ZL, Brockman MA, Ueno T. 2013. Attenuation of multiple Nef functions in HIV-1 elite controllers. Retrovirology 10:1. doi: 10.1186/1742-4690-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mwimanzi P, Markle TJ, Ogata Y, Martin E, Tokunaga M, Mahiti M, Kuang XT, Walker BD, Brockman MA, Brumme ZL, Ueno T. 2013. Dynamic range of Nef functions in chronic HIV-1 infection. Virology 439:74–80. doi: 10.1016/j.virol.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 41.Alsahafi N, Ding S, Richard J, Markle T, Brassard N, Walker B, Lewis GK, Kaufmann DE, Brockman MA, Finzi A. 2016. Nef proteins from HIV-1 elite controllers are inefficient at preventing antibody-dependent cellular cytotoxicity. J Virol 90:2993–3002. doi: 10.1128/JVI.02973-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mujib S, Saiyed A, Fadel S, Bozorgzad A, Aidarus N, Yue FY, Benko E, Kovacs C, Emert-Sedlak LA, Smithgall TE, Ostrowski MA. 2017. Pharmacologic HIV-1 Nef blockade promotes CD8 T cell-mediated elimination of latently HIV-1-infected cells in vitro. JCI Insight 2:93684. doi: 10.1172/jci.insight.93684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Foley B, Leitner T, Apetrei C, Hahn B, Mizrachi I, Mullins J, Rambaut A, Wolinsky S, Korber B (ed). 2017. HIV sequence compendium 2017. Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, Los Alamos, NM. [Google Scholar]
- 44.Laeyendecker O, Brookmeyer R, Cousins MM, Mullis CE, Konikoff J, Donnell D, Celum C, Buchbinder SP, Seage GR III, Kirk GD, Mehta SH, Astemborski J, Jacobson LP, Margolick JB, Brown J, Quinn TC, Eshleman SH. 2013. HIV incidence determination in the United States: a multiassay approach. J Infect Dis 207:232–239. doi: 10.1093/infdis/jis659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shahid A, Olvera A, Anmole G, Kuang XT, Cotton LA, Plana M, Brander C, Brockman MA, Brumme ZL. 2015. Consequences of HLA-B*13-associated escape mutations on HIV-1 replication and Nef function. J Virol 89:11557–11571. doi: 10.1128/JVI.01955-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salazar-Gonzalez JF, Salazar MG, Keele BF, Learn GH, Giorgi EE, Li H, Decker JM, Wang S, Baalwa J, Kraus MH, Parrish NF, Shaw KS, Guffey MB, Bar KJ, Davis KL, Ochsenbauer-Jambor C, Kappes JC, Saag MS, Cohen MS, Mulenga J, Derdeyn CA, Allen S, Hunter E, Markowitz M, Hraber P, Perelson AS, Bhattacharya T, Haynes BF, Korber BT, Hahn BH, Shaw GM. 2009. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J Exp Med 206:1273–1289. doi: 10.1084/jem.20090378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tully DC, Ogilvie CB, Batorsky RE, Bean DJ, Power KA, Ghebremichael M, Bedard HE, Gladden AD, Seese AM, Amero MA, Lane K, McGrath G, Bazner SB, Tinsley J, Lennon NJ, Henn MR, Brumme ZL, Norris PJ, Rosenberg ES, Mayer KH, Jessen H, Kosakovsky Pond SL, Walker BD, Altfeld M, Carlson JM, Allen TM. 2016. Differences in the selection bottleneck between modes of sexual transmission influence the genetic composition of the HIV-1 founder virus. PLoS Pathog 12:e1005619. doi: 10.1371/journal.ppat.1005619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herbeck JT, Rolland M, Liu Y, McLaughlin S, McNevin J, Zhao H, Wong K, Stoddard JN, Raugi D, Sorensen S, Genowati I, Birditt B, McKay A, Diem K, Maust BS, Deng W, Collier AC, Stekler JD, McElrath MJ, Mullins JI. 2011. Demographic processes affect HIV-1 evolution in primary infection before the onset of selective processes. J Virol 85:7523–7534. doi: 10.1128/JVI.02697-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shankarappa R, Margolick JB, Gange SJ, Rodrigo AG, Upchurch D, Farzadegan H, Gupta P, Rinaldo CR, Learn GH, He X, Huang XL, Mullins JI. 1999. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J Virol 73:10489–10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zanini F, Brodin J, Thebo L, Lanz C, Bratt G, Albert J, Neher RA. 2015. Population genomics of intrapatient HIV-1 evolution. Elife 4:e11282. doi: 10.7554/eLife.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fischer W, Ganusov VV, Giorgi EE, Hraber PT, Keele BF, Leitner T, Han CS, Gleasner CD, Green L, Lo CC, Nag A, Wallstrom TC, Wang S, McMichael AJ, Haynes BF, Hahn BH, Perelson AS, Borrow P, Shaw GM, Bhattacharya T, Korber BT. 2010. Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing. PLoS One 5:e12303. doi: 10.1371/journal.pone.0012303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Henn MR, Boutwell CL, Charlebois P, Lennon NJ, Power KA, Macalalad AR, Berlin AM, Malboeuf CM, Ryan EM, Gnerre S, Zody MC, Erlich RL, Green LM, Berical A, Wang Y, Casali M, Streeck H, Bloom AK, Dudek T, Tully D, Newman R, Axten KL, Gladden AD, Battis L, Kemper M, Zeng Q, Shea TP, Gujja S, Zedlack C, Gasser O, Brander C, Hess C, Gunthard HF, Brumme ZL, Brumme CJ, Bazner S, Rychert J, Tinsley JP, Mayer KH, Rosenberg E, Pereyra F, Levin JZ, Young SK, Jessen H, Altfeld M, Birren BW, Walker BD, Allen TM. 2012. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog 8:e1002529. doi: 10.1371/journal.ppat.1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Siemieniuk RA, Beckthold B, Gill MJ. 2013. Increasing HIV subtype diversity and its clinical implications in a sentinel North American population. Can J Infect Dis Med Microbiol 24:69–73. doi: 10.1155/2013/230380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuang T, Markle T, Martin E, Le A, Baraki B, Chopera D, Jessen H, Kelleher A, Markowitz M, Rosenberg E, Walker B, Brumme Z, Brockman M. 2012. Characterization of HIV-1 Nef function in early and chronic infection, abstr 111 2012 Keystone Symposium on Frontiers in HIV Pathogenesis, Therapy and Eradication (X8). [Google Scholar]
- 55.Hocqueloux L, Avettand-Fenoel V, Jacquot S, Prazuck T, Legac E, Melard A, Niang M, Mille C, Le Moal G, Viard JP, Rouzioux C, on behalf of the AC32 (Coordinated Action on HIV Reservoirs) of the Agence Nationale de Recherches sur le Sida et les Hépatites Virales (ANRS). 2013. Long-term antiretroviral therapy initiated during primary HIV-1 infection is key to achieving both low HIV reservoirs and normal T cell counts. J Antimicrob Chemother 68:1169–1178. doi: 10.1093/jac/dks533. [DOI] [PubMed] [Google Scholar]
- 56.Depince-Berger AE, Vergnon-Miszczycha D, Girard A, Fresard A, Botelho-Nevers E, Lambert C, Del Tedesco E, Genin C, Pozzetto B, Lucht F, Roblin X, Bourlet T, Paul S. 2016. Major influence of CD4 count at the initiation of cART on viral and immunological reservoir constitution in HIV-1 infected patients. Retrovirology 13:44. doi: 10.1186/s12977-016-0278-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jones LE, Perelson AS. 2007. Transient viremia, plasma viral load, and reservoir replenishment in HIV-infected patients on antiretroviral therapy. J Acquir Immune Defic Syndr 45:483–493. doi: 10.1097/QAI.0b013e3180654836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blankson JN, Finzi D, Pierson TC, Sabundayo BP, Chadwick K, Margolick JB, Quinn TC, Siliciano RF. 2000. Biphasic decay of latently infected CD4+ T cells in acute human immunodeficiency virus type 1 infection. J Infect Dis 182:1636–1642. doi: 10.1086/317615. [DOI] [PubMed] [Google Scholar]
- 59.Watanabe D, Ibe S, Uehira T, Minami R, Sasakawa A, Yajima K, Yonemoto H, Bando H, Ogawa Y, Taniguchi T, Kasai D, Nishida Y, Yamamoto M, Kaneda T, Shirasaka T. 2011. Cellular HIV-1 DNA levels in patients receiving antiretroviral therapy strongly correlate with therapy initiation timing but not with therapy duration. BMC Infect Dis 11:146. doi: 10.1186/1471-2334-11-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Akari H, Arold S, Fukumori T, Okazaki T, Strebel K, Adachi A. 2000. Nef-induced major histocompatibility complex class I down-regulation is functionally dissociated from its virion incorporation, enhancement of viral infectivity, and CD4 down-regulation. J Virol 74:2907–2912. doi: 10.1128/JVI.74.6.2907-2912.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jia X, Singh R, Homann S, Yang H, Guatelli J, Xiong Y. 2012. Structural basis of evasion of cellular adaptive immunity by HIV-1 Nef. Nat Struct Mol Biol 19:701–706. doi: 10.1038/nsmb.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vittinghoff E, McCulloch CE. 2007. Relaxing the rule of ten events per variable in logistic and Cox regression. Am J Epidemiol 165:710–718. doi: 10.1093/aje/kwk052. [DOI] [PubMed] [Google Scholar]
- 63.Nemes S, Jonasson JM, Genell A, Steineck G. 2009. Bias in odds ratios by logistic regression modelling and sample size. BMC Med Res Methodol 9:56. doi: 10.1186/1471-2288-9-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Anmole G, Kuang XT, Toyoda M, Martin E, Shahid A, Le AQ, Markle T, Baraki B, Jones RB, Ostrowski MA, Ueno T, Brumme ZL, Brockman MA. 2015. A robust and scalable TCR-based reporter cell assay to measure HIV-1 Nef-mediated T cell immune evasion. J Immunol Methods 426:104–113. doi: 10.1016/j.jim.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 65.Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, Smith K, Lisziewicz J, Lori F, Flexner C, Quinn TC, Chaisson RE, Rosenberg E, Walker B, Gange S, Gallant J, Siliciano RF. 1999. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med 5:512–517. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 66.Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, Kovacs C, Gange SJ, Siliciano RF. 2003. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med 9:727–728. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- 67.Chun TW, Justement JS, Moir S, Hallahan CW, Maenza J, Mullins JI, Collier AC, Corey L, Fauci AS. 2007. Decay of the HIV reservoir in patients receiving antiretroviral therapy for extended periods: implications for eradication of virus. J Infect Dis 195:1762–1764. doi: 10.1086/518250. [DOI] [PubMed] [Google Scholar]
- 68.Chowers MY, Spina CA, Kwoh TJ, Fitch NJ, Richman DD, Guatelli JC. 1994. Optimal infectivity in vitro of human immunodeficiency virus type 1 requires an intact nef gene. J Virol 68:2906–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carl S, Greenough TC, Krumbiegel M, Greenberg M, Skowronski J, Sullivan JL, Kirchhoff F. 2001. Modulation of different human immunodeficiency virus type 1 Nef functions during progression to AIDS. J Virol 75:3657–3665. doi: 10.1128/JVI.75.8.3657-3665.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kaleebu P, French N, Mahe C, Yirrell D, Watera C, Lyagoba F, Nakiyingi J, Rutebemberwa A, Morgan D, Weber J, Gilks C, Whitworth J. 2002. Effect of human immunodeficiency virus (HIV) type 1 envelope subtypes A and D on disease progression in a large cohort of HIV-1-positive persons in Uganda. J Infect Dis 185:1244–1250. doi: 10.1086/340130. [DOI] [PubMed] [Google Scholar]
- 71.Vasan A, Renjifo B, Hertzmark E, Chaplin B, Msamanga G, Essex M, Fawzi W, Hunter D. 2006. Different rates of disease progression of HIV type 1 infection in Tanzania based on infecting subtype. Clin Infect Dis 42:843–852. doi: 10.1086/499952. [DOI] [PubMed] [Google Scholar]
- 72.Baeten JM, Chohan B, Lavreys L, Chohan V, McClelland RS, Certain L, Mandaliya K, Jaoko W, Overbaugh J. 2007. HIV-1 subtype D infection is associated with faster disease progression than subtype A in spite of similar plasma HIV-1 loads. J Infect Dis 195:1177–1180. doi: 10.1086/512682. [DOI] [PubMed] [Google Scholar]
- 73.Venner CM, Nankya I, Kyeyune F, Demers K, Kwok C, Chen PL, Rwambuya S, Munjoma M, Chipato T, Byamugisha J, Van Der Pol B, Mugyenyi P, Salata RA, Morrison CS, Arts EJ. 2016. Infecting HIV-1 subtype predicts disease progression in women of sub-Saharan Africa. EBioMedicine 13:305–314. doi: 10.1016/j.ebiom.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Myers LE, McQuay LJ, Hollinger FB. 1994. Dilution assay statistics. J Clin Microbiol 32:732–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cotton LA, Rahman MA, Ng C, Le AQ, Milloy MJ, Mo T, Brumme ZL. 2012. HLA class I sequence-based typing using DNA recovered from frozen plasma. J Immunol Methods 382:40–47. doi: 10.1016/j.jim.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 76.Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gaschen B, Kuiken C, Korber B, Foley B. 2001. Retrieval and on-the-fly alignment of sequence fragments from the HIV database. Bioinformatics 17:415–418. [DOI] [PubMed] [Google Scholar]
- 78.Larsson A. 2014. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30:3276–3278. doi: 10.1093/bioinformatics/btu531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guindon S, Delsuc F, Dufayard JF, Gascuel O. 2009. Estimating maximum likelihood phylogenies with PhyML. Methods Mol Biol 537:113–137. doi: 10.1007/978-1-59745-251-9_6. [DOI] [PubMed] [Google Scholar]
- 80.Tavare S. 1986. Some probabilistic and statistical problems in the analysis of DNA sequences. Lectures Math Life Sci 17:57–86. [Google Scholar]
- 81.Siepel AC, Halpern AL, Macken C, Korber BT. 1995. A computer program designed to screen rapidly for HIV type 1 intersubtype recombinant sequences. AIDS Res Hum Retroviruses 11:1413–1416. doi: 10.1089/aid.1995.11.1413. [DOI] [PubMed] [Google Scholar]
- 82.Storey JD, Tibshirani R. 2003. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.