

Liver X receptors (LXRs) are nuclear receptors that mediate cholesterol and fatty acid homeostasis. Importantly, as ligand-activated transcription factors, LXRs represent potential targets for the treatment of hypercholesterolemia and atherosclerosis. Here, we demonstrate that LXRα, one of the two LXR isoforms, restricts reactivation of latent gammaherpesvirus from peritoneal cells. As gammaherpesviruses are ubiquitous oncogenic agents, LXRs may represent a targetable host factor for the treatment of poorly controlled gammaherpesvirus infection and associated lymphomagenesis.
KEYWORDS: chronic infection, gammaherpesvirus, liver X receptor alpha, peritoneal B cells
ABSTRACT
Gammaherpesviruses are ubiquitous viruses that establish lifelong infections. Importantly, these viruses are associated with numerous cancers and lymphoproliferative diseases. While risk factors for developing gammaherpesvirus-driven cancers are poorly understood, it is clear that elevated viral reactivation from latency often precedes oncogenesis. Here, we demonstrate that the liver X receptor alpha isoform (LXRα) restricts gammaherpesvirus reactivation in an anatomic-site-specific manner. We have previously demonstrated that deficiency of both LXR isoforms (α and β) leads to an increase in fatty acid and cholesterol synthesis in primary macrophage cultures, with a corresponding increase in gammaherpesvirus replication. Interestingly, expression of fatty acid synthesis genes was not derepressed in LXRα-deficient hosts, indicating that the antiviral effects of LXRα are independent of lipogenesis. Additionally, the critical host defenses against gammaherpesvirus reactivation, virus-specific CD8+ T cells and interferon (IFN) signaling, remained intact in the absence of LXRα. Remarkably, using a murine gammaherpesvirus 68 (MHV68) reporter virus, we discovered that LXRα expression dictates the cellular tropism of MHV68 in the peritoneal cavity. Specifically, LXRα−/− mice exhibit reduced latency within the peritoneal B cell compartment and elevated latency within F4/80+ cells. Thus, LXRα restricts gammaherpesvirus reactivation through a novel mechanism that is independent of the known CD8+ T cell-based antiviral responses or changes in lipid synthesis and likely involves changes in the tropism of MHV68 in the peritoneal cavity.
IMPORTANCE Liver X receptors (LXRs) are nuclear receptors that mediate cholesterol and fatty acid homeostasis. Importantly, as ligand-activated transcription factors, LXRs represent potential targets for the treatment of hypercholesterolemia and atherosclerosis. Here, we demonstrate that LXRα, one of the two LXR isoforms, restricts reactivation of latent gammaherpesvirus from peritoneal cells. As gammaherpesviruses are ubiquitous oncogenic agents, LXRs may represent a targetable host factor for the treatment of poorly controlled gammaherpesvirus infection and associated lymphomagenesis.
INTRODUCTION
Gammaherpesviruses are ubiquitous pathogens that establish lifelong infections in ∼95% of the adult human population. There are two known human gammaherpesviruses, Epstein-Barr virus (EBV) and Kaposi’s sarcoma-associated herpesvirus (KSHV). Importantly, EBV and KSHV are associated with numerous lymphomas and other malignancies, including the body cavity-restricted primary effusion lymphoma (1). Furthermore, while risk factors for the development of gammaherpesvirus-driven cancers remain poorly understood, elevated viral reactivation often precedes oncogenesis (2–5). Thus, prevention of gammaherpesvirus-associated cancers requires a more thorough understanding of the virus-host interactions that dictate control of viral latency and reactivation. Due to the exquisite species specificity of human gammaherpesviruses, which significantly hinders identification of mechanisms that regulate viral reactivation in vivo, this study takes advantage of the murine gammaherpesvirus 68 (MHV68) model, a natural rodent pathogen that is closely related to EBV and KSHV (6–8) and represents a powerful animal model to define molecular and cellular mechanisms of the host that control chronic gammaherpesvirus infection.
Liver X receptors (LXRs) are transcription factors of the nuclear receptor superfamily. There are two isoforms of LXRs, LXRα and LXRβ, which are transcribed from independent genes. While LXRα and LXRβ share approximately 77% amino acid sequence identity, the expression patterns of the two isoforms differ greatly. LXRα is expressed in highly metabolic tissues, such as adipose tissue, liver, and intestine (9). Interestingly, LXRα is also expressed in some immune tissues and cell types, such as the spleen and thymus and macrophages (10). LXRβ, on the other hand, is expressed ubiquitously (10). When activated by endogenous oxysterol ligands, LXRs induce the transcription of target genes, many of which encode proteins involved in fatty acid synthesis and cholesterol efflux. Thus, activation of LXRs, through synthetic or endogenous ligands, typically results in elevated fatty acid synthesis and cholesterol efflux (11–14). In addition to regulation of lipid metabolism, LXRs play important roles in the immune responses to pathogens. As such, LXRs are protective against in vivo infection with the intracellular pathogens Listeria monocytogenes (15) and Mycobacterium tuberculosis (16). Interestingly, while many reports describe anti-inflammatory activity of LXRs, along with negative regulation of T cell proliferation and CD4+ T cell differentiation (17, 18), these effects are likely context dependent, as LXRs facilitate TH1 and TH17 CD4+ T cell differentiation during M. tuberculosis infection (16).
Much of what is known about LXR biology in the context of viral infection has been defined using in vitro studies of RNA viruses and artificial synthetic LXR agonists. Activation of LXRs with artificial agonists was found to inhibit replication of hepatitis C virus (19, 20), HIV (21–23), and Newcastle disease virus (24) in cell culture, likely through perturbations of cellular cholesterol content. Only a few in vivo studies have defined the effects of synthetic LXR agonists during virus infection. Interestingly, while synthetic LXR agonists increased viral titers and the severity of pathology in a coxsackievirus infection model of myocarditis (25), treatment of humanized mice with synthetic LXR agonists suppressed HIV-1 replication (23, 26). Importantly, it is not known whether individual LXR isoforms and endogenous LXR ligands affect viral infection of an intact host.
We recently used a genetic approach to demonstrate that expression of both LXR isoforms attenuated replication of MHV68 in primary macrophages (27). Consistent with this antiviral activity, the expression of LXRs was elevated following viral infection in a type I interferon (IFN)-dependent manner. In contrast to the known ability of LXRs to activate their target genes in hepatocytes, we discovered that macrophages utilize LXRs to repress expression of enzymes involved in lipid synthesis, resulting in decreased activity of lipogenic pathways that support gammaherpesvirus replication (27). While these data demonstrate an antiviral role for LXRs during in vitro infection of primary macrophages, the role of LXRs during chronic gammaherpesvirus infection of an intact host remains unknown.
This study utilized genetic approaches to define the extent to which expression of LXRα, one of the two known LXR isoforms, affects chronic gammaherpesvirus infection. Expression of LXRα restricted reactivation of MHV68 from peritoneal cells during long-term infection. Interestingly, deficiency of LXRα alone did not result in derepression of LXR-dependent genes involved in lipid regulation as we had observed in LXRα−/− LXRβ−/− macrophages in vitro. Furthermore, despite the elevated viral reactivation, LXRα−/− mice displayed normal virus-specific CD8+ T cell generation, recruitment, and antiviral cytokine production. Further, LXRα did not affect the ability of bone marrow-derived macrophages and latently infected peritoneal cells to respond to exogenous IFN treatment. Importantly, using a β-lactamase reporter virus (28), we identified a dramatic change in the peritoneal tropism of MHV68 in mice lacking expression of LXRα. Thus, LXRα-mediated restriction of viral reactivation likely operates through a novel antiviral mechanism that dictates the peritoneal tropism of MHV68.
RESULTS
LXRα restricts MHV68 reactivation from peritoneal cells, but not splenocytes, in chronically infected animals.
To determine the extent to which expression of LXRα affects chronic gammaherpesvirus infection of an intact host, C57BL/6J (BL6) and LXRα−/− mice were intraperitoneally infected with 500 PFU of MHV68. At 16, 28, and 42 days postinfection (dpi), the frequencies of peritoneal exudate cells (PECs) and splenocytes that were viral DNA positive or supported viral reactivation ex vivo were determined using limiting-dilution assays.
MHV68 latency is established upon the clearance of acute infection, and the number of latently infected cells and viral reactivation peak at 16 days postinfection. Expression of LXRα did not alter viral reactivation (Fig. 1A) or the viral latent reservoir (Fig. 1B) in the spleen or the peritoneum at 16 days postinfection, indicating that LXRα does not affect the establishment of latent gammaherpesvirus infection. Intriguingly, following the establishment of latent infection, LXRα−/− mice began to lose control of MHV68 reactivation from peritoneal cells, but not splenocytes (Fig. 1C) (∼6.7-fold; 28 days postinfection). Despite increased reactivation, there was no significant change in the frequency of latently infected cells in LXRα−/− mice (Fig. 1D), suggesting that LXRα specifically suppressed gammaherpesvirus reactivation. The ability of LXRα to suppress MHV68 reactivation from peritoneal cells was also observed in long-term-infected animals (Fig. 1E and F) (42 days postinfection).
FIG 1.
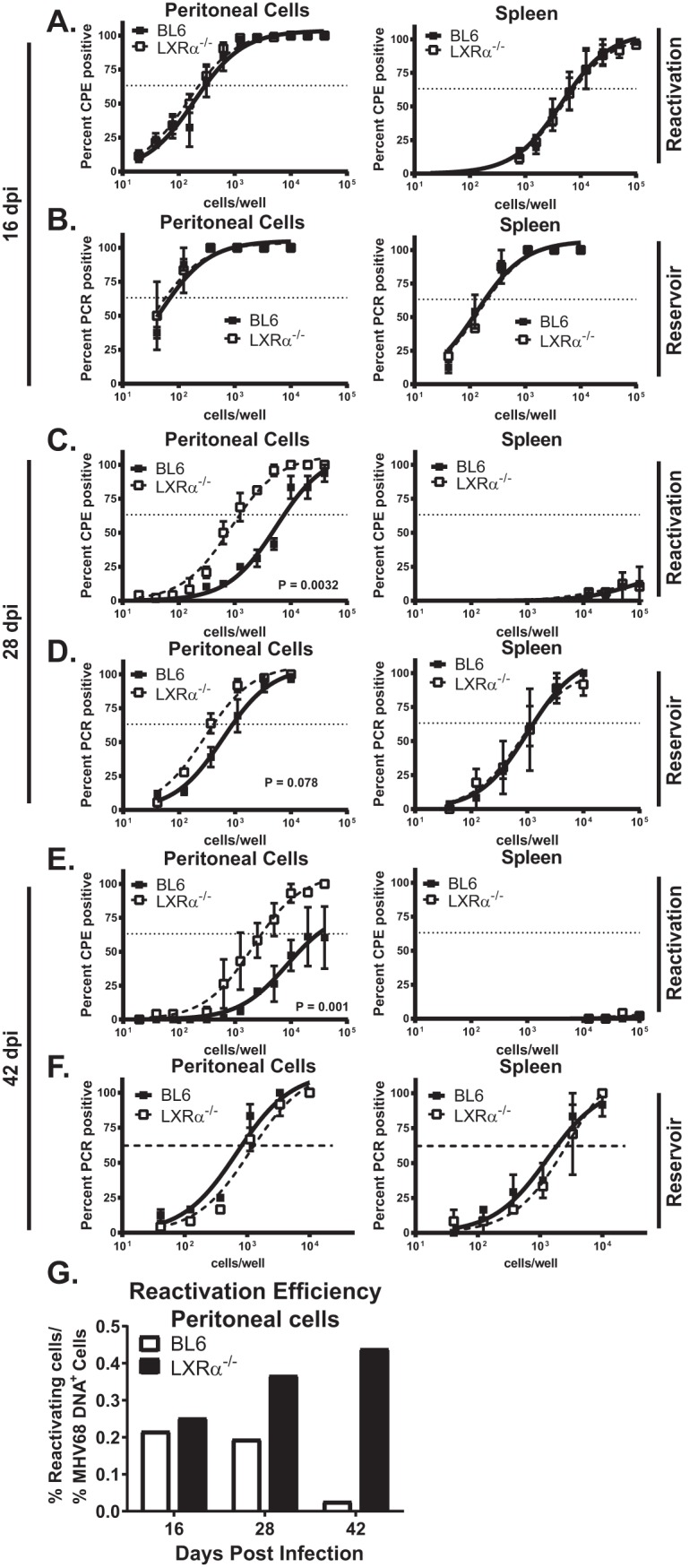
LXRα restricts MHV68 reactivation from peritoneal cells, but not splenocytes, in chronically infected animals. Mice of the indicated genotypes were intraperitoneally infected with 500 PFU wild-type MHV68. Mice were euthanized at 16 (A and B), 28 (C and D), or 42 (E and F) days postinfection, and splenocytes (right) or peritoneal exudate (left) cells were processed into single-cell suspensions. The cells were serially diluted and cocultured with MEFs to assess the frequency of cells reactivating latent virus (A, C, and E) or subjected to nested-PCR analysis to assess the frequency of cells harboring viral genomes (B, D, and F). The data are pooled from 2 to 4 independent experiments. The error bars represent standard errors of the mean. CPE, cytopathic effect. (G) The percentages of peritoneal cells positive for MHV68 DNA or supporting ex vivo reactivation were calculated for each time point using average frequencies of pooled data presented in panels A to F. Reactivation efficiency was defined as a ratio of percent reactivating to percent viral DNA-positive cells for each time point.
To calculate the efficiency of viral reactivation, the percentage of peritoneal cells positive for MHV68 DNA or supporting ex vivo reactivation was determined for each time point, using average frequencies presented in Fig. 1A to F. The ratio of percent reactivating to percent viral-DNA-positive cells demonstrated comparable efficiencies of MHV68 reactivation in BL6 and LXRα−/− mice at 16 days postinfection (Fig. 1G). In contrast, the efficiency of MHV68 reactivation from peritoneal cells was 2-fold and 16-fold higher in LXRα−/− mice at 28 and 42 days postinfection, respectively (Fig. 1G). Of note, persistent MHV68 replication was not observed at any of the time points that were examined. Taken together, the data showed that LXRα decreased the efficiency of gammaherpesvirus reactivation from peritoneal cells, but not splenocytes, during long-term infection.
LXRα deficiency does not result in increased expression of proviral lipogenic genes during chronic MHV68 infection.
We have previously demonstrated that expression of both LXR isoforms is stimulated by IFN signaling in cultured primary macrophages. Further, LXRs repressed the expression of cholesterol and fatty acid synthesis genes and activities of these proviral lipogenic pathways in the macrophage cultures (27). Having observed increased reactivation of MHV68 in peritoneal cells of chronically infected LXRα−/− mice, we next sought to determine whether LXRα expression is necessary to limit lipogenic gene expression in infected animals.
At 28 days postinfection of BL6 mice, we detected a modest increase in LXRα expression and a modest decrease in LXRβ expression compared to naive mice; however, these changes were not statistically significant (Fig. 2A and B). Interestingly, peritoneal cells from uninfected LXRα−/− mice displayed reduced LXRβ expression, suggesting that one LXR isoform may mediate homeostatic expression of the other. Importantly, the LXR target genes encoding fatty acid synthesis enzymes (FADS2, SCD2, and ACC) were not derepressed in the absence of LXRα (Fig. 2C to E). Instead, peritoneal cells from infected BL6 and LXRα−/− mice showed comparable expression of each of the genes. Interestingly, expression of FADS2 and ACC was reduced in the peritoneum of infected BL6 and LXRα−/− mice compared to naive controls (Fig. 2C and E), unveiling an interesting LXRα-independent virus-host interaction that limits lipogenic activity of the host at the site of long-term viral latency. In contrast, the expression of SCD2 did not differ between uninfected and infected mice (Fig. 2D). Taken together, these data indicate that LXRα is not required to restrict lipogenic gene expression in peritoneal cells and that the observed hyperreactivation of MHV68 is likely not a result of increased fatty acid synthesis in the absence of LXRα.
FIG 2.

LXRα deficiency does not result in increased expression of proviral lipogenic genes during chronic MHV68 infection. Mice of the indicated genotypes were intraperitoneally infected with 500 PFU wild-type MHV68 for 28 days or mock infected. Peritoneal cells were isolated and subjected to qRT-PCR analysis to assess expression of the indicated genes. Gapdh, glyceraldehyde-3-phosphate dehydrogenase. Data are pooled from two independent experiments. Each symbol represents an individual animal, means and standard errors of the mean are shown. *, P < 0.05; **, P < 0.01; ****, P < .0001.
MHV68-specific T cell responses remain intact in LXRα−/− mice.
Peritoneal reactivation of MHV68 is largely controlled by virus-specific CD8+ T cells (29, 30). Having observed a consistent increase in MHV68 reactivation from the peritoneal cells of LXRα−/− mice, we tested the hypothesis that LXRα is important for the generation and recruitment of MHV68-specific CD8+ T cells during long-term infection. MHV68-specific CD8+ T cells were detected using major histocompatibility complex (MHC) class I tetramers that incorporated either an MHV68 orf6 or orf61 immunodominant epitope (Fig. 3A) (31). At 28 days postinfection, BL6 and LXRα−/− mice exhibited similar frequencies and total numbers of tetramer-positive, MHV68-specific CD8+ cells in the peritoneum (Fig. 3B to E), indicating that the recruitment of MHV68-specific CD8+ T cells was not regulated by LXRα. Similar populations of MHV68-specific CD8 T cells were also observed in the spleens of infected BL6 and LXRα−/− mice (Fig. 3F to I), indicating that the generation of MHV68-specific CD8 T cells was LXRα independent. Thus, the generation and recruitment of MHV68-specific CD8+ T cells remained intact in the absence of LXRα.
FIG 3.

MHV68-specific T cell responses remain intact in LXRα−/− mice. Mice of the indicated genotypes were intraperitoneally infected with 500 PFU wild-type MHV68 or mock infected. At 28 days postinfection, the mice were euthanized and splenocytes and peritoneal cells were processed into single-cell suspensions. Cells were stained with α-CD3, α-CD8, and α-CD44 antibodies, and MHC class I tetramers were loaded with the immunodominant peptides from the viral ORF6 (B, C, F, and G) or ORF61 (D, E, H, and I) proteins. (A) Representative flow plots from stained peritoneal cells. (B to I) Frequencies and total numbers of virus-specific CD8+ T cells for peritoneal cells (B to E) and splenocytes (F to I). The data are pooled from two independent experiments. Each symbol represents an individual animal; means and standard errors of the mean are shown.
LXRα does not regulate expression of IFN-γ by MHV68-specific T cells.
IFN-γ is a critical factor that controls MHV68 reactivation during chronic infection (30, 32–35), and both CD8+ and CD4+ MHV68-specific effector T cells can express IFN-γ following recognition of viral peptides in the context of MHC class I or class II, respectively. Importantly, treatment of primary murine macrophage cultures or mice with artificial LXR agonists has been reported to induce IFN-γ expression, suggesting that IFN-γ may be an LXR target gene (36). Therefore, having observed comparable numbers of MHV68-specific CD8+ T cells in the peritoneum in BL6 and LXRα−/− mice, along with poorly controlled viral reactivation, we next tested the hypothesis that LXRα deficiency compromises the ability of antiviral T cells to express IFN-γ, a critical antiviral cytokine.
Ex vivo restimulation of peritoneal cells (Fig. 4A to C) and splenocytes (Fig. 4D and E) with the combination of immunodominant class I MHV68 peptides (ORF6487–495 and ORF61524–531) generated similar frequencies and numbers of IFN-γ–expressing CD8+ T cells, regardless of LXRα expression. Further, the mean fluorescent intensities (MFI) of IFN-γ staining were similar in BL6 and LXRα−/− mouse peritoneal CD8+ T cells (Fig. 4F), indicating that the amount of IFN-γ protein expressed on a per-cell basis was not affected by LXRα deficiency of MHV68-specific CD8+ T cells.
FIG 4.

LXRα does not regulate expression of IFN-γ by MHV68-specific CD8+ T cells. Mice of the indicated genotypes were intraperitoneally infected with 500 PFU wild-type MHV68 or mock infected. At 28 days postinfection, the mice were euthanized, and peritoneal cells were isolated. The peritoneal cells were stimulated with 10 μg/ml ORF6487–495 and ORF61524–531 immunodominant peptides in the presence of 10 μg/ml brefeldin A for 6 h at 37°C. Cells were stained for flow cytometric analysis with α-CD3 and α-CD8 and intracellularly with α-IFN-γ antibodies. (A) Representative flow plots illustrating IFN-γ production. (B to E) Summarized frequencies and total numbers of IFN-γ-producing CD8+ T cells from the peritoneal cavity (B and C) and spleen (D and E). (F) MFI of IFN-γ signal from IFN-γ+ peritoneal CD8+ T cells. The data are pooled from at least two independent experiments. Each symbol represents an individual animal; means and standard errors of the mean are shown.
In addition to CD8+ T cells, CD4+ T cells are important for the control of chronic MHV68 infection (37–39) and are capable of producing IFN-γ in response to the MHC class II peptide gp15067–83 as long as 8 months after infection (40). Stimulation with gp15067–83 produced a measurable IFN-γ response from splenic CD4+ T cells (see Fig. S1A and B in the supplemental material), however, expression of IFN-γ from stimulated peritoneal CD4+ T cells was minimal (see Fig. S1C and D). Importantly, there was no difference in IFN-γ expression in response to gp15067–83 stimulation between BL6 and LXRα−/− T cells. In summary, LXRα deficiency had no effect on the ability of MHV68-specific CD4 and CD8 T cells to express IFN-γ following exposure to immunodominant viral epitopes.
LXRα expression promotes neither signaling nor antiviral activity of IFN-γ.
LXRα had no effect on generation, recruitment, or expression of antiviral IFN-γ by MHV68-specific T cells (Fig. 3 and 4). However, because T cells are not infected by MHV68, IFN-γ must engage receptors on infected cells in order to exert its antiviral activity. We and others have shown that peritoneal macrophages host a significant proportion of latent MHV68 following intraperitoneal inoculation of the virus (41–43). Furthermore, LXRα is robustly expressed in macrophages (13). Thus, we aimed to determine whether LXRα was necessary for optimal IFN signaling in primary macrophages.
Treatment of BL6 or LXRα−/− bone marrow-derived macrophages with type I or type II IFN induced similar levels of STAT1 phosphorylation in both genotypes at 30 min postexposure (Fig. 5A). IFN signaling culminates in expression of interferon-stimulated genes (ISGs) that function as antiviral effectors. Expression of MNDA, an ISG that attenuates MHV68 replication (44), was also induced to similar levels following 7 h of treatment of primary macrophages with either IFN-β or IFN-γ (Fig. 5B), indicating that LXRα expression did not promote IFN signaling in a primary cell type relevant for MHV68 infection.
FIG 5.

LXRα expression promotes neither signaling nor antiviral activity of IFN-γ. (A and B) Bone marrow-derived macrophages were treated with 10 U/ml IFN-β, 1 U/ml IFN-γ, or vehicle control for 30 min (A) or 7 h (B). (A) Total and phosphorylated STAT1 proteins were measured by Western blotting in two biological repeats for each condition. (B) MNDA expression was measured by qRT-PCR. The results of two independent experiments were pooled, and the symbols represent biological replicates. (C) Peritoneal cells isolated from mice of the indicated genotypes at 28 days postinfection were subjected to limiting-dilution analyses to assess the frequency of viral reactivation. Cultures were treated with 5 U/ml IFN-γ or vehicle control (Veh) immediately following plating to assess the ability of exogenous IFN-γ to suppress MHV68 reactivation. The data were pooled from two independent experiments. (D) Serum was collected before infection and at 14 and 28 days postinfection. IFN-γ concentrations were determined by ELISA. The data are pooled from at least two independent experiments, and each symbol represents an individual animal. (E and F) Mice of the indicated genotypes were intraperitoneally infected with MHV68 as for Fig. 1 or mock infected. At 28 days postinfection, RNA was isolated from peritoneal cells immediately following harvest. Each symbol represents an individual animal. The data are representative of the results of two independent experiments (A) or pooled from two or more independent experiments (B to F). Error bars represent the standard error of the mean.
Because primary macrophages derived from naive animals may not fully recapitulate the biology of peritoneal macrophages in long-term-infected mice, the antiviral activity of exogenous IFN-γ was assessed in peritoneal cells isolated from mice at 28 days post-MHV68 infection. Peritoneal cells were treated with exogenous IFN-γ immediately following explantation and for the duration of ex vivo reactivation. Consistent with previous reports (45, 46), exogenous IFN-γ efficiently suppressed MHV68 reactivation from peritoneal cells isolated from latently infected mice, and this suppression was independent of LXRα expression (Fig. 5C). In summary, LXRα did not potentiate either IFN signaling in primary macrophages or the antiviral activity of IFN-γ during MHV68 reactivation.
LXRα does not promote overall IFN responses in the peritoneal cells of chronically infected mice.
MHV68-specific CD4+ and CD8+ T cells represent a significant, but not the only, source of IFN-γ during chronic infection. In order to measure systemic IFN-γ production, levels of IFN-γ in serum were assessed at 14 and 28 days postinfection. As previously reported (47), IFN-γ levels in serum dramatically increased at 14 days postinfection of BL6 mice and returned to near baseline levels by 28 days postinfection (Fig. 5D). Interestingly, unlike what was observed in MHV68-specific T cells, systemic IFN-γ levels were decreased at both 14 and 28 days postinfection in LXRα−/− mice.
Despite decreased IFN-γ levels in serum at 14 days postinfection, MHV68 latency and reactivation were well controlled in LXRα−/− mice during early latency (Fig. 1A and B). This was likely due to the overall significant elevation in IFN-γ expression at 14 days postinfection, which is likely far above the threshold required to control MHV68 reactivation. However, because much lower levels of IFN-γ circulate in BL6 mice once an infection becomes chronic, a further decrease in IFN-γ observed in the absence of LXRα could translate into increased MHV68 reactivation. In addition to IFN-γ, MHV68 reactivation during long-term infection is also controlled by type I IFN (48), with the latter represented by a family of at least 17 different cytokines. Further, we showed that type I and type II IFNs cooperate to induce expression of antiviral ISGs in MHV68-infected primary macrophages and during acute MHV68 infection (49).
Because expression of ISGs represents the combined signaling outcome of type I and type II IFNs, with both types of IFN produced in infected mice, expression of MNDA and MX-2 was measured in peritoneal cells, the site of poorly controlled MHV68 reactivation, at 28 days postinfection. The ISGs were chosen based on their demonstrated ability to restrict replication of MHV68 in vitro (44). There was no difference in the expression of MNDA or MX-2 in peritoneal cells of infected BL6 mice and LXRα−/− mice (Fig. 5E and F). Other ISGs identified by Liu et al. (44) were also measured; however, their expression level was below our limit of detection (data not shown). Thus, the combined in vivo signaling levels of IFN pathways were similar in the peritonea of long-term-infected BL6 and LXRα−/− mice, suggesting that LXRα-dependent restriction of MHV68 reactivation is mediated by a mechanism that is independent of the known cellular and molecular factors that control chronic gammaherpesvirus infection.
LXRα expression dictates latent viral tropism in peritoneal cells.
To address the possibility that LXRα expression may contribute to the tropism or latent gene expression of MHV68, BL6 and LXRα−/− mice were infected with a recombinant MHV68 generated by Nealy et al. in which the viral gene ORF73 is fused to β-lactamase (28). In mice infected with MHV68.ORF73βla, infected cells expressing the ORF73 product mLANA also exhibit β-lactamase activity, which can be detected via the use of the fluorescent β-lactamase substrate CCF2-AM. Consistent with the initial characterization of the MHV68.ORF73βla virus (28), we were able to detect a small yet distinct population of mLANA+ cells in bulk splenocytes at 28 days postinfection (Fig. 6A). Further, nearly all mLANA+ splenocytes from BL6 and LXRα−/− mice were B cells, as exhibited by expression of the B cell marker CD19 (Fig. 6A). Thus, in concordance with limiting-dilution viral assays (Fig. 1), LXRα expression did not affect splenic MHV68 latency.
FIG 6.

LXRα expression dictates latent viral tropism or gene expression in peritoneal cells. Mice of the indicated genotypes were intraperitoneally infected with MHV68.ORF73βla for 28 days. Splenocytes and peritoneal cells were isolated and stained for CD19 and F4/80 surface markers. The cells were then loaded with the fluorescent β-lactamase substrate CCF2-AM for 1 h at room temperature. (A and B) Bulk splenocytes (A) and peritoneal cells (B) were examined for mLANA expression via flow cytometry, as indicated by the fluorescent signal from cleaved CCF2. (C) mLANA− and mLANA+ cells were gated and examined for CD19 and F4/80 expression. Data from two independent experiments are summarized.
Excitingly, using the MHV68.ORF73βla virus, we were able to detect mLANA+ peritoneal cells at 28 days postinfection (Fig. 6B), which to our knowledge has not been demonstrated. Consistent with previous reports defining macrophages as the predominant peritoneal viral reservoir (41, 46), approximately 70% of mLANA+ peritoneal cells from BL6 mice stained positive for F4/80, a marker highly expressed on murine peritoneal macrophages. As expected, the majority of F4/80-negative peritoneal cells that expressed mLANA in BL6 mice were CD19+ (∼20% to 25%), consistent with viral tropism for peritoneal B-1 and B-2 B cells (41). Remarkably, mLANA+ peritoneal cells from LXRα−/− mice exhibited considerably different F4/80 and CD19 staining, with the vast majority of mLANA-expressing peritoneal cells staining positive for F4/80 and very little CCF2 signal in CD19+ B cells (Fig. 6B and C). Thus, LXRα was critical to support MHV68 latent gene expression in peritoneal B cells. These observations correlated with decreased frequency of MHV68 DNA-positive cells in sorted B2 and B1b B cells from LXRα−/− mice (data not shown). Further, decreased MHV68 presence in peritoneal B cells of LXRα−/− mice was not due to decreased abundance of these cells in the peritoneum in LXRα−/− mice (Fig. 6B and C; see Fig. S2 in the supplemental material). In summary, LXRα expression promoted latent MHV68 infection of peritoneal B cells.
DISCUSSION
In this study, we demonstrate a novel antiviral role for LXRα in the control of chronic gammaherpesvirus infection. This is, to our knowledge, the first time a genetic approach, rather than synthetic LXR agonists, was used to define the physiological role of an individual LXR isoform during virus infection of an intact host. We found that LXRα was necessary to suppress MHV68 reactivation specifically in the peritoneal cavity, an anatomic site that houses primary effusion lymphoma driven by KSHV in humans. Increased gammaherpesvirus reactivation is associated with increased risk of viral lymphomagenesis (2–5); thus, attenuation of gammaherpesvirus reactivation by LXRα makes this host factor a potential candidate in the suppression of gammaherpesvirus-driven malignancies. In support of this hypothesis, expression of LXRα, but not the related LXRβ, is decreased in EBV-transformed primary human B cells (50). Intriguingly, our data rule out LXRα-dependent changes in the expression of lipogenic enzymes, generation and recruitment of virus-specific T cells, and IFN signaling, suggesting that LXRα suppresses long-term MHV68 reactivation via a novel antiviral mechanism.
We have previously demonstrated that deficiency of both LXR isoforms results in elevated viral replication in primary macrophage cultures (27). This antiviral activity was ascribed to the underappreciated ability of LXRs to repress the expression of target genes, thereby limiting the activity of proviral fatty acid and cholesterol synthesis pathways. However, a single deficiency of LXRα was not sufficient to increase the expression of fatty acid synthesis enzymes in peritoneal cells (Fig. 2), indicating compensation by the remaining LXRβ, at least with regard to the regulation of peritoneal lipid homeostasis.
MHV68-specific CD8+ and CD4+ T cells are critical for the control of chronic MHV68 infection and reactivation (29, 30, 37, 51–53), and T cell deficiency is a well-defined risk factor for gammaherpesvirus-driven malignancies (8, 38, 54). To probe trans-acting LXRα mechanisms, we examined the generation of MHV68-specific CD8+ and CD4+ effector T cells and their recruitment to the peritoneal cavity and found no LXRα-dependent effects. We also found that MHV68-specific CD8+ T cells expressed similar levels of IFN-γ regardless of LXRα expression. Our finding is in contrast to a published study that showed increased IFN-γ expression in T cell lines following exposure to artificial LXR agonists (36). The difference in our findings could be the result of differences between the experimental systems (T cell lines in vitro versus primary T cells in vivo) or the off-target effects of synthetic LXR agonists. However, we also found a decrease in the circulating levels of IFN-γ in LXRα−/− MHV68-infected animals, suggesting that LXRα might facilitate IFN-γ expression in other cell types in vivo. Importantly, this decrease in serum IFN-γ did not translate into attenuated IFN responses in the peritoneal cells, suggesting that either similar levels of IFN-γ are produced in the peritoneal cavities of BL6 and LXRα−/− mice (levels that were also below the level of detection of commercially available enzyme-linked immunosorbent assays [ELISAs]; data not shown) or that increased production of cytokines from the type I IFN family compensated for the decreased levels of IFN-γ in the peritoneum in LXRα-deficient mice.
The peritoneal cavity harbors numerous cell types during MHV68 infection, including CD4+ and CD8+ T cells, B-1 and B-2 B cells, macrophages, NK cells, dendritic cells, and granulocytes (55). At 16 days post-intraperitoneal infection, macrophages represent the greatest proportion of latently infected cells in the peritoneal cavity, followed by B1 and B2 B cells (41). Gammaherpesvirus tropism for macrophages is not an artifact of the MHV68 animal model; instead, macrophages represent a physiologically relevant cell type for human and rodent gammaherpesvirus infection (41–43, 56–59). As the infection progresses into its long-term stage, MHV68 tropism gradually shifts to peritoneal B cells; however, macrophages continue to house a considerable proportion of latent MHV68 in the peritoneum in long-term-infected animals. Latently infected macrophages account for the majority of peritoneal reactivation in BL6 mice during early latency (16 days postinfection), as determined by macrophage and B cell depletion studies (46). While the cell types responsible for reactivating MHV68 at later times during chronic infection remain unknown, it is likely that macrophages continue to contribute to the viral reactivation from the peritoneal cells of long-term-infected BL6 mice.
Fascinatingly, through the use of an MHV68 reporter virus, we discovered a unique LXRα-dependent shift in peritoneal mLANA expression (Fig. 6). This finding likely reflects a shift in viral tropism within the peritoneal cavity, as most latently infected cells express mLANA, at least at some point during latency (28). The shift in viral tropism is further supported by our unpublished data demonstrating a reduced frequency of latently infected B-1b and B-2 B cells in the peritonea of LXRα−/− mice. Increased frequency of latent MHV68 infection of LXRα−/− peritoneal macrophages undoubtedly contributes to increased MHV68 reactivation, based on previous studies (46). However, a 15% increase in latently infected macrophages is unlikely to fully account for the 10-fold increase in the frequency of MHV68 reactivation observed in LXRα−/− peritoneal cells at 28 days postinfection, suggesting that multiple mechanisms contribute to increased viral reactivation. In one plausible scenario, LXRα may function in a macrophage-intrinsic manner to restrict MHV68 reactivation. We entertained the possibility that LXRα directly interacts with the MHV68 genome to suppress viral gene expression. However, our bioinformatics analyses failed to uncover high-fidelity LXR binding sites in the MHV68 genome (data not shown), suggesting that direct LXR-based suppression of viral gene expression is unlikely. However, this does not rule out LXRα-dependent regulation of host gene expression that suppresses reactivation. Another intriguing possibility is that latently infected peritoneal B cells produce a signal that restricts viral reactivation from macrophages, a novel cross talk that will be tested in future studies. LXR activation with synthetic agonists is already known to limit signaling downstream of tumor necrosis factor (TNF), interleukin 1 (IL-1), Toll-like receptor 3 (TLR3), and TLR4 (reviewed by Bensinger and Tontonoz [60]). It would be beneficial to determine if these signaling changes occur in vivo during chronic gammaherpesvirus infection when the cells are exposed to only the endogenous LXR ligands and the implications of such changes for gammaherpesvirus infection. Further, defining the LXRα-regulated cellular and cytokine milieu may offer a novel mechanistic insight into not only the antiviral activity of this host factor, but also into the ability of LXRα to attenuate Listeria (15) and Mycobacterium (16) infections.
MATERIALS AND METHODS
Ethics statement.
All experimental manipulations of mice were approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin (MCW) (AUA971) and adhered to the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and the American Veterinary Medical Association Guidelines on Euthanasia.
Animals.
All mouse strains used in this study were maintained at the MCW animal facility. BL6 mice were originally purchased from Jackson Laboratories (Bar Harbor, ME). LXRα−/− mice (C57BL/6 genetic background) were a kind gift from Qing Miao (MCW). At 6 to 10 weeks of age, mice were intraperitoneally inoculated with 500 PFU of MHV68 (WUMS) in 200 μl serum-free Dulbecco’s modified Eagle medium (DMEM) or mock infected. The mice were euthanized by CO2 inhalation from a compressed gas source in a nonovercrowded chamber as mandated by the NIH Guide for the Care and Use of Laboratory Animals and the American Veterinary Medical Association Panel on Euthanasia. Mice were bled at the indicated time points via the submandibular route, and serum was isolated using BD Microtainer blood collection tubes (Becton, Dickinson and Company, Franklin Lakes, NJ).
Limiting-dilution assays.
The frequency of cells reactivating MHV68 ex vivo and the frequency of cells harboring viral genomes were determined as previously described (61). Briefly, to determine the frequency of cells reactivating virus ex vivo, serial 2-fold dilutions of splenocytes or peritoneal cell suspensions harvested from infected mice were plated onto monolayers of mouse embryonic fibroblasts (MEFs) at 24 replicates per dilution. In order to control for any preformed infectious virus, 2-fold serial dilutions of mechanically disrupted splenocytes or PECs were plated as described above. Cytopathic effect was scored at 21 days postplating. Because primary MEFs were used to amplify the virus, the sensitivity of the limiting-dilution reactivation assay was a single PFU of MHV68. Because the endpoint of viral amplification in MEFs was measured, the limiting-dilution reactivation assay was not susceptible to variability of titers released from primary cells upon viral reactivation ex vivo. Persistent viral replication was assessed similarly, with 6 × 106 splenocytes or 2.4 × 106 peritoneal cells suspended in hypotonic solution (primary cell culture medium diluted 1:3 in water), frozen at −80°, thawed (to release pre-formed infectious particles from the cells), and subjected to limiting-dilution assays as described above.
Flow cytometry.
Splenocytes and peritoneal exudate cells were suspended in fluorescence-activated cell sorter (FACS) buffer, prestained with Fc block, and incubated with the indicated antibodies on ice. When necessary, following surface staining, cells were fixed and permeabilized with BD Cytofix/Cytoperm (Fisher Scientific, Hampton, NH) for the detection of intracellular antigens. Data acquisition was performed on an LSR II flow cytometer (BD Biosciences, Sparks, MD) and analyzed using FlowJo software (Tree Star, Ashland, OR). The following antibodies were purchased from BioLegend (San Diego, CA): CD3ε (17A2), CD4 (GK1.5), CD8a (53-6.7), IFN-γ (XMG1.2), and CD44 (IM7). Allophycocyanin-conjugated MHC class I tetramers specific for MHV68 epitopes Db/ORF6487–495 (AGPHNDMEI) and Kb/ORF61524–531 (TSINFVKI) were obtained from the NIH Tetramer Core Facility (Emory University, Atlanta, GA). Restimulation of virus-specific CD4+ and CD8+ T cells was performed by incubating PECs or splenocytes with 10 μg/ml ORF6 and ORF61 or 2.5 μg/ml gp150 peptides (Thermo Fisher Scientific, Waltham, MA) in the presence of 10 μg/ml brefeldin A for 6 h at 37°C. Unstimulated cells were incubated with brefeldin A alone.
ELISA.
IFN-γ concentrations were determined using the IFN-γ ELISA MAX Deluxe set (BioLegend, San Diego, CA) according to the manufacturer’s instructions, using Nunc MaxiSorp flat-bottom plates (Thermo Fisher Scientific, Waltham, MA). Serum was diluted 1:2 in assay diluent prior to analysis. Horseradish peroxidase (HRP) enzymatic activity was stopped by the addition of 1 N HCl, and absorbance was read at 450 nm on a model 1420 Victor 3V multilabel plate reader (PerkinElmer, Waltham, MA).
Western analyses.
Samples were collected in Laemmli buffer (0.1 M Tris, 4% SDS, 4 mM EDTA, 100 mM beta-mercaptoethanol, 3.2 M glycerol with 0.05% bromophenol blue), boiled for 10 min, and subjected to SDS-PAGE. The protein was transferred to a polyvinylidene difluoride membrane (Immobilon; Millipore, Billerica, MA) using a Transblot transfer cell semidry system (Bio-Rad, Hercules, CA). The membranes were blocked with 5% bovine serum albumin (BSA) or 5% nonfat milk in TBST buffer (150 mM NaCl, 10 mM Tris, and 0.2% Tween) for 1 hour at room temperature. The membranes were incubated with anti-pSTAT1 (1:500 in 1% BSA; clone D4A7; Cell Signaling Technology, Boston, MA), anti-STAT1 (1:10,000 in 5% nonfat milk; clone E-23; Santa Cruz Biotechnology, Santa Cruz, CA), or anti-β-actin (1:50,000 in 5% nonfat milk; clone 13E5; Cell Signaling Technology, Boston, MA) antibody at 4°C overnight. The membranes were washed three times for 10 min each time, followed by incubation with secondary HRP-conjugated antibodies (1:20,000; Jackson ImmunoResearch, West Grove, PA) for 1 hour at room temperature. The membranes were again washed three times for 10 min each time and then developed with Immobilon Western chemiluminescent HRP substrate (Millipore Sigma, Burlington, MA) and imaged with an Azure C600 imaging system (Azure Biosystems, Dublin, CA).
Primary cell culture.
Bone marrow was harvested from male and female mice that were between 3 and 10 weeks of age. Primary bone marrow-derived macrophages were generated as previously described (62). At least two independently derived batches of macrophages were used in each experiment. MEFs were generated from embryonic day 12 (E12) to E14 embryos and pooled at early passages.
Gene expression studies.
Total RNA was harvested, reverse transcribed, and analyzed by quantitative reverse transcription (qRT)-PCR as previously described (63). cDNA was assessed in triplicate, along with corresponding reactions minus reverse transcriptase, by real-time PCR using a CFX Connect system (Bio-Rad, Hercules, CA). Analysis was performed using primers listed in Table 1. These primers were validated for sensitivity and specificity using bioinformatics and wet laboratory approaches; the slope and linear range were established for each primer pair, and all the primers were utilized within their linear ranges.
TABLE 1.
Primers used for qRT-PCR analyses of gene expression
| Gene | Primer sequence (5′–3′) |
|
|---|---|---|
| Forward | Reverse | |
| LXRα | TAAGGGAGAGTCAACAGG | GGTCAACAAGGTCTTCAG |
| LXRβ | GCAGTTGGCACTAGAAG | GGTAGGCTGAGGTGTAA |
| ACC | GGAGCACCTCAAGCAGATATT | GGCTCTGACTTCTCCGTATTG |
| FADS2 | TCTCAGATCACCGAGGACTT | GGACAGGAGGAGAAAGAAGAAC |
| SCD2 | ACCTTCCTCACTCAGAGATACA | AAGCCTGGGAGGGATAAGA |
| MNDA | GGAGTGGACAATGGCACAACATCA | TGACGAAACTGTGGTCTCCACACA |
| MX-2 | AGCAGAGTGACACAAGCGAGAAGA | AGCCCTTCTGTCCCTGAATCACAA |
MHV68.ORF73βla detection.
BL6 and LXRα−/− mice were infected intraperitoneally with 500 PFU MHV68.ORF73βla. Mice were euthanized at 28 dpi, and then splenocytes and peritoneal cells were isolated and pooled from each group (4 or 5 mice/group). Approximately 2 × 107 cells from each group of mice were prestained with Fc block and then stained with αF4/80-allophycocyanin (APC)-Cy7 and αCD19-APC (BioLegend, San Diego, CA) for 30 min on ice. The cells were washed twice and then loaded with CCF2-AM (GeneBLAzer kit; ThermoFisher Scientific, Waltham, MA) at room temperature for 1 h. The cells were again washed twice and then suspended in FACS buffer and analyzed via flow cytometry.
Statistical analyses.
Statistical analyses were performed using Student's t test (Prism; GraphPad Software, Inc.).
Supplementary Material
ACKNOWLEDGMENTS
Funding for this study was provided by CA183593 and CA203923 (V.L.T.) and by 18PRE33960455 (P.T.L.).
We thank Kyle Stoltz for his support of this study. We thank the members of the Corbett and Cui laboratory for lively discussions of the study and Bonnie Dittel and Daisy Sahoo for their thoughtful insights.
P.T.L. and V.L.T. wrote the manuscript. P.T.L., C.N.J., and V.L.T. designed the experiments. P.T.L., C.N.J., E.J.D., and K.E.J. performed and analyzed experiments.
We declare no competing interests.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.02071-18.
REFERENCES
- 1.Cesarman E. 2014. Gammaherpesviruses and lymphoproliferative disorders. Annu Rev Pathol 9:349–372. doi: 10.1146/annurev-pathol-012513-104656. [DOI] [PubMed] [Google Scholar]
- 2.Campbell TB, Borok M, Gwanzura L, MaWhinney S, White IE, Ndemera B, Gudza I, Fitzpatrick L, Schooley RT. 2000. Relationship of human herpesvirus 8 peripheral blood virus load and Kaposi's sarcoma clinical stage. AIDS 14:2109–2116. [DOI] [PubMed] [Google Scholar]
- 3.Meerbach A, Wutzler P, Hafer R, Zintl F, Gruhn B. 2008. Monitoring of Epstein-Barr virus load after hematopoietic stem cell transplantation for early intervention in post-transplant lymphoproliferative disease. J Med Virol 80:441–454. doi: 10.1002/jmv.21096. [DOI] [PubMed] [Google Scholar]
- 4.Feng WH, Cohen JI, Fischer S, Li L, Sneller M, Goldbach-Mansky R, Raab-Traub N, Delecluse HJ, Kenney SC. 2004. Reactivation of latent Epstein-Barr virus by methotrexate: a potential contributor to methotrexate-associated lymphomas. J Natl Cancer Inst 96:1691–1702. doi: 10.1093/jnci/djh313. [DOI] [PubMed] [Google Scholar]
- 5.Orlandi E, Paulli M, Viglio A, Pagnucco G, Riboni R, Baldanti F, Lazzarino M. 2001. Epstein-Barr virus-positive aggressive lymphoma as a consequence of immunosuppression after multiple salvage treatments for follicular lymphoma. Br J Haematol 112:373–376. [DOI] [PubMed] [Google Scholar]
- 6.Efstathiou S, Ho YM, Hall S, Styles CJ, Scott SD, Gompels UA. 1990. Murine herpesvirus 68 is genetically related to the gammaherpesviruses Epstein-Barr virus and herpesvirus saimiri. J Gen Virol 71:1365–1372. doi: 10.1099/0022-1317-71-6-1365. [DOI] [PubMed] [Google Scholar]
- 7.Virgin HW, Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto AJ, Speck SH. 1997. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J Virol 71:5894–5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tarakanova VL, Suarez FS, Tibbetts SA, Jacoby M, Weck KE, Hess JH, Speck SH, Virgin HW. 2005. Murine gammaherpesvirus 68 infection induces lymphoproliferative disease and lymphoma in BALB b2 microglobulin deficient mice. J Virol 79:14668–14679. doi: 10.1128/JVI.79.23.14668-14679.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Repa JJ, Mangelsdorf DJ. 2000. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu Rev Cell Dev Biol 16:459–481. doi: 10.1146/annurev.cellbio.16.1.459. [DOI] [PubMed] [Google Scholar]
- 10.Bookout AL, Jeong Y, Downes M, Ruth TY, Evans RM, Mangelsdorf DJ. 2006. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell 126:789–799. doi: 10.1016/j.cell.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hong C, Tontonoz P. 2014. Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat Rev Drug Discov 13:433–444. doi: 10.1038/nrd4280. [DOI] [PubMed] [Google Scholar]
- 12.Repa JJ, Turley SD, Lobaccaro JA, Medina J, Li L, Lustig K, Shan B, Heyman RA, Dietschy JM, Mangelsdorf DJ. 2000. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science 289:1524–1529. [DOI] [PubMed] [Google Scholar]
- 13.Costet P, Luo Y, Wang N, Tall AR. 2000. Sterol-dependent transactivation of theABC1 promoter by the liver X receptor/retinoid X receptor. J Biol Chem 275:28240–28245. doi: 10.1074/jbc.M003337200. [DOI] [PubMed] [Google Scholar]
- 14.Schwartz K, Lawn RM, Wade DP. 2000. ABC1 gene expression and ApoA-I-mediated cholesterol efflux are regulated by LXR. Biochem Biophys Res Commun 274:794–802. doi: 10.1006/bbrc.2000.3243. [DOI] [PubMed] [Google Scholar]
- 15.Joseph SB, Bradley MN, Castrillo A, Bruhn KW, Mak PA, Pei L, Hogenesch J, O'Connell RM, Cheng G, Saez E, Miller JF, Tontonoz P. 2004. LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell 119:299–309. doi: 10.1016/j.cell.2004.09.032. [DOI] [PubMed] [Google Scholar]
- 16.Korf H, Vander BS, Romano M, Steffensen KR, Stijlemans B, Gustafsson JA, Grooten J, Huygen K. 2009. Liver X receptors contribute to the protective immune response against Mycobacterium tuberculosis in mice. J Clin Invest 119:1626–1637. doi: 10.1172/JCI35288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui G, Qin X, Wu L, Zhang Y, Sheng X, Yu Q, Sheng H, Xi B, Zhang JZ, Zang YQ. 2011. Liver X receptor (LXR) mediates negative regulation of mouse and human Th17 differentiation. J Clin Invest 121:658–670. doi: 10.1172/JCI42974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, Shih R, Parks JS, Edwards PA, Jamieson BD, Tontonoz P. 2008. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell 134:97–111. doi: 10.1016/j.cell.2008.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bocchetta S, Maillard P, Yamamoto M, Gondeau C, Douam F, Lebreton S, Lagaye S, Pol S, Helle F, Plengpanich W, Guerin M, Bourgine M, Michel ML, Lavillette D, Roingeard P, Le Goff W, Budkowska A. 2014. Up-regulation of the ATP-binding cassette transporter A1 inhibits hepatitis C virus infection. PLoS One 9:e92140. doi: 10.1371/journal.pone.0092140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zeng J, Wu Y, Liao Q, Li L, Chen X, Chen X. 2012. Liver X receptors agonists impede hepatitis C virus infection in an Idol-dependent manner. Antiviral Res 95:245–256. doi: 10.1016/j.antiviral.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 21.Jiang H, Badralmaa Y, Yang J, Lempicki R, Hazen A, Natarajan V. 2012. Retinoic acid and liver X receptor agonist synergistically inhibit HIV infection in CD4+ T cells by up-regulating ABCA1-mediated cholesterol efflux. Lipids Health Dis 11:69. doi: 10.1186/1476-511X-11-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanley TM, Viglianti GA. 2011. Nuclear receptor signaling inhibits HIV-1 replication in macrophages through multiple trans-repression mechanisms. J Virol 85:10834–10850. doi: 10.1128/JVI.00789-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dubrovsky L, Van DR, Senina S, Guendel I, Pushkarsky T, Sviridov D, Kashanchi F, Bukrinsky M. 2012. Liver X receptor agonist inhibits HIV-1 replication and prevents HIV-induced reduction of plasma HDL in humanized mouse model of HIV infection. Biochem Biophys Res Commun 419:95–98. doi: 10.1016/j.bbrc.2012.01.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sheng XX, Sun YJ, Zhan Y, Qu YR, Wang HX, Luo M, Liao Y, Qiu XS, Ding C, Fan HJ, Mao X. 2016. The LXR ligand GW3965 inhibits Newcastle disease virus infection by affecting cholesterol homeostasis. Arch Virol 161:2491–2501. doi: 10.1007/s00705-016-2950-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papageorgiou AP, Heggermont W, Rienks M, Carai P, Langouche L, Verhesen W, De Boer RA, Heymans S. 2015. Liver X receptor activation enhances CVB3 viral replication during myocarditis by stimulating lipogenesis. Cardiovasc Res 107:78–88. doi: 10.1093/cvr/cvv157. [DOI] [PubMed] [Google Scholar]
- 26.Ramezani A, Dubrovsky L, Pushkarsky T, Sviridov D, Karandish S, Raj DS, Fitzgerald ML, Bukrinsky M. 2015. Stimulation of liver X receptor has potent anti-HIV effects in a humanized mouse model of HIV infection. J Pharmacol Exp Ther 354:376–383. doi: 10.1124/jpet.115.224485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lange P, Schorl C, Sahoo D, Tarakanova V. 2018. Liver X receptors suppress activity of cholesterol and fatty acid synthesis pathways to oppose gammaherpesvirus replication. mBio 9:e01115-18. doi: 10.1128/mBio.01115-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nealy MS, Coleman CB, Li H, Tibbetts SA. 2010. Use of a virus-encoded enzymatic marker reveals that a stable fraction of memory B cells expresses latency-associated nuclear antigen throughout chronic gammaherpesvirus infection. J Virol 84:7523–7534. doi: 10.1128/JVI.02572-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Braaten DC, Sparks-Thissen RL, Kreher S, Speck SH, Virgin HW. 2005. An optimized CD8+ T-cell response controls productive and latent gammaherpesvirus infection. J Virol 79:2573–2583. doi: 10.1128/JVI.79.4.2573-2583.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tibbetts SA, Van Dyk L, Speck SH, Virgin HW. 2002. Immune control of the number and reactivation phenotype of cells latently infected with a gamma-herpesvirus. J Virol 76:7125–7132. doi: 10.1128/JVI.76.14.7125-7132.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freeman ML, Lanzer KG, Cookenham T, Peters B, Sidney J, Wu TT, Sun R, Woodland DL, Sette A, Blackman MA. 2010. Two kinetic patterns of epitope-specific CD8 T-cell responses following murine gammaherpesvirus 68 infection. J Virol 84:2881–2892. doi: 10.1128/JVI.02229-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Evans AG, Moser JM, Krug LT, Pozharskaya V, Mora AL, Speck SH. 2008. A gammaherpesvirus-secreted activator of V beta 4(+) CD8(+) T cells regulates chronic infection and immunopathology. J Exp Med 205:669–684. doi: 10.1084/jem.20071135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dutia BM, Clarke CJ, Allen DJ, Nash AA. 1997. Pathological changes in the spleens of gamma interferon receptor-deficient mice infected with murine gammaherpesvirus: a role for CD8 T cells. J Virol 71:4278–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sparks-Thissen RL, Braaten DC, Hildner K, Murphy TL, Murphy KM, Virgin HW. 2005. CD4 T cell control of acute and latent murine gammaherpesvirus infection requires IFN gamma. Virology 338:201–208. doi: 10.1016/j.virol.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 35.Dal Canto AJ, Virgin HW, Speck SH. 2000. Ongoing viral replication is required for gammaherpesvirus 68-induced vascular damage. J Virol 74:11304–11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Q, Ma X, Chen Y, Zhang L, Jiang M, Li X, Xiang R, Miao R, Hajjar DP, Duan Y, Han J. 2014. Identification of interferon-gamma as a new molecular target of liver X receptor. Biochem J 459:345–354. doi: 10.1042/BJ20131442. [DOI] [PubMed] [Google Scholar]
- 37.Sparks-Thissen RL, Braaten DC, Kreher S, Speck SH, Virgin HW. 2004. An optimized CD4 T-cell response can control productive and latent gammaherpesvirus infection. J Virol 78:6827–6835. doi: 10.1128/JVI.78.13.6827-6835.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liang X, Crepeau RL, Zhang W, Speck SH, Usherwood EJ. 2013. CD4 and CD8 T cells directly recognize MHV-68 immortalized cells and prevent tumor outgrowth. J Virol 87:6051–6054. doi: 10.1128/JVI.00375-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Usherwood EJ, Ross AJ, Allen DJ, Nash AA. 1996. Murine gammaherpesvirus-induced splenomegaly: a critical role for CD4 T cells. J Gen Virol 77:627–630. doi: 10.1099/0022-1317-77-4-627. [DOI] [PubMed] [Google Scholar]
- 40.Flano E, Woodland DL, Blackman MA, Doherty PC. 2001. Analysis of virus-specific CD4(+) T cells during long-term gammaherpesvirus infection. J Virol 75:7744–7748. doi: 10.1128/JVI.75.16.7744-7748.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rekow MM, Darrah EJ, Mboko WP, Lange PT, Tarakanova VL. 2016. Gammaherpesvirus targets peritoneal B-1 B cells for long-term latency. Virology 492:140–144. doi: 10.1016/j.virol.2016.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H, Ikuta K, Sixbey JW, Tibbetts SA. 2008. A replication-defective gammaherpesvirus efficiently establishes long-term latency in macrophages but not B cells in vivo. J Virol 82:8500–8508. doi: 10.1128/JVI.00186-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weck KE, Kim SS, Virgin HW, Speck SH. 1999. Macrophages are the major reservoir of latent murine gammaherpesvirus 68 in peritoneal cells. J Virol 73:3273–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu SY, Sanchez DJ, Aliyari R, Lu S, Cheng G. 2012. Systematic identification of type I and type II interferon-induced antiviral factors. Proc Natl Acad Sci U S A 109:4239–4244. doi: 10.1073/pnas.1114981109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steed A, Barton ES, Tibbetts SA, Popkin DL, Lutzke ML, Rochford R, Virgin HW. 2006. Gamma interferon blocks gammaherpesvirus reactivation from latency. J Virol 80:192–200. doi: 10.1128/JVI.80.1.192-200.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steed A, Buch T, Waisman A, Virgin HW. 2007. Interferon gamma blocks gammaherpesvirus reactivation from latency in a cell type specific manner. J Virol 81:6134–6140. doi: 10.1128/JVI.00108-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barton ES, White DW, Cathelyn JS, Brett-McClellan KA, Engle M, Diamond MS, Miller VL, Virgin HW. 2007. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature 447:326–329. doi: 10.1038/nature05762. [DOI] [PubMed] [Google Scholar]
- 48.Barton ES, Lutzke ML, Rochford R, Virgin HW. 2005. Alpha/beta interferons regulate murine gammaherpesvirus latent gene expression and reactivation from latency. J Virol 79:14149–14160. doi: 10.1128/JVI.79.22.14149-14160.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mboko WP, Rekow MM, Ledwith MP, Lange PT, Schmitz KE, Anderson S, Tarakanova VL. 2016. IRF-1 and type I interferon cooperate to control acute gammaherpesvirus infection. J Virol 91:e01444-16. doi: 10.1128/JVI.01444-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yenamandra SP, Klein G, Kashuba E. 2009. Nuclear receptors and their role in Epstein-Barr virus induced B cell transformation. Exp Oncol 31:67–73. [PubMed] [Google Scholar]
- 51.Flano E, Woodland DL, Blackman MA. 1999. Requirement for CD4+ T cells in V beta 4+CD8+ T cell activation associated with latent murine gammaherpesvirus infection. J Immunol 163:3403–3408. [PubMed] [Google Scholar]
- 52.Ehtisham S, Sunil-Chandra NP, Nash AA. 1993. Pathogenesis of murine gammaherpesvirus infection in mice deficient in CD4 and CD8 T cells. J Virol 67:5247–5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McClellan JS, Tibbetts SA, Gangappa S, Brett KA, Virgin HW. 2004. Critical role of CD4 T cells in an antibody-independent mechanism of vaccination against gamma-herpesvirus latency. J Virol 78:6836–6845. doi: 10.1128/JVI.78.13.6836-6845.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Latour S, Winter S. 2018. Inherited immunodeficiencies with high predisposition to Epstein-Barr virus-driven lymphoproliferative diseases. Front Immunol 9:1103. doi: 10.3389/fimmu.2018.01103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kulinski JM, Darrah EJ, Broniowska KA, Mboko WP, Mounce BC, Malherbe LP, Corbett JA, Gauld SB, Tarakanova VL. 2015. ATM facilitates mouse gammaherpesvirus reactivation from myeloid cells during chronic infection. Virology 483:264–274. doi: 10.1016/j.virol.2015.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shimakage M, Kimura M, Yanoma S, Ibe M, Yokota S, Tsujino G, Kozuka T, Dezawa T, Tamura S, Ohshima A, Yutsudo M, Hakura A. 1999. Expression of latent and replicative-infection genes of Epstein-Barr virus in macrophage. Arch Virol 144:157–166. [DOI] [PubMed] [Google Scholar]
- 57.Gregory SM, Wang L, West JA, Dittmer DP, Damania B. 2012. Latent Kaposi's sarcoma-associated herpesvirus infection of monocytes downregulates expression of adaptive immune response costimulatory receptors and proinflammatory cytokines. J Virol 86:3916–3923. doi: 10.1128/JVI.06437-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rappocciolo G, Jenkins FJ, Hensler HR, Piazza P, Jais M, Borowski L, Watkins SC, Rinaldo CR. Jr.. 2006. DC-SIGN is a receptor for human herpesvirus 8 on dendritic cells and macrophages. J Immunol 176:1741–1749. [DOI] [PubMed] [Google Scholar]
- 59.Wang LX, Kang G, Kumar P, Lu W, Li Y, Zhou Y, Li Q, Wood C. 2014. Humanized-BLT mouse model of Kaposi's sarcoma-associated herpesvirus infection. Proc Natl Acad Sci U S A 111:3146–3151. doi: 10.1073/pnas.1318175111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bensinger SJ, Tontonoz P. 2008. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature 454:470. doi: 10.1038/nature07202. [DOI] [PubMed] [Google Scholar]
- 61.Weck KE, Barkon ML, Yoo LI, Speck SH, Virgin HW. 1996. Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J Virol 70:6775–6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tarakanova VL, Leung-Pineda V, Hwang S, Yang C-W, Matatall K, Basson M, Sun R, Piwnica-Worms H, Sleckman BP, Virgin HW. 2007. Gamma-herpesvirus kinase actively initiates a DNA damage response by inducing phosphorylation of H2AX to foster viral replication. Cell Host Microbe 1:275–286. doi: 10.1016/j.chom.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mboko WP, Mounce BC, Emmer J, Darrah E, Patel SB, Tarakanova VL. 2014. Interferon regulatory factor-1 restricts gammaherpesvirus replication in primary immune cells. J Virol 88:6993–7004. doi: 10.1128/JVI.00638-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.