

Marburg virus (MARV) is a virus similar to Ebola virus and also causes a hemorrhagic disease which is highly lethal. In contrast to EBOV, only a few vaccines have been developed against MARV, and researchers do not understand what kind of immune responses are required to protect from MARV. Here we show that antibodies directed against MARV after application of our vaccine protect in an animal system but fail to neutralize the virus in a widely used virus neutralization assay against MARV. This newly discovered activity needs to be considered more when analyzing MARV vaccines or infections.
KEYWORDS: ADCC, Marburg virus, antibody function, filovirus, immunization, rabies virus, vaccine
ABSTRACT
Marburg virus (MARV) is a filovirus related to Ebola virus (EBOV) associated with human hemorrhagic disease. Outbreaks are sporadic and severe, with a reported case mortality rate of upward of 88%. There is currently no antiviral or vaccine available. Given the sporadic nature of outbreaks, vaccines provide the best approach for long-term control of MARV in regions of endemicity. We have developed an inactivated rabies virus-vectored MARV vaccine (FILORAB3) to protect against Marburg virus disease. Immunogenicity studies in our labs have shown that a Th1-biased seroconversion to both rabies virus and MARV glycoproteins (GPs) is beneficial for protection in a preclinical murine model. As such, we adjuvanted FILORAB3 with glucopyranosyl lipid adjuvant (GLA), a Toll-like receptor 4 agonist, in a squalene-in-water emulsion. Across two different BALB/c mouse challenge models, we achieved 92% protection against murine-adapted Marburg virus (ma-MARV). Although our vaccine elicited strong MARV GP antibodies, it did not strongly induce neutralizing antibodies. Through both in vitro and in vivo approaches, we elucidated a critical role for NK cell-dependent antibody-mediated cellular cytotoxicity (ADCC) in vaccine-induced protection. Overall, these findings demonstrate that FILORAB3 is a promising vaccine candidate for Marburg virus disease.
IMPORTANCE Marburg virus (MARV) is a virus similar to Ebola virus and also causes a hemorrhagic disease which is highly lethal. In contrast to EBOV, only a few vaccines have been developed against MARV, and researchers do not understand what kind of immune responses are required to protect from MARV. Here we show that antibodies directed against MARV after application of our vaccine protect in an animal system but fail to neutralize the virus in a widely used virus neutralization assay against MARV. This newly discovered activity needs to be considered more when analyzing MARV vaccines or infections.
INTRODUCTION
Filoviruses are filamentous, enveloped viruses that can cause highly lethal hemorrhagic fever in both humans and nonhuman primates (NHPs) (1). Three major genera comprise the filovirus family: Ebolavirus, Marburgvirus, and the newly discovered Cuevavirus. While the Ebolavirus genus contains five species (Zaire ebolavirus [EBOV], Sudan ebolavirus [SUDV], Bundibugyo ebolavirus [BDBV], Reston ebolavirus [RESTV], and Taï Forest ebolavirus [TAFV]), the Marburgvirus genus contains only one, the eponymously named Marburg marburgvirus (MARV) (2). MARV is further subdivided on the basis of the different isolates, including Ci67, Musoke, and Angola, and of a more distinct lineage, Ravn virus (RAVV).
MARV was the first filovirus to be identified when it sickened laboratory workers handling tissue from infected nonhuman primates originating from Uganda in 1967 (1). MARV has since reemerged at least 8 times and has been imported to the United States and Europe by travelers who became infected in Africa (1). The MARV Angola subspecies emerged in 2004 and caused the largest MARV outbreak known to date, with a case fatality rate of 88% (3, 4).
The glycoprotein (GP) of filoviruses mediates attachment and entry of the viruses into target cells. In infected cells, the GP precursor protein is cleaved during proteolytic transport from the endoplasmic reticulum to the Golgi apparatus by host furin protease into two distinct subunits that associate via disulfide bonds (5). In the native MARV GP structure, three monomeric GP1-GP2 pairs come together to form the GP trimer on the viral surface. GP1 is shielded by two heavily glycosylated domains (the glycan cap and the mucin-like domain), which restricts access to the putative receptor binding site and facilitates viral immune evasion by epitope masking (6). The GP2 subunit contains part of the mucin-like domain, the transmembrane domain to anchor GP into the viral membrane, and the fusion machinery necessary to trigger viral entry into cells (7).
Recent studies in nonhuman primates have demonstrated that passive administration of polyclonal sera against MARV can provide effective postexposure therapy for MARV infection (8, 9). Monoclonal antibody (MAb) therapies are also currently being developed for postexposure prophylaxis (10, 11). However, postexposure prophylaxis alone is not enough to combat the threat of Marburg virus disease (MVD), which may spread quickly once an outbreak has occurred. Preventative treatment with vaccines is strongly needed, especially considering recent ring vaccination methods to strategically limit the spread of Ebola virus infection (12). Various MARV vaccine approaches are under way. MARV GP DNA-based vaccines are safe but have low rates of seroconversion against GP antigen in phase I clinical trials in humans (13, 14). MARV virus-like particles (3 doses plus adjuvant) fully protected cynomolgus macaques against MARV and heterologous Ravn virus (RAVV) lethal aerosol exposure (15). Preclinical work in nonhuman primates using either heterologous multivalent adenovirus type 26 (Ad26)-Ad35 prime-boost vaccination regimens or vesicular stomatitis virus (VSV) as a platform has shown protection against MARV (range, 75% to 100% protection) (16–18).
Despite these advances, potential mechanisms of vaccine-induced protection against MARV are still poorly understood, and there is still currently no approved antiviral or vaccine available to treat MARV disease. For a vaccine against MARV to be successful, it should provide long-lasting immunity. Exposures are spontaneous and unpredictable in regions of endemicity, so long-term immunity in at-risk populations would diminish spillover events to humans from the viral reservoir and could curb subsequent human-to-human transmission to greatly limit the spread of an epidemic. To understand the factors that influence long-term immunity, it is necessary to define the immune response required to achieve prolonged protection against MARV. A recent study reported that survivors of MARV infection develop multivariate CD4+ T cell responses but limited CD8+ T cell responses, suggesting that CD8+ T cells may not be required for a protective response against MARV (19). However, different vaccine platforms may invoke different mechanisms to elicit protection and may not necessarily need to mimic a natural infection (20). Interestingly, neutralizing antibody (nAb) responses in survivors of MARV infection are rare and diminish rapidly over time (19, 21).
In this study, we evaluated our inactivated bivalent rabies virus (RABV)-vectored MARV vaccine, FILORAB3, as a promising human vaccine candidate for Marburg virus and elucidated the mechanism of protection by determining the parameters of optimal vaccine efficacy. The vaccine is a chemically deactivated purified rabies virus virion that expresses and incorporates both RABV G and MARV GP in the virion. We have chosen to use RABV as our vector for MARV GP for the following reasons: (i) due to its relative rarity, preexisting immunity should not be a widespread problem; (ii) target populations should be susceptible to successful immunization; (iii) the RABV vaccine strain used (SAD-B19) is highly attenuated and contains a mutation that abolishes neurovirulence; (iv) rabies vaccine vectors replicate efficiently in Vero cells, which are qualified for production of vaccines for human use; (v) rabies virus replicates in the cytoplasm, so integration into the host genome is not a concern; (vi) killed rabies virus virions are replication deficient; (vii) recombination events involving the rabies virus genome are extremely rare; (viii) insertion of foreign genes into the rabies virus genome is stable; and (ix) the rabies virus genome has only five genes and the proteins that they encode are not immunosuppressive toward the host (22).
Here, we demonstrate in a preclinical murine model that vaccinated animals show Th1-biased seroconversion to both RABV and MARV glycoproteins. Furthermore, when the mice were immunized with a single dose of adjuvanted vaccine, they achieved full protection from lethal challenge with mouse-adapted MARV (ma-MARV). Although our vaccine elicited high titers of specific antibodies, there was no strong induction of neutralizing antibodies, even after challenge. As such, we explored the role of antibody-dependent cellular cytotoxicity (ADCC) in the protection against MARV challenge. For filoviruses as well as other viruses, neutralization in vitro does not necessarily correlate with protection in vivo. Nonneutralizing antibodies are known to confer protection by ADCC, phagocytosis, prevention of virus budding, and other mechanisms (10, 23–30). Through both an in vitro approach and an in vivo approach, we identified an important role for ADCC and other nonneutralizing antibody functions in vaccine-induced immunity by FILORAB3.
RESULTS
Generation of a rabies vaccine encoding a Marburg virus GP.
Recombinant rabies virus expressing MARV glycoprotein (i.e., FILORAB3) was constructed by inserting a gene composed of rabies virus transcriptional start and stop sequences flanking the codon-optimized MARV Angola strain GP between the nucleoprotein (N) and phosphoprotein (P) genes of BSNP333, an attenuated parental rabies virus vector derived from the SAD-B19 vaccine strain (31–34). Based on our previous studies showing that codon optimization can increase the level of foreign glycoprotein expression and incorporation into budding rabies virus virions (35, 36), we utilized a codon-optimized version of MARV GP (Angola strain). The vector BNSP333 also contains an arginine-to-glutamine mutation in amino acid position 333 of the rabies virus glycoprotein, which further reduces neurovirulence and thus increases its safety profile (37). For our studies, we generated both live, replication-competent, and chemically inactivated versionsj of the recombinant virus (Fig. 1). FILORAB3 was inactivated by treatment with β-propiolactone (BPL), an alkylating agent frequently used to inactivate viruses, including RABV (38).
FIG 1.

FILORAB3 vaccine constructs. Schematic of RABV vaccine constructs expressing the codon-optimized Angola strain MARV GP (FILORAB3) used for immunizations. MARV GP was inserted between the N and P genes of the negative-sense RNA genome of RABV. The mutation at position 333 in RABV G that attenuates neurovirulence is depicted. INAC, inactivated.
Infectious recombinant virus was recovered by transfecting into BSR mammalian cells the FILORAB3 cDNA along with support plasmids individually bearing each of the RABV genes under the control of a T7 promoter and a plasmid-expressing T7 polymerase, as previously described (7, 37). To evaluate the in vitro replication potential of live FILORAB3, BSR cells were infected in a multistep growth curve at a multiplicity of infection (MOI) of 0.1 with the initial passages of recovered live virus in parallel with the parental vector, BNSP333, and a previously developed recombinant rabies virus bearing the Ebola virus Mayinga strain glycoprotein (i.e., FILORAB1) (Fig. 2). Viral titers were assessed at several time points for a duration of 96 h postinfection. While the appearance of viral progeny was not different between the parental strain and recombinant virus, FILORAB3 did grow to a lower overall titer by the terminal time point of 96 h postinfection, similar to FILORAB1.
FIG 2.
Growth kinetics of recombinant RABV/MARV (FILORAB3). Multistep growth curve comparing the kinetics of live virus replication of FILORAB3 (red), the parental RABV vaccine vector (BNSP333; blue), and recombinant rabies virus expressing the codon-optimized Ebola virus glycoprotein (FILORAB1, green) in the recovery cell line (BSR cells). BSR cells were infected at a multiplicity of infection of 0.1, and supernatant was collected at 0, 24, 48, 72, and 96 h. Virus titers were measured by a focus-forming assay (y axis) and plotted through time (x axis).
Expression of MARV GP by recombinant RABV vaccine.
To confirm efficient coexpression of both the RABV and MARV glycoproteins in cells infected with recombinant virus, Vero cells were infected at an MOI of 0.1 with either live FILORAB3 or BNSP333 (control) for 48 h before immunofluorescence surface staining was performed. Monoclonal antibodies directed against RABV G appear in green, monoclonal antibodies directed against MARV GP appear in red, and the overlap of expression of both glycoproteins is indicated by yellow (Fig. 3A). Cells that were infected with FILORAB3 recombinant virus exhibited coexpression of both glycoproteins, which suggests that these envelope proteins are being properly expressed by the vaccine vector, folded, and trafficked to the surface.
FIG 3.

Vaccine vector characterization. (A) Vero CCL-81 cells were infected at an MOI of 0.1 with the BNSP333 parental rabies virus vaccine or recombinant FILORAB3 for 48 h before surface immunostaining with monoclonal antibodies directed against RABV G (green) and MARV GP (red). Yellow indicates an overlap in the expression of both glycoproteins. (B) (Left) Four micrograms each of purified inactivated FILORAB3 and control virions was loaded onto a denaturing 10% SDS-PAGE gel and stained with SYPRO Ruby to visualize the incorporated proteins. Full-length codon-optimized MARV GP and soluble MARV GP (2 μg) with transmembrane and cytoplasmic domain deletion (used for antibody capture in ELISAs) are indicated by the red box. FILORAB1 purified virions were included as a control for successful foreign glycoprotein incorporation. Critical RABV proteins are indicated. (Right) Confirmation of MARV GP incorporation into purified FILORAB3 virions by Western blot analysis. Two micrograms of purified inactivated FILORAB3 or control virions was loaded onto a 10% SDS-PAGE gel and transferred to a nitrocellulose membrane. The blot was probed with a cocktail of two mouse-derived monoclonal antibodies specific for either the GP1 or the GP2 subunit of MARV GP. Lysates from Vero cells infected with MARV were used as a positive control. As a negative control, mock-infected or Lassa virus-infected Vero cell lysate was used.
Incorporation of MARV GP into RABV virions.
For the inactivated vaccine to be immunogenic against both RABV G and MARV GP, both glycoproteins must be incorporated into budding virions. To analyze the incorporation of RABV G and MARV GP into purified virions, virus particles were isolated from the supernatant of infected Vero cells by filtration and concentration, followed by purification over a 20% sucrose cushion. The virus particles were resolved by SDS-PAGE and visualized by SYPRO Ruby staining. FILORAB3 purified virions showed incorporation of all essential RABV proteins (Fig. 3B, left) and in the same ratios as the parental virions (third versus first lanes after the marker lane). An additional protein of the expected size for MARV GP1 (170 kDa) was detected in FILORAB3 viral particles (highlighted in the red box in Fig. 3B, left) (5, 39, 40). GP2 could not be visualized on this gel because it migrated at a similar size as the RABV P protein. FILORAB1 was included as a positive control. Incorporation of the codon-optimized MARV GP appeared to occur but did so to a lesser extent than previously shown for EBOV GP.
To confirm expression of both subunits of the MARV glycoprotein, we analyzed purified virions by Western blotting (Fig. 3B, right) and probed with a cocktail of two monoclonal antibodies directed against both GP1 and GP2. Two proteins migrating at about 170 kDa and 40 kDa, consistent with the molecular weights of MARV GP1 and GP2, respectively, were detected (39, 40) (Fig. 3B, right, third lane). These results further confirm that MARV GP was incorporated into the virion and was cleaved and processed. Two proteins of similar size were detected in cell lysates from Vero E6 cells infected with wild-type (WT) MARV (fifth lane). No protein was detected in mock-infected Vero E6 cell lysates (sixth lane) or in lysates from cells infected with an unrelated virus (Lassa virus [LASV], seventh lane), indicating that the identified bands are in fact specific to FILORAB3 and Marburg virus protein incorporation.
Pathogenicity in vivo.
We know from previous studies in our labs that BNSP333 is apathogenic in adult mice but neurovirulent in suckling mice following intracranial (i.c.) inoculation (41). To assess whether inclusion of MARV GP into the BNSP333 vector would increase neurovirulence, newborn/suckling mice were i.c. inoculated with escalating doses of FILORAB3 (Fig. 4A). As a positive control, we used the BNSP vector, which lacks the attenuating mutation at amino acid position 333 in RABV G. Mice receiving FILORAB3 experienced significantly delayed lethality compared to control mice (P < 0.0001). The majority of mice in each FILORAB3 group (≥70%) survived up to at least day 10, whereas all control mice succumbed to infection by day 5. This suggests that in infected mice FILORAB3 retained the neurovirulence characteristic of the parental BNSP333 vector but did not exacerbate it (Fig. 4A). To further evaluate the safety of the vaccine, we exposed adult severe combined immunodeficient (SCID) mice to FILORAB3 or BNSP by intramuscular injection in the thigh of 6 log10 focus-forming units (FFU) (Fig. 4B) (42). As expected, all the control mice succumbed by day 20 postinfection, while all the mice receiving FILORAB3 survived, further demonstrating the safety of FILORAB3. Taken together, these data indicate that FILORAB3 possesses a good safety profile for use as a live vaccine in certain target species. Furthermore, the inactivated FILORAB3 vaccine for humans should be even safer since it does not have the capacity to replicate.
FIG 4.

Pathogenicity in vivo of FILORAB3. (A) Survival in newborn BALB/c mice (n = 8) intracranially challenged with escalating doses of live FILORAB3 compared to live parental rabies virus (BNSP). (B) Survival in adult SCID mice (n = 8) challenged intramuscularly with 6 log10 FFU of either FILORAB3, BNSP as a positive control, or PBS as a negative control. The log-rank (Mantel-Cox) test was used for comparison of survival curves to assess significant differences in survival.
FILORAB3 induces humoral immunity to both RABV and MARV in mice.
To analyze the immunogenicity of FILORAB3, groups of 5 C57BL/6 mice were immunized with 2 intramuscular doses, given 28 days apart, of either inactivated FILORAB3 plus adjuvant or the parental control vaccine (BNSP333) plus adjuvant (see Fig. 6A). Previous studies in NHPs with FILORAB1 indicated that a Toll-like receptor 4 (TLR-4) agonist, glucopyranosyl lipid adjuvant (GLA), in a squalene-in-water emulsion (GLA-SE) not only increases the humoral responses but also stimulates a Th1-biased humoral immune response that is considered beneficial for viral infection and protection against EBOV by RABV-based vectors (43). Therefore, we also included GLA-SE in our preclinical studies with FILORAB3 (44). Furthermore, GLA-SE has been shown to enhance the breadth and quality of humoral immune responses to influenza virus (35, 45–49).
FIG 6.

Humoral response to FILORAB3. (A) Experimental timeline for immunization of C57BL/6 mice (n = 5 per group). C57BL/6 mice were immunized intramuscularly in the gastrocnemius muscle with a total of 10 μg each of either FILORAB3 or BNSP333 inactivated viral particles adjuvanted with 5 μg of a TLR-4 agonist in 2% stable emulsion (GLA-SE). All mice were primed on day 0 and boosted once on day 28. (B) Sera recovered from immunized mice at day 42 postinoculation were diluted 1:50 and analyzed in a 3-fold serial dilution via an indirect ELISA to test for the relative quantities of both MARV GP-specific (top) and RABV G-specific (bottom) antibodies. The OD490 values were compared to those for antigen-specific monoclonal antibody positive-control samples (gray open circle lines). (C) EC50 values from total MARV (top) and RABV (bottom) IgG ELISA curves were analyzed. Statistical significance was performed using an unpaired t test with Welch’s correction to compare the FILORAB3- and BNSP333-immunized groups. The results are presented as mean values. *, P < 0.05. (D) IgG2 and IgG1 isotype responses in FILORAB3- and BNSP333-immunized mice were assessed by ELISA at day 42 postinoculation to evaluate Th1- versus Th2-biased humoral immunity. (E) Purified IgG derived from pooled sera of FILORAB3- or BNSP333-immunized mice was analyzed in an in vitro pseudotyped lentivirus luciferase assay to determine the titers of MARV GP- and RABV G-neutralizing antibodies compared to those of a positive-control monoclonal antibody known to neutralize either MARV or RABV pseudotyped virus in vitro (gray lines). Graphs are representative of average data from three independent experiments. The gray horizontal lines indicate the threshold for a 50% reduction in infection. Ab, antibody; HRP, horseradish peroxidase.
We collected final sera from the mice 42 days after the initial immunization and characterized the sera in an indirect enzyme-linked immunosorbent assay (ELISA) with soluble MARV GP (sMGP) (Fig. 5). Mice immunized with FILORAB3 plus adjuvant showed robust seroconversion toward MARV GP, while GP-specific titers were not detected in the negative controls (i.e., vector-immunized mice). Both the control and FILORAB3-immunized groups of mice showed strong seroconversion toward RABV G (Fig. 6B and C). To confirm a Th1-biased humoral response, we analyzed the isotype-specific antibody response by ELISA in mice receiving adjuvanted inactivated FILORAB3. Based on the relative titers of immunoglobulin G2c (IgG2c) to IgG1 in FILORAB3-immunized mice, it appears that the immune response is biased toward the production of Th1 antibodies (Fig. 6D).
FIG 5.

Characterization of recombinant soluble MARV GP capture antigen for indirect ELISA. (A) One microgram of purified soluble MARV GP antigen was loaded onto a denaturing SDS-PAGE gel for Western blot analysis. The blots were probed with anti-MARV GP1 or anti-MARV GP2 mouse monoclonal antibodies to confirm the presence of both subunits of MARV GP. (B) Fifty nanograms of soluble MARV GP antigen was used to coat the wells in a 96-well plate for an indirect ELISA to test the reactivity against MARV GP-specific sera or antibodies. A 1:500 starting dilution of serum from an NHP survivor of Marburg virus disease and a 1:50 starting dilution of an anti-GP2 monoclonal mouse antibody were analyzed in a 3-fold serial dilution in the ELISA. Pooled preimmune sera from naive BALB/c mice and NHPs were used as negative controls.
Neutralizing antibodies against MARV are not induced by FILORAB3 immunization.
Based on the high titers of MARV GP-specific antibodies elicited by our vaccine in mice, we tested whether these antibodies had the capacity to neutralize in vitro, thereby illuminating a potential mechanism of protection. To this end, we employed a lentivirus pseudotyped virus expressing a luciferase reporter gene (50, 51). The assay was performed with purified immunoglobulin G (IgG) derived from the final sera of mice in the previously described immunization study, and the virus neutralization percentage was standardized from the relative luminescence unit readout. Compared to a positive-control antibody (Fig. 6E, top, gray line) known to neutralize both retroviral and VSV pseudotypes containing MARV GP in vitro (52), purified IgG from the sera of FILORAB3-immunized mice did not elicit any detectable titers of neutralizing antibodies, which were comparable to the levels detected in sera from negative-control mice (i.e., background) (Fig. 6E, top). In contrast, both groups of sera elicited robust titers of RABV-neutralizing antibodies (compared to a positive-control human MAb; Fig. 6E, bottom, gray line), which is a known correlate of protection against rabies virus (49, 53–55).
FILORAB3 confers protection against ma-MARV challenge in mice (in the absence of neutralizing antibodies).
After we established that mice immunized with adjuvanted FILORAB3 had high titers of MARV GP-specific antibodies but insignificant titers of neutralizing antibodies, we analyzed the potential of the adjuvanted FILORAB3 vaccine to protect mice against lethal challenge with the mouse-adapted Marburg virus (ma-MARV). Groups of 10 BALB/c mice equally split by gender were immunized with either 1, 2, or 3 doses of adjuvanted FILORAB3 vaccine, according to the schedule defined in Fig. 7A (groups 5 to 7). One group of mice also received a single inoculation of live, replication-competent FILORAB3 (group 4), and another group received live, replication-competent BNSP333 as a negative control for the live virus vaccination (group 3). To assess survival throughout the course of the experiment, we included vehicle-infected mice that did not receive any challenge (group 1). As a positive control for lethality by ma-MARV, we included a group immunized with vehicle and challenged with a lethal dose of ma-MARV (group 2).
FIG 7.

Murine MARV challenge model. (A) Experimental timeline for immunization of BALB/c mice and the immunization groups included in the study. Groups of mice (n = 10 per group) were immunized intramuscularly in the gastrocnemius muscle with the indicated treatments. Mice receiving 3 doses of adjuvanted inactivated vaccine (group 7) were primed 61 days before challenge (day −61) and boosted both 54 and 40 days before challenge (day −54 and day −40, respectively). Mice receiving 2 doses of adjuvanted inactivated vaccine (group 6) were primed 54 days before challenge (day −54) and boosted 40 days before challenge (day −40). All other groups of mice were primed 40 days before challenge (day −40). (B) Survival of BALB/c mice challenged intraperitoneally (i.p.) with a lethal dose of ma-MARV (1,000 PFU). (C) Prechallenge humoral response in pooled mouse sera from each vaccine group after completion of the immunization schedule (day 0). The left and right panels show MARV GP- and RABV G-specific antibody titers, respectively (ELISA curves and the corresponding bar graph of EC50 values [top and bottom, respectively]), compared to those for the positive-control monoclonal antibody. (D) Bar graph of MARV GP-specific IgG1 and IgG2 isotype ELISA OD490 readings at the lowest antibody dilution (1:50) in prechallenge sera (day 0) for each vaccine group. (E) Postchallenge humoral response in pooled sera from survivor mice in the indicated vaccine groups after challenge with ma-MARV (day 28, necropsy). The left and right panels show MARV GP- and RABV G-specific antibody titers (bar graph of EC50 values), respectively, compared to those for positive-control monoclonal antibodies. The D’Agostino and Pearson normality test was performed to test for the normal distribution in each data set. Statistical significance for MARV GP antibody titers was performed using the nonparametric Kruskal-Wallis test and Dunn’s multiple-comparison test. For RABV G antibody titers, statistical significance was performed using ordinary one-way analysis of variance with Holm-Sidak’s multiple-comparison test. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; n.s., not significant. (F) Bar graph of MARV GP-specific IgG1 and IgG2 isotype ELISA OD490 readings at the lowest antibody dilution (1:150) in postchallenge sera (day 28) for each vaccine group. (G) Purified IgG derived from pooled mouse sera from the indicated vaccine groups was analyzed in an in vitro pseudotyped lentivirus luciferase assay to determine the titers of both prechallenge (day 0) and postchallenge (day 28) MARV GP- and RABV G-neutralizing antibodies compared to the titer of a positive-control monoclonal antibody known to neutralize either MARV or RABV pseudotyped virus in vitro. Graphs are representative of average data from three independent experiments. Gray horizontal line indicates the threshold for a 50% reduction in infection.
Whereas all negative-control vaccine groups (groups 1 to 3) as well as mice immunized with live FILORAB3 (group 4) succumbed to the infection by day 7 after challenge, we were able to achieve full protection against lethal ma-MARV challenge with just one inoculation of adjuvanted FILORAB3 (Fig. 7B). Linear regression analysis of the average optical density at 490 nm (OD490) value at the lowest antibody dilution (1:50) versus the percent survival for each group revealed that antibody titers correlated with survival (r2 = 0.7945). Analysis of prechallenge sera (from day −40) from these groups of mice by ELISA revealed that GP-specific seroconversion was achieved in all groups immunized with adjuvanted inactivated FILORAB3. Moreover, these titers were high compared to the background (Fig. 7C). Administering 2 and 3 doses of FILORAB3 increased the GP-specific titer in a dose-dependent fashion.
Mice immunized with live FILORAB3 (Fig. 7C, red line) seroconverted to MARV GP, but the GP ELISA titers were much lower than those in the inactivated vaccine groups. Live FILORAB3-immunized mice were fully susceptible to challenge, thereby suggesting that the immunogenicity from GP expression by the live, replication-competent vaccine is not able to control infection from ma-MARV, despite eliciting Th1-biased GP-specific antibodies. Thus, these data suggest not only that elicitation of a Th1-type response informs vaccine-induced survival but also that a threshold antibody response must be achieved. We have encountered this phenomenon in our previous vaccine study with FILORAB1 in NHPs. One protected NHP had a clear Th2-biased humoral response but was still protected against lethal challenge, presumably due to high prechallenge antibody titers (56).
As expected, all mice receiving either live or inactivated vaccine seroconverted toward RABV G, which occurred in a dose-dependent manner for the inactivated vaccine groups (Fig. 7C, right). The Th1 bias of the humoral response was confirmed, as measured by isotype ELISAs comparing the ratio of IgG2a to IgG1 at the lowest serum dilution (1:150) in these BALB/c mice (Fig. 7D). The postchallenge GP- and G-specific antibody titers in mice that survived ma-MARV challenge remained high, and GP-specific antibody titers increased in mice receiving 1, 2, or 3 doses of adjuvanted vaccine after challenge, as measured by determination of 50% effective concentration (EC50) values, indicating that vaccine-induced immunity can confer protection by controlling viral infection (Fig. 7E). Th1 skewing of the humoral response is maintained after challenge in survivors, as measured by isotype ELISAs comparing the ratio of IgG2a to IgG1 at the lowest serum dilution (1:450) (Fig. 7F). This suggests that a Th1 bias is important for a survival response toward MARV.
We also assessed the MARV- and RABV-neutralizing antibody titers in pooled serum samples both pre- and postchallenge in mice in this study. Consistent with the results of the immunogenicity study in C57BL/6 mice described above, BALB/c mice in this study immunized with either live or inactivated FILORAB3 did not elicit neutralizing antibody titers against MARV GP (Fig. 7G, top). In contrast, all mice receiving either live or inactivated FILORAB3 did elicit potent neutralizing antibodies against RABV G pre- and postchallenge, and the effect seemed to be dose dependent: the groups of mice receiving 2 and 3 doses of adjuvanted vaccine elicited increasingly higher titers of RABV G-specific nAbs (Fig. 7G, bottom).
Antibody-dependent cellular cytotoxicity.
Based on the strong immunogenicity of FILORAB3 against both RABV G and MARV GP, the negligible titers of neutralizing antibodies against MARV GP, and survival in mice following challenge with ma-MARV, we hypothesized that nonneutralizing antibodies might be important for vaccine-induced protection. To assess the capacity of antibodies elicited by vaccination to participate in ADCC effector functions, we developed an in vitro flow-based ADCC assay (Fig. 8). Briefly (as demonstrated in Fig. 9A), mouse 3T3 fibroblast target cells were transduced with mouse retrovirus expressing both an enhanced green fluorescent protein (EGFP) reporter gene and a MARV GP gene. These target cells were confirmed to express MARV GP on their surface by flow cytometry (Fig. 9B). Target cells were then incubated with 50 μg/ml of purified IgG derived from sera from immunized mice before the addition of primary effector NK cells purified from splenocytes from naive mice (Fig. 9C). After 4 h, the population of dead target cells over the total population of target cells was assessed and reported as the percent cytotoxicity (Fig. 9D).
FIG 8.
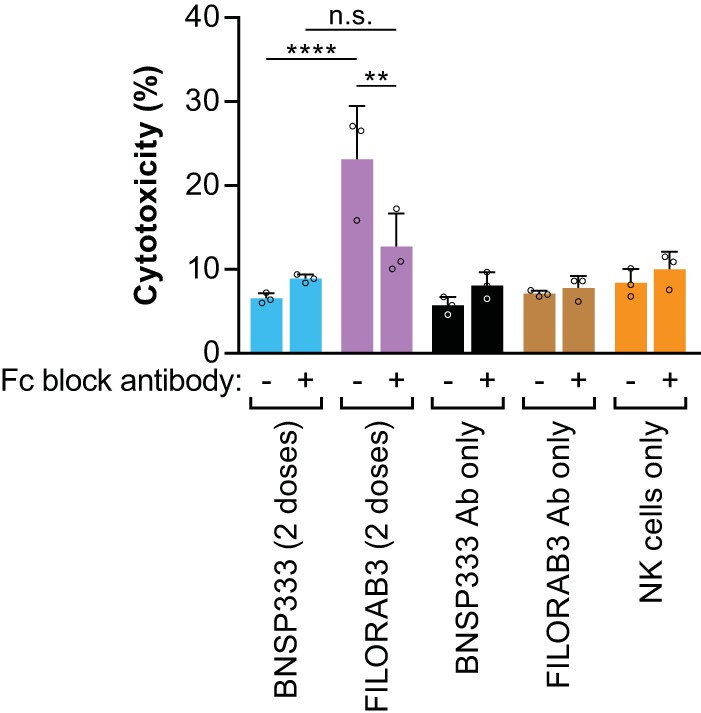
Evaluation of NK cell-mediated antibody-dependent cellular cytotoxicity (ADCC) effector function in an in vitro killing assay. Purified IgG derived from pooled sera from BALB/c mice immunized with either adjuvanted FILORAB3 (purple) or adjuvanted BNSP333 (blue) was incubated with 3T3-MARV cells with or without addition of an antibody cocktail to block the FcγRIII on the surface of NK cells. NK effector cells were subsequently added at an effector cell-to-target cell ratio of 5:1. Killing was measured by flow cytometry (reported as percent cytotoxicity). As negative controls, MARV-infected 3T3 cells were incubated with purified IgG alone in the absence of NK effector cells (black and brown bars) or with NK effector cells alone and no antibodies (orange bars). Statistical significance was performed using 2-way analysis of variance and post hoc analysis with Tukey’s multiple-comparison test. **, P < 0.01; ****, P < 0.0001; n.s., not significant. Data are representative of average values from triplicate values across 3 independent experiments.
FIG 9.

Characterization of in vitro NK cell-mediated ADCC assay. (A) Schematic depicting the experimental procedure and corresponding biological events for our in vitro mouse ADCC direct killing assay. IRES, internal ribosome entry site; EGFP, enhanced green fluorescent protein; FACS, fluorescence-activated cell sorting. (B) (Left) MARV GP expression in transduced mouse 3T3 target cells was confirmed by flow cytometry (green curve) using an anti-MARV GP mouse monoclonal antibody (MAb 3E10). 3T3 target cells expressing a nonspecific viral glycoprotein (LASV GPC) were stained with MAb 3E10 and assayed as a negative control. Vero CCL81 cells infected at an MOI of 0.1 for 48 h were fixed and stained with MAb 3E10 and assayed as a positive control for MARV GP expression. (Right) Bar graph derived from flow cytometry plots showing the geometric mean of the medium fluorescence intensity (MFI) for both GFP and MARV GP. (C) Phenotypic characterization by flow cytometry on magnetically purified NK cells enriched from splenocytes of naive BALB/c mice to determine the purity of NK cells in the effector cell population for the ADCC assay. CD3 and NKp46 biomarkers were used to identify the percentage of effector NK cells in the population. (D) Gating strategy on MARV-infected 3T3 cells for ADCC assay. Percent cytotoxicity was determined by the percentage of GFP+/PI+ cells in the total parental GFP+ cell population. The top row is a representative flow plot of killing in MARV-infected 3T3 target cells incubated with negative-control sera. The bottom row is a representative flow plot of killing in target cells incubated with purified IgG derived from pooled sera from BALB/c mice immunized with FILORAB3. (E) Overlapping PI histograms of MARV-infected 3T3 and LASV-infected 3T3 cells incubated with FILORAB3-purified IgG (1:100) showing the difference in cytotoxicity. Forty thousand target cells were used in the assay. SSC-A, side scatter area; FSC-A, forward scatter area; Q1, quadrant 1; Q2, quadrant 2.
At an effector cell-to-target cell ratio of 5:1, antibodies from mice immunized with adjuvanted, inactivated FILORAB3 demonstrated significantly more specific killing of target cells expressing MARV GP (P < 0.0001) than negative-control sera from BNSP333-immunized mice (Fig. 8). Furthermore, blocking the Fcγ receptor (FcγR) on the surface of NK cells abrogated the NK cell-mediated cytotoxicity to background killing levels, indicating that killing by NK cells in this assay is enhanced by GP-specific antibodies binding to the receptor and activating NK cells. While similar findings have been demonstrated with Ebola virus GP-specific antibodies from NHPs (25), this result adds to the growing body of evidence suggesting that nonneutralizing antibodies may be important for controlling MARV infection (57).
FcγR is important for in vivo protection in mice.
To test the in vivo relevance of Fcγ receptor-dependent effector mechanisms to confer protection, we utilized an Fcγ chain knockout (FcγKO) mouse model (43, 58, 59). This model has normal B and T cell compartments but does not express FcγI, -II, -III, or -IV on the surface of immune effector cells (i.e., macrophages, monocytes, NK cells) (43, 58, 59). Either wild-type BALB/c mice or FcγKO mice (on the BALB/c strain background) were immunized with 1 or 2 doses of adjuvanted FILORAB3 vaccine (groups 5 to 8). As positive controls, one group of WT mice and one group of knockout (KO) mice (groups 2 and 4) were mock immunized before challenge. As a study control, we included WT and KO mice that were immunized with vehicle but remained unchallenged. Each group consisted of 12 mice total (6 male and 6 female mice) (Fig. 10A).
FIG 10.

Evaluation of the in vivo relevance of the Fcγ receptor in protection against ma-MARV in a murine challenge model. (A) Experimental timeline for immunization of BALB/c mice and the immunization groups included in the study. Groups of wild-type or Fcγ knockout mice (n = 12 per group) were immunized intramuscularly in the gastrocnemius muscle with the indicated treatments. Mice receiving 2 doses of adjuvanted inactivated vaccine (groups 6 and 7) were primed 61 days before challenge (day −61) and boosted at both 54 and 40 days before challenge (day −54 and day −40, respectively). All other groups of mice were primed 40 days before challenge (day −40). (B) Survival of BALB/c mice challenged intraperitoneally (i.p.) with a lethal dose of ma-MARV (1,000 PFU). Statistical significance was performed using the log-rank (Mantel-Cox) test for comparison of survival curves (**, P < 0.01). (C) Prechallenge humoral response in pooled mouse sera from each vaccine group after completion of the immunization schedule (day 0). The left and right panels show MARV GP- and RABV G-specific antibody titers (bar graph of EC50 values), respectively, compared to the positive-control monoclonal antibody titers (gray bars). The D’Agostino and Pearson normality test was performed to test for the normal distribution in each data set. Statistical significance for both MARV GP and RABV G antibody titers was performed using the nonparametric Kruskal-Wallis test and Dunn’s multiple-comparison test (*, P < 0.05; **, P < 0.01; n.s., not significant). (D) Postchallenge humoral response in pooled sera from survivor mice in the indicated vaccine groups after challenge with ma-MARV (day 28, necropsy). (Left and middle) MARV GP- and RABV G-specific antibody titers, respectively, are represented by the EC50 values of the ELISA curves compared to those for positive-control monoclonal antibodies (gray bars). The D’Agostino and Pearson normality test was performed to test for the normal distribution in each data set. Tests for the statistical significance for MARV GP antibody titers were performed using the nonparametric Kruskal-Wallis test and Dunn’s multiple-comparison test (n.s., not significant). (Right) Bar graph of MARV GP-specific IgG1 and IgG2 isotype ELISA OD490 readings at the lowest antibody dilution (1:50) in postchallenge sera (day 28) for survivors in the indicated vaccine groups. (E) Pooled sera from mice from the indicated vaccine groups were analyzed in an in vitro pseudotyped lentivirus luciferase assay to determine the titers of both prechallenge (day 0) and postchallenge (day 28) MARV GP- and RABV G-neutralizing antibodies compared to those of a positive-control monoclonal antibody known to neutralize either MARV or RABV pseudotyped virus in vitro (gray lines). Graphs are representative of average data from three independent experiments. The gray horizontal lines indicate the threshold for a 50% reduction in infection.
Wild-type mice receiving 1 dose of adjuvanted vaccine demonstrated significantly better survival than FcγKO mice receiving 1 dose of adjuvanted vaccine (58.3% protection versus 0% protection, respectively; P = 0.0058) (Fig. 10B). To confirm that the difference in survival was not due to differential GP-specific antibody titers, we performed ELISA on prechallenge sera from these mice (day −40). We did not see a significant difference in either RABV G- or MARV GP-specific titers between these groups (P > 0.9999) (Fig. 10C). Survivors of challenge demonstrated an increase in GP-specific antibody titers, with no significant differences between the indicated groups (Fig. 10D). When we assayed for neutralizing antibodies in vitro, we found that neither of the single-dose groups elicited detectable titers of neutralizing antibodies (Fig. 10E, top left), findings that suggested that differences in their survival are tied to the lack of functional Fc receptors in the KO group. In WT and KO mice receiving 2 doses of the vaccine, serology testing by ELISA confirmed that the prechallenge serum (day −40) titers of RABV G and MARV GP were not significantly different (P > 0.9999) (Fig. 10C) and that there was no significant difference in survival between these groups (P > 0.9999) (Fig. 10B). The neutralizing antibody response elicited by both WT and KO mice receiving 2 doses of FILORAB3 was low, with a greater than 50% reduction in infection being achieved only at the lowest dilution of antibody (1:10), although the effect was insignificant considering the variability (standard deviation of the mean) (Fig. 10E). While we expected survival in the wild-type group, it was interesting that the 2-dose KO group showed survival. While the presence of a low titer of neutralizing antibodies in these groups could possibly have provided some protection against ma-MARV in the absence of Fc receptor function by limiting the spread of the virus, it was unlikely, since these GP-specific antibody titers were very low and our in vitro assay overestimated neutralizing antibody titers compared to those in the wild-type virus (50–52). Therefore, it is likely that other Fcγ-independent antibody effector mechanisms are involved in vaccine-induced immunity and protection, but it is clear from the results of this study that Fcγ receptor mechanisms have an important role in vaccine-induced protection.
DISCUSSION
We have described the generation, propagation, safety, immunogenicity, and protective efficacy of an inactivated recombinant rabies virus-vectored Marburg virus vaccine, FILORAB3, developed by successful incorporation of the codon-optimized version of the Marburg virus GP into RABV virions expressing native RABV G. Our results demonstrate that FILORAB3 induced strong humoral immunity in mice, as indicated by high titers of Th1-biased antibodies against MARV glycoprotein but negligible titers of MARV-neutralizing antibodies. The antibody response against MARV is consistent with features of natural Marburg virus infection in both humans and NHPs, whereby survivors experience Th1 skewing of the humoral response, marked by the rare occurrence of neutralizing antibodies that decrease rapidly over time (19, 21).
Based on its robust immunogenicity in vivo, we sought to evaluate the efficacy of FILORAB3 in a murine challenge model. From our previous immunogenicity and challenge studies in mice and NHPs with the recombinant RABV/Ebola virus vaccine candidate (FILORAB1), it was apparent that the quality of the antibody response has important consequences on the protection elicited by our vaccine (56). As such, we chose to use GLA-SE as the adjuvant in FILORAB3 (35, 46–48) with the goal of recapitulating a Th1-biased effect on the immune response. Two doses of the adjuvanted vaccine conferred 96% survival (combined efficacy from 2 challenge experiments) in mice against mouse-adapted MARV, while unprotected mice succumbed to the infection by day 7. Differences in survival between single-dose-immunized mice in the first and second mouse challenge studies could be due to apparent differences in the threshold of the antibody response. Matrajt et al. used modeling to show that above a certain response threshold, vaccinating more people with one dose of the influenza vaccine resulted in lower attack rates. However, below that required level of response, vaccinating fewer people with 2 doses is better for protection (60). A single dose of our FILORAB3 vaccine elicited a more variable response in the population, likely reflective of the relatively small number of subjects. After a single-dose vaccination, some animals had a response above the threshold required for protection and some had a response below the threshold. The varicella-zoster virus (VZV) vaccine showed 94.9% seroconversion after a single dose but only achieved 100% after 2 doses (61). In a preexposure setting, immunization with the human rabies vaccine must reach a threshold of at least 0.5 international units per ml of neutralizing activity in order to be protective. To achieve this protective threshold in 100% of the population, at least 3 doses of the rabies vaccine must be given (62). In our preclinical models, 2 doses of our FILORAB3 vaccine gave uniform results across both studies. Overall, 2 immunizations of vaccine may achieve higher variation in the epitope specificities of GP-specific antibodies (63), which can have positive implications for long-lasting immunity.
The prechallenge levels of antibody in mice immunized with inactivated vaccine were similar to the levels observed after challenge, indicating that vaccination established and maintained a crucial memory B cell response. Mice receiving a single inoculation of the live FILORAB3 had low titers of GP-specific antibodies and were not protected against lethal challenge, suggesting that antibodies play a major role in protection. Live virus vaccine antigenicity is dependent upon replication of the virus. It is possible that the live virus is highly attenuated upon peripheral administration and that the antigen load is not enough to induce a protective response. It is also possible that the immune response mounted against RABV G might quickly block the spread of the vector (at least in intramuscular immunization) and therefore prevent a potent IgG immune response against MARV GP. This is corroborated by our data, which show that the RABV G immune response with live FILORAB3 is lower than the corresponding response with killed vaccine. Furthermore, live attenuated viral vectors encoding foreign glycoprotein are known to induce potent cellular immune responses because they engage the endogenous pathway or cross-priming to present epitopes via major histocompatibility complex type I to CD8+ T cells (64, 65). Since live FILORAB3 does not confer protection in mice, it suggests that cytotoxic T cells may not play a major role in vaccine-induced protection against MARV or at least are not sufficient to control MARV infection in the absence of an appropriate antibody response.
In pursuit of defining the antibody-mediated mechanisms of protection by FILORAB3, we concluded that neutralizing antibodies do not play a significant role in protection against survival. However, since we saw high titers of specific IgG after vaccination, we hypothesized that nonneutralizing antibodies are involved in protection. This hypothesis was also supported by the finding that the antibody levels correlated with survival (r2 = 0.7945). The functions of nonneutralizing antibodies have been described for other viruses. We know that a cocktail of exclusively nonneutralizing antibodies can protect NHPs against lethal Ebola virus challenge when administered as postexposure prophylaxis (66). ZMapp, first used in the West African Ebola outbreak as emergency postexposure prophylaxis, was developed to include a nonneutralizing antibody (c13C6) that binds to the tip of the viral glycoprotein, after it was shown that inclusion of this antibody resulted in better survival in guinea pigs and in NHPs than a cocktail of neutralizing antibodies only (67). Nonneutralizing antibodies to HIV and lymphocytic choriomeningitis virus GPs inhibit infection of dendritic cells and of macrophages and limit virus spread (66, 68). Various nonneutralizing functions of antibodies elicited by vaccination against HIV have been described previously in great detail (44, 69, 70), and the recent RV144 Thai vaccine trial showed a correlation between nonneutralizing antibodies and protection (71, 72).
To this end, we developed a novel, in vitro flow cytometry-based mouse ADCC assay to assess the ability of antibodies elicited by FILORAB3 to induce direct killing by NK cells (23). Purified IgG from the sera of mice immunized with 2 doses of adjuvanted FILORAB3 led to significantly higher levels of killing by NK cells than by purified IgG from the sera of mice immunized with the parental rabies vaccine alone plus adjuvant. When either GP-specific or control antibodies alone or NK cells alone were added to target cells expressing MARV GP, killing was significantly lower than that when both specific antibodies and NK cells were present in the system.
Furthermore, the cytotoxic effect could be abrogated to background levels of killing by the addition of mouse-specific FcγRIII-blocking antibodies. Taken together, these data demonstrate that the NK cell-mediated killing measured in this assay is dependent upon FcγRIII engagement with GP-specific antibodies. Furthermore, killing was antigen dependent, as GP-specific antibodies did not enhance killing beyond a background level for target cells expressing either a different viral glycoprotein (Lassa virus GPC) or no viral envelope (Fig. 9E). While the importance of Fcγ-dependent antibody-mediated immune responses has been described for Ebola virus (24–26), this result provides the first piece of evidence that ADCC may play a significant role in the protection against Marburg virus (24, 73). Other Fcγ-dependent mechanisms of protection, such as FcγRI -mediated phagocytosis or killing by macrophages or monocytes, could also be important and have been described in viral immunity for other viruses but were not tested within the scope of this study (28–30).
For filoviruses, an in vitro ADCC capacity is not necessarily an effective predictor of in vivo protection (10). Therefore, we sought to determine the in vivo relevance of Fcγ-dependent antibody-mediated mechanisms of protection in a knockout mouse model in the BALB/c mouse background. These knockout mice were developed by embryonic gene targeting to replace the gamma chain gene with a null allele. As a result, among other gamma chain-dependent immune effector functions, these mice have NK cells that lack ADCC function but are fertile and viable and have normal B and T cell compartments (43). We found no significant difference in the titers of GP-specific antibody induced between wild-type and KO mice receiving one dose of adjuvanted vaccine, but there was a significant difference in the survival between these groups. Since both prechallenge animals and survivors had negligible titers of GP neutralizing antibodies (consistent with previous in vitro murine models), we concluded that in the absence of these potently neutralizing antibodies, a functional Fcγ receptor is essential for survival in these mice. However, since KO mice receiving 2 doses of the vaccine showed survival indiscriminate from that of the corresponding WT group, it is apparent that Fcγ-independent mechanisms of protection are also involved in protection. Takada and colleagues described in vitro the phenomenon of GP-specific antibodies that participate in the budding inhibition of MARV (57). These antibodies are not classically neutralizing but can bind to GP on the surface of infected cells and prevent the budding of progeny virions (57). Nonneutralizing antibodies that fix complement can also be an essential part of the antibody repertoire induced by FILORAB3 vaccination. The classical complement pathway has been shown to impact the control of influenza virus infections (74–77) and has been shown to be able to directly lyse HIV virions (78, 79). It is possible that our vaccine elicits antibodies that can bind to GP on the surface of infected cells and that can recruit the C1q protein to initiate the classical complement cascade to lyse the infected cell or mediate opsonophagocytosis; however, complement added to our in vitro neutralization study did not decrease the infectivity of lentivirus pseudotyped virions (data not shown), so direct lysis of MARV virions is not a likely mechanism of action. Lastly, since our vaccine is adjuvanted with GLA-SE, which is known to elicit type II interferon responses by induction of neutrophils and CD8+ T cells (80, 81), the possibility cannot be excluded that vaccination elicits cytotoxic T lymphocytes that can kill virally infected cells during acute infection.
The studies described here demonstrate the potential for a RABV platform for the development of a safe and effective MARV vaccine. We have demonstrated the preclinical safety and efficacy of the vaccine against the Angola strain of ma-MARV in mice. While protection against the highly pathogenic Angola strain is thought to confer protection against other MARV strains and RAVN (82–87), this still needs to be assessed with FILORAB3 in animal models. Presently, the immunogenicity and protective efficacy of FILORAB3 need to be evaluated in NHPs to determine whether this candidate vaccine merits evaluation in humans, but preclinical results in mice, described in this paper, offer a promising outlook for the development of a human FILORAB3 vaccine. Additionally, further investigation into various other mechanisms of protection are warranted in order to understand the optimal parameters of long-lasting immunity induced by FILORAB3.
MATERIALS AND METHODS
cDNA construction of vaccine vectors.
We inserted codon-optimized Marburg virus glycoprotein gene GP (Angola strain) between the N and P genes of the parental BNSP333 rabies virus vector using the BsiWI and NheI restriction sites. Codon bias optimization for human codon use was carried out by GenScript, Inc. The resulting plasmid was designated BNSP333-coMARV-GP (FILORAB3), and the correct sequence of the plasmid was confirmed by sequencing using primers targeting the region between the N and P genes.
Recovery of recombinant vectors.
The X-tremeGENE 9 transfection reagent (Roche Diagnostics) in Opti-MEM medium was used to transfect full-length viral cDNA clones along with support plasmids bearing the RABV N, P, G, and L genes under the control of a T7 promoter and a plasmid expressing T7 RNA polymerase into BSR cells on 6-well plates as described previously (32–34). Successful recovery was determined by a rabies virus focus-forming assay. Briefly, at 7 days after transfection, supernatant from each transfected well of the 6-well plate was transferred to duplicate wells of a 12-well plate seeded with Vero cells. Forty-eight hours later, cells in the 12-well plate were fixed with 80% acetone and stained with a fluorescein isothiocyanate (FITC)-conjugated antibody against RABV N (Fujirebio Diagnostics, Inc.). Fluorescence microscopy was used to observe the appearance of viral foci, indicative of recovered, infectious recombinant RABV.
Sucrose purification and inactivation of the virus particles.
FILORAB3 was grown on a large scale by infecting Vero cells in a 2-stack plate at an MOI of 0.001. The supernatant was collected every 4 days for a total of 6 harvests. The titers of the harvests were determined using the rabies virus focus-forming assay, and harvests 4 to 6 were pooled and concentrated 9 times in a stirred 300-ml ultrafiltration cell (Millipore). Concentrated supernatant was then centrifuged for 2 h at 25,000 rpm through a 20% sucrose cushion using an SW32 Ti rotor (Beckman, Inc.) to pellet the virus particles. The virion pellets were resuspended in phosphate-buffered saline (PBS), and protein concentrations were determined using a bicinchoninic acid (BCA) assay kit (Pierce). The virus particles were chemically inactivated with β-propiolactone (BPL) at a dilution of 1:2,000 overnight at 4°C. The BPL in the virus preparation was inactivated the next day by hydrolysis at 37°C for 30 min. The absence of infectious particles was verified by inoculating Vero cells in a T25 vessel with 10 μg of BPL-inactivated virus for 2 passages. Inoculated cells were fixed and stained with FITC-conjugated anti-RABV N MAb and visualized by fluorescence microscopy for the presence of foci of infection.
Immunofluorescence testing of the vaccine.
Vero cells were plated onto 12-well plates with 3E5 cells with 15-mm-diameter circular coverslips inserted and then incubated overnight at 37°C. On the next day the wells were infected at an MOI of 0.01 in 500 μl of serum-free medium (OptiPro) per well with FILORAB3 or BNSP333, the contents of the wells were mixed by rocking, and then the plates were incubated at 34°C for 48 h. After 48 h, the cells were washed with 1 ml of 1× PBS and then fixed with 500 μl of 2% paraformaldehyde (PFA) diluted in PBS for 15 min at room temperature. PFA was removed by aspiration, and cells were washed 3 times with 1× PBS. One milliliter of blocking solution (4% fetal bovine serum [FBS] in PBS) was added to each well for 1 h at room temperature while the plate was on the shaker. Blocking solution was aspirated off, and then 500 μl of primary antibody diluted 1:250 in 2% FBS was added for 1 h while rocking. The cells were washed 4 times with 1× PBS, and then 500 μl of 1:300 secondary antibodies containing Cy2 and Cy3 dyes were added and the cells were incubated at room temperature for 45 min. The cells were washed 5 times with 1× PBS, and then the cells were mounted onto slides with a mounting solution containing DAPI (4′,6-diamidino-2-phenylindole), coverslips were placed face down onto the slides, and the slides were stored overnight at room temperature for viewing of the cells the next day.
Pathogenicity and immunogenicity studies.
(i) Animal ethics statement. This study was carried out in strict adherence to the recommendations described in the Guide for the Care and Use of Laboratory Animals (88) as described previously (15), as well as the guidelines of the National Institutes of Health, the Office of Animal Welfare, and the United States Department of Agriculture. All animal work was approved by the Institutional Animal Care and Use Committee (IACUC) at Thomas Jefferson University (animal protocols 00990, 01155, and 01647). All procedures were carried out under isoflurane anesthesia by trained personnel, under the supervision of veterinary staff. Mice were housed in cages, in groups of 5, under controlled conditions of humidity, temperature, and light (12-h light/12-h dark cycles). Food and water were available ad libitum.
(ii) Immunizations.
Two groups of 6- to 8-week-old C56BL/6 mice were immunized intramuscularly with 10 μg of virus particles and 5 μg of GLA-SE in a total volume of 100 μl (50 μl per hindlimb). The 2 groups were as follows: mice immunized with inactivated FILORAB3 and mice immunized with inactivated BNSP333. Each group consisted of 5 female mice. Mice receiving inactivated vaccine were given 2 doses on day 0 and day 28.
(iii) Pathogenicity experiments.
Groups of suckling mice (n = 8) were intracranially infected with escalating doses of FILORAB3 (2 log10, 4 log10, 5 log10 FFU) or 5 log10 FFU of parental rabies virus BNSP. Groups of adult SCID mice (n = 8) were intracranially infected with 6 log10 FFU of live FILORAB3 or parental rabies virus BNSP. The mice were monitored for signs of disease, such as ruffled fur, ataxia, and disorientation, and weighed until day 40. Mice that lost more than 20% of their original body weight were considered to have reached the endpoint and were euthanized.
All vaccine efficacy experiments involving the use of mouse-adapted MARV (GenBank accession number KM261523) were performed under biosafety level 4 (BSL-4) conditions at the U.S. National Institute of Allergy and Infectious Diseases (NIAID) Integrated Research Facility, Frederick, MD, and approved by the U.S. National Institute of Allergy and Infectious Diseases Division of Clinical Research, Animal Care and Use Committee. Samples removed from the BSL-4 environment for further analyses were inactivated by 5 mrad gamma irradiation (serum) or the TRIzol LS reagent in a 3:1 (vol/vol) ratio (whole blood). Seven groups of 10 mice each were vaccinated as follows: group 1, vehicle (PBS) and no ma-MARV exposure; group 2, vehicle (PBS); group 3, BNSP333 (rabies virus parental vaccine) at 5.69 log10 PFU of vaccine; group 4, FILORAB3 at 5.69 log10 PFU of vaccine; group 5, 10 μg inactivated FILORAB3 plus 5 μg GLA-SE at day −40 of vaccination; group 6, 10 μg inactivated FILORAB plus 5 μg GLA-SE at day −54 and day −40 of vaccination (prime-boost); and group 7, 10 μg inactivated FILORAB3 plus 5 μg GLA-SE at day −61, day −54, and day −40 of vaccination (prime-boost-boost). All groups except group 1 were exposed to 1,000 PFU of ma-MARV by intraperitoneal injection, and group 1 received PBS on day 0.
Production of HA-tagged MARV GP.
Subconfluent T175 flasks of 293T cells (a human kidney cell line) were transfected with a eukaryotic expression vector (pDisplay) encoding amino acids 1 to 643 of the head and stalk domains of codon-optimized MARV GP (Angola strain) fused to a C-terminal hemagglutinin (HA) peptide. Supernatant was collected at 48 h after transfection, clarified by centrifugation, and filtered through a 0.45-μm-pore-size filter before being loaded onto an equilibrated anti-HA agarose column (Pierce) containing a 2.5-ml agarose bed volume. The supernatant was allowed to bind to the column overnight at 4°C. On the next day, the column was washed with 10 bed volumes of Tris-buffered saline (TBS)–Tween 20 (TBST; TBS with 0.05% Tween 20) and 2 bed volumes of TBS, and bound MARV GP-HA was eluted with 5 ml of 250 μg/ml HA peptide in TBS. Fractions were collected and analyzed for the presence of co-MARV GP by Western blotting with monoclonal anti-HA antibody (Sigma) prepared in 5% bovine serum albumin (BSA)–TBST. Peak fractions were pooled and dialyzed against PBS in 10,000-molecular-weight cutoff (MWCO) dialysis cassettes (Thermo Scientific) to remove excess HA peptide. After dialysis, the protein was quantified by BCA assay and frozen in aliquots at −80°C. Further characterization was carried out by Western blotting with monoclonal mouse antibodies 3E10 and 5A2 to confirm the presence of both the GP1 and GP2 subunits, respectively.
RABV and MARV GP response by ELISA.
Sera from immunized mice were collected by retro-orbital eye bleed while the mice were under isoflurane anesthesia on days 0, 28, and 42, and samples were tested for immunogenicity by indirect ELISA using C-terminal HA-tagged soluble recombinant protein for antibody capture (Fig. 5). We tested individual mouse serum samples as well as purified immunoglobulin G from day 42 for the presence of total IgG specific to MARV GP and RABV G. To test for anti-MARV GP humoral responses, we produced soluble MARV GP (sMGP) as described above. Soluble MARV GP was resuspended in coating buffer (50 mM Na2CO3 [pH 9.6]) at a concentration of 0.5 μg/ml and then plated in 96-well ELISA MaxiSorp plates (Nunc) at 100 μl in each well. RABV G was also resuspended in coating buffer at a concentration of 0.5 μg/ml and then plated in 96-well ELISA MaxiSorp plates (Nunc) at 100 μl per well. After overnight incubation at 4°C, the plates were washed 3 times with PBS–Tween 20 (PBST; 0.05% Tween 20 in 1× PBS) and incubated for 1 h at room temperature with blocking buffer (5% dry milk powder in 1× PBST) in a volume of 250 μl per well. The plates were then washed 3 times with PBST and incubated overnight at 4°C with 3-fold or 4-fold serial dilutions of sera from immunized mice in PBS containing 0.5% BSA. The plates were washed 3 times the next day, followed by the addition of horseradish peroxidase-conjugated goat anti-mouse-IgG (H+L) secondary antibody (1:20,000; Jackson ImmunoResearch). After incubation for 2 h at room temperature, the plates were washed 3 times with PBST and 200 μl of o-phenylenediamine dihydrochloride (OPD) substrate (Sigma) was added to each well. The reaction was stopped by the addition of 50 μl of 3 M H2SO4 per well after 15 min. The optical density at 490 nm (OD490) was determined.
ADCC.
In the direct antibody-dependent cellular cytotoxicity (ADCC) assay, target cells were 3T3 mouse fibroblast cells made to express GP antigen on their surface by transduction with murine stem cell virus (MSCV) bearing the GP gene (MSCV-MARV GP) and the green fluorescent protein (GFP) reporter gene. Effector cells were primary NK cells derived from naive mouse splenocytes and purified by use of a Miltenyi magnetic activated cell sorting mouse NK cell isolation kit II. Purified NK cells were further enriched in culture with recombinant mouse IL-2 and IL-15 cytokines.
Procedurally, labeled target cells were seeded in a 96-well U-bottom plate and incubated for 30 min at 37°C with 50 μg/ml of purified IgG derived from sera from animals that had previously been immunized with FILORAB3 to allow binding of GP-specific antibodies to surface antigen. Nonspecific sera were used as negative controls. Subsequently, purified NK cells were added at an effector cell-to-target cell ratio of either 5:1 or 10:1 and incubated for 4 h at 37°C. Propidium iodide (PI; 35 μg/ml) viability dye was then added to these cells, which were analyzed by flow cytometry using a BD LSRFortessa flow cytometer. ADCC activity was measured as the percentage of target cells killed (GFP positive [GFP+]/PI positive [PI+]) out of the total GFP-positive target cell population (1).
Virus neutralization assay (VNA).
HIV lentivirus bearing a luciferase reporter gene was pseudotyped with either MARV GP or RABV G, and 10,000 infectious particles per well (as measured by quantitative PCR using an Applied Biological Materials Inc. [abm] lentiviral titer determination assay) were incubated for 30 min (in a total volume of 60 μl) at 37°C with dilutions of purified immunoglobulin from the sera from FILORAB3-immunized and control immunized mice in a 96-well U-bottom plate before infection of a monolayer of 293T target cells that had been seeded in a 96-well flat-bottom plate 24 h prior in 5% complete Dulbecco modified Eagle medium. At 48 h after incubation at 37°C, the target cells were lysed and spin clarified, and supernatant from these cells was measured for luciferase activity (in relative light units) by use of a FluoStar Omega fluorimeter in a luciferase assay based in 96-well white plates, using d-luciferin salt (Sigma) reconstituted in ATP-containing buffer to a concentration of 0.5 mM as the substrate. Positive luciferase activity indicated infectivity by pseudotyped virus, and the infectivity was normalized to the infectivity under control conditions where no antibody was added (i.e., maximum infectivity signal). Neutralization was reported as a percentage of infectivity, and potent neutralization activity was measured by 50% inhibitory concentration values.
Data availability.
The MARV GP sequence was deposited in GenBank under accession number MK375262.
ACKNOWLEDGMENTS
This work was funded in part through the U.S. National Institute of Allergy and Infectious Diseases (NIAID) Division of Intramural Research and the NIAID Division of Clinical Research. Battelle Memorial Institute’s prime contract with the NIAID is under contract no. HHSN272200700016I.
We thank Oscar Rojas, Isis Alexander, and Kristina Howard (Integrated Research Facility, NIH, Fort Detrick, MD) for hands-on animal work and management. We thank Jennifer Wilson (Thomas Jefferson University, Philadelphia, PA) for critical readings and editing of the manuscript and Jiro Wada for help preparing the illustrations.
K.R.H. performed this work as an employee of Battelle Memorial Institute. R.F.J. and M.J.S. are inventors on a U.S. provisional patent application (title, Multivalent vaccines for rabies virus and filoviruses). M.J.S. serves on the Scientific Advisory Board of IDT Biologika, Dessau, Germany. All other authors declare no competing interests.
REFERENCES
- 1.Mühlberger E, Hensley LL, Towner JS. 2017. Marburg- and ebolaviruses: from ecosystems to molecules In Current topics in microbiology and immunology. Springer, New York, NY. [Google Scholar]
- 2.Kuhn JH. 2017. Guide to the correct use of filoviral nomenclature. Curr Top Microbiol Immunol 411:447–460. doi: 10.1007/82_2017_7. [DOI] [PubMed] [Google Scholar]
- 3.Keshwara R, Johnson RF, Schnell MJ. 2017. Toward an effective Ebola virus vaccine. Annu Rev Med 68:371–386. doi: 10.1146/annurev-med-051215-030919. [DOI] [PubMed] [Google Scholar]
- 4.World Health Organization. 2018. Ebola virus disease. World Health Organization, Geneva, Switzerland http://www.who.int/news-room/fact-sheets/detail/ebola-virus-disease.
- 5.Volchkov VE, Volchkova VA, Stroher U, Becker S, Dolnik O, Cieplik M, Garten W, Klenk HD, Feldmann H. 2000. Proteolytic processing of Marburg virus glycoprotein. Virology 268:1–6. doi: 10.1006/viro.1999.0110. [DOI] [PubMed] [Google Scholar]
- 6.Geyer H, Will C, Feldmann H, Klenk HD, Geyer R. 1992. Carbohydrate structure of Marburg virus glycoprotein. Glycobiology 2:299–312. doi: 10.1093/glycob/2.4.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manicassamy B, Wang J, Rumschlag E, Tymen S, Volchkova V, Volchkov V, Rong L. 2007. Characterization of Marburg virus glycoprotein in viral entry. Virology 358:79–88. doi: 10.1016/j.virol.2006.06.041. [DOI] [PubMed] [Google Scholar]
- 8.Geisbert TW, Hensley LE, Geisbert JB, Leung A, Johnson JC, Grolla A, Feldmann H. 2010. Postexposure treatment of Marburg virus infection. Emerg Infect Dis 16:1119–1122. doi: 10.3201/eid1607.100159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dye JM, Herbert AS, Kuehne AI, Barth JF, Muhammad MA, Zak SE, Ortiz RA, Prugar LI, Pratt WD. 2012. Postexposure antibody prophylaxis protects nonhuman primates from filovirus disease. Proc Natl Acad Sci U S A 109:5034–5039. doi: 10.1073/pnas.1200409109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fusco ML, Hashiguchi T, Cassan R, Biggins JE, Murin CD, Warfield KL, Li S, Holtsberg FW, Shulenin S, Vu H, Olinger GG, Kim DH, Whaley KJ, Zeitlin L, Ward AB, Nykiforuk C, Aman MJ, Berry JD, Berry J, Saphire EO. 2015. Protective MAbs and cross-reactive MAbs raised by immunization with engineered Marburg virus GPs. PLoS Pathog 11:e1005016. doi: 10.1371/journal.ppat.1005016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mire CE, Geisbert JB, Borisevich V, Fenton KA, Agans KN, Flyak AI, Deer DJ, Steinkellner H, Bohorov O, Bohorova N, Goodman C, Hiatt A, Kim DH, Pauly MH, Velasco J, Whaley KJ, Crowe JE Jr, Zeitlin L, Geisbert TW. 2017. Therapeutic treatment of Marburg and Ravn virus infection in nonhuman primates with a human monoclonal antibody. Sci Transl Med 9:eaai8711. doi: 10.1126/scitranslmed.aai8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gsell P-S, Camacho A, Kucharski AJ, Watson CH, Bagayoko A, Nadlaou SD, Dean NE, Diallo A, Diallo A, Honora DA, Doumbia M, Enwere G, Higgs ES, Mauget T, Mory D, Riveros X, Oumar FT, Fallah M, Toure A, Vicari AS, Longini IM, Edmunds WJ, Henao-Restrepo AM, Kieny MP, Kéïta S. 2017. Ring vaccination with rVSV-ZEBOV under expanded access in response to an outbreak of Ebola virus disease in Guinea, 2016: an operational and vaccine safety report. Lancet Infect Dis 17:1276–1284. doi: 10.1016/S1473-3099(17)30541-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grant-Klein RJ, Van Deusen NM, Badger CV, Hannaman D, Dupuy LC, Schmaljohn CS. 2012. A multiagent filovirus DNA vaccine delivered by intramuscular electroporation completely protects mice from Ebola and Marburg virus challenge. Hum Vaccin Immunother 8:1703–1706. doi: 10.4161/hv.21873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarwar UN, Costner P, Enama ME, Berkowitz N, Hu Z, Hendel CS, Sitar S, Plummer S, Mulangu S, Bailer RT, Koup RA, Mascola JR, Nabel GJ, Sullivan NJ, Graham BS, Ledgerwood JE, Gordon I, Holman L, Mendoza F, Novik L, Saunders J, Zephir K, Desai N, Young S, Casazza J, Larkin B, Yamshchikov G, Vasilenko O, Gomez PL, Andrews C, Conan-Cibotti M, Wallace K, Stein J, Sheets R, Decederfelt H, Starling J, Renehan P. 2015. Safety and immunogenicity of DNA vaccines encoding Ebolavirus and Marburgvirus wild-type glycoproteins in a phase I clinical trial. J Infect Dis 211:549–557. doi: 10.1093/infdis/jiu511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dye JM, Warfield KL, Wells JB, Unfer RC, Shulenin S, Vu H, Nichols DK, Aman MJ, Bavari S. 2016. Virus-like particle vaccination protects nonhuman primates from lethal aerosol exposure with Marburgvirus (VLP vaccination protects macaques against aerosol challenges). Viruses 8:94. doi: 10.3390/v8040094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geisbert TW, Feldmann H. 2011. Recombinant vesicular stomatitis virus-based vaccines against Ebola and Marburg virus infections. J Infect Dis 204:S1075–S1081. doi: 10.1093/infdis/jir349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mire CE, Geisbert JB, Agans KN, Satterfield BA, Versteeg KM, Fritz EA, Feldmann H, Hensley LE, Geisbert TW. 2014. Durability of a vesicular stomatitis virus-based Marburg virus vaccine in nonhuman primates. PLoS One 9:e94355. doi: 10.1371/journal.pone.0094355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones SM, Feldmann H, Stroher U, Geisbert JB, Fernando L, Grolla A, Klenk HD, Sullivan NJ, Volchkov VE, Fritz EA, Daddario KM, Hensley LE, Jahrling PB, Geisbert TW. 2005. Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat Med 11:786–790. doi: 10.1038/nm1258. [DOI] [PubMed] [Google Scholar]
- 19.Stonier SW, Herbert AS, Kuehne AI, Sobarzo A, Habibulin P, Dahan CVA, James RM, Egesa M, Cose S, Lutwama JJ, Lobel L, Dye JM. 2017. Marburg virus survivor immune responses are Th1 skewed with limited neutralizing antibody responses. J Exp Med 214:2563–2572. doi: 10.1084/jem.20170161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arnon R, Ben-Yedidia T. 2003. Old and new vaccine approaches. Int Immunopharmacol 3:1195–1204. doi: 10.1016/S1567-5769(03)00016-X. [DOI] [PubMed] [Google Scholar]
- 21.Natesan M, Jensen SM, Keasey SL, Kamata T, Kuehne AI, Stonier SW, Lutwama JJ, Lobel L, Dye JM, Ulrich RG. 2016. Human survivors of disease outbreaks caused by Ebola or Marburg virus exhibit cross-reactive and long-lived antibody responses. Clin Vaccine Immunol 23:717–724. doi: 10.1128/CVI.00107-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bukreyev A, Skiadopoulos MH, Murphy BR, Collins PL. 2006. Nonsegmented negative-strand viruses as vaccine vectors. J Virol 80:10293–10306. doi: 10.1128/JVI.00919-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dunkel A, Shen S, LaBranche CC, Montefiori D, McGettigan JP. 2015. A bivalent, chimeric rabies virus expressing simian immunodeficiency virus envelope induces multifunctional antibody responses. AIDS Res Hum Retroviruses 31:1126–1138. doi: 10.1089/AID.2014.0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Q, Fan C, Li Q, Zhou S, Huang W, Wang L, Sun C, Wang M, Wu X, Ma J, Li B, Xie L, Wang Y. 2017. Antibody-dependent-cellular-cytotoxicity-inducing antibodies significantly affect the post-exposure treatment of Ebola virus infection. Sci Rep 7:45552. doi: 10.1038/srep45552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shedlock DJ, Bailey MA, Popernack PM, Cunningham JM, Burton DR, Sullivan NJ. 2010. Antibody-mediated neutralization of Ebola virus can occur by two distinct mechanisms. Virology 401:228–235. doi: 10.1016/j.virol.2010.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corti D, Misasi J, Mulangu S, Stanley DA, Kanekiyo M, Wollen S, Ploquin A, Doria-Rose NA, Staupe RP, Bailey M, Shi W, Choe M, Marcus H, Thompson EA, Cagigi A, Silacci C, Fernandez-Rodriguez B, Perez L, Sallusto F, Vanzetta F, Agatic G, Cameroni E, Kisalu N, Gordon I, Ledgerwood JE, Mascola JR, Graham BS, Muyembe-Tamfun JJ, Trefry JC, Lanzavecchia A, Sullivan NJ. 2016. Protective monotherapy against lethal Ebola virus infection by a potently neutralizing antibody. Science 351:1339–1342. doi: 10.1126/science.aad5224. [DOI] [PubMed] [Google Scholar]
- 27.Nimmerjahn F, Gordan S, Lux A. 2015. FcgammaR dependent mechanisms of cytotoxic, agonistic, and neutralizing antibody activities. Trends Immunol 36:325–336. doi: 10.1016/j.it.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 28.He W, Chen CJ, Mullarkey CE, Hamilton JR, Wong CK, Leon PE, Uccellini MB, Chromikova V, Henry C, Hoffman KW, Lim JK, Wilson PC, Miller MS, Krammer F, Palese P, Tan GS. 2017. Alveolar macrophages are critical for broadly-reactive antibody-mediated protection against influenza A virus in mice. Nat Commun 8:846. doi: 10.1038/s41467-017-00928-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamada DH, Elsaesser H, Lux A, Timmerman JM, Morrison SL, de la Torre JC, Nimmerjahn F, Brooks DG. 2015. Suppression of Fcgamma-receptor-mediated antibody effector function during persistent viral infection. Immunity 42:379–390. doi: 10.1016/j.immuni.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bournazos S, Ravetch JV. 2017. Fcgamma receptor function and the design of vaccination strategies. Immunity 47:224–233. doi: 10.1016/j.immuni.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fisher CR, Streicker DG, Schnell MJ. 2018. The spread and evolution of rabies virus: conquering new frontiers. Nat Rev Microbiol 16:241–255. doi: 10.1038/nrmicro.2018.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conzelmann KK, Schnell M. 1994. Rescue of synthetic genomic RNA analogs of rabies virus by plasmid-encoded proteins. J Virol 68:713–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schnell MJ, Mebatsion T, Conzelmann KK. 1994. Infectious rabies viruses from cloned cDNA. EMBO J 13:4195–4203. doi: 10.1002/j.1460-2075.1994.tb06739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mebatsion T, Schnell MJ, Cox JH, Finke S, Conzelmann KK. 1996. Highly stable expression of a foreign gene from rabies virus vectors. Proc Natl Acad Sci U S A 93:7310–7314. doi: 10.1073/pnas.93.14.7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson RF, Kurup D, Hagen KR, Fisher C, Keshwara R, Papaneri A, Perry DL, Cooper K, Jahrling PB, Wang JT, Ter Meulen J, Wirblich C, Schnell MJ. 2016. An inactivated rabies virus-based Ebola vaccine, FILORAB1, adjuvanted with glucopyranosyl lipid A in stable emulsion confers complete protection in nonhuman primate challenge models. J Infect Dis 214:S342–S354. doi: 10.1093/infdis/jiw231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kurup D, Wirblich C, Feldmann H, Marzi A, Schnell MJ. 2015. Rhabdovirus-based vaccine platforms against henipaviruses. J Virol 89:144–154. doi: 10.1128/JVI.02308-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Papaneri AB, Wirblich C, Cooper K, Jahrling PB, Schnell MJ, Blaney JE. 2012. Further characterization of the immune response in mice to inactivated and live rabies vaccines expressing Ebola virus glycoprotein. Vaccine 30:6136–6141. doi: 10.1016/j.vaccine.2012.07.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perrin P, Morgeaux S. 1995. Inactivation of DNA by beta-propiolactone. Biologicals 23:207–211. doi: 10.1006/biol.1995.0034. [DOI] [PubMed] [Google Scholar]
- 39.Mohan GS, Ye L, Li W, Monteiro A, Lin X, Sapkota B, Pollack BP, Compans RW, Yang C. 2015. Less is more: Ebola virus surface glycoprotein expression levels regulate virus production and infectivity. J Virol 89:1205–1217. doi: 10.1128/JVI.01810-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mittler E, Kolesnikova L, Hartlieb B, Davey R, Becker S. 2011. The cytoplasmic domain of Marburg virus GP modulates early steps of viral infection. J Virol 85:8188–8196. doi: 10.1128/JVI.00453-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blaney JE, Wirblich C, Papaneri AB, Johnson RF, Myers CJ, Juelich TL, Holbrook MR, Freiberg AN, Bernbaum JG, Jahrling PB, Paragas J, Schnell MJ. 2011. Inactivated or live-attenuated bivalent vaccines that confer protection against rabies and Ebola viruses. J Virol 85:10605–10616. doi: 10.1128/JVI.00558-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Warfield KL, Alves DA, Bradfute SB, Reed DK, VanTongeren S, Kalina WV, Olinger GG, Bavari S. 2007. Development of a model for marburgvirus based on severe-combined immunodeficiency mice. Virol J 4:108. doi: 10.1186/1743-422X-4-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. 1994. FcR gamma chain deletion results in pleiotrophic effector cell defects. Cell 76:519–529. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 44.Ackerman ME, Alter G. 2013. Opportunities to exploit non-neutralizing HIV-specific antibody activity. Curr HIV Res 11:365–377. doi: 10.2174/1570162X113116660058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Willet M, Kurup D, Papaneri A, Wirblich C, Hooper JW, Kwilas SA, Keshwara R, Hudacek A, Beilfuss S, Rudolph G, Pommerening E, Vos A, Neubert A, Jahrling P, Blaney JE, Johnson RF, Schnell MJ. 2015. Preclinical development of inactivated rabies virus-based polyvalent vaccine against rabies and filoviruses. J Infect Dis 212:S414–S424. doi: 10.1093/infdis/jiv251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clegg CH, Roque R, Perrone LA, Rininger JA, Bowen R, Reed SG. 2014. GLA-AF, an emulsion-free vaccine adjuvant for pandemic influenza. PLoS One 9:e88979. doi: 10.1371/journal.pone.0088979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arias MA, Van Roey GA, Tregoning JS, Moutaftsi M, Coler RN, Windish HP, Reed SG, Carter D, Shattock RJ. 2012. Glucopyranosyl lipid adjuvant (GLA), a synthetic TLR4 agonist, promotes potent systemic and mucosal responses to intranasal immunization with HIVgp140. PLoS One 7:e41144. doi: 10.1371/journal.pone.0041144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coler RN, Baldwin SL, Shaverdian N, Bertholet S, Reed SJ, Raman VS, Lu X, DeVos J, Hancock K, Katz JM, Vedvick TS, Duthie MS, Clegg CH, Van Hoeven N, Reed SG. 2010. A synthetic adjuvant to enhance and expand immune responses to influenza vaccines. PLoS One 5:e13677. doi: 10.1371/journal.pone.0013677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.World Health Organization. 2007. Rabies vaccines WHO position paper. Wkly Epidemiol Rec 82:425–436. [PubMed] [Google Scholar]
- 50.Kishishita N, Takeda N, Anuegoonpipat A, Anantapreecha S. 2013. Development of a pseudotyped-lentiviral-vector-based neutralization assay for chikungunya virus infection. J Clin Microbiol 51:1389–1395. doi: 10.1128/JCM.03109-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Montefiori DC. 2009. Measuring HIV neutralization in a luciferase reporter gene assay. Methods Mol Biol 485:395–405. doi: 10.1007/978-1-59745-170-3_26. [DOI] [PubMed] [Google Scholar]
- 52.Flyak AI, Ilinykh PA, Murin CD, Garron T, Shen X, Fusco ML, Hashiguchi T, Bornholdt ZA, Slaughter JC, Sapparapu G, Klages C, Ksiazek TG, Ward AB, Saphire EO, Bukreyev A, Crowe JE Jr.. 2015. Mechanism of human antibody-mediated neutralization of Marburg virus. Cell 160:893–903. doi: 10.1016/j.cell.2015.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moore SM, Hanlon CA. 2010. Rabies-specific antibodies: measuring surrogates of protection against a fatal disease. PLoS Negl Trop Dis 4:e595. doi: 10.1371/journal.pntd.0000595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McGettigan JP. 2010. Experimental rabies vaccines for humans. Expert Rev Vaccines 9:1177–1186. doi: 10.1586/erv.10.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Faul EJ, Aye PP, Papaneri AB, Pahar B, McGettigan JP, Schiro F, Chervoneva I, Montefiori DC, Lackner AA, Schnell MJ. 2009. Rabies virus-based vaccines elicit neutralizing antibodies, poly-functional CD8+ T cell, and protect rhesus macaques from AIDS-like disease after SIV(mac251) challenge. Vaccine 28:299–308. doi: 10.1016/j.vaccine.2009.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blaney JE, Marzi A, Willet M, Papaneri AB, Wirblich C, Feldmann F, Holbrook M, Jahrling P, Feldmann H, Schnell MJ. 2013. Antibody quality and protection from lethal Ebola virus challenge in nonhuman primates immunized with rabies virus based bivalent vaccine. PLoS Pathog 9:e1003389. doi: 10.1371/journal.ppat.1003389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kajihara M, Marzi A, Nakayama E, Noda T, Kuroda M, Manzoor R, Matsuno K, Feldmann H, Yoshida R, Kawaoka Y, Takada A. 2012. Inhibition of Marburg virus budding by nonneutralizing antibodies to the envelope glycoprotein. J Virol 86:13467–13474. doi: 10.1128/JVI.01896-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith P, DiLillo DJ, Bournazos S, Li F, Ravetch JV. 2012. Mouse model recapitulating human Fcgamma receptor structural and functional diversity. Proc Natl Acad Sci U S A 109:6181–6186. doi: 10.1073/pnas.1203954109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bruhns P. 2012. Properties of mouse and human IgG receptors and their contribution to disease models. Blood 119:5640–5649. doi: 10.1182/blood-2012-01-380121. [DOI] [PubMed] [Google Scholar]
- 60.Matrajt L, Britton T, Halloran ME, Longini IM. Jr, 2015. One versus two doses: what is the best use of vaccine in an influenza pandemic? Epidemics 13:17–27. doi: 10.1016/j.epidem.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kosuwon P, Sutra S, Kosalaraksa P. 2004. Advantage of a two-dose versus one-dose varicella vaccine in healthy non-immune teenagers and young adults. Southeast Asian J Trop Med Public Health 35:697–701. [PubMed] [Google Scholar]
- 62.Bernard KW, Roberts MA, Sumner J, Winkler WG, Mallonee J, Baer GM, Chaney R. 1982. Human diploid cell rabies vaccine. Effectiveness of immunization with small intradermal or subcutaneous doses. JAMA 247:1138–1142. doi: 10.1001/jama.1982.03320330034022. [DOI] [PubMed] [Google Scholar]
- 63.Toellner KM, Sze DM, Zhang Y. 2018. What are the primary limitations in B-cell affinity maturation, and how much affinity maturation can we drive with vaccination? A role for antibody feedback. Cold Spring Harb Perspect Biol 10:a028795. doi: 10.1101/cshperspect.a028795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Publicover J, Ramsburg E, Rose JK. 2004. Characterization of nonpathogenic, live, viral vaccine vectors inducing potent cellular immune responses. J Virol 78:9317–9324. doi: 10.1128/JVI.78.17.9317-9324.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.de Vries RD, Rimmelzwaan GF. 2016. Viral vector-based influenza vaccines. Hum Vaccin Immunother 12:2881–2901. doi: 10.1080/21645515.2016.1210729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Howell KA, Brannan JM, Bryan C, McNeal A, Davidson E, Turner HL, Vu H, Shulenin S, He S, Kuehne A, Herbert AS, Qiu X, Doranz BJ, Holtsberg FW, Ward AB, Dye JM, Aman MJ. 2017. Cooperativity enables non-neutralizing antibodies to neutralize ebolavirus. Cell Rep 19:413–424. doi: 10.1016/j.celrep.2017.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qiu X, Wong G, Audet J, Bello A, Fernando L, Alimonti JB, Fausther-Bovendo H, Wei H, Aviles J, Hiatt E, Johnson A, Morton J, Swope K, Bohorov O, Bohorova N, Goodman C, Kim D, Pauly MH, Velasco J, Pettitt J, Olinger GG, Whaley K, Xu B, Strong JE, Zeitlin L, Kobinger GP. 2014. Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature 514:47–53. doi: 10.1038/nature13777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hangartner L, Zellweger RM, Giobbi M, Weber J, Eschli B, McCoy KD, Harris N, Recher M, Zinkernagel RM, Hengartner H. 2006. Nonneutralizing antibodies binding to the surface glycoprotein of lymphocytic choriomeningitis virus reduce early virus spread. J Exp Med 203:2033–2042. doi: 10.1084/jem.20051557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Holl V, Peressin M, Decoville T, Schmidt S, Zolla-Pazner S, Aubertin AM, Moog C. 2006. Nonneutralizing antibodies are able to inhibit human immunodeficiency virus type 1 replication in macrophages and immature dendritic cells. J Virol 80:6177–6181. doi: 10.1128/JVI.02625-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Holl V, Peressin M, Moog C. 2009. Antibody-mediated Fcgamma receptor-based mechanisms of HIV inhibition: recent findings and new vaccination strategies. Viruses 1:1265–1294. doi: 10.3390/v1031265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Excler JL, Ake J, Robb ML, Kim JH, Plotkin SA. 2014. Nonneutralizing functional antibodies: a new “old” paradigm for HIV vaccines. Clin Vaccine Immunol 21:1023–1036. doi: 10.1128/CVI.00230-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zolla-Pazner S. 2016. Non-neutralizing antibody functions for protection and control HIV in humans and SIV and SHIV in non-human primates. AIDS 30:2551–2553. doi: 10.1097/QAD.0000000000001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Singh K, Marasini B, Chen X, Spearman P. 2018. A novel Ebola virus antibody-dependent cell-mediated cytotoxicity (Ebola ADCC) assay. J Immunol Methods 460:10–16. doi: 10.1016/j.jim.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Terajima M, Cruz J, Co MD, Lee JH, Kaur K, Wrammert J, Wilson PC, Ennis FA. 2011. Complement-dependent lysis of influenza A virus-infected cells by broadly cross-reactive human monoclonal antibodies. J Virol 85:13463–13467. doi: 10.1128/JVI.05193-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rattan A, Pawar SD, Nawadkar R, Kulkarni N, Lal G, Mullick J, Sahu A. 2017. Synergy between the classical and alternative pathways of complement is essential for conferring effective protection against the pandemic influenza A(H1N1) 2009 virus infection. PLoS Pathog 13:e1006248. doi: 10.1371/journal.ppat.1006248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.O'Brien KB, Morrison TE, Dundore DY, Heise MT, Schultz-Cherry S. 2011. A protective role for complement C3 protein during pandemic 2009 H1N1 and H5N1 influenza A virus infection. PLoS One 6:e17377. doi: 10.1371/journal.pone.0017377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lilienthal GM, Rahmoller J, Petry J, Bartsch YC, Leliavski A, Ehlers M. 2018. Potential of murine IgG1 and human IgG4 to inhibit the classical complement and Fcgamma receptor activation pathways. Front Immunol 9:958. doi: 10.3389/fimmu.2018.00958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stoiber H, Clivio A, Dierich MP. 1997. Role of complement in HIV infection. Annu Rev Immunol 15:649–674. doi: 10.1146/annurev.immunol.15.1.649. [DOI] [PubMed] [Google Scholar]
- 79.Xu Y, Zhang C, Jia L, Wen C, Liu H, Wang Y, Sun Y, Huang L, Zhou Y, Song H. 2009. A novel approach to inhibit HIV-1 infection and enhance lysis of HIV by a targeted activator of complement. Virol J 6:123. doi: 10.1186/1743-422X-6-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dubois Cauwelaert N, Desbien AL, Hudson TE, Pine SO, Reed SG, Coler RN, Orr MT. 2016. The TLR4 agonist vaccine adjuvant, GLA-SE, requires canonical and atypical mechanisms of action for TH1 induction. PLoS One 11:e0146372. doi: 10.1371/journal.pone.0146372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Orr MT, Desbien AL, Cauwelaert ND, Reed SG. 2016. Mechanisms of activity of the combination TLR4 agonist and emulsion adjuvant GLA-SE. J Immunol 196(1 Suppl):75.2. [Google Scholar]
- 82.Daddario-DiCaprio KM, Geisbert TW, Geisbert JB, Stroher U, Hensley LE, Grolla A, Fritz EA, Feldmann F, Feldmann H, Jones SM. 2006. Cross-protection against Marburg virus strains by using a live, attenuated recombinant vaccine. J Virol 80:9659–9666. doi: 10.1128/JVI.00959-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Alves DA, Glynn AR, Steele KE, Lackemeyer MG, Garza NL, Buck JG, Mech C, Reed DS. 2010. Aerosol exposure to the Angola strain of Marburg virus causes lethal viral hemorrhagic fever in cynomolgus macaques. Vet Pathol 47:831–851. doi: 10.1177/0300985810378597. [DOI] [PubMed] [Google Scholar]
- 84.Cross RW, Fenton KA, Geisbert JB, Ebihara H, Mire CE, Geisbert TW. 2015. Comparison of the pathogenesis of the Angola and Ravn strains of Marburg virus in the outbred guinea pig model. J Infect Dis 212:S258–S270. doi: 10.1093/infdis/jiv182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Thi EP, Mire CE, Ursic-Bedoya R, Geisbert JB, Lee ACH, Agans KN, Robbins M, Deer DJ, Fenton KA, MacLachlan I, Geisbert TW. 2014. Marburg virus infection in nonhuman primates: therapeutic treatment by lipid-encapsulated siRNA. Sci Transl Med 6:250ra116. doi: 10.1126/scitranslmed.3009706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Geisbert TW, Geisbert JB, Leung A, Daddario-DiCaprio KM, Hensley LE, Grolla A, Feldmann H. 2009. Single-injection vaccine protects nonhuman primates against infection with Marburg virus and three species of Ebola virus. J Virol 83:7296–7304. doi: 10.1128/JVI.00561-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Towner JS, Khristova ML, Sealy TK, Vincent MJ, Erickson BR, Bawiec DA, Hartman AL, Comer JA, Zaki SR, Stroher U, Gomes da Silva F, del Castillo F, Rollin PE, Ksiazek TG, Nichol ST. 2006. Marburgvirus genomics and association with a large hemorrhagic fever outbreak in Angola. J Virol 80:6497–6516. doi: 10.1128/JVI.00069-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The MARV GP sequence was deposited in GenBank under accession number MK375262.