

Hepatitis B virus (HBV) is a ubiquitous human pathogen, and HBV infection is a major global health burden. Chronic HBV infection is associated with the development of liver diseases, including fulminant hepatitis, hepatic fibrosis, cirrhosis, and hepatocellular carcinoma. A currently approved vaccine can prevent HBV infection, and medications are able to reduce viral loads and prevent liver disease progression. However, current treatments rarely achieve a cure for chronic infection. Thus, it is important to gain insight into the mechanisms of HBV replication. In this study, we found that the host factor RBM24 is involved in pregenomic RNA (pgRNA) packaging and regulates HBV replication. These findings highlight a potential target for antiviral therapeutics of HBV infection.
KEYWORDS: HBV polymerase, HBV epsilon element, RBM24, interaction, pgRNA packaging
ABSTRACT
Encapsidation of pregenomic RNA (pgRNA) is a crucial step in hepatitis B virus (HBV) replication. Binding by viral polymerase (Pol) to the epsilon stem-loop (ε) on the 5′-terminal region (TR) of pgRNA is required for pgRNA packaging. However, the detailed mechanism is not well understood. RNA-binding motif protein 24 (RBM24) inhibits core translation by binding to the 5′-TR of pgRNA. Here, we demonstrate that RBM24 is also involved in pgRNA packaging. RBM24 directly binds to the lower bulge of ε via RNA recognition submotifs (RNPs). RBM24 also interacts with Pol in an RNA-independent manner. The alanine-rich domain (ARD) of RBM24 and the reverse transcriptase (RT) domain of Pol are essential for binding between RBM24 and Pol. In addition, overexpression of RBM24 increases Pol-ε interaction, whereas RBM24 knockdown decreases the interaction. RBM24 was able to rescue binding between ε and mutant Pol lacking ε-binding activity, further showing that RBM24 mediates the interaction between Pol and ε by forming a Pol-RBM24-ε complex. Finally, RBM24 significantly promotes the packaging efficiency of pgRNA. In conclusion, RBM24 mediates Pol-ε interaction and formation of a Pol-RBM24-ε complex, which inhibits translation of pgRNA and results in pgRNA packing into capsids/virions for reverse transcription and DNA synthesis.
IMPORTANCE Hepatitis B virus (HBV) is a ubiquitous human pathogen, and HBV infection is a major global health burden. Chronic HBV infection is associated with the development of liver diseases, including fulminant hepatitis, hepatic fibrosis, cirrhosis, and hepatocellular carcinoma. A currently approved vaccine can prevent HBV infection, and medications are able to reduce viral loads and prevent liver disease progression. However, current treatments rarely achieve a cure for chronic infection. Thus, it is important to gain insight into the mechanisms of HBV replication. In this study, we found that the host factor RBM24 is involved in pregenomic RNA (pgRNA) packaging and regulates HBV replication. These findings highlight a potential target for antiviral therapeutics of HBV infection.
INTRODUCTION
Hepatitis B virus (HBV) infection remains a huge threat to human health, resulting in severe liver diseases, such as fulminant hepatitis, hepatic fibrosis, cirrhosis, and hepatocellular carcinoma (1). HBV is a noncytopathic, hepatotropic virus that belongs to the Hepadnaviridae family, with a partially double-stranded, relaxed circular DNA (rcDNA) genome of 3.2 kb. After cell infection mediated by sodium taurocholate cotransporting polypeptide (NTCP) (2), the 3.2-kb rcDNA genome enters the nucleus and transforms into a nucleosome-decorated covalently closed circular DNA (cccDNA) minichromosome. This cccDNA acts as a transcription template to produce the four major mRNAs from different initiation sites, including the 3.5-kb pregenomic RNA (pgRNA)/preC RNA and 2.4-, 2.1-, and 0.7-kb subgenomic RNAs (3). In addition to encoding both the polymerase and core protein, pgRNA also serves as the template for reverse transcription and thus plays an essential role in HBV replication (4). A stem-loop structure (epsilon [ε]) at the 5′-terminal region (TR) of pgRNA is recognized by polymerase (Pol) to form a Pol-ε complex, followed by recruitment of the core protein to mediate pgRNA encapsulation. The Pol-ε interaction constitutes the first step of reverse transcription initiation to yield progeny viral rcDNA (5–7). Thus, the Pol-ε interaction initiates not only encapsidation but also reverse transcription of pgRNA (6, 7). The internal bulge structure of ε, the Pol central region of the terminal protein (TP) domain, and the N-terminal region of the reverse transcriptase (RT) domain are essential for HBV Pol-ε binding in vitro (8). In contrast, pgRNA packaging may require the entire Pol protein and all the structural elements of ε plus a closely spaced 5′ cap (9). In addition, some duck hepatitis B virus (DHBV) RNA mutations have no effect on Pol binding, although they abolish its function in pgRNA packaging and protein priming (10, 11). Overall, the structural elements and detailed mechanism of pgRNA encapsidation are not fully understood.
Some host factors have been identified as being involved in mediating the interaction between the Pol and pgRNA. The Hsp90 complex, which includes the 90-kDa heat shock protein (Hsp90) and several cochaperones, such as Hsp70, Hsp40, Hop/p60, and p23, was found to be required for HBV Pol-ε interaction in vitro and pgRNA packaging in vivo (8, 12). Nonetheless, pgRNA packaging could not be reproduced in vitro (11, 13). Furthermore, the HBV Pol-ε complex formed in vitro is inactive in protein priming in the presence of the Hsp90 complex (8). These results suggest that additional host factors may be required to establish a functional HBV Pol-ε interaction.
By regulating the stability and/or alternative splicing of mRNAs, RNA-binding protein 24 (RBM24) has been shown to have an important function in cardiovascular development and myogenesis (14–16). RBM24 contains a conserved RNA recognition motif (RRM) that consists of RNP1 and RNP2 submotifs. We previously reported that RBM24 is involved in hepatitis C virus (HCV) translation and replication by interacting with HCV RNA sequences (17). In addition, RBM24 plays dual roles in HBV replication, stabilizing HBV RNA and inhibiting core translation via binding to both 5′- and 3′-TR sequences (18). Its capacity to bind to 5′-TR sequences of pgRNA suggests that RBM24 may also participate in pgRNA encapsidation.
In this study, we show that RBM24 is able to interact with both ε and Pol. The RRM domain recognizes the lower bulge of ε via RNPs, whereas the alanine-rich domain (ARD) of RBM24 and the RT domain of Pol are essential for binding. RBM24 promotes Pol-ε interaction and enhances pgRNA packaging, and RBM24 is incorporated into the HBV capsid. Our results thus demonstrate that RBM24 is a novel host factor involved in pgRNA packing by mediating the interaction between Pol and ε.
RESULTS
RBM24 directly binds to ε of the HBV TR sequence in vitro.
RBM24 inhibits translation of the HBV core protein via RBM24-TR interaction (18). In this study, we further mapped the binding domain of the HBV TR via a pulldown assay. Purified recombinant RBM24 protein or RBM24 overexpression lysates were incubated with the biotin-labeled TR and a TR sequence with a deletion of the ε element (TRΔε); biotin-labeled p21-ARE (AU-rich element) and Saccharomyces cerevisiae tRNA served as the positive and negative controls, respectively. The RNA-protein complex pulled down by streptavidin beads was detected via Western blotting using an anti-RBM24 antibody (Fig. 1A). In the absence or presence of other cellular proteins, RBM24 was able to interact with TR RNA, as with the positive control p21-ARE, whereas RBM24 hardly bound to TRΔε or the negative control (yeast tRNA). These data indicated that the ε element was essential for the RBM24-TR interaction. To confirm the binding specificity between RBM24 and ε, RBM24 binding to biotin-labeled ε RNA was competitively inhibited by adding excess cold ε probe, and the result indicated that RBM24 specifically binds to ε (Fig. 1B). To further dissect the substructural domain of ε targeted by RBM24, ε RNAs with deletions of certain domains were synthesized and tested (Fig. 1C). The results indicated that RBM24 did not bind to the ε element with a deletion of the lower bulge (B loop, nucleotides [nt] 1860 to 1865) (19). Thus, RBM24 specifically recognizes ε RNA via the lower bulge.
FIG 1.

RBM24 specifically binds to HBV ε. HEK293T cells were transfected with 24 μg of pRBM24 in 100-mm dishes, and cell lysates containing RBM24 protein were harvested at 48 hpt. Recombinant human RBM24 (rhRBM24) proteins were purified in vitro. (A) Lysates (400 μg) of cells transfected with pRBM24 or 50 pM rhRBM24 were incubated with 2 μg of biotin-labeled p21-ARE (positive control), yeast tRNA (negative control), the TR, or the TRΔε fragment, followed by pulldown with streptavidin beads and assessment by Western blotting. IB, immunoblotting. (B) Lysates (400 μg) of cells transfected with pRBM24 or 50 pM rhRBM24 were incubated with 2 μg of biotin-labeled p21-ARE, yeast tRNA, or the ε RNA fragment. An excess amount of cold unlabeled ε RNA (0×, 10×, or 20×) was added to compete with the binding of RBM24 to the biotin-labeled ε fragment, followed by pulldown with streptavidin beads and assessment by Western blotting. (C) The apical loop and internal bulge structural elements were deleted from full-length ε [FL(ε)] to generate FLΔA and FLΔB, respectively. Lysates (400 μg) of cells transfected with pRBM24 or 50 pM rhRBM24 were incubated with 2 μg of biotin-labeled ε, FLΔA, or FLΔB, followed by pulldown with streptavidin beads and assessment by Western blotting. (D) The mutation bulge structural elements, B1-6, ApaB, B1-4, B35, B2346, B15, B1A, B1U, B1G, B5A, B5C, and B5G were constructed, Lysates (400 μg) of cells transfected with pRBM24 or 50 pM rhRBM24 were incubated with 2 μg of biotin-labeled ε mutations. followed by pulldown with streptavidin beads and assessment by Western blotting.
To further determine if the lower bulge acted only as a structural element or if its specific nucleotide sequences were required for RBM24 binding, we constructed base substitution mutants in this region, mainly mutants which had previously been reported (12). The mutant B1-6 (5′→3′) had all 6 nucleotides of the bulge replaced and completely lost the activity to bind RBM24 (Fig. 1D), indicating that some of the bulge sequences may specifically be required for RBM24-ε binding. The mutants ApaB (having substitutions at positions 1, 2, 4, and 5), B1-4, and B35 also lost the ability to bind RBM24, while the B2346 mutant still displayed RBM24-binding activity, which demonstrated that the first and fifth positions may be critical for RBM24 binding. Further investigation showed that the single nucleotide substitution mutants B1A, B1U, B1G, B5A, and B5G still have detectable RBM24-binding activity, and the mutant B5C retained low residual binding. However, changes at the first and fifth positions (B15) completely abolished RBM24 binding (Fig. 1D). Thus, the first and fifth positions of the lower bulge are critical for RBM24-ε binding.
RBM24 binds to Pol in an RNA/ε-independent manner.
As both RBM24 and HBV Pol bind to ε of the 5′-TR, it is interesting to analyze whether RBM24 interacts with Pol. Accordingly, HepG2 cells were cotransfected with the pHA-Pol and pFlag-RBM24 or pFlag-ΔRNP1/2 plasmids. An immunofluorescence (IF) assay clearly showed that both wild-type RBM24 and truncated RBM24 with a deletion of two RNA recognition domains (ΔRNP1/2) colocalized with Pol in the cytoplasm (Fig. 2A). Therefore, RBM24 may interact with Pol in an RNA-binding domain-independent manner.
FIG 2.

Interaction between RBM24 and Pol is independent of RNA/ε. (A) HepG2 cells were cotransfected with plasmids pHA-Pol and pFlag-RBM24, pFlag-ΔRNP1/2, or the control vector. The cells were immunostained with an anti-HA antibody and an anti-Flag antibody at 48 hpt. Nuclei were stained with Hoechst 33258. Higher-magnification images of the selected areas are also shown. Bars, 10 μm for panels a1 to a4, b1 to b4, c1 to c4, and d1 to d4 and 2 μm for panels a5, b5, c5, and d5. (B) HEK293T cells were cotransfected with HA-Pol, pFlag-RBM24, and pCMV-HE or the empty vector and lysed at 48 hpt. Cell lysates were pretreated with RNase A and DNase I or left untreated and subjected to a co-IP assay. An anti-Flag antibody was used for co-IP. The precipitates were analyzed by Western blotting using anti-Flag and anti-HA antibodies. Nucleic acid was detected by agarose gel electrophoresis. Actin served as a loading control. (C) HEK293T cells were cotransfected with pHA-Pol and pFlag-ΔRNP1/2 or the empty vector. The cells were lysed and subjected to a co-IP assay at 48 hpt. An anti-Flag antibody was used for the co-IP. The precipitates were analyzed by Western blotting using anti-Flag and anti-HA antibodies. Actin served as a loading control.
The observed interaction between RBM24 and Pol was further assessed by an immunoprecipitation assay. Both wild-type RBM24 (Fig. 2B) and ΔRNP1/2 (Fig. 2C) were immunoprecipitated with Pol. However, the addition of ε RNA transcribed from pCMV-HE (19) did not enhance this interaction between RBM24 and Pol (Fig. 2B, lanes 5 and 6). RNase A and DNase I did not abolish the interaction between Pol and RBM24 (Fig. 2B, lanes 8 and 9), indicating that RBM24 interacts with Pol in an RNA/ε-independent manner. Nuclease treatment reduced the amounts of interaction between Pol and RBM24 (Fig. 2B, lanes 8 and 9), implying that nuclear acid helps the interaction between Pol and RBM24.
The ARD of RBM24 interacts with the reverse transcriptase domain of HBV polymerase.
We then mapped interaction domains in RBM24 and Pol. In a coimmunoprecipitation (co-IP) assay with RBM24, only the RT and RT plus RNase H (RH) regions of Pol were observed as interacting partners (Fig. 3B), indicating that RBM24 interacts with the RT region of Pol, which has been recognized as a domain important for Pol-ε interaction (20). However, Pol bound to the ARD but not the RRM region of RBM24 (Fig. 3C), which also supports the notion that RBM24 interacts with Pol in an RNA/ε-independent manner. Furthermore, the coimmunoprecipitation assay showed that the ARD region interacts with the RT region (Fig. 3D). Altogether, RBM24 interacts with Pol via the ARD and the RT domain.
FIG 3.

The alanine-rich domain of RBM24 interacts with the reverse transcriptase domain of HBV polymerase. (A) Schematic illustrations of truncated Pol constructs and truncated RBM24 constructs. (B to D) HEK293T cells were cotransfected with the indicated plasmids and lysed at 48 hpt, followed by a co-IP assay using an anti-Flag antibody. The precipitates were analyzed by Western blotting using anti-Flag and anti-HA antibodies. Actin served as a loading control.
RBM24 mediates Pol-ε interaction and formation of the Pol-RBM24-ε complex.
RBM24 binds to both ε and Pol, suggesting that RBM24, Pol, and ε may associate together and even form a complex. To examine this possibility, an immunofluorescence assay was performed to determine colocalization among pgRNA, Pol, and RBM24. In brief, pgRNA was mainly located in the cytoplasm (Fig. 4A, panel b5) and colocalized with RBM24 and Pol (Fig. 4A, panel c5). However, pgRNA did not colocalize with ΔRNP1/2 and Pol (Fig. 4A, panel d5). Thus, the RNPs of RBM24 are essential for colocalization among RBM24, Pol, and pgRNA. For confirmation, we further analyzed colocalization among RBM24, Pol, and pgRNA in pairs by measuring the product of the difference from the mean (PDM) using merged channel velocity software (Fig. 4B) (21), whereby the positive PDM channel represents colocalization. According to the results, pgRNA and RBM24 (Fig. 4B, panel c7), pgRNA and Pol (Fig. 4B, panel c10), and RBM24 and Pol (Fig. 4B, panel c13) were significantly detected in positive PDM channels, demonstrating that pgRNA, RBM24, and Pol mutually colocalize with each other. In contrast, pgRNA and ΔRNP1/2 (Fig. 4B d8) and pgRNA and Pol (Fig. 4B, panel d11) were in negative PDM channels, whereas ΔRNP1/2 and Pol were still detected in the positive PDM channel (Fig. 4B, panel d13), showing that pgRNA did not colocalize with ΔRNP1/2 and Pol. These results suggest that RBM24 mediates the interaction between Pol and pgRNA through its RNP domain.
FIG 4.

RBM24 promotes Pol-ε interaction and formation of the Pol-RBM24-ε complex. (A) HepG2 cells were cotransfected with P−C− (the HBV C-null and Pol-null construct that was used to express the pgRNA), pHA-Pol, and pFlag-RBM24 or pFlag-ΔRNP1/2. At 48 hpt, the cells were fixed and subjected to IF staining and RNA FISH. An anti-HA antibody and an anti-Flag antibody were used as primary antibodies for the staining of Pol and RBM24, respectively. A probe targeting the 5′ end of pgRNA was used for pgRNA staining. Nuclei were stained with Hoechst 33258. Higher-magnification images of the selected area are also shown. Bars, 10 μm for panels a1 to a5, b1 to b5, c1 to c5, and d1 to d5 and 2 μm for panels a6, b6, c6, and d6. (B) The product of the difference from the mean (PDM) for the two channels (pgRNA/Pol, pgRNA/RBM24, or RBM24/Pol) in panels c5 and d5 was analyzed. Volocity software generated a positive (c7, c10, c13, d7, d10, and d13) and a negative (c8, c11, c14, d8, d11, and d14) PDM channel. (C to E) HEK293T cells transfected with the indicated plasmids or siRNA were lysed, followed by a biotin-ε pulldown assay using Dynabeads M-280 streptavidin. The input or pulled-down proteins were detected by Western blotting. Actin served as a loading control.
The interaction among RBM24, Pol, and pgRNA was further analyzed by a biotin pulldown assay (Fig. 4C to E). Consistent with data from a previous study (19), wild-type Pol harvested from cell lysates recognized and bound to ε of HBV pgRNA (Fig. 4C, lane 1), whereas mutant Pol (mutPol) (amino acids [aa] 1 to 175 and 300 to 775), which lacks the ε-binding site but retains the RT region (9), was not pulled down by biotin-ε (Fig. 4C, lane 2). Biotin-ε pulled down not only Pol and RBM24 separately (Fig. 4D, lanes 1, 3, and 5) but also both RBM24 and Pol at the same time (Fig. 4D, lane 4). Notably, overexpression of RBM24 increased the amount of Pol pulled down by biotin-ε compared with pcDNA3.1 (Fig. 4D, lanes 3 and 4), and the amount of Pol pulled down was significantly decreased after RBM24 was knocked down (Fig. 4D, lanes 5 and 6). Interesting, biotin-ε still pulled down mutPol, which lacks ε-binding activity, when RBM24 was overexpressed (Fig. 4D, lanes 7 to 10), although overexpression of ΔRNP1/2 did not enhance the amount of Pol pulled down by biotin-ε (Fig. 4E, lanes 1 and 2). These results further support that RBM24 meditates Pol-ε interaction and formation of the Pol-RBM24-ε complex.
The observed interaction between RBM24 and the RT domain of Pol (Fig. 3B) suggests that the former possibly mediates Pol-ε interaction through the RT domain. To address this, the binding activities of ε and Pol, TP, spacer (SP), RT, or RH were evaluated after knockdown of RBM24. In the control small interfering RNA (siRNA) (siNC) group, ε obviously pulled down Pol, TP, and the RT domain, which was consistent with data from a previous report (20) (Fig. 5A and B). Knockdown of RBM24 significantly decreased the amounts of Pol and the RT domain precipitated by biotin-ε. Thus, RBM24 might interact with RT and mediate the Pol-ε interaction. Further analyses showed that the RT1 motif of RT was not involved in the interaction between RT and RBM24, as RT with the RT1 deletion was still precipitated by RBM24 (Fig. 5C). Moreover, both RT-ε binding and RTΔRT1-ε binding were reduced by RBM24 knockdown (Fig. 5D), indicating that the RT domain but not the RT1 motif is required for RBM24 to mediate the RT-ε interaction. Additionally, RBM24 is not involved in the interaction between TP and ε, as the degree to which the TP domain was pulled down by biotin-ε was same as that of the control group (Fig. 5B).
FIG 5.
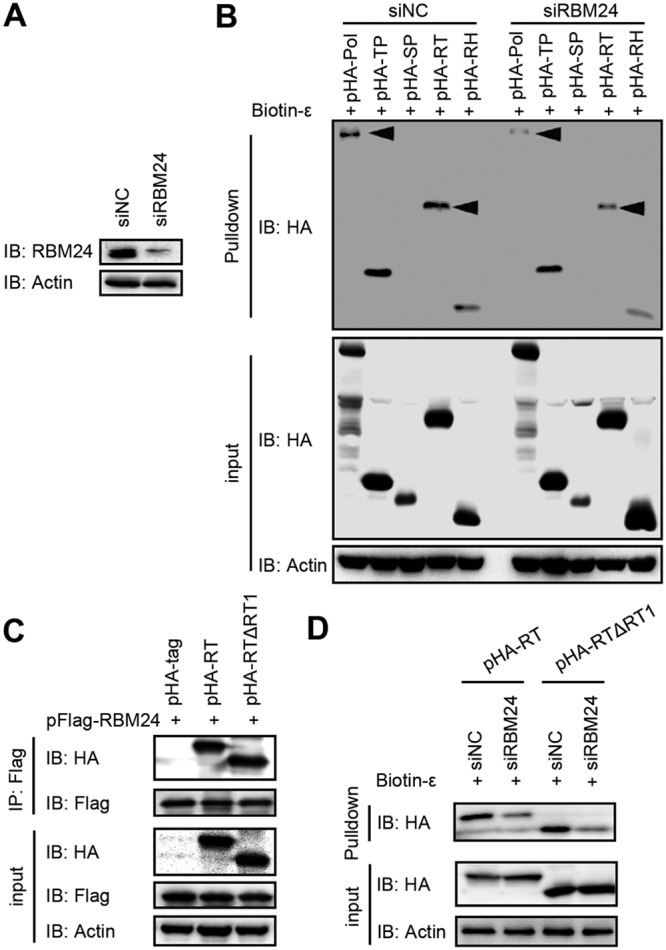
RBM24 mediates Pol-ε interaction via the RT domain. HEK293T cells were transfected with the indicated siRNAs or plasmids. (A) The level of RBM24 was detected by Western blotting. (B) Cell lysates were subjected to a biotin-ε pulldown assay. The input or pulled-down proteins were detected by Western blotting. Actin served as a loading control. (C) Cell lysates were subjected to co-IP with an anti-Flag antibody. The precipitates were detected by Western blotting. (D) Cell lysates were subjected to a biotin-ε pulldown assay. The input or pulled-down proteins were detected by Western blotting. Actin served as a loading control.
RBM24 promotes pgRNA packaging.
As the interaction between Pol and ε triggers pgRNA packaging, we further analyzed whether RBM24 has any effect on pgRNA packaging by detecting the levels of HBV capsid and packaged pgRNA (Fig. 6A and E). Both knockdown and overexpression of RBM24 decreased the amounts of HBV DNAs and total HBV RNAs (Fig. 6A), as reported previously (18). Knockdown of RBM24 increased the amounts of core protein and capsids, while significantly decreased amounts of packaged pgRNA and minus-strand DNA [(−)DNA] were observed (Fig. 6A, lanes 1 and 2). RBM24 overexpression reduced the amounts of core protein and capsids but increased pgRNA and (−)DNA packaging into capsids (Fig. 6A, lanes 3 and 4). Furthermore, knockdown of RBM24 decreased the pgRNA packaging efficiency (ratio of packaged pgRNAs in the siRBM24 and siNC groups) (Fig. 6B, lanes 1 and 2), whereas overexpression of RBM24 significantly increased the pgRNA packaging efficiency (ratio of packaged pgRNAs in the pRBM24 and pcDNA3.1 groups) (Fig. 6B, lanes 3 and 4). The packaging efficiency was further normalized with the core protein level, and the result showed that knockdown of RBM24 decreased while overexpression of RBM24 increased the pgRNA packaging efficiency (Fig. 6B, lines 1′, 2′, 3′, and 4′). These findings suggest that RBM24 enhances HBV pgRNA packaging. To confirm the above-described results, we assessed the level of pgRNA packaged into capsids by a coimmunoprecipitation assay using an anticore antibody. The relative pgRNA packaging efficiency was normalized to the level of pgRNA in capsids (C) to the 5% input total pgRNA (T). As shown in Fig. 6C and D, silencing of RBM24 decreased the pgRNA packaging efficiency, whereas overexpression of RBM24 increased it. RBM24 also promoted pgRNA packaging when a core-null HBV expression plasmid (pHBV1.3ΔC) and an exogenous core expression plasmid (pHA-core) were used (Fig. 6E). Taken together, these results indicate that RBM24 is involved in pgRNA packaging, which was further supported by (−)DNA results (Fig. 6A and E).
FIG 6.

RBM24 promotes pgRNA packaging. HepG2 cells were cotransfected with the indicated plasmids or siRNA. (A) Cell lysates were subjected to native agarose gel electrophoresis followed by transfer to a nitrocellulose membrane for the detection of core/capsid using anti-HBc or to a nylon membrane for the detection of encapsidated pgRNA and (−)DNA using a biotin-labeled HBV plus-strand-specific riboprobe (nt 1890 to 2306) against the 5′ end of pgRNA and HBV minus-strand-specific riboprobes, respectively. HBV transcripts were detected by Northern blotting. (B) Relative band intensities for encapsidated pgRNA from three independent experiments were quantified using NIH ImageJ software, and the results are presented as a percentage of the RNA in the siNC control or further normalized with the level of core. (C) Capsids in cell lysates were immunoprecipitated with an anticore antibody, and total pgRNA from a 5% input (T) and encapsidated pgRNA from the immunoprecipitated capsid (C) were detected by Northern blotting. The levels of RBM24 and core were detected by Western blotting using anti-RBM24 and anticore antibodies, respectively. Actin served as a loading control. (D) Relative band intensities for encapsidated pgRNA from three independent experiments were quantified using NIH ImageJ software, and the results are presented as a percentage of the RNA in the siNC control. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (E) Cells were cotransfected with pHBV1.3ΔC, pHA-core, and siNC or siRBM24, pcDNA3.1, or pRBM24; lysed; and subjected to the assay described above for panel A.
RBM24 is incorporated into capsids.
RBM24 mediates Pol-ε interaction by binding to Pol and ε, suggesting that RBM24 may be incorporated into viral nucleocapsids. Due to the low endogenous expression level of RBM24, HepAD38 cells were transfected with pHA-RBM24, and cell lysates were subjected to OptiPrep density gradient analysis. As shown in Fig. 7A, isolated fractions were subjected to an enzyme-linked immunosorbent assay (ELISA) for detecting HBsAg to identify nucleocapsid/virus fractions. As shown in Fig. 7A, HBsAg was mainly distributed among fractions 7, 8, and 9, treated either with or without RNase A. However, HBsAg was not detected in all factions, demonstrating that nonencapsidated proteins had been digested by proteinase K. This result verifies that nucleocapsids/virions were mainly present in fractions 7, 8, and 9. Moreover, the core protein was primarily detected in fractions 7 to 10 (Fig. 7A, panel a), which contained the nucleocapsid/virus fractions. Overexpression of RBM24 did not change the location of the core fraction, whereas RBM24 was found in fractions 1 to 12, either treated with RNase A to remove the cellular RNA complexes that may be associated with RBM24 or without RNase A (Fig. 7A, panels b and c). Nonetheless, when the cell lysates were further treated with proteinase K to remove nonencapsidated proteins, RBM24 was still detected in fractions 8 and 9, containing nucleocapsids/virions. Isolated fractions were further subjected to a dot blot assay for HBV DNA detection. The peak of RBM24 strongly overlapped that of the core protein and HBV DNA (Fig. 7A). These results suggest that RBM24 is incorporated into the HBV capsids.
FIG 7.

RBM24 is incorporated into the capsid. (A) pHA-RBM24 was transfected into HepAD38 cells, and cell lysates were subjected to an OptiPrep density gradient and treated without or with RNase A or RNase A plus proteinase K (Pro. K). The gradients were fractionated into 15 fractions. For each fraction, HA-RBM24 and the core protein were detected by Western blotting, HBV DNA was detected by dot blotting, and HBsAg was detected by an ELISA. OD450, optical density at 450 nm. (B) HepG2 cells were cotransfected with pHY106 and pHA-RBM24. At 48 hpt, the cells were fixed and subjected to IF staining and RNA FISH. An anti-HA antibody and an anti-Flag antibody were used as primary antibodies for identifying Pol and RBM24, respectively. A probe targeting the 5′ end of pgRNA was used for identifying pgRNA. Nuclei were stained with Hoechst 33258. (C) The PDM for the two channels (pgRNA/core, pgRNA/RBM24, or RBM24/core) was analyzed. Volocity software generated a positive (a1, a4, and a7) and a negative (a2, a5, and a8) PDM channel. (D) HBV virions were purified from serum of patients with HBV infection. A total of 50 μg of HBV virions (lines 4 and 5) or 100 μg of HepG2 cell lysates (lines 4 and 5) was digested with or without proteinase K, and the samples were resolved by SDS-PAGE. Purified rhRBM24 was loaded as a control. RBM24, the positive control eIF4E, and core were detected by Western blotting using the anti-RBM24 antibody, anti-eIF4E antibody, and anticore antibody.
For confirmation, we further examined the localization of RBM24, core protein, and pgRNA (Fig. 7B). The results showed that pgRNA and RBM24 (Fig. 7C, panel a1), pgRNA and core (Fig. 7C, panel a4), and RBM24 and core (Fig. 7C, panel a7) were significantly detected in positive PDM channels. These results indicate that RBM24 colocalizes with the core protein and pgRNA, which was consistent with the data shown in Fig. 6A.
To further assess whether endogenous RBM24 is incorporated into capsids, we detected the existence of endogenous RBM24 in HBV virions purified from serum of patients with chronic HBV infection. RBM24 and the positive control, eukaryotic translation initiation factor 4E (eIF4E), were still clearly detectable in HBV virions after proteinase K treatment (Fig. 7D, line 5), which means that endogenous RBM24 is incorporated into capsids.
DISCUSSION
The binding of Pol to ε, which is located at the 5′ end of pgRNA, triggers pgRNA packaging into the nucleocapsid and the initiation of reverse transcription (6, 7). However, the detailed mechanism has yet to be fully elucidated. Here, we identify that the novel cellular factor RBM24 mediates interaction between HBV Pol and ε and promotes pgRNA packaging. RBM24 binds to the lower loop of ε (B loop) via its RNP domain while interacting with the RT domain of Pol through the ARD in an RNA/ε-independent manner. RBM24 mediates Pol-ε interaction and is packaged into the capsid together with pgRNA.
In HBV infection, Pol must first bind to ε on the 5′ end of pgRNA to become competent for pgRNA packaging and reverse transcription (22–25). Biochemical studies of the interaction between HBV Pol and ε RNA have been hampered by a lack of appropriate in vitro systems. It has been reported that the TP and RT domains of Pol are required for ε binding (26–29), although it remains to be determined whether these sequences are directly involved in RNA binding. In fact, purified HBV RT showed little or no ε-binding activity (8). The Hsp90 chaperone system and ATP hydrolysis might assist in RT folding and further enhance RT-ε binding in both HBV and DHBV (8, 12, 24). Hsp90 interacts only with Pol (30) and not RNA, and the addition of the Hsp90 complex results in only 30% of HBV Pol binding to ε (8), suggesting that additional host factors may be required for Pol-ε interaction. As an RNA-binding protein, RBM24, which has been shown to target the 3′-TR and 5′-TR of HBV RNA (18), was found in our study to directly bind to both ε and Pol, forming a complex. Knockdown of RBM24 significantly decreased binding between ε and Pol or RT, which highlights the fact that endogenous RBM24 plays an essential role as a bridge in Pol-ε binding. Thus, RBM24 mediates Pol-ε interaction.
It has been generally accepted that a conformational change in Pol is a precondition for Pol-pgRNA binding, as is the case for the Hsp90 chaperone to mediate Pol-ε binding (8). The C-terminal region (TP3) of TP, the conserved cysteines in the spacer and RT, and the RT1 motif in the RT domain of Pol are all required for ε binding (20). mutPol with a deletion of the junction between TP and RT (positions 176 to 299 [retains the T3 and RT1 regions]), which lacks ε-binding activity, was explained as causing misfolding of Pol (9). RBM24 rescues the binding activity between mutPol and ε (Fig. 4D), suggesting that the Pol-RBM24 interaction might induce a mutPol conformation change for ε binding. We thus hypothesize a model in which RBM24 binds directly to ε via RNP domains and interacts with the RT domain of Pol via the ARD of RBM24, mediating the interaction between Pol and pgRNA. RBM24 binding to Pol induces a conformational transition of Pol together with other host factors, such as Hsp90, and further forms a more stable complex of Pol-RBM24-ε, which recruits the core protein and forms the capsid/virion for reverse transcription of pgRNA and HBV DNA synthesis. This hypothesis requires further investigation using purified Pol in vitro.
pgRNA serves as both a template for reverse transcription and the mRNA template for translation of the Pol and core proteins. Pol may function as a switch for template selection by binding to ε at the 5′ end of pgRNA, which suppresses the translation of core and results in pgRNA packaging for reverse transcription (31, 32). In addition to viral Pol, a host factor directly involved in pgRNA packaging has not yet been reported. The DDX3 DEAD-box RNA helicase is incorporated into nucleocapsids in an HBV Pol-dependent manner but does not affect pgRNA encapsidation (33, 34). We have demonstrated that overexpression of RBM24 inhibits translation of core (18), and in the present study, RBM24 knockdown markedly decreased the pgRNA encapsidation efficiency, whereas overexpression of RBM24 notably increased it (Fig. 6), suggesting that RBM24 is involved in pgRNA packaging. Moreover, the observation of an increasing interaction between Pol and ε mediated by RBM24 (Fig. 4) demonstrated that RBM24 helps the Pol-ε complex recruit the core protein and initiate encapsidation of pgRNA into nucleocapsids. This was further supported by the fact that RBM24 also increases the production of minus-strand DNA (Fig. 6). Regardless, it remains unknown whether RBM24 is involved in reverse transcription of pgRNA and DNA synthesis, which is under investigation. Pol-ε binding also recruits core protein and then initiates capsid assembly. The enhancement of Pol-ε binding by RBM24 implies that RBM24 may facilitate the interaction between Pol and core protein. Further investigation should be carried out.
Taken together, we are the first to demonstrate that a novel host factor, RBM24, with RNA-binding activity targets ε and Pol and mediates the Pol-ε interaction, which can promote pgRNA packaging. Inhibition of Pol-RBM24-ε formation and viral encapsidation might be a new strategy for developing new anti-HBV drugs.
MATERIALS AND METHODS
Cell culture and transfection.
HepG2 cells (catalog number HLCL-007; ATCC); HepAD38 cells, a generous gift from Christoph Seeger (Fox Chase Cancer Center, PA, USA) (35); and HEK293T cells (catalog number HEMCL-032; ATCC) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 2 mM glutamine (catalog number 12100-046; Gibco), 10% fetal bovine serum (FBS) (catalog number 10099-141; Gibco), and 100 U/ml penicillin-streptomycin (catalog number 15140-122; Gibco) at 37°C in a 5% CO2 atmosphere. Plasmids and siRNAs were transfected into HepG2 cells using Lipofectamine 3000 reagent (catalog number L3000-015; Invitrogen) or into HepAD38 and HEK293T cells using Lipofectamine 2000 reagent (catalog number 11668-019; Invitrogen) according to the manufacturer’s instructions. The following siRNAs were used: control siRNA (siNC) (catalog number SI03650318; Qiagen) and RBM24 siRNA (siRBM24) (catalog number SI03030195; Qiagen).
Plasmids.
The HBV replication-competent clone pHY106 (HBV genome genotype A, subtype adw2 [GenBank accession number AF305422.1]) and wild-type RBM24 protein expression plasmids pRBM24, pHA-RBM24, and pFlag-RBM24 were used as described previously (18). Gene expression cassettes for RBM24 lacking both RNP1 and RNP2 were amplified and cloned into pXJ40-Flag to generate pFlag-RBM24ΔRNP1/ΔRNP2 (pFlag-ΔRNP1/2). The RRM domain or alanine-rich domain (ARD) of RBM24 was amplified and subcloned into pXJ40-Flag to generate the truncated RBM24 constructs pFlag-RRM and pFlag-ARD. The open reading frame (ORF) of HBV Pol was amplified from pHY106 and subcloned into pXJ40-HA to generate pHA-Pol. The terminal protein (TP), spacer (SP), RT, and/or RNase H (RH) domain of Pol were amplified and subcloned into pXJ40-HA to generate the truncated Pol constructs pHA-TP, pHA-SP, pHA-RT, pHA-RH, pHA-TP+SP, and pHA-RT+RH. The truncated Pol fragment lacking RNA-binding activity was amplified and subcloned into pXJ40-HA to generate pHA-mutPol (deletion of amino acids 176 to 299) (9). pCMV-HE was kindly provided by Jianming Hu (Penn State University College of Medicine, PA, USA) (9). pHBV1.3ΔC, a core-null HBV expression plasmid, was kindly provided by Haitao Guo (Indiana University School of Medicine, IN, USA) (36). P−C− pgRNA, a Pol- and core-null HBV expression plasmid, was kindly provided by Wang-Shick Ryu (Yonsei University, Seoul, South Korea) (34). All primers used in this study are listed in Table 1.
TABLE 1.
Oligonucleotides


Western blotting.
Western blotting was performed as described previously (18). The following antibodies were used: antiactin (catalog number 66009-1; ProteinTech), anti-RBM24 (catalog number ab94567; Abcam), anti-HBc (catalog number B0586; Dako), rabbit antihemagglutinin (anti-HA) (catalog number 3724S; Cell Signaling Technology), mouse anti-HA (catalog number H9658; Sigma), rabbit anti-Flag (catalog number 2368S; Cell Signaling Technology), mouse anti-Flag (catalog number F1804; Sigma), streptavidin-horseradish peroxidase (HRP) (catalog number CT353; U-cytech), anti-mouse secondary antibodies (catalog number 115-035-146; Jackson), and anti-rabbit secondary antibodies (catalog number 111-035-003; Jackson).
Coimmunoprecipitation assay.
HEK293T cells transfected with the indicated plasmids were lysed with immunoprecipitation (IP) buffer at 48 h posttransfection (hpt). Cell lysates containing 800 μg total protein were incubated with protein G beads precoated with mouse anti-Flag (catalog number F1804; Sigma-Aldrich) or a nonspecific mouse IgG control antibody (catalog number I5381; Sigma), as described previously (37). Whole-cell lysates and precipitates were detected by Western blotting.
Streptavidin pulldown assay.
A streptavidin pulldown assay was performed as described previously (18, 38). In brief, total protein prepared from HEK293T cells transfected with the indicated plasmids or purified recombinant human RBM24 (rhRBM24) proteins was incubated with biotin-RNA fragments or unlabeled cold RNA fragments, followed by pulldown using Dynabeads M-280 streptavidin (catalog number 11205D; Invitrogen) and Western blotting.
Immunofluorescence staining and RNA fluorescence in situ hybridization.
HepG2 cells transfected with the indicated plasmids were subjected to immunofluorescence (IF) staining and confocal laser scanning microscopy as described previously (39). Anti-HA (catalog number 3724S; Cell Signaling Technology) and anti-DYKDDDDK (catalog number F1804-1MG; Sigma) antibodies were used as primary antibodies, and Alexa Fluor 568-conjugated (catalog number A21202; Life Technologies) and Alexa Fluor 633-conjugated (catalog number A21050; Life Technologies) antibodies were used as secondary antibodies. For RNA fluorescence in situ hybridization (FISH), cells on coverslips were fixed in 4% paraformaldehyde for 10 min and permeabilized with 70% ethanol for 1 h. The coverslips were incubated in hybridization buffer containing a probe targeting the 5′ end of pgRNA (5′-Alexa Fluor 488-CGTAAAGAGAGGTGCGCCCCGTGGT-3′; Sangon Biotech). Finally, fluorescent molecules targeting antigens of interest or immunofluorescence antibodies were incorporated as described previously (40–42). The colocalization signals were analyzed by the Volocity 5.2 enhanced colocalization tool. The product of the difference from the mean (PDM) channels was calculated for each voxel from the two channels analyzed. The positive PDM channel is represented as colocalization pixels; the negative PDM channel is represented as pixels that have no colocalization.
HBV pgRNA packaging assay.
HBV pgRNA packaging was analyzed as described previously (9, 43). In brief, cells transfected with the indicated plasmids or siRNA were lysed, and the supernatants were harvested, followed by native agarose gel electrophoresis. HBV capsids were transferred to a nitrocellulose (NC) membrane for blotting with a primary antibody recognizing HBcAg/capsid (catalog number ab7841; Abcam). Another duplicate was transferred to a nylon membrane (Hybond-N+ membrane; GE) under the same conditions, and encapsidated pgRNA was detected using a biotin-labeled HBV plus-strand-specific riboprobe (nt 1890 to 2306) against the 5′ end of pgRNA; encapsidated minus-strand DNA [(−)DNA] was detected using biotin-labeled HBV minus-strand-specific riboprobes (nt 1890 to 2306). The membranes were incubated with streptavidin-HRP (catalog number CT353; U-cytech) and SuperSignal West Pico chemiluminescent substrate (catalog number 34080; Thermo). HBV pgRNA packaging was further analyzed using an immunoprecipitation assay as described previously (29, 44). In brief, capsids were immunoprecipitated with an anti-HBc antibody (catalog number B0586; Dako). Encapsidated RNA and total RNA were extracted from immunoprecipitated capsids and whole-cell lysates, respectively, using TRIzol reagent (catalog number 15596-018; Invitrogen) according to the manufacturer’s instructions and subjected to Northern blotting.
Northern blotting and ELISA.
To detect HBV RNAs, Northern blotting was performed as described previously (45, 46). An ELISA was performed as described previously to detect HBsAg (43).
Density gradient analysis.
HepAD38 cells were transfected with pHA-RBM24, and cell lysates were subjected to 10 to 50% OptiPrep (catalog number D1556; Sigma) separation by ultracentrifugation for 45 min at 55,000 rpm using an SW55 swing-out rotor (Beckman Instruments), as described previously (33, 34). The gradients were fractionated into 15 fractions; 50 μl of each fraction was analyzed by Western blotting for the detection of core and HA-RBM24, and 50 μl of each fraction was analyzed by dot blotting for the detection of HBV DNA.
Purification of HBV virions.
Virions from serum of HBV patients were prepared as described previously (47). Briefly, patient serum was centrifuged to remove cell debris and precipitated with polyethylene glycol 8000 (PEG 8000). The precipitated virions were resuspended in TNE (10 mM Tris, pH 8.0, 100 mM NaCl, 1 mM EDTA) buffer and then digested with DNase I. The digested virions were subjected to 10 to 50% OptiPrep (catalog number D1556; Sigma) separation by ultracentrifugation. The virions from the core protein peak fraction were reprecipitated with PEG 8000 and lysed by resuspension in TNE buffer. The resuspended virions were then treated with 0.5 μg/μl proteinase K (catalog number 19133; Qiagen) to remove nonencapsidated proteins. The reactions were detected by Western blotting. These experiments were approved by the Institutional Review Board (IRB) (WIVH02201801) at Wuhan Institute of Virology, Chinese Academy of Sciences (CAS).
Statistical analysis.
Data were analyzed using a two-tailed unpaired t test. Statistical significance was set at the P values indicated in the legend of Fig. 6.
ACKNOWLEDGMENTS
We are grateful to Christoph Seeger for providing HepAD38 cells, Jianming Hu for providing the pCMV-HE plasmid, and Haitao Guo for providing the pHBV1.3ΔC plasmid. We are grateful to Ding Gao, Anna Du, and Juan Min (Core Facility and Technical Support, Wuhan Institute of Virology) for excellent technical support.
This work was supported by grants from the National Basic Research Priorities Program of China (2013CB911100), the National Natural Science Foundation of China (31621061 and 31770180), and the Youth Innovation Promotion Association, CAS (number 2016303).
REFERENCES
- 1.Liang TJ, Block TM, McMahon BJ, Ghany MG, Urban S, Guo JT, Locarnini S, Zoulim F, Chang KM, Lok AS. 2015. Present and future therapies of hepatitis B: from discovery to cure. Hepatology 62:1893–1908. doi: 10.1002/hep.28025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, Fu L, Song M, Chen P, Gao W, Ren B, Sun Y, Cai T, Feng X, Sui J, Li W. 2012. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tong S, Revill P. 2016. Overview of hepatitis B viral replication and genetic variability. J Hepatol 64:S4–S16. doi: 10.1016/j.jhep.2016.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nassal M. 2015. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 64:1972–1984. doi: 10.1136/gutjnl-2015-309809. [DOI] [PubMed] [Google Scholar]
- 5.Hamilton PJ, de Marshall TF, Anderson J, Fuglsang H. 1974. Observer variation in clinical onchocerciasis. Trans R Soc Trop Med Hyg 68:187–189. [DOI] [PubMed] [Google Scholar]
- 6.Tavis JE, Perri S, Ganem D. 1994. Hepadnavirus reverse transcription initiates within the stem-loop of the RNA packaging signal and employs a novel strand transfer. J Virol 68:3536–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nassal M, Rieger A. 1996. A bulged region of the hepatitis B virus RNA encapsidation signal contains the replication origin for discontinuous first-strand DNA synthesis. J Virol 70:2764–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu J, Flores D, Toft D, Wang X, Nguyen D. 2004. Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase function. J Virol 78:13122–13131. doi: 10.1128/JVI.78.23.13122-13131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones SA, Clark DN, Cao F, Tavis JE, Hu J. 2014. Comparative analysis of hepatitis B virus polymerase sequences required for viral RNA binding, RNA packaging, and protein priming. J Virol 88:1564–1572. doi: 10.1128/JVI.02852-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beck J, Nassal M. 1997. Sequence- and structure-specific determinants in the interaction between the RNA encapsidation signal and reverse transcriptase of avian hepatitis B viruses. J Virol 71:4971–4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu J, Lin L. 2009. RNA-protein interactions in hepadnavirus reverse transcription. Front Biosci (Landmark Ed) 14:1606–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu J, Boyer M. 2006. Hepatitis B virus reverse transcriptase and epsilon RNA sequences required for specific interaction in vitro. J Virol 80:2141–2150. doi: 10.1128/JVI.80.5.2141-2150.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin L, Hu J. 2008. Inhibition of hepadnavirus reverse transcriptase-epsilon RNA interaction by porphyrin compounds. J Virol 82:2305–2312. doi: 10.1128/JVI.02147-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang T, Lin Y, Liu J, Zhang ZG, Fu W, Guo LY, Pan L, Kong X, Zhang MK, Lu YH, Huang ZR, Xie Q, Li WH, Xu XQ. 2016. Rbm24 regulates alternative splicing switch in embryonic stem cell cardiac lineage differentiation. Stem Cells 34:1776–1789. doi: 10.1002/stem.2366. [DOI] [PubMed] [Google Scholar]
- 15.Grifone R, Xie X, Bourgeois A, Saquet A, Duprez D, Shi DL. 2014. The RNA-binding protein Rbm24 is transiently expressed in myoblasts and is required for myogenic differentiation during vertebrate development. Mech Dev 134:1–15. doi: 10.1016/j.mod.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 16.Jiang Y, Zhang M, Qian Y, Xu E, Zhang J, Chen X. 2014. Rbm24, an RNA-binding protein and a target of p53, regulates p21 expression via mRNA stability. J Biol Chem 289:3164–3175. doi: 10.1074/jbc.M113.524413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cao H, Zhao K, Yao Y, Guo J, Gao X, Yang Q, Guo M, Zhu W, Wang Y, Wu C, Chen J, Zhou Y, Hu X, Lu M, Chen X, Pei R. 30 January 2018. RNA binding protein 24 regulates the translation and replication of hepatitis C virus. Protein Cell doi: 10.1007/s13238-018-0507-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yao Y, Yang B, Cao H, Zhao K, Yuan Y, Chen Y, Zhang Z, Wang Y, Pei R, Chen J, Hu X, Zhou Y, Lu M, Wu C, Chen X. 2018. RBM24 stabilizes hepatitis B virus pregenomic RNA but inhibits core protein translation by targeting the terminal redundancy sequence. Emerg Microbes Infect 7:86. doi: 10.1038/s41426-018-0091-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones SA, Boregowda R, Spratt TE, Hu J. 2012. In vitro epsilon RNA-dependent protein priming activity of human hepatitis B virus polymerase. J Virol 86:5134–5150. doi: 10.1128/JVI.07137-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones SA, Hu J. 2013. Hepatitis B virus reverse transcriptase: diverse functions as classical and emerging targets for antiviral intervention. Emerg Microbes Infect 2:e56. doi: 10.1038/emi.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. 2004. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J 86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu J, Anselmo D. 2000. In vitro reconstitution of a functional duck hepatitis B virus reverse transcriptase: posttranslational activation by Hsp90. J Virol 74:11447–11455. doi: 10.1128/JVI.74.24.11447-11455.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang GH, Seeger C. 1993. Novel mechanism for reverse transcription in hepatitis B viruses. J Virol 67:6507–6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu J, Toft DO, Seeger C. 1997. Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J 16:59–68. doi: 10.1093/emboj/16.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirsch RC, Lavine JE, Chang LJ, Varmus HE, Ganem D. 1990. Polymerase gene products of hepatitis B viruses are required for genomic RNA packaging as well as for reverse transcription. Nature 344:552–555. doi: 10.1038/344552a0. [DOI] [PubMed] [Google Scholar]
- 26.Cao F, Badtke MP, Metzger LM, Yao E, Adeyemo B, Gong Y, Tavis JE. 2005. Identification of an essential molecular contact point on the duck hepatitis B virus reverse transcriptase. J Virol 79:10164–10170. doi: 10.1128/JVI.79.16.10164-10170.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Badtke MP, Khan I, Cao F, Hu J, Tavis JE. 2009. An interdomain RNA binding site on the hepadnaviral polymerase that is essential for reverse transcription. Virology 390:130–138. doi: 10.1016/j.virol.2009.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stahl M, Beck J, Nassal M. 2007. Chaperones activate hepadnavirus reverse transcriptase by transiently exposing a C-proximal region in the terminal protein domain that contributes to epsilon RNA binding. J Virol 81:13354–13364. doi: 10.1128/JVI.01196-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim S, Lee J, Ryu WS. 2009. Four conserved cysteine residues of the hepatitis B virus polymerase are critical for RNA pregenome encapsidation. J Virol 83:8032–8040. doi: 10.1128/JVI.00332-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu J, Seeger C. 1996. Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc Natl Acad Sci U S A 93:1060–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ryu DK, Kim S, Ryu WS. 2008. Hepatitis B virus polymerase suppresses translation of pregenomic RNA via a mechanism involving its interaction with 5′ stem-loop structure. Virology 373:112–123. doi: 10.1016/j.virol.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 32.Beck J, Nassal M. 2007. Hepatitis B virus replication. World J Gastroenterol 13:48–64. doi: 10.3748/wjg.v13.i1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim S, Wang H, Ryu WS. 2010. Incorporation of eukaryotic translation initiation factor eIF4E into viral nucleocapsids via interaction with hepatitis B virus polymerase. J Virol 84:52–58. doi: 10.1128/JVI.01232-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang H, Kim S, Ryu WS. 2009. DDX3 DEAD-box RNA helicase inhibits hepatitis B virus reverse transcription by incorporation into nucleocapsids. J Virol 83:5815–5824. doi: 10.1128/JVI.00011-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, King RW. 1997. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother 41:1715–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y, Nie H, Mao R, Mitra B, Cai D, Yan R, Guo JT, Block TM, Mechti N, Guo H. 2017. Interferon-inducible ribonuclease ISG20 inhibits hepatitis B virus replication through directly binding to the epsilon stem-loop structure of viral RNA. PLoS Pathog 13:e1006296. doi: 10.1371/journal.ppat.1006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang B, Liu XJ, Yao Y, Jiang X, Wang XZ, Yang H, Sun JY, Miao Y, Wang W, Huang ZL, Wang Y, Tang Q, Rayner S, Britt WJ, McVoy MA, Luo MH, Zhao F. 2018. WDR5 facilitates human cytomegalovirus replication by promoting capsid nuclear egress. J Virol 92:e00207-18. doi: 10.1128/JVI.00207-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang F, Bi J, Xue X, Zheng L, Zhi K, Hua J, Fang G. 2012. Up-regulated long non-coding RNA H19 contributes to proliferation of gastric cancer cells. FEBS J 279:3159–3165. doi: 10.1111/j.1742-4658.2012.08694.x. [DOI] [PubMed] [Google Scholar]
- 39.Xu S, Pei R, Guo M, Han Q, Lai J, Wang Y, Wu C, Zhou Y, Lu M, Chen X. 2012. Cytosolic phospholipase A2 gamma is involved in hepatitis C virus replication and assembly. J Virol 86:13025–13037. doi: 10.1128/JVI.01785-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S. 2008. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5:877–879. doi: 10.1038/nmeth.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shulla A, Randall G. 2015. Spatiotemporal analysis of hepatitis C virus infection. PLoS Pathog 11:e1004758. doi: 10.1371/journal.ppat.1004758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramanan V, Trehan K, Ong ML, Luna JM, Hoffmann HH, Espiritu C, Sheahan TP, Chandrasekar H, Schwartz RE, Christine KS, Rice CM, van Oudenaarden A, Bhatia SN. 2016. Viral genome imaging of hepatitis C virus to probe heterogeneous viral infection and responses to antiviral therapies. Virology 494:236–247. doi: 10.1016/j.virol.2016.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao K, Wu C, Yao Y, Cao L, Zhang Z, Yuan Y, Wang Y, Pei R, Chen J, Hu X, Zhou Y, Lu M, Chen X. 2017. Ceruloplasmin inhibits the production of extracellular hepatitis B virions by targeting its middle surface protein. J Gen Virol 98:1410–1421. doi: 10.1099/jgv.0.000794. [DOI] [PubMed] [Google Scholar]
- 44.Jeong JK, Yoon GS, Ryu WS. 2000. Evidence that the 5′-end cap structure is essential for encapsidation of hepatitis B virus pregenomic RNA. J Virol 74:5502–5508. doi: 10.1128/JVI.74.12.5502-5508.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pei R, Qin B, Zhang X, Zhu W, Kemper T, Ma Z, Trippler M, Schlaak J, Chen X, Lu M. 2014. Interferon-induced proteins with tetratricopeptide repeats 1 and 2 are cellular factors that limit hepatitis B virus replication. J Innate Immun 6:182–191. doi: 10.1159/000353220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou M, Zhao K, Yao Y, Yuan Y, Pei R, Wang Y, Chen J, Hu X, Zhou Y, Chen X, Wu C. 2017. Productive HBV infection of well-differentiated, hNTCP-expressing human hepatoma-derived (Huh7) cells. Virol Sin 32:465–475. doi: 10.1007/s12250-017-3983-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ludgate L, Ning X, Nguyen DH, Adams C, Mentzer L, Hu J. 2012. Cyclin-dependent kinase 2 phosphorylates S/T-P sites in the hepadnavirus core protein C-terminal domain and is incorporated into viral capsids. J Virol 86:12237–12250. doi: 10.1128/JVI.01218-12. [DOI] [PMC free article] [PubMed] [Google Scholar]