

Human enteroviruses of species A (EV-A), including EV-A71, are the leading cause of hand-foot-and-mouth disease (HFMD) and may also cause severe neurological manifestations. We investigated the circulation and molecular evolution of EV-A in Madagascar, focusing particularly on the recently described EV-A71 genogroup F. Eight different types, collected mostly in 2011, were identified, highlighting the local circulation and diversity of EV-A. Comparative genome analysis revealed evidence of frequent genetic exchanges between the different types of isolates. The three EV-A71 isolates had different evolutionary histories in terms of recombination. The engineering and characterization of recombinants involving progenitors belonging to different EV-A types indicated a high degree of permissiveness for genetic exchange in EV-A. No outbreak of disease due to EV-A has yet been reported in Madagascar, but the diversity, circulation, and evolution of these viruses justify the surveillance of EV-A circulation to prevent possible HFMD outbreaks due to emerging strains.
KEYWORDS: RNA virus, viral evolution, emergence, enterovirus, picornavirus, recombination
ABSTRACT
Human enteroviruses of species A (EV-A) are the leading cause of hand-foot-and-mouth disease (HFMD). EV-A71 is frequently implicated in HFMD outbreaks and can also cause severe neurological manifestations. We investigated the molecular epidemiological processes at work and the contribution of genetic recombination to the evolutionary history of EV-A in Madagascar, focusing on the recently described EV-A71 genogroup F in particular. Twenty-three EV-A isolates, collected mostly in 2011 from healthy children living in various districts of Madagascar, were characterized by whole-genome sequencing. Eight different types were identified, highlighting the local circulation and diversity of EV-A. Comparative genome analysis revealed evidence of frequent recent intra- and intertypic genetic exchanges between the noncapsid sequences of Madagascan EV-A isolates. The three EV-A71 isolates had different evolutionary histories in terms of recombination, with one isolate displaying a mosaic genome resulting from recent genetic exchanges with Madagascan coxsackieviruses A7 and possibly A5 and A10 or common ancestors. The engineering and characterization of recombinants generated from progenitors belonging to different EV-A types or EV-A71 genogroups with distantly related nonstructural sequences indicated a high level of permissiveness for intertypic genetic exchange in EV-A. This permissiveness suggests that the primary viral functions associated with the nonstructural sequences have been highly conserved through the diversification and evolution of the EV-A species. No outbreak of disease due to EV-A has yet been reported in Madagascar, but the diversity, circulation, and evolution of these viruses justify surveillance of EV-A circulation and HFMD cases to prevent possible outbreaks due to emerging strains.
IMPORTANCE Human enteroviruses of species A (EV-A), including EV-A71, are the leading cause of hand-foot-and-mouth disease (HFMD) and may also cause severe neurological manifestations. We investigated the circulation and molecular evolution of EV-A in Madagascar, focusing particularly on the recently described EV-A71 genogroup F. Eight different types, collected mostly in 2011, were identified, highlighting the local circulation and diversity of EV-A. Comparative genome analysis revealed evidence of frequent genetic exchanges between the different types of isolates. The three EV-A71 isolates had different evolutionary histories in terms of recombination. The engineering and characterization of recombinants involving progenitors belonging to different EV-A types indicated a high degree of permissiveness for genetic exchange in EV-A. No outbreak of disease due to EV-A has yet been reported in Madagascar, but the diversity, circulation, and evolution of these viruses justify the surveillance of EV-A circulation to prevent possible HFMD outbreaks due to emerging strains.
INTRODUCTION
Human enteroviruses of species A (EV-A), which belong to the Picornaviridae family, are a leading cause of hand-foot-and-mouth disease (HFMD) (1, 2). HFMD is a self-limited febrile illness characterized by mouth ulcers, a spotty rash, and blisters on the palms and soles. Nineteen types of EV-A viruses are known to infect humans (e.g., coxsackievirus [CV] A4 to A7, A10, and A14 and EV-A120). The viruses most frequently implicated in HFMD outbreaks are CV-A6, CV-A16, and EV-A71 (3, 4). EV-A71 can also cause severe neurological manifestations, such as meningitis, encephalitis, meningoencephalitis, Guillain-Barré syndrome, and polio-like acute flaccid paralysis, particularly in children (5). The most severe cases may result in death due to cardiopulmonary failure. Major HFMD outbreaks associated with EV-A71 have regularly been reported in the Asia-Pacific region, in China, Malaysia, Taiwan, Vietnam, and Cambodia (6–8), making EV-A71 a major health concern in these countries. EV-A71 infections have been reported regularly in European countries, but outbreaks have been infrequent over the last 20 years (9–11).
EV-A viruses are nonenveloped viruses with a single-stranded (+) RNA genome. This genome consists of a single open reading frame (ORF) flanked by two untranslated regions (UTRs) at its 5′ and 3′ ends (here referred to as the 5' UTR and 3' UTR) (12). The ORF contains three coding regions (P1 to P3). P1 encodes the four viral capsid proteins (VP1 to VP4). P2 and P3 encode nonstructural proteins involved in intracellular viral replication. The error proneness of the viral RNA-dependent RNA polymerase drives the evolution of EV genomes through point mutations. In addition, recombination between EVs is also a major genetic mechanism in the emergence of viral lineages (13–16). Recombination frequently occurs between EV-B or EV-C viruses (16–21). Recombination is also involved in the evolution of EV-A viruses, including EV-A71 (22–27), and has been shown to be associated with the emergence of different (sub)lineages of EV-A71 (28).
Molecular epidemiology studies based on genomic sequence 1D encoding the VP1 protein (here referred to as 1DVP1) led to the classification of EV-A71 strains into phylogenetic lineages or genogroups A to G. Genogroups B and C have been implicated in most of the reported outbreaks worldwide over the last few decades. These two canonical genogroups have been further subdivided into subgenogroups B0 to B5 and C1 to C5. Phylogeographic studies of genogroups B and C revealed transmissions between Asia and Europe (29). Genogroup A includes the prototype strain (BrCr) isolated in 1969 and a few recent isolates from China (30). Genogroups D and G were recently reported in India (31). Genogroup E has been isolated exclusively in Africa (Central African Republic, Cameroon, and Senegal) (32–34), and genogroup F has been reported only in Madagascar (30). The EV-A71 genogroups D, E, F, and G were identified in patients with acute flaccid paralysis and healthy individuals but were not reported in any specific outbreaks. The rare identification of the EV-A71 genogroups D, E, F, and G raises important questions about their genetic origins, their links to the canonical genogroups B and C, and their pathogenicity.
We have published several reports on the diversity and evolution of cocirculating human enteroviruses of species C (EV-C) in Madagascar, including pathogenic circulating recombinant vaccine-derived polioviruses (cVDPVs) (35–37). The diversity and complexity of the recombinant EV-C forms, particularly for the cVDPVs that have been circulating for only a short period (2 to 3 years), suggested that recombination is frequent and that a given recombinant form has a short life span (38, 39). This may reflect the considerable diversity of EV-C types circulating in the population and the high frequency of intra- and intertypic recombination events (35, 36, 39). In addition, our previous studies suggested that new recombinant pathogenic cVDPV lineages emerge through the acquisition of sequences from EV-C, such as CV-A viruses (40, 41). We therefore investigated the occurrence of similar intertypic recombination events between EV-A strains circulating in Madagascar. In particular, we investigated the underlying molecular processes and the contribution of genetic recombination to the evolutionary history of EV-A in Madagascar by analyzing EV-A isolates circulating on the island over short time periods and by engineering recombinants, focusing specifically on EV-A71 from genogroup F.
RESULTS
Phylogenetic relationships between EV-A71 genogroup F and heterotypic EV-A strains circulating locally and worldwide.
We explored the diversity of EV-A in Madagascar through the whole-genome sequencing of 23 EV-A isolates from different regions of the island. Most were isolated from the stools of healthy children in 2011 (30, 37). These viruses were typed on the basis of full 1DVP1 sequences (42), and eight different EV-A types were identified (Table 1).
TABLE 1.
Molecular typing of enterovirus A isolates collected in Madagascar
| Isolatea | Locationb | EV type | Identity to the VP1 prototype sequence |
|
|---|---|---|---|---|
| Nucleotide (%) | Amino acid (%) | |||
| MAD9745-MDG11 | Antsiranana | CV-A4 | 84.2 | 98.4 |
| MAD2940-MDG11 | Mahatalaky | CV-A5 | 82.4 | 95.3 |
| MAD3144-MDG11 | Mahatalaky | CV-A5 | 82.7 | 95.6 |
| MAD2628-MDG11 | Toliara | CV-A6 | 81.8 | 93.8 |
| MAD9828-MDG11 | Antsiranana | CV-A6 | 82.7 | 93.4 |
| MAD2986-MDG11 | Mahatalaky | CV-A7 | 82.0 | 95.6 |
| MAD3101-MDG11 | Mahatalaky | CV-A7 | 82.1 | 95.6 |
| MAD3127-MDG11 | Mahatalaky | CV-A7 | 82.1 | 94.6 |
| MAD3179-MDG11 | Mahatalaky | CV-A7 | 82.0 | 95.3 |
| MAD3956-MDG11 | Mahajanga | CV-A7 | 81.6 | 94.9 |
| MAD9700-MDG11 | Antsiranana | CV-A7 | 82.3 | 94.6 |
| MAD9750-MDG11 | Antsiranana | CV-A7 | 82.1 | 95.3 |
| MAD3995-MDG11 | Mahajanga | CV-A10 | 76.2 | 92.0 |
| MAD9506-MDG11 | Mahajanga | CV-A10 | 76.1 | 91.6 |
| MAD9802-MDG11 | Antsiranana | CV-A10 | 76.1 | 93.0 |
| MAD9856-MDG11 | Antsiranana | CV-A10 | 77.0 | 93.0 |
| MAD9937-MDG11 | Antsiranana | CV-A10 | 76.9 | 96.6 |
| MAD72-MDG07 | Anjozorobe | CV-A14 | 84.2 | 99.0 |
| MAD2718-MDG11 | Toliara | CV-A14 | 84.7 | 99.0 |
| MAD7842-MDG10 | Tsaratanana | EV-A71 | 80.8 | 95.0 |
| MAD72341-MDG04c | Mahatalaky | EV-A71 | 82.4 | 95.3 |
| MAD3126-MDG11c | Mahatalaky | EV-A71 | 80.3 | 93.9 |
| MAD2741-MGD11d | Toliara | EV-A120 | ||
Isolates were mostly from healthy children, with the exception of those in italics, which were obtained from two patients with acute flaccid paralysis. The laboratory name is followed by the country code (MDG) and the last two numbers of the year of isolation (e.g., 11 for 2011).
Map available in Fig. 6.
Previously published (30).
Previously published (72).
The complete genomes of Madagascan EV-A isolates were compared with a set of 810 publicly available genomes from 21 different EV-A types. We also included the full genome sequences of two representative EV-A71 isolates from genogroup E (Central African Republic and Cameroon) and one EV-A71 isolate from subgenogroup C1 from Cameroon. The initial data set was downsized to 153 representative sequences (see Table S1 in the supplemental material) for comprehensive phylogenetic analyses with the 5′ UTR, P1, P2, and P3 regions. The phylogenies reconstructed by the maximum likelihood (ML) method are shown in Fig. 1.
FIG 1.

Maximum likelihood analysis of the nucleotide sequences of different genomic regions of the enterovirus A species. A sequence alignment including the complete genomes of 153 virus strains from 21 types of enterovirus A was constructed and split into four partition data sets corresponding to the capsid-encoding region P1 (A), the 5′ untranslated region (5′ UTR) (B), and the nonstructural protein-encoding regions P2 (C) and P3 (D). Phylograms were reconstructed by the maximum likelihood (ML) method. Bootstrap values of ≥70% are indicated at key nodes. For clarity, consistent clusters of strains are boxed with the name of the corresponding EV-A type or EV-A71 genogroup. Isolate naming is according to the following scheme: EV type, laboratory name, country code (e.g., MDG), and the last two numbers of the year of isolation. Madagascan sequences are highlighted in red. Types of isolates are indicated on the right (colored vertical lines and type names). The two main clusters of P2 and P3 sequences are indicated as clades I and II. Scale bars indicate the ML of substitution per nucleotide position. Gen, genogroup; Sgen, subgenogroup.
In the P1 capsid-encoding region, all sequences clustered to form a bootstrap (BST)-supported monophyletic cluster assigned to their respective type affiliations (BST value of 100%) with ≥76.2% nucleotide identity and ≥91.4% amino acid identity per type cluster (Fig. 1A). Within the EV-A71 type cluster, all EV-A71 sequences segregated into consistent subclusters (BST value of ≥99%) corresponding to the 1DVP1-based genogroups A, B, C, E, and F. Most EV-A isolates from Madagascar formed a distinct subcluster within their respective type cluster (BST value of ≥95%, ≥82.2% nucleotide identity, and ≥97.7% amino acid identity for each Madagascar subcluster).
The phylograms inferred from the 5′ UTR, P2, and P3 sequences displayed several bootstrap-supported topological incongruences (BST value of ≥90%) (Fig. 1B to D) relative to the phylogram inferred from P1 sequences (Fig. 1A). The clustering of EV-A71 genogroups A and E and of individual subgenogroups B and C (B0 to B5 and C1 to C5) was conserved in all trees, but incongruent clustering was observed for EV-A71 genogroup F and other Madagascan EV-A types. The 5′ UTR sequences of most Madagascan isolates segregated into clusters with sequences of other isolates of the same EV type (Fig. 1B), some of which were related to sequences from isolates collected in China. On the trees inferred from the P2 (Fig. 1C) and P3 (Fig. 1D) sequences, the EV-A71 isolates of subgenogroups B and C were scattered over different, distant parts of the tree and were clustered with other EV-A viruses. These patterns are consistent with the previously described recombinant forms of genogroups B and C (22, 24, 26, 28, 43). All but two of the EV-A isolates from Madagascar clustered in a large clade (Fig. 1C and D, clade I), including EV-A71 genogroups E and F and CV-A5 and CV-A10 sequences from China recovered in 2009 and 2013 (22). Only two CV-A10 isolates from Madagascar (MAD3995 and MAD9506) had P2 and P3 sequences distantly related to those of other Madagascan isolates. They clustered in a second large clade (clade II) including EV-A71 sequences from subgenogroups B3 and C4 (BST values of 96% and 95% for the P2 and P3 phylograms, respectively).
Overall, our data showing that the P1 structural genomic regions of most EV-A isolates from Madagascar constitute distinct subclusters within their type clusters suggest that these isolates have been circulating and evolving locally in the island population for a relatively long period of time. In addition, the clustering of the sequences of most of the P2 and P3 nonstructural genomic regions of EV-A isolates from Madagascar, including those of EV-A71 genogroup F, strongly suggests that a local long-term evolutionary process involving intertypic recombination events has been at work.
Recombination in Madagascan virus isolates and the evolution of EV-A71 genogroup F.
Recombination was investigated by comparing the full-length genomic sequences of all Madagascan EV-A isolates, EV-A71 genogroup E, the EV-A71 prototype strain BrCr, and other isolates displaying evidence of genetic relationships in the phylogenetic trees described above. Similarity analyses showed that the Madagascan EV-A isolates had signatures of genetic exchange in the P2 and P3 regions, as indicated by genomic fragments with nucleotide identify of ≥95% (Fig. 2A and Fig. S1).
FIG 2.

Genomic structure of mosaic recombinant genomes of enterovirus A isolates. (A) Similarity plot analyses of different EV-A genomic sequences, with the genomic sequence of EV-A71 MAD7842-MDG10 as a reference. A scheme indicating the genomic organization of EV and the structural and nonstructural regions is shown. Similarity plot profiles are color coded according to EV type or the genogroup of EV-A71. Numbers of isolates for each type and genogroup are indicated in brackets. The nucleotide sequence identity threshold indicating evolution through genetic exchanges in the noncoding and nonstructural coding regions was set at 95%. Additional similarity plots with this and other EV-A genomes as references are shown Fig. S1 in the supplemental material. (B) Putative genomic crossovers with recombinant partners and positions in the nonstructural protein-coding regions of a set of EV-A genomes. The consistency of recombination events was assessed with seven different statistical methods implemented in a recombination detection program (RDP4) (44). Genomes for which significant evidence of recombination (P value of <0.001) was obtained with 7 methods (****), 6 methods (***), 5 methods (**), and 4 methods (*) are indicated. The nucleotide positions of the recombinant parts are indicated by dots if determined by the program (99% confidence interval) or by arrows if undetermined. Detailed values obtained with the program are presented in Table S2.
Genetic exchanges and their boundaries were considered validated if supported by at least four of the seven statistical methods used in the recombination detection program RDP4 (44). Intratypic exchanges were observed for CV-A7 isolates, and multiple intertypic exchanges between CV-A4, -A5, -A6, -A7, and -A10, EV-A71, and EV-A120 isolates were detected (Fig. 2B and Table 2 for statistical data).
TABLE 2.
RDP analysis of recombination events in enterovirus A genomes
| Enterovirus isolate | Location(s) (nt [99% CI])a | Genetic partner(s) | Methodb |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| RDP | GeneConv | Boot scan | MaxChi | Chimaera | SiScan | 3Seq | Consistencyc | |||
| EV-A71 MAD7842 | ||||||||||
| Segment 1 | 3474 (3388–3481), 6102 (ND) | CV-A5 MAD2940, CV-A5 MAD3144 | 2.286 × 10−07 | 7.754 × 10−11 | 1.262 × 10−24 | 2.591 × 10−18 | 1.558 × 10−04 | ** | ||
| Segment 2 | 6750 (6733–6770), 7407 (ND) | CV-A5 MAD2940, CV-A5 MAD3144 | 1.369 × 10−11 | 1.119 × 10−11 | 6.617 × 10−13 | 3.237 × 10−14 | 1.231 × 10−14 | 5.837 × 10−17 | *** | |
| Segment 3 | 3613 (3505–3614), 6780 (6139–6812) | CV-A7 MAD9750 | 1.006 × 10−68 | 1.839 × 10−31 | 4.170 × 10−67 | 5.542 × 10−31 | 1.595 × 10−14 | 1.616 × 10−26 | 2.784 × 10−10 | **** |
| Segment 4 | 4259 (4203–4274), 6804 (6727–6794) | CV-A10 MAD9856, CV-A10 MAD9937 | 4.798 × 10−101 | 1.201 × 10−66 | 1.487 × 10−100 | 1.493 × 10−31 | 1.180 × 10−11 | 9.371 × 10−32 | 1.625 × 10−113 | **** |
| CV-A4 MAD9745 | 6367 (6355–6370), 3′ end | CV-A6 MAD9828 | 1.405 × 10−78 | 3.209 × 10−62 | 1. 518 × 10−79 | 1.285 × 10−22 | 2.330 × 10−12 | 1.108 × 10−22 | 5.766 × 10−39 | **** |
| CV-A6 MAD2628 | 3465 (3100–3628), undetermined 3′ end | CV-A5 MAD2940, CV-A5 MAD3144 | 1.520 × 10−17 | 7.330 × 10−10 | 5.802 × 10−04 | 8.753 × 10−60 | * | |||
| CV-A6 MAD9828 | 6368 (6356–6371), undetermined 3′ end | CV-A4 MAD9745 | 7.029 × 10−79 | 1.604 × 10−62 | 7.592 × 10−80 | 6.429 × 10−23 | 1.165 × 10−12 | 2.828 × 10−27 | 2.883 × 10−39 | **** |
| EV-A120 MAD2741 | 4332 (4064–4458), undetermined 3′ end | CV-A5 MAD2940, CV-A5 MAD3144 | 6.113 × 10−26 | 3.894 × 10−37 | 2.647 × 10−09 | 9.706 × 10−17 | 2.337 × 10−32 | ** | ||
| CV-A7 MAD2986d | 4893 (4882–4901), 6395 (6020–6401) | CV-A7 MAD3127, CV-A10 MAD9856, CV-A10 MAD9937 | 3.063 × 10−84 | 2.951 × 10−62 | 3.209 × 10−85 | 1.165 × 10−28 | 1.423 × 10−26 | 6.910 × 10−24 | 7.500 × 10−82 | **** |
| CV-A7 MAD9700 | 5389 (5325–5440), undetermined 3′ end | CV-A6 MAD2628 or CV-A10 MAD9802 | 9.236 × 10−97 | 5.343 × 10−65 | 8.703 × 10−100 | 4.469 × 10−31 | 5.502 × 10−17 | 3.669 × 10−35 | 9.393 × 10−102 | **** |
| CV-A10 MAD9856e | ||||||||||
| Segment 1 | 4383 (4294–4397), 6977 (6853–7019) | CV-A7 MAD975 | 1.874 × 10−39 | 1.758 × 10−37 | 2.582 × 10−54 | 2.850 × 10−24 | 4.376 × 10−20 | 2.060 × 10−40 | *** | |
| Segment 2 | 4263 (4169–4274), 6765 (6748–6812) | EV-A71 MAD7842 | 2.692 × 10−78 | 4.698 × 10−43 | 4.429 × 10−80 | 8.704 × 10−27 | 1.103 × 10−07 | 4.585 × 10−27 | 2.405 × 10−75 | **** |
| CV-A10 MAD9802 | 4830 (4785–4945), undetermined 3′ end | CV-A7 MAD9700 | 1.932 × 10−62 | 1.756 × 10−86 | 1.179 × 10−76 | 2.011 × 10−32 | 4.555 × 10−28 | 4.513 × 10−57 | *** | |
Nucleotide localization of the genomic segments implicated in recombination. In some instances, the region is shown in place of a nucleotide position. The 99% confidence interval (CI) is shown as the nucleotide segment from X to Y. ND, not determined.
The seven tests for recombination were implemented in the RDP4 program (44).
Asterisks indicate the number of methods used to determine statistical evidence of a recombination event: ****, 7 methods; ***, 6 methods; **, 5 methods; *, 4 methods. Genomes in which statistical evidence of a recombination event was obtained with fewer than 4 methods were not indicated.
Also valid for CV-A7 MAD3179 and MAD3101.
Also valid for CV-A10 MAD9937.
The genome of the EV-A71 genogroup F isolate MAD7842 displayed significant evidence of evolution through recombination with genomic fragments of CV-A5 isolates (MAD2940 or MAD3144) in the 2C protein-encoding region and the 3′ part of the 3D region (Fig. 2 and Table 2). A large part of the genome of this isolate, encoding proteins 2B to 3D, presented significant evidence of recombination with the genomes of CV-A10 (MAD9856 and MAD9937) and CV-A7 (MAD9750) isolates. We cannot exclude the possibility that these recombination events involved unknown recent common ancestors.
The high degree of similarity between the nucleotide sequences of defined genomic fragments encoding nonstructural proteins of certain cocirculating Madagascan EV-A isolates provides strong evidence for the occurrence of recent and frequent intra- and intertypic genetic exchanges leading to mosaic recombinant genomes.
Comparative analysis of the evolutionary history and temporal origin of EV-A71 genogroup F for two different genomic regions.
The evolutionary history of the capsid-encoding region of EV-A71 was investigated by performing Bayesian analysis with an EV-A71 1DVP1 sequence data set (Fig. 3A and B and Table S1). The mean evolution rate was 3.97 × 10−3 (95% highest posterior density [HPD], 3.47 × 10−3 to 4.45 × 10−3) substitutions per site year. The most recent common ancestor of all EV-A71 lineages was dated to 1901 (95% HPD, 1870 to 1931). The three EV-A71 sequences sampled in Madagascar between 2004 and 2011 (genogroup F) shared a common ancestor that arose in 1995 (node N10; 95% HPD, 1990 to 2000; posterior probability [PP] = 1) (Fig. 3B). Interestingly, the isolates of genogroup F and those of the cosmopolitan genogroup B diverged from a common ancestor in 1946 (node N2; 95% HPD, 1937 to 1955; PP = 0.98). The isolates of the other newly described EV-A71 genogroups D and G (reported in India) and genogroup E (reported in sub-Saharan Africa) diverged at a similar time from another common ancestor (node N3; PP = 0.95) in 1950 (95% HPD, 1935 to 1964).
FIG 3.

Evolutionary history of EV-A71 genogroup F, as assessed by maximum clade credibility phylogenetic analysis. (A) Patterns of evolution were reconstructed for EV-A71 from the nucleotide sequences encoding the capsid protein 1DVP1 and for a segment of the P3 (nonstructural protein-encoding) region, as indicated on the schematic diagram of the EV genome. (B) Maximum clade credibility tree based on the capsid protein 1DVP1 sequence, including EV-A71 of various geographic origins from all described EV-A71 (sub)genogroups. (C) Maximum clade credibility tree based on the nonstructural P3 region specifically including EV-A71 strains within genogroup F and EV-A sequences belonging to clade I (Fig. 1D). The maximum clade credibility trees were inferred by Bayesian Markov chain Monte Carlo analysis, with a discrete model, to determine the virus corresponding to tree branches. Tips are scaled according to the date of virus sampling (timescale on the x axis). The main lineages are color coded according to EV-A71 (sub)genogroup and EV-A type. Time to the most recent common ancestor (tMRCA) and highest posterior probability density (HPD) intervals (blue lines) are indicated for the main nodes. The size of the circles at the nodes is proportional to posterior probability (PP).
We investigated the evolutionary history of lineages genetically related to recombinant EV-A71 genogroup F (clade I in the above ML trees) (Fig. 1C and D) by Bayesian analysis of a segment of the P3 region (nucleotide positions 5200 to 6200). We found no evidence of a recombination breakpoint common to different recombinant lineages within this segment (Fig. 2 and data not shown). The mean evolution rate was 6.54 × 10−3 (95% HPD, 4.30 × 10−3 to 8.90 × 10−3) substitutions per site year. The three EV-A71 genogroup F sequences had different evolutionary histories (Fig. 3C). The P3 segment of the EV-A71 isolate MAD7842 had a common direct ancestry (PP = 1) with segments of six other heterotypic virus strains sampled in Madagascar and was assigned to EV-A types CV-A7 and CV-A10. The common ancestor of this cluster was dated to 2007 (95% HPD, 2005 to 2009) and was assigned to CV-A7 with a probability of 0.84. The other putative EV types were EV-A71 (P = 0.15) and CV-A10 (P = 0.01). This phylogenetic pattern suggested that the EV-A71 genogroup F isolate MAD7842 acquired all or part of its P3 genomic region through recombination with a CV-A7 strain. The other two EV-A71 genogroup F sequences had different unresolved evolutionary patterns and were temporally distant from the EV-A71 isolate MAD7842 cluster. According to P3 sequences, the origin of the genogroup F lineage was dated to 1994 (95% HPD, 1988 to 2001), consistent with the 1DVP1-based phylogeny (1995; 95% HPD, 1990 to 2000) (compare Fig. 3B and C).
Overall, Bayesian phylogenetic analysis indicated a recent divergence of the Madagascan EV-A71 genogroup F isolates (common ancestor dating back to 1995) and a genetic link between this genogroup and the cosmopolitan genogroup B (common ancestor dating back to 1946). The nonstructural P3 sequences of the Madagascan EV-A71 isolates followed a different evolutionary history. This pattern confirmed the existence of active evolution through frequent intertypic recombination events in Madagascar, with a recent event dating back to about 3 years before isolation.
Conservation of functional compatibility among genetic domains of EV-A71 and other EV-A viruses.
We investigated the propensity for recombination of EV-A71 and other EV-A viruses by constructing recombinant genomes with 5′ and 3′ halves originating from different EV types. We combined the 5' UTR-P1-2A genomic region of a given EV-A (strain X) with the 2BC-P3-3′ UTR region of another isolate (strain Y) from the same or another phylogenetic clade. We selected the progenitors from clade I (EV-A71 genogroup F, isolate MAD72341; CV-A7, isolate MAD9750) and clade II (EV-A71 subgenogroup C4, isolate SEP06; CV-A10, isolate MAD3995) (Fig. 4). A recombinant of the X and Y strains (X/Y) and its converse counterpart Y/X were constructed by a fusion-PCR-based procedure developed for the engineering of chimeric genomes (45). The transcription of the viral chimeric cDNA was controlled with the T7 RNA polymerase promoter, and genomic RNAs and viruses were produced in human 293T/T7 cells expressing this polymerase following the transfection of viral cDNAs. We engineered recombination breakpoints at the junction of viral proteins 2A and 2B because this genomic stretch is subject to frequent recombination in natural isolates (Fig. 2A and Fig. S1). We produced the following chimeric X/Y genomes: EV-A71(C4)/CV-A10, EV-A71(F)/CV-A10, EV-A71(C4)/CV-A7, and EV-A71(F)/CV-A7; their converse YX genomes were also produced. We also used the full-length genomes of the four primary progenitors to transfect cells to obtain infectious virus controls.
FIG 4.

Comparative plaque phenotypes of the progenitor and chimeric viruses. Plaque phenotype (A to D) was assessed for the progenitor and chimeric viruses on Vero E6 cells or RD cells for viruses with the 5′ half of the CV-A10 genome. Negative and positive controls (coxsackievirus B3) are presented (E). Schematic diagrams of the progenitor and chimeric genomes are shown. Virus names and genomes are color coded according to genetic background and affiliation to the nonstructural CDS clade I (red) or clade II (blue) (Fig. 1C and D). Titers of viral suspensions are indicated.
All of the generated chimeric genomes yielded viable recombinant viruses after the transfection of 293T/T7 cells with viral cDNA and one passage in sensitive cell lines, as indicated by cytopathic effects (CPEs) in infected cells. The nucleotide sequences of the recombinant genomes were checked by Illumina high-throughput sequencing. The recombinant genomes displayed 0- to 3-nucleotide (nt) differences relative to the progenitor sequences (Table 3). These differences were located mostly in the VP1 coding sequence. We hypothesized that these mutations were introduced during gene amplification or selected from the genome pool present in the original stock of progenitor viruses. We cannot rule out the possibility that some of these substitutions were selected to adapt viral functions to a given recombinant.
TABLE 3.
Nucleotide and amino acid substitutions in the reconstituted progenitors and engineered chimeric viruses
| Chimeric virusa | Genome location | Position (nt) | Nucleotide sequence in: |
Amino acid substitution (progenitor vs progeny) | ||
|---|---|---|---|---|---|---|
| Original progenitorb | Progenyc |
|||||
| Reconstituted progenitor | Chimera | |||||
| EV-A71(C4)/CVA7 | VP1 | 2871 | C | G | G | E145Q |
| 2A | 3333 | A | G | A | K2E | |
| EV-A71(C4)/CVA10 | VP1 | 2871 | C | G | G | E145Q |
| 2A | 3333 | A | G | A | K2E | |
| EV-A71(F)/CVA7 | VP1 | 2886 | C | G | G | P148R |
| EV-A71(F)/CVA10 | VP2 | 1234 | T | T | C | |
| VP1 | 2886 | C | G | G | P148R | |
| CVA7/EV-A71(C4) | VP1 | 2776 | A | G | A | I112M |
| 2877 | C | C | T | T146I | ||
| 2882 | C | T | C | L148F | ||
| CVA7/EV-A71(F) | 2776 | A | G | A | I112M | |
| 2877 | C | C | T | T146I | ||
| 2882 | C | T | C | L148F | ||
No nucleotide substitutions were found between the original or reconstituted progenitors and the progeny chimeric viruses CVA10/EV-A7(C4) and CVA10/EV-A71(F).
Original progenitor virus used for reconstructing the progenitor or engineering the chimeric viruses.
Nucleotide sequence of the reconstituted progenitor or engineered chimeric viruses after genome transfection and a single passage in cells.
The phenotypic characteristics of progenitors and chimeric viruses were assessed through plaque assays and replication kinetics in Vero E6 cells. Viruses with the 5′ half of CV-A10 genomes were analyzed in rhabdomyosarcoma (RD) cells, the only cells in which they replicate efficiently. Most of the progenitors and recombinant viruses studied formed turbid plaques of various sizes (Fig. 4A to D), contrasting with the round plaques generally produced by coxsackievirus B3 (EV-B) (Fig. 4E). Titers of viral stocks were assessed by determining the 50% tissue culture infective dose (TCID50) per milliliter and the number of PFU per milliliter. The differences between these two values for a given stock varied between viruses, suggesting differences in the efficiency with which clear-cut CPEs were induced in infected cells. The plaques of some of the recombinant viruses were similar in size and appearance to those of the donor of the 5′ half of the genome (Fig. 4D, CV-A7/X). In other recombinants, the presence of a certain 3′ half of the genome modified the plaque phenotype, e.g., EV-A71(C4)/CV-A7 versus A71(C4)/SEP06, and EV-A71(F)/CV-A7 versus EV-A71(F)/MAD7234 in Fig. 4A and B, respectively.
We then compared the growth kinetics of the progenitor and recombinant viruses (Fig. 5A to D). Progenitor viruses and most of the chimeric viruses induced complete cell lysis within 24 to 48 h of infection. All but one [EV-A71(C4)/CV-A7] of the chimeric viruses used had growth kinetics similar to the kinetics of the progenitors, with only minor differences in virus yields and genome copy numbers. The EV-A71(C4)/CV-A7 chimeric virus induced only partial CPE, consistent with its tiny plaques (Fig. 5A) and its titer and genome copy number, which differed from those of its EV-A71 progenitor.
FIG 5.

Replication kinetics of the progenitor and chimeric viruses. Replication kinetics of the progenitor (black) and chimeric (gray and white) viruses were analyzed in triplicate following infection of Vero E6 cells or RD cells. RD cells were used for viruses carrying the 5′ half of the CV-A10 genome. Infected cells and supernatants were collected at 0, 6, 16, 24, and 48 h postinfection (hpi). Virus production is expressed as the number of genome copies and TCID50, as indicated. The data are presented are mean values ± standard deviations. The occurrence of a cytopathic effect and complete cell lysis are indicated. Virus names and genomes are color coded according to genetic background and affiliation to the nonstructural CDS clade I (red) or clade II (blue), as indicated in the legend of Fig. 4.
Overall, these results indicate a high degree of conservation and compatibility of viral functions through genetic exchanges between EV-A71 genogroups C4 and F and CV-A7 and CV-A10, allowing intertypic recombination. In certain cases, recombination can lead to phenotypic modifications.
DISCUSSION
Characterization of a set of EV-A isolates from Madagascar demonstrated the diversity of viral types cocirculating throughout this country and provided evidence of local evolutionary processes characterized by frequent recent intertypic genetic exchanges leading to mosaic recombinant genomes. EV-A71 isolates from the recently discovered genogroup F were subject to a similar active evolution process, strongly suggesting that EV-A strains have been circulating and interacting genetically with each other for many years on this island. The construction and characterization of viable recombinants suggested that EV-A evolution has kept primary viral functions compatible for intertypic recombination.
At least eight of the known EV-A types were cocirculating all over the island (Fig. 6) during the short period considered here (Table 1), highlighting the diversity of the EV-A strains circulating in the population. Similar levels of EV-A diversity have been reported in Asia and Europe for clinical specimens, stools from refugees, and wastewater (1, 22, 46–49). Such high levels of diversity have already been reported in Madagascar for EV species B and C (35, 36, 50). The phylogenetic analysis of the capsid-encoding region P1, based on 810 available published EV-A genomes, revealed that all reported EV-A isolates from Madagascar belonged to distinct lineages different from the other EV-A isolates circulating worldwide. Furthermore, the phylogenetic links between their nonstructural genomic regions suggested that most of the Madagascan isolates shared a recent common ancestor, resulting in a robust clade of related sequences (clade I) (Fig. 1C and D). The phylogenetic analysis of whole virus genomes from different Madagascan districts provided further evidence of the extensive circulation of EV-A isolates throughout the country, constituting a dynamic local EV-A ecosystem.
FIG 6.
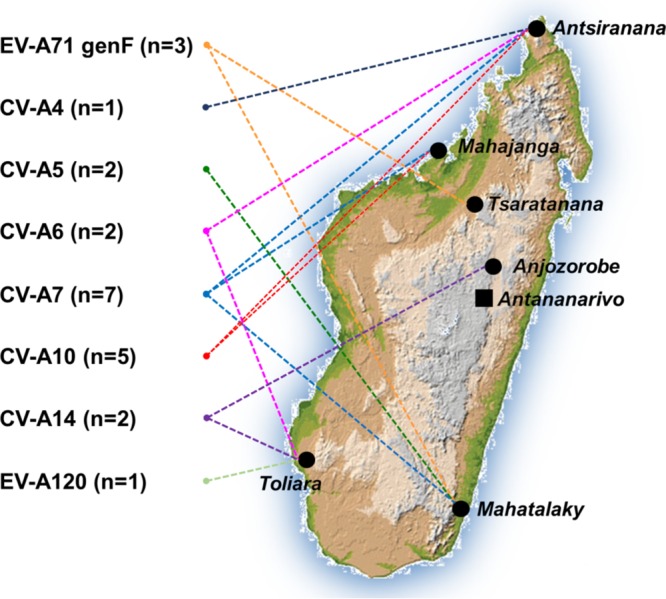
Circulation of enterovirus A isolates in different geographic areas of Madagascar. The Madagascan isolates used in this study are color coded by type and shown on the map of the island (map from Wikimedia Commons Atlas of the World [https://commons.wikimedia.org/wiki/Atlas_of_the_world]).
However, despite the insularity of the EV-A ecosystem in Madagascar, it displays relationships with other EV-A strains from other countries. Our Bayesian phylogenies indicated a common ancestry for the genes encoding the VP1 capsid proteins of EV-A71 genogroups F and B in the 1940s even though EV-A71 genogroup F has only ever been reported in Madagascar. The nonstructural regions of a number of EV-A isolates recovered from Madagascar are related to those of Chinese CV-A5 and -A10 strains (22) and the recently reported African EV-A71 genogroup E (32–34). In addition, the two CV-A10 isolates in clade II (MAD3995 and MAD9506) were collected in the Mahajanga district and were distantly related to the other virus isolates collected in Madagascar. The Mahajanga district is a port of entry and destination for people originating from the Comoros Islands, Europe, India, and, more recently, China, so these CV-A10 isolates may belong to a recently imported lineage. Virus importations may, therefore, contribute to the diversity of the local EV-A ecosystem. Only a few EV-A sequences from Africa are available in databases. Given the proximity of this continent to Madagascar, we cannot rule out the possibility that the discovery of new isolates in Africa in the future will modify our view of the insular nature and molecular evolution of isolates from Madagascar.
The phylogenetic incongruences between structural and nonstructural genes suggest that multiple intertypic recombination events have occurred within the Madagascan EV-A ecosystem. Recombination has already been documented in the evolutionary history of EV-A, and the recombinant forms described here are consistent with those described in previous studies (22, 24, 28, 49). Most EV-A strains from Madagascar appear to have mosaic genomes combining fragments with nucleotide sequences displaying a high degree of identity (>95%) to two to four different viral isolates. EV-A71 isolates from genogroup F have been subject to a similar active evolution process because the three EV-A71 strains studied had different patterns of recombination. In particular, the EV-A71 MAD7842 isolate had a striking mosaic genome that emerged following several recombination events with partners related to the cocirculating CV-A7 and, possibly, CV-A5 and CV-A10 viruses. Previous studies in Madagascar reported similar genome plasticity and mosaicism within EV-C species (38) and provided evidence of local evolutionary processes, with frequent recent intertypic genetic exchanges leading to mosaic recombinant genomes. Present data are consistent with a model of modular evolution in which the diversification of the capsid genes determining EV type and genogroup is dissociated from that of the nonstructural genes (21, 41, 51–55). Previous studies have highlighted the occurrence of much lower rates of recombination (by a factor of about five) in EV-A71 than in other species or types, such as CV-A2, -A4, or -A10, and suggested a high degree of conservation of EV-A71 among enterovirus types and species (16, 24, 26, 28, 43). However, this study and other recent studies analyzing the genetic features of EV-A71 and other EV-A strains circulating in Africa, Taiwan, and elsewhere suggested that EV-A71 and cocirculating EV-A strains may evolve more frequently through intertypic recombination in certain contexts (49, 56).
We evaluated these possible intertypic genetic exchanges and the genetic features limiting recombination further by engineering eight different intertypic chimeric viruses from four EV progenitors. These progenitors belonged to three different types and two different EV-A71 genogroups (F and C4) and had closely or distantly related nonstructural sequences (Fig. 1, clades I and II). All chimeric combinations and counterparts gave rise to viable infectious viruses, highlighting the tremendous genome plasticity of EV-A. The primary viral functions of most of the chimeric viruses were similar to those of their progenitors. These findings merit confirmation through the engineering of other kinds of EV-A recombinants, but the implications of our findings are already far-reaching. Indeed, they suggest that despite long-term diversification, the primary viral functions associated with the structural and nonstructural genomic regions of EV-A species have been conserved and remain compatible for intra- and intertypic recombination. Both characteristics, conservation of viral functions and compatibility for recombination, may have remained closely linked during evolution, as proposed for EV species (55).
Only two of the engineered chimeras, EV-A71(F)/CV-A7 and EV-A71(C4)/CV-A7, gave rise to viruses causing weaker cytopathic effects and smaller plaques than their progenitors. These data indicate that, in certain cases, intertypic recombination can lead to phenotypic modifications. By engineering intratypic recombinants of EV-A71 subgenogroups B3 and C2, Phuektes et al. showed that the capsid-encoding region (P1) has the strongest effect on phenotype and that the P2/P3 regions have a more moderate effect (57). These phenotypic differences may be associated with changes in the pathogenicity of recombinants in infected hosts. However, we cannot exclude the possibility that recombinants displaying no differences relative to their progenitors in cultured cells have different virulence characteristics in infected hosts. Recombination has been shown to modify EV-C pathogenicity in animals without affecting the capacity of the virus to replicate in cultured cells (40, 41, 58).
The EV-A types detected in Madagascar have been reported in diverse countries in sporadic cases and outbreaks of disease, including febrile illness, herpangina, HFMD, and more severe diseases, such as encephalitis and polio-like syndrome (59, 60). No outbreak linked to EV-A has yet been reported on the island. However, phylogenetic analysis revealed extensive circulation of EV-A in the island population. In particular, our findings provide evidence for the evolutionary diversification of EV-A71 genogroup F in Madagascar (Fig. 1, 2, and 6) due to mutation and frequent genetic exchanges with other EV types. Virus migration also plays an important role, as shown by the recent importation of CV-A10 and the phylogenetic links between EV-A isolates from Madagascar and virus isolates from other countries. The phenotypes of the engineered recombinants indicate that local isolates can evolve through intertypic genetic exchanges with distantly related EV-A. All these data suggest that surveillance of human EV-A and HFMD cases in Madagascar should be implemented to detect possible HFMD and encephalitis outbreaks due to emerging strains.
MATERIALS AND METHODS
Ethics statement.
Field missions were performed in 2011, with the collection of stool specimens from healthy children under the age of 5 years. The study protocol was reviewed and approved by the National Research Ethics Committee (Madagascar Ethics Committee agreements 011-MSANP/CE and 046-MSANP/CE). Written informed consent was obtained from the parents or legal guardians of the children enrolled in the study.
Isolation of enterovirus A in Madagascar.
EV-A was isolated on human rhabdomyosarcoma (RD) cells from stool specimens treated with chloroform, as described in the standard procedures of the laboratory manual for the WHO Global Polio Laboratory Network (61). Stool specimens were sampled in 2011 from healthy children or, in a few cases, from children with acute flaccid paralysis. Isolates with cytopathic effects on RD cells were primarily typed by molecular methods through partial 1DVP1 sequencing, as previously described (50, 62).
Whole-genome sequencing.
Whole-genome sequences were determined for EV-A from Madagascar and for previously reported EV-A71 isolates from genogroup F (MAD72341 and MAD3126), genogroup E (CAF-NMA-03-008 and CAE-C08-146), and subgenogroup C1 (CAE-C08-041) (30, 32, 34). RNA genomes were extracted with a High Pure Virus RNA kit (Roche), according to the manufacturer’s instructions. Complete genome sequences were determined by genome walking. The terminal sequences of the viral genomes were confirmed by rapid amplification of the cDNA ends (RACE) with a 5′/3′ RACE kit (Roche). Sanger sequencing was performed with a BigDye Terminator kit on an ABI 3730xl 96-capillary DNA Analyser (Applied Biosystems). Alternatively, viral genome sequences were obtained by the Illumina high-throughput technique with generic primers specific for EV-A (63).
Data collection and nucleotide sequence data sets.
Complete genome sequences for all enteroviruses from species A were retrieved from the GenBank database (to March 2016). Wrongly annotated sequences, partial genomes, coding sequences (CDS) including internal termination codons, and sequences without a fully specified date (year) and country of origin were discarded. A sequence alignment including the complete genomes of 810 virus strains from 21 enterovirus types was generated with ClustalW implemented in BioEdit, version 7.2.3, software (http://www.mbio.ncsu.edu/bioedit/bioedit.html) and split into four partitions corresponding to the 5′ UTR, P1, P2, and P3 genomic regions. For more accurate investigation of the phylogenetic relationships by maximum likelihood (ML) analysis, the initial global data sets were downsized to 153 sequences representing genetic clusters and various outbreaks at different time periods and locations (see Table S1 in the supplemental material).
Maximum likelihood and neighbor-joining phylogenetic analysis.
Phylograms based on the sequence partitions corresponding to the 5′ UTR and P1, P2, and P3 regions were reconstructed with different methods. Maximum likelihood (ML) trees were generated with PhyML software (64), with the general time-reversible plus invariant site and gamma distribution model (GTR plus I and γ) and 500 bootstrap replicates for the estimation of node consistency. The ML best-fit model for nucleotide substitution was validated statistically with JModel Test software (65, 66). The resulting ML trees were annotated with FigTree, version 1.4.2, software (http://tree.bio.ed.ac.uk/). Neighbor-joining (NJ) trees were generated with MEGA6 software (67), using the Kimura two-parameter evolutionary model and 1,000 bootstrap replicates for the estimation of node consistency.
Recombination analysis.
The degree of nucleotide sequence identity between complete genome sequences was analyzed with SimPlot, version 3.5.1, software (68). The pairwise similarity between all sequences in a multiple sequence alignment was calculated with a 200-nt window moved along the sequence in 20-nt steps. The consistency of recombination events was evaluated with seven different statistical methods implemented in RDP4 software (44). In some cases, we checked that the recombinant genome sequences did not result from fortuitous cross-sequencing of viral mixtures present in isolates by carrying out control Illumina high-throughput sequencing following reverse transcription-PCR (RT-PCR) with generic primers specific for EV-A (63).
Sequence data for Bayesian phylogenetic analyses and tree reconstruction.
For the inference of coalescent times for the EV-A71 isolates from genogroups A to G, a sample of 190 1DVP1 sequences, including the three EV-A71 sequences (MAD7842, MAD3126, and MAD72341), was randomly selected from the sets of sequences used in our earlier analyses (29, 30). We subsampled sequences of strains recovered between 1963 and 2015 from the seven EV-A71 genogroups and all subgenogroups of genogroups B and C to obtain this 1DVP1 sequence data set. We demonstrated that including more sequences in the data set had no effect on the coalescent times calculated from the genealogy.
The sequence samples are described in Table S1. The EV-A71 genealogy was inferred with the Bayesian Markov chain Monte Carlo (MCMC) methods available in BEAST, version 1.8.2, software (69). Phylogenetic inference was performed with the general time reversible (GTR) substitution model with gamma rate heterogeneity across sites. Coalescent times were inferred under a relaxed molecular clock model (uncorrelated log-normal distribution across lineages). A Bayesian Skyline prior was chosen as the tree prior, with 10 different population size intervals and a piecewise linear model (70). The tree was reconstructed with a Markov chain Monte Carlo process of 120 million states, sampled every 12,000 states. A second sequence data set (n = 30 taxa) was selected from the ML phylogeny inferred from the P3 sequences. This sample included 27 taxa genetically related to the three EV-A71 genogroup F isolates in a monophyletic cluster. The phylogenetic analysis was performed as described above, except that a discrete trait analysis with a Bayesian stochastic search variable selection (BSSVS) procedure was included to investigate the changes in EV types across lineages. The MCMC process was performed with 50 million states and a sampling frequency of 1/5,000.
Cell culture.
Monkey kidney epithelial Vero E6 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with GlutaMAX (Gibco 31966-021) and supplemented with 5% fetal calf serum. Human rhabdomyosarcoma RD cells (WHO) were cultured in DMEM (T15-106; PAA) supplemented with 2 mM l-glutamine and 5% fetal calf serum. Human embryonic kidney 293T/T7 cells (Pierre Charneau, Institut Pasteur, Paris, France), which constitutively express the T7 RNA polymerase, were cultured in minimal essential medium (MEM) (M5650; Sigma) supplemented with 2 mM l-glutamine and 10% fetal calf serum. All cell lines were cultured at 37°C under a humid atmosphere containing 5% CO2.
Engineering of chimeric genomes.
Chimeric genomes were engineered with a previously described fusion-PCR-based procedure (45). The RNA genomes of the progenitor viruses were denatured (5 min at 65°C). Then they were subjected to full-length reverse transcription (30 min at 50°C) with a Maxima H Minus First Strand cDNA synthesis kit (Thermo Scientific) with specific reverse primers (20 pmol) containing a 5′ poly(T) tail (n = 24) followed by the last 24 complementary nucleotides of the considered genome (a full list of primers is given in Table S2). Then full-length cDNA genomes were amplified using a Phusion High-Fidelity DNA polymerase (Thermo scientific) with the previous reverse primers and a forward primer containing a T7 promoter and the first 25 nt of the virus genome (25 pmol each). The amplification conditions were as follows: 30 s at 98°C, a preamplification step (7 s at 98°C, 20 s at 63°C, and 3.5 min at 72°C), and 20 cycles of 7 s at 98°C, 20 s at 65°C, and 3.5 min at 72°C. After a final elongation for 5 min at 72°C, the progenitor T7 promoter/cDNAs were purified with a QIAquick PCR purification kit (Qiagen). The full-length T7 promoter/cDNAs of the progenitor viruses were used both to regenerate progenitor viruses through cell transfection and as a template to produce overlapping segments for chimeric genome construction. Overlapping segments were produced by nested PCR with a Phire Hot Start Pol II kit (Thermo Scientific) and overlapping primers (10 pmol each) containing 50 nt of nonpriming 5′ tails designed to match the sequence of one progenitor genome upstream or downstream from the 2A/2B breakpoint, followed by 25 nt matching the second progenitor genome. The amplification conditions were as follows: 30 s at 98°C, followed by 30 cycles of 5 s at 98°C, 5 s at 60°C, and 40 s at 72°C. After a final elongation phase for 3 min at 72°C, the amplified overlapping segments were subjected to electrophoresis in an agarose gel, purified with a QIAquick gel purification kit (Qiagen), and quantified with a NanoDrop spectrophotometer (Thermo Scientific).
Equimolar amounts of purified overlapping amplicons were mixed (10-μl final volume) and ligated with Phire Hot Start Pol II (Thermo Scientific) in a final volume of 50 μl without added primers. The fusion conditions were 30 s at 98°C followed by 10 cycles of 5 s at 98°C, 1 min at 56°C, and 3 min at 72°C. The forward and reverse primers previously used for full-length genome amplification (20 pmol each) were then added to the fusion mixture, and PCR resumed, with heating for 30 s at 98°C followed by 20 cycles of 5 s at 98°C, 5 s at 65°C, and 3 min at 72°C. After a final elongation for 7 min at 72°C, the T7 promoter/chimeric genome amplicons were purified with a QIAquick PCR Purification kit (Qiagen) and quantified with a NanoDrop spectrophotometer (Thermo Scientific).
Recovery of chimeric viruses.
Human embryonic kidney 293T/T7 cells (Pierre Charneau, Institut Pasteur, Paris, France), which constitutively express the T7 RNA polymerase, were transfected with 1 μg of purified T7 promoter/cDNAs from progenitor or chimeric genomes with FuGENE HD transfection reagent (Promega). Transfected cells were incubated at 37°C until complete cell lysis. Cell supernatants were collected and clarified by centrifugation, and 200 μl of the resulting suspension was passaged once on 80% confluent Vero E6 or RD cells grown in T-25 flasks incubated at 37°C. When Vero E6 or RD cell lysis was complete, flasks were frozen and thawed three times. Cell supernatants were collected, clarified by centrifugation, and stored at −20°C until use. Virus titers were determined in microplates by calculating the 50% tissue culture infective dose (TCID50) per milliliter according to the Kärber method. All recovered progeny virus genomes were sequenced with Illumina high-throughput sequencing technology.
Viral plaque assay.
Vero E6 or RD cell monolayers in six-well culture plates (2 × 106 per well) were washed twice and infected with 500 μl of 10-fold serial dilutions of viral stock suspension per well. Viral adsorption was allowed to proceed for 2 h, and the cells were then washed, covered with 2.4% (vol/vol) Avicel RC-581 (FMC BioPolymers, Brussels, Belgium) and 2× culture medium (2× DMEM supplemented with 40 mM Tricine, 2.2 g/liter NaHCO3, 50 mM MgCl2, 4 mM l-glutamine, 4% fetal bovine serum [FBS]) and incubated at 37°C under a humidified atmosphere containing 5% CO2. After a week, cells were fixed by incubation for 1 h with one volume of 4% paraformaldehyde. The overlays were removed from the fixed cells, which were washed with phosphate-buffered saline (PBS) and stained with crystal violet. Coxsackievirus B3 (CV-B3), an EV-B virus, was used as a positive control for plaque assays. CV-B3-infected cells were fixed and stained 3 days postinfection.
Growth kinetics.
Growth kinetics assays were performed in 96-well culture plates seeded overnight with 1 × 104 Vero E6 or RD cells per well. The cells were washed and infected in three independent replicates at a multiplicity of infection of 1. Virus adsorption was allowed to proceed for 2 h. The free viral inoculum was then removed, and the cells were washed twice and incubated at 37°C. Infected cells were frozen at different times postinfection. Plates were subjected to three freeze-thaw cycles, and each supernatant was then collected and clarified by centrifugation. Virus titers were determined in microplates by calculating the 50% tissue culture infective dose (TCID50) according to the Kärber method. The number of viral genome copies from each supernatant was evaluated by reverse transcription-quantitative PCR (RT-qPCR), as previously described (71).
Accession number(s).
The sequencing data generated during this study are publicly available from the EMBL-EBI database under accession numbers LT719044 to LT719068 (Table S1).
Supplementary Material
ACKNOWLEDGMENTS
We thank Pierre Charneau for providing the 293T/T7 cells and Julie Sappa for help with preparing the English manuscript.
This work was supported by the Institut Pasteur, a Program Transversal de Recherche PTR484 grant, and an Action Concertée des Instituts Pasteur ACIP A22-16 and Roux Howard Cantarini postdoctoral fellowship (R.V.) (https://www.pasteur.fr/fr) and by the Fondation Total grant S-CM15010-05B (https://www.foundation.total/fr).
The funders had no role in study design, data collection and interpretation, or in the decision to submit the work for publication.
R.V., R.R., J.-M.H., and F.D. initiated and designed this study; R.V., R.R., M.-L.J., M.B., S.R., S.A., J.R., B.B., and J.-L.B. collected data and performed experiments; R.V., J.-L.B., and F.D. wrote the first draft of the manuscript. All authors revised the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01667-18.
REFERENCES
- 1.Mirand A, Henquell C, Archimbaud C, Ughetto S, Antona D, Bailly JL, Peigue-Lafeuille H. 2012. Outbreak of hand, foot and mouth disease/herpangina associated with coxsackievirus A6 and A10 infections in 2010, France: a large citywide, prospective observational study. Clin Microbiol Infect 18:E110–E118. doi: 10.1111/j.1469-0691.2012.03789.x. [DOI] [PubMed] [Google Scholar]
- 2.Tapparel C, Siegrist F, Petty TJ, Kaiser L. 2013. Picornavirus and enterovirus diversity with associated human diseases. Infect Genet Evol 14:282–293. doi: 10.1016/j.meegid.2012.10.016. [DOI] [PubMed] [Google Scholar]
- 3.Pallansch M, Oberste M, Whitton J. 2013. Enteroviruses: polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses, p 839–893. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Rancaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 1 Lippincott Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 4.Wang J, Teng Z, Cui X, Li C, Pan H, Zheng Y, Mao S, Yang Y, Wu L, Guo X, Zhang X, Zhu Y. 2018. Epidemiological and serological surveillance of hand-foot-and-mouth disease in Shanghai, China, 2012–2016. Emerg Microbes Infect 7:8. doi: 10.1038/s41426-017-0011-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Solomon T, Lewthwaite P, Perera D, Cardosa MJ, McMinn P, Ooi MH. 2010. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect Dis 10:778–790. doi: 10.1016/S1473-3099(10)70194-8. [DOI] [PubMed] [Google Scholar]
- 6.Chong P, Liu CC, Chow YH, Chou AH, Klein M. 2015. Review of enterovirus 71 vaccines. Clin Infect Dis 60:797–803. doi: 10.1093/cid/ciu852. [DOI] [PubMed] [Google Scholar]
- 7.Duong V, Mey C, Eloit M, Zhu H, Danet L, Huang Z, Zou G, Tarantola A, Cheval J, Perot P, Laurent D, Richner B, Ky S, Heng S, Touch S, Sovann L, van Doorn R, Tan Tran T, Farrar JJ, Wentworth DE, Das SR, Stockwell TB, Manuguerra JC, Delpeyroux F, Guan Y, Altmeyer R, Buchy P. 2016. Molecular epidemiology of human enterovirus 71 at the origin of an epidemic of fatal hand, foot and mouth disease cases in Cambodia. Emerg Microbes Infect 5:e104. doi: 10.1038/emi.2016.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McMinn P, Lindsay K, Perera D, Chan HM, Chan KP, Cardosa MJ. 2001. Phylogenetic analysis of enterovirus 71 strains isolated during linked epidemics in Malaysia, Singapore, and Western Australia. J Virol 75:7732–7738. doi: 10.1128/JVI.75.16.7732-7738.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Midgley SE, Nielsen AG, Trebbien R, Poulsen MW, Andersen PH, Fischer TK. 2017. Co-circulation of multiple subtypes of enterovirus A71 (EV-A71) genotype C, including novel recombinants characterised by use of whole genome sequencing (WGS), Denmark 2016. Euro Surveill 22:30565. doi: 10.2807/1560-7917.ES.2017.22.26.30565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mirand A, Schuffenecker I, Henquell C, Billaud G, Jugie G, Falcon D, Mahul A, Archimbaud C, Terletskaia-Ladwig E, Diedrich S, Huemer HP, Enders M, Lina B, Peigue-Lafeuille H, Bailly JL. 2010. Phylogenetic evidence for a recent spread of two populations of human enterovirus 71 in European countries. J Gen Virol 91:2263–2277. doi: 10.1099/vir.0.021741-0. [DOI] [PubMed] [Google Scholar]
- 11.van der Sanden S, Koopmans M, Uslu G, van der Avoort H, Dutch Working Group For Clinical V. 2009. Epidemiology of enterovirus 71 in the Netherlands, 1963 to 2008. J Clin Microbiol 47:2826–2833. doi: 10.1128/JCM.00507-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Racaniello VR. 2013. Picornaviridae: the viruses and their replication, p 795–838. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Rancaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 1 Lippincott Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 13.Lukashev AN, Lashkevich VA, Ivanova OE, Koroleva GA, Hinkkanen AE, Ilonen J. 2003. Recombination in circulating enteroviruses. J Virol 77:10423–10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oprisan G, Combiescu M, Guillot S, Caro V, Combiescu A, Delpeyroux F, Crainic R. 2002. Natural genetic recombination between co-circulating heterotypic enteroviruses. J Gen Virol 83:2193–2200. doi: 10.1099/0022-1317-83-9-2193. [DOI] [PubMed] [Google Scholar]
- 15.Santti J, Hyypia T, Kinnunen L, Salminen M. 1999. Evidence of recombination among enteroviruses. J Virol 73:8741–8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simmonds P, Welch J. 2006. Frequency and dynamics of recombination within different species of human enteroviruses. J Virol 80:483–493. doi: 10.1128/JVI.80.1.483-493.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arita M, Zhu SL, Yoshida H, Yoneyama T, Miyamura T, Shimizu H. 2005. A Sabin 3-derived poliovirus recombinant contained a sequence homologous with indigenous human enterovirus species C in the viral polymerase coding region. J Virol 79:12650–12657. doi: 10.1128/JVI.79.20.12650-12657.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown B, Oberste MS, Maher K, Pallansch MA. 2003. Complete genomic sequencing shows that polioviruses and members of human enterovirus species C are closely related in the noncapsid coding region. J Virol 77:8973–8984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burns CC, Shaw J, Jorba J, Bukbuk D, Adu F, Gumede N, Pate MA, Abanida EA, Gasasira A, Iber J, Chen Q, Vincent A, Chenoweth P, Henderson E, Wannemuehler K, Naeem A, Umami RN, Nishimura Y, Shimizu H, Baba M, Adeniji A, Williams AJ, Kilpatrick DR, Oberste MS, Wassilak SG, Tomori O, Pallansch MA, Kew O. 2013. Multiple independent emergences of type 2 vaccine-derived polioviruses during a large outbreak in northern Nigeria. J Virol 87:4907–4922. doi: 10.1128/JVI.02954-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Combelas N, Holmblat B, Joffret ML, Colbere-Garapin F, Delpeyroux F. 2011. Recombination between poliovirus and coxsackie A viruses of species C: a model of viral genetic plasticity and emergence. Viruses 3:1460–1484. doi: 10.3390/v3081460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang P, Faase JA, Toyoda H, Paul A, Wimmer E, Gorbalenya AE. 2007. Evidence for emergence of diverse polioviruses from C-cluster coxsackie A viruses and implications for global poliovirus eradication. Proc Natl Acad Sci U S A 104:9457–9462. doi: 10.1073/pnas.0700451104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo WP, Lin XD, Chen YP, Liu Q, Wang W, Wang CQ, Li MH, Sun XY, Shi M, Holmes EC, Zhang YZ. 2015. Fourteen types of co-circulating recombinant enterovirus were associated with hand, foot, and mouth disease in children from Wenzhou, China. J Clin Virol 70:29–38. doi: 10.1016/j.jcv.2015.06.093. [DOI] [PubMed] [Google Scholar]
- 23.Huang SC, Hsu YW, Wang HC, Huang SW, Kiang D, Tsai HP, Wang SM, Liu CC, Lin KH, Su IJ, Wang JR. 2008. Appearance of intratypic recombination of enterovirus 71 in Taiwan from 2002 to 2005. Virus Res 131:250–259. doi: 10.1016/j.virusres.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 24.Lukashev AN, Shumilina EY, Belalov IS, Ivanova OE, Eremeeva TP, Reznik VI, Trotsenko OE, Drexler JF, Drosten C. 2014. Recombination strategies and evolutionary dynamics of the Human enterovirus A global gene pool. J Gen Virol 95:868–873. doi: 10.1099/vir.0.060004-0. [DOI] [PubMed] [Google Scholar]
- 25.Oberste MS, Penaranda S, Maher K, Pallansch MA. 2004. Complete genome sequences of all members of the species Human enterovirus A. J Gen Virol 85:1597–1607. doi: 10.1099/vir.0.79789-0. [DOI] [PubMed] [Google Scholar]
- 26.van der Sanden S, van Eek J, Martin DP, van der Avoort H, Vennema H, Koopmans M. 2011. Detection of recombination breakpoints in the genomes of human enterovirus 71 strains isolated in the Netherlands in epidemic and non-epidemic years, 1963–2010. Infect Genet Evol 11:886–894. doi: 10.1016/j.meegid.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 27.Yoke-Fun C, AbuBakar S. 2006. Phylogenetic evidence for inter-typic recombination in the emergence of human enterovirus 71 subgenotypes. BMC Microbiol 6:74. doi: 10.1186/1471-2180-6-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McWilliam Leitch EC, Cabrerizo M, Cardosa J, Harvala H, Ivanova OE, Koike S, Kroes AC, Lukashev A, Perera D, Roivainen M, Susi P, Trallero G, Evans DJ, Simmonds P. 2012. The association of recombination events in the founding and emergence of subgenogroup evolutionary lineages of human enterovirus 71. J Virol 86:2676–2685. doi: 10.1128/JVI.06065-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hassel C, Mirand A, Lukashev A, TerletskaiaLadwig E, Farkas A, Schuffenecker I, Diedrich S, Huemer HP, Archimbaud C, Peigue-Lafeuille H, Henquell C, Bailly JL. 2015. Transmission patterns of human enterovirus 71 to, from and among European countries, 2003 to 2013. Euro Surveill 20:30005. doi: 10.2807/1560-7917.ES.2015.20.34.30005. [DOI] [PubMed] [Google Scholar]
- 30.Bessaud M, Razafindratsimandresy R, Nougairede A, Joffret ML, Deshpande JM, Dubot-Peres A, Heraud JM, de Lamballerie X, Delpeyroux F, Bailly JL. 2014. Molecular comparison and evolutionary analyses of VP1 nucleotide sequences of new African human enterovirus 71 isolates reveal a wide genetic diversity. PLoS One 9:e90624. doi: 10.1371/journal.pone.0090624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saxena VK, Sane S, Nadkarni SS, Sharma DK, Deshpande JM. 2015. Genetic diversity of enterovirus A71, India. Emerg Infect Dis 21:123–126. doi: 10.3201/eid2101.140743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bessaud M, Pillet S, Ibrahim W, Joffret ML, Pozzetto B, Delpeyroux F, Gouandjika-Vasilache I. 2012. Molecular characterization of human enteroviruses in the Central African Republic: uncovering wide diversity and identification of a new human enterovirus A71 genogroup. J Clin Microbiol 50:1650–1658. doi: 10.1128/JCM.06657-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fernandez-Garcia MD, Kebe O, Fall AD, Dia H, Diop OM, Delpeyroux F, Ndiaye K. 2016. Enterovirus A71 genogroups C and E in children with acute flaccid paralysis, West Africa. Emerg Infect Dis 22:753–755. doi: 10.3201/eid2204.151588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sadeuh-Mba SA, Bessaud M, Massenet D, Joffret ML, Endegue MC, Njouom R, Reynes JM, Rousset D, Delpeyroux F. 2013. High frequency and diversity of species C enteroviruses in Cameroon and neighboring countries. J Clin Microbiol 51:759–770. doi: 10.1128/JCM.02119-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rakoto-Andrianarivelo M, Guillot S, Iber J, Balanant J, Blondel B, Riquet F, Martin J, Kew O, Randriamanalina B, Razafinimpiasa L, Rousset D, Delpeyroux F. 2007. Co-circulation and evolution of polioviruses and species C enteroviruses in a district of Madagascar. PLoS Pathog 3:e191. doi: 10.1371/journal.ppat.0030191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rakoto-Andrianarivelo M, Rousset D, Razafindratsimandresy R, Chevaliez S, Guillot S, Balanant J, Delpeyroux F. 2005. High frequency of human enterovirus species C circulation in Madagascar. J Clin Microbiol 43:242–249. doi: 10.1128/JCM.43.1.242-249.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Razafindratsimandresy R, Joffret ML, Rabemanantsoa S, Andriamamonjy S, Heraud JM, Delpeyroux F. 2013. Reemergence of recombinant vaccine-derived polioviruses in healthy children, Madagascar. Emerg Infect Dis 19:1008–1010. doi: 10.3201/eid1906.130080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bessaud M, Joffret ML, Holmblat B, Razafindratsimandresy R, Delpeyroux F. 2011. Genetic relationship between cocirculating Human enteroviruses species C. PLoS One 6:e24823. doi: 10.1371/journal.pone.0024823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joffret ML, Jegouic S, Bessaud M, Balanant J, Tran C, Caro V, Holmblat B, Razafindratsimandresy R, Reynes JM, Rakoto-Andrianarivelo M, Delpeyroux F. 2012. Common and diverse features of cocirculating type 2 and 3 recombinant vaccine-derived polioviruses isolated from patients with poliomyelitis and healthy children. J Infect Dis 205:1363–1373. doi: 10.1093/infdis/jis204. [DOI] [PubMed] [Google Scholar]
- 40.Jegouic S, Joffret ML, Blanchard C, Riquet FB, Perret C, Pelletier I, Colbere-Garapin F, Rakoto-Andrianarivelo M, Delpeyroux F. 2009. Recombination between polioviruses and co-circulating coxsackie A viruses: role in the emergence of pathogenic vaccine-derived polioviruses. PLoS Pathog 5:e1000412. doi: 10.1371/journal.ppat.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muslin C, Joffret M-L, Pelletier I, Blondel B, Delpeyroux F. 2015. Evolution and emergence of enteroviruses through intra- and inter-species recombination: plasticity and phenotypic impact of modular genetic exchanges in the 5′ untranslated region. PLoS Pathog 11:e1005266. doi: 10.1371/journal.ppat.1005266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oberste MS, Maher K, Kilpatrick DR, Pallansch MA. 1999. Molecular evolution of the human enteroviruses: correlation of serotype with VP1 sequence and application to picornavirus classification. J Virol 73:1941–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bible JM, Iturriza-Gomara M, Megson B, Brown D, Pantelidis P, Earl P, Bendig J, Tong CY. 2008. Molecular epidemiology of human enterovirus 71 in the United Kingdom from 1998 to 2006. J Clin Microbiol 46:3192–3200. doi: 10.1128/JCM.00628-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. 2015. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol 1:vev003. doi: 10.1093/ve/vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bessaud M, Delpeyroux F. 2012. Development of a simple and rapid protocol for the production of customized intertypic recombinant polioviruses. J Virol Methods 186:104–108. doi: 10.1016/j.jviromet.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 46.Apostol LN, Suzuki A, Bautista A, Galang H, Paladin FJ, Fuji N, Lupisan S, Olveda R, Oshitani H. 2012. Detection of non-polio enteroviruses from 17 years of virological surveillance of acute flaccid paralysis in the Philippines. J Med Virol 84:624–631. doi: 10.1002/jmv.23242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bottcher S, Neubauer K, Baillot A, Rieder G, Adam M, Diedrich S. 2015. Stool screening of Syrian refugees and asylum seekers in Germany, 2013/2014: identification of Sabin-like polioviruses. Int J Med Microbiol 305:601–606. doi: 10.1016/j.ijmm.2015.08.008. [DOI] [PubMed] [Google Scholar]
- 48.Harvala H, Calvert J, Van Nguyen D, Clasper L, Gadsby N, Molyneaux P, Templeton K, McWilliams Leitch C, Simmonds P. 2014. Comparison of diagnostic clinical samples and environmental sampling for enterovirus and parechovirus surveillance in Scotland, 2010 to 2012. Euro Surveill 19:20772 https://www.eurosurveillance.org/content/10.2807/1560-7917.ES2014.19.15.20772. [DOI] [PubMed] [Google Scholar]
- 49.Cobbin JCA, Britton PN, Burrell R, Thosar D, Selvakumar K, Eden JS, Jones CA, Holmes EC. 2018. A complex mosaic of enteroviruses shapes community-acquired hand, foot and mouth disease transmission and evolution within a single hospital. Virus Evol 4:vey020. doi: 10.1093/ve/vey020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caro V, Guillot S, Delpeyroux F, Crainic R. 2001. Molecular strategy for “serotyping” of human enteroviruses. J Gen Virol 82:79–91. doi: 10.1099/0022-1317-82-1-79. [DOI] [PubMed] [Google Scholar]
- 51.Gaunt E, Harvala H, Osterback R, Sreenu VB, Thomson E, Waris M, Simmonds P. 2015. Genetic characterization of human coxsackievirus A6 variants associated with atypical hand, foot and mouth disease: a potential role of recombination in emergence and pathogenicity. J Gen Virol 96:1067–1079. doi: 10.1099/vir.0.000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Holmblat B, Jegouic S, Muslin C, Blondel B, Joffret ML, Delpeyroux F. 2014. Nonhomologous recombination between defective poliovirus and coxsackievirus genomes suggests a new model of genetic plasticity for picornaviruses. mBio 5:e01119-14. doi: 10.1128/mBio.01119-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koonin EV, Dolja VV. 2014. Virus world as an evolutionary network of viruses and capsidless selfish elements. Microbiol Mol Biol Rev 78:278–303. doi: 10.1128/MMBR.00049-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lowry K, Woodman A, Cook J, Evans DJ. 2014. Recombination in enteroviruses is a biphasic replicative process involving the generation of greater-than genome length “imprecise” intermediates. PLoS Pathog 10:e1004191. doi: 10.1371/journal.ppat.1004191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lukashev AN. 2005. Role of recombination in evolution of enteroviruses. Rev Med Virol 15:157–167. doi: 10.1002/rmv.457. [DOI] [PubMed] [Google Scholar]
- 56.Lee KM, Gong YN, Hsieh TH, Woodman A, Dekker NH, Cameron CE, Shih SR. 2018. Discovery of enterovirus A71-like nonstructural genomes in recent circulating viruses of the enterovirus A species. Emerg Microbes Infect 7:111. doi: 10.1038/s41426-018-0107-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Phuektes P, Chua BH, Sanders S, Bek EJ, Kok CC, McMinn PC. 2011. Mapping genetic determinants of the cell-culture growth phenotype of enterovirus 71. J Gen Virol 92:1380–1390. doi: 10.1099/vir.0.029371-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bessaud M, Joffret ML, Blondel B, Delpeyroux F. 2016. Exchanges of genomic domains between poliovirus and other cocirculating species C enteroviruses reveal a high degree of plasticity. Sci Rep 6:38831. doi: 10.1038/srep38831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hu YF, Yang F, Du J, Dong J, Zhang T, Wu ZQ, Xue Y, Jin Q. 2011. Complete genome analysis of coxsackievirus A2, A4, A5, and A10 strains isolated from hand, foot, and mouth disease patients in China revealing frequent recombination of human enterovirus A. J Clin Microbiol 49:2426–2434. doi: 10.1128/JCM.00007-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Puenpa J, Vongpunsawad S, Osterback R, Waris M, Eriksson E, Albert J, Midgley S, Fischer TK, Eis-Hübinger AM, Cabrerizo M, Gaunt E, Simmonds P, Poovorawan Y. 2016. Molecular epidemiology and the evolution of human coxsackievirus A6. J Gen Virol 97:3225–3231. doi: 10.1099/jgv.0.000619. [DOI] [PubMed] [Google Scholar]
- 61.World Health Organization. 2004. Polio laboratory manual, 4th ed. World Health Organization, Geneva, Switzerland.
- 62.Oberste MS, Maher K, Williams AJ, Dybdahl-Sissoko N, Brown BA, Gookin MS, Penaranda S, Mishrik N, Uddin M, Pallansch MA. 2006. Species-specific RT-PCR amplification of human enteroviruses: a tool for rapid species identification of uncharacterized enteroviruses. J Gen Virol 87:119–128. doi: 10.1099/vir.0.81179-0. [DOI] [PubMed] [Google Scholar]
- 63.Joffret ML, Polston PM, Razafindratsimandresy R, Bessaud M, Heraud JM, Delpeyroux F. 2018. Whole genome sequencing of enteroviruses species A to D by high-throughput sequencing: application for viral mixture. Front Microbiol 9:2339. doi: 10.3389/fmicb.2018.02339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 65.Darriba D, Taboada GL, Doallo R, Posada D. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 67.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC. 1999. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 73:152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Drummond AJ, Rambaut A, Shapiro B, Pybus OG. 2005. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol 22:1185–1192. doi: 10.1093/molbev/msi103. [DOI] [PubMed] [Google Scholar]
- 71.Volle R, Nourrisson C, Mirand A, Regagnon C, Chambon M, Henquell C, Bailly JL, Peigue-Lafeuille H, Archimbaud C. 2012. Quantitative real-time RT-PCR assay for research studies on enterovirus infections in the central nervous system. J Virol Methods 185:142–148. doi: 10.1016/j.jviromet.2012.06.019. [DOI] [PubMed] [Google Scholar]
- 72.Razafindratsimandresy R, Joffret ML, Delpeyroux F, Heraud JM. 2014. First full genome sequence of a human enterovirus a120, isolated in Madagascar. Genome Announc 2:e00568-14. doi: 10.1128/genomeA.00568-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.