

Vaccinia virus was used as vaccine against smallpox and was instrumental in the successful eradication of that disease. Although smallpox vaccination is no longer in place in the overall population, the use of vaccinia virus in the development of viral vector-based vaccines has become popular. Nonetheless, different vaccinia virus strains are known and induce different immune responses. To look into this, we compared immune responses triggered by mouse infections with the nonreplicative MVA strain, the attenuated Lister strain, or the virulent WR strain. We observed that the WR strain was capable of downmodulating mouse cell responses, whereas the highly attenuated MVA strain induced high levels of cell-mediated immunity. Infections by the intermediately attenuated Lister strain induced cell responses that were intermediary between those induced by WR and MVA. We propose that the virulence of a vaccinia virus strain is directly proportional to its ability to downmodulate specific compartments of antiviral cell responses.
KEYWORDS: Lister strain, MVA, vaccinia virus, Western Reserve strain, mouse model, modulation of the host immune response
ABSTRACT
Vaccinia virus (VACV) is a notorious virus for a number of scientific reasons; however, most of its notoriety comes from the fact that it was used as a vaccine against smallpox, being ultimately responsible for the eradication of that disease. Nonetheless, many different vaccinia virus strains have been obtained over the years; some are suitable to be used as vaccines, whereas others are virulent and unsuitable for this purpose. Interestingly, different vaccinia virus strains elicit different immune responses in vivo, and this is a direct result of the genomic differences among strains. In order to evaluate the net result of virus-encoded immune evasion strategies of vaccinia viruses, we compared antiviral immune responses in mice intranasally infected by the highly attenuated and nonreplicative MVA strain, the attenuated and replicative Lister strain, or the virulent WR strain. Overall, cell responses elicited upon WR infections are downmodulated compared to those elicited by MVA and Lister infections, especially in determined cell compartments such as macrophages/monocytes and CD4+ T cells. CD4+ T cells are not only diminished in WR-infected mice but also less activated, as evaluated by the expression of costimulatory molecules such as CD25, CD212, and CD28 and by the production of cytokines, including tumor necrosis factor alpha (TNF-α), gamma interferon (IFN-γ), interleukin-4 (IL-4), and IL-10. On the other hand, MVA infections are able to induce strong T-cell responses in mice, whereas Lister infections consistently induced responses that were intermediary between those induced by WR and MVA. Together, our results support a model in which the virulence of a VACV strain is proportional to its potential to downmodulate the host’s immune responses.
IMPORTANCE Vaccinia virus was used as vaccine against smallpox and was instrumental in the successful eradication of that disease. Although smallpox vaccination is no longer in place in the overall population, the use of vaccinia virus in the development of viral vector-based vaccines has become popular. Nonetheless, different vaccinia virus strains are known and induce different immune responses. To look into this, we compared immune responses triggered by mouse infections with the nonreplicative MVA strain, the attenuated Lister strain, or the virulent WR strain. We observed that the WR strain was capable of downmodulating mouse cell responses, whereas the highly attenuated MVA strain induced high levels of cell-mediated immunity. Infections by the intermediately attenuated Lister strain induced cell responses that were intermediary between those induced by WR and MVA. We propose that the virulence of a vaccinia virus strain is directly proportional to its ability to downmodulate specific compartments of antiviral cell responses.
INTRODUCTION
Vaccinia virus (VACV) is the prototypic member of the Poxviridae family, which encompasses complex DNA viruses that replicate in the cell cytoplasm of many different hosts. VACV is an emblematic virus for a number of scientific and virological reasons; nonetheless, the virus has earned most of its notoriety from the fact that it was used as a vaccine against smallpox and, therefore, was directly responsible for the eradication of that disease. This was only possible due to the genomic and antigenic similarities between variola virus and VACV, so people infected by the latter became immunogenically protected against the former. Cowpox virus, another close relative of variola virus and VACV, was originally utilized by Edward Jenner on his early quest for protection against smallpox. Later on, cowpox was replaced by VACV as a smallpox immunogen, albeit exactly how this happened is a subject of debate. Equally uncertain is the biological origin of VACV, although studies have suggested that the virus may have been derived from a horsepox-like virus ancestor (1). As a vaccine against smallpox, VACV was distributed all over the world and was cultivated in the skin of horses, cattle, and sheep as well as in embryonated chicken eggs, depending on the locality, resulting in the appearance of different strains as viruses evolved and adapted to different biological settings. Different strains were given different names, reflecting the country/locality and/or health agency in/by which the virus was propagated (2, 3).
The highly attenuated modified vaccinia virus Ankara (MVA) strain was obtained after passing the chorioallantoid vaccinia virus Ankara (CVA) strain approximately 570 times in primary chicken embryo fibroblasts (CEFs). As a result of the adaptation process, the virus lost about 30 kb of its DNA and became unable to replicate in most mammalian cells (4, 5). Sequencing of the virus’ genome and comparison to other VACV strains revealed that DNA losses included genes related to host immune regulation, immune evasion, and host range (6, 7). Nonetheless, the block in the MVA replication cycle occurs at relatively late stages of virion assembly and maturation, and therefore, the virus expresses early, intermediate, and late viral genes as well as any recombinant gene placed under the control of such promoters, natural or synthetic (8, 9). Due to these characteristics, MVA is considered extremely safe and was used as a vaccine during the smallpox eradication campaign (5). The VACV Lister strain (VACV-LST), developed at the Lister Institute in the United Kingdom, is a vaccine strain that was used throughout the globe during the smallpox vaccination years. In fact, VACV-LST is considered the most widely distributed smallpox vaccine at that time, being used in the Americas, Europe, Africa, and Asia (2, 10). Like MVA, VACV-LST is significantly attenuated and apparently caused fewer adverse events than other smallpox vaccines available at the time of the smallpox eradication campaign (10). Different from MVA, however, VACV-LST is able to fully replicate within humans and other mammalian hosts. The VACV Western Reserve (VACV-WR) strain originated from repeatedly passing the VACV New York City Board of Health (NYCBH) strain in rabbits, mice, and diverse cell cultures. Adaptation to these hosts rendered VACV-WR highly neuropathogenic to mice and able to replicate to high titers in different mammalian tissues, making it unsuitable to be used as a vaccine (2, 11). Nevertheless, VACV-WR became the model virus for most studies concerning aspects of VACV and poxvirus biology.
Based on the success of the smallpox eradication program, the relative ease of making recombinant VACVs, the large genome capacity of all poxviruses, and their ability to accommodate heterologous genes, the idea of using recombinant VACVs to protect against heterologous pathogens grew over the past decades. The excellent safety record of some VACV vaccine strains, especially MVA, has turned them into natural candidates in the development of recombinant viral vectors. Indeed, MVA-based vectored vaccines against important infectious diseases have been described (12), including HIV (13–15), malaria (16), tuberculosis (17, 18), and Ebola virus (19), and therapeutic anticancer vaccines have also been described (20–22).
The ability of poxviruses to modulate, evade, and counteract host immune responses is largely recognized, and many proteins encoded by VACV and other poxviruses are known to affect particular compartments of host immunity, including the interferon (IFN) system, cytokine and chemokine signaling, complement, and more (23). Despite all these immune evasion mechanisms, poxvirus infections induce both innate and adaptive immune responses in hosts. Studies aiming to dissect the individual contribution of each arm of the immune system to protection against poxvirus infection revealed that CD4+ T-cell-dependent humoral responses are key for virus clearance, whereas CD8+ T-cell responses mediate protection upon reinfection (24). Nevertheless, how much do the immune evasion mechanisms encoded by VACV and other poxviruses contribute globally to the downmodulation of the host immune response to them? The individual contributions of poxvirus-encoded immune evasion-related proteins have been extensively studied by employing, for instance, deletion mutants; however, the combined effect of all encoded mechanisms of immune interference is far less understood. In order to look into this, we investigated mouse immune responses triggered by three different VACV strains presenting various degrees of virulence: the nonreplicative MVA strain, the replicative vaccine strain VACV-LST, and the virulent VACV-WR strain. We observed that monocyte/macrophage (CD14+ cells) and CD4+ T-cell activation seem to be significantly downmodulated in animals infected with VACV-WR but not in MVA-infected mice, whereas CD8+ T-cell downmodulation by any strain is apparently less obvious. In most experiments, infection with the intermediately virulent VACV-LST strain rendered an intermediate downmodulation profile between those observed in MVA- and VACV-WR-infected animals. Thus, our results revealed a pattern of global downmodulation of immune responses that is directly proportional to the virulence of each VACV strain.
RESULTS
Clinical signs in infected mice.
All animals infected intranasally (i.n.) with VACV-WR developed clinical signs such as weight loss, piloerection, arched backs, swelling in the face, and conjunctivitis, starting on days 6 and 7 postinfection (p.i.). About 30% of WR-infected animals died on days 10 and 11 p.i. As for the VACV-LST- or MVA-infected animals, no clinical signs were observed, and all mice were physically similar to control animals inoculated with phosphate-buffered saline (PBS) until day 14, when they were euthanized. Animals inoculated intraperitoneally (i.p.) presented a similar clinical pattern, according to each virus, albeit signs of infection in animals that developed disease seemed to be delayed in relation to those in animals inoculated through the nasal route. Such observations are congruent with those of many other similar studies (for an example, see reference 25).
Anti-VACV antibody production in infected animals.
To verify the production of anti-VACV antibodies, sera from intranasally inoculated animals were subjected to an enzyme-linked immunosorbent assay (ELISA). VACV-specific antibodies were detected in the sera of infected animals from all groups, compared to the absorbance values obtained for the mock-infected (uninfected) control group. Animals infected with VACV-WR showed a significantly higher level of production of antibodies than all other groups. Infection with VACV-LST induced an intermediate pattern of humoral response, with antibody levels that were higher than antibody levels in MVA-inoculated animals but lower than what was observed in mice infected with VACV-WR. MVA-inoculated mice generated anti-VACV antibody levels that were higher than those of the uninfected controls; however, antibody amounts in this group were significantly smaller than those obtained from animals infected with replicative VACVs (Fig. 1).
FIG 1.
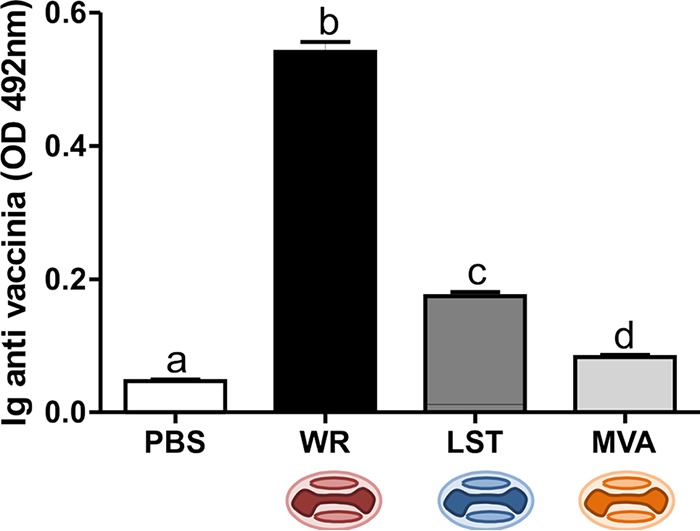
Anti-VACV antibody generation upon mouse infection with different VACV strains. Anti-vaccinia virus total antibodies were measured by an ELISA in serum of mice inoculated with PBS, VACV-WR, MVA, or VACV-LST. Bars represent the means ± standard errors of the values obtained for each group of mice (n = 7). Different letters indicate statistical differences (P < 0.05) between groups. For better visualization and orientation, colorful virus cartoons represent virus-infected groups in all figures (red, VACV-WR; blue, VACV-LST; orange, MVA). OD, optical density.
Analysis of splenic immune cell subsets during VACV infection.
Analyses of the mean frequencies (percentages) of T and B lymphocytes, NK cells, and monocytes were performed on splenocytes of mice 14 days after infection through the intranasal route. Although there were no significant differences in the frequencies of total T lymphocytes (CD3+) among all groups, the CD4+ and CD8+ T-cell subsets showed different frequencies between groups. Animals infected with VACV-WR presented a marked decrease in the frequency of CD4+ T lymphocytes and an increased frequency of CD8+ T cells compared to the uninfected control group and the MVA group (Table 1). In order to see whether these phenomena could be observed in animals inoculated through a different route of infection, we infected mice intraperitoneally using same amount of viruses as described above. In this case, route-dependent differences were detected when results were compared to those for mice infected through the nasal cavity (Fig. 2). In this case, CD4+ T-cell levels in VACV-infected mice, regardless of the virus strain, were similar to those of the uninfected controls (Fig. 2a). As for CD8+ T cells, a significant decrease was detected in MVA-infected mice, whereas infections with the replicative VACVs showed no difference in relation to the uninfected controls (Fig. 2b).
TABLE 1.
Mean frequencies of T (CD3+, CD4+, and CD8+) and B (CD19+) lymphocytes, NK cells (CD3− CD49+), and monocytes (CD14+) on splenocytes from noninfected mice or animals intranasally infected by vaccinia virus
| Group | Frequency (%) of cell phenotypea |
|||||
|---|---|---|---|---|---|---|
| CD3+ | CD4+ | CD8+ | CD19+ | CD3− CD49+ | CD14+ | |
| PBS | 43.7 | 27.0 (bc) | 13.9 (a) | 50.0 | 4.5 | 2.5 (a) |
| WR | 41.4 | 23.2 (a) | 17.3 (c) | 48.7 | 4.3 | 1.8 (b) |
| LST | 41.1 | 25.4 (ab) | 15.7 (bc) | 49.1 | 4.5 | 2.2 (ab) |
| MVA | 41.5 | 29.2 (c) | 14.4 (ab) | 47.9 | 4.1 | 2.4 (a) |
Different letters indicate statistical differences (P < 0.05) between groups (n = 7).
FIG 2.
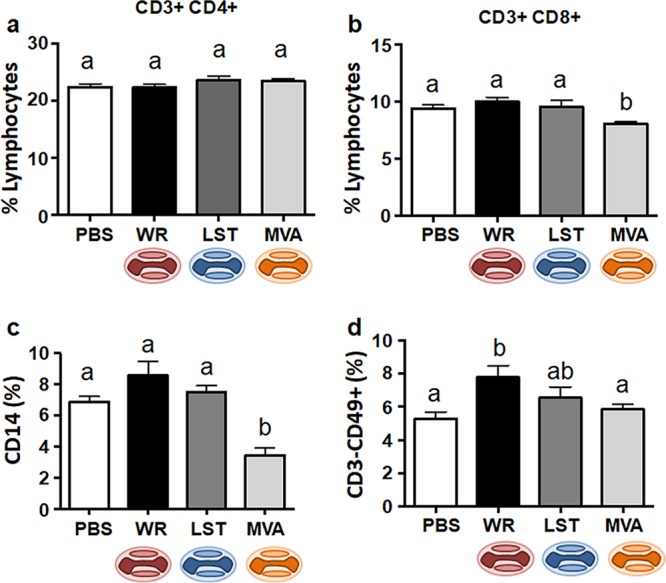
Mean frequencies of T lymphocytes, macrophages/monocytes, and NK cells on splenocytes from noninfected mice or animals intraperitoneally infected by vaccinia virus. Splenocytes from mice euthanized at 14 days p.i. were analyzed by flow cytometry, and lymphocytes were gated based on forward scatter (FSC) and side scatter (SSC). (a and b) Fluorochrome-labeled antibodies were used to detect the surface markers CD3, CD4 (CD4+ T lymphocytes) (a), CD8 (CD8+ T lymphocytes) (b), and CD49 (NK cells) (d). (c) Granulocytes were gated based on FSC and SSC, and macrophages/monocytes were identified according to the presence of the CD14 surface marker. Bars represent the means ± standard errors of each cell phonotype frequency within the gated population. Different letters indicate statistical differences (P < 0.05) between groups (n = 7 per group). For better visualization and orientation, colorful virus cartoons represent virus-infected groups in all figures (red, VACV-WR; blue, VACV-LST; orange, MVA).
Animals infected with VACV-WR through the nasal route showed a significant decrease in the frequency of macrophages compared to uninfected and MVA-infected mice (Table 1). The frequencies of B and NK cells were not statistically different among groups (Table 1). These results may suggest an ability of VACV-WR to suppress specific compartments of the adaptive immune response in infected hosts, something that has been described previously during infections with circulating and virulent strains of VACV (26). The global frequencies of T cells, B cells, NK cells, and monocytes/macrophages in the MVA-inoculated group were not different from those observed in the uninfected mice when infection was done through the intranasal route. (Table 1). Finally, cell frequencies in the VACV-LST group were intermediate between those seen in VACV-WR- and MVA-infected animals, independently of the route of infection. For instance, the observed CD4+ T-lymphocyte frequencies in VACV-LST-infected animals were higher than the frequencies in VACV-WR-infected mice but lower than the frequencies in the MVA-infected and mock-infected groups. Nevertheless, CD4+ T-cell frequencies in the VACV-LST group were statically different only compared to the MVA-infected animals (Table 1). Similar results were obtained when macrophages and monocytes (CD14+ cells) were analyzed. On the other hand, CD8+ T-cell frequencies in the VAVC-LST group were higher than those observed for the MVA- or mock-infected animals but were lower than those of the VACV-WR group after intranasal infection.
A different picture appeared when animals were infected through the intraperitoneal route. In this case, animals infected with replicative VACVs showed amounts of macrophages in the spleen that were not statistically different from those in uninfected controls, whereas MVA-infected animals produced fewer detected CD14+ cells (Fig. 2c). As for the NK cells, infection with VACV-WR produced an increased amount of CD3− CD49+ cells in the spleen, whereas the other infections yielded results that were not significantly different from those for the mock-infected group (Fig. 2d). Total frequencies of B cells in the VACV-inoculated groups were not different from those observed in the uninfected mice (not shown).
Immunophenotyping of macrophages, NK cells, and lymphocytes.
To further investigate how the different VACV strains modulate the immune system, we analyzed the activation profiles of several cell subsets in the spleens of infected mice. This was done through the evaluation of the presence of costimulatory molecules as activation markers. For example, the expression of CD80 and CD86 was evaluated as being indicative of CD14+ cell activation. Analyses of costimulatory molecules on monocytes/macrophages were performed by calculating the mean fluorescence intensity (MFI) in antibody-labeled cell preparations. The average expression level of CD86 was lower in macrophages from animals infected with VACV-WR than in the group infected with MVA when viruses were inoculated through the intranasal route of infection (Fig. 3a). Although not statistically supported, the MFI value for CD14+ CD86+ cells in VACV-LST-infected animals was intermediary between those in VACV-WR- and MVA-infected mice. There was no statistical difference among all groups regarding the expression of the CD80 molecule (data not shown).
FIG 3.
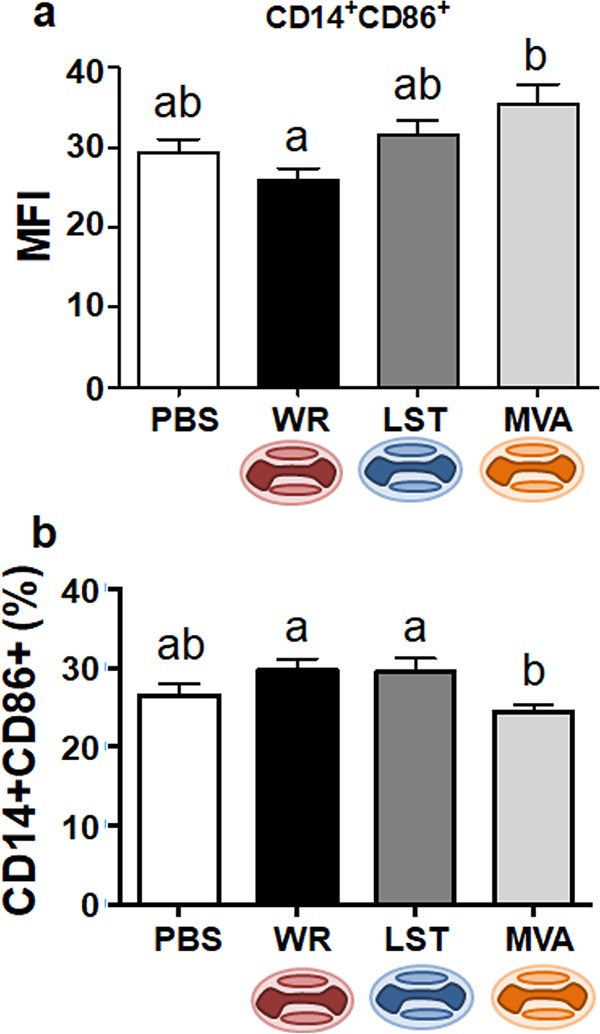
Expression of the CD86 activation marker in monocytes/macrophages from VACV-infected or uninfected mice. Splenocytes from mice euthanized at 14 days p.i. were incubated with fluorochrome-labeled antibodies to detect the expression of CD14 and CD86 molecules on cell surfaces by flow cytometry. Splenocytes were from intranasally (a) or intraperitoneally (b) infected mice. Bars represent the means ± standard errors of the mean fluorescence intensity (MFI) values of costimulatory molecules on macrophages (a) or the means ± standard errors of each cell phenotype frequency within the gated population (b). Different letters indicate statistical differences (P < 0.05) between groups (n = 7). For better visualization and orientation, colorful virus cartoons represent virus-infected groups in all figures (red, VACV-WR; blue, VACV-LST; orange, MVA).
As observed for the total cell counts, the activation of CD14+ cells in animals inoculated intraperitoneally showed a different pattern. MVA-infected mice produced fewer CD14+ CD86+ cells than mice infected with the replicative VACVs (Fig. 3b). Nonetheless, CD14+ CD86+ results for the infected groups were not statistically different from those seen in the uninfected control group. At this point, it was clear that patterns of immune modulation were different for the same viruses when inoculated through the two different routes of infection. The observed differences could be explained by the different virological and immunological dynamics in the two infection models. Orthopoxviruses use the lungs as a primary replication organ both in natural infections as well as after experimental inoculations (25, 27–31), whereas intraperitoneal infection is a rather artificial route of virus entry. Because these viruses do not use the abdominal cavity as a port to the host, they may have not evolved strategies to cope with the particularities of the host immune system at this site and consecutive lymphoid tissues. For this reason, we decided to focus solely on analyses of immune responses after intranasal infections with the different VACV strains.
Macrophages/monocytes, as well as dendritic cells (DCs), are key players in the activation of adaptive immune responses, working as antigen-presenting cells (APCs). Moreover, monocytes/macrophages have been shown to be essential in the control of Ectromelia virus (ECTV) infection in mice (ECTV is a close relative of VACV within the Orthopoxvirus genus) (32). Considering that we observed a virus-driven modulation of macrophage activation during infection through the intranasal route, we decided to analyze macrophage-lymphocyte linking molecules in this model of infection. The CD28 costimulatory molecule is a CD80 and CD86 ligand on T cells and is an effective costimulatory signal for T-cell activation (33). Indeed, decreased CD86 expression correlated with diminished CD28 expression on CD4+ (Fig. 4a) and CD8+ (Fig. 5a) lymphocytes in VACV-WR-infected animals. On the other hand, MVA-inoculated mice showed no alterations in the frequency of CD4+ CD28+ or CD8+ CD28+ cells compared to the uninfected group. As for the VACV-LST-infected mice, an intermediary pattern between the VACV-WR and MVA groups was, once again, observed (Fig. 4a and Fig. 5a).
FIG 4.
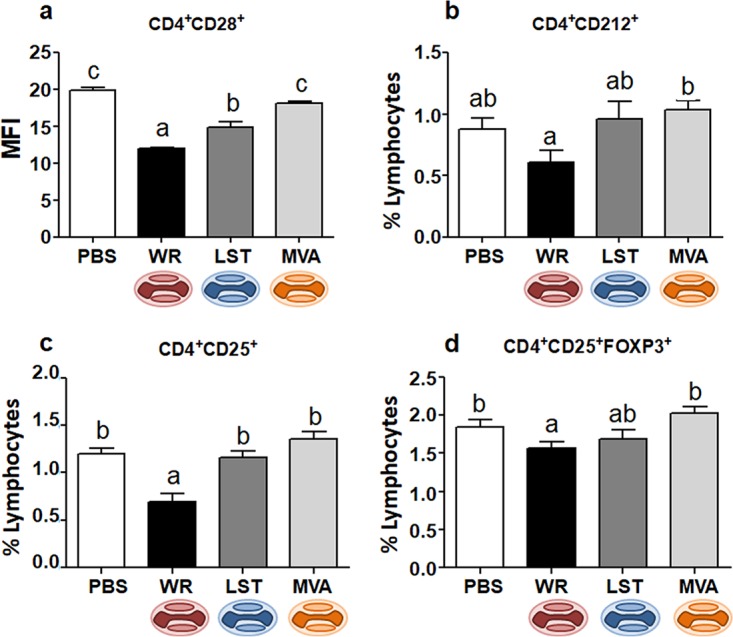
Expression of activation markers and Foxp3 in CD4+ T cells from VACV-infected or uninfected mice. Mice were infected intranasally, and splenocytes obtained at 14 days p.i. were incubated with fluorochrome-labeled antibodies to detect the expression of CD28 (a), CD212 (b), CD25 (c and d), and Foxp3 (d) in CD4+ T cells. In panel a, bars represent the means ± standard errors of the mean fluorescence intensity (MFI) values of costimulatory molecules on CD4+ lymphocytes; in panels b to d, bars represent the means ± standard errors of each cell phenotype frequency compared to the total number of lymphocytes. Different letters indicate statistical differences (P < 0.05) between groups (n = 7). For better visualization and orientation, colorful virus cartoons represent virus-infected groups in all figures (red, VACV-WR; blue, VACV-LST; orange, MVA).
FIG 5.
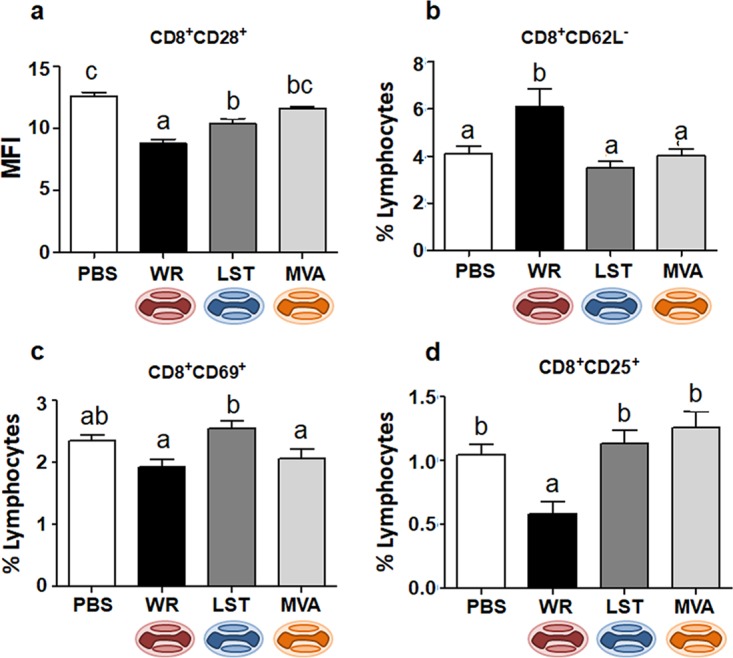
Expression of activation markers in CD8+ T cells from VACV-infected or uninfected mice. Mice were infected intranasally, and splenocytes obtained at 14 days p.i. were incubated with fluorochrome-labeled antibodies to detect the expression of CD28 (a), CD62L (b), CD69 (c), and CD25 (d) in CD8+ T cells. In panel a, bars represent the means ± standard errors of the mean fluorescence intensity (MFI) values of costimulatory molecules on CD8+ lymphocytes; in panels b to d, bars represent the means ± standard errors of each cell phenotype frequency compared to the total number of lymphocytes. Different letters indicate statistical differences (P < 0.05) between groups (n = 7). For better visualization and orientation, colorful virus cartoons represent virus-infected groups in all figures (red, VACV-WR; blue, VACV-LST; orange, MVA).
Next, we decided to further evaluate the activation status of other specific immune cell subsets in the animals infected through the nasal route. When we analyzed CD4+ T cells expressing the interleukin-12 (IL-12) receptor (CD212), we found a reduced frequency in animals infected with VACV-WR compared to the group of mice infected with MVA (Fig. 4b). The expression of CD212 in CD4+ T cells of VACV-WR-infected animals was also apparently decreased compared to the same cells from VACV-LST-infected or uninfected mice, although this was not statistically supported. The general profiles of CD8+ CD212+ T cells among all groups were similar to those observed for CD4+ CD212+ cells; however, there was no statistical difference between groups (data not shown). Next, we examined the expression of other immune activation cell surface markers. In all groups, the expression of CD69, a type C lectin expressed in recently activated cells, was not significantly altered on NK or CD4+ T cells of infected mice (data not shown). As for CD8+ T cells, only infection with VACV-LST induced an expression of CD69 that was statistically different from those in the other groups (Fig. 5c).
Finally, we further evaluated the presence of two other activation markers in T cells from VACV-infected and uninfected mice. First, we looked for the presence of CD62L, an L-selectin that is present on naive T cells and is lost upon T-cell activation. Next, we looked at CD25, the alpha chain of the IL-2 receptor, which is highly expressed upon T-cell activation (34). Compared to all other groups, VACV-WR-infected animals presented an increase in the frequency of CD8+ CD62L− cells, suggesting an augmented recruitment of naive T cells that became negative for this L-selectin (Fig. 5b). On the other hand, mice from the VACV-WR group showed decreases in the frequencies of both CD4+ (Fig. 4c) and CD8+ (Fig. 5d) T cells expressing CD25, and this tendency was also observed in animals infected through the peritoneum, although this was not statistically supported in the latter case (data not shown). A decrease in regulatory T-cell (CD4+ CD25+ Foxp3+) frequencies was observed after intranasal infection with VACV-WR compared to the other groups (Fig. 4d), although no statistical differences were observed between VACV-WR- and VACV-LST-infected mice. As described above, T cells from MVA-infected mice presented no alterations in CD25 and Foxp3 expression compared to the uninfected group, whereas cells from the VACV-LST group presented an intermediate activation profile between the VACV-WR and MVA/uninfected groups, although this was not always statistically supported.
Lymphocyte cytokine production analysis.
To investigate the production of cytokines by T lymphocytes from mice intranasally infected by different VACV strains, harvested splenocytes were stimulated with viral antigens (UV-inactivated VACV-WR) and analyzed by flow cytometry employing intracellular cytokine staining (ICS) assays. The frequency of tumor necrosis factor alpha (TNF-α)-producing CD4+ and CD8+ T lymphocytes in VACV-WR-infected animals was consistently lower than that of cells from uninfected animals and was also lower than that in mice infected with MVA and VACV-LST. In the case of CD4+ T cells producing TNF-α, however, there was no statistically significant differences between the VACV-WR- and VACV-LST-infected groups, and both replicative VACV strains seem to have induced a decrease in TNF-α production in CD4+ T cells in relation to uninfected animals and MVA-infected mice (Fig. 6a). When we looked at the production of other cytokines (gamma interferon [IFN-γ], IL-4, and IL-10) by T cells from VACV-infected or uninfected mice, similar patterns were detected. In most cases, frequencies of T cells producing cytokines were lower in splenocytes from VACV-WR-infected animals than in animals infected with other VACVs and mock-infected controls (Fig. 6b to d). In some cases, such differences were not statistically supported; nonetheless, a global pattern can be clearly observed: VACV-WR seems to induce a decrease in the production of cytokines in CD4+ and CD8+ T cells compared to the other virus-infected groups, with the exception of IL-4 CD8+ T cells (Fig. 6c). MVA inoculation, on the other hand, seems to exert an upregulation of cytokine production by T cells compared to uninfected controls or VACV-WR-infected mice, although in some cases, this was not statistically supported. Once more, infection with VACV-LST generates a profile that is intermediary between those observed in MVA- and VACV-WR-infected mice (Fig. 6).
FIG 6.
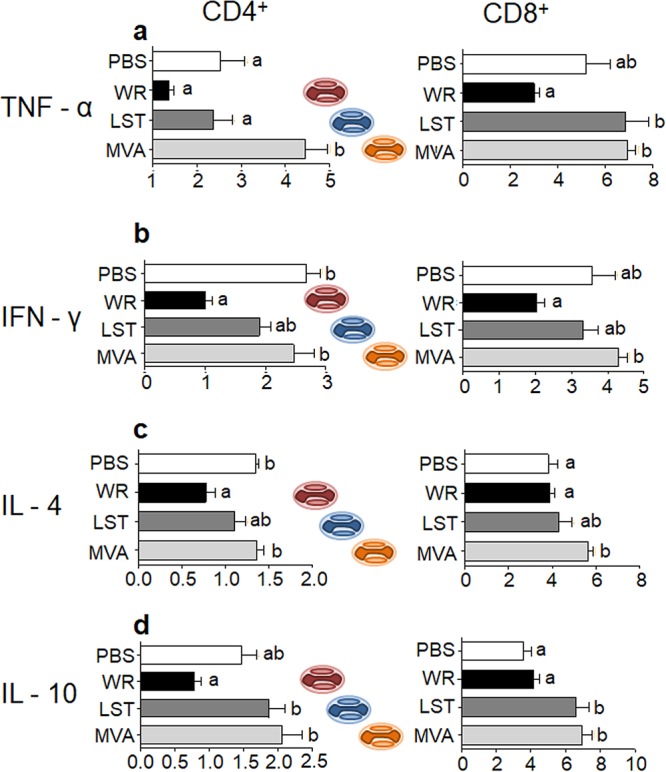
Cytokine production by CD4+ and CD8+ T cells after stimulation by viral antigens. Mice were infected intranasally, and splenocytes obtained at 14 days p.i. were incubated for 18 h in the presence of the stimulus (UV-inactivated VACV-WR). After incubation, cells were stained with fluorochrome-labeled antibodies to detect the expression of CD4 or CD8 cell surface molecules and intracytoplasmic TNF-α, IFN-γ, IL-4, and IL-10 by flow cytometry. Bars represent the means ± standard errors of the frequencies of cells producing cytokines in relation to the total number of CD4+ or CD8+ cells. Different letters indicate statistical differences (P < 0.05) between groups (n = 7). For better visualization and orientation, colorful virus cartoons represent virus-infected groups in all figures (red, VACV-WR; blue, VACV-LST; orange, MVA).
DISCUSSION
Poxviruses, and particularly orthopoxviruses, are known to encode a multitude of immune evasion mechanisms. The number of evasion strategies encoded in an individual poxvirus is directly proportional to the large size of its genome, which is able to accommodate many genes that code for proteins that interfere with a diverse array of the host’s immune responses. The mechanisms by which each of the immune evasion-related proteins interfere with the host’s immune response as well as the consequent impact of this protein on immunity against poxviruses have been evaluated in studies employing deletion mutants in which the specific gene coding for the immune evasion-related protein was removed (reviewed in reference 23). Nonetheless, this is not a practical approach when one wants to study the global effect of all encoded immune evasion-related proteins in a single virus population. Comparison of published genomes could be useful to this end, but inferences based solely on such information are limited. In the work of Meisinger-Henschel and colleagues, as an example, the authors introduced the six major deletions found in the MVA genome into to the CVA parental virus genome. As a result, the mutated CVA strain still did not develop the attenuation and restricted host range profile of MVA, suggesting that the MVA phenotype is a cooperative effect of gene deletions, point mutations outside the major deletion areas, and existing genes (35). In fact, the global way by which a virus interacts with its host in order to achieve replicative success is not the result of the expression of a single gene product, but rather, it is the net result of the sum of many host and virus factors interacting simultaneously during the virus replication cycle.
One possible way to glance into the combined effect of the many immunity evasion proteins encoded by a single virus is to study the immune responses induced by different strains of the same virus species presenting different virulence profiles and different genome characteristics (or gene contents). Thus, we opted to evaluate the modulation of the host’s immune responses upon mouse infection with three different VACV strains: the attenuated and nonreplicative MVA strain, the attenuated and replicative VACV-LST strain, and the virulent VACV-WR strain.
In our studies, we used the intranasal (i.n.) model of infection, through which BALB/c mice are highly susceptible when infected by virulent VACV strains (28). As a comparison system, we also infected animals intraperitoneally (i.p.) by employing the same infective doses and conditions as those used for the i.n. infections. VACV-WR i.n. infection results in acute lung infection, where the virus initially replicates. Thereafter, the virus disseminates to various organs, including brain, spleen, and liver (36–38). MVA, on the other hand, cannot multiply as efficiently in the lungs, and virus titers decay rapidly. Even when inoculated at high doses, MVA does not spread efficiently to other tissues. When inoculated through the i.p. route, however, MVA is able to reach organs such as the liver, spleen, and ovaries as efficiently as VACV-WR (39). As for VACV-LST, the virus does not multiply in mouse cells as readily as VACV-WR, but when administered in high doses, it can disseminate efficiently and reach organs such as kidneys and heart (40). Upon i.p. inoculation, orthopoxviruses readily reach systemic lymphoid organs, were they initially multiply, and later on, they are able to spread to other tissues and organs (27, 39). Such descriptions are compatible with the results that we observed in our infection experiments, in which we saw high mortality levels and intense clinical signs in VACV-WR-infected mice but not in animals infected with the attenuated VACV strains. Abdalrhman and colleagues (40) showed that VACV-WR-infected animals developed high titers of virus in the lungs and serum neutralizing antibodies on the 14th day of infection. On the other hand, they only found considerable antibody titers in VACV-LST-infected mice in the first week after infection, and the production of neutralizing antibodies was detected only when high viral doses were used. In the same study, MVA was found in the lungs only in the first 2 days of infection and did not induce the production of neutralizing antibodies. Ramirez et al. compared VACV-WR and MVA infections in mice and showed that WR induces a more expressive humoral response and neutralizing antibodies, whereas MVA triggered significant levels of anti-vaccinia virus IgG only when inoculated intraperitoneally at high doses (39, 41). MVA administered transdermally yielded nondetectable or very small amounts of IgG, while MVA administered intramuscularly (i.m.) resulted in higher levels of specific IgG (42). Likewise, MVA administered in mice through percutaneous inoculation elicited specific antibody responses, and animals were protected from a consecutive VACV lethal challenge, at levels comparable to or higher than those with subcutaneous or i.m. inoculation (43). The diminished humoral response observed in MVA-infected mice compared to animals infected by replicative VACV strains is a direct result of the inability of MVA to multiply productively in mouse cells and, hence, decreased exposure of viral antigens to the host’s immune system.
As mentioned above, the qualitative and quantitative aspects of the host immune response to a given pathogen are intrinsically dependent on the route of entry. Examples of studies that have look into this are abundant, but some are particularly illustrative. The mouse CD8+ T-cell response upon intradermal infection with human herpesvirus 1, for instance, is strictly CD4+ T-cell dependent; however, the same response is mostly CD4+ T-cell independent when inoculation occurs via the intraocular route of infection (44). Likewise, different routes of infection result in different disease parameters and immune responses in macaques inoculated with simian immunodeficiency virus (45). This phenomenon is not restricted to viruses and has been documented in model infections by other pathogens, such as Leishmania donovani, for which a strong Th1 response was induced upon subcutaneous infections, whereas inoculation through other routes tended to generate Th2-prone responses (46). As expected, the immune responses to VACV and other poxviruses are also intimately dependent on the route of virus inoculation. The cytotoxic-T-lymphocyte (CTL) responses to VACV-WR inoculated intraperitoneally are largely dependent on CD4+ T cells, and infections in CD4+ T-cell-deficient mice result in poor expansion of virus-specific CD8+ T-cell clones. On the other hand, intranasal infections by VACV-WR resulted in robust virus-specific CD8+ T-cell responses in the presence or absence of CD4+ T-helper cells, especially when higher viral loads were employed (30). Moreover, the authors of that study observed that intranasal VACV infections resulted in higher expression levels of antiviral cytokines such as type I IFN (IFN-I) and TNF-α, and they suggested that this is caused by the mucosal microenvironment found by the viruses when they reach the lungs, an environment that is mostly absent when they are inoculated through the peritoneum and reach primarily lymphoid organs. This may help to explain why our results showed different trends in immune responses and possible evasion strategies when infections using the two routes of infection are compared. Upon reaching the lung, VACVs can readily infect pulmonary epithelial cells as well as alveolar macrophages and DCs (31). Indeed, it has been demonstrated that VACVs have a strong tropism toward APCs (27, 47). Therefore, although VACVs distribute systemically after multiplying in the primary organ, they are prone to encountering a much larger population of APCs in the lungs than in lymphoid tissues. Thus, a possible evasion mechanism based on downmodulation of CD14+ cells going to the spleen, for example, would be more evident in animals infected through the nasal route of infection than in animals infected through the intraperitoneal route (Table 1 and Fig. 2c). Because professional APCs are essential for the three phases of the mounting T-cell response, cell expansion, contraction, and memory cell formation, through canonical signaling (signals 1 and 2), the observed differences in subset cell populations verified when mice were infected through the nose or the peritoneum could be related to different APC dynamics in each case (Fig. 2 to 4). Based on such an assumption, and considering that the respiratory route is the natural entry port for orthopoxviruses, we decided to focus on the evaluation of immune responses that developed after animals were infected through the i.n. route. Indeed, because the i.p. route is a rather artificial route of entry, is not logical to imagine that viruses would have evolved immune evasion mechanisms directed to this immune microenvironment.
When we analyzed the anti-VACV cell responses by looking at splenocytes of VACV-infected mice after i.n. infection, one particular aspect stood out: VACV-WR-infected mice had fewer CD4+ T lymphocytes and monocytes/macrophages than uninfected controls or mice infected with the attenuated VACV strains. On the other hand, animals infected with VACV-WR presented more cytotoxic T cells at day 14 p.i. than any other group. These results were similar to those observed in peripheral blood mononuclear cells (PBMCs) from human patients naturally infected with circulating VACV in Brazil. In that case, individuals presented more CD14+ cells, fewer B cells, and similar T-cell frequencies compared to uninfected individuals (26). The differences between that study and the results observed in our mouse infections may be related to the different hosts, different stages of infection (not specified in the human study), different viral strains (humans were infected by zoonotic VACVs in rural Brazil), and different routes of infection (human infection occurs naturally through direct contact with lesions in sick cows). Nonetheless, the cellular activation patterns in mouse and human studies were strikingly similar. Like individuals infected with zoonotic VACVs, animals infected with VACV-WR presented, in general, reductions in the levels of activated cells, particularly in the activation status of CD14+ cells and CD4+ T cells. MVA-inoculated animals, however, presented cell frequencies and activation patterns that were similar to those of the uninfected controls, whereas mice infected with VACV-LST elicited intermediary patterns between those of VACV-WR- and MVA-infected/uninfected animals.
The lower frequency of CD4+ T cells expressing the receptor for the cytokine IL-12 in animals infected with VACV-WR through the i.n. route may be related to the lower percentage of monocytes/macrophages and lower expression level of the costimulatory molecule CD86 in the spleens from these animals. APCs have been described as one of the preferential targets of VACV infection (27, 31, 47, 48), and if they are less present and less activated, CD4+ cells would receive weaker stimulation via IL-12 and, consequently, decrease their IFN-γ production. Indeed, as seen in our results, infection by VACV-WR is marked by a lower percentage of CD4 lymphocytes producing this cytokine (Fig. 6b), which constitutes an important pathway to maintain the Th1 response and to control intracellular pathogens. Once again, this is similar to the results seen in humans, in which numbers of monocytes and activated CD4+ T cells are also decreased in patients infected with circulating virus samples compared to uninfected patients (26). Likewise, we observed a lower frequency of CD28+ T lymphocytes in VACV-WR-infected animals. These mice also showed a reduction in CD25-expressing T cells, which may further explain the decreased activation of lymphocytes and, possibly, lower level of IL-2 production. This may also justify the lower frequency of regulatory T cells in VACV-WR-infected animals, as a high dependence on IL-2 has been described for these cells (49). On the other hand, the VACV-LST group presented an intermediate percentage of regulatory T cells and a similar percentage of activated T lymphocytes in relation to the MVA and PBS control groups. MVA-infected mice showed the same frequency of activated T lymphocytes as the PBS group, suggesting an inability of this virus strain to suppress this particular compartment of the host’s immune system. Indeed, MVA was shown to robustly induce CCL2 chemokine expression in a Toll-like receptor 2 (TLR-2)-dependent fashion and rapidly attract activated leukocytes (50).
The higher frequency of CD8+ CD62L− lymphocytes observed in animals infected by VACV-WR through the i.n. route is a direct outcome of the high level of spread of this virus to other organs beyond the initial exposure site (36–38). Thus, there is a greater circulation of antigens and consequent sensitization of naive lymphocytes in draining lymph nodes. As a result, the stimulated cells lose this selectin and migrate toward the affected tissues in an attempt to control the infection. It is important to mention that, upon VACV-WR mouse infection, we have not seen a marked downmodulation of either the total number of CD8+ T cells or the expression of activation markers after VACV-specific stimulation (with the exception of CD25). In the case of the MVA infections, CD8+ T-cell numbers and activation patterns were frequently similar to those observed in mock-infected animals. Indeed, strikingly poor CD8+ T-cell immunogenicity, considering either epitope-specific responses or the total number of activated CD8+ T cells irrespective of specificity, has been described for BALB/c mice inoculated with MVA (51).
After VACV infection, NK and T cells produce TNF-α and IFN-γ, which are important proinflammatory cytokines involved in the buildup of an effective adaptive antiviral immune response (52). Compared to all other groups in our study, considering the i.n. route of infection, the production of those cytokines by CD4+ and CD8+ T cells from animals infected with VACV-WR seems to be importantly downmodulated. On the other hand, MVA-inoculated mice produced a larger amount of virus-responsive T cells producing TNF-α and IFN-γ, although only CD4+ T cells expressed cytokine levels that were above the noninfected control threshold. Indeed, Ramirez and coworkers observed that MVA-infected mice produced more TNF-α than cells from VACV-WR-infected animals, although in that case, the authors used the i.p. route of infection (39). BALB/c mice are more susceptible to VACV-WR infections when the virus is given through the i.n. route than when it is given through the i.p. route of infection (25). As demonstrated by our results, the production of TNF-α and IFN-γ by T cells after VACV-WR infection is severely compromised, and this may explain the host’s inability to control and successfully clear the infection. Nonetheless, the level of IL-4 production by T cells in MVA-infected mice was higher than that in VACV-WR-infected animals. Because IL-4 stimulates B-cell activation and induces the differentiation of naive T cells into Th2 cells, these results are not consistent with the fact that we observed a much more intense anti-VACV humoral response in VACV-WR-infected mice. This needs to be further explored. Finally, we evaluated the production of the IL-10 anti-inflammatory cytokine after VACV infection. There have been reports that some pathogens are able to modulate the host immune responses by stimulating or mimicking IL-10 (53). We observed that mice infected with VACV-WR presented reduced levels of IL-10 compared to uninfected controls and to animals inoculated with MVA or VACV-LST, which presented robust production of IL-10.
In conclusion, our results showed that the virulent VACV-WR strain is able to induce a marked downmodulation of the host’s immune response upon mouse infections through the i.n. route, a natural entry route for orthopoxviruses. Such modulation is particularly evident for some specific compartments of the host’s immunity, including CD4+ T lymphocytes and CD14+ cell responses. On the other hand, mice infected with the attenuated, nonreplicative MVA strain presented immune responses that were quite similar to those seen in uninfected animals. Finally, the attenuated but replicative VAV-LST strain induced immune responses that were frequently intermediary between those seen for the other VACVs upon infection. Such results may support the proposition of a model in which the in vivo virulence of a given VACV strain is directly proportional to its potential to downmodulate the host’s immune responses (Fig. 7). Genomic and proteomic analyses would be useful to support such assumptions; however, the global way by which a virus interacts with its host in order to achieve replicative success is not the result of the expression of a single gene product, but rather, it is likely the net result of many host and virus factors interacting simultaneously during the virus replication cycle.
FIG 7.
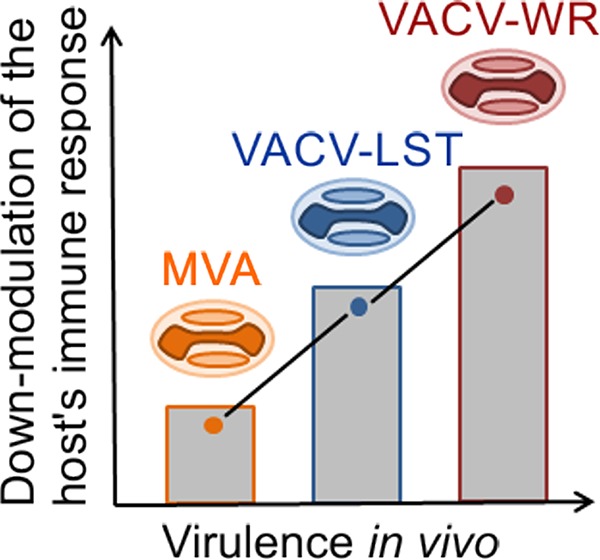
Model of the correlation between the virulence of vaccinia virus strains and their ability to downmodulate the host’s immune responses in vivo. MVA, modified vaccinia virus Ankara strain; VACV-LST, vaccinia virus Lister strain; VACV-WR, vaccinia virus Western Reserve strain. The graph represents a qualitative correlation only. Not all immune compartments are equally modulated. For better visualization and orientation, colorful virus cartoons represent virus-infected groups in all figures (red, VACV-WR; blue, VACV-LST; orange, MVA).
MATERIALS AND METHODS
Cells and viruses.
African green monkey cells (BSC-40 cells; ATCC, USA) and primary chicken embryo fibroblast (CEF) cells were grown at 37°C with 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Gibco, NY, USA) and 2 mM l-glutamine (Invitrogen Gibco, NY, USA). CEFs were prepared as described previously (54).
VACV strains employed in this study were MVA, VACV-WR, and VACV-LST, kindly provided by B. Moss (National Institutes of Health, USA), C. Jungwirth (Universität Wurzburg, Germany), and the Instituto Butantã (Brazil), respectively. VACV-WR and VACV-LST were grown and titrated in BSC-40 cells, whereas MVA was propagated and titrated on CEFs. All viruses were purified in sucrose cushions.
Infection of mice.
Mice were obtained from the Centro de Bioterismo (CEBIO) (Instituto de Ciências Biológicas, UFMG, Belo Horizonte, Brazil) and maintained in our experimental animal facility throughout the experiments. They were kept in ventilated cages with food and water ad libitum. All protocols involving animal experimentation were approved by the Committee of Ethics for Animal Experimentation (CETEA) at UFMG, under permit 9/2009. The CETEA-UFMG is affiliated with the National Council of Animal Experimentation Control (CONCEA).
Groups of 6-week-old BALB/c mice, composed of seven males each, were anaesthetized by intraperitoneal injection of ketamine and xylazine (70 mg and 12 mg/kg of body weight in phosphate-buffered saline [PBS], respectively) and inoculated by intranasal (i.n.) route using 5 × 103 PFU of VACV-WR or VACV-LST or 108 PFU of MVA in 10 μl of PBS. Negative controls received only PBS. The same amounts of viruses were used in experiments using the intraperitoneal route of infection, and inoculations were carried out as described previously (39). The VACV-WR infective dose was determined according to methods described previously by Ferreira and coworkers (28), who had previously established the 50% lethal dose (LD50) values for the very same sample. As strain Lister is also replicative, we opted to use the same dose. Because the MVA strain is unable to fully replicate in mice, animals were inoculated with a higher dose, as previously described (25). Considering that there was some mortality in the group of animals infected with VACV-WR, an excess of 3 animals (totaling 10 animals) was used in each experiment so that at the end of the 14th day, at least 7 animals would still be alive and available for the experiment assessments.
Splenocyte preparation and immunophenotyping.
Fourteen days after infection, mice were anaesthetized and euthanized, and blood and spleens were collected. Spleens were macerated, and after erythrocyte lysis (ACK [ammonium-chloride-potassium] lysing buffer), the remaining cells were resuspended in RPMI medium. Next, 5 × 105 cells were stained with fluorescence-labeled antibodies (BD Pharmingen, NJ, USA) specific for cell surface molecules diluted in PBS–0.5% bovine serum albumin (BSA). FOXP3 intracellular staining was performed according to the kit manufacturer’s directions (eBioscience, CA, USA). After 30 min of incubation at 4°C in the dark, cells were washed twice with PBS–0.5% BSA, fixed with fluorescence-activated cell sorter (FACS) fix solution, and stored at 4°C in the dark. A FACS LSR Fortessa or a FACSCalibur instrument (Becton, Dickinson, NJ, USA) was used for flow cytometry, and further analyses were performed using FlowJo software (TreeStar Inc., OR, USA).
Intracellular cytokine staining assay.
To detect intracellular cytokines, 107 spleen cells were stimulated overnight with UV-inactivated VACV-WR (5 × 105 PFU/ml) and then incubated for 4 h at 37°C with brefeldin A (Sigma, MO, USA) at 1 mg/ml. Cells were then washed in FACS buffer and stained with anti-CD4 and anti-CD8 antibodies (BD Pharmingen, NJ, USA) for 30 min at 4°C in the dark. After fixation and permeabilization with FACS buffer containing 0.5% saponin, cells were stained with mouse anti-TNF-α, -IFN-γ, -IL-4, and -IL-10 (BD Pharmingen, NJ, USA) for a further 30 min at room temperature. Finally, cells were washed once with FACS buffer containing 0.5% saponin and twice in FACS buffer alone. Cell preparations were fixed in FACS fix solution and stored at 4°C in the dark. Results were acquired using a FACSCalibur instrument (Becton, Dickinson, NJ, USA) and analyzed with FlowJo software (TreeStar Inc., OR, USA).
Enzyme-linked immunosorbent assay.
Serum samples obtained from infected animals were used in an enzyme-linked immunosorbent assay (ELISA). In short, 96-well plates (Falcon, MO, USA) were coated overnight with 100 ng/well of antigenic surface proteins from the WR strain in sodium carbonate buffer (0.5 M; pH 9.6) at 4°C. Plates were washed with PBS containing 0.05% Tween 20 and blocked for 2 h at 37°C with PBS containing 0.25% casein. Plates were incubated overnight at 4°C with mouse serum samples in a 1/20 dilution in PBS-casein. Plates were washed again and incubated with goat anti-mouse horseradish peroxidase (HRP)-labeled immunoglobulins [IgM, IgG, and IgA(H+L)] (Southern Biotechnology Associates, Birmingham, AL) for 1 h at 37°C. The plates were then washed five times and incubated in the dark with H2O2 in the presence of orthophenylene-diamine (OPD; Sigma, St. Louis, MO, USA) in sodium citrate buffer (0.1 M; pH 5.0) for 15 min. Reactions were stopped by the addition of 30 μl of 2 N H2SO4 to the mixture. Color development was measured at 492 nm in an automatic ELISA reader (ASYS Expert Plus; Biochrom, Cambridge, UK). Results were obtained by averaging the duplicates of each serum dilution in individual mice. Each score is shown as the mean ± standard error of the mean (SEM) of data from groups of 7 animals.
Statistical analysis.
For comparison between groups (noninfected versus infected mice), data were subjected to analysis of variance (ANOVA) and a Tukey posttest. To analyze the results obtained within the same group, stimulated or not, in the experiments for intracellular cytokine assays, parametric Student’s t test was used. In both tests, P values of less than 0.05 were considered significantly different. Statistical analyses were performed using Prism 5 software (GraphPad Software).
ACKNOWLEDGMENTS
We thank T. M. G. Pinho for technical help.
The work was supported by FAPEMIG and CNPq grants and by the Instituto Nacional de Ciência e Tecnologia de Vacinas (INCTV) (National Institute of Science and Technology of Vaccines). F. G. da Fonseca was a CNPq fellowship recipient during the development of this work. The Post-Graduation programs at UFMG are supported by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). L. F. D. de Freitas and R. P. Rocha were/are CAPES fellowship recipients.
We declare no conflict of interest. Funding sources had no role in the experiment design, interpretation of results, or writing or submission of the article.
REFERENCES
- 1.Tulman ER, Delhon G, Afonso CL, Lu Z, Zsak L, Sandybaev NT, Kerembekova UZ, Zaitsev VL, Kutish GF, Rock DL. 2006. Genome of horsepox virus. J Virol 80:9244–9258. doi: 10.1128/JVI.00945-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jacobs BL, Langland JO, Kibler KV, Denzler KL, White SD, Holechek SA, Wong S, Huynh T, Baskin CR. 2009. Vaccinia virus vaccines: past, present and future. Antiviral Res 84:1–13. doi: 10.1016/j.antiviral.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qin L, Favis N, Famulski J, Evans DH. 2015. Evolution of and evolutionary relationships between extant vaccinia virus strains. J Virol 89:1809–1824. doi: 10.1128/JVI.02797-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mayr A, Stickl H, Muller HK, Danner K, Singer H. 1978. The smallpox vaccination strain MVA: marker, genetic structure, experience gained with the parenteral vaccination and behavior in organisms with a debilitated defence mechanism. Zentralbl Bakteriol B 167:375–390. (In German.) [PubMed] [Google Scholar]
- 5.Stickl H, Hochstein-Mintzel V, Mayr A, Huber HC, Schäfer H, Holzner A. 1974. MVA vaccination against smallpox: clinical tests with an attenuated live vaccinia virus strain (MVA). Dtsch Med Wochenschr 99:2386–2392. (In German.) doi: 10.1055/s-0028-1108143. [DOI] [PubMed] [Google Scholar]
- 6.Antoine G, Scheiflinger F, Dorner F, Falkner FG. 1998. The complete genomic sequence of the modified vaccinia Ankara strain: comparison with other orthopoxviruses. Virology 244:365–396. doi: 10.1006/viro.1998.9123. [DOI] [PubMed] [Google Scholar]
- 7.Meyer H, Sutter G, Mayr A. 1991. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. J Gen Virol 72:1031–1038. doi: 10.1099/0022-1317-72-5-1031. [DOI] [PubMed] [Google Scholar]
- 8.Blanchard TJ, Alcami A, Andrea P, Smith GL. 1998. Modified vaccinia virus Ankara undergoes limited replication in human cells and lacks several immunomodulatory proteins: implications for use as a human vaccine. J Gen Virol 79:1159–1167. doi: 10.1099/0022-1317-79-5-1159. [DOI] [PubMed] [Google Scholar]
- 9.Sutter G, Moss B. 1992. Nonreplicating vaccinia vector efficiently expresses recombinant genes. Proc Natl Acad Sci U S A 89:10847–10851. doi: 10.1073/pnas.89.22.10847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenthal SR, Merchlinsky M, Kleppinger C, Goldenthal KL. 2001. Developing new smallpox vaccines. Emerg Infect Dis 7:920–926. doi: 10.3201/eid0706.010602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qin L, Upton C, Hazes B, Evans DH. 2011. Genomic analysis of the vaccinia virus strain variants found in Dryvax vaccine. J Virol 85:13049–13060. doi: 10.1128/JVI.05779-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kreijtz JH, Gilbert SC, Sutter G. 2013. Poxvirus vectors. Vaccine 31:4217–4219. doi: 10.1016/j.vaccine.2013.06.073. [DOI] [PubMed] [Google Scholar]
- 13.Garcia F, Bernaldo de Quiros JC, Gomez CE, Perdiguero B, Najera JL, Jimenez V, Garcia-Arriaza J, Guardo AC, Perez I, Diaz-Brito V, Jiménez V, García-Arriaza J, Guardo AC, Pérez I, Díaz-Brito V, Conde MS, González N, Alvarez A, Alcamí J, Jiménez JL, Pich J, Arnaiz JA, Maleno MJ, León A, Muñoz-Fernández MA, Liljeström P, Weber J, Pantaleo G, Gatell JM, Plana M, Esteban M. 2011. Safety and immunogenicity of a modified pox vector-based HIV/AIDS vaccine candidate expressing Env, Gag, Pol and Nef proteins of HIV-1 subtype b (MVA-b) in healthy HIV-1-uninfected volunteers: a phase I clinical trial (RISVAC02). Vaccine 29:8309–8316. doi: 10.1016/j.vaccine.2011.08.098. [DOI] [PubMed] [Google Scholar]
- 14.Guimaraes-Walker A, Mackie N, McCormack S, Hanke T, Schmidt C, Gilmour J, Barin B, McMichael A, Weber J, Legg K, Babiker A, Hayes P, Gotch F, Smith C, Dally L, Dorrell L, Cebere I, Kay R, Winstone N, Moore S, Goonetilleke N, Fast P, IAVI-006 Study Group. 2008. Lessons from IAVI-006, a phase I clinical trial to evaluate the safety and immunogenicity of the pTHr.HIVA DNA and MVA.HIVA vaccines in a prime-boost strategy to induce HIV-1 specific T-cell responses in healthy volunteers. Vaccine 26:6671–6677. doi: 10.1016/j.vaccine.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 15.Sandstrom E, Nilsson C, Hejdeman B, Brave A, Bratt G, Robb M, Cox J, Vancott T, Marovich M, Stout R, Aboud S, Bakari M, Pallangyo K, Ljungberg K, Moss B, Earl P, Michael N, Birx D, Mhalu F, Wahren B, Biberfeld G, HIV Immunogenicity Study 01/02 Team. 2008. Broad immunogenicity of a multigene, multiclade HIV-1 DNA vaccine boosted with heterologous HIV-1 recombinant modified vaccinia virus Ankara. J Infect Dis 198:1482–1490. doi: 10.1086/592507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Webster DP, Dunachie S, Vuola JM, Berthoud T, Keating S, Laidlaw SM, McConkey SJ, Poulton I, Andrews L, Andersen RF, Bejon P, Butcher G, Sinden R, Skinner MA, Gilbert SC, Hill AV. 2005. Enhanced T cell-mediated protection against malaria in human challenges by using the recombinant poxviruses FP9 and modified vaccinia virus Ankara. Proc Natl Acad Sci U S A 102:4836–4841. doi: 10.1073/pnas.0406381102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McShane H, Pathan AA, Sander CR, Keating SM, Gilbert SC, Huygen K, Fletcher HA, Hill AV. 2004. Recombinant modified vaccinia virus Ankara expressing antigen 85a boosts BCG-primed and naturally acquired antimycobacterial immunity in humans. Nat Med 10:1240–1244. doi: 10.1038/nm1128. [DOI] [PubMed] [Google Scholar]
- 18.Nicol MP, Grobler LA. 2010. MVA-85a, a novel candidate booster vaccine for the prevention of tuberculosis in children and adults. Curr Opin Mol Ther 12:124–134. [PubMed] [Google Scholar]
- 19.Tapia MD, Sow SO, Lyke KE, Haidara FC, Diallo F, Doumbia M, Traore A, Coulibaly F, Kodio M, Onwuchekwa U, Sztein MB, Wahid R, Campbell JD, Kieny MP, Moorthy V, Imoukhuede EB, Rampling T, Roman F, De Ryck I, Bellamy AR, Dally L, Mbaya OT, Ploquin A, Zhou Y, Stanley DA, Bailer R, Koup RA, Roederer M, Ledgerwood J, Hill AVS, Ballou WR, Sullivan N, Graham B, Levine MM. 2016. Use of CHAd3-EBO-Z Ebola virus vaccine in Malian and US adults, and boosting of Malian adults with MVA-BN-Filo: a phase 1, single-blind, randomised trial, a phase 1b, open-label and double-blind, dose-escalation trial, and a nested, randomised, double-blind, placebo-controlled trial. Lancet Infect Dis 16:31–42. doi: 10.1016/S1473-3099(15)00362-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amato RJ, Stepankiw M. 2012. Clinical efficacy of TroVax in the treatment of progressive castration-resistant prostate cancer. Clin Med Insights Oncol 6:67–73. doi: 10.4137/CMO.S7654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corona Gutierrez CM, Tinoco A, Navarro T, Contreras ML, Cortes RR, Calzado P, Reyes L, Posternak R, Morosoli G, Verde ML, Rosales R. 2004. Therapeutic vaccination with MVA E2 can eliminate precancerous lesions (CIN 1, CIN 2, and CIN 3) associated with infection by oncogenic human papillomavirus. Hum Gene Ther 15:421–431. doi: 10.1089/10430340460745757. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Hernandez E, Gonzalez-Sanchez JL, Andrade-Manzano A, Contreras ML, Padilla S, Guzman CC, Jimenez R, Reyes L, Morosoli G, Verde ML, Rosales R. 2006. Regression of papilloma high-grade lesions (CIN 2 and CIN 3) is stimulated by therapeutic vaccination with MVA E2 recombinant vaccine. Cancer Gene Ther 13:592–597. doi: 10.1038/sj.cgt.7700937. [DOI] [PubMed] [Google Scholar]
- 23.Smith GL, Benfield CT, Maluquer de Motes C, Mazzon M, Ember SW, Ferguson BJ, Sumner RP. 2013. Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. J Gen Virol 94:2367–2392. doi: 10.1099/vir.0.055921-0. [DOI] [PubMed] [Google Scholar]
- 24.Xu R, Johnson AJ, Liggitt D, Bevan MJ. 2004. Cellular and humoral immunity against vaccinia virus infection of mice. J Immunol 172:6265–6271. doi: 10.4049/jimmunol.172.10.6265. [DOI] [PubMed] [Google Scholar]
- 25.Earl PL, Americo JL, Moss B. 2017. Insufficient innate immunity contributes to the susceptibility of the castaneous mouse to orthopoxvirus infection. J Virol 91:e01042-17. doi: 10.1128/JVI.01042-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Silva Gomes JA, de Araujo FF, de Souza Trindade G, Quinan BR, Drumond BP, Ferreira JM, Mota BE, Nogueira ML, Kroon EG, Santos Abrahao J, Côrrea-Oliveira R, da Fonseca FG. 2012. Immune modulation in primary vaccinia virus zoonotic human infections. Clin Dev Immunol 2012:974067. doi: 10.1155/2012/974067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramirez JC, Finke D, Esteban M, Kraehenbuhl JP, Acha-Orbea H. 2003. Tissue distribution of the Ankara strain of vaccinia virus (MVA) after mucosal or systemic administration. Arch Virol 148:827–839. doi: 10.1007/s00705-003-0006-z. [DOI] [PubMed] [Google Scholar]
- 28.Ferreira JM, Drumond BP, Guedes MI, Pascoal-Xavier MA, Almeida-Leite CM, Arantes RM, Mota BE, Abrahao JS, Alves PA, Oliveira FM, Ferreira PC, Bonjardim CA, Lobato ZI, Kroon EG. 2008. Virulence in murine model shows the existence of two distinct populations of Brazilian vaccinia virus strains. PLoS One 3:e3043. doi: 10.1371/journal.pone.0003043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abrahão JS, Guede MI, Trindade GS, Fonseca FG, Campos RK, Mota BF, Lobato ZI, Silva-Fernandes AT, Rodrigues GO, Lima LS, Ferreira PC, Bonjardim CA, Kroon EG. 2009. One more piece in the VACV ecological puzzle: could peridomestic rodents be the link between wildlife and bovine vaccinia outbreaks in Brazil? PLoS One 4:e7428. doi: 10.1371/journal.pone.0007428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu Z, Molloy MJ, Usherwood EJ. 2016. CD4(+) T-cell dependence of primary CD8(+) T-cell response against vaccinia virus depends upon route of infection and viral dose. Cell Mol Immunol 13:82–93. doi: 10.1038/cmi.2014.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altenburg AF, van de Sandt CE, Li BWS, MacLoughlin RJ, Fouchier RAM, van Amerongen G, Volz A, Hendriks RW, de Swart RL, Sutter G, Rimmelzwaan GF, de Vries RD. 2017. Modified vaccinia virus Ankara preferentially targets antigen presenting cells in vitro, ex vivo and in vivo. Sci Rep 7:8580. doi: 10.1038/s41598-017-08719-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karupiah G, Buller RM, Van Rooijen N, Duarte CJ, Chen J. 1996. Different roles for CD4+ and CD8+ T lymphocytes and macrophage subsets in the control of a generalized virus infection. J Virol 70:8301–8309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Linsley PS, Ledbetter JA. 1993. The role of the CD28 receptor during T-cell responses to antigen. Annu Rev Immunol 11:191–212. doi: 10.1146/annurev.iy.11.040193.001203. [DOI] [PubMed] [Google Scholar]
- 34.Fazekas de St Groth B, Smith AL, Higgins CA. 2004. T cell activation: in vivo veritas. Immunol Cell Biol 82:260–268. doi: 10.1111/j.0818-9641.2004.01243.x. [DOI] [PubMed] [Google Scholar]
- 35.Meisinger-Henschel C, Späth M, Lukassen S, Wolferstätter M, Kachelriess H, Baur K, Dirmeier U, Wagner M, Chaplin P, Suter M, Hausmann J. 2010. Introduction of the six major genomic deletions of modified vaccinia virus Ankara (MVA) into the parental vaccinia virus is not sufficient to reproduce an MVA-like phenotype in cell culture and in mice. J Virol 84:9907–9919. doi: 10.1128/JVI.00756-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reading PC, Smith GL. 2003. A kinetic analysis of immune mediators in the lungs of mice infected with vaccinia virus and comparison with intradermal infection. J Gen Virol 84:1973–1983. doi: 10.1099/vir.0.19285-0. [DOI] [PubMed] [Google Scholar]
- 37.Smee DF, Bailey KW, Sidwell RW. 2001. Treatment of lethal vaccinia virus respiratory infections in mice with cidofovir. Antivir Chem Chemother 12:71–76. doi: 10.1177/095632020101200105. [DOI] [PubMed] [Google Scholar]
- 38.Turner GS. 1967. Respiratory infection of mice with vaccinia virus. J Gen Virol 1:399–402. doi: 10.1099/0022-1317-1-3-399. [DOI] [PubMed] [Google Scholar]
- 39.Ramirez JC, Gherardi MM, Esteban M. 2000. Biology of attenuated modified vaccinia virus Ankara recombinant vector in mice: virus fate and activation of B- and T-cell immune responses in comparison with the Western Reserve strain and advantages as a vaccine. J Virol 74:923–933. doi: 10.1128/JVI.74.2.923-933.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abdalrhman I, Gurt I, Katz E. 2006. Protection induced in mice against a lethal orthopox virus by the Lister strain of vaccinia virus and modified vaccinia virus Ankara (MVA). Vaccine 24:4152–4160. doi: 10.1016/j.vaccine.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 41.Ramirez JC, Gherardi MM, Rodriguez D, Esteban M. 2000. Attenuated modified vaccinia virus Ankara can be used as an immunizing agent under conditions of preexisting immunity to the vector. J Virol 74:7651–7655. doi: 10.1128/JVI.74.16.7651-7655.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knitlova J, Hajkova V, Voska L, Elsterova J, Obrova B, Melkova Z. 2014. Development of eczema vaccinatum in atopic mouse models and efficacy of MVA vaccination against lethal poxviral infection. PLoS One 9:e114374. doi: 10.1371/journal.pone.0114374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meseda CA, Atukorale V, Kuhn J, Schmeisser F, Weir JP. 2016. Percutaneous vaccination as an effective method of delivery of MVA and MVA-vectored vaccines. PLoS One 11:e0149364. doi: 10.1371/journal.pone.0149364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rajasagi NK, Kassim SH, Kollias CM, Zhao X, Chervenak R, Jennings SR. 2009. CD41 T cells are required for the priming of CD81 T cells following infection with herpes simplex virus type 1. J Virol 83:5256–5268. doi: 10.1128/JVI.01997-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stevceva L, Tryniszewska E, Hel Z, Nacsa J, Kelsall B, Washington Parks R, Franchini G. 2001. Differences in time of virus appearance in the blood and virus-specific immune responses in intravenous and intrarectal primary SIVmac251 infection of rhesus macaques; a pilot study. BMC Infect Dis 1:9. doi: 10.1186/1471-2334-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaur S, Kaur T, Garg N, Mukherjee S, Raina P, Athokpam V. 2008. Effect of dose and route of inoculation on the generation of CD4+ Th1/Th2 type of immune response in murine visceral leishmaniasis. Parasitol Res 103:1413–1419. doi: 10.1007/s00436-008-1150-x. [DOI] [PubMed] [Google Scholar]
- 47.Chahroudi A, Chavan R, Koyzr N, Waller EK, Silvestri G, Feinberg MB. 2005. Vaccinia virus tropism for primary hematolymphoid cells is determined by restricted expression of a unique virus receptor. J Virol 79:10397–10407. doi: 10.1128/JVI.79.16.10397-10407.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jelinkova A, Benda R, Danes L. 1973. Electron microscope study of the course of poxvirus officinale infection in cultures of alveolar macrophages. Acta Virol 17:124–129. [PubMed] [Google Scholar]
- 49.Fontenot JD, Rudensky AY. 2005. A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. Nat Immunol 6:331–337. doi: 10.1038/ni1179. [DOI] [PubMed] [Google Scholar]
- 50.Lehmann MH, Torres-Dominguez LE, Price PJ, Brandmuller C, Kirschning CJ, Sutter G. 2016. Ccl2 expression is mediated by type I IFN receptor and recruits NK and T cells to the lung during MVA infection. J Leukoc Biol 99:1057–1064. doi: 10.1189/jlb.4MA0815-376RR. [DOI] [PubMed] [Google Scholar]
- 51.Russell TA, Tscharke DC. 2014. Strikingly poor CD8+ T-cell immunogenicity of vaccinia virus strain MVA in BALB/c mice. Immunol Cell Biol 92:466–469. doi: 10.1038/icb.2014.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abboud G, Tahiliani V, Desai P, Varkoly K, Driver J, Hutchinson TE, Salek-Ardakani S. 2016. Natural killer cells and innate interferon gamma participate in the host defense against respiratory vaccinia virus infection. J Virol 90:129–141. doi: 10.1128/JVI.01894-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kurilla MG, Swaminathan S, Welsh RM, Kieff E, Brutkiewicz RR. 1993. Effects of virally expressed interleukin-10 on vaccinia virus infection in mice. J Virol 67:7623–7628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hernandez R, Brown DT. 2010. Growth and maintenance of chick embryo fibroblasts (CEF). Curr Protoc Microbiol Appendix 4:4I. doi: 10.1002/9780471729259.mca04is17. [DOI] [PubMed] [Google Scholar]