

The ability of herpes simplex virus 1 (HSV-1) to periodically reactivate from latency results in virus transmission and recurrent disease. The incidence of reactivation from latency is increased by chronic or acute stress. Stress increases the levels of corticosteroids, which bind and activate the glucocorticoid receptor (GR). Since GR activation is an immediate early response to stress, we tested whether the GR influences productive infection and the promoter that drives infected cell protein 0 (ICP0) expression. Pretreatment of cells with a GR-specific antagonist (CORT-108297) significantly reduced virus replication. Although the GR had little effect on ICP0 promoter activity alone, the Krüppel-like transcription factor 15 (KLF15) cooperated with the GR to stimulate promoter activity in transfected cells. In transfected or infected cells, the GR and KLF15 occupied ICP0 sequences important for transactivation. Collectively, these studies provide insight into how stress can directly stimulate productive infection and viral gene expression.
KEYWORDS: HSV-1, KLF15, glucocorticoid receptor, stress response, viral gene expression, productive infection
ABSTRACT
Following acute infection, herpes simplex virus 1 (HSV-1) establishes lifelong latency in neurons. Physical, emotional, and chemical stresses are linked to increasing the incidence of reactivation from latency, but the mechanism of action is not well understood. In general, stress increases corticosteroid levels, leading to activation of the glucocorticoid receptor (GR), a pioneer transcription factor. Consequently, we hypothesized that stress-mediated activation of the GR can stimulate productive infection and viral gene expression. New studies demonstrated that the GR-specific antagonist (CORT-108297) significantly reduced HSV-1 productive infection in mouse neuroblastoma cells (Neuro-2A). Additional studies demonstrated that the activated GR and Krüppel-like transcription factor 15 (KLF15) cooperatively transactivated the infected cell protein 0 (ICP0) promoter, a crucial viral regulatory protein. Interestingly, the synthetic corticosteroid dexamethasone and GR or KLF15 alone had little effect on ICP0 promoter activity in transfected Neuro-2A or Vero cells. Chromatin immunoprecipitation (ChIP) studies revealed that the GR and KLF15 occupied ICP0 promoter sequences important for transactivation at 2 and 4 h after infection; however, binding was not readily detected at 6 h after infection. Similar results were obtained for cells transfected with the full-length ICP0 promoter. ICP0 promoter sequences lack a consensus “whole” GR response element (GRE) but contain putative half-GREs that were important for dexamethasone induced promoter activity. The activated GR stimulates expression of, and interacts with, KLF15; consequently, these data suggest KLF15 and the GR form a feed-forward loop that activates viral gene expression and productive infection following stressful stimuli.
IMPORTANCE The ability of herpes simplex virus 1 (HSV-1) to periodically reactivate from latency results in virus transmission and recurrent disease. The incidence of reactivation from latency is increased by chronic or acute stress. Stress increases the levels of corticosteroids, which bind and activate the glucocorticoid receptor (GR). Since GR activation is an immediate early response to stress, we tested whether the GR influences productive infection and the promoter that drives infected cell protein 0 (ICP0) expression. Pretreatment of cells with a GR-specific antagonist (CORT-108297) significantly reduced virus replication. Although the GR had little effect on ICP0 promoter activity alone, the Krüppel-like transcription factor 15 (KLF15) cooperated with the GR to stimulate promoter activity in transfected cells. In transfected or infected cells, the GR and KLF15 occupied ICP0 sequences important for transactivation. Collectively, these studies provide insight into how stress can directly stimulate productive infection and viral gene expression.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) infection leads to serious recurrent eye infections in more than 400,000 individuals annually, including herpetic stromal keratitis (HSK) (1). HSK is characterized by corneal scarring and neovascularization, which can ultimately lead to blindness. Long-term oral acyclovir treatment only reduces HSK recurrences by 41% because most cases are the result of reactivation from latency (2). Many cases of HSV-1-induced recurrent encephalitis are also due to reactivation from latency (3). Identifying the factors that drive reactivation from latency may lead to novel therapeutic strategies designed to interfere with reactivation from latency.
A primary site for lifelong latent infections is sensory neurons in trigeminal ganglia (TG) following infection of the oral, ocular, or nasal cavity (4). Abundant viral protein expression and infectious virus are not readily detected during latency. In contrast, abundant expression of the virus-encoded latency-associated transcript occurs in latently infected sensory neurons. The ability of HSV-1 to reactivate from latency is crucial for virus transmission and recurrent disease. Increased physical, emotional, and chemical stressors correlate with a higher incidence of reactivation from latency in humans (5, 6). Addition of the synthetic corticosteroid dexamethasone (DEX) was reported to accelerate explant-induced reactivation from latency (7) and productive infection (8). Stressful stimuli, in general, increase levels of corticosteroids, which then enter a cell and bind to the glucocorticoid receptor (GR) or mineralocorticoid receptor (MR) (9). The MR or GR corticosteroid complex enters the nucleus, specifically binds a glucocorticoid response element (GRE), alters chromatin confirmation, and activates transcription (9, 10).
DEX-inducible cellular factors in TG neurons were previously identified during early phases of bovine herpesvirus 1 (BoHV-1) reactivation from latency (11). A subset of these cellular genes encode transcription factors, including four Krüppel-like transcription factors (KLF), including KLF4, KLF6, KLF15, and PZLF (promyelocytic leukemia zinc finger). A recent study demonstrated KLF15 and GR cooperate to transactivate the bovine herpesvirus 1 (BoHV-1) immediate early transcription unit 1 (IEtu1) promoter, which drives IE expression of infected cell protein 0 (ICP0) and ICP4 (12). In response to stress, the GR and KLF15 regulate gene expression dynamics and integrate signals by a positive feed-forward loop (13–15). KLF members and specificity protein 1 (Sp1) belong to the same superfamily of transcription factors, and these family members bind GC-rich sequences (16, 17). Certain KLF family members can activate some promoters via Sp1 binding sites, which are present in many HSV-1 promoters. Since viral genes necessary for productive infection are not abundantly expressed in latently infected sensory neurons, it is reasonable to suggest that cellular transcription factors, including GR and KLF family members, stimulate viral transcription during early stages of reactivation from latency.
In this study, we tested whether DEX stimulated productive infection and whether the GR plus DEX transactivated the ICP0 promoter. A GR antagonist (CORT-108297) reduced productive infection in mouse neuroblastoma cells (Neuro-2A). Additional studies indicated that the GR and KLF15 cooperated to stimulate ICP0 promoter activity. Conversely, KLF4, KLF6, and PZLF had little to no effect on ICP0 promoter activity alone or in combination with the GR and DEX treatment. Chromatin immunoprecipitation (ChIP) studies demonstrated that GR and KLF5 associated with the ICP0 promoter, both in cells transfected with the promoter and in HSV-1-infected cells.
RESULTS
The GR antagonist CORT-108297 impairs productive infection.
To test whether the GR influences HSV-1 productive infection, the GR antagonist CORT-108297 was used. CORT-108297 was chosen for these studies because it is a GR-specific antagonist that has no effect on the progesterone receptor or mineralocorticoid receptor and has no known off-target effects (18, 19). Neuro-2A cells were used for these studies because they have neuronal-like properties, and when growth factors are removed, these cells withdraw from the cell cycle and differentiate into dopamine-like neurons (20). Neuro-2A cell cultures were treated with increasing concentrations of CORT-108297 for 24 h in order to assess cytotoxic effects of this antagonist. Cytotoxicity was not observed at the higher concentrations of CORT-108297 (10, 25, or 50 μM [Fig. 1A]). At 1 μM CORT-108297, the GR antagonist slightly enhanced cell growth relative to the dimethyl sulfoxide (DMSO) control.
FIG 1.
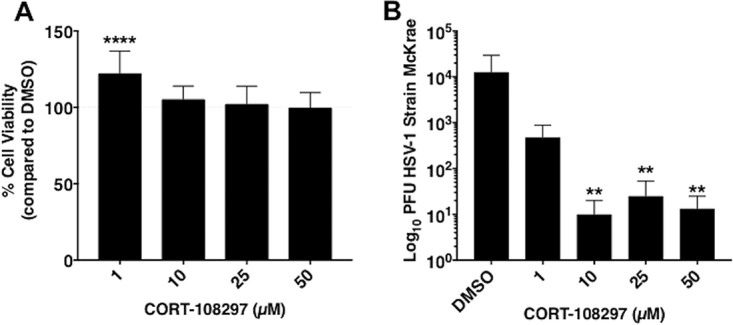
Effect of CORT-108297 on cell growth and productive infection. (A) Neuro-2A cells were grown in 24-well dishes that contained MEM plus 5% FBS and then cultures were treated with increasing levels of the GR-specific antagonist CORT-108297 or DMSO for 24 h. Cell viability was determined as described in Materials and Methods. Data are shown as mean percent viability versus the DMSO control in quadruplicate wells. (B) Neuro-2A cells were treated with the respective concentrations of CORT-108297 or DMSO for 2 h prior to infection with HSV-1 (MOI of 1). Twenty-four hours later, total virus was measured by plaque assays as described in Materials and Methods. All analyses were performed and graphs constructed using GraphPad Prism (version 7.0d). Ordinary one-way ANOVA with no correction for multiple comparisons was performed on all data sets. **, P < 0.005; ****, P < 0.0001.
Next, Neuro-2A cell cultures were treated with increasing concentrations of CORT-108297 for 2 h prior to infection with wild-type (wt) HSV-1 (strain McKrae). The same concentration of CORT-108297 was reapplied to cultures following infection and allowed to incubate for 24 h. Cells were harvested and then total virus was measured by plaque assay. The levels of infectious virus decreased more than 2 logs when Neuro-2A cell cultures were incubated with 10, 25, or 50 μM CORT-108297 relative to cultures treated with the DMSO vehicle (Fig. 1B). Even at 1 μM CORT-108297, a 1-log decrease in virus replication was observed. In summary, this study provided evidence that inhibiting the GR significantly reduced virus replication in Neuro-2A cells.
KLF15 cooperates with the activated GR to transactivate ICP0 promoter activity.
To test whether the GR can regulate ICP0 promoter activity, dual-luciferase assays were performed using a construct that contains the full-length (FL) ICP0 promoter upstream of the firefly luciferase gene. Previous studies demonstrated that the FL ICP0 promoter construct is regulated by heat stress-induced factors and there are numerous transcription factor binding sites in this promoter (21) (Fig. 2A). We also examined KLF family members that are induced during BoHV-1 reactivation from latency (11) because certain KLF family members cooperate with the GR to stimulate the BoHV-1 immediate early transcription unit 1 (IEtu1) promoter (12). The FL ICP0 construct was cotransfected into Neuro-2A or Vero cells with the GR, a KLF family member, and a Renilla luciferase gene driven by a minimal thymidine kinase (TK) promoter. These studies were performed in minimal essential medium (MEM) plus 2% stripped fetal bovine serum (FBS) in the presence or absence of DEX. FBS was passed through a column containing “activated” charcoal, removing hormones, lipid-based molecules, certain growth factors, and cytokines, yielding stripped FBS; however, this process does not remove salts, glucose, and most amino acids. A commercially available corticosterone-specific enzyme-linked immunosorbent assay (ELISA; Enzo Life Sciences) demonstrated that corticosterone levels in 10% FBS were 3,517.4 pg/ml but only 192.7 pg/ml in 2% stripped serum. Corticosterone was measured because mouse serum cortisol and corticosterone are closely correlated in dynamics under different physiological or stressful stimuli (22). Further evidence that FBS contains functional corticosteroids was the finding that nearly all Neuro-2A cells incubated with MEM plus 10% FBS contain GR in the nucleus; conversely, most Neuro-2A cells contained GR in the cytoplasm when incubated with 2% stripped FBS (23). Hence, it was important to use stripped FBS to evaluate the effect DEX has on ICP0 promoter activity.
FIG 2.

ICP0 promoter activation by GR and KLF family members. (A) Schematic of full-length ICP0 promoter (FL ICP0) and deletion constructs used for this study. Locations of certain transcription factor binding sites in the FL ICP0 promoter are indicated below the ICP0 constructs. Neuro-2A (B, D, and F) or Vero (C, E, and G) cells were cultured in 2% stripped FBS after transfection with the full-length ICP0 promoter construct. Shown is luciferase activity at 40 h after cells were cotransfected with the FL ICP0 promoter (0.5 μg DNA), vector expressing the mouse GR (1.0 μg of DNA), and/or vector expressing human KLF15 (B and C), KLF4 (D and E), or KLF6 (F and G) (0.5 μg of DNA). Empty vector plasmid was added to certain samples to equalize the total amount of DNA in each transfection. Twenty-four hours following transfection, certain cultures were treated with DEX for 14 h (10 μM). The results are the means from 3 independent experiments. Statistical analysis was performed as described in Materials and Methods. *, P < 0.05; **, P < 0.005; ****, P < 0.0001. ns, not significant.
Strikingly, GR+DEX+KLF15 increased ICP0 promoter activity 9-fold in Neuro-2A cells and 25-fold in Vero cells relative to basal activity of the ICP0 promoter (Fig. 2B and C). Even when KLF15+GR was cotransfected with the ICP0 promoter (no DEX treatment), promoter activity was increased approximately 12-fold in Vero cells. GR+DEX increased ICP0 promoter activity only 2- to 3-fold, but this effect was significantly higher than with the GR or DEX alone (Fig. 2B to G). KLF15 alone increased ICP0 promoter activity 2- and 5-fold in Neuro-2A and Vero cells, respectively (Fig. 2B and C).
In contrast to the effects that GR+DEX+KLF15 had on ICP0 promoter activity, GR+DEX and KLF4 or KLF6 transactivated the FL ICP0 promoter only 5-fold in Vero cells (Fig. 2E and G). In Neuro-2A cells, KLF4 and KLF6 had little effect on the FL ICP0 promoter regardless of the presence of GR or DEX relative to GR+DEX+KLF15 (Fig. 2D and F). Two other KLF family members, KLF9 and PLZF, also showed little to no increase in ICP0 promoter activity, either alone or with GR and DEX (data not shown). Collectively, these studies indicated that KLF15 and the activated GR cooperated to transactivate the FL ICP0 promoter if DEX was added to cultures.
KLF15 and GR mediated transactivation of ICP0 promoter deletion mutants.
ICP0 promoter deletion mutants (Fig. 2A) were used to localize sequences important for cooperative transactivation by the GR and KLF15. GR+DEX+KLF15 transactivated the −635 ICP0 promoter slightly more than the FL ICP0 promoter in Neuro-2A cells (Fig. 3A). Conversely, in Vero cells GR+DEX+KLF15 transactivated the −635 ICP0 promoter less efficiently than the FL ICP0 construct (Fig. 3B). However, the differences observed between the −635 construct versus the FL ICP0 promoter were not significantly different in either cell type. Relative to the FL ICP0 or −635 deletion promoter, the −458, −281, −163, and −95 promoters exhibited significantly less promoter activity when cotransfected with GR+ DEX+KLF15 in Neuro-2A or Vero cells. However, GR+DEX+KLF15 transactivated the −458, −281, −163, and −95 ICP0 promoter constructs approximately 5-fold, suggesting that more than one domain in the ICP0 promoter was necessary for maximal transactivation by GR+DEX+KLF15 (Fig. 3). Consistent with the FL ICP0 promoter, none of the deletion mutants were transactivated more than 2- to 3-fold by GR+DEX in both cell types (Fig. 3).
FIG 3.

Localization of ICP0 promoter sequences that are transactivated by the GR and KLF15. Neuro-2A (A) and Vero (B) cells were cultured in 2% stripped FBS after transfection with the FL ICP0 promoter or designated ICP0 deletion mutant fused to the firefly luciferase gene. Luciferase activity was measured 40 h after cells were cotransfected with the designated ICP0 construct (0.5 μg of DNA), vector expressing the mouse GR (1.0 μg of DNA), and/or vector expressing human KLF15 (0.5 μg of DNA) as indicated. Empty vector plasmid was added to certain samples to maintain the same amount of DNA in each transfection. Twenty-four hours following transfection, designated cultures were treated with DEX for 14 h (10 μM). The results are the means from 3 independent experiments. Statistical analysis was performed as described in Materials and Methods. Unpaired t test was performed between FL ICP0 promoter cotransfected with GR+DEX+KLF15 and indicated samples. *, P < 0.05; **, P < 0.005. N.S., not significant.
A half-GRE in the ICP0 promoter was important for transactivation by the GR and KLF15.
Based on the results in Fig. 3, the primary GR/KLF15-responsive region (GR/KLF15 RR) appears to span −635 to −458 of the ICP0 promoter (Fig. 4A). Analysis of the FL ICP0 promoter sequences failed to identify a consensus “whole” GRE with dyad symmetry (Fig. 4B). However, 5 potential half-GREs that contain either the 5′ core ACA or 3′ core sequence TGT were identified: 1 within the GR/KLF15 RR and the other 4 sites downstream of these sequences (Fig. 4B). A GR monomer can bind certain half-GREs, leading to promoter activation, and most contain the ACA core (24) (Fig. 4B). The 5′ potential half-GRE within the −800 to −458 fragment is near two Sp1 binding sites and other GC-rich sequences, which KLF15 could potentially interact with (17, 25). To test whether these half-GREs are important, we generated ICP0 promoter constructs in which GRE 1 (Δ5' GRE) or all five half-GREs were mutated (GRE null). For all of these mutants, the core ACA or GTG core sequence was deleted. In Neuro-2A cells, GR+DEX+KLF15 transactivated the Δ5′ GRE and GRE null mutants approximately 10-fold (Fig. 4C). In contrast, the −635 construct was transactivated 16-fold by GR+DEX+KLF15, which was significantly different than the mutant constructs. In both mutant constructs, we observed 3-fold activation by GR+DEX and 5- to 7-fold activation by KLF15, which were similar to the case with the −635 ICP0 promoter construct.
FIG 4.

Role of half-GREs in ICP0 promoter activation in transfected cells. (A) Schematic of ICP0 promoter that includes the location of the GR/KLF15 responsive region (GR/KLF15 RR), the locations of half-GREs, and sequences amplified by the respective PCR primers used in this study. Locations of transcription factor binding sites in the ICP0 promoter are indicated below the −635 ICP0 promoter construct. (B) DNA sequence of GRE consensus and partial GREs in the ICP0 promoter. Nucleotides above the consensus GRE indicate changes that still result in GR binding and transactivation, and “N” refers to any base. The putative half-GREs in the ICP0 promoter correspond to the numbered half-GREs in panel A. The Δ5′ GRE mutant has the TGT core sequence motif deleted. The GRE null mutant has deletions of all of the TGT or ACA core motifs present in the five half-GREs. (C to E) Neuro-2A cells were cultured in 2% stripped FBS after transfection with the −635 ICP0 promoter or half-GRE mutant promoters fused to the firefly luciferase gene. Shown is luciferase activity at 40 h after cells were cotransfected with the designated ICP0 promoter (0.5 μg of DNA), vector expressing a mouse GR (1.0 μg of DNA), and/or vector expressing human KLF15 (0.5 μg of DNA). Empty vector plasmid was added to certain samples to maintain the same amount of DNA in each transfection. Twenty-four hours following transfection, certain cultures were treated with DEX for 14 h (10 μM) and/or RU486 (10 μM). Dual-luciferase activity was determined as described in Materials and Methods. The results are the means from 3 independent experiments. Statistical analysis was performed as described in Materials and Methods. Unpaired t test was performed between the −635 ICP0 construct cotransfected with GR+DEX+KLF15 and indicated samples. **, P < 0.005; ***, P < 0.0005.
To further examine the effect of DEX and the GR on ICP0 promoter activation, we tested whether RU486, a GR and progesterone receptor antagonist (26, 27), influenced transactivation. RU486, not CORT-108297, was used because this inhibitor has historically been used to examine the effects of the GR on transcriptional activation (9, 10, 26, 27). When cultures were treated with RU486, transactivation of the −635 ICP0 promoter construct (Fig. 4C) or the FL ICP0 promoter (data not shown) by GR+DEX+KLF15 was significantly reduced by RU486 treatment. Conversely, the effect GR+DEX had on the wt −635 promoter was not significantly affected by RU486. With respect to the Δ5′ GRE mutant (Fig. 4D) and the ΔGRE null (Fig. 4E) mutants, RU486 treatment did not significantly reduce the effect that GR+DEX+KLF15 had on transactivation. These studies suggested that the Δ5′ GRE in the −635 ICP0 construct influenced GR+DEX+KLF15-mediated transactivation. It should also be pointed out that mutating all 5 half-GREs did not eliminate the ability of GR+DEX+KLF15-transactivated ICP0 promoter activity, further suggesting that additional ICP0 sequences are necessary for maximal transactivation.
The GR and KLF15 interact with ICP0 promoter sequences in transfected Neuro-2A cells.
To test whether the GR and KLF15 were recruited to the ICP0 promoter, chromatin immunoprecipitation (ChIP) studies were performed in transfected Neuro-2A and Vero cells. Cells were transfected with the −635 promoter construct, followed by treatment with DEX or a vehicle; following ChIP with the desired antibodies, ICP0 promoter sequences were amplified with the −635 primer set (see Fig. 4A for sequences amplified by these specific primers). Specific PCR products were not readily detected from ChIPs of cells transfected with the −635 promoter when IPs were performed using the control IgG (Fig. 5), which was similar to the background. Interestingly, increased occupancy of the GR to the −635 ICP0 promoter construct was observed in Neuro-2A cells following DEX treatment (Fig. 5A). Furthermore, cotransfection of Neuro-2A cells with KLF15 and the GR exhibited a significant increase of GR occupancy to the −635 ICP0 promoter when DEX was added. In Neuro-2A cells transfected with KLF15, occupancy of the −635 ICP0 promoter by KLF15 was readily detected; however, GR+DEX+KLF15 reduced KLF15 occupancy of the −635 ICP0 promoter.
FIG 5.

Association of GR and KLF15 with the ICP0 promoter construct. Neuro-2A (A and B) and Vero (C and D) cells were cultured in 2% stripped FBS following transfection with the FL ICP0 promoter and either mouse GR, human KLF15, or empty vector and treated with DEX 24 h later. Cells were cross-linked with 16% paraformaldehyde 48 h following transfection and harvested for ChIP studies. ChIP was performed, as described in Materials and Methods, using the GR antibody, KLF15 antibody, or a control isotype IgG. PCR was performed using the −635 primers, and the DNA was run on a 1% agarose gel to separate DNA fragments and stained with ethidium bromide. Bands were quantified using Image Lab software and graphed as percent input. These results are the means from 3 independent experiments. Samples designated by an asterisk(s) show statistically significant differences from isotype control IgG by the t test (*, P < 0.05; **, P < 0.005).
In Vero cells, occupancy of the −635 promoter by the GR was readily detected following cotransfection with GR+DEX and GR+DEX+KLF15; however, KLF15 did not significantly increase GR occupancy (Fig. 5C). Occupancy of the −635 promoter by KLF15 was readily detected when Vero cells were transfected with KLF15 and occupancy was slightly increased when Vero cells were cotransfected with GR+DEX+KLF15 (Fig. 5D). In Vero or Neuro-2A cells transfected with the −635 or −95 construct, GR and KLF15 was not associated with the promoter when PCR was performed with the −95 primer set shown in Fig. 4A (data not shown). Hence, the GR and KLF15 were recruited to the −635 ICP0 promoter when DEX was added to cultures and promoter occupancy occurred upstream of the −95 primer set.
The GR and KLF15 interact with ICP0 promoter sequences during early stages of infection.
To investigate if the GR and KLF15 bound ICP0 sequences during productive infection, ChIP assays were performed. Vero cells were mock infected or infected with HSV-1 and then treated with the control vehicle or DEX. ChIP was performed using the R0 primers (Fig. 4A) following IP with the GR, KLF15 antibody, or a nonspecific IgG as an isotype control. We have consistently found that the GR occupied the ICP0 promoter at 2 h after infection when DEX was added to cultures. At 4 h after infection the GR occupied ICP0 promoter sequences in the absence of DEX and occupancy was significantly increased by DEX treatment. Strikingly, GR occupancy was not readily detected at 6 (Fig. 6A), 8, or 16 h after infection (data not shown).
FIG 6.

Association of GR and KLF15 with viral DNA during productive infection. Vero cells were infected with HSV-1 (MOI of 1) and then cells were incubated in 2% stripped FBS for the indicated times. ChIPs were performed using the GR antibody (A), KLF15 antibody (B), or nonspecific IgG as an isotope control (A and B). PCR was performed using the R0 ICP0 primers and DNA separated through a 1% agarose gel; DNA was then stained with ethidium bromide. Bands were quantified using Image Lab software and graphed as percent input. These results are the means from 3 independent experiments. Samples designated by an asterisk show statistically significant differences from isotype control IgG by the t test (*, P < 0.05; **, P < 0.005). HPI, hours postinfection.
KLF15 consistently occupied ICP0 promoter sequences at 2 and 4 h after infection in the absence of DEX treatment (Fig. 6B). Only at 4 h after infection was there a significant increase of KLF15 occupancy following DEX treatment. At 6, 8, or 16 h after infection, we observed a slight increase in the amplification of ICP0 promoter sequences that were immunoprecipitated by the IgG isotype control, which correlated with a dramatic increase of viral DNA due to viral DNA replication (data not shown). These studies indicated that the GR and KLF15 occupied ICP0 promoter sequences during early stages of productive infection and GR occupancy was enhanced by DEX treatment.
DISCUSSION
In this study, we obtained evidence that a GR specific antagonist (CORT-108297) reduced virus shedding in productively infected Neuro-2A cells, indicating that the activated GR stimulated productive infection. This result was consistent with a previous study that demonstrated pretreating human gingival fibroblasts with DEX prior to HSV-1 infection increased GR steady-state levels and viral yields (8). The HSV-1 genome contains numerous GRE consensus binding sites (data not shown), suggesting that the activated GR has several properties that could be important for stimulating viral productive infection and gene expression following a stressful stimulus. For example, the activated GR can stimulate transcription from certain GREs more than 5 kb from a given promoter (28). Furthermore, the activated GR can bind a subset of GREs in silent chromatin (29, 30) and induce a nuclease-hypersensitive site, which culminates in transcription (9), all properties consistent with the GR being coined as a “pioneer transcription factor” (31). One of the GREs in the viral genome is within the HSV-1 origin of replication (oriL), but not oriS, and this GRE stimulates replication of a plasmid in neuronal-like cells (32). Interestingly, mutagenesis of the GRE in oriL reduces viral pathogenesis during acute infection and impairs reactivation from latency (33).
Previous studies demonstrated that DEX treatment induced KLF15 expression in many TG neurons (11, 12), suggesting that this is part of the general stress response. KLF15, like other KLF family members, resembles the Sp1 transcription factor family, and both families interact with GC-rich motifs (17, 34, 35). However, KLF15, but not the other KLF family members examined, efficiently cooperated with the activated GR to stimulate ICP0 promoter activity. Notably, the GR and KLF15 regulate gene expression dynamics and integrate signals by a feed-forward loop in response to stress (13–15). The salient features of a feed-forward loop are that a primary factor (GR in this case) stimulates expression of a second factor (KLF15), and then the GR associates with KFL15 to stimulate transcription of additional genes (13–15). GR and KLF15 activate expression of genes in specific pathways, for example, genes that regulate adipogenesis (36) and amino acid-metabolizing enzymes (14, 15). We previously demonstrated that KLF15 by itself transactivated the FL ICP0 promoter (35). However, the conditions of these studies were dramatically different. For example, 10% FBS, not 2% stripped FBS, was used for these studies. As mentioned above, stripped FBS lacks corticosteroids and other growth factors, indicating that in the presence of 10% FBS the GR and potentially other transcription factors were activated. Furthermore, the transfection reagent used for the previous study was TransIT Neural (Mirus Bio, Madison, WI), which is not available now. TransIT Neural transfection reagent is more efficient at transfecting Neuro-2A cells than the TransIT-X2 transfection reagent or Lipofectamine transfection reagents (data not shown). Hence, the use of 2% stripped FBS was pivotal in demonstrating that the activated GR cooperated with KLF15 to transactivate the ICP0 promoter.
Recent studies demonstrated that the GR and KLF15 cooperate to transactivate the BoHV-1 IEtu1 promoter (12). Relative to HSV-1, regulation of BoHV-1 ICP0 and ICP4 coding has several significant differences. First, the IEtu1 promoter drives expression of two IE mRNAs, IE/2.9 and IE/4.2, from an alternatively spliced primary mRNA, and these transcripts are subsequently translated into bICP0 and bICP4, respectively (37, 38). Second, the IEtu1 promoter contains two consensus “whole” GREs that when mutated inhibit the ability of DEX or DEX+KLF15 to stimulate promoter activity (12, 23). A small fragment encompassing the wt IEtu1 GREs can confer DEX induction to a heterologous promoter; this was not the case for sequences comprising the GR/KLF15 RR (data not shown). Finally, an E promoter drives expression of the E/2.6 transcript that is translated into bICP0. The HSV-1 FL ICP0 promoter does not contain a consensus GRE, nor does GR+DEX efficiently transactivate the FL ICP0 promoter, unless KLF15 is included in the transfection. Finally, the FL ICP0 promoter contains several TAATGARAT motifs, whereas there is only one such motif in the IEtu1 promoter.
We (12) and others (15) demonstrated that KLF15 and the GR stably interact with each other, suggesting that the association of these two transcription factors has functional significance. We postulate that at least two mechanisms exist where the KLF15/GR complex can stimulate stress-activated transcription: (i) the KLF15/GR complex is recruited to consensus GREs via specific binding by the GR, as seen with the IEtu1 promoter, or (ii) KLF15 mediates binding of the KLF15/GR complex to the ICP0 promoter, which lacks a consensus GRE. The second scenario does not preclude GR from influencing recruitment to ICP0 promoter sequences, perhaps via a half-GRE. Furthermore, these scenarios do not exclude the possibility that novel transcriptional coactivators mediate these two scenarios. Several KLF15 binding sites have been reported (39–42), suggesting that sequence requirements for cooperative transactivation by KLF15 and the GR will be difficult to identify by merely analyzing the sequence. Studies designed to understand how the GR/KLF15 complex regulates transactivation of the IEtu1 promoter relative to the ICP0 promoter are in progress.
MATERIALS AND METHODS
Cells and virus.
Mouse neuroblastoma cells (Neuro-2A) and monkey kidney cells (Vero) were grown in minimal essential medium (MEM) supplemented with penicillin (10 U/ml), streptomycin (100 μg/ml), l-glutamine (2 mM), and 10% FBS. Some experiments replaced 10% FBS with 2% stripped FBS.
A virulent wt HSV-1 strain (McKrae) was obtained from Steven Wechsler.
Inhibition of productive infection by glucocorticoid receptor.
Confluent Neuro-2A cell monolayers were pretreated with increasing concentrations of CORT-108297 for 2 h at 37°C and 5% CO2. Plates were then infected with HSV-1 strain McKrae at a multiplicity of infection (MOI) of 1 and kept at 37°C and 5% CO2 with rocking for 1 h. After incubation, plates were washed with phosphate-buffered saline (PBS) and medium containing CORT-108297 or DMSO was added. Twenty-four hours after infection, total virus was determined via plaque assays. In brief, total virus was collected following three cycles of freezing and thawing (−80 to 37°C). Dilutions of supernatant from infected Neuro-2A cells were plated on Vero cell monolayers in quadruplicate and plaque assays performed. SeaPlaque agar (1.2%; Lonza Group Ltd., Houston, TX) in PBS was diluted 1:1 in MEM and overlaid on infected wells. Plates were monitored for plaque development and stained using crystal violet. Plaques were counted, and data are shown as means ± standard deviations (SD).
Cytotoxicity of CORT-108297 was assayed by incubating Neuro-2A cell monolayers with the respective concentrations of CORT-108297 for 24 h in 24-well plates. Viability was assessed using the CellTiter-Glo luminescent cell viability assay (G8461; Promega, Madison, WI) according to the manufacturer’s instructions. Plates were read using a SpectraMax M3 microplate reader (Molecular Devices, San Jose, CA). Data are shown as means ± SD for quadruplicate wells.
Statistics.
All analyses were performed and graphs constructed using GraphPad Prism (version 7.0d). Ordinary one-way analysis of variance (ANOVA) or t test with no correction for multiple comparisons was performed on all data sets.
Transfection.
Prior to transfection, Neuro-2A or Vero cells were washed with PBS, and antibiotic-free medium with 2% stripped FBS was added. Cells were transfected using TransIT-X2 transfection reagent (Mirus, Madison, WI), following the manufacturer’s guidelines, with the designated ICP0 promoter construct driving a firefly luciferase gene and a plasmid containing a Renilla luciferase gene with a minimal thymidine kinase (TK) promoter. Additionally, cells were transfected with plasmids expressing either mouse glucocorticoid receptor (GR) or human Krüppel-like transcription factor 15 (KLF15). The mouse GR expression vector was obtained from Joseph Cidlowski, NIH. The KLF15 expression vector was obtained from Deborah Otteson (University of Houston). To maintain an equal concentration in each transfection reaction, empty plasmid was added to each reaction, sufficient to balance total DNA. Following transfection, cells were maintained in MEM containing 2% stripped FBS. Twenty-four hours following transfection, cells were treated with water-soluble DEX at a 10 μM final concentration. Forty-eight hours following transfection, cells were washed with PBS and harvested.
Dual-luciferase assay.
Cells were transfected with ICP0 promoter constructs, and the GR and KLF15 were harvested 48 h following transfection with a passive lysis buffer. A dual-luciferase assay was performed on cell lysates using a commercially available kit (Promega; E1910), and luminescence was measured using a GloMax 20/20 luminometer (Promega; E5331). Luciferase activity was normalized by comparing levels of Renilla luciferase gene controlled by a simple TATA box, cotransfected with the previously mentioned plasmids (50 ng of DNA).
Chromatin immunoprecipitation.
Neuro-2A or Vero cells were grown in 100-mm dishes and transfected with the desired constructs or infected with HSV-1 (MOI of 1), and after transfection or infection, MEM with 2% stripped FBS was added to cultures. Cells were cross-linked with formaldehyde and harvested for ChIP, which was performed as described previously (12, 23), using 5 μg of GR antibody (Cell Signaling, Danvers, MA; catalogue number 3660S) or KLF15 antibody (Abcam, Cambridge, MA; catalogue number ab2647). In brief, DNA was purified using phenol-chloroform and precipitated with ethanol. PCR was performed using primers described below, run on 1% agarose gels, and stained with ethidium bromide for visualization. DNA bands were quantified using Image Lab software and presented as percent input. Input samples represent 6.7% of cell lysate.
Plasmids and primer sets.
The full-length ICP0 promoter (−800 to +150) and the −95 deletion construct (KOS sequences) were previously described and provided by the late Priscilla Schafer (21). Further deletion constructs (−635, −486, −281, and −168) were synthesized by Genscript and cloned into pGL3-Basic Vector at HindIII and SacI restriction enzyme sites. The half-GRE mutants (Fig. 4A and B) were also synthesized in pGL3-Basic Vector at HindIIII and SacI restriction enzyme sites.
Primer sets used for these studies are as follows: ICP0 −635, GGTAACTCCCGCCCAGTG (forward [F]) and GTGTTCCGCCAAAAAAGC (reverse [R]); ICP0 −95, GCCATTGGGGGAATCGTC (F) and TGTGGTGATGCGGAGAGG (R); and ICP0-R0, CGCTTCCCGGTATGGTAATTAGAAAC (F) and CGTGTGTTCCGCCAAAAAAGCAATTAGC (R). ICP0-R0 primers were previously described (43) and were designed using strain 17 sequences. The ICP0 −635 and ICP0 −95 primers were designed using KOS sequences, as all of the ICP0 promoter constructs are derived from KOS sequences (21).
ACKNOWLEDGMENTS
This research was supported by grants from the National Institute of Neurological Disorders and Stroke of the NIH (R21NS102290), the USDA-NIFA Competitive Grants Program (13-01041 and 16-09370), and the Oklahoma Center for Respiratory and Infectious Diseases (National Institutes of Health Centers for Biomedical Research Excellence Grant P20GM103648) and by funds from the Sitlington Endowment.
REFERENCES
- 1.Liesegang TJ. 1999. Herpes simplex virus epidemiology and ocular importance. Cornea 18:127–143. doi: 10.1097/00003226-199903000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Shimeld C, Efstathiou S, Hill T. 2001. Tracking the spread of a lacZ-tagged herpes simplex virus type 1 between the eye and the nervous system of the mouse: comparison of primary and recurrent infection. J Virol 75:5252–5262. doi: 10.1128/JVI.75.11.5252-5262.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamada S, Kameyama T, Nagaya S, Hashizume Y, Yoshida M. 2003. Relapsing herpes simplex encephalitis: pathological confirmation of viral reactivation. J Neurol Neurosurg Psychiatry 74:262–264. doi: 10.1136/jnnp.74.2.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perng G-C, Jones C. 2010. Towards an understanding of the herpes simplex virus type 1 latency-reactivation cycle. Interdiscip Perspect Infect Dis 2010:1–18. doi: 10.1155/2010/262415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glaser R, Kiecolt-Glaser JK, Speicher CE, Holliday JE. 1985. Stress, loneliness, and changes in herpesvirus latency. J Behav Med 8:249–260. doi: 10.1007/BF00870312. [DOI] [PubMed] [Google Scholar]
- 6.Padgett DA, Sherida JF, Dorne J, Berntson GG, Candelora J, Glaser R. 1998. Social stress and the reactivation of latent herpes simplex virus type 1. Proc Natl Acad Sci U S A 95:7231–7235. doi: 10.1073/pnas.95.12.7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Du T, Zhou G, Roizman B. 2012. Induction of apoptosis accelerates reactivation from latent HSV-1 in ganglionic organ cultures and replication in cell cultures. Proc Natl Acad Sci U S A 109:14616–14621. doi: 10.1073/pnas.1212661109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Erlandsson AC, Bladh L-C, Stierna P, Yucel-Lindberg T, Hammersten O, Modeer T, Harmenberg J, Wikstrom A-C. 2002. Herpes simplex virus type 1 infection and glucocorticoid treatment regulate viral yield, glucocorticoid receptor and NF-kB levels. J Endocrinol 175:165–176. doi: 10.1677/joe.0.1750165. [DOI] [PubMed] [Google Scholar]
- 9.Oakley RH, Cidlowski JA. 2013. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol 132:1033–1044. doi: 10.1016/j.jaci.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Funder JW. 1997. Glucocorticoids and mineralocorticoid receptors: biology and clinical relevance. Annu Rev Med 48:231–240. doi: 10.1146/annurev.med.48.1.231. [DOI] [PubMed] [Google Scholar]
- 11.Workman A, Eudy J, Smith L, Frizzo da Silva L, Sinani D, Bricker H, Cook E, Doster A, Jones C. 2012. Cellular transcription factors induced in trigeminal ganglia during dexamethasone-induced reactivation from latency stimulate bovine herpesvirus 1 productive infection and certain viral promoters. J Virol 86:2459–2473. doi: 10.1128/JVI.06143-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El-Mayet FS, Sawant L, Thungunutla P, Jones C. 2017. Combinatorial effects of the glucocorticoid receptor and Krüppel-like transcription factor 15 on bovine herpesvirus 1 transcription and productive infection. J Virol 91:e00904-17. doi: 10.1128/JVI.00904-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mangan S, Alon U. 2003. Structure and function of the feed-forward loop network motif. Proc Natl Acad Sci U S A 100:11980–11985. doi: 10.1073/pnas.2133841100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sasse S, Zuo Z, Kadiyala V, Zhang L, Pufall MA, Jain MK, Phang TL, Stormo GD, Gerber AN. 2015. Response element composition governs correlations between binding site affinity and transcription in glucocorticoid receptor feed-forward loops. J Biol Chem 290:19756–19769. doi: 10.1074/jbc.M115.668558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sasse SK, Mailloux CM, Barczak AJ, Wang Q, Altonsy MO, Jain MK, Haldar SM, Gerber AN. 2013. The glucocorticoid receptor and KLF15 regulate gene expression dynamics and integrate signals through feed-forward circuitry. Mol Cell Biol 33:2104–2115. doi: 10.1128/MCB.01474-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Black AR, Black JD, Azizkhan-Clifford J. 2001. Sp1 and Kruppel-like transcription factor family of transcription factors in cell growth and cancer. J Cell Physiol 188:143–160. doi: 10.1002/jcp.1111. [DOI] [PubMed] [Google Scholar]
- 17.Kaczynski J, Cook T, Urrutia R. 2003. Sp1- and Kruppel-like transcription factors. Genome Biol 4:206–208. doi: 10.1186/gb-2003-4-2-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clark R, Ray NC, Williams K, Blaney P, Ward S, Crackett PH, Hurley C, Dyke HJ, Clark DE, Lockey P, Devos R, Wong M, Porres SS, Bright CP, Jenkins RE, Belanoff J. 2008. 1H-pyrazolo[3, 4-g]hexahydro-isoquinolines as selective glucocorticoid receptor antagonists with high functional activity. Bioorg Med Chem Lett 18:1312–1317. doi: 10.1016/j.bmcl.2008.01.027. [DOI] [PubMed] [Google Scholar]
- 19.Morgan BP, Swick AG, Hargrove DM, LaFlamme JA, Moynihan MS, Carroll RS, Martin KA, Lee E, Decosta D, Bordner J. 2002. Discovery of potent, nonsteroidal, and highly selective glucocorticoid receptor antagonists. J Med Chem 45:2417–2424. doi: 10.1021/jm0105530. [DOI] [PubMed] [Google Scholar]
- 20.Tremblay R, Sikorska M, Sandhu JK, Lanthier P, Ribecco-Lutkiewicz M, Bani-Yaghoub M. 2010. Differentiation of mouse Neuro-2A cells into dopamine neurons. J Neurosci Methods 186:60–67. doi: 10.1016/j.jneumeth.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 21.Kushnir AS, Davido DJ, Schaffer PA. 2010. Role of nuclear factor Y in stress-induced activation of the herpes simplex virus type 1 ICP0 promoter. J Virol 84:188–200. doi: 10.1128/JVI.01377-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gong S, Miao Y-L, Jiao G-Z, Sun M-J, Li H, Lin J, Luo M, Tan J-H. 2015. Dynamics and correlation of serum cortisol and corticostrone under different physiological or stressful conditions in mice. PLoS One 10:e0117503. doi: 10.1371/journal.pone.0117503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kook I, Henley C, Meyer F, Hoffmann F, Jones C. 2015. Bovine herpesvirus 1 productive infection and the immediate early transcription unit 1 are stimulated by the synthetic corticosteroid dexamethasone. Virology 484:377–385. doi: 10.1016/j.virol.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 24.Schiller B, Chodankar R, Watson LC, Stallcup MR, Yamamoto KR. 2014. Glucocorticoid receptor binds half sites as a monomer and regulates specific target genes. Genome Biol 15:418. doi: 10.1186/s13059-014-0418-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J, Yang Y, Jiang B, Zhang X, Zou Y, Gong Y. 2010. SP1 and KLF15 regulate basal transcription of the human LRP5 gene. BMC Genet 11:12–19. doi: 10.1186/1471-2156-11-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pandit S, Geissler W, Harris G, Sitlani A. 2002. Allosteric effects of dexamethasone and RU486 on glucocorticoid receptor-DNA interactions. J Biol Chem 277:1538–1543. doi: 10.1074/jbc.M105438200. [DOI] [PubMed] [Google Scholar]
- 27.Schulz M, Eggert M, Baniahmad A, Dostert A, Heinzel T, Renkawitz R. 2002. RU486-induced glococorticoid receptor agonism is controlled by the receptor N terminus and by corepressor binding. J Biol Chem 277:26238–26243. doi: 10.1074/jbc.M203268200. [DOI] [PubMed] [Google Scholar]
- 28.Polman JA, Welten JE, Bosch DS, de Jonge RT, Balog J, van der Maarel SM, de Kloet ER, Datson NA. 2012. A genome-wide signature of glucocorticoid receptor binding in neuronal PC12 cells. BMC Neurosci 13:118–125. doi: 10.1186/1471-2202-13-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.John S, Sabo PJ, Thurman RE, Sung MH, Biddie SC, Johnson TA, Hager GL, Stamatoyannopoulos JA. 2011. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet 43:264–268. doi: 10.1038/ng.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perlman T. 1992. Glucocorticoid receptor DNA-binding specificity is increased by the organization of DNA in nucleosomes. Proc Natl Acad Sci U S A 89:3884–3888. doi: 10.1073/pnas.89.9.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zaret KS, Carrol JS. 2011. Pioneer transcription factors: establishing competence for gene expression. Genes Dev 25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hardwicke MA, Schaffer PA. 1997. Differential effects of nerve growth factor and dexamethasone on herpes simplex virus type 1 oriL- and oriS-dependent DNA replication in PC12 cells. J Virol 71:3580–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balliet JW, Schaffer PA. 2006. Point mutations in herpes simplex virus type 1 oriL, but not in oriS, reduce pathogenesis during acute infection of mice and impair reactivation from latency. J Virol 80:440–450. doi: 10.1128/JVI.80.1.440-450.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bieker JJ. 2001. Kruppel-like factors: three fingers in many pies. J Biol Chem 276:34355–34358. doi: 10.1074/jbc.R100043200. [DOI] [PubMed] [Google Scholar]
- 35.Sinani D, Cordes E, Workman A, Thunuguntia P, Jones C. 2013. Stress induced cellular transcription factors expressed in trigeminal ganglionic neurons stimulate the herpes simplex virus type 1 (HSV-1) infected cell protein 0 (ICP0) promoter. J Virol 87:1183–1192. doi: 10.1128/JVI.02783-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asada M, Rauch A, Shimizu H, Maruyama H, Miyaki S, Shibamori M, Kawasome H, Ishiyama H, Tuckermann J, Asahara H. 2011. DNA-binding dependent glucocorticoid receptor activity promotes adipogenesis via Kruppel-like factor 15 gene expression. Lab Invest 91:203–215. doi: 10.1038/labinvest.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wirth UV, Fraefel C, Vogt B, Vlcek C, Paces V, Schwyzer M. 1992. Immediate-early RNA 2.9 and early RNA 2.6 of bovine herpesvirus 1 are 3′ coterminal and encode a putative zinc finger transactivator protein. J Virol 66:2763–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wirth UV, Vogt B, Schwyzer M. 1991. The three major immediate-early transcripts of bovine herpesvirus 1 arise from two divergent and spliced transcription units. J Virol 65:195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Uchida S, Tanaka Y, Ito H, Saitoh-Ohara F, Inazawa J, Yokoyama KK, Sasaki S, Marumo F. 2000. Transcriptional regulation of the CLC-K1 by myc-associated zinc finger protein, a novel zinc finger repressor. Mol Cell Biol 20:7319–7331. doi: 10.1128/MCB.20.19.7319-7331.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takeda K, Yahagi N, Aita Y, Murayama Y, Sawada Y, Piao X, Toya N, Oya Y, Shikama A, Takarada A, Masuda Y, Nishi M, Kuobota M, Izumida Y, Yamamoto T, Sekiya M, Matsuzaka T, Nakagawa Y, Urayama O, Kawakami Y, Iizuka Y, Gotoda T, Itaka K, Kataoka K, Nagai R, Kadowaki T, Yamada N, Lin Y, Jain MK, Shimano H. 2016. KLF15 enables switching between lipogenesis aand gluconeogenesis during fasting. Cell Rep 16:2373–2386. doi: 10.1016/j.celrep.2016.07.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Du X, Rosenfield RL, Qin K. 2009. KLF15 is a transcriptional regulator of the human 17beta-hydroxysteroid dehydrogenase type 5 gene. A potential link between regulation of testosterone production and fat stores in women. J Clin Endocrinol Metab 94:2594–2601. doi: 10.1210/jc.2009-0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Otteson DC, Lai H, Liu Y, Zack DJ. 2005. Zinc-finger domains of the transcriptional repressor KLF15 binds multiple sites in rhodopsin and IRBP promoters including the CRS-1 and G-rich elements. BMC Mol Biol 6:15. doi: 10.1186/1471-2199-1186-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whitlow ZW, Kristie TM. 2009. Recruitment of the transcriptional coactivator HCF-1 to viral immediate-ealy promoters during intiation of reactivation from latency of herpes simplex virus type 1. J Virol 83:9591–9595. doi: 10.1128/JVI.01115-09. [DOI] [PMC free article] [PubMed] [Google Scholar]