

Herpes simplex virus (HSV) is responsible for significant morbidity in humans due to its ability to cause oral and genital lesions, ocular disease, and encephalitis. While antivirals can attenuate the severity and frequency of disease, there is no vaccine or cure. Understanding the molecular details of HSV latency and reactivation is key to the development of new therapies. One of the difficulties in studying HSV latency has been the need to rely on establishment of latent infections in animal models. While rodent primary neuron culture models have shown promise, they yield relatively small numbers of latently infected neurons for biochemical and molecular analyses. Here we present the use of a human central nervous system (CNS)-derived conditionally proliferating cell line that can be differentiated into mature neurons and latently infected with HSV-1. This model shows promise as a scalable tool to study molecular and biochemical aspects of HSV-1 latency and reactivation in human neurons.
KEYWORDS: latency, neuron
ABSTRACT
Lund human mesencephalic (LUHMES) cells are human embryonic neuronal precursor cells that can be maintained as proliferating cells due to the expression of a tetracycline-regulatable (Tet-off) v-myc transgene. They can be differentiated to postmitotic neurons by the addition of tetracycline, glial cell-derived neurotrophic factor (GDNF), and dibutyryl cAMP. We demonstrate that these cells can be infected with herpes simplex virus 1 (HSV-1) at a multiplicity of infection (MOI) of 3 with the majority of cells surviving. By 6 days postinfection, there is a loss of lytic gene transcription and an increase in the numbers of neurons that express the latency-associated transcripts (LATs). Importantly, the virus can then be reactivated by the addition of a phosphoinositide 3-kinase inhibitor, which has previously been shown to reactivate HSV-1 in rat neuron cultures. While rodent primary culture neuron systems have been described, these are limited by their lack of scalability, as it is difficult to obtain more than 500,000 neurons to employ for a given experiment. Several recent papers have described a human dorsal root ganglion (DRG) neuron culture model and human induced pleuripotent stem cell (iPSC) neuron culture models that are scalable, but they require that the presence of an antiviral suppression be maintained following HSV-1 infection. The human LUHMES cell model of HSV-1 infection described here may be especially useful for studying HSV-1 latency and reactivation on account of its scalability, its amenability to maintenance of latency without the continual use of antiviral inhibitors, and its latent gene expression profile which mirrors many properties observed in vivo, importantly, the heterogeneity of cells expressing the LATs.
IMPORTANCE Herpes simplex virus (HSV) is responsible for significant morbidity in humans due to its ability to cause oral and genital lesions, ocular disease, and encephalitis. While antivirals can attenuate the severity and frequency of disease, there is no vaccine or cure. Understanding the molecular details of HSV latency and reactivation is key to the development of new therapies. One of the difficulties in studying HSV latency has been the need to rely on establishment of latent infections in animal models. While rodent primary neuron culture models have shown promise, they yield relatively small numbers of latently infected neurons for biochemical and molecular analyses. Here we present the use of a human central nervous system (CNS)-derived conditionally proliferating cell line that can be differentiated into mature neurons and latently infected with HSV-1. This model shows promise as a scalable tool to study molecular and biochemical aspects of HSV-1 latency and reactivation in human neurons.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is a ubiquitous human virus that establishes a lifelong latent infection within peripheral nerve ganglia. Initial infection typically occurs within the orolabial epithelia, where the virus is taken up by peripheral nerve termini and transported to the nuclei of the neurons in the peripheral ganglia. After entering the neurons, a lytic infection proceeds in some neurons, whereas latency is established in others. Latency is characterized by the circularization of the genomes and restricted expression of viral lytic genes. Periodically, the latent genomes reactivate and progeny virions migrate in an anterograde direction to the site of the initial infection, where they either are shed asymptomatically or produce cold sores. The mechanisms of regulation of latency and reactivation are complex and incompletely understood. New systems are needed to better understand the detailed molecular processes behind these phases of infection in order to identify targets for better therapeutic approaches.
HSV-1 latency has been studied primarily in the ganglia of mice and rabbits that have been experimentally infected (1). The reason for this is that most cell lines (including those of neuronal origin) do not support establishment of latency and instead progress to lytic infection and cell death. While animal models have been useful in studying many aspects of HSV-1 latency and reactivation, a major drawback has been the low numbers of latently infected cells that are available for biochemical or molecular analyses. For example, neurons comprise roughly only 10% of the cells in a ganglion, and under the best conditions only 10% to 20% of the neurons in a ganglion are latently infected following infection of the mouse or rabbit (1). Therefore, the use of techniques such as mass spectrometry to identify proteins interacting with the latent genomes or photoactivatable ribonucleoside-enhanced cross-linking and immunoprecipitation (PAR-CLIP) to study microRNA (miRNA) interactions has been out of reach.
Over the years, there have been some initial successes at establishing cell models of HSV-1 quiescence or latency, though only recently have some of these in vitro models gained traction. Some of the earliest models involved the use of primary fibroblast lines along with the inclusion of either inhibitors of viral replication or HSV mutants that were conditionally restricted in replication (2). These models proved useful for elucidating many aspects of HSV-1 interactions with nuclear structures and transcriptional control of viral genes (3, 4) but were criticized for requiring artificial blocks to viral replication as a means of establishing latency, which can feasibly occlude or misrepresent some important aspects by which latency is naturally established and maintained.
Two additional in vitro models of HSV-1 quiescence or latency employ tumor cell lines that can be differentiated into neurons. The first was rat pheochromocytoma cells (PC12) that can be differentiated by the addition of nerve growth factor (NFG) (5, 6). The quiescent HSV-1 could be reactivated by thermal stress, trichostatin A (TSA), or forskolin, and this model was used to look at the dependence of HSV-1 replication and reactivation on ICP0 and VP16 (7, 8). The second model was human neuroblastoma cells (SH-SY5Y) that can be differentiated by treatment with retinoic acid. These cells can establish a latent HSV-1 infection, which can subsequently be reactivated (9, 10). While both of these models have shown promise, concerns have been raised regarding the need for inhibitors of HSV-1 replication during the maintenance phase of latency and the fact that these cells are derived from tumors that have alterations in their genomes.
A promising model of latency was developed by Wilcox et al. using primary dorsal root ganglion (DRG) neurons from embryonic rats (11–13). While this model also required the use of acyclovir (ACV) to suppress initial lytic replication, the acyclovir could be removed and HSV-1 reactivated. The use of primary neuronal cell cultures was largely abandoned until relatively recently, with the development of rat embryonic superior cervical ganglion (SCG) cultures (14, 15), as well as primary adult mouse trigeminal ganglion (TG) and dorsal root ganglion (DRG) cultures (16–18). One advantage of the SCG model is that these autonomic ganglia are a less heterogeneous neuronal population, containing primarily TrkA+ neurons (19). Importantly, the adult mouse TG and DRG cultures are the only primary culture systems that do not require acyclovir to maintain latency efficiently. These culture systems have been very valuable in providing details regarding the role of NGF signaling in promoting HSV reactivation (14), analyzing the different sensory neuron populations that are permissive for lytic versus latent infections (16), and dissecting the regulation of how reactivation stimuli translate to epigenetic remodeling of the latent chromatin (20). However, two significant limitations of these models are that (i) large numbers of animals are required to obtain just 106 neurons for a given experiment and (ii) while HSV can establish latency in rodent neurons, these cells are not ideal for probing virus-host interactions that might be human neuron specific. While recent advances in human embryonic stem cell (21) and human induced pleuripotent stem cell (iPSC) cultures (22, 23) have shown initial promise, their differentiation is time-consuming and scalability difficult.
Illustrating the latter point above, our recent interest in noncoding RNAs, especially in the latency region of the HSV-1 genome, prompted us to search for a scalable human neuronal culture system that would allow biochemical and molecular studies of virus-host interactions important for latency and reactivation in neurons. We sought a model that might provide 107 to 108 cells per experiment in the host background most relevant for understanding the protein and RNA interactions of HSV-1 (especially those of miRNAs and long noncoding RNAs [lncRNAs]) during latency.
Lund human mesencephalic (LUHMES) neuronal cells are a subclone of the tetracycline-controlled, v-myc-overexpressing human mesencephalon-derived cell line MESC2.10 (24), characterized at and originating from Lund University (Lund, Sweden) (25). The source material for MESC2.10 was derived from 8-week-old human ventral mesencephalic tissue. Karyotyping of the cell line showed a normal set of chromosomes and female phenotype (26). An extensive characterization of this cell line’s timeline of differentiation, expression of mature neuronal markers, neurite outgrowth, and electrophysiological properties has been previously reported (27), and the cells have been widely used by the neuroscience community (28–30).
Here we present the characterization of LUHMES cells for their ability to support HSV-1 latency and reactivation. We have confirmed that LUHMES cells can be efficiently differentiated into postmitotic cells that express markers of mature neurons by 5 days postdifferentiation. We demonstrate that (i) LUHMES cells can be efficiently infected with HSV-1, (ii) by 6 days postinfection (p.i.), relative lytic gene expression decreases and the number of cells expressing the latency-associated transcript (LAT) increases, (iii) a stable latent or quiescent state of infection can be established with only 48 h of treatment with acyclovir at the time of infection (its continual presence is not required to maintain latency), and (iv) reactivation can be efficiently induced by the addition of a phosphoinositide 3-kinase (PI3K) inhibitor. In addition, we provide evidence that while >87% of the LUHMES cells harbor HSV-1 genomes following infection at a multiplicity of infection (MOI) of 3, only a proportion of them express detectable LAT, similar to the observation in latently infected ganglia (31, 32). Supported by these observations, we present the LUHMES cell model of HSV-1 infection as a physiologically relevant platform for molecular and biochemical analyses of HSV latency and reactivation.
RESULTS
The LUHMES cell line has been previously characterized in the literature as having properties of fully differentiated dopaminergic neurons and thus has been developed as a valuable tool for studying neurological diseases such as Parkinson’s disease (27). Since LUHMES cells have been engineered to express v-myc in a tetracycline-regulatable (Tet-off) system, they retain a proliferative capacity that makes them useful for high-throughput, large-scale studies, as cells can be grown to the desired number and then differentiated by addition of tetracycline to shut off the v-myc promoter. Morphological characteristics of mitotically active LUHMES cultures display properties of terminally fated neurons as can be seen from neurite-like projections (Fig. 1A). We have confirmed that LUHMES cultures can be readily grown as mitotic cells and that they can be differentiated to postmitotic cells expressing mature neuron markers by 5 days after medium change with addition of tetracycline, glial cell-derived neurotrophic factor (GDNF), and dibutyryl cAMP, which corresponds to the timeline reported previously (27). For the experiments presented here, LUHMES cells were plated onto coverslips, differentiated, and analyzed by immunofluorescence for neuron-specific markers. Following 7 days of differentiation, neuronal filament projections up to 200 μm long could be seen (Fig. 1B and C). Like others (27) we found that the differentiated cells contain neurofilament H (200 kDa)-positive intermediate filaments (Fig. 1D to F), βIII-tubulin (Fig. 1G), and synaptophysin (Fig. 1H and I).
FIG 1.

Conditionally immortalized proliferating LUHMES cells can be uniformly differentiated into postmitotic neurons. (A to C) Bright-field micrographs showing LUHMES neuronal cultures in the undifferentiated proliferative phase (A) and then following 7 days of differentiation (B), with the boxed inset magnified (C). (D to I) Immunofluorescence micrographs showing that differentiated LUHMES cells are positive for neuronal-specific markers. Green, neurofilament H (NF-H) (D to F) and synaptophysin (Syn) (H and I); red, βIII-tubulin (G). (I) Magnified image, with arrows indicating synaptophysin-positive punctate staining and intermediate III vimentin filaments in purple. Nuclei stained with DAPI are shown in all immunofluorescence images. Scale bars, 20 μm.
We next investigated whether cultures of LUHMES neurons are permissive for HSV-1 infection. We previously demonstrated that HSV-1-infected LUHMES cells expressed ICP4 at 2 to 4 h postinfection (33). Here we sought to investigate whether LUHMES cells supported a productive infection and also to determine the infection kinetics. Proliferating LUHMES cells were plated onto 6-well dishes and cultured for 3 days, at which time the medium was changed to induce differentiation. Following 5 days of differentiation, LUHMES cells were infected with HSV-1 17syn+ at an MOI of 3 or were mock infected, and cells were harvested at day 1 to 4 p.i. for Western blotting. We found the HSV-1 immediate early (IE) protein ICP4 and the early (E) protein ICP8 to be abundantly expressed by 24 h postinfection and to remain stable over the time course we studied (Fig. 2A). We also assessed the HSV-1 genome copy number over a longer time course of infection (15 days) by quantitative PCR (qPCR) using primers and probe spanning the UL30 gene. For this purpose, LUHMES cells were plated at 25,000 cells/well of 24-well plates, cultured for 3 days, and, following 5 days of differentiation, infected in the presence of 50 μM acyclovir with 17syn+ at an MOI of 3. At 48 h p.i., the medium was removed and replaced with fresh medium without acyclovir. Beginning at 3 days p.i., cells were harvested for HSV-1 genome content analysis. We found the HSV-1 genome copy number to remain constant over the course of the 15 days following infection (Fig. 2B). From this same time course, we also harvested RNA for determining HSV-1 transcript levels. Using PCR primers spanning the HSV-1 latency-associated transcript (LAT) intron, we found that the LAT intron is most abundant at 3 to 5 days postinfection and then decreases in abundance until day 8 p.i., where it remains easily detectable at relatively constant levels thereafter (Fig. 2C). The results show little well-to-well variation in transcript levels (two independent wells for each day are shown in Fig. 2C), indicating that the system may be amenable to high-throughout studies. We also measured HSV-1 transcript levels from three different kinetic classes of lytic genes: the immediate early ICP4 gene, the early thymidine kinase (TK) gene, and the late glycoprotein C (gC) gene (Fig. 2D). ICP4 transcripts are highly expressed at days 3 and 5 p.i. and then decrease to low levels and remain steady for the remainder of the 15-day infection (Fig. 2D). The expression of the early TK gene is most abundant at day 3 p.i., dropping off at 5 days and remaining very low during the course of infection (Fig. 2D). Transcription of the HSV-1 late gC gene is only minimally detectable at day 3 p.i. but is suppressed during the remaining course of the 15-day infection, consistent with a latent infection (Fig. 2D). Assay of the culture supernatants revealed that infectious virus is detected only at days 1 and 2 p.i., indicating that the production of infectious virions (Table 1) decreases a day or so before lytic RNA levels diminish.
FIG 2.

Differentiated LUHMES cultures are permissive for HSV-1 infection. (A) Infection of 5-day-postdifferentiated LUHMES cells with 17syn+ at an MOI of 3 results in robust expression of the viral immediate early (ICP4) and early (ICP8) proteins as shown by Western blotting (numbers indicate days p.i.; M, mock control). (B) HSV-1 genome copy number was measured by qPCR over a 15-day time course of 17syn+ infection of LUHMES cells and found to be maintained during the course of the infection. (C) The production of the HSV-1 latency-associated transcript (LAT) was measured by RT-qPCR and found to be abundantly expressed during the course of the experiment, with highest expression seen early following infection (day 3 to 5 p.i.), tapering off around day 8 p.i., and then maintained at levels fairly constant for the remainder of the experiment. (D) Three additional HSV-1 transcripts representing different kinetic phases of the viral life cycle were measured by RT-qPCR: ICP4 (immediate early), TK (early), and the late gene gC. HSV-1 transcript levels were calculated following normalization to GAPDH and to no-RT controls and are represented as fold change over levels in mock-infected cells (where the mock-infected cell value was set to 1).
TABLE 1.
Infectious virus detected in culture supernatants following HSV-1 infection
| Day postinfection | Infectious virus (PFU × 103) |
|---|---|
| 1 | 16 |
| 2 | 2 |
| 3 | 0 |
| 4 | 0 |
| 5 | 0 |
| 6 | 0 |
| 7 | 0 |
| 8 | 0 |
| 9 | 0 |
| 10 | 0 |
| 11a | 0 |
| 12 | 0 |
| 13 | 28 |
Wortmannin was added at day 11 to induce reactivation.
In order to characterize HSV-1 transcript localization in the LUHMES neurons, we performed RNAScope in situ hybridization (34) for representative latent (LAT in red) and lytic (ICP4 in green) transcripts over a 13-day period following infection (Fig. 3). Mitotically active LUHMES cells were plated onto fibronectin/poly-l-ornithine-coated coverslips at 25,000 cells per well of 24-well plates, and following 3 days of growth, differentiation was induced as described in Materials and Methods. At 5 days postdifferentiation, LUHMES cells were infected with HSV-1 strain 17syn+ at an MOI of 3 in the presence of 50 μM ACV. Based on the time course of detection of viral transcripts in the previous reverse transcription-PCR (RT-PCR) analyses (Fig. 2D), RNAScope analysis was performed at 1, 4, 6, and 13 days p.i. (Fig. 3B). Both ICP4 and the LATs were highly abundant during day 1 to day 4 p.i. Interestingly, by day 6 p.i., the majority of the neurons were positive for the LATs, with very few neurons displaying evidence of lytic transcription. This quiescent period of HSV-1 infection was noted throughout the remainder of the 13-day experiment, where the large majority of cells expressed only the LAT. Therefore, the RNAScope and RT-PCR data (Fig. 2D) suggest that a quiescent or latent state is established in the LUHMES cells 5 days after infection with HSV-1 (Fig. 3A).
FIG 3.
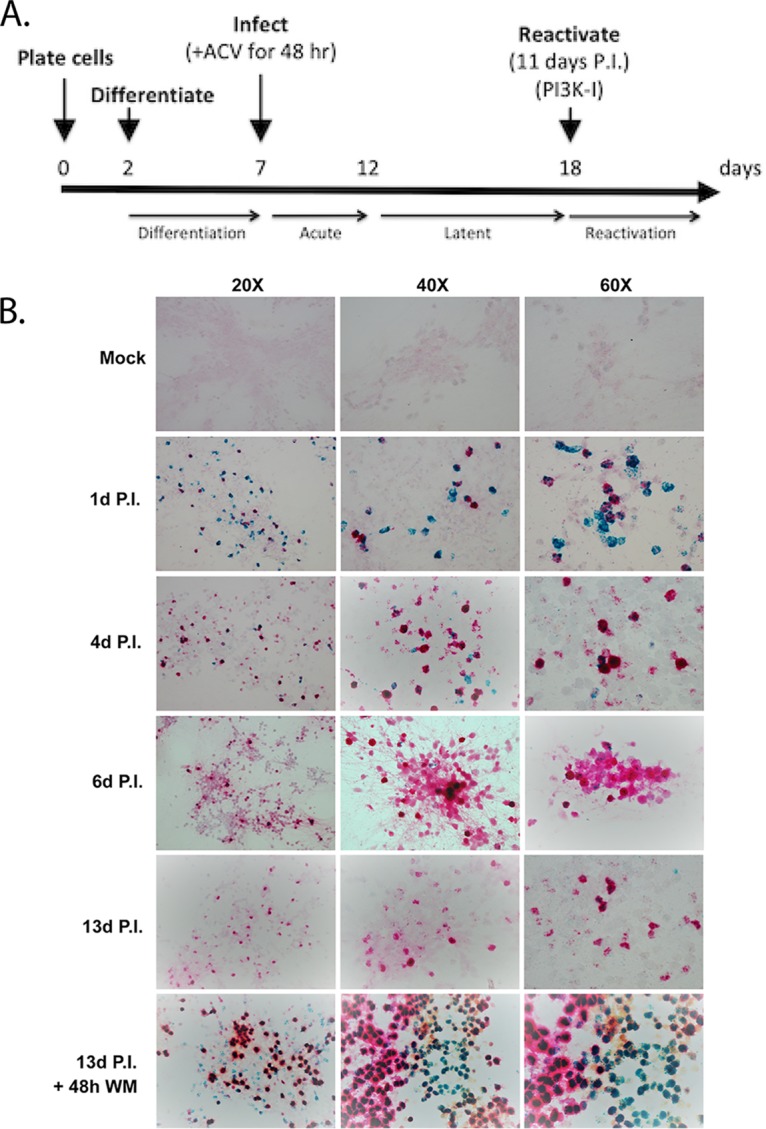
Establishment of a timeline for HSV-1 latency and reactivation by in situ detection of HSV-1 LATs and ICP4 RNA transcripts. (A) Experimental timeline of neuronal differentiation, infection, latency, and reactivation. (B) LUHMES neuronal cultures were plated and differentiated on coverslips for 5 days and then infected with 17syn+ at an MOI of 3 in the presence of 50 μM acyclovir (ACV). Forty-eight hours later, the medium was changed to medium without ACV and infection allowed to proceed, with harvesting of coverslips for RNAScope analysis on the indicated days. RNAScope analysis was performed per the manufacturer’s recommendation, and probes for LAT and ICP4 transcripts were designed by ACD. HSV-1 LATs were detected from the C2 channel (red) and the HSV-1 lytic transcripts (ICP4) were detected from the C1 channel (green). Reactivation was induced by incubation of day 11 (p.i.) cultures (labeled as 13d P.I. + 48h WM) for 48 h with the PI3K pathway inhibitor wortmannin (1 μM final concentration).
Based on the findings from other groups that activation of the PI3K pathway is important for initiation and maintenance of HSV-1 latency in neuronal cultures (14), we employed the nonspecific PI3K inhibitor wortmannin to induce reactivation. At 11 days p.i., 1 μM wortmannin was added to the LUHMES differentiation medium, and coverslips were harvested for RNAScope analysis at 24 and 48 h. Detection of ICP4 transcripts was seen at 24 h p.i. (data not shown), but the most robust activation of the lytic transcripts was seen by 48 h after exposure of the neurons to wortmannin (Fig. 3B). The ICP4 transcripts are detected in both LAT-positive and LAT-negative cells, though we cannot say at this point whether the cells were expressing the LAT at the time of reactivation or whether this occurred afterwards. In addition, we harvested supernatants each day over the course of the experiment to titrate for infectious virus. No infectious virus was detected in supernatants from 3 to 13 days p.i.; however, HSV-1 was detected in the supernatants at 24 and 48 h after Wortmannin treatment (Table 1). It should be noted that while no infectious virus was detected during the 3 to 13 days p.i. in this experiment, we have detected infectious virus in some wells during this time frame in other experiments (data not shown), indicating some low-level occurrence of spontaneous reactivation. In summary, this experiment indicates that the latent HSV-1 infection in the differentiated LUHMES cells can be efficiently induced to reactivate.
LUHMES neuronal cultures were also assessed for HSV-1 antigen expression and localization by immunofluorescence following a timeline of infection similar to that described for the RNAScope experiment. LUHMES cells were plated onto coverslips and infected with HSV-1 17syn+ at an MOI of 3 in the presence of 50 μM ACV. Following 48 h, the medium was replaced without additional ACV as described for the RNAScope experiments. Coverslips were harvested during the acute infection phase (days 1 to 4 p.i.) and during latency (days 6 and 7 p.i.). LUHMES cells were analyzed for expression of the HSV-1 lytic proteins ICP4 and ICP8 as well as for the neuron-specific microtubule βIII-tubulin. As can be seen by abundant expression of the ICP4 and ICP8 proteins by 24 h p.i. (Fig. 2A), LUHMES neurons are susceptible to HSV-1 infection and support the viral life cycle. Immunofluorescence experiments support the Western blot results (Fig. 2A), with a large proportion of neuronal nuclei expressing ICP4 following 24 h of infection with HSV-1 17syn+ (Fig. 4A to D). ICP8 expression is punctate, indicative of prereplicative sites being present during the acute phase of infection (Fig. 4I). ICP4 expression was analyzed up to 7 days p.i. Between day 4 p.i. (ICP4 [Fig. 4E]) and days 6 through 7 p.i. (ICP4 [Fig. 4J and K] and ICP8 [Fig. 4L]), both ICP4 and ICP8 expression become considerably less abundant as HSV-1 genomes entered latency.
FIG 4.

Lytic HSV-1 proteins are abundantly expressed in LUHMES neuronal cultures following acute infection and are reduced during latency. Differentiated, postmitotic LUHMES neurons were infected with 17syn+ at an MOI of 3 and analyzed by immunofluorescence for expression of viral lytic proteins over a time course of 7 days. (A to E) During the acute infection, ICP4 (shown in green) is abundantly expressed in almost all the neurons as large well-developed centers in the nucleus (1 to 4 days p.i.). (F) Mock-infected LUHMES cells. (G to I) Punctate, nuclear, chromatin-associated ICP8 (shown in green) is also evident during the acute infection (2 to 3 days p.i.). (J to L) By 6 to 7 days p.i., a dramatic reduction in both ICP4 (J and K) and ICP8 (L) expression is seen. Scale bars represent 20 μm. Neurofilaments are stained for βIII-tubulin (red); nuclei are stained with DAPI (blue).
Finally, we investigated the efficiency of infection and establishment of viral genomes in the neuronal nuclei of LUHMES cells following infection with HSV-1 by fluorescent in situ hybridization (FISH). LUHMES cells were plated, differentiated, and infected at an MOI of 3 on coverslips as described in Materials and Methods. At 24 h p.i., mock- and HSV-1-infected coverslips were harvested for FISH. Nuclear localization of HSV-1 genomes in LUHMES cells was clearly evident by DAPI (4′,6′-diamidino-2-phenylindole) staining coincident with detection of the Alexa Fluor 594 signal indicative of HSV-1 probe hybridization to viral genomes (Fig. 5A). Seven fields of 20×-magnified LUHMES neuronal cells processed for FISH were counted. At 24 h p.i., the majority (87% ± 15.9%) of nuclei were positive for viral genomes (Fig. 5B).
FIG 5.

HSV-1 genomes were characterized by FISH. LUHMES cells were plated and differentiated on coverslips as described in Materials and Methods and infected with 17syn+ at an MOI of 3 in the presence of 50 μM ACV. Coverslips were harvested at 24 h p.i. and neuronal cultures analyzed for the presence of HSV-1 genomes using a biotin-labeled HSV-1 probe followed by amplification with HRP-streptavidin and Alexa Fluor 594 tyramide (AF594). (A) The majority of neuronal nuclei display evidence of harboring HSV-1 genomes (compare mock with 24 h p.i.). Genomes are shown in magenta and DAPI-stained nuclei in blue; scale bar, 20 μm. (B) Total nuclei (DAPI) and replication centers (AF594) were quantified by counting each from 7 fields of cells, and the data are represented as average counts per field. The proportion of neuronal nuclei containing at least one genome-positive signal was 87.4% ± 15.9%.
DISCUSSION
Here we describe a scalable human neuronal culture system for the study of HSV latency and reactivation. This novel system offers several significant advantages. (i) LUHMES cells are derived from human neuronal precursors and differentiate into postmitotic neurons that express mature neuronal markers. As a human neuronal line, these cells should be useful in evaluating interactions between viral and host proteins and RNAs that may be species and cell type specific, for example interactions between HSV miRNAs and human RNA targets. (ii) LUHMES cells can be rapidly and efficiently differentiated to express mature neuronal markers evident by 5 days postdifferentiation. This differentiation process is more rapid than that with many other human neuron models, such as iPSCs (35). (iii) LUHMES cells can be efficiently infected with HSV-1, and at an MOI of 3, greater than 87% of the cells are infected. (iv) A quiescent state of latency can be established as early as 6 days postinfection. (v) Unlike many in vitro neuronal models such as human iPSCs and primary neuron cultures derived from rodent embryonic tissues, which require antiviral pressure during the establishment of latency (and often to maintain latency), LUHMES cells require only an initial treatment with acyclovir for the first 48 h of infection. (vi) Latent infections of LUHMES cells is scalable, and the undifferentiated cells can be propagated rapidly and plated into large flasks or dishes and then differentiated. It is therefore straightforward to obtain 107 to 108 or more latently infected, differentiated neurons for a given experiment. This opens the door to performing more cell number-intensive biochemical and molecular assays, such as mass spectrometry and cross-linking immunoprecipitation procedures such as PAR-CLIP, that until now have not been feasible.
The field has set a high bar for in vitro models of HSV quiescent or latent infection. The classic definition of latency is (i) the presence of HSV genomes within a cell (or ganglion) that can be maintained without the presence of inhibitors, (ii) undetectable levels of lytic proteins, (iii) little or no lytic gene expression, (iv) expression of the HSV-1 LATs, (v) the absence of detectable infectious virus, and (vi) ability of the latent genomes to be reactivated. With the advent of more sensitive techniques for measuring transcription, it is now appreciated that even in vivo, the latent infection is more dynamic than originally thought (for a review, see reference 36) and that abundant LAT expression is not present in the majority of neurons harboring latent HSV genomes. With this perspective, the data presented here are consistent with the ability of the LUHMES cells to support a latent infection that meets even the classic and more stringent definition. We demonstrate that the levels of lytic transcripts drop to very low levels by 6 to 8 days p.i. as measured by either RT-qPCR or RNAScope in situ hybridization. No infectious virus can be detected after 3 days p.i., and a large proportion of the cells can be reactivated following the addition of a PI3K inhibitor. From the RT-qPCR analyses (Fig. 2D), the levels of IE and E transcripts are at a low baseline by day 8 postinfection, and we consistently observe that these levels remain low between days 8 and 14. It should be noted that variable increases in IE and E transcripts occur after day 14, which could represent some increased level of spontaneous reactivation.
One of the original hallmarks of latency was the expression of the LATs. While it is now well established that during latency not all cells express the LATs (31), in vivo models have clearly shown that the number of LAT-expressing cells increases as latency is established (37). Therefore, it was initially surprising to us to see a decrease in total LAT transcript levels by RT-qPCR analysis between day 3 and day 8 as latency was established (Fig. 2C). However, when we examined LAT intron accumulation by RNAScope analysis in situ (Fig. 3B), the proportion of cells with abundant LAT intron clearly increased as latency was established, consistent with the observations in vivo. Therefore, it is our interpretation that the higher levels of primary LAT transcript detected at day 3 p.i. by RT-qPCR reflects abundant LAT transcript expression as a late transcript during the initial stages of acute infection, in a subset of cells. However, when LAT intron accumulation is examined, it clearly increases with time.
In the studies presented here, the differentiated LUHMES cells were infected in the presence of ACV for 48 h p.i. While the cells can be infected without ACV and a latent infection is still established, a certain proportion of the cells show evidence of cytopathic effect (CPE) and die (data not shown). Therefore, it is our sense that the acyclovir treatment enhances the efficiency of the establishment of latency in these cultures, but unlike in the case of other culture systems, the acyclovir treatment does not need to be maintained for extended periods of time.
LUHMES neuronal cells have great potential as an in vitro model system for studying HSV-1 latency, but some caveats should be considered. While cell culture models are attractive for performing molecular and biochemical analyses, they are not a substitute for in vivo models. Cell culture models lack the context of the in vivo infection with respect to the heterogeneity of cell type and tissue compartments involved, the initial presentation of virus to the neuron, and also the lack of an acquired immune response. For this reason, findings from HSV studies in the LUHMES model or any in vitro model should be validated in the mouse and/or rabbit model and, where possible, in human tissue samples.
A related point is that HSV infects many subtypes of neurons, and there are data that strongly suggest that neuronal subtype plays an important role in directing the outcome of HSV-1 infection (16, 38). The reductionist drive that pushes for the exploitation of homogeneous in vitro models often fails to address the importance of heterogeneous populations of cells for the outcome of infections in vivo. The LUHMES cell model presented here provides one model to study HSV-1 latency in vitro. It should be clearly stated that the LUHMES cells are central nervous system (dopaminergically fated) neurons. Therefore, the data obtained from these cells will need to be validated and compared in peripheral neurons. While one group has had success in producing sensory neurons from human iPSCs (23), this model is currently difficult to establish, and the number of cells available for analysis are limiting.
Finally, an important observation from this study is that even though >87% of the LUHMES cells infected with HSV-1 harbor latent genomes as assessed by FISH (Fig. 5A), RNAScope RNA in situ hybridization for the LAT intron reveals that at day 13 p.i. only a proportion of the neurons express detectable LAT (Fig. 3B). This is a significant finding in that while the LUHMES cells are a homogeneous cell line, LAT intron accumulation occurs in only a proportion of the latent cells, similar to what is observed in vivo. This observation further reflects the parallels between the latent infection in LUHMES cells and in vivo latency and also suggests that the LUHMES cells may provide a means to study the regulation of this LAT accumulation.
In conclusion, LUHMES cells represent a very useful and tractable human neuronal model to study a number of aspects of HSV latency that have been out of reach due to difficulties in obtaining sufficient numbers of latently infected cells.
MATERIALS AND METHODS
Cell culture.
Lund human mesencephalic (LUHMES) cells were obtained from the ATCC (catalog no. CRL-2927) and cultured according to supplier’s recommendations and as previously described (27). All of the experiments in this study were conducted with cells at a passage of 2 to 5 from the ATCC stock. Briefly, cell culture flasks and dishes were sequentially coated with poly-l-ornithine hydrobromide (Sigma, catalog no. P3655) overnight at room temperature followed by fibronectin (Sigma, catalog no. F2006) overnight at 37°C, rinsed with phosphate-buffered saline (PBS), and allowed to fully air dry at room temperature before cell plating. LUHMES cells were maintained in proliferation medium (Dulbecco modified Eagle medium [DMEM]-F12; ATCC, catalog no. 30-2006) containing 1% N2 supplement (ThermoFisher Scientific, catalog no. A1370701), 1× penicillin-streptomycin-glutamine solution (ThermoFisher Scientific, catalog no. 10378016), and recombinant human fibroblast growth factor (FGF)-basic (Peprotech, catalog no. 100-18B; reconstituted at 100 μg/ml in 10 mM Tris [pH 7.6] plus 0.1% bovine serum albumin [BSA]) added to proliferation medium immediately before use at a 40-ng/ml final concentration. LUHMES cells were passaged upon reaching no more than 75% confluence. For induction of differentiation, cells were switched to DMEM-F12 medium plus N2 supplement containing these additional components: tetracycline hydrochloride at a 1-μg/ml final concentration (Sigma, catalog no. T7660), N6,2′-O-dibutyryladenosine 3′,5′-cyclic monophosphate sodium salt at a 1 mM final concentration (Sigma, catalog no. D0627), and recombinant human glial cell-derived neurotrophic factor (GDNF) at a 2-ng/ml final concentration (R&D Systems, catalog no. 212-GD-010). All experiments were conducted with LUHMES neuronal cells following at least 5 days after induction of differentiation.
Infection, establishment of latency, and reactivation.
LUHMES cells were grown and differentiated on poly-l-ornithine/fibronectin-coated coverslips as follows. First, 25,000 cells per well were seeded onto 24-well plates and allowed to proliferate for 2 to 3 days, at which time the culture medium was changed to medium containing differentiation-inducing factors (see “Cell culture” above). Following 5 days of differentiation, postmitotic neurons were pretreated with 50 μM acyclovir (ACV) for 2 h and then infected with dilutions of stocks from clarified lysates of HSV-1 17syn+ in complete medium at an MOI of 3 with ACV added to the virus inoculum and overlay medium, and 48 h later, the medium was changed without addition of ACV. The neurons were continuously cultured for up to 15 days postinfection (p.i.) with medium changes every 2 days. Coverslips were harvested each day for analysis of LAT and ICP4 transcripts and for HSV-1 genomes in situ. By tracking the timing of increased LAT and decreased lytic ICP4 transcripts, a period of latency or quiescence was determined to occur at around day 6 p.i. Reactivation was induced at day 11 p.i. by incubating infected neurons for 48 h with 1 μM wortmannin (Sigma, catalog no. W1628). Neurons were also analyzed by immunofluorescence for the presence of HSV-1-specific proteins and for determination of neuronal markers.
Immunofluorescence.
Differentiated LUHMES cells were fixed at various times postinfection for analysis of HSV-1 proteins in situ. Two fixation methods were employed, depending on the antigen of interest. For ICP4 and neurofilament staining, neurons were washed with PBS and fixed by brief submersion in supercooled methanol (MeOH) on dry ice followed by incubation in MeOH for 30 min at −20°C. For ICP8 immunofluorescence, neurons were permeabilized with cytoskeleton (CSK) buffer [100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 10 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES), pH 6.8] and fixed with 4% formaldehyde in PBS as described below. For immunofluorescence, neurons were incubated in blocking buffer (5% goat serum, 0.1% NP-40, 20 mM Tris-HCl [pH 7.4], 150 mM sodium chloride) for 30 min at room temperature and then incubated with primary antibodies for 1 h at room temperature at the following dilutions in antibody dilution and wash (ADW) buffer (20 mM Tris-HCl [pH 7.4], 150 mM NaCl, 0.1% NP-40): anti-HSV-1 ICP4 hybridoma at 1:100, anti-HSV-1 ICP8 mouse monoclonal antibody (MAb) 11E2 (Abcam, catalog no. ab20194) at 1:200, Synaptophysin mouse ascites (Sigma, catalog no. S-5768, clone SVP-38) at 1:200, anti-βIII-tubulin rabbit polyclonal antibody (PAb) (Abcam, catalog no. ab18207) at 1:500; anti-neurofilament 200 mouse MAb (Sigma, catalog no. N0142) at 1:2,000, and anti-vimentin rabbit MAb (Abcam, catalog no. ab92547) at 1:200. Coverslips were washed 3 times for 5 min each with ADW buffer and incubated with secondary antibodies diluted in ADW buffer. Secondary antibodies used in this study were Alexa Fluor 594 goat anti-rabbit (ThermoFisher Scientific, catalog no. A11072; 1:2,000) and Alexa Fluor 488 goat anti-mouse (ThermoFisher Scientific, catalog no. A11029; 1:1,000). Coverslips were washed 3 times for 5 min each with ADW buffer and mounted onto slides with ProLong Diamond antifade mountant with DAPI (ThermoFisher Scientific, catalog no. P36962). Immunofluorescence was conducted using a Nikon Eclipse E600 microscope with epifluorescence, and photography was with a QImaging Exi Aqua Monochrome digital camera.
Western blotting.
Twenty-four-well plates of LUHMES cells were harvested for Western blotting of HSV-1 antigens as previously described (26). LUHMES cells were infected with 17syn+ at an MOI of 3 or mock infected and were harvested on the indicated days by direct lysis in 1× NuPAGE lithium dodecyl sulfate (LDS) sample buffer (ThermoFisher Scientific, catalog no. NP0007) and loaded onto 4 to 12% Bis-Tris NuPAGE gels (ThermoFisher Scientific) with NuPAGE morpholinepropanesulfonic acid (MOPS) SDS running buffer (ThermoFisher Scientific, catalog no. NP000102). Following transfer, polyvinylidene difluoride (PVDF) membranes were incubated with the following antibodies: anti-HSV-1 ICP8 mouse MAb 11E2 (Abcam, catalog no. ab20194) at a 1:10,000 dilution, anti-HSV-1 ICP4 hybridoma at a 1:500 dilution, and anti-βIII-tubulin rabbit PAb (Abcam, catalog no. ab18207) at a 1:1,000 dilution. Secondary antibody detection was performed with Pierce goat anti-rabbit poly-horseradish peroxidase (poly-HRP) (ThermoFisher Scientific, catalog no. 32260) or Pierce goat anti-mouse poly-HRP (ThermoFisher Scientific, catalog no. 32230) at a 1:5,000 dilution, developed with a chemiluminescent substrate (Pierce ECL Western blotting substrate, catalog no. 32209), and imaged on a GE ImageQuant LAS4000 instrument.
RT-qPCR.
RNA was extracted from mock- or HSV-1 17syn+-infected LUHMES cells with TRIzol reagent followed by spin column purification (ThermoFisher Scientific, catalog no. 12183555). RNA samples were then treated with Turbo DNase for 30 min at 37°C, followed by incubation with DNase inactivation reagent for 5 min at room temperature (ThermoFisher Scientific Turbo DNA-free kit, catalog no. AM1907). Reverse transcription (RT) was performed in 20-μl reaction mixtures using the RETROscript kit (ThermoFisher Scientific, catalog no. AM1710) with either random decamers for GAPDH (glyceraldehyde-3-phosphate dehydrogenase), ICP4, TK, and gC or gene-specific priming for analysis of LAT expression. RT reaction mixtures were prepared per the manufacturer’s protocol, and reactions were performed for the lytic transcripts by incubating 1 μg of RNA with Moloney murine leukemia virus (MMLV) RT for 60 min at 42°C, followed by inactivation at 92°C for 10 min. A primer spanning the HSV-1 LAT intron (5′-GTG GTC GGA CGG GTA AGT AA-3′) was used for gene-specific RT as follows. First, 5× annealing buffer (100 mM Tris-HCl [pH 8.3], 50 mM KCl) was prepared, diluted to 1× in a mixture containing 20 μM primer and 20 U RNasin, and incubated for 10 min at 37°C. To this mixture, 1 μg RNA was added and heated for 3 min at 80°C, and then primer annealing was performed for 45 min at 50°C. cDNA was generated by adding MMLV RT, deoxynucleoside triphosphates (dNTPs), and RT buffer in a final reaction volume of 25 μl and incubated at 50°C for 45 min. RT enzyme was inactivated by incubation at 92°C for 10 min and qPCR performed using the following Applied Biosystems TaqMan assays: human GAPDH (catalog no. 4331182) and Assay ID s02786624_g1. The ABI TaqMan primer and probe sequences for the HSV-1 LAT intron, ICP4, TK, and gC are listed in Table 2. For analysis of HSV-1 genome copies, TaqMan assays were conducted using primers and probe spanning the UL30 gene (Table 2).
TABLE 2.
HSV-1 real-time PCR primers and probesa
| Target gene | Forward primer | Reverse primer | Probe |
|---|---|---|---|
| LAT intron | CGCCCCAGAGGCTAAGG | GGGCTGGTGTGCTGTAACA | CCACGCCACTCGCG |
| ICP4 | CACGGGCCGCTTCAC | GCGATAGCGCGCGTAGA | CCGACGCGACCTCC |
| TK | CACGCTACTGCGGGTTTATATAGAC | GGCTCGGGTACGTAGACGATAT | CACCACGCAACTGC |
| gC | CCTCCACGCCCAAAAGC | GGTGGTGTTGTTCTTGGGTTTG | CCCCACGTCCACCCC |
| UL30 | AGAGGGACATCCAGGACTTTGT | CAGGCGCTTGTTGGTGTAC | ACCGCCGAACTGAGCA |
All primer and probe sequences are written 5′ to 3′.
RNAScope analysis.
Gene expression analysis for latency and lytic transcripts was conducted by harvesting coverslips of differentiated LUHMES cells following infection with HSV-1 17syn+ on the indicated days. RNAScope (Advanced Cell Diagnostics [ACD], Inc.) analysis was conducted per the manufacturer’s recommendations using probes designed against the 17syn+ genome spanning a region of the LAT transcript (315651-C2) or the ICP4 transcript (406031-C1). At the indicated time p.i., coverslips were washed with PBS and neurons were fixed by incubation with 4% formaldehyde in PBS for 30 min at room temperature, washed 2 times with PBS, and dehydrated through an ethanol series as recommended by the manufacturer. Coverslips were stored in 100% ethanol at −20°C until processed for in situ hybridization. For hybridization, neurons were rehydrated and digested with protease III (from the kit) diluted 1:15 in PBS at room temperature for 10 min, followed by two PBS washes. Probes were used at a 1:50 ratio of LAT (C2) to ICP4 (C1) and were incubated with neurons at 40°C for 2 h in a hybridization oven and washed with 1× wash buffer (from the kit) 2 times for 2 min each at room temperature, and slides were stored overnight at room temperature in 5× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) buffer. Detection was performed as described in the manufacturer’s protocol, with both red and green signals allowed to develop for 10 min at room temperature, followed by mounting onto slides with VectaMount (Vector Labs). Bright-field images were captured using a Nikon Eclipse E600 microscope outfitted with a Nikon Digital Sight DS-U3 camera.
FISH.
For fluorescence in situ hybridization (FISH), at the indicated time p.i., neurons were permeabilized and fixed as follows. Medium was aspirated from coverslips and neurons incubated in ice-cold cytoskeleton (CSK) buffer (100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 10 mM PIPES, pH 6.8) for 30 s on ice, followed by incubation in ice-cold CSK buffer containing 0.4% Triton X-100 for 30 s on ice, and then rinsed with ice-cold CSK buffer. Neurons were then fixed with 4% formaldehyde in PBS for 10 min at room temperature, washed 4 times with ice-cold 70% ethanol, and stored at −20°C until processed for in situ hybridization. For DNA hybridization, neurons were dehydrated through an ethanol series for 2 min each at room temperature (70%, 85%, and 100%) and then air dried for 15 min at room temperature. RNase treatment was performed at 37°C for 30 min (1.25mg/ml RNase A diluted in 2× SSC buffer) followed by blocking of endogenous biotin (avidin/biotin blocking kit; ThermoFisher Scientific, catalog no. 004303) for 20 min at room temperature. Neurons were dehydrated again through an ethanol series for 2 min each at room temperature (85%, 95%, and 100%) and allowed to air dry at room temperature for 15 min. Denaturation was performed by incubating coverslips at 95°C for 11 min in prewarmed 2× SSC–70% formamide solution, followed by immediate dehydration through an ice-cold ethanol series for 2 min each (70%, 85%, 95%, and 100%) and air drying at room temperature for 15 min. The DNA hybridization probe was generated by random-primed labeling of a HindIII-linearized plasmid containing the HSV-1 genome region spanning the UL30 3′end through UL31 and the 3′500 bp of UL32 (pBS-KS) with incorporation of biotin-16-dUTP (Roche, catalog no. 11585649910). The probe was diluted to 2 ng/μl in hybridization solution with 50% formamide (2× hybridization buffer [4× SSC and 20% dextran sulfate; 1× prepared by addition of an equal volume of formamide), added to coverslips, and incubated overnight at 37°C in a humid chamber with paper towels soaked in 2× SSC–50% formamide solution. The following day, coverslips were washed with 2× SSC–50% formamide 3 times for 10 min each at 39°C, 2× SSC for 10 min at 39°C, 2× SSC for 10 min at room temperature, and 1× SSC for 10 min at room temperature and rinsed three times with PBS. The Alexa Fluor 594 tyramide SuperBoost kit (ThermoFisher Scientific, catalog no. B40935) was used for signal amplification following the manufacturer’s protocol. Briefly, coverslips were blocked (component A from the kit) for 1 h at room temperature and then incubated with HRP-streptavidin (component B from the kit) for 1 h at room temperature. Following PBS washes, tyramide working solution was added to coverslips and incubated for 5 min at room temperature. Coverslips were mounted onto slides with ProLong Diamond antifade mountant with DAPI and imaged as described in “Immunofluorescence” above. Analysis and calculation of HSV-1 genomes present in each neuronal nucleus was performed using NIS-Elements basic research (Nikon BR3.2) software interfaced with a Nikon Eclipse E600 microscope using edge detection and object count dialog 618 options. Seven fields of LUHMES cells at a magnification of ×20 were analyzed by counting individual DAPI-stained and Alexa Fluor 594-positive neurons.
ACKNOWLEDGMENTS
This work was supported in part by grants R01 AI48633 and R21 AI112382 from the NIH.
We acknowledge C. Fisher and A. Ishov for helpful comments and advice and NanoVir, LLC, for use of their equipment. We thank E. Barrozo and D. Phelan for helpful comments on the manuscript.
REFERENCES
- 1.Wagner EK, Bloom DC. 1997. Experimental investigation of herpes simplex virus latency. Clin Microbiol Rev 10:419–443. doi: 10.1128/CMR.10.3.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Preston CM, Russell J, Harris RA, Jamieson DR. 1994. Herpes simplex virus latency in tissue culture cells. Gene Ther 1:S49–S50. [PubMed] [Google Scholar]
- 3.Preston CM, Nicholl MJ. 2008. Induction of cellular stress overcomes the requirement of herpes simplex virus type 1 for immediate-early protein ICP0 and reactivates expression from quiescent viral genomes. J Virol 82:11775–11783. doi: 10.1128/JVI.01273-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Everett RD, Murray J, Orr A, Preston CM. 2007. Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J Virol 81:10991–11004. doi: 10.1128/JVI.00705-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Danaher RJ, Jacob RJ, Chorak MD, Freeman CS, Miller CS. 1999. Heat stress activates production of herpes simplex virus type 1 from quiescently infected neurally differentiated PC12 cells. J Neurovirol 5:374–383. doi: 10.3109/13550289909029478. [DOI] [PubMed] [Google Scholar]
- 6.Danaher RJ, Jacob RJ, Miller CS. 1999. Establishment of a quiescent herpes simplex virus type 1 infection in neurally-differentiated PC12 cells. J Neurovirol 5:258–267. doi: 10.3109/13550289909015812. [DOI] [PubMed] [Google Scholar]
- 7.Danaher RJ, Cook RK, Wang C, Triezenberg SJ, Jacob RJ, Miller CS. 2013. C-terminal trans-activation sub-region of VP16 is uniquely required for forskolin-induced herpes simplex virus type 1 reactivation from quiescently infected-PC12 cells but not for replication in neuronally differentiated-PC12 cells. J Neurovirol 19:32–41. doi: 10.1007/s13365-012-0137-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller CS, Danaher RJ, Jacob RJ. 2006. ICP0 is not required for efficient stress-induced reactivation of herpes simplex virus type 1 from cultured quiescently infected neuronal cells. J Virol 80:3360–3368. doi: 10.1128/JVI.80.7.3360-3368.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shipley MM, Mangold CA, Kuny CV, Szpara ML. 2017. Differentiated human SH-SY5Y cells provide a reductionist model of herpes simplex virus 1 neurotropism. J Virol 91:e00958-17. doi: 10.1128/JVI.00958-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shipley MM, Mangold CA, Szpara ML. 2016. Differentiation of the SH-SY5Y human neuroblastoma cell line. J Vis Exp 17:53193. doi: 10.3791/53193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilcox CL, Johnson EM. 1988. Characterization of nerve growth factor-dependent herpes simplex latency in neurons in vitro. J Virol 62:393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilcox CL, Smith RL, Freed CR, Johnson EM Jr. 1990. Nerve growth factor-dependence of herpes simplex virus latency in peripheral sympathetic and sensory neurons in vitro. J Neurosci 10:1268–1275. doi: 10.1523/JNEUROSCI.10-04-01268.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith RL, Escudero JM, Wilcox CL. 1994. Regulation of the herpes simplex virus latency-associated transcripts during establishment of latency in sensory neurons in vitro. Virology 202:49–60. doi: 10.1006/viro.1994.1321. [DOI] [PubMed] [Google Scholar]
- 14.Camarena V, Kobayashi M, Kim JY, Roehm P, Perez R, Gardner J, Wilson AC, Mohr I, Chao MV. 2010. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host Microbe 8:320–330. doi: 10.1016/j.chom.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kobayashi M, Kim JY, Camarena V, Roehm PC, Chao MV, Wilson AC, Mohr I. 2012. A primary neuron culture system for the study of herpes simplex virus latency and reactivation. J Vis Exp 2:3823. doi: 10.3791/3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bertke AS, Swanson SM, Chen J, Imai Y, Kinchington PR, Margolis TP. 2011. A5-positive primary sensory neurons are nonpermissive for productive infection with herpes simplex virus 1 in vitro. J Virol 85:6669–6677. doi: 10.1128/JVI.00204-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bertke AS, Ma A, Margolis MS, Margolis TP. 2013. Different mechanisms regulate productive herpes simplex virus 1 (HSV-1) and HSV-2 infections in adult trigeminal neurons. J Virol 87:6512–6516. doi: 10.1128/JVI.00383-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maroui MA, Calle A, Cohen C, Streichenberger N, Texier P, Takissian J, Rousseau A, Poccardi N, Welsch J, Corpet A, Schaeffer L, Labetoulle M, Lomonte P. 2016. Latency entry of herpes simplex virus 1 is determined by the interaction of its genome with the nuclear environment. PLoS Pathog 12:e1005834. doi: 10.1371/journal.ppat.1005834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson AC, Mohr I. 2012. A cultured affair: HSV latency and reactivation in neurons. Trends Microbiol 20:604–611. doi: 10.1016/j.tim.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cliffe AR, Arbuckle JH, Vogel JL, Geden MJ, Rothbart SB, Cusack CL, Strahl BD, Kristie TM, Deshmukh M. 2015. Neuronal stress pathway mediating a histone methyl/phospho switch is required for herpes simplex virus reactivation. Cell Host Microbe 18:649–658. doi: 10.1016/j.chom.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pourchet A, Modrek AS, Placantonakis DG, Mohr I, Wilson AC. 2017. Modeling HSV-1 latency in human embryonic stem cell-derived neurons. Pathogens 6:E24. doi: 10.3390/pathogens6020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.D'Aiuto L, Prasad KM, Upton CH, Viggiano L, Milosevic J, Raimondi G, McClain L, Chowdari K, Tischfield J, Sheldon M, Moore JC, Yolken RH, Kinchington PR, Nimgaonkar VL. 2015. Persistent infection by HSV-1 is associated with changes in functional architecture of iPSC-derived neurons and brain activation patterns underlying working memory performance. Schizophr Bull 41:123–132. doi: 10.1093/schbul/sbu032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zimmer B, Ewaleifoh O, Harschnitz O, Lee YS, Peneau C, McAlpine JL, Liu B, Tchieu J, Steinbeck JA, Lafaille F, Volpi S, Notarangelo LD, Casanova JL, Zhang SY, Smith GA, Studer L. 2018. Human iPSC-derived trigeminal neurons lack constitutive TLR3-dependent immunity that protects cortical neurons from HSV-1 infection. Proc Natl Acad Sci U S A 115:E8775–E8782. doi: 10.1073/pnas.1809853115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lotharius J, Brundin P. 2002. Impaired dopamine storage resulting from alpha-synuclein mutations may contribute to the pathogenesis of Parkinson's disease. Hum Mol Genet 11:2395–2407. doi: 10.1093/hmg/11.20.2395. [DOI] [PubMed] [Google Scholar]
- 25.Lotharius J, Falsig J, van Beek J, Payne S, Dringen R, Brundin P, Leist M. 2005. Progressive degeneration of human mesencephalic neuron-derived cells triggered by dopamine-dependent oxidative stress is dependent on the mixed-lineage kinase pathway. J Neurosci 25:6329–6342. doi: 10.1523/JNEUROSCI.1746-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paul G, Christophersen NS, Raymon H, Kiaer C, Smith R, Brundin P. 2007. Tyrosine hydroxylase expression is unstable in a human immortalized mesencephalic cell line–studies in vitro and after intracerebral grafting in vivo. Mol Cell Neurosci 34:390–399. doi: 10.1016/j.mcn.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 27.Scholz D, Poltl D, Genewsky A, Weng M, Waldmann T, Schildknecht S, Leist M. 2011. Rapid, complete and large-scale generation of post-mitotic neurons from the human LUHMES cell line. J Neurochem 119:957–971. doi: 10.1111/j.1471-4159.2011.07255.x. [DOI] [PubMed] [Google Scholar]
- 28.Ratcliffe LE, Vazquez Villasenor I, Jennings L, Heath PR, Mortiboys H, Schwartzentruber A, Karyka E, Simpson JE, Ince PG, Garwood CJ, Wharton SB. 2018. Loss of IGF1R in human astrocytes alters complex I activity and support for neurons. Neuroscience 390:46–59. doi: 10.1016/j.neuroscience.2018.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hollerhage M, Moebius C, Melms J, Chiu WH, Goebel JN, Chakroun T, Koeglsperger T, Oertel WH, Rosler TW, Bickle M, Hoglinger GU. 2017. Protective efficacy of phosphodiesterase-1 inhibition against alpha-synuclein toxicity revealed by compound screening in LUHMES cells. Sci Rep 7:11469. doi: 10.1038/s41598-017-11664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiang W, Schlachetzki JC, Helling S, Bussmann JC, Berlinghof M, Schaffer TE, Marcus K, Winkler J, Klucken J, Becker CM. 2013. Oxidative stress-induced posttranslational modifications of alpha-synuclein: specific modification of alpha-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol Cell Neurosci 54:71–83. doi: 10.1016/j.mcn.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 31.Mehta A, Maggioncalda J, Bagasra O, Thikkavarapu S, Saikumari P, Valyi-Nagy T, Fraser NW, Block TM. 1995. In situ DNA PCR and RNA hybridization of herpes simplex virus sequences in trigeminal ganglia of latently infected mice. Virology 206:633–640. doi: 10.1016/S0042-6822(95)80080-8. [DOI] [PubMed] [Google Scholar]
- 32.Chen XP, Mata M, Kelley M, Glorioso JC, Fink DJ. 2002. The relationship of herpes simplex virus latency associated transcript expression to genome copy number: a quantitative study using laser capture microdissection. J Neurovirol 8:204–210. doi: 10.1080/13550280290049642. [DOI] [PubMed] [Google Scholar]
- 33.Edwards TG, Bloom DC, Fisher C. 2018. The ATM and Rad3-Related (ATR) protein kinase pathway is activated by herpes simplex virus 1 and required for efficient viral replication. J Virol 92:e01884-17. doi: 10.1128/JVI.01884-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang F, Flanagan J, Su N, Wang LC, Bui S, Nielson A, Wu X, Vo HT, Ma XJ, Luo Y. 2012. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J Mol Diagn 14:22–29. doi: 10.1016/j.jmoldx.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.D'Aiuto L, Williamson K, Dimitrion P, McNulty J, Brown CE, Dokuburra CB, Nielsen AJ, Lin WJ, Piazza P, Schurdak ME, Wood J, Yolken RH, Kinchington PR, Bloom DC, Nimgaonkar VL. 2017. Comparison of three cell-based drug screening platforms for HSV-1 infection. Antiviral Res 142:136–140. doi: 10.1016/j.antiviral.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bloom DC. 2016. Alphaherpesvirus latency: a dynamic state of transcription and reactivation. Adv Virus Res 94:53–80. doi: 10.1016/bs.aivir.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 37.Margolis TP, Sedarati F, Dobson AT, Feldman LT, Stevens JG. 1992. Pathways of viral gene expression during acute neuronal infection with HSV-1. Virology 189:150–160. doi: 10.1016/0042-6822(92)90690-Q. [DOI] [PubMed] [Google Scholar]
- 38.Margolis TP, Imai Y, Yang L, Vallas V, Krause PR. 2007. Herpes simplex virus type 2 (HSV-2) establishes latent infection in a different population of ganglionic neurons than HSV-1: role of latency-associated transcripts. J Virol 81:1872–1878. doi: 10.1128/JVI.02110-06. [DOI] [PMC free article] [PubMed] [Google Scholar]