

We analyzed genome sequences from 6 baboon species and found evidence of ancient and recent hybridization among divergent species.
Abstract
Recent studies suggest that closely related species can accumulate substantial genetic and phenotypic differences despite ongoing gene flow, thus challenging traditional ideas regarding the genetics of speciation. Baboons (genus Papio) are Old World monkeys consisting of six readily distinguishable species. Baboon species hybridize in the wild, and prior data imply a complex history of differentiation and introgression. We produced a reference genome assembly for the olive baboon (Papio anubis) and whole-genome sequence data for all six extant species. We document multiple episodes of admixture and introgression during the radiation of Papio baboons, thus demonstrating their value as a model of complex evolutionary divergence, hybridization, and reticulation. These results help inform our understanding of similar cases, including modern humans, Neanderthals, Denisovans, and other ancient hominins.
INTRODUCTION
The increasing availability of genomic data across the tree of life has begun to challenge traditional concepts and assumptions regarding the genetics and population biology of phylogenetic differentiation and speciation (1, 2). Reconstruction of the history of closely related lineages suggests that cladogenesis (differentiation from a common ancestor that produces one or more new species) is often not as straightforward as assumed by traditional models of speciation (3, 4). Evolving lineages may exchange functionally important genetic information while remaining phenotypically distinct and diagnosable (5, 6). Comparing genomic data across such closely related, divergent but still interfertile lineages provides new insight into cladogenesis in general and the nature, rate, and consequences of genomic evolution in particular.
Baboons (order Primates; family Cercopithecidae; genus Papio) are large-bodied, geographically widespread Old World monkeys (OWMs; here, we use the term “baboon” to refer only to species in the genus Papio). Their diversity provides an opportunity to investigate the genomic, morphological, and behavioral aspects of an evolutionary radiation in a broadly successful and adaptable primate. Extensive work in both natural and captive populations has produced considerable insight into baboon morphology, physiology, neurobiology, and behavior that provides additional context for comparative evolutionary analyses (7–11). Mitochondrial DNA (mtDNA) diversity suggests that this evolutionary radiation began about 2 million years (Ma) ago (12), approximately the same time as the radiation of our own genus, Homo, and in the same sub–Saharan African environment (13). However, unlike Homo, which is now reduced to a single surviving species (Homo sapiens), Papio still includes six extant lineages, the products of successive speciation events. These six species are morphologically and behaviorally distinct (10–12) and have broad, adjoining but nonoverlapping geographic ranges across sub–Saharan Africa and southwest Arabia (Fig. 1; see Supplementary Text for explanation of the taxonomy used here). The morphological and behavioral traits that define each species are unambiguous (Fig. 1) and expressed quite homogeneously over large geographic distances (10, 11, 14).
Fig. 1. Papio baboon species.

(A) The appearance and current distribution of each baboon species, and the locations of three well-documented active hybrid zones are also shown. x1: hybrid zone between P. hamadryas and P. anubis (19, 28), x2: hybrid zone between P. cynocephalus and P. anubis (17, 26), x3: hybrid zone between P. kindae and P. ursinus (18). Drawings of each species by S. Nash. (B) Distinguishing features of Papio species. Body mass data from (16, 59) and unpublished data from J.P.-C., J.R., and C.J.J.
Despite these phenotypic differences, genetic evidence reveals an underlying complexity to baboon evolutionary history. Maternally inherited mtDNA yields a phylogeny that includes at least seven major haplogroups whose distribution is discordant with the relationships implied by phenotypic comparisons (12, 15). Since migration among baboon social groups and populations is generally sex-biased, with males usually (but not always) the dispersing sex (16), population relationships based on maternally inherited mtDNA will not necessarily correspond to population relationships based on nuclear DNA or phenotype. Furthermore, baboon species produce fertile hybrid offspring in the wild and can form long-lasting hybrid zones (17–19) despite substantial species-specific differences in body size, secondary sexual characteristics, and social systems (11).
This constellation of genetic and phenotypic observations raises a number of questions regarding the history of differentiation and genetic exchange among baboons. Is baboon hybridization a recent phenomenon, or does the discordance between the mtDNA phylogeny and phenotypic similarity result in part from ancient episodes of admixture and evolutionary reticulation? Has admixture been limited to the extant lineages within Papio, or as has been suggested for hominins (20, 21), have now extinct “ghost” lineages also played a role? The current study investigates baboon genomic diversity to provide a detailed reconstruction of the history of evolutionary differentiation among the six extant species and new insights regarding the processes, correlates, and consequences of genomic differentiation. These results, for a clade in which multiple lineages and active hybrid zones are available for direct observation, provide important insights relevant to other cases of complex differentiation and admixture, including that of ancestral modern humans and our extinct relatives such as Neanderthals, Denisovans, and other early human lineages.
RESULTS
We produced a whole-genome reference assembly (Panu_3.0; GenBank accession GCA_000264685.2) for an olive baboon (Papio anubis), the baboon species most commonly used in biomedical research (fig. S1 and tables S1 and S2) (7). To investigate genetic differentiation in the genus, we analyzed whole-genome sequences from 16 additional individuals, 2 to 4 individuals representing each of the six species within Papio, and a gelada (Theropithecus gelada), a member of a closely related genus that serves as an outgroup (fig. S2 and table S3). This diversity panel produced >54.6 million single-nucleotide variants (SNVs), of which >42.4 million are variable within Papio (fig. S3 and table S4). To develop a second independent perspective on genome differentiation, we identified novel Alu insertions, a type of genetic variation that results from a fundamentally different mutational mechanism. Unexpectedly, we found a dramatically elevated number of recent Alu insertions in the baboons (and in rhesus macaques) relative to human and other primate genomes (Fig. 2 and table S5). There are 192,889 full-length AluY elements in the P. anubis genome. The rate of accumulation of lineage-specific AluY insertions has therefore been more than fourfold higher (Fig. 2) in baboons and rhesus macaques than in hominoids (humans, chimpanzees, or orangutans) and about threefold higher than in the African green monkey (genus Chlorocebus), another OWM (22).
Fig. 2. Comparison of Alu mobilization rates in selected primate genomes.
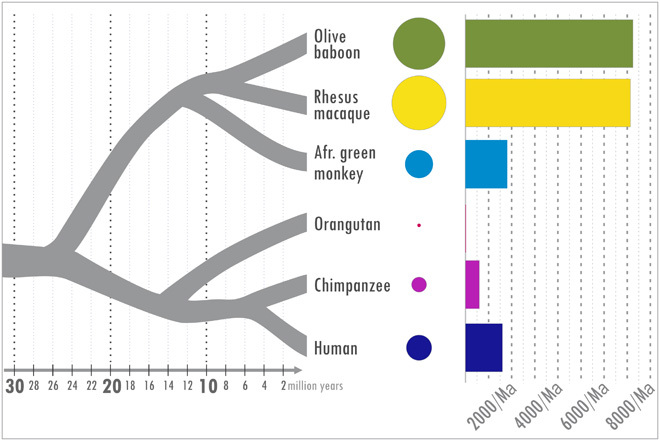
Only Alu elements specific to each lineage are included. The size of the circle corresponds to the number of near full-length lineage-specific AluY elements in that species. The bars on the right show the estimated number of insertions per million years for each lineage. For baboon (Panu_3.0), rhesus macaque (Mmul_8.0.1), African green monkey (chlSab2), chimpanzee (Pan_tro3), and human (GRCh38/hg38), AluY sequences were retrieved computationally by cross comparisons using the most recent available assemblies. Orangutan estimates are from P_pygmaeus2.0.2 (60). The number of lineage-specific AluY elements is similar in rhesus macaque and baboon, and more than twice that in the African green monkeys, despite a longer period of independent evolution for the African green monkeys.
Our phylogenetic analyses provide several new insights into baboon population and genomic history. Maximum likelihood (ML) analyses of concatenated SNVs show that individual baboons cluster correctly with their conspecifics while separating the six extant species into distinct northern and southern clades (Fig. 3 and fig. S4A). In contrast, Bayesian analysis of the same SNV data suggests that P. kindae is sister to the northern clade rather than to P. cynocephalus and P. ursinus (fig. S4B). The existence of multiple hybrid zones and documented discrepancies between relationships based on mtDNA and on phenotypes (Fig. 1) (12, 15) argue that male-driven admixture and/or incomplete lineage sorting (ILS) have influenced genetic relationships among these species. When we used a polymorphism-aware phylogenetic approach, PoMo (23, 24), we again obtained a basal north-south divergence, with P. kindae placed in the southern group. However, the relationships among the three southern species differ from the ML result (Fig. 3). PoMo also infers much longer terminal branch lengths for P. ursinus and P. papio than for other lineages. Simulations (fig. S5 and table S6) show that admixture among divergent lineages can affect inferred branch lengths and that lineages that have experienced admixture will exhibit artificially shorter branch lengths due to allele sharing across lineages. This suggests that the other four lineages may have been more affected by admixture than P. ursinus and P. papio, which is consistent with the fact that these two species are found at the extreme southern and western reaches of baboon distributions, respectively (Fig. 1).
Fig. 3. Phylogenetic relationships among baboon species.
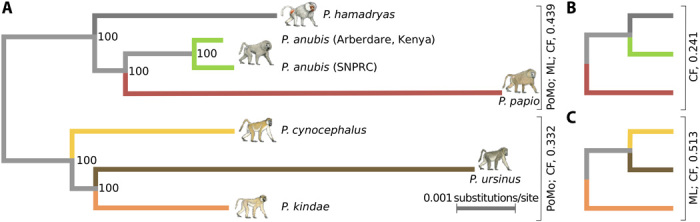
(A) Phylogeny generated using the polymorphism-aware phylogenetic method (PoMo) (23, 24). This topology for the three northern species is also supported by ML analysis of concatenated SNVs and by 43.9% of informative gene trees filtered to exclude any coding sequence genes [scaled concordance factor (CF) of 0.439, greater than the other two alternatives]. The topology shown for the three southern clade species is supported by the PoMo analysis and has a scaled CF score of 0.332. (B) One alternative topology for the northern species, supported by a scaled CF of 0.241. (C) One alternative topology for the southern species, supported by ML analysis of concatenated SNVs and a scaled CF score of 0.513, i.e., a larger proportion of gene trees that are devoid of coding genes than the other two alternative trees.
To explicitly test for admixture among the six extant baboon species, we performed an analysis using f-statistics, followed by modeling using coalescent hidden Markov methods (Fig. 4A, table S7, and fig.S6). The best-fitting model (see Materials and Methods) indicates that the history of P. kindae includes an ancient admixture event involving a lineage related to extant P. ursinus (52% contribution to extant P. kindae) and an unsampled lineage (possibly extinct) belonging to the northern clade (48% contribution). The f-statistics suggest that extant P. papio is closely related to P. anubis, but received ~10% genetic input from an ancestral northern lineage also not yet sampled, possibly extinct.
Fig. 4. Evolutionary and demographic history for Papio baboons.
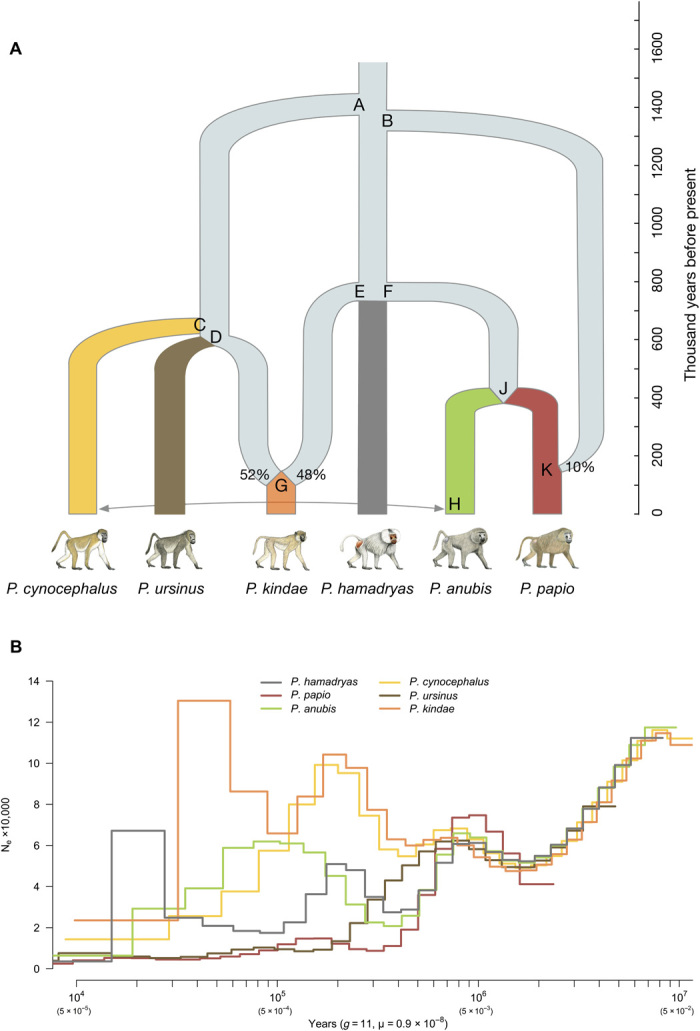
(A) Analyses using f-statistics indicate that P. kindae was formed via input from both a southern clade lineage and a northern clade lineage, with contributions estimated to be 52 and 48%. P. papio is inferred to have been produced through 10% introgression from an unidentified ancient northern lineage into a population related to P. anubis. Dates for divergence and admixture events were inferred through CoalHMM, and internal nodes representing those divergence or admixture events are labeled A through K. Our analyses of asymmetric haplotype sharing also inferred admixture from P. cynocephalus into P. anubis approximately 21 generations ago. (B) Reconstruction of baboon demographic history using PSMC methods. A prolonged bottleneck was observed in the lineage ancestral to P. papio beginning ~400 thousand years (ka) ago, while the populations ancestral to P. hamadryas and P. anubis increased between ~280 and ~160 ka ago. After diverging, P. anubis followed an upward trend whereas P. hamadryas declined. At ~400 ka ago, Ne for P. ursinus diverged from estimates for the populations ancestral to P. cynocephalus and P. kindae, and underwent a species-specific prolonged bottleneck. At ~300 ka ago, the Ne reconstructed for both P. cynocephalus and P. kindae increased, peaking ~150 ka ago before experiencing a subsequent decline. PSMC methods are not always reliable for the most recent time periods.
Our results also shed new light on the historical dynamics of hybridization between P. anubis (a northern clade species) and P. cynocephalus (a southern clade species), which has previously been reported in southern Kenya near Amboseli National Park (17). Behavioral observations and microsatellite-based analyses support recent introgression from P. anubis into P. cynocephalus since the 1980s (25, 26). Our analysis of genome-wide haplotype block sharing indicates that a P. anubis individual from the Aberdare region of Kenya, more than 200 km north of Amboseli, is also admixed with P. cynocephalus, carrying ~546 Mb of nuclear DNA derived from P. cynocephalus (fig. S7). If we assume that this resulted from a single admixture event, then it is estimated to have occurred about 21 generations (~220 years) ago. However, other more complex explanations are also possible. The second individual from the P. anubis Aberdare population also carries P. cynocephalus haplotypes, but these shared genomic segments are fewer and shorter and likely result from more ancient introgression. Consistent with other studies (27), our findings suggest that there have been multiple episodes of gene flow involving these two species over a considerable time span and that the effects of past hybridization extend far beyond the current hybrid zone. This complexity may well be representative of the complexity of other known baboon hybrid zones (10, 12, 15, 18, 19, 28).
Motivated by the results from the f-statistics and haplotype sharing, we conducted two additional tests across the Papio diversity panel to examine the hypothesis of ancient admixture using independent methods. Alu insertion polymorphisms are valuable phylogenetic characters because the polarity of specific mutational changes can be unambiguously established for any given genomic segment (fig. S8) (29). The haplotype that carries a novel Alu insertion is derived from the orthologous haplotype lacking the Alu repeat, and reversals are rare. Majority-rule Dollo parsimony analyses of the baboons using novel Alu insertions once again revealed a north-south difference. However, the descendent lineages are poorly resolved, exhibiting apparent homoplasy (fig. S8). In a phylogeny constructed using characters with well-defined polarity, such homoplasy would not be expected unless a radiation of species experienced substantial ILS and/or gene flow among divergent lineages (30).
We next examined differences in the evolutionary history of different segments across the baboon genome. We divided the reference genome into 808 discrete gene-free (putatively neutral) regions. Using BUCKy (31) and the SNV genotypes from the diversity panel, we performed Bayesian concordance analysis (BCA). Individual animals again, as expected, cluster by species. The basal north-south divergence is supported, but the concordance factors (CFs) for relationships within each of those two geographic clades are low (Fig. 3). P. hamadryas is most frequently sister to an anubis-papio clade, but the two other possible topologies [(papio-(ham-anubis)) and (anubis-(ham-papio))] do not appear at equal frequency (fig. S9), as would be expected under ILS. Similarly, P. kindae is most frequently sister to a cynocephalus-ursinus clade, matching the ML results but not f-statistics or PoMo results. Again, the two minor BCA topologies are not found in equal proportions (fig. S9). Together, the Alu insertion and BCA results support the conclusion that reticulation rather than ILS without reticulation has influenced baboon genomic divergence (Table 1).
Table 1. Summary of diverse data types and analytical approaches used to investigate the phylogeny of baboon species.
| Data type | Analytical method | Primary clustering | Northern clade result | Southern clade result |
| SNVs | ML phylogeny | North versus south | hamad-[papio-anubis] | kinda-[cyno-ursinus] |
| SNVs | Bayesian phylogeny | North plus kinda versus two south |
kinda-[hamad-[papio-anubis]] | [cyno-ursinus] |
| SNVs | Polymorphism-aware phylogeny | North versus south | hamad-[papio-anubis] | cyno-[kinda-ursinus] |
| SNVs | f-statistics | North versus south | hamad-[papio-anubis] | cyno-[kinda-ursinus] |
| Alu insertion polymorphisms | Majority-rule Dollo parsimony | North versus south | Unresolved phylogeny with north-south split |
Unresolved phylogeny with north-south split |
| Locus-specific trees for segments without coding sequences |
Concordance factors | North versus south | hamad-[papio-anubis] | kinda-[cyno-ursinus] |
The timing of lineage divergences and admixture events was estimated using a coalescent hidden Markov model (CoalHMM; figs. S10 to S15 and tables S8 and S9) (32, 33). Using an estimated mutation rate of 0.9 × 10-8 per base pair per generation and a generation time of 11 years [see Materials and Methods and (11, 34)], we obtain the results presented in Fig. 4A. To reconstruct demographic history, we generated pairwise sequential Markovian coalescent (PSMC) plots (35), assuming the generation time and mutation rate cited above (Fig. 4B). With the exception of P. papio, which has a truncated plot, the remaining five species are very similar in effective population size (Ne) from 4 Ma ago up until ~1.4 Ma ago, supporting the conclusion that all baboon species share the same demographic history (that is, were effectively one lineage) before ~1.4 Ma ago. All Ne plots show an upward trend after ~1.5 Ma ago, but the species-specific increases occur at different rates, possibly corresponding to population growth and dispersion once ecological conditions allowed demographic expansion (14). Given the paleontological evidence for a southern origin of this genus (36), we speculate that the more pronounced apparent decline in Ne for northern clade species relative to the southern lineages ~700,000 to 800,000 years ago may reflect dispersal-related bottlenecks as the geographic range of baboons was extended to the north. Similarly, the CoalHMM suggests that the north-south admixture that produced extant P. kindae occurred about 100,000 years ago, and the PSMC results suggest an increase in Ne for P. kindae about this time.
To examine potential functional consequences of baboon admixture, we investigated 2201 suitable genic regions (local genomic segments that contain one annotated protein-coding gene each and exhibit sufficient phylogenetic signal to support one particular phylogenetic tree over all alternative trees). We identified individual loci that exhibit phylogenetic relationships (gene trees) concordant or discordant with the consensus species-level phylogeny that separates the three northern species from the three southern species. Cluster 1 contains 1143 genic regions with phylogenies closely matching that result (fig. S16). Cluster 2 consists of 629 genic regions for which P. cynocephalus carries haplotypes that are not closely related to other southern clade haplotypes (fig. S17). The genes in these regions are enriched for the gene ontology (GO) terms “learning and memory” (P = 0.012), “cognition” (P = 0.012), “head development” (P = 0.014), and “brain development” (P = 0.017), as well as several GO categories related to reproduction (see table S10). Cluster 3 includes 429 genic regions displaying phylogenetic relationships among southern clade species consistent with the phylogeny in Fig. 3a. However, the cluster 3 haplotypes from the northern clade P. anubis are more closely related to haplotypes from the southern clade, while haplotypes in northern clade P. papio generally form the sister to all other baboon haplotypes (fig. S18). The genes found in cluster 3 regions are enriched for GO terms related to the ontogenetic development of several organ systems (kidney, heart, circulatory, and endocrine systems, all significantly enriched with P < 0.03) (table S10). We note that the two species exhibiting the clearest gene tree discrepancies relative to the species-level phylogeny (i.e., species carrying haplotypes that apparently crossed species boundaries) are P. anubis and P. cynocephalus, a northern clade and a southern clade species, respectively, that actively hybridize today in southern Kenya (17) and exhibit evidence of nuclear DNA swamping (12).
DISCUSSION
Like our own genus Homo, the ancestral stock of Papio baboons began diverging into multiple lineages within sub–Saharan Africa by about 1.5 Ma ago. Like early Homo, baboon species today differ in body size and morphology (9, 11, 13). We report here that multiple baboon lineages have experienced episodes of admixture, some involving genetic exchange among lineages that persist today while other episodes involved extinct lineages. In contrast to Homo, Papio today includes six surviving differentiated lineages (phylogenetic species), providing a unique context for the investigation of genetic and phenotypic consequences of both ancient and modern interspecies hybridization.
No one simple dichotomously branching tree accurately reflects all aspects of genomic differentiation among extant baboon species. However, our provisional scenario for baboon genome evolution, presented in Fig. 4A, does establish the context for further explorations of baboon biology. We observed a markedly increased rate of recent Alu insertion mobilization in the baboons relative to human and other hominoids (Fig. 2). Previous studies suggest that hybridization between divergent lineages can generate increases in the rate of novel insertions of repetitive elements (37, 38). Another topic of broad interest is the origin of reproductive isolation among incipient species (1). One expectation for the genus Papio is that, given the timing of the radiation and the degree of morphological and behavioral differentiation among species, incipient barriers to gene flow may be evident between some pairs of species. Studies of the present-day hybrid zone between northern clade P. anubis and southern clade P. cynocephalus find no readily apparent barriers to reproduction between these species (17, 26). However, studies of captive P. anubis × P. cynocephalus hybrids document significantly elevated frequencies of craniodental anomalies in hybrids, especially hybrid males, indicating some degree of genetic incompatibility (39). Field studies of the hybrid zone between P. ursinus and P. kindae describe a deficit of hybrid individuals carrying Y chromosomes from P. ursinus and mtDNA from P. kindae compared to the converse (18). This suggests that when hybridization began between these two forms, some type of barrier (premating or postmating) reduced the frequency or fertility of matings by male P. ursinus with female P. kindae, while the converse mating type was more successful (18). Last, P. anubis and P. hamadryas differ substantially in their social organization and social structure (11, 28, 40). Among anubis baboons, both males and females are polygamous. Hamadryas societies are multilevel, with “harem”-like, one-male breeding units (OMUs) as basal social entities. In these OMUs, the single adult male defends exclusive access to one or more adult females. Other differences in sex-specific dispersal and social relationships are also observed (11). Despite the dramatic differences in social systems, these species hybridize in the wild (28). Hybrid males can achieve substantial reproductive success, at least in groups consisting mainly of hybrids (19). There is no clear evidence for a barrier to gene flow between the species, although the geographic distribution of phenotypically recognizable hybrids is narrow.
The demonstration that hybridization among modern humans, Neanderthals, and Denisovans had enduring effects on the modern human gene pool has raised questions about the demographic processes, as well as the genomic and phenotypic consequences, of admixture among primate lineages separated on the order of hundreds of thousands of years (20, 21). Baboons have such a history but can still be studied in present-day hybrid zones and therefore constitute an important context for future research. Potential areas of study include the effects of genetic variation on neurotransmitter function and its impact on species-level differences in social relationships and social behavior (41, 42). The presence of hybrid zones between species pairs separated by different genetic distances (e.g., the distant P. anubis/P. cynocephalus versus the much closer P. anubis/P. hamadryas) makes it feasible to investigate the effects of increasing genetic differentiation. Access to well-characterized captive research colonies of baboons provides further opportunity for innovative studies concerning developmental, metabolic, and neurobiological consequences of interbreeding among divergent lineages (7, 39, 43, 44).
MATERIALS AND METHODS
Sequencing of the reference genome sample
The sequence data for the olive baboon (P. anubis) whole-genome assembly were generated through different methods over time, as the dominant sequencing technologies evolved. The earliest data were generated using the Sanger technology, followed by Roche 454 FLX data. Later, Illumina short-read data (both paired-end and mate-pair reads) were generated using the Genome Analyzer IIx first and then the HiSeq 2000 platforms later. Last, Pacific Biosciences RSII data were also produced. All the Sanger, Roche 454, and Illumina read data were generated from a single female olive baboon of Kenyan ancestry [animal ID 1X1155, National Center for Biotechnology Information (NCBI) BioSample SAMN02981400; Southwest National Primate Research Center, San Antonio, TX]. The PacBio read data were generated from a single olive baboon (animal ID 20111; assigned three BioSample numbers: SAMN03165174, SAMN03165175, and SAMN03165176) from the same research colony. The depth of genome coverage used in the assembly was as follows: Sanger 2.5×, Roche 454 4.5×, Illumina 85×, and PacBio RSII 12×. All reads have been deposited in NCBI under BioProject PRJNA54005.
Genome assembly
The genome assembly processes used are shown in fig. S1. The initial olive baboon genome assembly, Pham_1.0, used only Sanger and Roche 454 data. This assembly is no longer available at NCBI but can still be accessed at the University of California, Santa Cruz (UCSC). To avoid confusion, we emphasize that although listed under Baboon (hamadryas) and named papHam1 on the USCS genome browser and at the Ensembl Pre! Site (http://pre.ensembl.org/Papio_hamadryas/Info/Index), this first version of the assembly was derived not from a hamadryas baboon but from the female olive baboon identified above. The analyses reported here only used the later improved assemblies, Panu_2.0 and Panu_3.0.
Panu_2.0 (named Panu_2.0 in NCBI and papAnu2.0 in Ensembl and UCSC; GenBank accession GCA_000264685.1) was produced from the available Sanger, Roche 454, and Illumina reads, derived from the same female olive baboon used for Pham_1.0. Assembly analyses used the GAC (Genomic Analysis Cluster) compute facilities at the Baylor College of Medicine Human Genome Sequencing Center (HGSC). Sanger and Roche 454 reads were first assembled using CABOG version 6.1 with parameter settings of utgErrorRate = 0.02, ovlErrorRate = 0.07, cnsErrorRate = 0.07, cgwErrorRate = 0.12, and unitigger = bog. Two sets of 100–base pair (bp) Illumina read data, 2 billion reads from a 240-bp insert paired-end library, and 500 million reads from a 2.5-kb insert mate-pair library were mapped to the CABOG assembly using BWA with default parameters. The scaffolds of this initial CABOG-generated assembly were improved on the basis of the read mapping locations using Atlas-Link version 1.0 (https://www.hgsc.bcm.edu/software), with the minimum required links (min_link) set at four for the 240-bp library and three for the 2.5-kb library. The Atlas-GapFill version 1.0 process (https://www.hgsc.bcm.edu/software) was then performed to fill gaps between contigs within scaffolds by extracting local read pairs and aligning the local assemblies of these pairs to the gaps.
The assembled contigs and scaffolds of Panu_2.0 were placed on baboon chromosomes by mapping to the rhesus macaque (Macaca mulatta) genome assembly (GCF_000002255.3, mmul_051212, rhemac2) using Mummer3 (parameters = nucmer -l 12 -c 65 -g 1000 -b 1000; delta-filter -1 -l 500; show-coords -cl -L 500). It should be noted that chromosome organization is largely conserved between rhesus macaque and baboon (45). A baboon scaffold was split when it did not have continuous alignment on the macaque genome and if the potential breakpoint was validated by low clone coverage in the baboon data (low coverage defined as clone coverage from the 2.5-kb Illumina library of <5×). A set of 323 scaffolds (a total of 217 Mb) were identified this way and therefore split. The N50 of the contigs in the Panu_2.0 assembly is 40.3 kb, and the N50 of the scaffolds is 529 kb. The total length of the Panu_2.0 assembly is 2.95 Gb with 55.1 Mb of gaps. Because the scaffolds for Panu_2.0 (and Panu_3.0) have been mapped onto baboon chromosomes, this genome assembly is presented in public databases (NCBI, UCSC, and Ensembl genome browsers) as chromosome-associated sequences rather than as sets of independent scaffolds and superscaffolds.
Last, we improved the Panu_2.0 assembly through two additional methods. First, a small number of differences between the baboon and rhesus macaque genomes were identified using fluorescence in situ hybridization (FISH) mapping of probes containing human BAC sequences. These scaffolds were refined to be consistent with the FISH results from the baboon genome. Last, a total of 12× whole-genome coverage was produced on the PacBio RSII platform, with half of the reads >7 kb. These data were mapped to the Panu_2.0 assembly and two-thirds (67%) of the 118,928 gaps within scaffolds were closed using PBJelly software (46). The base quality of the assembly was polished using the Pilon program (47) and the available Illumina data.
This final assembly (Panu_3.0) has a contig N50 of 149.8 kb and, due to the mapping of these scaffolds to chromosomes, it has near whole chromosome length superscaffolds. The gap filling with PBJelly added only 10.98 Mb to the assembly (0.37% of the Panu_2.0 assembly length), but closed a large number of gaps, reducing the number of contigs from 198,931 to 118,251. The Panu_3.0 assembly was tested against available baboon EST (Expressed Sequence Tag) sequence datasets to quantify extent of coverage (i.e., completeness). Of the 144,708 Sanger EST sequences available at the time of testing, 99.98% were successfully mapped to the assembly. Among the total ESTs, 98.77% mapped with >90% of their length and 97.48% mapped at >95% of length. Seven finished BAC clones were mapped to the Panu_2.0 assembly. The genomic coverage in the BACs was high, with 98 to 100% of the BAC sequence in the assembly. The assembled contigs and scaffolds were aligned linearly to the finished BACs, suggesting that misassemblies are rare. Within Panu_3.0, only 3.2% of the sequence falls in unscaffolded contigs.
Sequence variation across the diversity panel
DNA was obtained from 16 animals representing all six species of Papio baboons and the gelada, T. gelada (table S3). Of the 16 individuals, 9 were wild animals sampled in the field, and the remaining samples were obtained from captive colonies. The species identity of each sampled animal was determined from its external phenotype. The integrity of subsequent sequence data files was confirmed by comparing the mtDNA sequences obtained through whole-genome analysis to other mtDNA sequences from baboons of known species and geographic location (12). All such species assignments were confirmed and validated. All these diversity samples were sequenced to an average read depth of 30.7× using the Illumina HiSeq 2000 sequencing platform (100-bp paired-end reads), with the one exception that the T. gelada sample was sequenced on the Illumina HiSeq X platform.
We used BWA-MEM version 0.7.12-r1039 (https://arxiv.org/abs/1303.3997) to align the Illumina reads to the baboon reference assembly Panu3.0/papAnu3 and generate BAM (Binary Alignment Map) files (fig S2). Picard MarkDuplicates version 1.105 (http://broadinstitute.github.io/picard/) was used to identify and mark duplicate reads. Variants were called using GATK version 3.3-0 following best practices for that version (https://software.broadinstitute.org/gatk/best-practices/). In brief, indels were realigned using IndelRealigner. HaplotypeCaller was used to generate gVCFs for each sample. Joint genotype calling was performed on all samples using GenotypeGVCFs to generate a VCF file. GATK hard filters (SNPs: “QD < 2.0 || FS > 60.0 || MQ < 40.0 || MQRankSum < −12.5 || ReadPosRankSum < −8.0”; Indels: “QD < 2.0 || FS > 200.0 || ReadPosRankSum < −20.0”) (https://software.broadinstitute.org/gatk/documentation/article?id=2806) were applied, and all variant calls that failed the filters were removed.
To perform functional annotations through WGSA (Whole Genome Sequence Annotator) (48), the SNVs identified in the baboon diversity panel were transferred to the human genome (hg19) using liftOver and treated as human SNVs. All annotation resources available for version 0.5 were used for this analysis, including five functional prediction scores, eight conservation scores, allele frequencies from four large-scale resequencing studies, and variants in four disease-related databases, among others.
Alternative phylogenetic analyses
Given the clear discordances between mitochondrion-based and phenotype-based phylogenies (12, 15), we performed an extensive series of phylogenetic analyses using different data types and analytical approaches. Our goal was to develop robust conclusions regarding population history that are supported by multiple datasets and analyses.
Phylogenetic analysis of concatenated whole-genome SNV data
The dataset for this analysis consisted of SNV calls for the 15 baboons from the diversity panel (table S4), plus one gelada (T. gelada). From the variant call file (vcf) produced by GATK, only SNV positions surviving filtering steps conducted with vcffilter from vcflib (https://github.com/ekg/vcflib; settings: -f "QUAL > 20 & DP > 10 & MQ > 30 & QD > 20") were used for further analysis (24,588,548 SNVs). SNVs were extracted from the filtered vcf file with bcftools from SAMtools 1.2 (settings: bcftools query -f ’%CHROM\t%POS\t%REF\t%ALT [\t%SAMPLE =%GT]\n’). The resulting table was converted into individual FASTA sequences using a custom Python script. Individual cases where a baboon exhibited more than one different nonreference allele at the same site were recorded as ambiguous. Merging all FASTA sequences into a single file provided a multiple sequence alignment of all individuals and all concatenated SNVs. Positions in the alignment where no information was given for at least one species were removed. A total of 22,433,604 SNVs remained for analysis. Model selection using the Bayesian information criterion in IQ-TREE 1.3.13 (49) revealed the TVM+ASC+G model as the best-fit model for this dataset. Phylogenetic trees were reconstructed with ML and Bayesian approaches using IQ-TREE and MrBayes 3.2.6 (50), respectively. IQ-TREE settings: TVM+ASC+G model, 1000 ultrafast bootstraps; MrBayes settings: TVM+G, 100,000 generations and 10% burnin.
Polymorphism-aware phylogenetic model analysis of whole-genome data
To estimate species-level phylogeny while allowing for current and possible ancient polymorphism, we applied the PoMo model (23) implemented in IQ-TREE (49) to the baboon diversity Panu_2.0 SNV data together with fourfold degenerate sites of the orthologous gene set. Briefly, the PoMo model represents the evolution of an individual nucleotide site within a given fixed species-level phylogeny as a continuous time Markov chain along that phylogeny. Rather than considering only four states (four alternative nucleotides) for a given genomic position, PoMo allows for polymorphism within species by expanding the state space in the Markov chain to include heterozygous nucleotide compositions, assuming two nucleotides per site, in addition to the traditional four nucleotide states. Mutation (e.g., using the HKY model) introduces new nucleotides. The Moran model was used to describe genetic drift or changes in allele frequencies over time. PoMo generates a single species tree, but does allow for ILS. Additional details are available in (23, 24).
Simulation study comparing methods
We analyzed the robustness of the PoMo results to admixture between differentiating lineages using the baboon phylogeny as the assumed context. We defined the input phylogeny as that obtained by modeling potential admixture among the six baboon species through f-statistics (see below). Total branch lengths for each lineage were set as inferred from baboon data, and Watterson’s θ within species was set to 0.0025. We tested the ability of PoMo to accurately reconstruct the phylogeny for P. kindae by varying the proportion of admixture into the P. kindae lineage from a northern clade species from 0 (no admixture) to 80%. We simulated 1000 genes (1000 bp per gene) on five chromosomes and created 1000 gene trees using MSMS (51). We next concatenated the sequence data for five chromosomes and all gene trees in each species. We then used both PoMo and the HKY model to generate phylogenies and compared their ability to reconstruct the correct species-level phylogeny.
Analyses of admixture through f-statistics
Admixture graphs (52) model the ancestry of a set of samples in the form of a directed acyclic graph where edges capture drift along ancestral lineages, leaves represent the samples, and inner nodes represent either most recent common ancestral populations or admixture events where a new population is created as a mixture of two other populations. Hence, these graphs can capture more complex histories than simple tree phylogenies, but only simple forms of gene flow. Admixture graphs model all gene flow as admixture events and cannot easily model periods of continuous gene flow. Admixture graphs are parameterized by edge lengths (the amount of drift that occurred on a given ancestral lineage) and admixture proportions (how much of an ancestral admixed population was derived from one donor population rather than another). Properties of an admixture graph can be captured by so-called f-statistics (52), and these can be estimated from genomic data. This makes it possible to compare the statistics predicted by a graph, F, with statistics estimated from data, f. Graph parameters are estimated by minimizing the distance between F and f. We have implemented an R package (https://github.com/mailund/admixture_graph) for inferring graphs and graph parameters from vectors of observed statistics and applied this approach to the baboon data.
We used qpDstats from the ADMIXTOOLS package to compute estimates of f with corresponding Z values for all quartets (W, X, Y, Z), where W was rhesus macaque and X, Y, and Z were all combinations of baboon samples from three different species. The bulk of our analyses of f-statistics used sequence diversity and SNV data based on mapping reads to the Panu_2.0 genome assembly. Once the improved Panu_3.0 assembly was complete, which closed thousands of gaps in scaffolds but increased the total sequence length of the assembly by only 0.37%, we retested the likelihoods of the inferred phylogenetic relationships among lineages and the inferred history of admixture using SNVs called based on Panu_3.0. All conclusions regarding species phylogeny and admixture events that were based on Panu_2.0 data were confirmed when tested using SNV and diversity information based on read mapping against Panu_3.0. This is likely because the upgrade from Panu_2.0 to Panu_3.0 added little to the total sequence length.
Grouping of samples
When estimating f-statistics, we can either pool samples from the same population together or compute at the level of individual samples. Pooling samples would potentially give a better estimate of population allele frequencies, but can mask within-population differences that might be informative about recent gene flow. We therefore chose to estimate the statistics at the individual sample level. For this analysis, we estimated the f4-statistics for all triplets of baboon samples combined with rhesus macaque. These were computed using the qpDstats tool from the ADMIXTOOLS package (https://github.com/DReichLab/AdmixTools). Inferring admixture graphs with each sample as a leaf is computationally intractable. The number of possible graphs grows superexponentially with the number of leaves, and our brute-force approach to exploring the graph space only scales to a small number of leaves. We therefore chose to keep the graphs at the species level. This way, samples from the same species are expected to have the same relationship to all other species. Parameters are estimated from the combined set of individual samples within each species.
We inspected the estimated f-statistics to ensure that the species grouping matched with similar vectors of statistics. In general, samples from the same species had very similar f-statistics compared to all other samples. The single exception was P. anubis sample 30877 from the Aberdare region of Kenya that has a different profile from the other olive baboons. This sample shows evidence of a recent admixture with P. cynocephalus. To focus on the ancient admixture events only, we removed this sample from the admixture graph analysis; thus, the species “anubis” in the following refers only to the remaining P. anubis samples.
Fitting admixture graphs
A given graph topology specifies a polynomial of edge lengths and admixture proportions as the expected value for each f-statistic. These polynomials are linear with respect to the edge lengths only, and so we stored them as rows in a matrix of polynomials of admixture proportions only. To measure the fit between a graph and the observed statistics, we defined a cost function: a weighted sum of squared errors between graph predictions F and statistics f. The weights are reciprocals of the SDs of the statistics f, given by ADMIXTOOLS (as the Z values divided by f). We fitted the parameters of the graph using a mix of analytic and numerical optimization. After fixing the admixture proportions, the polynomial equations of predictions F are linear and thus solvable analytically. To optimize the admixture proportions, we used the Nelder-Mead package (https://cran.r-project.org/web/packages/neldermead/index.html).
Exploring the space of admixture graph topologies
We explored the space of graph topologies with a brute-force approach, bounding the number of allowed admixture events. Because of the large number of possible graphs, we could not exhaustively explore all the topologies including all the species (six baboon species and the rhesus macaque outgroup). Instead, we used various heuristics.
We could explore all the trees with seven leaves. For admixture graphs with a single admixture event, we could only explore all the graphs with six leaves; hence, we built all the graphs with one baboon species missing and then reinserted the missing species into the graph (such that inserting it cannot add a second admixture event). For additional admixture events, we took a greedy approach and explored all the graphs reachable by adding a new admixture event to the best graphs with one less event (although we have no guarantee that the optimal graph with n events is necessarily an extension in this way of the optimal graph with n - 1 events).
Estimating graph parameters and testing models
We developed a Metropolis-Hastings MCMC (Markov Chain Monte Carlo) to sample the posterior of graph parameters given observed f-statistics. By sampling the posterior of graph parameters, we obtained estimates of admixture proportions and the uncertainty in these estimates. In addition, we can use the posterior samples in an importance sampler to obtain the likelihood of the observed statistics given a graph topology by integrating over the parameters of the graph.
Sampling graph parameters
Let D denote the observed data (the estimated f-statistics), T a given graph topology, and θ the graph parameters of graph T (edge lengths and admixture proportions). The purpose of the MCMC is to sample over the posterior distribution of graph parameters given the observed data and the graph topology, i.e., sample from p(θ|D, T). Since we can compute the likelihood of a parameter point, p(D|θ, T), up to a normalization factor, we can use the Metropolis-Hastings algorithm to construct an MCMC that samples over the posterior distribution. We transformed the parameter space to make the proposal distribution symmetric (thus, the acceptance probability for the Metropolis-Hastings step is simply the posterior ratio) and to ensure that the parameters are all legal for their interpretation in the graph framework. Edge lengths, which must be positive, were log-transformed, and admixture proportions, which must fall within the unit interval, were transformed with the inverse normal cumulative distribution function. After transformation, we used an adaptive algorithm to construct the proposal distribution based on correlations in the previously sampled variables. For each graph, we ran three independent chains with 10,000 steps and we checked convergence by comparing the distributions from the independent chains. Since edge lengths are measured in terms of drift, they do not have a simple interpretation as time parameters. We therefore considered them as nuisance parameters in the MCMC analysis. We used them to test convergence of the Markov chains, but in the results presented here, we focused on estimates of admixture proportions.
Calculating graph topology likelihoods
To compare two graph topologies, we can compute the topology likelihood p(D|T) for a given topology T. Using this likelihood estimate, we can compare two topologies using the Bayes factor , which captures the relative support the data provides for one topology over another: If, e.g., , we would consider T1 the more likely topology unless a priori topology T2 was at least 10 times more likely than T1.
Given data D and topology T, the likelihood of T is computed by integrating over all the graph parameters for the topology p(D|T) = ∫p(D, θ|T) dθ = ∫p(D|θ, T) p(θ|T) dθ. This integral can be approximated by sampling from the prior distribution of graph parameters and computing the mean likelihood, , where θi ~ p(θ|T), but this estimator has a large variance since most parameters drawn from the prior distribution have a very low likelihood. Instead, we used the samples from the posterior distribution to estimate the likelihood using an importance sampler.
Now, because , we can estimate , where θi ~ p(θ|D, T).
Identification of admixture through asymmetric allele sharing
To further investigate possible admixture among baboon species, we identified asymmetries in informative site patterns within the 15 baboon diversity samples. This approach detects tracts of recent introgression between the different baboon lineages. Nucleotide sites where a derived variant is shared by two species to the exclusion of another are informative of the underlying gene tree. The two informative site patterns, which group together species that are not the most closely related species, arise from either recurrent mutations or ILS. These patterns are expected to show equal frequencies if the mutation rate is constant across the tree and if there has been no asymmetric admixture between a non-sister species and the two sister species tested. A strong and consistent asymmetry in the frequency of the two site patterns supporting alternative gene trees may thus result from asymmetric admixture. To identify tracts of recent admixture, we counted informative site patterns in 1-Mb windows along the alignments of individual genomes from all species trios of baboons. We computed the 0.99 quantile of counts of the two alternative site patterns across 1-Mb windows (the two site patterns that support gene trees with a topology different from the species tree). We then call admixture tracts as consecutive 1-Mb bins where the count of one alternative site pattern was above the 0.99 quantile of the other alternative site pattern.
Polymorphic Alu detection and characterization
A computational analysis was performed to identify full-length lineage specific AluY sequences in the olive baboon (Panu_3.0), rhesus macaque (Mmul_8.0.1), African green monkey (chlSab2), orangutan (P_pygmaeus 2.0.2), chimpanzee (Pan_tro3), and human (GRCH38/hg38) reference genomes, as previously described by Steely et al. (53).
Initial screen for polymorphic Alu insertions
Two methods were used to identify 494 informative Alu insertions, a subset of young elements currently polymorphic within the genus Papio. The first method used BLAT to align AluY sequences obtained from the P. anubis assembly (Panu_2.0) against M. mulatta (RheMac2) and H. sapiens (GRCh38/hg38). Insertions present in P. anubis yet absent from the other two assemblies were subjected to polymerase chain reaction (PCR) analysis to determine whether they were polymorphic across baboon species. A total of 187 loci were retained after this analysis.
Computational analysis of diversity samples
In our second method, whole-genome sequence data generated from six of the diversity panel baboons (16098, 28547, 28755, 34472, 34474, and 97124) were aligned to P. anubis genome (Panu_2.0), as previously described by Jordan et al. (54).
Primer design
Oligonucleotide primers for locus-specific PCR were designed as reported in (53).
Sanger sequencing
Following PCR analysis of candidate Alu insertion polymorphisms on the DNA panel of baboons, a small number of loci required Sanger sequencing for clarification. The first category included eight loci ascertained from either a yellow or kinda baboon genome, computationally absent from the reference genome Panu_2.0, but for which PCR results indicated a filled site, or Alu present amplicon size, for reference DNA sample 27861. These were classified as possible “false-negative” events (supposed to be absent but were not), and PCR fragments were sequenced from 27861 and the ascertained individual to confirm the existence of a shared insertion event. The second category included nine loci in which the Alu present PCR product was either larger or smaller than the predicted filled size amplicon, or displayed both size bands, in one or more Papio species, but not all. All applicable PCR fragments were sequenced to confirm that the ascertained Alu element of interest was present and to determine what the extra sequence contained. Four PCR fragments per locus were gel-purified using a Wizard SV gel purification kit (Promega Corporation, Madison, WI, USA, catalog A9282) according to the manufacturer’s instruction. Cycle sequencing was performed, and resulting products were cleaned by standard ethanol precipitation. Capillary electrophoresis was performed on an ABI 3130xl Genetic Analyzer (Applied Biosystems Inc., Foster City, CA) and evaluated using ABI software Sequence Scanner v1.0. Sequence alignment figures were constructed in BioEdit, and a consensus sequence for each locus was determined from the multiple forward and reverse Sanger sequences obtained for each locus (53).
Phylogenetic analysis
A phylogenetic tree was created using the larger dataset of polymorphic Alu elements as characters. If an Alu insertion was fixed present (homozygous) at a particular locus, it was coded as “1”. If the insertion was fixed absent at a particular locus, it was coded as “0”. Insertions that were found to be heterozygous in an individual were coded as “1,0”. If a locus could not be resolved through PCR, then it was coded as a “?”. Mesquite 3.04 (53, 54) was used to create a Dollo parsimony matrix with all characters set to the character type Dollo.up. A heuristic search was completed using PAUP* 4.0a147 (54) with a total of 10,000 bootstrap replicates. The majority-rule tree does not include bootstrap values below 50% for any branches, but does include values for consistency index, retention index, and homoplasy index. A neighbor-joining phylogenetic tree was also created using the same dataset to illustrate the overall topology. The trees were produced in PAUP* and visualized using FigTree 1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/).
Reconstructing phylogeny and admixture from gene tree analyses
Locus-specific phylogeny from loci not containing protein-coding genes
To infer baboon phylogeny from a series of independent putatively neutral loci (gene trees), we identified and excluded bases with any of the following characteristics: genic regions, based on refGene annotations accessed via the refGene table of UCSC’s Genome Browser; bases with coverage <7× for any individual in the diversity panel; bases with any missing genotype; bases with read depth above the 95th percentile; CpG sites; bases within 3 bp of an indel; repetitive DNA as designated by RepeatMasker (using the open-3-3-0 version of RepeatMasker with sensitive setting RepBase library release 20110920) and Tandem Repeats Finder (period of 12 or less); and bases within 100 bp of a phastCons element. For the bases that passed filtration, we concatenated nearby loci separated by gaps of less than 1 kb using BEDtools. We then retained only loci of size 1 to 100 kb for further analyses to maximize information content while still reducing the chance of unappreciated recombination.
For each locus, we extracted the sequence for all baboon diversity panel individuals, replacing heterozygous sites with their corresponding IUPAC (International Union of Pure and Applied Chemistry) codes. To get an outgroup sequence for each locus, we compared the baboon reference genome (Panu_2.0) to the rhesus macaque reference genome sequence (rheMac2) using megablast, retaining hits with e values less than 1 × 10-100 and at least 95% identity. Only loci with a single rhesus match were retained. All analyses used custom scripts and BEDtools, SAMtools, and VCFtools. We aligned the sequences at each locus with Muscle using default parameters.
We inferred a gene tree for each locus using MrBayes 3.2.1 (50), setting the outgroup to the rhesus sequence. For each alignment, we used a GTR+G model of molecular evolution, and we ran MrBayes twice for 1,000,000 generations, sampling every 100. We assessed convergence by checking statistics in MrBayeslog (LnL, PSRF, and average SD of split frequencies <0.01) and by using Tracer v.1.5 to estimate the effective sample size as >200 and to compare the performance of the independent analyses. After checking for convergence, we summarized the posterior distribution of trees after removing the first 25% of generations.
Filtration for phylogenetic information content and BCA
We used the program mbsum from BUCKy v 1.4.2 to summarize the posterior tree output from MrBayes for each locus (31, 55). As is expected with a recent radiation, many loci had limited phylogenetic information content, decreasing the signal-to-noise ratio and increasing concordance analysis runtime. To quickly remove loci with limited signal, we filtered loci based on the frequency of the highest supported tree in the MrBayes tally of tree topologies. A locus with no information is expected to output a flat distribution of random topologies, each appearing once. In contrast, a locus with strong signal will have the same topology occur multiple times. We removed loci if the most frequently supported topology occurred in fewer than 10% of trials. We then ran the main BUCKy program with default parameters to infer CF summary statistics that describe the proportion of trees that contain a particular clade.
Genic tree analysis of sequences containing annotated protein-coding genes
We also analyzed 3267 chromosomal segments that each contain one annotated protein-coding gene. Segments were selected based on refGene tables within the UCSC Genome Browser. The inference of local tree topologies was performed as for the loci discussed above that do not contain protein-coding genes. We began with 3267 genic segments, but following filtering for phylogenetic signal, length, and other criteria, we obtained final results for 2201. For each of these genic segments, we computed pairwise Euclidean distances using the Kendall and Colijn metric (56) and then performed PCA (Principal Component Analysis) on this distance matrix to group trees into six clusters based on tree similarity. We chose to further investigate the first three clusters. We performed a GO overrepresentation test using as a reference list the genes for all trees that survived filtering. Results of this for GO biological processes and molecular functions are reported in table S10.
CoalHMM trees
CoalHMMs (32, 33, 57) exploit the Markov approximation to the sequential coalescent process (58). These models can be constructed to capture various demographic scenarios in the ancestry of a set of sampled chromosomes. We have constructed a new model to infer parameters relevant for chromosomes from an admixed population and one or two populations related to the donor populations of the admixture event. Denote the populations in the model A, B, and C, where C is descended from the admixed population and A and B are related to the two donor populations. Parameters of the model include divergence times between all pairs of populations and the admixture proportions for the admixed population.
Kinda admixture
We first consider the most probable graph with a single admixture event. As an example, we consider P. kindae as the admixed population, C. We have two choices for population A, P. cynocephalus and P. ursinus, and two choices for population B, P. hamadryas and P. papio. With these choices, since each of the populations considered for A and B has two samples, we could choose independent chromosomes to compare for replication of results. With these species, we can estimate the time parameters for specific evolutionary events: (i) the north-south clade split, (ii) the split between hamadryas/papio and the species that hybridized to become P. kindae, (iii) the cynocephalus/ursinus split, (iv) the split between ursinus and the other species that hybridized to become P. kindae, and (v) the time of the admixture that formed P. kindae. Not all of these time points can be estimated by all triplets of species, but all can be estimated from at least two sets of species triplets, giving us four independent estimates.
Simulation test of goodness of fit and debiasing estimates
To examine which parameters are likely to be biased, and by how much, we simulated data with parameters in a grid of time points around the estimated points and estimated the parameters from these simulated data. Figure S12 shows the results with the estimated time points and the admixture proportions together with simulated data, where the simulated values are shown as black points and the corresponding estimated parameters are shown as red error bars (these error bars are wider since we used smaller datasets for the simulated data for computational reasons).
Dating divergence and admixture events within the CoalHMM
The CoalHMM produces a phylogenetic tree with relative dates for nodes within the tree scaled to nucleotide substitutions. We first assumed that the initial divergence of northern and southern clades of baboons occurred 2.0 Ma ago, as suggested by analyses of mtDNA divergences (12). The ratios of inferred lineage-specific nucleotide substitutions were then used to calculate absolute dates for nodes in the CoalHMM tree. As an alternative approach to dating, we used the estimated baboon mutation rate of 0.9 × 10-8 per base pair per generation (see section S2). With a generation time of 11 years, the absolute date for each node within the CoalHMM tree was calculated using the branch lengths determined through coalescent modeling, assuming the indicated mutation rate and generation time.
Supplementary Material
Acknowledgments
We acknowledge the contributions of the sequence production staff of the Human Genome Sequencing Center: K. A. Abraham, H. A. Akbar, S. A. Ali, U. A. Anosike, P. A. Aqrawi, F. A. Arias, T. A. Attaway, R. A. Awwad, C. B. Babu, D. B. Bandaranaike, P. B. Battles, A. B. Bell, B. B. Beltran, D. B. Berhane-Mersha, C. B. Bess, C. B. Bickham, T. B. Bolden, K. Cardenas, K. C. Carter, M. Cavazos, A. Chandrabose, S. Chao, D. C. Chau, A. C. Chavez, R. Chu, K. C. Clerc-Blankenburg, A. Cockrell, M. C. Coyle, A. Cree, M. D. Dao, M. L. Davila, L. D. Davy-Carroll, S. D. Denson, S. Dugan, V. Ebong, S. Elkadiri, S. F. Fernandez, P. F. Fernando, N. Flagg, L. F. Forbes, G. Fowler, C. F. Francis, L. F. Francisco, Q. F. Fu, R. Gabisi, R. G. Garcia, T. Garner, T. G. Garrett, S. G. Gross, S. G. Gubbala, K. Hawkins, B. Hernandez, K. H. Hirani, M. H. Hogues, B. H. Hollins, L. J. Jackson, M. J. Javaid, J. C. Jayaseelan, A. J. Johnson, B. J. Johnson, J. J. Jones, V. J. Joshi, D. Kalra, J. K. Kalu, N. K. Khan, L. Kisamo, L. L. Lago, Y. Lai, F. L. Lara, T.-K. Le, F. L. Legall-Iii, S. L. Lemon, L. Lewis, J. L. Liu, Y.-S. Liu, D. L. Liyanage, P. London, J. L. Lopez, L. L. Lorensuhewa, E. Martinez, R. M. Mata, T. M. Mathew, T. Matskevitch, C. M. Mercado, I. M. Mercado, K. M. Morales, M. M. Morgan, M. M. Munidasa, L. N. Nazareth, I. N. Newsham, D. N. Ngo, L. N. Nguyen, P. Nguyen, T. N. Nguyen, N. N. Nguyen, M. Nwaokelemeh, M. O. Obregon, G. O. Okwuonu, F. O. Ongeri, C. O. Onwere, I. O. Osifeso, A. P. Parra, S. P. Patil, A. P. Perez, Y. P. Perez, C. P. Pham, E. Primus, L.-L. Pu, M. P. Puazo, J. Q. Quiroz, S. Richards, J. R. Rouhana, M. R. Ruiz, S.-J. Ruiz, N. S. Saada, J. S. Santibanez, M. S. Scheel, S. Scherer, B. S. Schneider, D. S. Simmons, I. S. Sisson, E. S. Skinner, N. Tabassum, L.-Y. Tang, A. Taylor, R. T. Thornton, J. T. Tisius, G. T. Toledanes, Z. T. Trejos, K. U. Usmani, R. V. Varghese, S. V. Vattathil, V. V. Vee, D. W. Walker, G. W. Weissenberger, C. W. White, K. Wilczek-Boney, A. W. Williams, K. Wilson, I. Woghiren, J. W. Woodworth, R. W. Wright, Y.-Q. Wu, Y. Xin, Y. Zhang, Y. Z. Zhu, and X. Zou. The biomaterials for the DNA sequencing of the reference P. anubis baboon and several of the diversity panel of baboons were provided by the Southwest National Primate Research Center, San Antonio, TX, which was supported by a grant from the NIH Office of Research Infrastructure Programs (P51-OD011133). The research reported here complied with governmental and IACUC regulations and guidelines. J.R. is also affiliated with the Wisconsin National Primate Research Center, Madison, WI. C.K. is also affiliated with the Institut für Populationsgenetik, Vetmeduni Vienna, Austria, and D.S. is newly affiliated with Eötvös Lorand University Budapest and Max Perutz Laboratories Vienna. Funding: The sequencing and analysis activities at the Human Genome Sequencing Center, Baylor College of Medicine, were supported by NIH (NHGRI) grants U54-HG003273 and U54-HG006484 to R.A.G. and GAC grant 1 S10 RR026605 to J. G. Reid. This research was also supported by NIH grant R01-GM59290 to M.A.B.; grants from the Austrian Science Fund (FWF-P24551 and FWF-W1225) and Vienna Science and Technology Fund (WWTF-MA16-061) to C.K.; grants from the Wellcome Trust (WT108749/Z/15/Z) and EMBL to B.A., F.J.M., and M.M.; grants VEGA 1/0719/14 and APVV-14-0253 to T. Vinar (Consortium Member); MINECO/FEDER grant, NIH U01-MH106874 grant, Howard Hughes International Early Career award, and Obra Social “La Caixa” award to T.M.-B.; NSF grants BNS83-03506 to J.P.-C.; NSF1029302 to J.P.-C., J.R., and C.J.J.; BNS96-15150 to J.P.-C., C.J.J., and T.D.; and National Geographic Society and Leakey Foundation grants to J.P.-C. and C.J.J. E.E.E. is an investigator of the Howard Hughes Medical Institute. This work was supported, in part, by U.S. NIH grant HG002385 to E.E.E. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The raw read data, sample metadata, and other information for this genome assembly project are available under Bioproject PRJNA260523 at www.ncbi.nlm.nih.gov. Additional information concerning RNA sequencing data is available at the Nonhuman Primate Reference Transcriptome Project (http://nhprtr.org/). Further information regarding SNV and indel variation is available as a track on the UCSC browser (https://hgsc.bcm.edu/non-human-primates/baboon-genome-project). Additional data related to this paper may be requested from the authors.
Full membership of the Baboon Genome Analysis Consortium:
Bronwen Aken1, Nicoletta Archidiacono2, Georgios Athanasiadis3, Mark A. Batzer4, Thomas O. Beckstrom4, Christina Bergey5,6, Konstantinos Billis1, Andrew Burrell5, Oronzo Capozzi2, Claudia R. Catacchio2, Jade Cheng3, Laura A. Cox7,8, Huyen H. Dinh9, Todd Disotell5, HarshaVardhan Doddapaneni9, Evan E. Eichler10,11, James Else12, Richard A. Gibbs9,13, Matthew W. Hahn14, Yi Han9, R. Alan Harris9,13, John Huddleston10, Shalini N. Jhangiani9, Clifford J. Jolly5, Vallmer E. Jordan4, Anis Karimpour-Fard15, Miriam K. Konkel32, Gisela H. Kopp16,17, Viktoriya Korchina9, Carolin Kosiol18, Maximillian Kothe19, Christie L. Kovar9, Lukas Kuderna20, Sandra L. Lee9, Kalle Leppälä3, Xiaoming Liu21, Yue Liu9, Thomas Mailund3, Tomas Marques-Bonet20,22,23,33, Alessia Marra-Campanale2, Fergal J. Martin1, Christopher E. Mason24, Marc de Manuel Montero20, Matthieu Muffato1, Kasper Munch3, Shwetha Murali9, Donna M. Muzny9,13, Angela Noll19, Kymberleigh A. Pagel25, Antonio Palazzo2, Jera Pecotte7, Vikas Pejaver25, Jane Phillips-Conroy26, Lenore Pipes24, Veronica Searles Quick15, Predrag Radivojac25, Archana Raja10, Brian J. Raney27, Muthuswamy Raveendran9, Karen Rice7, Mariano Rocchi2, Jeffrey Rogers9,13, Christian Roos19, Mikkel Heide Schierup3, Dominik Schrempf28, James M. Sikela15, Roscoe Stanyon29, Cody J. Steely4, Gregg W. C. Thomas14, Jenny Tung30, Mario Ventura2, Tauras P. Vilgalys30, Tomas Vinar31, Jerilyn A. Walker4, Lutz Walter19, Kim C. Worley9,13, and Dietmar Zinner16.
1European Molecular Biology Laboratory, European Bioinformatics Institute, Hinxton, UK. 2Department of Biology, University of Bari, Bari, Italy. 3Bioinformatics Research Centre, Aarhus University, Aarhus, Denmark. 4Department of Biological Sciences, Louisiana State University, Baton Rouge, LA, USA. 5Department of Anthropology, New York University, New York, NY, USA. 6Department of Biological Sciences, Notre Dame University, South Bend, IN, USA. 7Southwest National Primate Research Center, Texas Biomedical Research Institute, San Antonio, TX, USA. 8Department of Genetics, Texas Biomedical Research Institute, San Antonio, TX, USA. 9Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX, USA. 10Department of Genome Sciences, University of Washington, Seattle, WA, USA. 11Howard Hughes Medical Institute, University of Washington, Seattle, WA, USA. 12Department of Pathology and Laboratory Medicine and Yerkes Primate Research Center, Emory University, Atlanta, GA, USA. 13Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA. 14Department of Biology, Indiana University, Bloomington, IN, USA. 15Department of Biochemistry and Molecular Genetics, University of Colorado Anschutz Medical Campus, Denver, CO, USA. 16Cognitive Ethology Laboratory, German Primate Center, Leibniz Institute for Primate Research, Göttingen, Germany. 17Department of Biology, University of Konstanz, Konstanz, Germany. 18Centre of Biological Diversity, School of Biology, St. Andrews, UK. 19Primate Genetics Laboratory, German Primate Center, Leibniz Institute for Primate Research, Göttingen, Germany. 20Institute of Evolutionary Biology (UPF-CSIC), PRBB, Barcelona, Spain. 21School of Public Health, University of Texas Health Science Center, Houston, TX, USA. 22Catalan Institution of Research and Advanced Studies (ICREA), Barcelona, Spain. 23CNAG-CRG, Centre for Genomic Regulation, Barcelona Institute of Science and Technology, Barcelona, Spain. 24Department of Physiology and Biophysics, Weill Cornell Medical College, New York, NY, USA, and HRH Prince Alwaleed Bin Talal Bin Abdulaziz Alsaud Institute of Computational Biomedicine, Weill Cornell Medicine, New York, NY, USA. 25Department of Computer Science and Informatics, Indiana University, Bloomington, IN, USA. 26Department of Neuroscience, Washington University School of Medicine, St. Louis. MO, USA, and Department of Anthropology, Washington University, Seattle, WA, USA. 27Genomics Institute, University of California, Santa Cruz, CA, USA. 28Institut für Populationsgenetik, Veterinärmedizinische Universität Wien, Vienna, Austria. 29Department of Biology, University of Florence, Florence, Italy. 30Department of Evolutionary Anthropology, Duke University, Durham, NC, USA. 31Faculty of Mathematics, Physics and Informatics, Comenius University, Bratislava, Slovakia. 32Department of Genetics and Biochemistry, Clemson University, Clemson, SC, USA. 33Institut Catala de Paleontologia Miquel Crusafont, Universitat Autonoma de Barcelona, Barcelona, Spain.
Consortium Member Contributions:
Designed the study and supervised the analysis: J. Rogers*, K. C. Worley, and R. A. Gibbs. Managed or supervised the sequence production: D. M. Muzny*, C. L. Kovar, H. H. Dinh, and Y. Han. Managed or supervised the preparation of sequencing libraries: H. Doddapaneni*, S. Lee, and D. M. Muzny. Produced the assembly: K. C. Worley*, Y. Liu, S. Murali, and R. A. Harris. Project and data management: D. M. Muzny*, M. Raveendran, R. A. Harris, K. C. Worley, S. N. Jhangiani, V. Korchina, C. Kovar. Genome annotation: B. Aken*, F. J. Martin, M. Muffato, K. Billis, and X. Liu. Alu repeat analysis: M. A. Batzer*, J. A. Walker, M. K. Konkel, V. E. Jordan, C. J. Steely, and T. O. Beckstrom. SNV and indel analysis: R. A. Harris and M. Raveendran. Admixture and phylogenetic analysis: T. Mailund, M. H. Schierup, K. Leppälä, J. Cheng, K. Munch, and G. Athanasiadis. Phylogenetic and population analysis: C. Bergey, A. Burrell, A. Noll, D. Schrempf, C. Kosiol, G. H. Kopp, G. Athanasiadis, K. Munch, J. Phillips-Conroy, M. Kothe, T. Disotell, J. Tung, J. Rogers, C. J. Jolly, D. Zinner, and C. Roos. Cytogenetics and assembly validation: M. Rocchi*, R. Stanyon, E. E. Eichler, N. Archidiacono, A. Palazzo, and O. Capozzi. Gene family analysis: M. W. Hahn*, J. Sikela*, G. W. C. Thomas, V. Searles Quick, A. Karimpour-Fard, and L. Walter. Methylation analysis: J. Tung* and T. P. Vilgalys. Positive selection analysis: C. Kosiol*, T. Vinar*, and B. J. Raney. Posttranslational modifications: P. Radivojac*, K. A. Pagel, and V. Pejaver. Segmental duplication analysis: E. E. Eichler*, M. Ventura, A. Raja, C. Catacchio, A. Marra-Campanale, and J. Huddleston. Copy number variation: T. Marques-Bonet*, L. Kuderna, and M. d. M. Montero. Transcriptome analysis: C. E. Mason* and L. Pipes. Provided essential biomaterials: K. Rice, J. Pecotte, J. Phillips-Conroy, C. J. Jolly, J. Rogers, J. Else, and L. A. Cox. Provided text and/or figures: D. Zinner, C. Roos, T. Mailund, K. Leppälä, E. E. Eichler, G. Athanasiadis, J. Cheng, K. Munch, C. Kosiol, C. Bergey, A. Burrell, M. K. Konkel, J. A. Walker, M. A. Batzer, and J. Tung. Wrote the paper: J. Rogers*, C. J. Jolly, J. Tung, M. Hahn, D. Zinner, C. Roos, T. Marques-Bonet, and K. C. Worley. *Group leader.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaau6947/DC1
Section S1. Rationale for baboon taxonomy and nomenclature
Section S2. Rationale for mutation rate used in PSMC analyses
Section S3. Sequencing and assembly of olive baboon genome
Section S4. Annotation and gene content of the baboon genome
Section S5. Identification of SNVs and small indels within baboon species
Section S6. Validation of species identity within the diversity panel
Section S7. Lineage-specific Alu insertion in OWMs and hominoids
Section S8. Alternative methods for constructing phylogenetic trees
Section S9. Identification of admixture through asymmetric allele sharing
Section S10. Polymorphic AluY insertions across Papio species
Section S11. Bayesian concordance analyses of gene trees devoid of coding sequences
Section S12. CoalHMMs of admixture trees and events
Section S13. Locus-specific phylogenetic trees for chromosomal segments containing annotated genes
Fig. S1. Panu3.0 genome assembly process.
Fig. S2. Workflow used in variant calling pipeline.
Fig. S3. Details regarding SNV calls.
Fig. S4. Maximum likelihood and Bayesian phylogenetic trees based on SNV data.
Fig. S5. Test of phylogeny reconstruction using PoMo.
Fig. S6. Identification of admixture using f-statistics.
Fig. S7. Evidence for admixture from haplotyping sharing.
Fig. S8. A cladogram of Papio individuals from the diversity panel.
Fig. S9. Bayesian concordance analysis.
Fig. S10. Bootstrap analysis of timing of divergence events.
Fig. S11. Confidence intervals for baboon admixture proportions.
Fig. S12. Results for simulated admixture analysis.
Fig. S13. Results for correction factor adjustment of admixture history.
Fig. S14. Model used to estimate specific divergence and admixture history.
Fig. S15. Unbiased estimates dating divergences and admixture events.
Fig. S16. Phylogeny representing cluster 1 genic regions.
Fig. S17. Phylogeny representing cluster 2 genic regions.
Fig. S18. Phylogeny representing cluster 3 genic regions.
Table S1. Assembly statistics.
Table S2. Annotation of baboon genome assemblies.
Table S3. DNA samples used for diversity analysis.
Table S4. SNV variation among 15 Papio baboons and a gelada.
Table S5. Full-length AluY insertions and lineage-specific insertions in primate genomes.
Table S6. Effect of admixture on branch lengths measured in substitutions per site.
Table S7. Bayes factors comparing alternate phylogenies.
Table S8. Divergence time estimates across triplets.
Table S9. Admixture proportion estimates across triplets.
Table S10. GO terms associated with genes falling in clusters 1 to 3 of genic regions.
REFERENCES AND NOTES
- 1.Seehausen O., Butlin R. K., Keller I., Wagner C. E., Boughman J. W., Hohenlohe P. A., Peichel C. L., Saetre G.-P., Bank C., Brännström Å., Brelsford A., Clarkson C. S., Eroukhmanoff F., Feder J. L., Fischer M. C., Foote A. D., Franchini P., Jiggins C. D., Jones F. C., Lindholm A. K., Lucek K., Maan M. E., Marques D. A., Martin S. H., Matthews B., Meier J. I., Möst M., Nachman M. W., Nonaka E., Rennison D. J., Schwarzer J., Watson E. T., Westram A. M., Widmer A., Genomics and the origin of species. Nat. Rev. Genet. 15, 176–192 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Wolf J. B. W., Ellegren H., Making sense of genomic islands of differentiation in light of speciation. Nat. Rev. Genet. 18, 87–100 (2017). [DOI] [PubMed] [Google Scholar]
- 3.D. Otte, J. A. Endler, Speciation and Its Consequences (Sinauer Associates, Inc., 1989). [Google Scholar]
- 4.E. Mayr, Systematics and the Origin of Species (Columbia Univ. Press, 1942). [Google Scholar]
- 5.M. L. Arnold, Divergence with Genetic Exchange (Oxford Univ. Press, 2015). [Google Scholar]
- 6.Mallet J., Besansky N., Hahn M. W., How reticulated are species? Bioessays 38, 140–149 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cox L. A., Comuzzie A. G., Havill L. M., Karere G. M., Spradling K. D., Mahaney M. C., Nathanielsz P. W., Nicolella D. P., Shade R. E., Voruganti S., VandeBerg J. L., Baboons as a model to study genetics and epigenetics of human disease. ILAR J. 54, 106–121 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kammerer C. M., Cox L. A., Mahaney M. C., Rogers J., Shade R. E., Sodium-lithium countertransport activity is linked to chromosome 5 in baboons. Hypertension 37, 398–402 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Jolly C. J., A proper study for mankind: Analogies from the Papionin monkeys and their implications for human evolution. Am. J. Phys. Anthropol. 116 (suppl. 33), 177–204 (2001). [DOI] [PubMed] [Google Scholar]
- 10.C. J. Jolly, in Species, Species Concepts and Primate Evolution, W. H. Kimbel, L. B. Martin, Eds. (Plenum Press, 1993), pp. 67–107. [Google Scholar]
- 11.L. Swedell, in Primates in Perspective, C. J. Campbell, A. Fuentes, K. C. MacKinnon, S. K. Bearder, R. M. Stumpf, Eds. (Oxford Univ. Press, ed. 2, 2011). [Google Scholar]
- 12.Zinner D., Wertheimer J., Liedigk R., Groeneveld L. F., Roos C., Baboon phylogeny as inferred from complete mitochondrial genomes. Am. J. Phys. Anthropol. 150, 133–140 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antón S. C., Potts R., Aiello L. C., Human evolution. Evolution of early Homo: An integrated biological perspective. Science 345, 1236828 (2014). [DOI] [PubMed] [Google Scholar]
- 14.D. Zinner, U. Buba, S. Nash, C. Roos, in Primates of Gashaka, V. Sommer, C. Roos, Eds. (Springer, 2011), pp. 267–306. [Google Scholar]
- 15.Zinner D., Keller C., Nyahongo J. W., Butynski T. M., de Jong Y. A., Pozzi L., Knauf S., Liedigk R., Roos C., Distribution of mitochondrial clades and morphotypes of baboons Papio spp. (Primates: Cercopithecidae) in eastern Africa. J. East African Nat. Hist. 104, 143–168 (2015). [Google Scholar]
- 16.Fischer J., Kopp G. H., Dal Pesco F., Goffe A., Hammerschmidt K., Kalbitzer U., Klapproth M., Maciej P., Ndao I., Patzelt A., Zinner D., Charting the neglected West: The social system of Guinea baboons. Am. J. Phys. Anthropol. 162 (suppl. 63), 15–31 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Charpentier M. J. E., Fontaine M. C., Cherel E., Renoult J. P., Jenkins T., Benoit L., Barthès N., Alberts S. C., Tung J., Genetic structure in a dynamic baboon hybrid zone corroborates behavioural observations in a hybrid population. Mol. Ecol. 21, 715–731 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Jolly C. J., Burrell A. S., Phillips-Conroy J. E., Bergey C., Rogers J., Kinda baboons (Papio kindae) and grayfoot chacma baboons (P. ursinus griseipes) hybridize in the Kafue river valley, Zambia. Am. J. Primatol. 73, 291–303 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Bergman T. J., Phillips-Conroy J. E., Jolly C. J., Behavioral variation and reproductive success of male baboons (Papio anubis × Papio hamadryas) in a hybrid social group. Am. J. Primatol. 70, 136–147 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Pääbo S., The diverse origins of the human gene pool. Nat. Rev. Genet. 16, 313–314 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Vernot B., Tucci S., Kelso J., Schraiber J. G., Wolf A. B., Gittelman R. M., Dannemann M., Grote S., McCoy R. C., Norton H., Scheinfeldt L. B., Merriwether D. A., Koki G., Friedlaender J. S., Wakefield J., Pääbo S., Akey J. M., Excavating Neandertal and Denisovan DNA from the genomes of Melanesian individuals. Science 352, 235–239 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Warren W. C., Jasinska A. J., García-Pérez R., Svardal H., Tomlinson C., Rocchi M., Archidiacono N., Capozzi O., Minx P., Montague M. J., Kyung K., Hillier L. D. W., Kremitzki M., Graves T., Chiang C., Hughes J., Tran N., Huang Y., Ramensky V., Choi O.-W., Jung Y. J., Schmitt C. A., Juretic N., Wasserscheid J., Turner T. R., Wiseman R. W., Tuscher J. J., Karl J. A., Schmitz J. E., Zahn R., O’Connor D. H., Redmond E., Nisbett A., Jacquelin B., Müller-Trutwin M. C., Brenchley J. M., Dione M., Antonio M., Schroth G. P., Kaplan J. R., Jorgensen M. J., Thomas G. W. C., Hahn M. W., Raney B. J., Aken B., Nag R., Schmitz J., Churakov G., Noll A., Stanyon R., Webb D., Thibaud-Nissen F., Nordborg M., Marques-Bonet T., Dewar K., Weinstock G. M., Wilson R. K., Freimer N. B., The genome of the vervet (Chlorocebus aethiops sabaeus). Genome Res. 25, 1921–1933 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Maio N., Schrempf D., Kosiol C., PoMo: An allele frequency-based approach for species tree estimation. Syst. Biol. 64, 1018–1031 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schrempf D., Minh B. Q., De Maio N., von Haeseler A., Kosiol C., Reversible polymorphism-aware phylogenetic models and their application to tree inference. J. Theor. Biol. 407, 362–370 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Alberts S. C., Altmann J., Immigration and hybridization patterns of yellow and anubis baboons in and around Amboseli, Kenya. Am. J. Primatol. 53, 139–154 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Tung J., Charpentier M. J. E., Garfield D. A., Altmann J., Alberts S. C., Genetic evidence reveals temporal change in hybridization patterns in a wild baboon population. Mol. Ecol. 17, 1998–2011 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Wall J. D., Schlebusch S. A., Alberts S. C., Cox L. A., Snyder-Mackler N., Nevonen K. A., Carbone L., Tung J., Genomewide ancestry and divergence patterns from low-coverage sequencing data reveal a complex history of admixture in wild baboons. Mol. Ecol. 25, 3469–3483 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Phillips-Conroy J. E., Jolly C. J., Brett F. L., Characteristics of hamadryas-like male baboons living in anubis baboon troops in the Awash hybrid zone, Ethiopia. Am. J. Phys. Anthropol. 86, 353–368 (1991). [DOI] [PubMed] [Google Scholar]
- 29.Ray D. A., Xing J., Salem A.-H., Batzer M. A., SINEs of a nearly perfect character. Syst. Biol. 55, 928–935 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Mendes F. K., Hahn M. W., Gene tree discordance causes apparent substitution rate variation. Syst. Biol. 65, 711–721 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Larget B. R., Kotha S. K., Dewey C. N., Ané C., BUCKy: Gene tree/species tree reconciliation with Bayesian concordance analysis. Bioinformatics 26, 2910–2911 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Dutheil J. Y., Ganapathy G., Hobolth A., Mailund T., Uyenoyama M. K., Schierup M. H., Ancestral population genomics: The coalescent hidden Markov model approach. Genetics 183, 259–274 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mailund T., Dutheil J. Y., Hobolth A., Lunter G., Schierup M. H., Estimating divergence time and ancestral effective population size of Bornean and Sumatran orangutan subspecies using a coalescent hidden Markov model. PLOS Genet. 7, e1001319 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tung J., Archie E. A., Altmann J., Alberts S. C., Cumulative early life adversity predicts longevity in wild baboons. Nat. Commun. 7, 11181 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H., Durbin R., Inference of human population history from individual whole-genome sequences. Nature 475, 493–496 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.N. G. Jablonski, S. Frost, in Cenozoic Mammals of Africa, L. Werdelin, W. J. Sanders, Eds. (University of California Press, 2010), pp. 393–428. [Google Scholar]
- 37.Dennenmoser S., Sedlazeck F. J., Iwaszkiewicz E., Li X.-Y., Altmüller J., Nolte A. W., Copy number increases of transposable elements and protein-coding genes in an invasive fish of hybrid origin. Mol. Ecol. 26, 4712–4724 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Neill R. J. W., O’Neill M. J., Graves J. A. M., Undermethylation associated with retroelement activation and chromosome remodelling in an interspecific mammalian hybrid. Nature 393, 68–72 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Ackermann R. R., Schroeder L., Rogers J., Cheverud J. M., Further evidence for phenotypic signatures of hybridization in descendant baboon populations. J. Hum. Evol. 76, 54–62 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.H. Kummer, in Primates: Studies in Adaptation and Variability, P. C. Jay, Ed. (Holt, Rinehart & Winston, 1968), pp. 293–312. [Google Scholar]
- 41.Bergey C. M., Phillips-Conroy J. E., Disotell T. R., Jolly C. J., Dopamine pathway is highly diverged in primate species that differ markedly in social behavior. Proc. Natl. Acad. Sci. U.S.A. 113, 6178–6181 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jolly C. J., Phillips-Conroy J. E., Kaplan J. R., Mann J. J., Cerebrospinal fluid monoaminergic metabolites in wild Papio anubis and P. hamadryas are concordant with taxon-specific behavioral ontogeny. Int. J. Primatol. 29, 1549–1566 (2008). [Google Scholar]
- 43.Kammerer C. M., Sparks M. L., Rogers J., Effects of age, sex, and heredity on measures of bone mass in baboons (Papio hamadryas). J. Med. Primatol. 24, 236–242 (1995). [DOI] [PubMed] [Google Scholar]
- 44.Kochunov P., Glahn D. C., Fox P. T., Lancaster J. L., Saleem K., Shelledy W., Zilles K., Thompson P. M., Coulon O., Mangin J. F., Blangero J., Rogers J., Genetics of primary cerebral gyrification: Heritability of length, depth and area of primary sulci in an extended pedigree of Papio baboons. Neuroimage 53, 1126–1134 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moore C. M., Janish C., Eddy C. A., Hubbard G. B., Leland M. M., Rogers J., Cytogenetic and fertility studies of a rheboon, rhesus macaque (Macaca mulatta) × baboon (Papio hamadryas) cross: Further support for a single karyotype nomenclature. Am. J. Phys. Anthropol. 110, 119–127 (1999). [DOI] [PubMed] [Google Scholar]
- 46.English A. C., Richards S., Han Y., Wang M., Vee V., Qu J., Qin X., Muzny D. M., Reid J. G., Worley K. C., Gibbs R. A., Mind the gap: Upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLOS ONE 7, e47768 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walker B. J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., Cuomo C. A., Zeng Q., Wortman J., Young S. K., Earl A. M., Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLOS ONE 9, e112963 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu X., White S., Peng B., Johnson A. D., Brody J. A., Li A. H., Huang Z., Carroll A., Wei P., Gibbs R., Klein R. J., Boerwinkle E., WGSA: An annotation pipeline for human genome sequencing studies. J. Med. Genet. 53, 111–112 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nguyen L.-T., Schmidt H. A., von Haeseler A., Minh B. Q., IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ronquist F., Teslenko M., van der Mark P., Ayres D. L., Darling A., Höhna S., Larget B., Liu L., Suchard M. A., Huelsenbeck J. P., MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ewing G., Hermisson J., MSMS: A coalescent simulation program including recombination, demographic structure and selection at a single locus. Bioinformatics 26, 2064–2065 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patterson N., Moorjani P., Luo Y., Mallick S., Rohland N., Zhan Y., Genschoreck T., Webster T., Reich D., Ancient admixture in human history. Genetics 192, 1065–1093 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Steely C. J., Baker J. N., Walker J. A., Loupe C. D. III, Analysis of lineage-specific Alu subfamilies in the genome of the olive baboon, Papio anubis. Mob DNA 9, 10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jordan V. E., Walker J. A., Beckstrom T. O., Steely C. J., McDaniel C. L., St. Romain C. P.; Baboon Genome Analysis Consortium, Worley K. C., Phillips-Conroy J., Jolly C. J., Rogers J., Konkel M. K., Batzer M. A., A computational reconstruction of Papio phylogeny using Alu insertion polymorphisms. Mob DNA 9, 13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ané C., Larget B., Baum D. A., Smith S. D., Rokas A., Bayesian estimation of concordance among gene trees. Mol. Biol. Evol. 24, 412–426 (2007). [DOI] [PubMed] [Google Scholar]
- 56.Kendall M., Colijn C., Mapping phylogenetic trees to reveal distinct patterns of evolution. Mol. Biol. Evol. 33, 2735–2743 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mailund T., Halager A. E., Westergaard M., Dutheil J. Y., Munch K., Andersen L. N., Lunter G., Prüfer K., Scally A., Hobolth A., Schierup M. H., A new isolation with migration model along complete genomes infers very different divergence processes among closely related great ape species. PLOS Genet. 8, e1003125 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McVean G. A. T., Cardin N. J., Approximating the coalescent with recombination. Philos. Trans. R. Soc. Lond. B Biol. Sci. 360, 1387–1393 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.E. Delson, C. J. Terranova, W. L. Jungers, E. J. Sargis, N. G. Jablonski, P. C. Dechow, Body Mass in Cercopithecidae (Primates, Mammalia): Estimation and Scaling in Extinct and Extant Taxa (Anthropological Papers of the American Museum of Natural History, 2000).
- 60.Locke D. P., Hillier L. W., Warren W. C., Worley K. C., Nazareth L. V., Muzny D. M., Yang S.-P., Wang Z., Chinwalla A. T., Minx P., Mitreva M., Cook L., Delehaunty K. D., Fronick C., Schmidt H., Fulton L. A., Fulton R. S., Nelson J. O., Magrini V., Pohl C., Graves T. A., Markovic C., Cree A., Dinh H. H., Hume J., Kovar C. L., Fowler G. R., Lunter G., Meader S., Heger A., Ponting C. P., Marques-Bonet T., Alkan C., Chen L., Cheng Z., Kidd J. M., Eichler E. E., White S., Searle S., Vilella A. J., Chen Y., Flicek P., Ma J., Raney B., Suh B., Burhans R., Herrero J., Haussler D., Faria R., Fernando O., Darré F., Farré D., Gazave E., Oliva M., Navarro A., Roberto R., Capozzi O., Archidiacono N., Della Valle G., Purgato S., Rocchi M., Konkel M. K., Walker J. A., Ullmer B., Batzer M. A., Smit A. F. A., Hubley R., Casola C., Schrider D. R., Hahn M. W., Quesada V., Puente X. S., Ordoñez G. R., López-Otín C., Vinar T., Brejova B., Ratan A., Harris R. S., Miller W., Kosiol C., Lawson H. A., Taliwal V., Martins A. L., Siepel A., RoyChoudhury A., Ma X., Degenhardt J., Bustamante C. D., Gutenkunst R. N., Mailund T., Dutheil J. Y., Hobolth A., Schierup M. H., Ryder O. A., Yoshinaga Y., de Jong P. J., Weinstock G. M., Rogers J., Mardis E. R., Gibbs R. A., Wilson R. K., Comparative and demographic analysis of orang-utan genomes. Nature 469, 529–533 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.J. Cracraft, in Speciation and Its Consequences, D. Otte, J. A. Endler, Eds. (Sinauer Associates, Inc., 1989). [Google Scholar]
- 62.Coyne J. A., Orr H. A., The evolutionary genetics of speciation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 353, 287–305 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.E. Mayr, Populations, Species and Evolution (Belknap Press of Harvard Univ. Press, 1963). [Google Scholar]
- 64.Cracraft J., Species concepts and speciation analysis. Curr. Ornithol. 1, 159–187 (1983). [Google Scholar]
- 65.C. Groves, Primate Taxonomy (Smithsonian Institution Press, 2001). [Google Scholar]
- 66.Grubb P., Butynski T. M., Oates J. F., Bearder S. K., Disotell T. R., Groves C. P., Struhsaker T. T., Assessment of the diversity of African primates. Int. J. Primatol. 24, 1301–1357 (2003). [Google Scholar]
- 67.Boissinot S., Alvarez L., Giraldo-Ramirez J., Tollis M., Neutral nuclear variation in Baboons (genus Papio) provides insights into their evolutionary and demographic histories. Am. J. Phys. Anthropol. 155, 621–634 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Frost S. R., Marcus L. F., Bookstein F. L., Reddy D. P., Delson E., Cranial allometry, phylogeography, and systematics of large-bodied papionins (primates: Cercopithecinae) inferred from geometric morphometric analysis of landmark data. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 275, 1048–1072 (2003). [DOI] [PubMed] [Google Scholar]
- 69.Ségurel L., Wyman M. J., Przeworski M., Determinants of mutation rate variation in the human germline. Annu. Rev. Genomics Hum. Genet. 15, 47–70 (2014). [DOI] [PubMed] [Google Scholar]
- 70.Elango N., Lee J., Peng Z., Loh Y.-H. E., Yi S. V., Evolutionary rate variation in Old World monkeys. Biol. Lett. 5, 405–408 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Newman T. K., Jolly C. J., Rogers J., Mitochondrial phylogeny and systematics of baboons (Papio). Am. J. Phys. Anthropol. 124, 17–27 (2004). [DOI] [PubMed] [Google Scholar]
- 72.O’Leary N. A., Wright M. W., Brister J. R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D., Astashyn A., Badretdin A., Bao Y., Blinkova O., Brover V., Chetvernin V., Choi J., Cox E., Ermolaeva O., Farrell C. M., Goldfarb T., Gupta T., Haft D., Hatcher E., Hlavina W., Joardar V. S., Kodali V. K., Li W., Maglott D., Masterson P., McGarvey K. M., Murphy M. R., O’Neill K., Pujar S., Rangwala S. H., Rausch D., Riddick L. D., Schoch C., Shkeda A., Storz S. S., Sun H., Thibaud-Nissen F., Tolstoy I., Tully R. E., Vatsan A. R., Wallin C., Webb D., Wu W., Landrum M. J., Kimchi A., Tatusova T., DiCuccio M., Kitts P., Murphy T. D., Pruitt K. D., Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aken B. L., Achuthan P., Akanni W., Amode M. R., Bernsdorff F., Bhai J., Billis K., Carvalho-Silva D., Cummins C., Clapham P., Gil L., García Girón C., Gordon L., Hourlier T., Hunt S. E., Janacek S. H., Juettemann T., Keenan S., Laird M. R., Lavidas I., Maurel T., McLaren W., Moore B., Murphy D. N., Nag R., Newman V., Nuhn M., Ong C. K., Parker A., Patricio M., Riat H. S., Sheppard D., Sparrow H., Taylor K., Thormann A., Vullo A., Walts B., Wilder S. P., Zadissa A., Kostadima M., Martin F. J., Muffato M., Perry E., Ruffier M., Staines D. M., Trevanion S. J., Cunningham F., Yates A., Zerbino D. R., Flicek P., Ensembl 2017. Nucleic Acids Res. 45, D635–D642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aken B. L., Ayling S., Barrell D., Clarke L., Curwen V., Fairley S., Fernandez Banet J., Billis K., García Girón C., Hourlier T., Howe K., Kähäri A., Kokocinski F., Martin F. J., Murphy D. N., Nag R., Ruffier M., Schuster M., Tang Y. A., Vogel J.-H., White S., Zadissa A., Flicek P., Searle S. M. J., The Ensembl gene annotation system. Database 2016, baw093 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zinner D., Groeneveld L. F., Keller C., Roos C., Mitochondrial phylogeography of baboons (Papio spp.): Indication for introgressive hybridization? BMC Evol. Biol. 9, 83 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Robinson D. F., Foulds L. R., Comparison of phylogenetics trees. Math. Biosci. 53, 131–147 (1981). [Google Scholar]
- 77.Kuhner M. K., Felsenstein J., A simulation comparison of phylogeny algorithms under equal and unequal evolutionary rates. Mol. Biol. Evol. 11, 459–468 (1994). [DOI] [PubMed] [Google Scholar]
- 78.Xue C., Raveendran M., Harris R. A., Fawcett G. L., Liu X., White S., Dahdouli M., Deiros D. R., Below J. E., Salerno W., Cox L., Fan G., Ferguson B., Horvath J., Johnson Z., Kanthaswamy S., Kubisch H. M., Liu D., Platt M., Smith D. G., Sun B., Vallender E. J., Wang F., Wiseman R. W., Chen R., Muzny D. M., Gibbs R. A., Yu F., Rogers J., The population genomics of rhesus macaques (Macaca mulatta) based on whole-genome sequences. Genome Res. 26, 1651–1662 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaau6947/DC1
Section S1. Rationale for baboon taxonomy and nomenclature
Section S2. Rationale for mutation rate used in PSMC analyses
Section S3. Sequencing and assembly of olive baboon genome
Section S4. Annotation and gene content of the baboon genome
Section S5. Identification of SNVs and small indels within baboon species
Section S6. Validation of species identity within the diversity panel
Section S7. Lineage-specific Alu insertion in OWMs and hominoids
Section S8. Alternative methods for constructing phylogenetic trees
Section S9. Identification of admixture through asymmetric allele sharing
Section S10. Polymorphic AluY insertions across Papio species
Section S11. Bayesian concordance analyses of gene trees devoid of coding sequences
Section S12. CoalHMMs of admixture trees and events
Section S13. Locus-specific phylogenetic trees for chromosomal segments containing annotated genes
Fig. S1. Panu3.0 genome assembly process.
Fig. S2. Workflow used in variant calling pipeline.
Fig. S3. Details regarding SNV calls.
Fig. S4. Maximum likelihood and Bayesian phylogenetic trees based on SNV data.
Fig. S5. Test of phylogeny reconstruction using PoMo.
Fig. S6. Identification of admixture using f-statistics.
Fig. S7. Evidence for admixture from haplotyping sharing.
Fig. S8. A cladogram of Papio individuals from the diversity panel.
Fig. S9. Bayesian concordance analysis.
Fig. S10. Bootstrap analysis of timing of divergence events.
Fig. S11. Confidence intervals for baboon admixture proportions.
Fig. S12. Results for simulated admixture analysis.
Fig. S13. Results for correction factor adjustment of admixture history.
Fig. S14. Model used to estimate specific divergence and admixture history.
Fig. S15. Unbiased estimates dating divergences and admixture events.
Fig. S16. Phylogeny representing cluster 1 genic regions.
Fig. S17. Phylogeny representing cluster 2 genic regions.
Fig. S18. Phylogeny representing cluster 3 genic regions.
Table S1. Assembly statistics.
Table S2. Annotation of baboon genome assemblies.
Table S3. DNA samples used for diversity analysis.
Table S4. SNV variation among 15 Papio baboons and a gelada.
Table S5. Full-length AluY insertions and lineage-specific insertions in primate genomes.
Table S6. Effect of admixture on branch lengths measured in substitutions per site.
Table S7. Bayes factors comparing alternate phylogenies.
Table S8. Divergence time estimates across triplets.
Table S9. Admixture proportion estimates across triplets.
Table S10. GO terms associated with genes falling in clusters 1 to 3 of genic regions.