

Abstract
Three-dimensional (3D) brain organoid culture has become an essential tool for investigating human brain development and modeling neurological disorders during the past few years. Given the specific regionalization during brain development, it is important to produce distinct brain organoids that reproduce different brain regions and their interaction. The authors’ laboratory recently established the platform to generate brain organoids resembling the medial ganglionic eminence (MGE), a specific brain region responsible for interneurogenesis, and found when fusing with organoid resembling the cortex, the fused organoids enabled modeling of interneuron migration in the brain. This unit describes four basic protocols that have been successfully applied in the authors’ laboratory, covering the generation of embryonic body (EB) with neuroectodermal fate, the production of MGE organoids (hMGEOs) and cortical organoids (hCOs), and the fusion of the two organoids.
Keywords: Human brain organoid, fusion, interneurons, excitatory neurons, human pluripotent stem cells (hPSCs)
INTRODUCTION
The use of brain organoids is essential for study of human brain development. One of the critical goals since the discovery of human brain organoids is to generate organoids representing the sub-structures in developing brain. Towards this goal, organoids recapitulating the organization and development of hippocampus, cerebellum, midbrain, and hypothalamus have been reported (Jo et al., 2016; Muguruma et al., 2015; Qian et al., 2016; Sakaguchi et al., 2015). In the cerebral cortex, glutamatergic excitatory neurons and GABAergic inhibitory neurons (interneurons) are the major components of the neuronal circuits. Excitatory neurons are produced in the dorsal pallium of developing cortex, while interneurons are mainly generated from the medial ganglionic eminence (MGE) of the subpallium. Approaches to produce brain organoids specifically resembling MGE/subpallium were established recently, and fusion of MGE/subpallium organoid with cortical organoid enabled the investigation of human interneuron migration (Bagley et al., 2017; Birey et al., 2017; Xiang et al., 2017). This unit describes a protocol for generation of human brain organoids to model MGE and cortex development, and for production of fused MGE-cortical organoids.
This unit begins with a method for inducing anterior neuroectodermal fates from human pluripotent stem cells (hPSCs), including human embryonic stem cells (hESCs) or human inducted pluripotent stem cells (hiPSCs), in a three-dimensional (3D) culture condition (Basic Protocol 1), followed by protocols for producing brain organoid resembling the cortex (cortical organoids [hCOs]; Basic Protocol 2) or the MGE (MGE organoids [hMGEOs]; Basic Protocol 3), and the protocol for fusing hCO and hMGEO together in order to establish a model for interneuron migration (Basic Protocol 4).
NOTE: All procedures are performed in a Class II biological hazard flow hood with standard aseptic technique, and cells are cultured in a humidified 37 °C incubator with 5% CO2.
NOTE: The use of hESCs must follow the regulations by Embryonic Stem Cell Research Oversight Committee (ESCRO) of the institution.
BASIC PROTOCOL 1: INDUCTION OF ANTERIOR NEUROECTODERMAL FATE IN ORGANOIDS
To generate brain organoids, the first step is to induce anterior neuroectodermal fate in organoids, a common process for both hCOs and hMGEOs. This protocol is to achieve anterior neuroectodermal differentiation in 3D cultures. hPSCs are dissociated into single cell suspension and seeded to ultra-low attachment tissue culture plates to allow embryoid body (EB) formation. To minimize un-directed differentiation, dual SMAD inhibition and canonical Wnt inhibition are adopted during this period.
Materials
hESCs or hiPSCs
Matrigel (BD, Cat No. 354230)
mTeSR1 (Stem Cell Technologies, Cat No. 05875)
DMEM/F-12 (Life Technologies, Cat No. 11330057)
Neural induction medium (see recipe)
Dispase (5 U/ml, Stem Cell Technologies, Cat No. 07913)
Accutase (1 × solution, Stem Cell Technologies, Cat No. AT104)
Knockout serum replacement (Life Technologies, Cat No. 10828028)
MEM-NEAA (Life Technologies, Cat No. 11140050)
Glutamax supplement (Life Technologies, Cat No. 35050)
β-Mercaptoethanol (Sigma, Cat No. M7522)
LDN-193189 (Sigma, Cat No. SML0559)
SB431542 (Abcam, Cat No. ab120163)
XAV939 (Sigma, Cat No. X3004)
Y-27632 compound (Stem Cell Technologies, Cat No. 72304)
Heat-inactivated FBS (Life Technologies, Cat No. 16140071)
Stereomicroscope
6-well tissue culture plate (Corning, Cat No. 353046)
U-bottom ultra-low-attachment 96-well plate (Corning, Cat No. CLS7007-24EA)
10 ml pipette
5 ml pipette
15 ml conical centrifuge tube
50 ml conical centrifuge tube
Maintain hPSCs in feeder-free condition
hPSCs are ready for passaging when approaching ~80% confluence. Typically, passaging is performed once a week. Before passaging the cells, a new tissue culture plate is coated with Matrigel.
Thaw 100 μl Matrigel on ice, and dilute with 6 ml cold DMEM/F12 medium.
Immediately add 1 ml diluted Matrigel to each well of 6-well tissue culture plate.
Place the plate in incubator for 2 hours before use.
Remove differentiated colonies under stereomicroscope by scrapping using 200 μl pipette tip.
Aspirate medium from the well, add 1 ml DMEM/F12 medium to the well, then add 166 μl Dispase.
Place the plate back to the incubate for 7 min.
Aspirate DMEM/F12 medium containing Dispase, and wash cells 2 times with 1 ml of DMEM/F12 medium.
Add 1 ml of DMEM/F12 medium to the well, scrap down the colonies vertically and horizontally using 1 ml pipette tip.
Transfer 100 μl DMEM/F12 medium containing the colony pieces from the well to 15 ml centrifuge tube and centrifuge for 3 min at 800 rpm.
Aspirate supernatant and gently re-suspend cells in 2 ml mTeSR1 medium.
-
Add the cell suspension to 1 well of the freshly-coated 6-well tissue culture plate from step 4.
Diluted Matrigel was removed before adding the cell suspension.
Depending on the growth properties of different cell lines, the dilution ratio during passaging may be adjusted.
Extra wells are plated 1 week before EB formation. Normally, 1 well of 6-well plate can produce ~2 million single cells after 1 week’s growth.
Return the plate to incubator and perform daily medium change with 2 ml mTeSR1/well starting from 48 hours after passaging the cells.
EB formation and neural induction
On the day of cell passaging (normally 1 week after step 12 as described above), colony should remain as un-differentiated state. Remove any differentiated colonies under stereomicroscope if needed.
Aspirate medium from the well, wash cells once with 1 ml of DMEM/F12 medium.
-
Add 1 ml of Accutase to each well of 6-well tissue culture plate, and return the plate to the incubator.
Compared with trypsin, Accutase will yield single cell suspension with higher viability for hESCs/hiPSCs.
Check cells under microscope, colonies should be dissociated to single cells after 10 min incubation.
Using 1 ml tip, pipette up and down 3 to 4 times gently to detach cells from the well and obtain cell single cell suspension.
Transfer cell suspension to 15 ml tube, which has 5 ml DMEM/F12 medium added in advance.
Centrifuge the tube for 3 min at 1100 rpm at RT.
Aspirate supernatant, and re-suspend cells with 1 ml of neural induction medium by pipetting up and down 3 to 4 times gently using 1 ml tip.
Take 10 μl cell suspension, and mix with 10 μl of trypan blue solution.
Load 10 μl cell-trypan blue mixture to cell counting slide. Count and calculate live cell concentration.
-
Calculate dilution ratio and dilute cells in neural induction media. Final cell concentration should be 60,000 live cells/ml (9,000 live cells/150 μl media). Add Y-27632 (final concentration 50 μM) and heat-inactivated FBS (final concentration 5% [v/v]) to the cell suspension and gently mix.
Compared with its usage in monolayer culturing, higher concentration (50 μM) of Y-27632 was used for EB formation.
Whether or not to add FBS is cell line-dependent. Pre-testing may be needed to confirm whether FBS is required to form EBs.
-
Add 150 μl of single cell suspension into each well of ultra-low attachment 96-well plate. Place plate in the incubator. This is day 0 of the differentiation.
On day 1, a central EB is formed in each well.
On day 2, remove 75 μl medium from each well carefully without disturbing the EB at the bottom.
Add 150 μl of neural induction medium supplemented with 50 μM Y-27632 to each well, and return the plate to incubator.
-
On day 4, 6, and 8, remove 100 μl medium from each well, and add 150 μl neural induction medium to each well.
On day 10, EBs will be transferred to spinning culture (see basic protocol 2 and 3).
BASIC PROTOCOL 2: GENERATION OF CORTICL ORGANOIDS
After neural induction, EBs are allowed to differentiate without any further treatment of regionalization factors. This approach leads to the generation of hCOs, which mainly contain both excitatory and inhibitory neurons, and resemble the cortical organization in embryonic brain.
Materials
hPSC-derived EBs (Basic Protocol 1)
Neural differentiation medium minus vitamin A (see recipe)
Neural differentiation medium (see recipe)
Ultra-low-attachment 6-well plate (Corning, Cat No. CLS3471-24EA)
Orbital shaker (IKA, Cat No. KS260)
25 ml pipette
10 ml pipette
5 ml pipette
Expansion of neural progenitor cells (NPC)
-
On day 10, transfer EBs from ultra-low-attachment 96-well plate to ultra-low-attachment 6-well plate using 5 ml pipette.
Transfer should be performed cautiously so that EBs are not damaged.
Normally transfer less than 8 EBs to each well of ultra-low-attachment 6-well plate.
Make a little tilt of the plate to collect the EBs to one corner of each well. Remove neural induction medium from the 6-well plate.
Add 3 ml neural differentiation medium minus vitamin A to each well.
Place the plate on orbital shaker inside the incubator and start spinning culture at 80 rpm.
On day 12, 14 and 16, remove medium from the well, and add 3 ml neural differentiation medium minus vitamin A to each well.
Neural maturation and long term culture
-
1. On day 18, remove medium from the well, and add 3 ml neural differentiation medium.
BDNF and ascorbic acid are supplemented to facilitate long term neural maturation.
Return the plate to incubator for spinning culture.
-
Change neural differentiation medium every 4 days.
For cell lines showing poor proliferation rate, 20 ng/ml FGF2 could be added to neural differentiation medium for 4 to 6 days (until day 22 to day 24). The requirement of FGF2 for growth is cell line-dependent and should be optimized for each specific cell line used.
BASIC PROTOCOL 3: GENERATION OF MGE ORGANOIDS
In contrast to the spontaneous differentiation in hCO culture, in hMGEO protocol EBs are cultured with ventral patterning medium to induce MGE fate by activating the sonic hedgehog (SHH) signaling pathway (Maroof et al., 2013). Unlike hCOs, hMGEOs mainly produce interneurons during long-term culturing.
Materials
hPSC-derived EBs (Basic Protocol 1)
MGE patterning medium (see recipe)
Neural differentiation medium (Basic Protocol 2)
Ultra-low-attachment 6-well plate (Corning, Cat No. CLS3471-24EA)
Orbital shaker (IKA, Cat No. KS260)
25 ml pipette
10 ml pipette
5 ml pipette
MGE patterning
-
On day 10, transfer EBs from ultra-low-attachment 96-well plate to ultra-low-attachment 6-well plate using 5 ml pipette.
Transfer should be performed cautiously so that EBs are not damaged.
Normally transfer less than 8 EBs to each well of ultra-low-attachment 6-well plate.
Make a little tilt of the plate to collect the EBs to one corner of a well. Remove neural induction medium from the 6-well plate.
Add 3 ml MGE patterning medium to each well.
Place the plate on orbital shaker inside the incubator and start spinning culture at 80 rpm.
On day 12, 14 and 16, remove medium from the well, and add 3 ml MGE patterning medium to each well.
Neural maturation and long term culture
-
On day 18, remove medium from the well, and add 3 ml neural differentiation medium.
BDNF and ascorbic acid are supplemented to facilitate long term neural maturation.
Return the plate to incubator for spinning culture.
-
Change neural differentiation medium every 4 days.
No extra growth factor (e.g. FGF2) is needed for hMGEO protocol.
BASIC PROTOCOL 4: FUSION OF BRAIN ORGANOIDS TO MODEL INTERNEURON MIGRATION
The generation of regionally specified brain organoids can enable the study of region-specific biological processes during human brain development. For instance, fusing hMGEO and hCO together models the tangential migration of interneurons from MGE to cortex.
Materials
Neural differentiation medium (Basic Protocol 2)
Ultra-low-attachment 6-well plate (Corning, Cat No. CLS3471-24EA)
U-bottom ultra-low-attachment 96-well plate (Corning, Cat No. CLS7007-24EA)
Orbital shaker (IKA, Cat No. KS260)
25 ml pipette
10 ml pipette
Viral labeling (optional)
If not using reporter cell lines, organoids need to be subjected to suitable labeling (e.g. AAV1 or lentivirus) before organoid fusing for easier visualization of neuronal migration.
-
On day 18, transfer 1 hMGEO or hCO from ultra-low-attachment 6-well plate to 1 well of ultra-low-attachment 96-well plate using 25 ml pipette.
It is also possible to transduce cells on day 0 after single cell dissociation. However, transduction condition needs optimization for day 0 transduction by users.
Wait 30 sec until the organoid sink to the bottom of the well.
Remove the medium from the well using 200 μl pipette tip.
-
Add 150 μl neural differentiation medium containing diluted virus.
Optimal viral titer for efficient labeling should be tested for different viruses and viral stocks.
For lentivirus, protamine sulfate (1 μg/ml) should be supplemented to the medium.
-
24 hours after transduction, remove the medium form the well using 200 μl pipette tip.
Remove the medium as much as possible but do not damage the organoid on the bottom.
Add 200 μl neural differentiation medium to the well and remove. Repeat this step again in order to wash away remaining virus.
Add 200 μl neural differentiation medium to the well. Organoid is now ready for fusion.
Organoid fusion
-
On day 18, transfer 1 hMGEO and 1 hCO from ultra-low-attachment 6-well plate to one well of ultra-low-attachment 96-well plate using 25 ml pipette.
If viral labeling is performed, following the steps described above to prepare organoids for fusion. Then, transfer 1 hMGEO and 1 hCO to 1 well of ultra-low-attachment 96-well plate for fusion.
Wait 30 sec until both hMGEO and hCO sink to the bottom of the well.
-
Remove the medium from the well using 200 μl pipette tip.
Remove the medium as much as possible, but remain some to cover the organoids on the bottom.
Add 200 μl neural differentiation medium to the well.
Place the plate in the incubator and culture without spinning.
2 days after fusion, cautiously take out the plate and remove 150 μl medium from the well without disturbing the organoids on the bottom.
-
Slowly add 200 μl neural differentiation medium.
Add medium along the wall of the well cautiously, and try not to disturb the organoids on the bottom.
Return the plate to the incubator and culture without spinning.
-
3 days after fusion, transfer the fused organoid with the medium to 1 well of ultra-low-attachment 6-well plate using 25 ml pipette.
Add 3 ml medium neural differentiation medium to the 1 well of the 6-well plate before transferring.
1 well of 6-well plate can contain 2 fused organoids for long-term culture. Do not put too many fused organoids to the same well.
To allow efficient organoid development and to avoid physical damage of fused organoids, 6-well plate (but not 96-well plate or 24-well plate) is used here.
Place the plate on orbital shaker inside the incubator for spinning culture at 80 rpm.
REAGENTS AND SOLUTIONS
Neural induction medium
DMEM/F-12 (Life Technologies, Cat No. 11330057)
Knockout serum replacement (Life Technologies, Cat No. 10828028) 15% (v/v)
MEM-NEAA (Life Technologies, Cat No. 11140050) 1% (v/v)
Glutamax supplement (Life Technologies, Cat No. 35050) 1% (v/v)
β-Mercaptoethanol (Sigma, Cat No. M7522) 100 μM
LDN-193189 (Sigma, Cat No. SML0559) 100 nM
SB431542 (Abcam, Cat No. ab120163) 10 μM
XAV939 (Sigma, Cat No. X3004) 2 μM
Store up to 10 days at 4 °C
Neural differentiation medium minus vitamin A
DMEM/F-12 (Life Technologies, Cat No. 11330057)
Neurobasal medium (Life Technologies, Cat No. 2110349)
Insulin (Sigma, Cat No. I9278) 0.025% (v/v)
MEM-NEAA (Life Technologies, Cat No. 11140050) 0.5% (v/v)
Glutamax supplement (Life Technologies, Cat No. 35050) 1% (v/v)
Penicillin/Streptomycin (Life Technologies, Cat No. 15140-122) 1% (v/v)
N2 supplement (Life Technologies, Cat No. 17502-048) 0.5% (v/v)
B27 supplement without vitamin A (Life Technologies, Cat No. 12587010) 1% (v/v)
β-Mercaptoethanol (Sigma, Cat No. M7522) 50 μM
Store up to 8 days at 4 °C
MGE patterning medium
DMEM/F-12 (Life Technologies, Cat No. 11330057)
Dextrose (Sigma, Cat No. G7021) 0.15 (w/v)
β-Mercaptoethanol (Sigma, Cat No. M7522) 100 μM
N2 supplement (Life Technologies, Cat No. 17502-048) 1% (v/v)
B27 supplement without vitamin A (Life Technologies, Cat No. 12587010) 2% (v/v)
Recombinant SHH (R&D Systems, Cat No. 464-SH-200) 100 ng/ml
Purmorphamine (Stem Cell Biotech, Cat No. 72204) 1 μM
Store up to 8 days at 4 °C
Neural differentiation medium
DMEM/F-12 (Life Technologies, Cat No. 11330057)
Neurobasal medium (Life Technologies, Cat No. 2110349)
Insulin (Sigma, Cat No. I9278) 0.025% (v/v)
MEM-NEAA (Life Technologies, Cat No. 11140050) 0.5% (v/v)
Glutamax supplement (Life Technologies, Cat No. 35050) 1% (v/v)
Penicillin/Streptomycin (Life Technologies, Cat No. 15140-122) 1% (v/v)
N2 supplement (Life Technologies, Cat No. 17502-048) 0.5% (v/v)
B27 supplement (Life Technologies, Cat No. 17504-044) 1% (v/v)
β-Mercaptoethanol (Sigma, Cat No. M7522) 50 μM
BDNF (Peprotech, Cat No. 450-02) 20 ng/ml
Ascorbic acid (Sigma, Cat No. A92902) 200 μM
Optional: FGF2 (Millipore, Cat No. GF003AF) 20 ng/ml
Store up to 2 weeks at 4 °C
COMMENTARY
Background Information
Self-renewing and pluripotency features of hPSCs have greatly facilitated modeling the development of human nervous system and various neurological disorders (Mertens et al., 2016). After the first neural culture was produced from hESCs (Zhang et al., 2001) as monolayer, the field has continuously evolved towards the goal of producing 3D neural cultures. The earliest neural culture in a 3D condition dates back to 2005. Subsequently, the methods have improved to combine the EB-derived neural rosette culture with the serum-free condition to efficiently produce rosette structures and neurons (Eiraku et al., 2008; Watanabe et al., 2005). In order to facilitate the neuroectoderm differentiation, dual-SMAD inhibition approach was devised (Chambers et al., 2009), which largely minimized the un-directed differentiation and shorten the differentiation time. Embedding hPSC-derived EB in Matrigel has produced various brain regions in the absence of serum or inductive signals, leading to the discovery of cerebral organoid, the first entirely 3D human mini-brain culture (Lancaster et al., 2013). Soon after, introduction of TGFβ and Wnt inhibitors to the 3D culture enabled the generation of a more regionalized mini-brain culture-forebrain organoid (Kadoshima et al., 2013). These pioneering studies facilitated the initiation of a new era for human neural culture - brain organoids.
The use of human brain organoids has provided critical and unique opportunities to study human brain development (Bershteyn et al., 2017; Li et al., 2017). We and others have used single cell RNA sequencing (scRNA-seq) to dissect lineage specification in the developing brain organoids (Quadrato et al., 2017; Xiang et al., 2017). Cortical spheroids also facilitated the study of astrocyte genesis in 3D culture (Pasca et al., 2015). During brain development neural tube undergoes various regionalization. In order to reproduce this process, efforts have been made to specify different brain regions in the organoids. Additionally, different regions of the brain are structurally and functionally inter-connected, and one of the essential goals in the field has been to produce various substructures of the brain and study their interactions. Towards this goal, early in 2017, three groups reported the fusion/assembly of brain organoids/spheroids resembling the MGE/suppallium and cortex regions and reproduced the migration of cortical interneurons in 3D cultures (Bagley et al., 2017; Birey et al., 2017; Xiang et al., 2017). These findings collectively emphasized the importance of generating region-specific organoids and their interaction in understanding the step-wise human brain developmental processes.
To achieve MGE/subpallium fate, all the three groups utilized ventral patterning factors such as SHH and purmorphamine in our study (Xiang et al., 2017), and the smoothened agonist SAG used by the other two groups (Bagley et al., 2017; Birey et al., 2017; Xiang et al., 2017). While we and Birey et al. do not use Matrigel as a supportive matrix for organoid development and perform static organoid fusion, Bagely et al. embed brain organoids inside Matrigel. There are also other variations in the details of the protocols reported by these three groups (e.g. cell seeding strategy, timeline for neural induction and patterning, spinning or static culture conditions, et al.). Nevertheless, all three groups followed the same central principle for the production of MGE/subpallium organoids and for the modeling of interneuron migration, namely the activation of the SHH signaling pathway and the fusion of MGE/subpallium organoids with cortical organoids to recapitulate tangential migration of interneurons.
Critical Parameters and Troubleshooting
Initial quality control
The quality of hESCs or hiPSCs is critical for successful generation of hMGEOs and hCOs. The majority of the cells under culturing should remain un-differentiated before starting the protocol. While maintained under feeder-free condition, hESCs or hiPSCs may show certain spontaneous differentiation. This issue can be circumvented by reducing the plating density when passaging the cells, and by mechanically removing the differentiated cells under stereomicroscope before making single cells. A complete single cell suspension is essential for efficient EB formation after plating (typical suspension as shown in Figure 1A). For all the cell lines we tested, colonies became dissociated into single cells after 10 min of Accutase treatment. Nevertheless, researchers should check the cell condition under microscopy and decide to extend or decrease the treatment period if needed. We also noticed the efficiency of EB formation is cell-line dependent. Heat-inactivated FBS is supplemented to the medium for the first 2 days in order to aid EB formation. For some cell lines, however, FBS supplement is not necessary during this period. In addition, as reported by others (Lancaster et al., 2017), the number of cells seeded may also affect the development of brain organoid later. We generally seed 9,000 live cells per well to form EB. For different cell lines used, this plating number may be tested and optimized.
Figure 1.
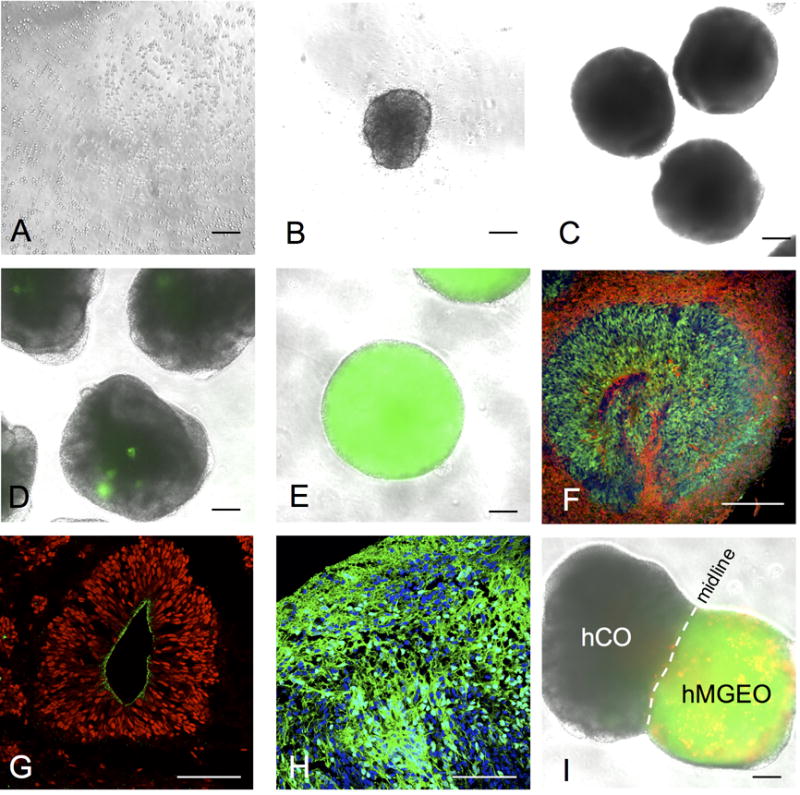
Different stages of human brain organoid culture. (A) Single cell suspension plated to ultra-low-attachment 96-well plate on day 0. (B) A EB formed in one well of ultra-low-attachment 96-well plate on day 1. (C) EBs transferred in to ultra-low-attachment 6-well plate on day 10. (D and E) On day 18, random clusters NKX2-1-GFP+ cells (green) are observed in hCOs (D), whereas hMGEOs typically show widespread NKX2-1-GFP+ cells (green) inside the organoids (E). (F) hCOs typically show PAX6+ (green) ventricular-like area and differentiated NeuN+ neurons (red) are located on the basal side. Image is from day 40 hCO cryosection. (G) SOX2+ cells (red) typically located in ventricular-like area in hCOs and the apical surface is N-cadherin+ (green). Image is from day 40 hCO cryosection. (H) NKX2-1+ cells (green) are typically enriched in hMGEOs. Image is from day 70 hMGEO cryosection. Scale bars in A-G represent 200 μm, and in H and L represent 100 μm. (I) Image of fused organoid 4 days after fusion. NKX2-1-GFP+ hMGEO was labeled with hSyn-RFP by lentivirus transduction before the fusion. Note some RFP+ cells already passed the midline at this stage. Scale bars represent 200 μm in A-E and I, and 100 μm in G and H.
Maintaining the organoid culture
After EB formation, medium change should always be performed cautiously so that the early EBs and later the regionalized brain organoids are not damaged. Matrigel embedding is not required in this protocol. However, other researchers have found dissolved Matrigel helps organoid development (Kadoshima et al., 2013; Qian et al., 2016). For long term culture of the organoids, adding dissolved Matrigel may be an option to consider. Adding extra growth factors (e.g. FGF2 or EGF) is not required to hMGEO production. However, some cell lines (e.g. H1 hESC line) may display low proliferation rate under hCO protocol. In such cases, we found adding FGF2 helped the growth of organoids. Therefore, researchers may test the requirement for extra growth factors (e.g. FGF2 and EGF) in the specific cell lines. Although variations may exist for different hESC or hiPSC lines in their capabilities to form brain organoids, as also observed in other’s studies (Lancaster et al., 2017), we found that once the condition is optimized for the specific cell line used, the productions of hMGEOs and hCOs remain consistent between different batches, especially for hMGEO protocol where neural differentiation is tightly guided by stepwise signaling controls.
Fusion control
Since the shape of each brain organoid may vary, sometimes hMGEO and hCO make contact each other via small contact area after they are transferred together. This will decrease the probability for successful fusion. In such cases, re-suspending the organoids by gentle pipetting using 200 μl tip to make hMGEO and hCO contact in a large contact area. Generally, increasing the contact surface between hMGEO and hCO benefits an efficient fusion. After returning the plate to static culture, any disturbance should be avoided during the first 48 hours of fusion. Extra caution is also needed during organoid transferring, because fused organoid may become separated if transferring is not performed gently. Transfer can be performed using either a 25 ml pipette or a cut 1 ml pipette tip, depending on the size of fused organoids. The key is to make sure that the fused organoid can flow through the opening of the pipette/cut-tip freely.
Anticipated Results
On day 0 of organoid production, a complete single cell suspension should be loaded to each well (Figure 1A). EBs will be formed inside each well within 24 hours after plating (Figure 1B). During the 10-day neural induction, EBs will gradually gain size and show smooth surface (Figure 1C). hMGEOs will be enriched with MGE-like progenitors by the end of ventral patterning period, whereas hCOs barely produce such progenitors at the same stage (Figure 1D and 1E). During further culture, hCOs will develop typical cellular organization as observed in embryonic brain. The ventricular-like area will be enriched with PAX6+ and SOX2+ progenitors, with the apical surface labeled with N-cadherin+ cells and the basal side labeled with differentiated NeuN+ neurons (Figure 1F and 1G). In hMGEOs, NKX2-1+ cells will be widely distributed inside the organoid (Figure 1H), resembling the developing MGE of the brain. Fusion of hMGEO and hCO can be performed after the period of NPC expansion in hCO and ventral patterning in hMGEO, and fused organoid can be further cultured with spinning (Figure 1I).
Time Considerations
Generally, fresh-thawed hESCs or hiPSCs need to recover for ~2 passages before making single cells, where cells are passaged once a week. Once single cells are plated, it will take 10 days to achieve efficient neuroectodermal induction, 8 days for ventral patterning in hMGEO protocol, and 8 days for NPC expansion in hCO protocol, respectively. The total time period required for neural maturation and further culture depends on the researcher’s goals in studying specific developmental stage. Generally, 1 month’s old hMGEOs and hCOs will be enriched with various neural progenitors. After ~2.5 months’ culture, both hMGEOs and hCOs will mainly contain differentiated neurons and glial cells (Xiang et al., 2017). Similarly, although fusion of regionally specified organoid only takes 3 days, the further culture as fused organoids also depends on the researcher’s specific purpose. We generally observe robust interneuron migration from hMGEO to hCO within 2~3 weeks after fusion, but the functional maturation and integration of migrated interneurons may take longer time.
Significance Statement.
Three-dimensional (3D) brain organoid culture is useful for investigating human brain development and disease modeling. The brain is composed of highly regionalized but connected structures, and one of critical goals in the field is to generate regionally specified brain organoid and model their interactions. Recently, three research groups, including the authors’ laboratory, reported the generation of regionally specified brain organoids, specifically distinct organoids resembling the developing human dorsal cortex (hCOs) or medial ganglionic eminence (hMGEOs). By fusing these two developmental distinct organoids together, researchers are able to model migration of inhibitory neurons, a process critical for the development of the brain. In this unit, we describe detailed protocols to generate and fuse human brain organoids.
Acknowledgments
I.-H. P. was partly supported by NIH (GM111667-01, 7R01AA025080-02, 7R01CA203011-2), CSCRF (12-SCB-YALE-11, 13-SCB-YALE-06), and KRIBB/KRCF research initiative program (NAP-09-3). We thank Dr. Andrew G. Elefanty for sharing HES-3 NKX2-1GFP/w human ES cell line and Dr. Stewart A. Anderson for sharing cortical interneuron differentiation protocol.
LITERATURE CITED
- Bagley JA, Reumann D, Bian S, Levi-Strauss J, Knoblich JA. Fused cerebral organoids model interactions between brain regions. Nature Methods. 2017;14:743. doi: 10.1038/nmeth.4304. + [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bershteyn M, Nowakowski TJ, Pollen AA, Di Lullo E, Nene A, Wynshaw-Boris A, Kriegstein AR. Human iPSC-Derived Cerebral Organoids Model Cellular Features of Lissencephaly and Reveal Prolonged Mitosis of Outer Radial Glia. Cell stem cell. 2017 Jan 9; doi: 10.1016/j.stem.2016.1012.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birey F, Andersen J, Makinson CD, Islam S, Wei W, Huber N, Fan HC, Metzler KRC, Panagiotakos G, Thom N, et al. Assembly of functionally integrated human forebrain spheroids. Nature. 2017;545:54–59. doi: 10.1038/nature22330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nature biotechnology. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiraku M, Watanabe K, Matsuo-Takasaki M, Kawada M, Yonemura S, Matsumura M, Wataya T, Nishiyama A, Muguruma K, Sasai Y. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell stem cell. 2008;3:519–532. doi: 10.1016/j.stem.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Jo J, Xiao Y, Sun AX, Cukuroglu E, Tran HD, Goke J, Tan ZY, Saw TY, Tan CP, Lokman H, et al. Midbrain-like Organoids from Human Pluripotent Stem Cells Contain Functional Dopaminergic and Neuromelanin-Producing Neurons. Cell stem cell. 2016;19:248–257. doi: 10.1016/j.stem.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoshima T, Sakaguchi H, Nakano T, Soen M, Ando S, Eiraku M, Sasai Y. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. P Natl Acad Sci USA. 2013;110:20284–20289. doi: 10.1073/pnas.1315710110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Corsini NS, Wolfinger S, Gustafson EH, Phillips AW, Burkard TR, Otani T, Livesey FJ, Knoblich JA. Guided self-organization and cortical plate formation in human brain organoids. Nature biotechnology. 2017;35:659–666. doi: 10.1038/nbt.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Muffat J, Omer A, Bosch I, Lancaster MA, Sur M, Gehrke L, Knoblich JA, Jaenisch R. Induction of Expansion and Folding in Human Cerebral Organoids. Cell stem cell. 2017;20:385–396. doi: 10.1016/j.stem.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroof AM, Keros S, Tyson JA, Ying SW, Ganat YM, Merkle FT, Liu B, Goulburn A, Stanley EG, Elefanty AG, et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell stem cell. 2013;12:559–572. doi: 10.1016/j.stem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens J, Marchetto MC, Bardy C, Gage FH. Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nat Rev Neurosci. 2016;17:424–437. doi: 10.1038/nrn.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muguruma K, Nishiyama A, Kawakami H, Hashimoto K, Sasai Y. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Rep. 2015;10:537–550. doi: 10.1016/j.celrep.2014.12.051. [DOI] [PubMed] [Google Scholar]
- Pasca AM, Sloan SA, Clarke LE, Tian Y, Makinson CD, Huber N, Kim CH, Park JY, O’Rourke NA, Nguyen KD, et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Methods. 2015;12:671–678. doi: 10.1038/nmeth.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian XY, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, Yao B, Hamersky GR, Jacob F, Zhong C, et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell. 2016;165:1238–1254. doi: 10.1016/j.cell.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadrato G, Nguyen T, Macosko EZ, Sherwood JL, Min Yang S, Berger DR, Maria N, Scholvin J, Goldman M, Kinney JP, et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature. 2017;545:48–53. doi: 10.1038/nature22047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi H, Kadoshima T, Soen M, Narii N, Ishida Y, Ohgushi M, Takahashi J, Eiraku M, Sasai Y. Generation of functional hippocampal neurons from self-organizing human embryonic stem cell-derived dorsomedial telencephalic tissue. Nature communications. 2015;6:8896. doi: 10.1038/ncomms9896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Kamiya D, Nishiyama A, Katayama T, Nozaki S, Kawasaki H, Watanabe Y, Mizuseki K, Sasai Y. Directed differentiation of telencephalic precursors from embryonic stem cells. Nat Neurosci. 2005;8:288–296. doi: 10.1038/nn1402. [DOI] [PubMed] [Google Scholar]
- Xiang Y, Tanaka Y, Patterson B, Kang YJ, Govindaiah G, Roselaar N, Cakir B, Kim KY, Lombroso AP, Hwang SM, et al. Fusion of Regionally Specified hPSC-Derived Organoids Models Human Brain Development and Interneuron Migration. Cell stem cell. 2017;21:383–398 e387. doi: 10.1016/j.stem.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SC, Wernig M, Duncan ID, Brustle O, Thomson JA. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nature biotechnology. 2001;19:1129–1133. doi: 10.1038/nbt1201-1129. [DOI] [PubMed] [Google Scholar]