

Abstract
Background
The sample ascertainment bias due to complex population structures remains a major challenge in genome-wide investigations of complex traits. In this study we derived the high-resolution population structure and levels of autozygosity of 377 Lipizzan horses originating from five different European stud farms utilizing the SNP genotype information of the high density 700 k Affymetrix Axiom™ Equine genotyping array. Scanning the genome for overlapping runs of homozygosity (ROH) shared by more than 50% of horses, we identified homozygous regions (ROH islands) in order to investigate the gene content of those candidate regions by gene ontology and enrichment analyses.
Results
The high-resolution population network approach revealed well-defined substructures according to the origin of the horses (Austria, Slovakia, Croatia and Hungary). The highest mean genome coverage of ROH (SROH) was identified in the Austrian (SROH = 342.9), followed by Croatian (SROH = 214.7), Slovakian (SROH = 205.1) and Hungarian (SROH = 171.5) subpopulations. ROH island analysis revealed five common islands on ECA11 and ECA14, hereby confirming a closer genetic relationship between the Hungarian and Croatian as well as between the Austrian and Slovakian samples. Private islands were detected for the Hungarian and the Austrian Lipizzan subpopulations. All subpopulations shared a homozygous region on ECA11, nearly identical in position and length containing among other genes the homeobox-B cluster, which was also significantly (p < 0.001) highlighted by enrichment analysis. Gene ontology terms were mostly related to biological processes involved in embryonic morphogenesis and anterior/posterior specification. Around the STX17 gene (causative for greying), we identified a ROH island harbouring the genes NR4A3, STX17, ERP44 and INVS. Within further islands on ECA14, ECA16 and ECA20 we detected the genes SPRY4, NDFIP1, IMPDH2, HSP90AB1, whereas SPRY4 and HSP90AB1 are involved in melanoma metastasis and survival rate of melanoma patients in humans.
Conclusions
We demonstrated that the assessment of high-resolution population structures within one single breed supports the downstream genetic analyses (e.g. the identification of ROH islands). By means of ROH island analyses, we identified the genes SPRY4, NDFIP1, IMPDH2, HSP90AB1, which might play an important role for further studies on equine melanoma. Furthermore, our results highlighted the impact of the homeobox-A and B cluster involved in morphogenesis of Lipizzan horses.
Electronic supplementary material
The online version of this article (10.1186/s12864-019-5564-x) contains supplementary material, which is available to authorized users.
Keywords: Lipizzan horse; Runs of homozygosity (ROH); ROH islands; NetView; Melanoma; HOXB cluster, selection signature
Background
The Lipizzan horse breed is globally one of the best-documented horse population, as pedigree records can be traced back to the known founder animals born in the early eighteenth century. The founder population, described in detail by Zechner et al. [1], Druml and Sölkner [2] and Druml et al. [3], comprises 456 animals, whereas the major part of horses originated from the former imperial stud farm of Lipica founded in the year 1580. Since the First World War the stud book of Lipizzan horses is closed and presently conservation breeding strategies are applied by eleven state stud farms located in nine European countries. National Lipizzan breeding herds have limited population sizes; therefore, maintenance of genetic diversity has been in the focus of state stud farms over decennia.
Numerous genetic approaches and methods have been applied to characterise the gene pool, the genetic diversity and the population structure of the Lipizzan breed. Comprehensive pedigree data (max. 31 generations, generation equivalent 19.1) were used to estimate inbreeding coefficients and effective population sizes by Zechner et al. [1], Sölkner and Druml [2] and Druml et al. [3]. Furthermore, microsatellite markers were employed to describe diversity measures, genetic distances and population structure by Achmann et al. [4]. Kavar et al. [5, 6] investigated mtDNA maternal diversity; Kasarda et al. [7] estimated genetic relatedness between Old Kladruber, Slovenian and Slovakian Lipizzans, whilst Wallner et al. [8] highlighted the patrilinear structure conducting haplotype-based analyses of the Y-chromosome. Most of these scientific publications arose from a multilateral research project, which is described in the review by Dovc et al. [9]. Based upon this scientific project further research was conducted focusing on the inheritance of melanoma, vitiligo and greying [10–13].
In this study we used the SNP genotype information of the Affymetrix Axiom™ Equine genotyping array [14] to analyse the high-resolution population structure of Lipizzan horses originating from five different European stud farms, which differ in breeding history and breeding objectives. To ascertain the high-resolution population structure of the gene pool, we applied a recently described three-step procedure as presented in Druml et al. [15], which includes individual levels of admixture and genomic inbreeding (ROH) in the final population network visualization. After the assessment of the high-resolution population structure, we identified overlapping homozygous regions (ROH islands) within the entire population and respective subpopulations, and conducted a gene ontology and enrichment analysis of annotated genes located within ROH islands. We demonstrate that the combination of different approaches elaborate insights in population structure and underlying differences in levels of autozygosity and distribution of ROH islands.
Results
Genetic diversity and admixture analysis
Principal Component analysis (PCA) as presented in Fig. 1a) revealed that the first three Principal Components (PCs) accounted for 35% of the total genetic variance. Based upon the visualization of the first three PCs the Austrian population is characterized by lower pairwise genetic distances and a distinct clustering. The other three subpopulations (Croatia, Hungary and Slovakia) appear to be highly interrelated simultaneously expressing a high level of genetic diversity, whilst a subset of the Austrian samples shares similarities with Croatian, Slovakian and Hungarian horses. FST analysis recapitulated these findings, as nearest relationships were documented for Austrian, Croatian and Slovakian samples (pairwise FST from 0.025 to 0.045), whereas the Hungarian subpopulation exhibited higher genetic differentiation (pairwise FST from 0.068 to 0.092) (Additional file 1).
Fig. 1.

Visualization of the dataset on the first 3 Principal Components (PCs) together explaining 35% of the variation. a On the left plot of PC1 and PC2; on the right plot of PC1 and PC3. b Graphical representation of individual cluster membership coefficients for Admixture runs increasing K = 2–8 for the Austrian, Croatian, Hungarian and Slovakian sample of Lipizzan horses
The first level (K = 2) of model-based clustering using the programme Admixture separated the Austrian population from the other Lipizzan samples (Fig. 1b). At the second level of clustering K = 3 the Hungarian sample were allocated in a distinct cluster. Further increasing K to 4 and 5 the Austrian subpopulation was further sub-structured, whilst at K = 6 the Slovakian sample formed a distinct cluster. At the additional levels of K = 7 and K = 8 the identified subpopulations were subsequently sub-structured. The visualization of the cross-validation (CV) error for each K, increasing K from 2 to 10, did not result in an optimal number of clusters (Additional file 2).
High-resolution network visualisation
Using a high-resolution network graph we combined individual levels of SROH (see the results below) and individual levels of admixture (K = 8). The network analysis improved the assessment of the population structure, compared to PCA and Admixture, by clearly separating the horses according to their origin, simultaneously highlighting four population outliers (Fig. 2). The horses of the stud farm Piber characteristically were connected by short and thick links, representing higher co-ancestry and lower genetic distance. The network highlighted two sub-clusters with higher levels of admixture, which were linked to Slovakian and Hungarian Lipizzans (marked with arrows in Fig. 2). According to pedigree information these cross-link animals were foreign bred mares, which were integrated into the Piber breeding population and thus are characterized by a lower co-ancestry level (longer and thinner links) and smaller SROH values (smaller node sizes).
Fig. 2.

High-resolution network visualisation of Lipizzan horses from the stud farms Piber (Austria), Topol’čianky (Slovakia), Đakovo and Lipik (Croatia) and Szilvasvárad (Hungary). a On the top network the horses (nodes) are colored by stud farm. On the lower network (b) nodes are containing individual cluster membership coefficients from Admixture analysis. Each node represents an individual and the size of node is in relation to SROH, link lengths between nodes illustrate the genetic distance between individuals, thickness of links are proportional to genetic relationship. Two arrows pinpoint sub-clusters in the Austrian sample
The Croatian sample was separated from the Austrian sample, connected directly by one cross-link animal. The network analysis identified three sub-clusters within the Croatian sample. According to pedigree information, the cluster on the right involved horses from the stud farm Lipik, whilst the left cluster represented the core population from the stud farm Đakovo. The accumulation in the middle of the Croatian sample included horses from Đakovo, which showed high levels of admixture with horses from Lipik, as well as with Slovakian and Hungarian Lipizzans, respectively. The Hungarian sample was characterised by the highest genetic distances between the samples and by the lowest SROH values, indicating genealogical separation between Hungarian and Austrian Lipizzans. The Slovakian sample showed genetic relationships in both directions – to the Austrian and to the Croatian samples. These relationships were illustrated by manifold cross-link animals, which had small SROH values and higher degree of admixture connecting both clusters.
Runs of homozygosity
Including the entire sample of Lipizzan horses in the ROH analysis the mean number of ROH (NROH) comprised 202.07 segments at a mean genome length covered by ROH (SROH) of 297.01 Mb (+ − 119.85), resulting in a mean ROH length (LROH) of 1.42 Mb (Table 1).
Table 1.
Mean values, standard deviation (SD), minimum and maximum values for the variables SROH (in Mb), NROH, LROH (in Mb) and FROH for the entire Lipizzan sample, the Austrian population sample of the stud farm Piber, the population of the Slovakian stud farm of Topol’čianky, the Croatian and the Hungarian sample
| Total sample | n | mean | SD | Min. | Max. |
|---|---|---|---|---|---|
| SROH | 377 | 297.01 | 119.85 | 1.91 | 550.21 |
| NROH | 202.07 | 51.96 | 3.00 | 333.00 | |
| LROH | 1.42 | 0.43 | 0.56 | 2.45 | |
| FROH | 0.13 | 0.05 | 0.00 | 0.25 | |
| Piber/Austria | |||||
| SROH | 254 | 342.88 | 105.43 | 3.35 | 550.21 |
| NROH | 211.85 | 46.00 | 6.00 | 333.00 | |
| LROH | 1.59 | 0.41 | 0.56 | 2.45 | |
| FROH | 0.15 | 0.05 | 0.00 | 0.25 | |
| Topol’čianky/Slovakia | |||||
| SROH | 55 | 205.05 | 76.75 | 30.38 | 359.72 |
| NROH | 178.58 | 50.81 | 48.00 | 301.00 | |
| LROH | 1.06 | 0.17 | 0.63 | 1.35 | |
| FROH | 0.09 | 0.03 | 0.01 | 0.16 | |
| Lipik, Đakovo/Croatia | |||||
| SROH | 45 | 214.66 | 85.99 | 81.31 | 401.22 |
| NROH | 186.87 | 47.38 | 96.00 | 320.00 | |
| LROH | 1.11 | 0.22 | 0.85 | 1.62 | |
| FROH | 0.10 | 0.04 | 0.04 | 0.18 | |
| Szilvasvárad/Hungary | |||||
| SROH | 23 | 171.53 | 112.58 | 1.91 | 402.80 |
| NROH | 158.39 | 83.30 | 3.00 | 131.00 | |
| LROH | 0.98 | 0.22 | 0.64 | 1.44 | |
| FROH | 0.07 | 0.05 | 0.00 | 0.18 | |
ROH parameters varied between samples. Highest estimates of SROH and NROH were identified for horses originating from the stud farm of Piber/Austria (SROH = 342.88 Mb; NROH = 211.85), followed by the Croatian horses (SROH = 214.66 Mb; NROH = 186.87) and Slovakian horses (SROH = 205.06 Mb; NROH = 187.58). Lowest SROH and NROH were found in the Hungarian sample (SROH = 171.53 Mb; NROH = 158.39) (Table 1). Five Austrian Lipizzan horses reached the highest SROH with more than 500 Mb, whereas the smallest SROH were detected in three Hungarian Lipizzan horses (1.91 Mb to 20 Mb).
Inbreeding as defined by FROH reached a mean value of 13.2% (+ − 5.3%) within the entire sample, ranging from 15.3% (+ − 4.7%) in the Piber population to 7.6% (+ − 5.0%) in the Hungarian sample (Table 1). To differentiate old and recent inbreeding we calculated FROH based upon different ROH length classes (Table 2). The values for FROH < 2Mb were highest in the Austrian (6.7%) and lowest in the Hungarian sample (5.6%). To quantify recent inbreeding we calculated FROH considering ROHs longer than 10 MB. Recent inbreeding was absent in the Slovakian sample and tended to zero in the Hungarian sample, whereas the highest value of 1.1% was observed for the Austrian sample.
Table 2.
Distribution of ROHs and FROH (in percent) in different length classes (0.5–1 Mb, 1–2 Mb, 2–4 Mb, 4–6 Mb, 6–8 Mb, 8–10 Mb, > 10 Mb) respectively cumulative length classes (< 1 Mb, < 2 Mb, < 4 Mb, < 6 Mb, < 8 Mb, < 10 Mb, total FROH) for the Austrian, Croatian, Hungarian, Slovakian samples and the entire data set (All)
| Distribution ROH in length classes | Austria | Croatia | Hungary | Slovakia | All |
|---|---|---|---|---|---|
| 0.5–1 Mb | 22.7 | 37.7 | 41.0 | 40.0 | 28.1 |
| 1–2 Mb | 21.3 | 31.1 | 32.1 | 32.3 | 24.8 |
| 2–4 Mb | 23.9 | 22.2 | 19.4 | 21.4 | 23.1 |
| 4–6 Mb | 12.9 | 5.7 | 5.2 | 4.8 | 10.4 |
| 6–8 Mb | 7.6 | 2.2 | 1.2 | 0.9 | 5.6 |
| 8–10 Mb | 4.2 | 0.4 | 0.4 | 0.5 | 3.0 |
| > 10 Mb | 7.4 | 0.6 | 0.6 | 0.0 | 5.1 |
| FROH per length class | |||||
| FROH 0.5 -1Mb | 3.5 | 3.6 | 3.1 | 3.7 | 3.5 |
| FROH 1-2Mb | 3.3 | 3 | 2.5 | 3 | 3.1 |
| FROH 2-4Mb | 3.7 | 2.2 | 1.5 | 2 | 3.1 |
| FROH 4-6Mb | 2 | 0.6 | 0.4 | 0.4 | 1.5 |
| FROH 6-8Mb | 1.2 | 0.2 | 0.1 | 0.1 | 0.8 |
| FROH 8-10Mb | 0.6 | 0 | 0 | 0 | 0.4 |
| FROH > 10Mb | 1.1 | 0.1 | 0 | 0 | 0.8 |
| FROH per length class (cum.) | |||||
| FROH < 1Mb | 3.5 | 3.6 | 3.1 | 3.7 | 3.5 |
| FROH < 2Mb | 6.7 | 6.5 | 5.6 | 6.6 | 6.6 |
| FROH < 4Mb | 10.4 | 8.7 | 7.1 | 8.6 | 9.7 |
| FROH < 6Mb | 12.4 | 9.3 | 7.5 | 9 | 11.2 |
| FROH < 8Mb | 13.5 | 9.5 | 7.6 | 9.1 | 12 |
| FROH < 10Mb | 14.2 | 9.5 | 7.6 | 9.1 | 12.5 |
| FROH total | 15.3 | 9.6 | 7.6 | 9.1 | 13.2 |
ROH patterns as revealed by the plot NROH versus SROH indicate for the majority of the Piber population a strong right shift towards a higher SROH and an increased variance of SROH (Additional file 3). The samples of Slovakia and Croatia were distributed along the diagonal, whereas in the left corner of the plot a cluster of 11 horses with the lowest ROH parameters can be observed, indicating out-breeding. These horses were also clearly identified as cross-link animals between stud farms within the high-resolution population network (Fig. 2).
Concerning the distribution of ROH segments of different length categories, the Austrian Lipizzan population is characterized by the lowest proportion of ROH smaller than 2 Mb (21.3%), which varied in the other samples between 31.1 and 32.3% (Table 2). At the same time the Austrian horses had the highest proportion of ROHs longer than 6 Mb (19.2%). In the other samples this ROH category was present at an amount from 1.4 to 3.2%. In the Austrian sample 7.4% of ROHs were longer than 10 Mb, whereas in the other samples this category was underrepresented at a percentage of < 0.6%.
ROH islands
Within the entire dataset, we identified three ROH islands on ECA11 and two islands on ECA14, which were shared by more than 50% of the horses (Table 3). The overlapping homozygous region on ECA11:24.13–24.81 Mb was shared by 61.3% of animals and harboured the genes COPZ2, CBX1, SNX11, HOXB1, HOXB2, HOXB3, HOXB5, HOXB6, HOXB7, HOXB8, HOXB13, TTLL6. Figure 3a illustrates this island and the position of the homeobox-B cluster (HOXB) for each sample separately. These overlapping homozygous regions were nearly identical in position and length in all samples and the frequency varied between 59.2% (Slovakia) and 72.7% (Hungary).
Table 3.
ROH islands, shared by more than 50% (ROH freq.) of the entire sample of Lipizzan horses and the annotated genes in corresponding regions
| Chr. | Begin | End | Length (kb) | ROH freq. | Known genes |
|---|---|---|---|---|---|
| 11 | 24,134.125 | 24,816.254 | 682.1 | 0.613 | COPZ2, CBX1, SNX11, HOXB1, HOXB2, HOXB3, HOXB5, HOXB6, HOXB7, HOXB8, HOXB13, TTLL6 |
| 11 | 30,677.562 | 30,685.590 | 8.0 | 0.501 | – |
| 11 | 31,015.821 | 31,942.748 | 926.9 | 0.666 | C11H17orf67, DGKE, COIL, SCPEP1, AKAP1, MSI2 |
| 14 | 34,988.691 | 34,999.535 | 10.8 | 0.501 | – |
| 14 | 34,651.534 | 34,935.341 | 283.8 | 0.514 | SPRY4, NDFIP1 |
Fig. 3.

a Plot of the ROH islands containing the HOXB cluster at ECA11:24.13–24.81 Mb for each single sample. In Figure b) the island on ECA11:31.01–31.94 Mb including the genes C11H17orf67, DGKE, COIL, SCPEP1, AKAP1, MSI2 is shown
The second ROH island on ECA11 (position 31,015.821 - 31,942.748) was up to 927 kb long in Austrian and Croatian samples and was shared by 72.8 and 62.6% of the horses, respectively (Fig. 3b). The Slovakian Lipizzans had two overlapping homozygous segments in this region. Within the Hungarian sample, 47.0% of horses shared a much smaller island, which was directly located in the Musashi RNA binding protein 2 (MSI2).
We identified a ROH island on ECA25 around the STX17 (syntaxin-17) gene responsible for grey coat colour, shared by 46.2% of horses within the entire sample, containing the genes NR4A3, STX17, ERP44 and INVS. This ROH island was embedded in the centre of a 1.38 Mb long homozygous region at position ECA25:5.69–7.07 Mb present in 36.6% of Lipizzan horses. The frequency was below our expectations. From a previous study, it is known that the STX17 genotype frequencies for homozygous G/G horses reached up to 67.3% in Lipizzans [13]. Scanning this region for ROHs shorter than 500 kb, we applied a smaller boundary (80 kb window length, min. 20 SNPs), and detected a 399.9kb long ROH island at position ECA25:6394.110-6794.044 centred round the STX17 gene that was shared by 66.1% of the entire sample (Fig. 4). Across the subpopulations the frequencies for this ROH island was highest in the Austrian sample (71.2%) and lowest in the Croatian sample (51.1%).
Fig. 4.

ROH island around the STX17 gene (begin and end illustrated by red vertical reference lines) at ECA25:6.394.110–6.794.044. Subpopulations are presented in different colours: Austria = blue, Slovakia = brown, Croatia = red, Hungary = green
Considering the Austrian sample we found 16 ROH islands, which were located on ECA3 (2 islands), ECA5, ECA7 (2 islands), ECA8, ECA11 (3 islands), ECA14, ECA16 (4 islands), ECA18 and ECA20 (Table 4). The ROH islands ranged in length between 20.2 kb and 953.3 kb, whereas the longest fragment on ECA11 at 31.01–31.94 Mb was shared by the highest number of individuals (72,8%), followed by the ROH island on ECA11 at position 24.13–24.81 Mb, which was observed in 61,1% of animals. Within the Austrian sample eight ROH islands on ECA3, ECA5, ECA8, ECA11, ECA14, ECA16, ECA18 and ECA20 were private.
Table 4.
ROH islands, shared by more than 50% (ROH freq.) of the Lipizzan horses from the Austrian stud farm Piber and the annotated genes in corresponding regions
| Chr. | Begin | End | Length (kb) | ROH freq. | Known genes |
|---|---|---|---|---|---|
| 3 | 118,669.793 | 118,769.903 | 100.1 | 0.508 | UVSSA, MAEA |
| 3 | 118,809.979 | 118,893.880 | 83.9 | 0.506 | CTBP1 |
| 5 | 48,702.703 | 49,211.103 | 508.4 | 0.530 | CHD1L, FMO5, PRKAB2 |
| 7 | 50,434.281 | 50,606.416 | 172.1 | 0.507 | – |
| 7 | 50,636.963 | 50,733.100 | 96.1 | 0.508 | – |
| 8 | 93,409.518 | 93,605.837 | 196.3 | 0.538 | CTDP1 |
| 11 | 24,134.125 | 24,816.254 | 682.1 | 0.611 | COPZ2, CBX1,SNX11, HOXB1, HOXB2, HOXB3, HOXB5, HOXB6, HOXB7, HOXB8, HOXB13, TTLL6 |
| 11 | 30,607.721 | 30,687.266 | 79.6 | 0.526 | – |
| 11 | 31,015.821 | 31,942.748 | 926.9 | 0.728 | C11H17orf67, DGKE, COIL, SCPEP1, AKAP1, MSI2 |
| 14 | 41,779.891 | 42,365.786 | 585.9 | 0.546 | FSTL4 |
| 16 | 37,870.208 | 38,823.469 | 953.3 | 0.542 | C16H3orf84, KLHDC8B, CCDC71, LAMB2, USP19, QARS, QRICH1, IMPDH2, NDUFAF3, DALRD3, ARIH2, WDR6, P4HTM, IP6K2, NCKIPSD, CELSR3, TMEM89, UQCRC1, SLC26A6, UCN2, PFKFB4, SHISA5, TREX1, ATRIP, CCDC51, PLXNB1, NME6, ECATH-3, ECATH-2 |
| 16 | 39,675.098 | 39,695.300 | 20.2 | 0.507 | – |
| 16 | 39,890.148 | 39,920.855 | 30.7 | 0.507 | CCDC12 |
| 16 | 40,077.806 | 40,159.633 | 81.8 | 0.514 | TMIE, ALS2CL |
| 18 | 635.232 | 1235.687 | 600.5 | 0.528 | ARHGEF4, PLEKHB2 |
| 20 | 43,065.487 | 43,166.223 | 100.8 | 0.507 | CAPN11, SLC29A1, HSP90AB1, SLC35B2, NFKBIE, TCTE1 |
Within the Slovakian sample we detected six ROH islands on ECA11 (3), ECA14, ECA16 and ECA22, whereas the islands on ECA11 and ECA16 overlapped with corresponding islands of the Austrian Lipizzans (Table 5). The ROH islands on ECA22:47.90–48.48 Mb and ECA14:34.65–35.08 Mb were also identified in the Croatian sample.
Table 5.
ROH islands, shared by more than 50% (ROH freq.) of the Lipizzan horses from the Slovakian stud farm Topol’čianky and the annotated genes in corresponding regions
| Chr. | Begin | End | Length (kb) | ROH freq. | Known genes |
|---|---|---|---|---|---|
| 11 | 24,140.999 | 24,793.573 | 652.6 | 0.598 | COPZ2, CBX1,SNX11, HOXB1, HOXB2, HOXB3, HOXB5, HOXB6, HOXB7, HOXB8, HOXB13, TTLL6 |
| 11 | 31,000.312 | 31,347.338 | 347.0 | 0.552 | C11H17orf67, DGKE |
| 11 | 31,507.758 | 31,942.748 | 435.0 | 0.567 | AKAP1, MSI2 |
| 14 | 34,651.534 | 35,086.649 | 435.1 | 0.609 | SPRY4, NDFIP1, GNPDA1, RNF14 |
| 16 | 39,675.098 | 39,775.712 | 100.6 | 0.507 | SETD2 |
| 22 | 47,905.031 | 48,488.839 | 583.8 | 0.576 | CDH4, TAF4, LSM14B, PSMA7, SS18L1, MTG2, HRH3, OSBPL2, ADRM1, LAMA5, CABLES2, RBBP8NL, GATA5 |
Within the Croatian sample six ROH islands on ECA4, ECA7, ECA11 (2 islands), ECA14, ECA22, which varied in length between 129.1 kb and 1.14 Mb, were identified. The longest overlapping homozygous region on ECA14:34.05–35.18 Mb was shared by 72.6% of horses and contained nine annotated genes (Table 6).
Table 6.
ROH islands, shared by more than 50% (ROH freq.) of the Lipizzan horses from the Croatian stud farms Lipik and Đakovo and the annotated genes in corresponding regions
| Chr. | Begin | End | Length (kb) | ROH freq. | Known genes |
|---|---|---|---|---|---|
| 4 | 58,110.547 | 58,620.749 | 510.2 | 0.529 | HOXA1, HOXA2, HOXA3, HOXA5, HOXA6, HOXA7, HOXA9, HOXA10, HOXA11, EVX1, HIBADH |
| 7 | 52,014.500 | 52,143.583 | 129.1 | 0.532 | – |
| 11 | 24,144.431 | 24,816.254 | 671.8 | 0.592 | COPZ2, CBX1,SNX11, HOXB1, HOXB2, HOXB3, HOXB5, HOXB6, HOXB7, HOXB8, HOXB13, TTLL6 |
| 11 | 31,032.580 | 31,942.748 | 910.2 | 0.626 | C11H17orf67, DGKE, COIL, SCPEP1, AKAP1, MSI2 |
| 14 | 34,051.957 | 35,189.508 | 1137.5 | 0.726 | ARHGAP26, FGF1, SPRY4, NDFIP1, GNPDA1, RNF14, PCDH12, PCDH1, DELE1 |
| 22 | 47,901.830 | 48,624.259 | 722.4 | 0.549 | TAF4, CDH4, LSM14B, PSMA7, SS18L1, MTG2, HRH3, OSBPL2, ADRM1, LAMA5, CABLES2, RBBP8NL, GATA5, SLCO4A1, NTSR1 |
The island on ECA4 at position 58.11–58.62 Mb was also present in the Hungarian Lipizzans and contained among other annotated genes the homeobox-A cluster (HOXA). Furthermore, an island on ECA22 at position 47.90–48.62 Mb was exclusively detected within the Croatian and Slovakian sample.
Within the Hungarian Lipizzans six ROH islands on ECA4, ECA8, ECA11, ECA22, ECA23, ECA30 were identified, varying in length between 497.4 kb and 699.2 kb (Table 7). Four ROH islands on ECA8, ECA22, ECA23 and ECA30 were specific for the Hungarian sample. On ECA8 also a private island of the Austrian sample was documented (Fig. 5).
Table 7.
ROH islands, shared by more than 50% (ROH freq.) of the Lipizzan horses from the Hungarian stud farms Szilvasvárad and the annotated genes in corresponding regions
| Chr. | Begin | End | Length (kb) | ROH freq. | Known genes |
|---|---|---|---|---|---|
| 4 | 58,107.774 | 58,605.189 | 497.4 | 0.522 | HOXA1, HOXA2, HOXA3, HOXA5, HOXA6, HOXA7, HOXA9, HOXA10, HOXA11, EVX1 |
| 8 | 22,121.802 | 22,651.589 | 529.8 | 0.602 | TMEM120B, RHOF, SETD1B, HPD, PSMD9, WDR66, BCL7A, MLXIP, IL31, LRRC43, B3GNT4, DIABLO, CLIP1 |
| 11 | 24,134.125 | 24,833.366 | 699.2 | 0.727 | COPZ2, CBX1,SNX11, HOXB1, HOXB2, HOXB3, HOXB5, HOXB6, HOXB7, HOXB8, HOXB13, TTLL6, CALCOCO2 |
| 22 | 45,518.067 | 46,096.341 | 578.3 | 0.522 | ZNF831, EDN3, PHACTR3 |
| 23 | 27,658.254 | 28,189.490 | 531.2 | 0.522 | UHRF2, GLDC, KDM4C |
| 30 | 4781.530 | 5465.214 | 683.7 | 0.522 | KIF26B, SMYD3 |
Fig. 5.

ROH distribution on ECA8 for the entire Lipizzan sample containing one private ROH island for the Hungarian sample (blue line) and one ROH island for the Austrian sample (red line) in the line plot below
Gene ontology and enrichment analysis
Gene ontology and enrichment analysis highlighted within all Lipizzan samples the homeobox-B cluster, where the highest significance levels (p < 0.001) were reached for the terms GO:0048704~embryonic skeletal system morphogenesis and GO:0009952~anterior/posterior pattern specification (Additional file 4). Further the term GO:0043565~sequence-specific DNA binding related to molecular functions reached a significance level of p < 0.001 for the entire Lipizzan sample. Three out of 20 genes located in the identified ROH islands were listed in OMIM (Online Mendelian Inheritance in Man) database (DGKE - 615,008~Hemolytic uremic syndrome; HOXB1–614744~Facial paresis; SPRY4–615266~Hypogonadotropic hypogonadism), but none of the identified genes were listed in OMIA (Online Mendelian Inheritance in Animals) database for horses.
In the Austrian Lipizzan population (Additional file 5) GO analysis confirmed the aforementioned terms in biological processes (GO:0048704 and GO:0009952 containing the HOXB cluster), and highlighted additionally the five terms GO:0021570~rhombomere 4 development, GO:0021612~facial nerve structural, GO:0006183~GTP biosynthetic process, GO:0006950~response to stress and GO:0071353~cellular response to interleukin-4, related to biological processes.
The Slovakian Lipizzans (Additional file 6) where characterized by three specific terms related to biological processes: GO:0048864~stem cell development, GO:0001525~angiogenesis, GO:0006368~transcription elongation from RNA polymerase II promoter.
GO analysis revealed 12 identical terms, related to biological processes for the Hungarian and Croatian samples and additionally two sample specific terms (GO:0006355~regulation of transcription, DNA-templated; GO:0001578~microtubule bundle formation) for the Hungarian and three terms for the Croatian sample (GO:0007156~homophilic cell adhesion via plasma membrane adhesion molecules; GO:0050890~cognition; GO:0001759~organ induction) (Additional files 7 and 8). All these 12 terms related to biological processes are based upon the HOXB- and HOXA-clusters. The highest significance levels (p < 0.001) were found in concordance with the other samples for GO:0009952~anterior/posterior pattern specification and GO:0048704~embryonic skeletal system morphogenesis. Summarizing the results from gene ontology analysis of these two samples a high number of terms were highlighted for different aspects of embryonic morphogenesis (f.e. GO:0009953~dorsal/ventral pattern formation, GO:0009954~proximal/distal pattern formation, GO:0008584~male gonad development, GO:0060065~uterus development, GO:0021615~glossopharyngeal nerve morphogenesis).
Single SNP analysis
Two islands in the entire sample and six islands in the Austrian Lipizzans were remarkable short (8.0–101.1 kb length) and matched in size with single genes. One short island on ECA3:118.67–118.77 Mb (length 101.1kb) directly overlapped with the genes UVSSA (UV-stimulated scaffold protein A) and MAEA (macrophage erythroblast attacher) and was shared by 50.8% of the Austrian horses. 76 kb upstream a second ROH island (ECA3:118.80–118.89 Mb, length 83.9kb) was centred on the CTBP1 gene (C-terminal binding protein 1), that was shared by 50.2% of Austrian Lipizzans. To examine levels of homozygosity for SNPs located within the three named genes, we extracted the following SNPs from the entire sample: UVSSA: AX-104358355 Pos.:118,712.729, AX-104277358 Pos.:118,716.414, AX-103191894 Pos.:118,724.527, AX-104669126 Pos.:118,727.858; MAEA: AX-104046583 Pos.:118,750.308, AX-104041685 Pos.:118,752.167; CTBP1: AX-103592395 Pos.:118,874.036. The four SNPs within UVSSA revealed homozygosity for 78.2% of individuals. For the two SNPs located in MAEA 90.5% of animals were homozygous in the Austrian population and 78.5% of the horses were homozygous for these SNPs within the entire sample. At the SNP located in CTBP1 all horses of the entire sample were homozygous.
Discussion
The high-resolution population network (Admixture and Netview analysis) of the Lipizzan horse breed revealed that the single stud farms represent differentiated subpopulations, whereas the highest genetic distances were observed between the Austrian and the Hungarian/Croatian samples. The Slovakian Lipizzans clustered between the Austrian and the Croatian Lipizzans. These findings are in concordance with Achmann et al. [4], who derived a comparable structure for 561 Lipizzans from seven European countries. In the present study the Austrian and Slovakian horses were characterized by a lower genetic distances and higher pairwise relationship coefficients compared to the Croatian and Hungarian animals. This can be explained by the limited census of breeding animals in the Slovakian stud farm Topol’čianky, and the limited level of introgression of horses into the Austrian breeding herd. Due to the obligatory performance test of stallions in the Spanish riding school, in the Austrian stud farm Piber only foreign mares can be used to increase genetic diversity, whereas in Hungary, Slovakia and Croatia an exchange of breeding stallions is commonly applied. At the same time the Croatian and Hungarian breeding stock has been bigger than the Slovakian one.
Generally genetic distances between horses, illustrated in the high-resolution network visualization, was relatively equally distributed within the breeding herds of Piber and Topol’čianky and no outstanding contribution of single animals (key-contributor) were detected, revealing the efforts of a conservation breeding program with narrow sex ratios and moderate selection intensity. Between Piber, Szilvasvárad and Topol’čianky cross-link animals were found, which showed markedly smaller mean genome-wide coverage of ROH and a higher degree of admixture. A comparable “outbreeding-effect” was detected in a previous study in the Haflinger breed, where outcrosses between horses from different countries/stud books were characterised by low SROH and the lack of ROHs > 6 Mb [15].
All ROH parameters observed in our four subpopulations, reached highest values in the Austrian Lipizzans, whereas the lowest values were found in the Hungarian sample. The values of FROH generally were in concordance with the findings of pedigree analyses from Zechner et al. [1] and did not exceed 15%. Genomic inbreeding, as described by FROH in the Lipizzan breed (FROH = 0.13) was lower than expected from long-term stud farm breeding at small census and it is comparable to values found in the Austrian Haflinger (0.13), the Slovenian Haflinger (0.12) and the Bosnian Mountain Horse (0.14) [15, 16]. Higher levels of FROH were identified in Shagya Arabians (FROH = 0.16) and Purebred Arabians (FROH = 0.18), two breeds characterized by early closure of stud books [15].The length of ROHs is expected to correlate to ancient and recent inbreeding due to number of recombination events. Thus, recent inbreeding events result in longer ROHs as only a few identical by descent (IBD) segments can be broken down by repeated meiosis. According to Browning and Browning [17] and Thompson [18] ROHs of a length of 16.6, 10.0 and 5.0 Mb are assumed to origin from common ancestors back in the 3rd, 5th and 10th generations (6, 10 and 20 meioses respectively). Considering a mean generation interval of 10 years in Lipizzan horses the origin of ROHs in length class of < 2 Mb can be dated to the foundation period of this breed (foundation time 1580 AC). We observed FROH values calculated based upon ROHs shorter than 2 Mb ranging from 5.6 to 7.1%. FROH > 10Mb was absent in Slovakian Lipizzans and tended toward zero in Croatian and Hungarian horses, indicating minimal recent inbreeding within the past five generations [18, 19]. In the Austrian population FROH > 10Mb reached 1.1%. These results demonstrated that it is feasible to minimize the increase of inbreeding in populations limited in size and closed stud books. The ROH profile illustration for the Austrian Lipizzan revealed a strong right shift and a higher variance typical for consanguineous and bottlenecked populations according to Ceballos et al. [20]. The Slovakian sample was characterized by a similar profile, but at a much lower extend, whereas the Croatian and Hungarian samples showed the effects of small census [20].
The identification of overlapping homozygous regions confirmed a closer genetic relationship between the Hungarian and Croatian samples as well as between the Austrian and Slovakian samples, which was also highlighted within the high-resolution population network. Private islands were detected for the Hungarian sample on ECA8, ECA14, ECA23 and ECA30 and for the Austrian sample on ECA3, ECA5, ECA8, ECA16 and ECA20. Within the Austrian Lipizzans an overlapping 953.3kb long homozygous region on ECA16:37.87–38.82 Mb was found, that contained among 29 annotated genes the two genes ECATH-3 and ECATH-2. Both were described as functional genes encoding Cathelicidin-derived antimicrobial peptides, involved in the peptide-based host defence of neutrophils and epithelia in horses [21, 22]. GO analysis for the Austrian Lipizzan gene list highlighted the term GO:0006950~response to stress for the gene UCN2 (urocortin 2, a member of the corticotropin-releasing hormone family) on ECA16 and the gene HSP90AB1 (Equus caballus heat shock protein 90 alpha family class B member 1) on ECA20, whereas the latter was assigned to four Equus caballus species by Vidale et al. [23]. HSP90AB1 is considered to play a role in the age-dependent metabolism of chondrocytes in horses [24].
All subpopulations shared a homozygous region on ECA11, nearly identical in position and length (mean length 676.4 kb) at 24.13–24.81 Mb containing the annotated genes COPZ2, CBX1, SNX11, HOXB1, HOXB2, HOXB3, HOXB5, HOXB6, HOXB7, HOXB8, HOXB13, TTLL6 (and CALCOCO2, additional in the Hungarian sample). For the Croatian/Hungarian Lipizzans an additional island on ECA4:58.10–58.60 Mb was confirmed containing the homeobox-A cluster. Enrichment analysis highlighted for all samples highly significant levels (p < 0.001) for the homeobox-B cluster, mostly related to biological processes involved in embryonic morphogenesis and anterior/posterior specification (GO:0009952, GO:0048704, GO:0009953, GO:0009954). Significant levels for GO terms based upon the HOXA cluster (GO:0060065~uterus development, GO:0009954~proximal/distal pattern formation, GO:0007338~single fertilization, GO:0008584~male gonad development) were exclusively documented for Croatian and Hungarian Lipizzans. The island on ECA11 including the HOXB cluster and highly significant GO terms related to morphogenesis were recently also documented in the Posavina horse by Grilz-Seger et al. [16]. Zhang et al. [25] listed HOXB13 in a recent selection signature study in horses. HOX genes play a fundamental role for morphological diversity in animals and for the control of axial morphology along the anterior-posterior body axis. As these gene clusters are directly assigned to axial body segments in numerical order, they can control single segments during embryogenesis concerning their position, segmentation and further differentiation [26].
On ECA11:31.01–31.94 another common homozygous region was found in all samples, whereas the frequency in Hungarian Lipizzans was with 47% slightly below the threshold. This ROH island, nearly identical in length (926.9/910.2 kb) in the Austrian and Croatian Lipizzans, harboured the genes C11H117orf67, DGKE, COIL, SCPEP1, AKAP1, MSI2 and was partially overlapping with two smaller islands of the Slovakian population. The Hungarian sample had a 244.4 long overlapping homozygous region within the Musashi RNA binding protein 2 (MSI2), which is involved in stem cell proliferation [27], haematopoiesis and can promote aggressive myeloid leukaemia in humans [28]. Four of these genes (COIL, SCPEP1, AKAP1, MSI2) were listed among other 121 genes that underwent positive selection during the domestication process of the horse [29].
In general the majority of ROH islands were medium sized (between 100 and 900 kb) to small sized (8–100 kb) according to the classification of Pemperton et al. [30]. Only in the Croatian sample one island on ECA14 was longer than 1 Mb. Most of the short ROH islands did not exceed the length of the contained gene, e.g. the island on ECA3:118.66–118.76 Mb, containing the two genes UV-stimulated scaffold protein A (UVSSA) and macrophage erythroblast attacher (MAEA). UVSSA is involved into the repair process of DNA damaged by ultraviolet (UV) sun rays. These two genes were directly followed by a 83 kb long island containing the gene C-terminal-binding protein 1 (CTBP1), a transcriptional co-repressor. Pemperton et al. [30] emphasized the investigation of short ROHs (< 100 kb), as they reflect homozygosity for ancient haplotypes contributing to local linkage disequilibrium (LD) patterns. Intermediary ROHs longer than 100 kb up to 2.5 Mb are deemed to be the result from background relatedness in populations with limited census. Furthermore, Pemperton et al. [30] stated that by the commonly applied windows approach as implemented in PLINK [31], ancient haplotypes and small ROHs due to high recombination rate might remain undetected. With the 500 kb applied boundary we were able to detect very small ROH islands, but SNP extraction revealed, that the frequencies of animals sharing those islands were underestimated, exemplarily demonstrated for the genes MAEA, UVSSA, CTBP1 and the STX17 locus.
Grey coat color, caused by a 4.6 kb duplication in intron 6 of Syntaxin 17 (STX17), is an essential part of the breeding objective in Lipizzan horses and it has been under selection since the past 150 years. The STX17 mutation is embedded into 352 kb long haplotype [13, 32].The frequency of the identified ROH island (ECA25:6,39-6,79) harbouring the genes STX17, NR4A3, ERP44 and INVS, was in concordance with genotype frequencies cited in Pielberg et al. [13], and ranged from 51.1% (Croatia) to 71.2% (Austria) among the respective subpopulations. In the Austrian breeding program only grey horses are used for reproduction, whilst the Slovakian, Hungarian and Croatian stud farms also permit solid colored horses. As a consequence the proportion of homozygous grey G/G horses is higher in the Austrian population.
The allele status of STX17 influences the progression of greying by age, grade of speckling (pigmentation among grey hair), grade of vitiligo and incidences and grade of melanoma [10–13]. The incidence of melanoma in Lipizzans was reported to affect 80% of the animals older than 15 years [10, 13]. Mostly melanomas in Lipizzans are benign and do not metastasize [33]. A previous study revealed, that the genes STX17, NR4A3, ERP44 and INVS, which are located in the aforementioned ROH island also were expressed in melanoma tissues [13]. Pielberg et al. [13] observed an over-expression of NR4A3 (nuclear receptor subfamily 4, group A, member 3), which is influenced by the dominant G-allele. The underlying molecular processes are not clearly understood [34]. Seltenhammer et al. [33] investigated factors retarding metastasis in affected grey Lipizzan horses and revealed common features with blue nevi in humans and identified potential markers for equine melanoma (S-100, PCNA, HMB-45, Ki-67, T-311, CD44). We identified within the entire Lipizzan sample an island on ECA14 containing the genes SPRY4 (Sprouty4) and NDFI1 (Nedd4-family interacting protein 1). SPYR4 is a member of the SPYR proteins, which are negative regulators of growth factor signaling pathways. Thus, SPRY4 inhibits transformed cell growth, migration and invasion, and epithelial-mesenchymal transition [35]. Shaverdashvili et al. [36] provided evidence in human melanoma for an inverse relationship between the expression of SPYR4 and Membrane-type 1 Matrix Metalloproteinase (MT1-MMP), one important driver of melanoma metastasis. Inhibition of MT1-MMP resulted in increased expression of SPYR4 and was correlated with the survival of melanoma patients in humans. NDFIP1 also acts as a tumour suppressor by repressing cell proliferation and was reported to be down-regulated in human uveal melamoma [37]. In a previous study Jiang et al. [34] investigated the ERK pathway and studied the effects of BRAF, RAS, GNAQ, GNA11, KIT on melanoma development in grey horses, whereas an association between STX17 and an activation of the ERK pathway was shown, but no activation of the aforementioned genes were observed. In the Austrian Lipizzan sample we identified an island containing the gene HSP90AB1. Metri et al. [38] identified HSP90AB1 as a main discriminator between metastatic and primary melanoma in humans, which also was correlated with survival rate of melanoma patients. In our data GO analysis revealed an enrichment of HSP90AB1 in the terms GO:0069550-reponse to stress and GO:0071353-cellular response to interleukin-4.
Conclusions
The enhanced high-resolution Netview approach results in a generally understandable visualisation, which is able to simultaneously present fundamental metrics of genetic diversity, coancestry and relatedness on population and individual level. This approach thus provides a high practical impact for conventional and conservation breeding management. The analyses of ROH islands based upon the conventional ROH size boundary of > = 500 kb have proven to be a valuable approach to screen for signatures of selection, also able to detect short homozygous overlapping regions. In order to avoid an underestimation of the frequencies of very short ROHs the underlying size boundary should be adapted in regions of special interest. The investigation of subpopulations within one single breed allows a fine-scale analysis and we demonstrated that slightly different breeding objectives have a high impact on genomic regions and gene content. ROH island analyses identified several annotated genes (SPRY4, NDFIP1, HSP90AB1 and IMPDH2), which may play a role for further studies on equine melanoma.
Methods
Ethics statement
This study was discussed and approved by the institutional Commission for Ethics and Animal Welfare, University of Veterinary Medicine, Vienna, protocol number: ETK-06/05/2015, in accordance with GSP guidelines and national legislation. The Lipizzan state stud farms Piber (Austria), Topol’čianky (Slovakia), Lipik, Đakovo (Croatia) and Szilvasvárad (Hungary) granted the permission to take hair samples from their horses.
Sampling
The horses included in this study originated from five European state stud farms, which are located in four different countries. The Austrian and the Slovakian samples represent the entire breeding populations of the stud farm of Piber (Austria, 254 horses) and Topol’čianky (Slovakia, 55 horses). The Croatian Lipizzans were sampled from the state stud farms Lipik (8 horses) and Đakovo (37 horses), whilst 23 Lipizzan horses were collected from the Hungarian state stud farm Szilvasvárad.
The hair samples were taken between the years 2013 and 2017 and the selected horses were born between 1989 and 2014. For Austria and Slovakia we were able to collect the whole stud farm populations. The breeding stock of the Croatian stud farms comprises together approximately 120 breeding animals, the number in the Hungarian state stud farm is on a similar level.
SNP genotyping
The single nucleotide polymorphism (SNP) genotypes for the 377 horses were determined using the Affymetrix Axiom™ Equine genotyping array [14]. The chromosomal position of the SNPs was derived from EquCab2.0 reference genome. We excluded SNPs positioned on the sex chromosomes (X: 28,017 SNPs and Y: 1 SNP), SNPs without known chromosomal position (30,864 SNPs) and SNPs with more than 10% missing genotypes, resulting in a total of 611,914 SNPs for each horse. For the analysis of population structure and genetic diversity we excluded SNPs with a minor allele frequency (MAF) less than 0.01, resulting in 492,719 SNPs per horse.
Genetic diversity and ROH analysis
In order to illustrate the population structure we applied Principal Component Analysis (PCA) on basis of the genetic relationship matrix (G) with pairwise identities by state (IBS) between horses as provided by PLINK v.1.7 [31]. The PCA was performed using R platform, whilst pairwise FST values were calculated with the R package Geneland (https://rdrr.io/cran/Geneland).
Population clustering and admixture analysis were determined using the program Admixture 1.23 [39]. We ran Admixture for 100 iterations increasing K from 2 to 10. Convergence between independent runs at the same K was monitored by comparing the resulting log-likelihood scores (LLs) following 100 iterations, and was inferred from stabilized LLs with less than 1 LL unit of variation between runs. Cross validation (CV) error estimation for each K was performed to determine the optimal number of clusters. Admixture results were visualized with the program Distruct 1.1 [40] and integrated in the high-resolution network following the procedure of Druml et al. [15].
ROH were determined with an overlapping window approach implemented in PLINK v1.7 [31] based on the settings: minimum SNP density = one SNP per 50 kb; maximum gap length = 100 kb; minimum length of homozygous segment > 500 kb (including more than 80 homozygous SNPs); one heterozygote and two missing genotypes were permitted within each segment.
The total number of ROH (NROH), average length of ROH (LROH) and the sum of all ROH segments (SROH) were summarized according to respective subpopulations. To analyze ROH length classes, ROHs were divided into following seven length categories: 0.5–1, 1–2, > 2–4, > 4–6, > 6–8, > 8–10 and > 10 Mb. The genomic inbreeding coefficients (FROH) were calculated by use of following formular:
,whereas the length of the autosomal genome (LAUTO), was set to 2243 GB. Statistical analyses and graphical representations of specific genomic regions and distribution of ROH segments were performed using the R-package detectROHs (www.r-project.org)and SAS v.9.1. [41].
High-resolution population networks
In order to ascertain the high-resolution population structure of Lipizzan horses, we performed a network visualization based upon the aforementioned IBS derived relationship matrix (G), following the description of Druml et al. [15]. The different components involved in the so-called NetView approach are described in detail by Neuditschko et al. [42] and Steining et al. [43]. Briefly, we computed genetic distances by subtracting pairwise relationships from 1 and applied the algorithm in its default setting (number of k nearest neighbors k-NN = 10). To illustrate the genetic relatedness between neighboring horses, we associated the thickness of edges (connecting lines) with the proportion of the genetic distance, whilst thicker edges corresponding to lower genetic distances. To identify highly inbred and outcrossed horses within the respective population networks we scaled the node size of each horse based on the individual SROH. The node colors of each horse represent the individual level of admixture at the selected number of K-clusters, whilst the node border colors illustrate the origin of the horses.
Gene characterization in ROH islands
The distribution of ROH segments across the genome was visualized using the R-package detect ROHs (www.r-project.org). Putative ROH islands were determined based upon overlapping homozygous regions, which were shared by more than 50% of the horses. ROH islands were calculated for the entire data sample and respective subpopulations. Genes located in ROH regions were identified with the map viewer of the equine ensemble database EquCab2.0 (www.ensembl.org) and Gene Ontology (GO) terms and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways of annotated genes were analyzed using the open source Database for Annotation, Visualization, and Integrated Discovery (DAVID) v6.8 package (https://david.ncifcrf.gov) using the Equus caballus annotation file as background [44].
Additional files
Pairwise FST values of the four Lipizzan subpopulations (Austrian, Croatia, Hungary, Slovakia). (DOC 41 kb)
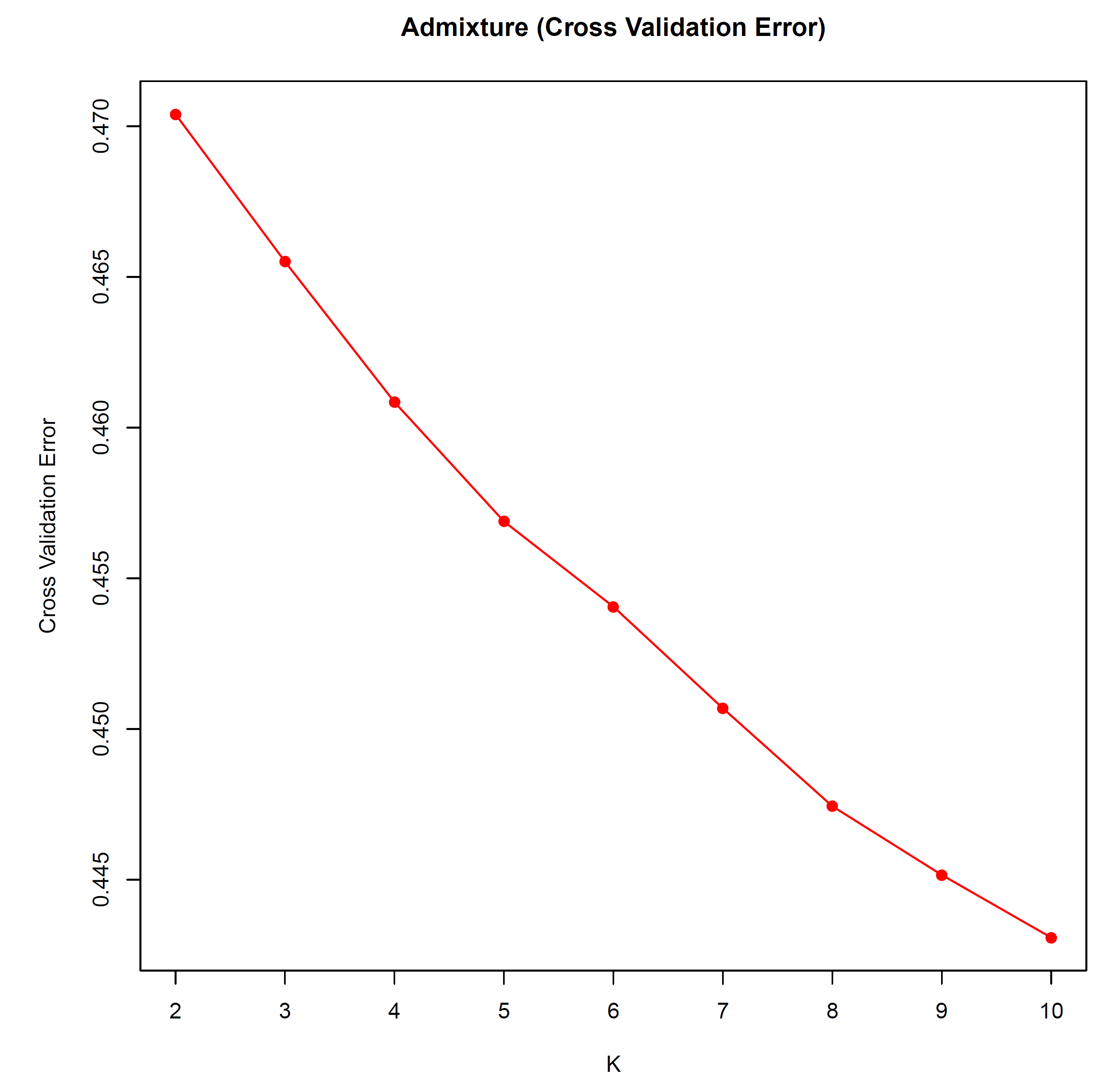{kind=link}
Graphical representation of Cross validation error to determine an optimal number of K clusters. (JPG 454 kb)
Profile plot of SROH versus NROH. Individuals are illustrated as dots (Đakovo/Croatia = blue; Lipik/Croatia = red; Piber/Austria = green; Szilvasvárad/Hungary = brown; Topol’čianky/Slovakia = purple). (DOCX 191 kb)
Gene Ontology (GO) terms and KEGG pathways based on annotated genes embedded in ROH islands for the entire Lipizzan sample. (DOC 44 kb)
Gene Ontology (GO) terms and KEGG pathways based on annotated genes embedded in ROH islands for the Lipizzan horses from the Austrian stud farm Piber. (DOC 47 kb)
Gene Ontology (GO) terms and KEGG pathways based on annotated genes embedded in ROH islands for the Lipizzan horses from the Slovak national stud farm of Topol’čianky. (DOC 48 kb)
Gene Ontology (GO) terms and KEGG pathways based on annotated genes embedded in ROH islands for the Lipizzan horses from the Croatian state stud farms Lipik/Đakovo. (DOC 61 kb)
Gene Ontology (GO) terms and KEGG pathways based on annotated genes embedded in ROH islands for the Lipizzan horses from the Hungarian state stud farm Szilvasvárad. (DOC 62 kb)
Acknowledgements
The authors thank the Lipizzan state studs Piber, Đakovo, Lipik, Topol’čianky and Szilvasvárad for providing the hair samples.
Funding
This work was financially supported by the Austrian Research Promotion Agency (FFG) [Contract number 843464] and the Federal Ministry for Sustainability and Tourism (BMNT) [Contract number 101332]. Austrian Research Promotion Agency (FFG) financially supported data collection and data analyses and Federal Ministry for Sustainability and Tourism (BMNT) supported financially the interpretation of data and the writing of the manuscript.
Availability of data and materials
The data that support the findings of this study are available from project consortium FFG project number 843464; Veterinary University Vienna, Xenogenetik and five European state stud farms, but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request and with permission of project consortium, FFG project number 843464; Veterinary University Vienna; Xenogenetik and five European state stud farms.
Abbreviations
- ADRM1
Proteasomal ubiquitin receptor ADRM1
- AKAP1
A-kinase anchoring protein 1
- ALS2CL
ALS2 C-terminal-like protein
- ARHGAP26
Rho GTPase activating protein 26
- ARHGEF4
Rho guanine nucleotide exchange factor 4
- ARIH2
Ariadne RBR E3 ubiquitin protein ligase 2
- ATRIP
ATR interacting protein
- B3GNT4
UDP-GlcNAc:betaGal beta-1,3-N-acetylglucosaminyltransferase 4
- BCL7A
BAF chromatin remodeling complex subunit BCL7A
- bp
Base pair
- BRAF
B-Raf proto-oncogene, serine/threonine kinase
- C11H17orf67
Chromosome 11 C17orf67 homolog
- C16H3orf84
Chromosome 16 H17orf84 homolog
- CABLES2
CDK5 and ABL1 enzyme substrate 2
- CAPN11
Calpain-11
- CBX1
Chromobox 1
- CCDC12
Coiled-coil domain-containing protein 12
- CCDC51
Coiled-coil domain containing 51
- CCDC71
Coiled-coil domain containing 71
- CDH
Cadherin
- CELSR3
Cadherin EGF LAG seven-pass G-type receptor 3
- CHD1L
Chromodomain Helicase DNA Binding Protein 1 Like
- CLIP1
CAP-Gly domain containing linker protein 1
- COIL
Coilin
- COPZ2
Coatomer Protein Complex Subunit Zeta 2
- CTBP1
C-Terminal Binding Protein 1
- CTDP1
CTD Phosphatase Subunit 1
- CV
Cross validation
- DALRD3
DALR anticodon binding domain containing 3
- DAVID
Database for Annotation, Visualization and Integrated Discovery
- DELE1
DAP3 binding cell death enhancer 1
- DGKE
Diacylglycerol kinase epsilon
- DIABLO
Diablo IAP-binding mitochondrial protein
- ECA11
Equine chromosome 11
- ECA14
Equine chromosome 14
- ECA16
Equine chromosome 16
- ECA18
Equine chromosome 18
- ECA20
Equine chromosome 20
- ECA25
Equine chromosome 25
- ECA3
Equine chromosome 3
- ECA30
Equine chromosome 30
- ECA5
Equine chromosome 5
- ECA8
Equine chromosome 8
- ECATH-2
Myeloid cathelicidin 2
- ECATH-3
Myeloid cathelicidin 3
- EDN3
Endothelin 3
- EquCab2.0
Equus caballus reference genome version 2
- ERK pathway
Extracellular-signal-regulated kinase
- ERP44
Endoplasmic reticulum protein 44
- EVX1
Even-skipped homeobox 1
- FGF1
Fibroblast growth factor 1
- FMO5
Flavin Containing Monooxygenase 5
- FROH
Inbreeding coefficient based upon ROH
- FST
Fixation coefficient
- FSTL4
Follistatin like 4
- G
Genetic relationship matrix
- GATA5
Transcription factor GATA-5
- GLDC
Glycine decarboxylase
- GNA1
Glucosamine 6-phosphate N-acetyltransferase
- GNAQ
G protein subunit alpha q
- GNPDA1
Glucosamine-6-phosphate deaminase 1
- GO
Gene ontology
- HIBADH
3-hydroxyisobutyrate dehydrogenase, mitochondrial
- HOXA1
Homeobox A1
- HOXA10
Homeobox A10
- HOXA11
Homeobox A11
- HOXA2
Homeobox A2
- HOXA3
Homeobox A3
- HOXA5
Homeobox A5
- HOXA6
Homeobox A6
- HOXA7
Homeobox A7
- HOXA9
Homeobox A9
- HOXB
Homeobox B
- HOXB1
Homeobox B1
- HOXB13
Homeobox B13
- HOXB2
Homeobox B2
- HOXB3
Homeobox B3
- HOXB5
Homeobox B5
- HOXB6
Homeobox B6
- HOXB7
Homeobox B7
- HOXB8
Homeobox B8
- HPD
4-hydroxyphenylpyruvic acid dioxygenase
- HRH3
Histamine H3 receptor
- HSP90AB1
Equus caballus heat shock protein 90 alpha family class B member 1
- IBS
Identity by state
- IL31
Interleukin 31
- IMPDH2
Inosine monophosphate dehydrogenase 2
- INVS
Inversin
- IP6K2
Inositol hexakisphosphate kinase 2
- K
Cluster
- kb
Kilobase
- KDM4C
Lysine demethylase 4C
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- KIF26B
Kinesin family member 26B
- KIT
KIT proto-oncogene, receptor tyrosine kinase
- KLHDC8B
Kelch domain containing 8B
- LAMA5
Laminin subunit alpha-5
- LAMB2
Laminin subunit beta 2
- LD
Linkage disequilibrium
- LROH
ROH length
- LRRC43
Leucine rich repeat containing 43
- LSM14B
LSM family member 14B
- MAEA
Macrophage Erythroblast Attacher
- MAF
Minor allele frequency
- Mb
Megabase
- MLXIP
MLX interacting protein
- MSI2
Musashi RNA binding protein 2
- mtDNA
Mitochondrial deoxyribonucleic acid
- MTG2
Mitochondrial ribosome-associated GTPase 2
- NCKIPSD
NCK interacting protein with SH3 domainM
- NDFIP1
Nedd4 family interacting protein 1
- NDUFAF3
NADH:Ubiquinone oxidoreductase complex assembly factor 3
- NFKBIE
NF-kappa-B inhibitor epsilon
- NME6
NME/NM23 nucleoside diphosphate kinase 6
- NR4A3
Nuclear receptor subfamily 4 group A member 3
- NROH
mean number of ROHs
- OMIA
Online Mendelian Inheritance in Animals
- OMIM
Online Mendelian Inheritance in Man
- OSBPL2
Oxysterol-binding protein-related protein 2
- P4HTM
Prolyl 4-hydroxylase, Transmembrane
- PC
Principal component
- PCA
Principal component analysis
- PCDH1
Protocadherin 1
- PCDH12
Protocadherin 12
- PFKFB4
6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 4
- PHACTR3
Phosphatase and actin regulator 3
- PLEKHB2
Pleckstrin homology domain-containing family B member 2
- PLXNB1
Plexin B1
- PRKAB2
Protein kinase AMP-activated non-catalytic subunit beta 2
- PSMA7
Proteasome subunit alpha type-7
- PSMD9
Proteasome 26S subunit, non-ATPase 9
- QARS
Glutaminyl-TRNA synthetase
- QRICH1
Glutamine rich 1
- RAS
Resistance to audiogenic seizures
- RBBP8NL
RBBP8 N-terminal-like protein
- RHOF
Ras homolog family member F, filopodia associated
- RNF14
Ring finger protein 14
- ROH
Runs of homozygosity
- SCPEP1
Serine carboxypeptidase 1
- SD
Standard deviation
- SETD1B
SET domain containing 1B, histone lysine methyltransferase
- SETD2
Histone-lysine N-methyltransferase SETD2
- SHISA5
Shisa family member 5
- SLC26A6
Solute carrier family 26 member 6
- SLC29A1
Solute carrier family 29 member 1
- SLC35B2
Solute carrier family 35 member B2
- SMYD3
SET and MYND domain containing 3
- SNP
Single nucleotide polymorphism
- SNX11
Sorting nexin 11
- SPRY4
Sprouty4
- SROH
mean genome coverage of ROH
- SS18L1
SS18L1 subunit of BAF chromatin remodeling complex
- STX17
Syntaxin 17
- TAF4
Transcription initiation factor TFIID subunit 4
- TCTE1
T-complex-associated-testis-expressed 1
- TMEM120B
Transmembrane protein 120B
- TMEM89
Transmembrane protein 89
- TMIE
Transmembrane inner ear expressed protein
- TREX1
Three prime repair exonuclease 1
- TTLL6
Tubulin tyrosine ligase like 6
- UCN2
Urocortin 2
- UHRF2
Ubiquitin like with PHD and ring finger domains 2
- UQCRC1
Ubiquinol-cytochrome c reductase core protein 1
- USP19
Ubiquitin specific peptidase 19
- UV
ultraviolet
- UVSSA
UV Stimulated Scaffold Protein A
- WDR6
WD repeat domain 6
- WDR66
WD repeat domain 66
- ZNF831
Zinc finger protein 831
Authors’ contributions
GGS, TD, MN performed statistical analyses, GGS and TD wrote the paper and designed the study, TD, MD and MH were responsible for data collection and GB and TD were responsible for funding acquisition and project administration. All authors read and approved the final manuscript.
Ethics approval
This study was discussed and approved by the institutional Commission for Ethics and Animal Welfare, University of Veterinary Medicine, Vienna, protocol number: ETK-06/05/2015, in accordance with GSP guidelines and national legislation.
Consent for publication
The Lipizzan state stud farms Piber (Austria), Topol’čianky (Slovakia), Lipik, Đakovo (Croatia) and Szilvasvárad (Hungary) granted the permission to take hair samples from their horses.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Gertrud Grilz-Seger, Email: grilz.seger@vetmeduni.ac.at.
Thomas Druml, Email: thomas.druml@vetmeduni.ac.at.
Markus Neuditschko, Email: markus.neuditschko@agroscope.admin.ch.
Max Dobretsberger, Email: maximilian.dobretsberger@vetmeduni.ac.at.
Michaela Horna, Email: michaela.horna@post.sk.
Gottfried Brem, Email: gottfried.brem@vetmeduni.ac.at.
References
- 1.Zechner P, Sölkner J, Bodo I, Druml T, Baumung R, Achmann R, Marti E, Habe F, Brem G. Analysis of diversity and population structure in the Lipizzan horse breed based on pedigree information. Livest Prod Sci. 2002;77:137–46.
- 2.Druml T, Sölkner J. Die Gründerpopulation der Lipizzanerrasse und deren Zuchtgeschichte anhand von Genanteilen. In: Brem G, editor. Lipizzaner im Spiegel der Wissenschaft. Wien: Verlag der Österreichischen Akademie der Wissenschaften; 2012. pp. 153–193. [Google Scholar]
- 3.Druml T, Horna M, Grilz-Seger G, Dobretsberger M, Brem G. Association of body shape with amount of Arabian genetic contribution in the Lipizzan horse. Arch Anim Breed. 2018;61:79–85. doi: 10.5194/aab-61-79-2018. [DOI] [Google Scholar]
- 4.Achmann R, Curik I, Dovč P, Kavar T, Bodo I, Habe F, Marti E, Sölkner J, Brem G. Microsatellite diversity, population subdivision and gene flow in the Lipizzan horse. Anim Genet. 2004;35:285–292. doi: 10.1111/j.1365-2052.2004.01157.x. [DOI] [PubMed] [Google Scholar]
- 5.Kavar T, Brem G, Habe F, Sölkner J, Dovč P. History of Lipizzan horse maternal lines as revealed by mtDNA analysis. Genet Sel Evol. 2002;34:635–548. doi: 10.1186/1297-9686-34-5-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kavar T, Habe F, Brem G, Dovč P. Mitochondrial D-loop sequence variation among the 16 maternal lines of the Lipizzan horse breed. Anim Genet. 1999;30:423–430. doi: 10.1046/j.1365-2052.1999.00557.x. [DOI] [PubMed] [Google Scholar]
- 7.Kasarda R, Vostrý L, Moravčíková N, Vostrá-Vydrová H, Dovč P, Kadlečík O. Detailed insight into genetic diversity of the old Kladruber horse substructure in comparison to the Lipizzan breed. Acta Agr Scand, Section A — Anim Science. 2016;66:67–74. [Google Scholar]
- 8.Wallner B, Vogl C, Shukla P, Burgstaller JP, Druml T, Brem G. Identification of genetic variation on the horse Y chromosome and the tracing of male founder lineages in modern breeds. PLoS One. 2013;8:e6001. doi: 10.1371/journal.pone.0060015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dovč P, Kavar T, Sölkner H, Achmann R. Development of the Lipizzan horse breed. Repro Dom Anim. 2006. 10.1111/j.1439-0531.2006.00726.x. [DOI] [PubMed]
- 10.Seltenhammer MH, Simhofer H, Scherzer S, Zechner P, Curik I, et al. Equine melanoma in a population of 296 grey Lipizzan horses. Equine Vet J. 2003;35:153–157. doi: 10.2746/042516403776114234. [DOI] [PubMed] [Google Scholar]
- 11.Curik I, Seltenhammer M, Sölkner J: Quantitative genetic analysis of melanoma and grey level in Lipizzan horses. Proceedings of the 7th world congress on genetics applied to livestock production 2002; CD-ROM:communication no. 05-09, august 19–23, Montpellier, France.
- 12.Curik I, Druml T, Seltenhammer M, Sundström E, Rosengren Pielberg G, Andersson L, Sölkner J. Complex inheritance of melanoma and pigmentation of coat and skin in Grey horses. PLoS Genet. 2013;9:e1003248. doi: 10.1371/journal.pgen.1003248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pielberg RG, Golovko A, Sundstro¨m E, Curik I, Lennartsson J. A cis-acting regulatory mutation causes premature hair graying and susceptibility to melanoma in the horse. Nat Genet. 2008;40:1004–1009. doi: 10.1038/ng.185. [DOI] [PubMed] [Google Scholar]
- 14.Schaefer RJ, Schubert M, Bailey E, Bannasch DL, Barrey E, Bar-Gal GK, Brem G, Brooks SA, Distl O, Fries R. Developing a 670k genotyping array to tag ~2M SNPs across 24 horse breeds. BMC Genomics. 2017;18:565–583. doi: 10.1186/s12864-017-3943-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Druml T, Neuditschko M, Grilz-Seger G, Horna M, Ricard A, Mesaric M, Cotman M, Pausch H, Brem G. Population networks associated with runs of homozygosity reveal new insights into the breeding history of the Haflinger horse. J Hered. 2018;109:384–392. doi: 10.1093/jhered/esx114. [DOI] [PubMed] [Google Scholar]
- 16.Grilz-Seger G, Mesarič M, Cotman M, Neuditschko M, Druml T, Brem G. Runs of homozygosity and population history of three horse breeds with small population size. J Equine Vet Sci. 2018;71:27–34. doi: 10.1016/j.jevs.2018.09.004. [DOI] [Google Scholar]
- 17.Browning SR, Browning BL. High-resolution detection of identity by descent in unrelated individuals. Am J Hum Genet. 2010;86:526–539. doi: 10.1016/j.ajhg.2010.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thompson EA. Identity by descent: variation in meiosis, across genomes, and in populations. Genetics. 2013;194. 10.1534/genetics.112.148825. [DOI] [PMC free article] [PubMed]
- 19.Arias JA, Keehan M, Fisher P, Coppieters W, Spelman R. A high density linkage map of the bovine genome. BMC Genet. 2009;10. 10.1186/1471-2156. [DOI] [PMC free article] [PubMed]
- 20.Ceballos FC, Joshi PK, Clark DW, Ramsay M, Wilson J. Runs of homozygosity: window into population history and trait architecture. Nat Rev Genet. 2018;19:220e34. doi: 10.1038/nrg.2017.109. [DOI] [PubMed] [Google Scholar]
- 21.Scocchi M, Bontempo D, Boscolo S, Tomasinsig L, Giulotto E, Zanetti M. Novel cathelicidins in horse leukocytes. FEBS Lett. 1999;457:459–464. doi: 10.1016/S0014-5793(99)01097-2. [DOI] [PubMed] [Google Scholar]
- 22.Skerlavaj B, Scocchi M, Gennaro R, Risso A, Zanetti M. (2001): structural and functional analysis of horse cathelicidin peptides. Antimicrob Agents Chemother. 2001;45:715–722. doi: 10.1128/AAC.45.3.715-722.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vidale P, Piras FM, Nergadze SG, Bertoni N, Verini-Supplizi A, Adelson D, Guérin G, Giulotto E. Chromosomal assignment of six genes (EIF4G3, HSP90, RBBP6, IL8, TERT, and TERC) in four species of the genus Equus. Anim Biotechnol. 2011;22:119–123. doi: 10.1080/10495398.2011.575300. [DOI] [PubMed] [Google Scholar]
- 24.Boehm AK, Seth M, Mayr KG, Fortier LA. Hsp90 mediates insulin-like growth factor 1 and interleukin-1beta signaling in an age-dependent manner in equine articular chondrocytes. Arthritis Rheum. 2007;56:2335–2343. doi: 10.1002/art.22664. [DOI] [PubMed] [Google Scholar]
- 25.Zhang C, Ni P, Ahmad HI, Gemingguli M, Baizilaitibei A, Gulibaheti D, Fang Y, Wang H, Asif AR, Xiao C, Chen J, Ma Y, Liu X, Du Y, Zhao S. Detecting the population structure and scanning for signatures of selection in horses (Equus caballus) from whole-genome sequencing data. Evol Bioinforma. 2018;14:1–9. doi: 10.1177/1176934318775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pearson JC, Lemons D, McGinnis W. Modulating Hox gene functions during animal body patterning. Nat Rev Genet. 2005;6:893–904. doi: 10.1038/nrg1726. [DOI] [PubMed] [Google Scholar]
- 27.Wuebben EL, Mallanna SK, Cox JL, Rizzino A. Musashi2 is required for the self-renewal and pluripotency of embryonic stem cells. PLoS One. 2012;7. 10.1371/journal.pone.0034827. [DOI] [PMC free article] [PubMed]
- 28.Kharas MG, Lengner CJ, Al-Shahrour F, Bullinger L, Ball B, Zaidi S, Morgan K, Tam W, Paktinat M, Okabe R, Gozo M, Einhorn W, Lane SW, Scholl C, Frohling S, Fleming M, Ebert BL, Gilliland DG, Jaenisch R, Daley GQ. Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nature Med. 2010;16:903–908. doi: 10.1038/nm.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schubert M, Jónsson H, Chang D, Sarkissian CD, Ermini L, Ginolhac A, et al. Prehistoric genomes reveal the genetic foundation and cost of horse domestication. PNAS. 2014. 10.1073/pnas.1416991111. [DOI] [PMC free article] [PubMed]
- 30.Pemperton TJ, Absher D, Feldman MW, Myers RM, Rosenberg NA, Li JZ. Genomic patterns of homozygosity in worldwide human populations. Am J Hum Genet. 2012;91:275e92. doi: 10.1016/j.ajhg.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sundström E, Imsland F, Mikko S, Wade C, Sigurdsson S, Pielberg GR, Golovko A, Curik I, Seltenhammer M, Sölkner J, Lindblad-Toh K, Andersson L. Copy number expansion of the STX17 duplication in melanoma tissue from Grey horses. BMC Genomics. 2012;13:365–378. doi: 10.1186/1471-2164-13-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seltenhammer MH, Heere-Ress E, Brandt S, Druml T, Jansen B, Pehamberger H, Niebauer GW. Comparative histopathology of grey-horse-melanoma and human malignant melanoma. Pigment Cell Res. 2004;17:674–681. doi: 10.1111/j.1600-0749.2004.00192.x. [DOI] [PubMed] [Google Scholar]
- 34.Jiang L, Campagne C, Sundström E, Sousa P, Imran S, Seltenhammer M, Pielberg G, Olsson MJ, Egidy G, Andersson L, Golovko A. Constitutive activation of the ERK pathway in melanoma and skin melanocytes in Grey horses. BMC Cancer. 2014;14:857–868. doi: 10.1186/1471-2407-14-857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tennis MA, Van Scoyk MM, Freeman SV, Vandervest KM, Nemenoff RA, Winn RA. Sprouty-4 inhibits transformed cell growth, migration and invasion, and epithelial-mesenchymal transition, and is regulated by Wnt7A through PPARgamma in non-small cell lung cancer. Mol Cancer Res. 2010;8:833–843. doi: 10.1158/1541-7786.MCR-09-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shaverdashvili K, Zhang K, Osman I, Honda K, Jobava R, Bedogni B. MT1-MMP dependent repression of the tumor suppressor SPRY4 contributes to MT1-MMP driven melanoma cell motility. Oncotarget. 2015;6:33512–33522. doi: 10.18632/oncotarget.5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peng J, Liu H, Liu C. MiR-155 promotes uveal melanoma cell proliferation and invasion by regulating NDFIP1 expression. Technol Cancer Res Treat. 2017;16:1160–1167. doi: 10.1177/1533034617737923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Metri R, Mohan A, Nsengimana J, Pozniak J, Molina-Paris C, Newton-Bishop J, Bishop D, Chandra N. Identification of a gene signature for discriminating metastatic from primary melanoma using a molecular interaction network approach. Sci Rep. 2017;7:17314. doi: 10.1038/s41598-017-17330-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosenberg NA. Distruct: a program for the graphical display of population structure. Mol Ecol Notes. 2004;4:137–138. doi: 10.1046/j.1471-8286.2003.00566.x. [DOI] [Google Scholar]
- 41.SAS Institute. 2009. SAS version 9.1. Cary (NC): SAS Institute, Inc.
- 42.Neuditschko M, Khatkar MS, Raadsma HW. Net view: a high-definition network-visualization approach to detect fine scale population structures from genome-wide patterns of variation. PLoS One. 2012. 10.1371/journal.pone.0048375. [DOI] [PMC free article] [PubMed]
- 43.Steinig EJ, Neuditschko M, Khatkar MS, Raadsma HW, Zenger KR. Netview p: a network visualization tool to unravel complex population structure using genome-wide SNPs. Mol Ecol Res. 2015;16:216–227. doi: 10.1111/1755-0998.12442. [DOI] [PubMed] [Google Scholar]
- 44.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Pairwise FST values of the four Lipizzan subpopulations (Austrian, Croatia, Hungary, Slovakia). (DOC 41 kb)
Graphical representation of Cross validation error to determine an optimal number of K clusters. (JPG 454 kb)
Profile plot of SROH versus NROH. Individuals are illustrated as dots (Đakovo/Croatia = blue; Lipik/Croatia = red; Piber/Austria = green; Szilvasvárad/Hungary = brown; Topol’čianky/Slovakia = purple). (DOCX 191 kb)
Gene Ontology (GO) terms and KEGG pathways based on annotated genes embedded in ROH islands for the entire Lipizzan sample. (DOC 44 kb)
Gene Ontology (GO) terms and KEGG pathways based on annotated genes embedded in ROH islands for the Lipizzan horses from the Austrian stud farm Piber. (DOC 47 kb)
Gene Ontology (GO) terms and KEGG pathways based on annotated genes embedded in ROH islands for the Lipizzan horses from the Slovak national stud farm of Topol’čianky. (DOC 48 kb)
Gene Ontology (GO) terms and KEGG pathways based on annotated genes embedded in ROH islands for the Lipizzan horses from the Croatian state stud farms Lipik/Đakovo. (DOC 61 kb)
Gene Ontology (GO) terms and KEGG pathways based on annotated genes embedded in ROH islands for the Lipizzan horses from the Hungarian state stud farm Szilvasvárad. (DOC 62 kb)
Data Availability Statement
The data that support the findings of this study are available from project consortium FFG project number 843464; Veterinary University Vienna, Xenogenetik and five European state stud farms, but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request and with permission of project consortium, FFG project number 843464; Veterinary University Vienna; Xenogenetik and five European state stud farms.