

INTRODUCTION
The US Food and Drug Administration (FDA)'s Critical Path Initiative (CPI) was launched in 2004 and aimed at accelerating the stagnating product development pipeline. Subsequently, in 2006, the Critical Path Opportunities List (CPOL) identified specific priorities to facilitate the CPI vision. Since then, the FDA's Center for Drug Evaluation and Research directed considerable efforts to achieve the goals of CPI and CPOL. Collaborations with Public–Private Partnerships (PPPs) helped address several CPOL priorities to yield meaningful results to benefit public health.
THE CRITICAL PATH
In 2004, the US Food and Drug Administration (FDA) acknowledged a growing gap between the rate of basic science discovery and the translation of these discoveries into the development of medical products. To address this gap, the FDA instituted the Critical Path Initiative (CPI), which called for increased efforts to catalyze innovation in product development through the launch of several initiatives.1 These efforts aim to modernize product quality and manufacturing standards, develop novel approaches to assess safety and effectiveness, build nonclinical and in silico predictive models, and develop novel clinical trials and analyses methodologies. When taken together, these tools, standards, and approaches aim to assess safety, efficacy, quality, and performance of FDA‐regulated products (collectively termed regulatory science). These efforts strive to streamline medical product development and accelerate the translation of scientific discovery into commercial products.
The CPI further emphasized that a joint effort between the research community, industry, and FDA scientists was essential to realize the CPI vision.1 To this end, the FDA convened both external stakeholders and FDA scientists to identify research priorities that could guide the FDA to bring focus to specific unmet public health needs. This effort resulted in the Critical Path Opportunities List (CPOL) published in 2006.2 The stakeholders identified six top‐priority focus areas related to the safety, efficacy, quality, and performance of FDA regulated products:
Topic 1: Better evaluation tools;
Topic 2: Streamlining clinical trials;
Topic 3: Harnessing bioinformatics;
Topic 4: Moving manufacturing into the 21st century;
Topic 5: Developing products to address urgent public health needs;
Topic 6: At‐risk populations: Pediatrics.
Each of these topics includes a range of specific “opportunities” outlining unmet research area needs, for a total of 76. These opportunities range from broad regulatory science development efforts to targeted research that would address specific gaps in public health in a variety of therapeutic areas, and altogether, aim to advance drug development along the critical path.2
Following the publications of CPI and CPOL, the FDA's Centers including the Center for Drug Evaluation and Research (CDER) sought to collaborate with academic institutions, industry, patients groups, nonprofit institutions, foundations, and government agencies to foster the development of tools, standards, and approaches to enhance drug development.1, 3 Several Public–Private Partnerships (PPPs) have formed over the past decade and helped inform the modernization of CDER's regulatory processes and drug development efforts. This article highlights some key deliverables from PPP efforts aimed at the CPI's and CPOL's vision.
PPPs AND CDER ENGAGEMENT
PPPs and precompetitive research
A PPP is a collaboration between multiple stakeholder organizations, including at least one nonprofit or 501(c)(3) organization, to achieve a shared goal that is beyond the capability of any one stakeholder. In drug development, a PPP may be established upon emergence and identification of a public health regulatory or drug development need and can include global collaborations that engage international government entities, academia, industry, patient advocacy groups, nonprofit institutions, and professional organizations. Once established, the PPP members conduct “precompetitive research,” whereby stakeholders, who may be competitors or have different business interests, share resources and nonproprietary data to develop knowledge, insights, and strategies to address the identified unmet need. The resulting deliverables benefit all stakeholders and can take many forms, such as best practices, scientific recommendations, patient registries, databases, disease models, training modules, and drug development tools (DDT) advancement. Once completed, the PPP deliverables are shared in the public domain to be broadly applied towards streamlining drug development and increasing the efficiency of regulatory decision‐making.
CDER's engagement with PPPs
CDER staff are often invited to participate in PPPs as regulatory partners to facilitate, inform, provide a regulatory perspective, and to help external groups understand CDER's current thinking on specific topics. To facilitate consistency in CDER staff engagement in PPP discussions, and to avoid any conflicts of interest or appearance of undue influence, CDER has adopted a formal process for staff participation in PPPs and consortia.4 The PPP activities with which CDER staff are engaged must be science‐driven, aimed to improve public health, compliant with federal laws and policy, and structured to uphold the principles of transparency, fairness, inclusiveness, and scientific rigor. CDER staff cannot discuss confidential commercial information and nonpublic regulatory information with the PPP participants.
Currently, there are 30 PPPs with CDER engagement. The activities of these PPPs encompass many therapeutic areas that focus on both disease specific regulatory science gaps as well as broader‐based methodologies (Table 1).
Table 1.
PPPs with CDER engagement
| PPP name | Abr. | Year established | PPP focus |
|---|---|---|---|
| Global Language of Business | GS‐1 | 1973 | Manage pharmaceutical barcode and supply chain |
| Institute for Safe Medication Practices | ISMP | 1975 | Advance real world evidence generation through public access to Sentinel Infrastructure |
| Product Quality Research Institute | PQRI | 1996 | Improve drug product quality, manufacturing, and regulation |
| International Life Sciences Institute, Health and Environmental Sciences Institute ‐ Developmental and Reproductive Toxicology Committee | HESI-ReproTox | 2005 | Advance science related to development and reproductive toxicology |
| National Institute for Pharmaceutical Technology & Education | NIPTE | 2005 | Advance drug product quality through research and education |
| Biomarker Consortium | BC | 2006 | Develop biomarkers across multiple therapeutic areas |
| Cardiac Safety Research Consortium | CSRC | 2006 | Develop of methodologies and medical products for evaluation of cardiotoxicity |
| Predictive Safety Testing Consortium | PSTC | 2006 | Develop biomarkers and tools for evaluation of drug‐induced toxicity |
| Clinical Trials Transformation Initiative | CTTI | 2007 | Improve the quality and efficiency of clinical trials |
| International Serious Adverse Events Consortium | iSAEC | 2007 | Develop genetic biomarkers for drug‐induced adverse events |
| Coalition Against Major Diseases Consortium | CAMD | 2008 | Advance technologies for neurodegenerative disease medical product development |
| International Life Sciences Institute, Health and Environmental Sciences Institute – Cardiac Safety Committee | HESI -CSC | 2008 | Develop preclinical and clinical models for evaluation of cardiotoxicity |
| Patient Reported Outcomes Consortium | PRO | 2008 | Develop patient reported outcomes |
| PROTECT Initiative | PROTECT | 2008 | Prevent medication overdoses in pediatric patients |
| RX‐360 ‐ International Supply Chain Consortium | RX-360 | 2009 | Improve safety and efficiency of the pharmaceutical supply chain |
| Analgesic Clinical Trial Translations, Innovations, Opportunities and Networks Initiative | ACTTION | 2010 | Optimize analgesic clinical trials |
| Critical Path to TB Drug Regimens Consortium | CPTR | 2010 | Advance tuberculosis drug development |
| Electronic Patient Reported Outcomes Consortium | ePRO | 2010 | Develop electronic patient reported outcomes |
| Polycystic Kidney Disease Outcomes Consortium | PKDOC | 2010 | Develop polycystic kidney disease biomarkers |
| SmartTots | SmartTots | 2010 | Improve safety of pediatric use of anesthetics and sedatives |
| Coalition for Accelerating Standards and Therapies | CFAST | 2012 | Develop therapeutic area data standards |
| Kidney Health Initiative | KHI | 2012 | Advance kidney health and patient safety |
| Multiple Sclerosis Outcomes Assessment Consortium | MSOAC | 2012 | Develop patient reported outcomes for multiple sclerosis |
| TransCelerate Biopharma, Inc | TransCelerate | 2012 | Accelerate the research and development of innovative new therapies |
| Innovation in Medical Evidence Development and Surveillance Program | IMEDS | 2013 | Advance science tools for post‐market evidence generation and surveillance |
| Accelerating Medicines Partnership | AMP | 2014 | Identify and validate biological targets of disease for drug development |
| Duchenne Regulatory Science Consortium | D-RSC | 2015 | Develop tools and protocols for Duchenne muscular dystrophy clinical trials |
| International Neonatal Consortium | INC | 2015 | Develop neonatal therapeutics |
| Critical Path for Parkinson's | CPP | 2015 | Defining best practices and protocols for Parkinson's disease clinical trials |
| Global Pediatric Clinical Trials Network Pre‐Launch Consortium | PTC | 2015 | Develop global pediatric clinical trial network |
PPP websites are hyperlinked above. Publicly available PPP deliverables on the PPP website are not referenced in the text.
PPP DELIVERABLES AND THE CRITICAL PATH OPPORTUNITIES LIST
CDER engagement with PPP partners in the precompetitive space has contributed to development of key deliverables that include a wide array of drug development tools, databases, education/training modules, and best practice recommendations that aim to address unmet needs. To identify the CPOL deliverables resulting from the PPP efforts, all PPP initiatives and deliverables between 2006 and July 2016 were catalogued from publicly available sources (Figure 1). Next, each of these PPP initiatives and deliverables were aligned to the topics identified in the 2006 CPOL (Figure 2). This assessment revealed that these PPPs have addressed specific gaps from all six topics identified in the CPOL. The following section highlights some key contributions from PPPs which included CDER engagement, addressing each of six CPOL topics.
Figure 1.
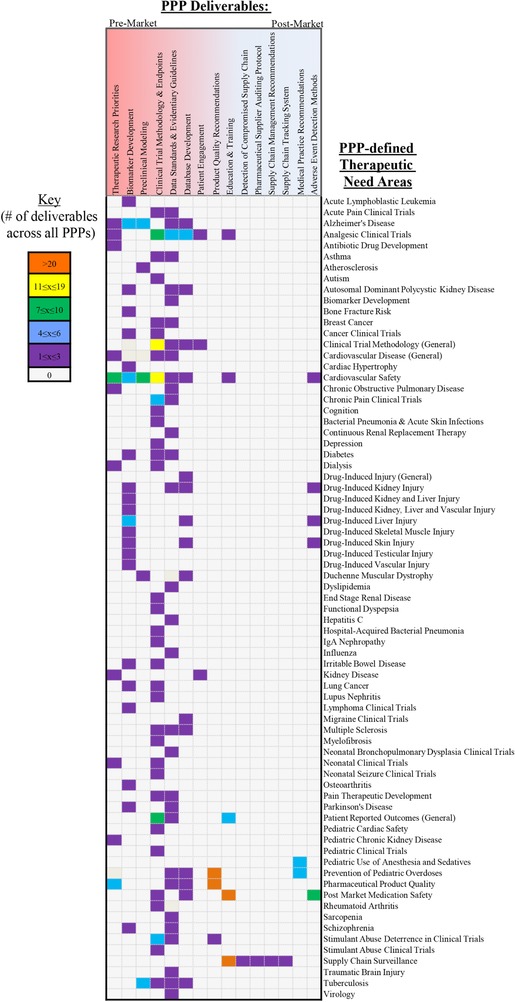
Therapeutic areas and needs addressed by PPPs: Ongoing and completed initiatives and deliverables (2006‐July 2016). Heat map of identified PPP initiatives and deliverables from publicly available sources arranged by therapeutic need areas.
Figure 2.
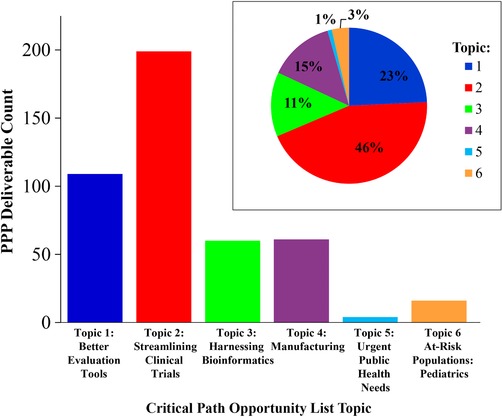
Critical path opportunities addressed by PPPs: ongoing and completed initiatives and deliverables by CPOL topics. Bar graph of the number of ongoing and completed PPP initiatives and deliverables identified in Figure 1 that address one of the six CPOL topics. Inset: Percentage of ongoing and completed PPP initiatives and deliverables per CPOL opportunity topic.
Topic 1: Better evaluation tools
The CPI identified the development and qualification of biomarkers for specific uses (e.g., safety, diagnostic, risk, prognostic, predictive, response, and monitoring) in product development as one of the key areas of focus. Furthermore, accurate evaluation of drug safety is highlighted in 6 of the 76 opportunities of the CPOL as a major bottleneck throughout drug development.1, 2 In 2005, CDER created a regulatory path and established a program for the qualification of DDTs that included qualification of biomarkers for specific contexts of use,5, 6, 7 and PPPs have advanced these efforts related to biomarker/DDT qualification. In 2007, the Predictive Safety Testing Consortium (PSTC) was the first to obtain qualification for a suite of seven predictive biomarkers for nephrotoxicity for use in nonclinical drug development through CDER's DDT qualification pathway.8, 9 Subsequently, both the European Medicines Agency (EMA) and Japan's Pharmaceuticals and Medical Devices Agency (PMDA) qualified PSTC's biomarkers in 2008 and 2010, respectively10, 11 (Table 2). PSTC's effort provided a model for future biomarker qualification efforts and was highlighted in a special edition of the journal Nature Biotechnology.5, 7, 12, 13, 14 Following these initial nonclinical efforts, three PPPs, PSTC, Biomarker Consortium (BC), and the European Innovative Medicines Initiative (IMI) Safer and Faster Evidence‐based Translation Consortium (SAFE‐T), initiated an international collaboration to translate PSTC's nonclinical nephrotoxicity biomarkers into clinical applications.15
Table 2.
Examples of PPP efforts contributing to drug development tool (DDT) qualification and letters of support
| PPP | Drug development tool | Qualification/letter of support | Year of DDT |
|---|---|---|---|
| CAMD | Quantitative model describing the natural history of the cognitive change in Alzheimer's disease to be used in dose selection, population inclusion, sample size estimates and study duration | FDA and EMA qualification opinion as ‘Fit for Purpose’ | 2013, 2013 |
| CAMD | Cerebrospinal fluid analytes as exploratory prognostic markers for enrichment in Alzheimer's disease clinical trials (Aβ1‐42, total‐tau, and phosphorylated‐tau) | FDA Letter of Support | 2015 |
| CAMD | Hippocampal Volume (HV) as exploratory prognostic marker for enrichment in Alzheimer's disease clinical trials | FDA Letter of Support and EMA Qualification | 2015, 2011 |
| CAMD/ CPP | Dopamine Transporter (DAT) as exploratory prognostic markers for enrichment in early stage Parkinson's disease clinical trials | FDA Letter of Support | 2015 |
| CPTR | Hollow Fiber Model as preclinical assay for TB drug development | EMA Qualification | 2015 |
| HESI/ CSRC/ Others | Circulating cardiac troponin for preclinical prediction drug‐induced cardiotoxicity | FDA Qualification | 2012 |
| PKDOC | Total Kidney Volume for predicting progression to polycystic kidney disease | FDA and EMA Qualification | 2016, 2015 |
| PSTC | Suite of seven proteins for preclinical prediction of drug‐induced kidney injury (urinary β2‐microblobulin, urinary total protein, urinary albumin, urinary KIM‐1, urinary clusterin, urinary cystatin c, and urinary trefoil factor 2) | FDA, EMA, and PMDA Qualification | 2008, 2008, 2010 |
| PSTC | Two additional proteins for preclinical prediction of drug‐induced kidney injury (urinary osteopontin, lipocalin 2) | FDA and EMA Letter of Support | 2014, 2014 |
| PSTC | Suite of four proteins for clinical prediction of drug‐induced skeletal muscle injury (myosin light chain 3, skeletal muscle troponin I, fatty acid binding protein 3, and creatine kinase‐muscle type) | FDA and EMA Letter of Support | 2015, 2015 |
| SAFE‐T | Four exploratory biomarkers for clinical safety assessment of the risk of drug‐induced liver injury progression (cytokeratin 18, total and hyperacetylated high mobility group protein B1, osteopontin, and macrophage colony‐stimulating factor 1 receptor) | FDA Letter of Support | 2016 |
For additional information on the FDA Fit‐for‐Purpose Initiative, please visit http://www.fda.gov/Drugs/DevelopmentApprovalProcess/ucm505485.htm.
CDER continues to partner with PPP stakeholders to further clarify the biomarker qualification process. These activities include hosting scientific meetings and public discussions, publication of a harmonized biomarker terminology glossary, and refinement of the evidentiary criteria for the analytical, clinical, and the statistical rigor needed for biomarker qualification for their respective contexts of use. A complete compilation of these activities can be found on the FDA Drug Development Tools website.16
PSTC continues to engage in discussions with the FDA and the EMA to qualify additional clinical safety biomarkers for drug‐induced liver, skeletal muscle, cardiac, vascular, kidney, testicular, and pancreatic injury. Kidney and skeletal muscle injury biomarkers efforts by the PSTC were recognized for their public health potential by both the FDA and EMA through the Letter of Support (LOS) initiative, a CDER initiative to enhance the visibility of promising biomarkers and encourage their further evaluation through public sharing of the issued LOS.17 An LOS was also issued to PSTC and SAFE‐T to encourage further study and development of soluble biomarkers of endothelial cell injury and inflammation as exploratory biomarkers for monitoring drug‐induced vascular injury (Table 2). Other PPPs involved in the development of drug‐safety biomarkers include the International Serious Adverse Events Consortium (iSAEC) and the Health and Environmental Sciences Institute Cardiac Safety Committee (HESI‐CSC). iSAEC is developing databases with DNA sequences and corresponding phenotypes through precompetitive sharing of clinical data by participating pharmaceutical companies. These DNA‐variant databases are used to identify genetic biomarkers to predict the risk of drug‐related serious adverse events. iSAEC's initial studies successfully identified genetic variants associated with drug‐related liver toxicity and serious skin rashes, and their white papers have been cited more than 200 times across PubMed publications.18, 19, 20, 21 HESI‐CSC contributed to the evidence‐base for nonclinical use of circulating cardiac troponins T and I as safety biomarkers for prediction of drug‐induced cardiotoxicity, which received FDA qualification in 2012 (Table 2).22
In addition to drug safety biomarkers, several PPPs are focusing their efforts on disease‐specific biomarkers to inform and bring greater efficiency to drug development in specific therapeutic areas. In 2015, the FDA qualified total kidney volume as a prognostic biomarker for the enrichment of clinical trials in autosomal dominant polycystic kidney disease, which was developed by the Polycystic Kidney Disease Outcomes Consortium (PKDOC) (Table 2).23 The Coalition Against Major Diseases (CAMD) is developing two prognostic biomarkers for use in Alzheimer's disease drug development. The cerebrospinal fluid (CSF) analytes, Aβ1‐42, total‐tau, and phospho‐tau, are being developed as prognostic biomarkers to identify patients with mild cognitive impairment (MCI) likely to progress to Alzheimer's disease.24 Second, CAMD assembled a standardized database to support the use of hippocampal volume as a prognostic biomarker for enrichment in clinical trials for Alzheimer's disease, which received EMA qualification in 2011 and an LOS from the FDA in 2015 (Table 2).25 Use of these biomarkers aims to enable Alzheimer's disease patient enrichment in early disease clinical trials.26 Furthermore, the Critical Path for Parkinson's (CPP) consortium was formed in December 2015 as an offshoot of CAMD to assume leadership of Parkinson's disease treatment efforts. Together with CAMD, CPP is developing dopamine transporter neuroimaging by single‐photon emission computed tomography (SPECT) as a prognostic biomarker for the enrichment of Parkinson's disease clinical trials, and received an FDA LOS in 2015 (Table 2).27
CDER also developed a platform for a collaborative discussion to foster additional communication with the scientific community regarding challenges in drug development, and innovative methodology and technology to address these, called Critical Path Innovation Meetings (CPIM). CPIMs are drug‐product‐independent and nonbinding. Since the CPIM inception and until July 2016, 30 CPIMs were held that included discussions addressing the broad scope of CPI and CPOL.28 PPPs have participated in and played an active role in these discussions.
Topic 2: Streamlining clinical trials
The CPOL emphasizes the need to reform the clinical trial system through integrating innovative, efficient, and streamlined trial designs. Of the six CPOL topics, PPPs are most heavily invested in the area of streamlining clinical trials (Figure 2). In addition to the development and qualification of biomarkers for various contexts of use in the clinical trials discussed above, PPPs are addressing this unmet need through development of new therapeutic area data standards, clinical trial methodologies and designs, and clinical outcome assessments (COA) in clinical trials (e.g., patient reported outcomes).
Therapeutic area data standards
Large amounts of data are generated throughout drug development and clinical use. These data are a rich source of information, with great potential for analysis and interpretation to inform future drug development efforts and regulatory decisions. However, to reliably analyze and extrapolate between studies, a standardized data format is essential for data acquisition, integration, and analysis. The Coalition for Accelerating Standards and Therapies (CFAST) is a partnership between several nonprofit organizations and the FDA that was initiated to create and maintain data standards, tools, and methods for conducting research in therapeutic areas. As of July 2016, the partnership has published data standards in 21 therapeutic areas. Many PPPs have collaborated with CFAST to produce therapeutic area data standards, including the Critical Path to TB Drug Regimens (CPTR), the Duchenne Regulatory Science Consortium (D‐RSC), the Kidney Health Initiative (KHI), the Multiple Sclerosis Outcome Assessments Consortium (MSOAC), PKDOC, and CAMD.
Implementation of data standards has enabled the development of PPP deliverables such as pooled databases for broader multistudy analyses that can identify signals and trends not apparent when evaluating an individual study. For example, in 2016 the CPTR generated the TB‐PACTS database (Tuberculosis (TB)‐Platform for Aggregation of Clinical TB Studies) to define TB data standards. TB‐PACTS is a data platform of curated and standardized phase III TB clinical trials investigating shorter TB treatment durations, including the REMoxTB, RIFAQUIN, and OFLUTUB clinical trials. This data platform can be leveraged to analyze TB clinical trial parameters across clinical trials, such as dose selection and new therapeutic combinations.29 This effort holds tremendous promise to advance TB drug development.
Clinical trial methodologies and design
The Clinical Trials Transformation Initiative (CTTI) aims to enhance clinical trial quality and efficiency and addresses complex clinical trial issues such as the informed consent process, factors that influence investigator decisions to participate in clinical trials, and implementation of central Institutional Review Boards (IRBs). CTTI developed recommendations to streamline Good Clinical Practice (GCP) training practices regarding frequency, format of training, and identifying key elements for training programs that included elements outlined in the investigator section of the International Conference on Harmonisation (ICH) E6 guidance.30, 31 In 2011, CTTI published a survey to catalog the breadth of monitoring practices used in clinical trials and developed recommendations on effective and efficient monitoring.32 Subsequently, these efforts informed the FDA guidance for industry, Oversight of Clinical Investigations: A Risk‐Based Approach to Monitoring, published in 2013 (Table 3). Additionally, CTTI generated a shared data platform for aggregated analysis of the clinical trial information housed on ClinicalTrials.gov, that has now been leveraged to identify trends in the reporting of clinical trials and methodology differences across many different therapeutic areas to help inform evidence‐based strategic plans for biomedical research.33
Table 3.
Examples of PPP‐informed guidances
| PPP | Regulatory institution | Guidance |
|---|---|---|
| ACTTION | European Medicines Agency | Clinical development of medicinal products intended for the treatment of pain |
| BC | Food and Drug Administration | Acute Bacterial Skin and Skin Structure Infections: Developing Drugs for Treatment |
| CPTR | Food and Drug Administration | Pulmonary Tuberculosis: Developing Drugs for Treatment |
| CSRC | International Conference on Harmonisation | ICH E14: Clinical Evaluation of QT/ QTc Interval Prolongation and Proarrhthmic Potential for Non‐Antiarrhythmic Drugs |
| CTTI | Food and Drug Administration | Oversight of Clinical Investigations: A Risk‐Based Approach to Monitoring |
| HESI | International Conference on Harmonization | ICH S7B: Non‐clinical Evaluation of the Potential for Delayed Ventricular Repolarization by QT Interval Prolongation |
| PQRI | International Conference on Harmonisation | ICH Q11: Development and Manufacture of Drug Substances |
| PQRI | Food and Drug Administration | Waiver of In Vivo Bioavailability and Bioavailability and Bioequivalence Studies for Immediate‐Release Solid Oral Dosage forms |
| PQRI | Food and Drug Administration | Established Conditions: Reportable CMC Changes for Approved Drug and Biologic Products |
| PQRI | Food and Drug Administration | Liposome Drug Products ‐ Chemistry, Manufacturing, and Controls; Human Pharmacokinetics and Bioavailability; and Labeling Documentation |
| PQRI | Food and Drug Administration | Dissolution Testing and Specification Criteria for Immediate‐Release Solid Oral Dosage Forms Containing Biopharmaceutics Classification Systems Class 1 and 3 Drugs |
| PQRI | Food and Drug Administration | Elemental Impurities in Drug Products |
For additional information on FDA guidances, please visit https://www.fda.gov/RegulatoryInformation/Guidances/Default.htm.
For additional information on ICH guidances, please visit http://www.ich.org/products/guidelines.html.
Several challenges exist in the design of clinical efficacy trials for analgesic medications. One of the key aims of the Analgesic, Anesthetic, and Addiction Clinical Trial Translations, Innovations, Opportunities and Networks (ACTTION) initiative is to streamline clinical trial methodology for the development of pain therapeutics. Much of their effort concentrates on defining clinically meaningful outcome measures across multiple types of pain trials, including acute and chronic disorders. ACTTION holds scientific meetings and develops white papers to define research priorities for the development of COAs, patient training systems to facilitate data collection of COAs, and recommendations of the COA instruments that should be used under certain clinical trial designs. One such white paper recommended clinical trial designs for confirmatory chronic pain trials that are now cited in the 2016 EMA draft guideline on Clinical Development of Medicinal Products Intended for the Treatment of Pain (Table 3).34
In addition to CTTI and ACTTION, several other PPP activities have markedly influenced clinical trial efficiency. In 2010, BC launched an adaptive phase II clinical trial of multiple drug regimens for breast cancer in the neoadjuvant setting, called Investigation of Serial Studies to Predict Your Therapeutic Response with Imaging and Molecular Analysis (I‐SPY‐2). The design of the I‐SPY‐2 trial employed molecular profiling to inform clinical trial arm and treatment decisions. The trial design evaluated 12 therapies with 10 molecular biomarkers and is now advancing two therapies to phase III trials in triple‐negative and in Human Epidermal Growth Factor Receptor 2 (HER‐2) positive/hormone receptor negative breast cancer patients.35, 36 The I‐SPY‐2 trial is regarded as the model for adaptive clinical trial design and has laid the groundwork for additional master protocols such as the LUNG‐MAP for lung cancer, and for drug development in pediatric populations.37, 38, 39 Additionally, the Cardiac Safety Research Consortium (CSRC) conducted the first multicenter registry‐based clinical trial with patient level randomization in the United States. The Study of Access Site for Enhancing Percutaneous Coronary Intervention (PCI) for Women (SAFE‐PCI for Women) clinical trial was designed to evaluate radial vs. femoral artery PCI access exclusively in women.40, 41 In 2013, SAFE‐PCI was acknowledged by the Transcatheter Cardiovascular Therapeutics (TCT) conference for its novel methodology.42
Patient‐reported outcomes
Patients are the ultimate stakeholders in drug development and outcomes from clinical trials. Patient engagement during clinical trials is highlighted in the CPOL, and is a priority for CDER. The DDT Qualification Program established by CDER in 2005 includes a regulatory pathway for the qualification of COA instruments. COA instruments are currently under review for a broad range of therapeutic areas.16 The Patient Reported Outcomes (PRO) Consortium is focused on the development and implementation of patient‐reported outcome instruments in clinical trials. The COAs under development with the PRO Consortium and MSOAC address irritable bowel syndrome, depression, asthma, nonsmall‐cell lung cancer, rheumatoid arthritis, functional dyspepsia, mild cognitive impairment due to Alzheimer's disease, and multiple sclerosis.
Another PPP with an emphasis on patient engagement is the electronic PRO (ePRO) Consortium. The ePRO Consortium is focused on implementation of technology allowing the electronic capture of patient‐reported outcomes. Overall, the ePRO Consortium aims to provide a collaborative, instructional, and informational venue to advance the science of clinical trial assessment. Additional examples of patient engagement‐related activities through PPPs include the Kidney Health Initiative (KHI) that recently created the Patient and Family Partnership Council (PFPC) to incorporate the patient perspective into their deliverables. Also, ACTTION has an ongoing working group called Community Patient Awareness about Clinical Trials (COMPAACT) to investigate and promote patient engagement in pain clinical trials. In 2015, CTTI developed best practice recommendations and tools for growing partnerships between patient advocacy groups and clinical trialists.
Topic 3: Harnessing bioinformatics
The development of mathematical models, application of innovative statistical methodologies, and use of computational analyses hold potential for reducing the size of animal and human trials while improving the interpretability of study results. In addition, in silico modeling approaches provide predictive capabilities to streamline drug development, from molecule selection and predictive toxicology to clinical trial design. Through the sharing of Alzheimer's disease clinical trials data by member companies, CAMD developed a quantitative disease progression model describing the natural history of the cognitive changes in Alzheimer's disease patients to inform dose selection, population inclusion, sample size estimates, and study duration.43 At the time of its development, the aforementioned DDT Qualification Program did not have a designated qualification pathway for such tools. In 2013, both the FDA and EMA awarded a “Fit for Purpose” designation to the CAMD quantitative model to serve as a longitudinal model to inform Alzheimer's disease clinical trial design (Table 2). The FDA subsequently developed the “Fit for Purpose” Initiative to provide a mechanism for regulatory acceptance of dynamic tools for use in drug development.16 CAMD also developed a clinical trial simulation tool to assist in optimizing clinical trial design for mild to moderate Alzheimer's disease by testing clinical trials' operating characteristics. The simulation tool can be used to model disease progression, drug effects, clinical trial dropout rates, placebo effect, and relevant sources of variability to inform and streamline clinical trial design.44
Both HESI‐CSC and the Cardiac Safety Research Consortium (CSRC) aim to advance cardiac safety evaluation for new and existing medical products through the development of nonclinical models and clinical methodologies, respectively. Until recently, the clinical assessment of the potential for drug‐induced arrhythmias was tested through the “Thorough QT Study” (TQT study), a test evaluating electrocardiographic QT intervals across multiple therapeutic doses. However, while highly sensitive, TQT studies demonstrated inadequate specificity and were considered resource‐intensive.45, 46, 47 Collaborations between CDER and CSRC resulted in a 2012 scientific discussion (termed the “Think‐Tank Meeting” at CSRC) that developed recommendations in the form of white papers for nonclinical and phase I assessments of the potential for drug‐induced arrhythmias.45, 46, 48 The resulting recommendations informed a December 2015 revision of the ICH E14 guidance on monitoring QT assessment during early dose escalation and safety studies as an alternative to TQT studies (Table 3).49, 50, 51 A similar meeting hosted by the HESI‐CSC aimed to define the relationship between drug‐induced cardiac ventricular repolarization and the rare, life‐threatening clinical event torsades de pointes. The results from this meeting informed the ICH S7B guidance on nonclinical cardiac safety assessments for evaluation of clinical risk of QT interval prolongation (Table 3).52, 53, 54 Both of these efforts emphasized the need for better predictive models for cardiac safety. As a result, an ongoing collaboration between international regulatory agencies, industry, academia, CDER, CSRC, and HESI‐CSC is developing a comprehensive in vitro proarrhythmia assay (CiPA). CiPA is a mechanism‐based approach that leverages ion channel databases and computationally reconstructs human ventricular cardiomyocyte action potentials to predict the effect of new therapeutics on myocyte electrophysiology. This system will incorporate parameters beyond QT prolongation into drug‐induced arrhythmia risk assessments to enable more accurate predictions of drug‐induced cardiac proarrhythmia potential in drug development.55, 56, 57
CSRC also collaborates with the FDA and Mortara Instruments to generate an electrocardiogram (ECG) warehouse for algorithm development and evaluation designed to detect arrhythmogenic potential of drugs. Currently, this database includes ECGs derived from moxifloxacin (the positive control in TQT studies), placebo data sets, and the congenital long QT syndrome data submitted to CSRC.58, 59 Thus far, proof‐of‐principle studies have demonstrated the value of the ECG Warehouse for developing algorithms that assess drug‐induced QT prolongation potential.60
Topic 4: Moving manufacturing into the 21st century
The Product Quality Research Institute (PQRI) and the National Institute for Pharmaceutical Technology and Education (NIPTE) are supporting efforts to promote commercial‐scale product quality and efficient manufacturing. PQRI addresses a wide range of manufacturing topics, including life‐cycle management to detect drifts in manufacturing process and approaches to shelf‐life estimation of pharmaceutical products.61, 62 Through their product‐quality research, PQRI have provided evidence‐based recommendations to the FDA regarding manufacturing standards, which have informed numerous regulatory guidances for a broad range of topics (Table 3).
The NIPTE consortium is a multiuniversity collaboration whose mission includes the advancement of drug quality. Recent topics undertaken by the consortium include defining Quality by Design principles in drug manufacturing, validation of model systems for biosimilarity analyses, understanding the nature of co‐crystallization, and studying nonlinear optical Stokes ellipsometric microscopy for rapid discrimination of active pharmaceutical ingredients. Additionally, NIPTE partnered with the FDA on an Excipient Properties Knowledge Base, which houses pharmaceutical excipients properties and contains models, methods, and best practices for use in the design of pharmaceutical products and processes.63 The publicly available FDA‐NIPTE database is hosted by PharmaHUB, an in silico platform for pharmaceutical product development and manufacturing, and is extensively used, with over 33,000 downloads since December 2006.64
Topic 5: Developing products to address urgent public health needs
One of the Topic 5 opportunities is improving antimicrobial product testing to enhance the efficiency of clinical trials for infectious diseases. As part of their focus on the development of new drug regimens for TB, the CPTR consortium is developing new testing methods and evaluation tools. To this end, CPTR endorsed the FDA‐approved liquid culture model and the hollow fiber model to facilitate TB drug development. The liquid culture model is a microbial culturing system that allows sputum to be readily tested for TB in resource‐poor areas where clinical trials often take place. The use of liquid culture as an accepted diagnostic device was subsequently included in the draft guidance for industry Pulmonary Tuberculosis: Developing Drugs for Treatment as an approach to measure baseline bacterial cultures and therapeutic efficacy (Table 3 ). This liquid culture device was used in clinical trials supporting the 2012 FDA approval of bedaquiline, a therapeutic drug to be used as part of combination therapy for multidrug‐resistant tuberculosis. This was the first drug for multidrug‐resistant TB to be approved by the FDA in decades.65 Additionally, CPTR endorsed an in vitro drug development tool called the hollow fiber model that mimics the natural TB disease state to inform dose and regimen selection.66 The FDA provided a companion editorial that the hollow fiber model could be useful in helping to select drug regimens for later‐stage clinical development.67 In 2015, this model was qualified by the EMA for the preclinical phases to provide preliminary proof of concept, to select pharmacodynamics targeted initial dose selection, and to assist in confirming dose regimens for later clinical trials in TB drug development (Table 2).
In addition to CPTR's efforts to improve antimicrobial product testing, CTTI and BC are engaged in other efforts to advance therapies for infectious diseases. CTTI is focusing on improving the conduct and feasibility of hospital‐acquired and ventilator‐associated bacterial pneumonia (HABP/VABP) clinical trials; specifically, informed consent, protocol design, choice of Institutional Review Board, and efficacy outcome measures.68 BC's efforts have contributed to the development and content validity of a patient‐reported outcome instrument for community‐acquired bacterial pneumonia (CABP) and acute bacterial skin and skin structure infections (ABSSSI) for use as a tool to assess how a patient feels, functions, and survives in antiinfective clinical trials. Through retrospective analysis, data interpretation, and synthesis of data from modern‐day clinical trials, relevant outcome measures were confirmed and informed the FDA guidances for use in drug development (Table 3).69
Topic 6: At‐risk populations: Pediatrics
PPPs including the International Neonatal Consortium (INC), the Global Pediatric Clinical Trials Network Pre‐Launch Consortium (PTC), SmartTots, and the PROTECT Initiative, are leading efforts to directly address the unique unmet needs of the pediatric populations. INC was established in 2015 and has set up working groups focused on neonatal clinical pharmacology, seizure trial protocols, neonatal databases, and bronchopulmonary dysplasia definitions.70 PTC was created in 2015 to leverage the convening power of the PPP model to bring the stakeholders together and build a unique framework for pediatric global clinical trials. PTC aims to create a first‐of‐its‐kind global clinical trial network for pediatric patients to facilitate clinical trials and data collection in this patient population to provide the evidence‐base to support pediatric drug development. The remaining pediatric PPPs, SmartTots, and PROTECT Initiative focus on postmarket pediatric medication use and safety, and are discussed below.
PPP DELIVERABLES AND POST‐MARKET SAFETY
The Critical Path Initiative in 2004 was a call to action to streamline the drug development pipeline, and as a result, the CPOL focuses primarily on premarket drug development. However, monitoring and evaluating medication safety throughout a product's lifecycle is also a critical part of the FDA mission. To this end, CDER engages with several PPPs that address unmet needs in the postmarket space, including real‐world evidence generation, medication error prevention, pharmaceutical supply chain monitoring, and drug safety in the pediatric population.
Real‐world evidence generation
The Innovation in Medical Evidence Development and Surveillance (IMEDS) system enables public and private sector entities to collaborate with multiple healthcare data partners and the analytic center utilized by the FDA through the Agency's Sentinel System.71 IMEDS offers several important advantages for partnering organizations. First, the large underlying distributed database offers privacy‐protected information about medical products used by millions of patients. The data are quality checked to FDA standards and formatted using the same common data model used by the FDA. Second, modular programs incorporate epidemiologic methods and computer software templates which are routinely used by the FDA. Third, years of collective experience with distributed drug safety analyses amassed by the analytic center and data partner staff provides critical context for new IMEDS users.
Finally, IMEDS ensures transparency, with detailed descriptions of analytic decisions and publication of results in sufficient detail to promote replication by others. In addition to providing access to existing capabilities developed for the Sentinel System, IMEDS partners can stimulate new methods and validation research as well as the development of complementary infrastructure that can benefit both regulators and those interested in drug development. IMEDS has also sponsored investigations of methodologic challenges affecting real‐world evidence generation such as identification and validation of health outcomes, the role of epidemiologic design choices, and controlling random and systematic error.72, 73, 74
Medication error prevention and safe medication use
ISMP is a membership‐based nonprofit organization that aims to alert and educate the patient and healthcare community on medication safety. In addition to monitoring the FDA Adverse Event Reporting System (FAERS) for medication errors, ISMP maintains its own FDA‐recognized voluntary medication error reporting system called the ISMP Medication Error Reporting Program (MERP). Through this effort, ISMP can identify and disseminate emerging medication safety concerns through numerous newsletters and alerts systems. This monitoring system has assisted in many repackaging or label changes and further prompted other FDA regulatory actions, such as the 2009 recall of Tylenol Infant Drops.75 Also, ISMP aims to prospectively minimize the potential for medication error through packaging and distribution recommendations, resident training programs, and consulting services for healthcare systems.
Supply chain maintenance
Two PPPs, GS‐1 and RX‐360, have taken measures to address the safety and efficiency of the pharmaceutical supply chain. GS‐1 is a business‐focused PPP that serves several industries including retail, food service, transportation, and healthcare. GS‐1 manages a global barcode and standards system for point‐to‐point supply chain monitoring to ensure product quality. For healthcare, these barcodes can be used to improve patient care through a number of applications, such as patient identification wristbands, serial numbers for patient records, product matching to patient data, and system tracking of medical instruments through use and decontamination.
RX‐360 is an international supply chain consortium with a goal to protect patient safety by sharing information and developing processes to improve the integrity and quality of the healthcare supply chain. RX‐360 provides best practice recommendations on quality and authenticity of the supplies and suppliers of drug components and finished products.76
Pediatric medication safety
SmartTots and the PROTECT Initiative are PPPs which were developed in response to medical needs in the practice of pediatric medicine. Following the publication of several studies that identified an association between exposure to pediatric anesthetics and cognitive deficits in animals, SmartTots was established to promote the awareness of this information and to drive research to develop evidence‐based recommendations for the clinical use of sedatives and anesthetics in infants and toddlers.77, 78, 79 In 2014, SmartTots and the FDA convened a group of experts to update the 2012 Consensus Statement for both parents and healthcare providers, which outlines the state of the pediatric anesthesia science and promoting awareness of the potential risks.80, 81 This statement was further endorsed by 19 pediatric and anesthesiologist societies, including the American Society of Anesthesiologists, American Academy of Pediatrics, and Society for Pediatric Anesthesia. In addition to being a scientific resource, SmartTots also supports research looking at long‐term neurocognitive changes following pediatric anesthetic exposure to develop best practices related to pediatric anesthesia and sedative use. For example, one study performed prospective neuropsychological assessment to determine the neurodevelopmental safety of early childhood anesthesia exposure in children who had a single exposure of anesthetics during inguinal hernia repair before the age of 36 months. The goal of this study was to examine if a single anesthesia exposure in otherwise healthy young children was associated with impaired neurocognitive development and abnormal behavior in later childhood.82
The PROTECT Initiative is a collaborative effort originating from the Center for Disease Control and Prevention (CDC) with the goal of preventing unintentional pediatric medication overdoses. The PROTECT Initiative created voluntary guidelines for standardization and clarification of dosing and volumetric measures displayed on over‐the‐counter liquid medications along with education programs to teach parents and caregivers about pediatric medication safety. Furthermore, the PROTECT Initiative developed safety packaging recommendations that were incorporated into repackaging of liquid Infants' and Children's Tylenol in 2011, which can be referenced on the PROTECT Initiative website.83
THE FUTURE OF CDER ENGAGEMENT WITH PPPs
The CPI provided a broad direction for change in the product development pipeline, and the CPOL offered an array of research opportunities to spur progress on CPI's multiple fronts. The challenging nature of these opportunities inspired several PPPs to explore the precompetitive space as a platform for stakeholder collaborations to make progress. Through information exchange, tools and standards development, and data sharing, much has been achieved over the past decade. CDER's role in these precompetitive PPP collaborations leverages the collective knowledge gained from regulatory review experience to advise stakeholders to achieve overlapping needs and public health goals.
This article highlights the efforts of CDER's PPP engagements to modernize drug development through innovative trial designs, drug development tools, new predictive models, and building on the informatics infrastructure to standardize data collection. These interactions have informed the development of numerous industry guidances and best practices in both preapproval and postapproval drug development settings. Taken together, the field of regulatory science is integrating basic scientific discoveries into actionable and meaningful public health products, and CDER's engagements with PPPs are influencing every step of the critical path, from early drug development to postmarket safety. Our experience demonstrates that the challenges of intellectual property and data sharing can be overcome and that PPP deliverables can be communicated publicly for broad application. As the products of these partnerships mature, it is incumbent upon all stakeholders to share the collective knowledge and impact from integrating the deliverables into drug development programs. Cataloguing and sharing these experiences and successes publicly will enable further learning and encourage the adoption of these novel approaches to achieve unmet public health needs. With continued partnership, multisector collaborations can continue to advance innovations in medical product development and the health of all Americans.
Acknowledgments
Article edits and comments were provided by Richardae Araojo, Patrick Archdeacon, Nancy Chang, Chekesha Clingman, Jacqueline Corrigan‐Curay, John Koerner, Allison Lin, David Martin, Susan McCune, Kristen Miller, Laurie Muldowney, Robert O'Neill, Jacqueline O'Shaughnessy, Elektra Papadopoulos, Melissa Robb, Jonas Santiago, Stephanie Shapley, Norman Stockbridge, and Joseph Toerner.
Conflict of Interest
The authors have nothing to disclose.
References
- 1. US Food and Drug Administration . Innovation or Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products. http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/CriticalPathInitiative/CriticalPathOpportunitiesReports/ucm113411.pdf 2017, June 9.
- 2. US Food and Drug Administration . Critical Path Opportunities List. http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/CriticalPathInitiative/CriticalPathOpportunitiesReports/UCM077258.pdf 2017, June 9.
- 3. Parekh, A. et al Catalyzing the Critical Path Initiative: FDA's progress in drug development activities. Clin. Pharmacol. Ther. 97, 221–233 (2015). [DOI] [PubMed] [Google Scholar]
- 4. US Food and Drug Administration . CDER Staff Participation in Public Private Partnerships and Consortia. https://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/ManualofPoliciesProcedures/UCM532571.pdf 2017, March 1.
- 5. Goodsaid, F.M. & Mendrick, D.L. Translational medicine and the value of biomarker qualification. Sci. Transl. Med. 2, 47ps4 (2010). [DOI] [PubMed] [Google Scholar]
- 6. Goodsaid, F. & Frueh, F. Biomarker qualification pilot process at the US Food and Drug Administration. AAPS J. 9, E105–108 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goodsaid, F. & Papaluca, M. Evolution of biomarker qualification at the health authorities. Nat. Biotechnol. 28, 441–443 (2010). [DOI] [PubMed] [Google Scholar]
- 8. Goodsaid, F.M. et al Novel biomarkers of acute kidney toxicity. Clin. Pharmacol. Ther. 86, 490–496 (2009). [DOI] [PubMed] [Google Scholar]
- 9. Goodsaid, F.M. , Frueh, F.W. & Mattes, W. Strategic paths for biomarker qualification. Toxicology 245, 219–223 (2008). [DOI] [PubMed] [Google Scholar]
- 10. Critical Path Institute . FDA and EMEA Conclude that New Renal Safety Biomarkers are Qualified for Specific Regulatory https://c-path.org/pdf/PSTC_nephro_VXDS_summary_final.pdf 2017, March 27.
- 11. Critical Path Institute . Japanese PMDA Announces First Biomarker Qualification. https://c-path.org/japanese-pmda-announces-first-biomarker-qualification
- 12. Sistare, F.D. , et al Towards consensus practices to qualify safety biomarkers for use in early drug development. Nat. Biotechnol. 28, 446–454 (2010). [DOI] [PubMed] [Google Scholar]
- 13. Warnock, D.G. & Peck, C.C. A roadmap for biomarker qualification. Nat. Biotechnol. 28, 444–445 (2010). [DOI] [PubMed] [Google Scholar]
- 14. Bonventre, J.V. , Vaidya, V.S. , Schmouder, R. , Feig, P. & Dieterle, F. Next‐generation biomarkers for detecting kidney toxicity. Nat. Biotechnol. 28, 436–440 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Foundation of the National Institutes of Health . The FNIH Biomarkers Consortium Launches Project to Improve Diagnosis of Kidney Injury. https://c-path.org/wp-content/uploads/2013/09/FNIH_biomarkers_consortium_kidney_safety_PR.pdf 2017, June 9.
- 16. Center for Drug Evaluation and Research . Drug Development Tools (DDT) Qualification Programs. https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugDevelopmentToolsQualificationProgram/default.htm 2017, March 27.
- 17. US Food and Drug Administration . Letter of Support Initiative. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/ucm434382.htm 2017, June 9.
- 18. Daly, A.K. et al HLA‐B*5701 genotype is a major determinant of drug‐induced liver injury due to flucloxacillin. Nat. Genet. 41, 816–819 (2009). [DOI] [PubMed] [Google Scholar]
- 19. Shen, Y. et al Genome‐wide association study of serious blistering skin rash caused by drugs. Pharmacogenomics J. 12, 96–104 (2012). [DOI] [PubMed] [Google Scholar]
- 20. Lucena, M.I. et al Susceptibility to amoxicillin‐clavulanate‐induced liver injury is influenced by multiple HLA class I and II alleles. Gastroenterology 141, 338–347 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Urban, T.J. et al Limited contribution of common genetic variants to risk for liver injury due to a variety of drugs. Pharmacogenet. Genomics. 22, 784–795 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hausner, E.A. et al Qualification of cardiac troponins for nonclinical use: a regulatory perspective. Regul. Toxicol. Pharmacol. 67, 108–114 (2013). [DOI] [PubMed] [Google Scholar]
- 23. Brosnahan, G.M. Volume progression in polycystic kidney disease. N. Engl. J. Med. 355, 733; author reply 734 (2006). [DOI] [PubMed] [Google Scholar]
- 24. Molinuevo, J.L. et al The clinical use of cerebrospinal fluid biomarker testing for Alzheimer's disease diagnosis: a consensus paper from the Alzheimer's Biomarkers Standardization Initiative. Alzheimers Dement. 10, 808–817 (2014). [DOI] [PubMed] [Google Scholar]
- 25. Isaac, M. & Gispen‐de, Wied C. CNS biomarkers: Potential from a regulatory perspective: Case study — Focus in low hippocampus volume as a biomarker measured by MRI. Eur. Neuropsychopharmacol. 25, 1003–1009 (2015). [DOI] [PubMed] [Google Scholar]
- 26. Carrillo, M.C. et al Research and standardization in Alzheimer's trials: reaching international consensus. Alzheimers Dement. 9, 160–168 (2013). [DOI] [PubMed] [Google Scholar]
- 27. Antonini, A. & Biundo, R. Parkinson disease: Can dopamine transporter imaging define early PD? Nat. Rev. Neurol. 10, 432–433 (2014). [DOI] [PubMed] [Google Scholar]
- 28. US Food and Drug Administration . Critical Path Innovation Meetings (CPIM). http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugInnovation/ucm395888.htm 2016, August 18.
- 29. Critical Path to TB Drug Regimens Consortium . Access the TB‐PACTS Platform. Critical Path Institute, 2016. [Google Scholar]
- 30. International Committee on Harmonisation . Guidance for Industry E6 Good Clinical Practice Consolidated Guidance http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm073122.pdf 2016, August 18.
- 31. Clinical Trial Transformation Initiative . CTTI Recommendations: Good Clinical Practice (GCP) Training for Investigators. https://www.ctti-clinicaltrials.org/sites/www.ctti-clinicaltrials.org/files/GCP-Recommendations.pdf 2017, June 9.
- 32. Morrison, B.W. et al Monitoring the quality of conduct of clinical trials: a survey of current practices. Clin. Trials. 8, 342–349 (2011). [DOI] [PubMed] [Google Scholar]
- 33. Tasneem, A. et al The database for aggregate analysis of ClinicalTrials.gov (AACT) and subsequent regrouping by clinical specialty. PLoS One 7, e33677 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dworkin, R.H. et al Research design considerations for confirmatory chronic pain clinical trials: IMMPACT recommendations. Pain 149, 177–193 (2010). [DOI] [PubMed] [Google Scholar]
- 35. Rugo, H.S. et al Adaptive randomization of veliparib‐carboplatin treatment in breast cancer. N. Engl. J. Med. 375, 23–34 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Park, J.W. et al Adaptive randomization of neratinib in early breast cancer. N. Engl. J. Med. 375, 11–22 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harrington, D. & Parmigiani, G. I‐SPY 2—A glimpse of the future of phase 2 drug development? N. Engl. J. Med. 375, 7–9 (2016). [DOI] [PubMed] [Google Scholar]
- 38. Herbst, R.S. et al Lung Master Protocol (Lung‐MAP)‐A biomarker‐driven protocol for accelerating development of therapies for squamous cell lung cancer: SWOG S1400. Clin. Cancer Res. 21, 1514–1524 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maryland Center for Excellence in Regulatory Science and Innovation (M‐CERSI) . Pediatric Master Protocols Workshop. http://pharmacy.umaryland.edu/centers/cersievents/master-protocols/agenda/ 2016, October 3.
- 40. Hess, C.N. et al Comparison of quality‐of‐life measures after radial versus femoral artery access for cardiac catheterization in women: Results of the Study of Access Site for Enhancement of Percutaneous Coronary Intervention for Women quality‐of‐life substudy. Am. Heart J. 170, 371–379 (2015). [DOI] [PubMed] [Google Scholar]
- 41. Rao, S.V. et al A registry‐based randomized trial comparing radial and femoral approaches in women undergoing percutaneous coronary intervention: the SAFE‐PCI for Women (Study of Access Site for Enhancement of PCI for Women) trial. JACC Cardiovasc. Interv. 7, 857–867 (2014). [DOI] [PubMed] [Google Scholar]
- 42. Duke Clinical Research Institute . TCT 2013: SAFE‐PCI featured at TCT for its groundbreaking research and operational innovations http://dcri.org/tct-2013-safe-pci-featured-tct-groundbreaking-research-operational-innovations/ 2017, June 9.
- 43. Romero, K. et al The future is now: model‐based clinical trial design for Alzheimer's disease. Clin. Pharmacol. Ther. 97, 210–214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rogers, J.A. et al Combining patient‐level and summary‐level data for Alzheimer's disease modeling and simulation: a beta regression meta‐analysis. J. Pharmacokinet. Pharmacodyn. 39, 479–498 (2012). [DOI] [PubMed] [Google Scholar]
- 45. Darpo, B. et al Cardiac Safety Research Consortium: can the thorough QT/QTc study be replaced by early QT assessment in routine clinical pharmacology studies? Scientific update and a research proposal for a path forward. Am. Heart J. 168, 262–272 (2014). [DOI] [PubMed] [Google Scholar]
- 46. Darpo, B. , et al The IQ‐CSRC prospective clinical phase 1 study: “Can early QT assessment using exposure response analysis replace the thorough QT study?” Ann. Noninvas. Electrocardiol. 19, 70–81 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Garnett, C.E. et al Methodologies to characterize the QT/corrected QT interval in the presence of drug‐induced heart rate changes or other autonomic effects. Am. Heart J. 163, 912–930 (2012). [DOI] [PubMed] [Google Scholar]
- 48. Rock, EP. et al Assessing proarrhythmic potential of drugs when optimal studies are infeasible. Am. Heart J. 157, 827–836 (2009). [DOI] [PubMed] [Google Scholar]
- 49. Darpo, B. , Garnett, C. , Keirns, J. & Stockbridge, N . Implications of the IQ‐CSRC Prospective Study: Time to Revise ICH E14. Drug Saf. 38, 773–780 (2015). [DOI] [PubMed] [Google Scholar]
- 50. Cavero, I. , Holzgrefe, H. & Clements, M. The prospective IQ‐CSRC trial: A prototype early clinical proarrhythmia assessment investigation for replacing the ICH E14 thorough QTc (TQT) study. J. Pharmacol. Toxicol. Methods. 80, 1–8 (2016). [DOI] [PubMed] [Google Scholar]
- 51. International Committee on Harmonisation . Final Concept Paper: E14 Q&As (R3): Revision of ICH E14 Q&As (R2). http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E14/E14_Q_A_R3__Final_Concept_Paper_9June_2015.pdf 2017, March 24.
- 52. Cavero, I. & Crumb, W. Moving towards better predictors of drug‐induced Torsade de Pointes. 2–3 November 2005, Crystal City, Virginia, USA. Expert Opin. Drug Saf. 5, 335–340 (2006). [DOI] [PubMed] [Google Scholar]
- 53. Bass, A.S. et al International Life Sciences Institute (Health and Environmental Sciences Institute, HESI) initiative on moving towards better predictors of drug‐induced torsades de pointes. Br. J. Pharmacol. 154, 1491–1501 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pierson, J.B. et al A public‐private consortium advances cardiac safety evaluation: achievements of the HESI Cardiac Safety Technical Committee. J. Pharmacol. Toxicol. Methods. 68, 7–12 (2013). [DOI] [PubMed] [Google Scholar]
- 55. Sager, P.T. , Gintant, G. , Turner, J.R. , Pettit, S. & Stockbridge, N. Rechanneling the cardiac proarrhythmia safety paradigm: a meeting report from the Cardiac Safety Research Consortium. Am. Heart J. 167, 292–300 (2014). [DOI] [PubMed] [Google Scholar]
- 56. Cavero, I. & Holzgrefe, H . Comprehensive in vitro Proarrhythmia Assay, a novel in vitro/in silico paradigm to detect ventricular proarrhythmic liability: a visionary 21st century initiative. Expert Opin. Drug Saf. 13, 745–758 (2014). [DOI] [PubMed] [Google Scholar]
- 57. Servick, K. A painstaking overhaul for cardiac safety testing. Science. 353, 976–977 (2016). [DOI] [PubMed] [Google Scholar]
- 58. Kligfield, P. & Green, C.L. The Cardiac Safety Research Consortium ECG database. J. Electrocardiol. 45, 690–692 (2012). [DOI] [PubMed] [Google Scholar]
- 59. Kligfield, P. et al The Cardiac Safety Research Consortium electrocardiogram warehouse: thorough QT database specifications and principles of use for algorithm development and testing. Am. Heart J. 160, 1023–1028 (2010). [DOI] [PubMed] [Google Scholar]
- 60. Green, C.L. et al Detection of QT prolongation using a novel electrocardiographic analysis algorithm applying intelligent automation: prospective blinded evaluation using the Cardiac Safety Research Consortium electrocardiographic database. Am. Heart J. 163, 365–371 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yu, L.X. et al Advancing product quality: A summary of the inaugural FDA/PQRI Conference. AAPS J. 17, 1011–1018 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yu, L.X. et al Advancing product quality: A summary of the second FDA/PQRI Conference. AAPS J. 18, 528–543 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Basu, P.K. et al NIPTE‐FDA Excipients Knowledge Base. https://pharmahub.org/resources/458 2016, November 15.
- 64. PharmaHUB . PharmaHUB Usage: Overview. https://pharmahub.org/usage/overview 2016, August 9.
- 65. Cox, E. & Laessig, K. FDA approval of bedaquiline—the benefit‐risk balance for drug‐resistant tuberculosis. N. Engl. J. Med. 371, 689–691 (2014). [DOI] [PubMed] [Google Scholar]
- 66. Romero, K. , Clay, R. & Hanna, D. Strategic regulatory evaluation and endorsement of the hollow fiber tuberculosis system as a novel drug development tool. Clin. Infect. Dis. 61 (Suppl 1), S5–9 (2015). [DOI] [PubMed] [Google Scholar]
- 67. Chilukuri, D. et al The hollow fiber system model in the nonclinical evaluation of antituberculosis drug regimens. Clin. Infect. Dis. 61 (Suppl 1), S32–3 (2015). [DOI] [PubMed] [Google Scholar]
- 68. Toerner, J.G. & Rubin, D. Advancing new antibacterial drug development for treatment of hospital‐acquired and ventilator‐associated bacterial pneumonia. Clin. Infect. Dis. 63 (Suppl 2), S37–38 (2016). [DOI] [PubMed] [Google Scholar]
- 69. Talbot, G.H. , Powers, J.H. , Hoffmann, S.C. , Biomarkers Consortium of the Foundation for the National Institutes of Health C‐A, Teams H‐VP. Developing outcomes assessments as endpoints for registrational clinical trials of antibacterial drugs: 2015 update from the Biomarkers Consortium of the Foundation for the National Institutes of Health. Clin. Infect. Dis. 62, 603–607 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Turner, M.A. et al The International Neonatal Consortium: collaborating to advance regulatory science for neonates. Pediatr. Res. 2016. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 71. Sentinel Coordinating Center . Sentinel. https://www.sentinelinitiative.org/ 2017, June 9.
- 72. Lanes, S. , Brown, J.S. , Haynes, K. , Pollack, M.F. & Walker, A.M. Identifying health outcomes in healthcare databases. Pharmacoepidemiol. Drug Saf. 24, 1009–1016 (2015). [DOI] [PubMed] [Google Scholar]
- 73. Gruber, S. et al Design and analysis choices for safety surveillance evaluations need to be tuned to the specifics of the hypothesized drug‐outcome association. Pharmacoepidemiol. Drug Saf. 25, 973–981 (2016). [DOI] [PubMed] [Google Scholar]
- 74. Gruber, S. & Tchetgen, E. Limitations of empirical calibration of p‐values using observational data. Stat. Med. 2016. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Practices IfSM . Some ISMP Accomplishments. https://www.ismp.org/about/accomplishments.aspx 2016, August 10.
- 76. RX‐360 . RX‐360 Resource Library. http://rx-360.org/resources/resource-library/ 2017, June 9.
- 77. Ikonomidou, C. et al Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 283, 70–74 (1999). [DOI] [PubMed] [Google Scholar]
- 78. Mellon, R.D. , Simone, A.F. & Rappaport, B.A. Use of anesthetic agents in neonates and young children. Anesth. Analg. 104, 509–520 (2007). [DOI] [PubMed] [Google Scholar]
- 79. SmartTots . Seminal Pediatric Anesthesia Research Studies. http://smarttots.org/resources/seminal-studies/ 2017, June 9.
- 80. Char, D. , Ramamoorthy, C. & Wise‐Faberowski, L. Cognitive dysfunction in children with heart disease: the role of anesthesia and sedation. Congenit. Heart Dis. 11, 221–229 (2016). [DOI] [PubMed] [Google Scholar]
- 81. SMARTTotes . Consensus Statement on the Use of Anesthetic and Sedative Drugs in Infants and Toddlers. http://smarttots.org/wp-content/uploads/2015/10/ConsensusStatementV910.5.2015.pdf 2016, August 9. [DOI] [PubMed]
- 82. Sun, L.S. et al Association between a single general anesthesia exposure before age 36 months and neurocognitive outcomes in later childhood. JAMA 315, 2312–2320 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Protect Initiative . Results and Accomplishments. https://www.cdc.gov/medicationsafety/protect/protect_initiative.html 2017, June 1.