

Abstract
Vaniprevir is an inhibitor of the hepatitis C virus (HCV) NS3/4A protease. The aim of these double‐blind, placebo‐controlled phase I studies was to evaluate the safety and pharmacokinetics of vaniprevir in healthy male volunteers. The primary objective for both studies was the safety and tolerability of vaniprevir. Single‐dose and steady‐state pharmacokinetics were also assessed. In both studies, there was no apparent relationship between the frequency or intensity of adverse events and vaniprevir dose. At single doses >20 mg, the plasma area under the curve (AUC)0–∞ and maximum concentration (Cmax) increased in a greater‐than‐dose‐proportional manner. The geometric mean ratios (GMRs; fed/fasted) were 1.22 and 0.79 for AUC0–∞ and Cmax, respectively. Following multiple doses, GMR accumulations for AUC0–12h and Cmax (day 14/day 1) ranged from 1.53 to 1.90 and from 1.41 to 1.92, respectively. These data support the use of vaniprevir with peginterferon and ribavirin in patients with HCV infection.
Study Highlights
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
✓ Vaniprevir is a selective inhibitor of the HCV NS3/4A protease that is approved in Japan for the treatment of chronic HCV infection in combination with peginterferon + ribavirin.
WHAT QUESTION DID THIS STUDY ADDRESS?
✓ This study evaluated the safety, tolerability, and PKs of single and multiple doses of vaniprevir in healthy volunteers.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
✓ Dose‐related increases in vaniprevir exposure occurred in a greater‐than‐dose‐proportional manner with both single and twice‐daily dosing. Maximum plasma concentrations were achieved within 1–3 h of dosing and plasma terminal t1/2 was 4–6 h at steady state. Safety assessments indicate that vaniprevir monotherapy was well‐tolerated in healthy volunteers.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE
✓ These data support the administration of twice‐daily vaniprevir 300 mg with peginterferon and ribavirin for the treatment of chronic HCV infection in Japanese patients.
Direct‐acting antiviral therapies have transformed the treatment of hepatitis C virus (HCV) infection. In this rapidly growing field, the first‐generation protease inhibitors boceprevir1, 2 and telaprevir3, 4 have been overtaken by all oral interferon‐free and ribavirin‐free treatment regimens.5, 6, 7, 8 Collectively, these treatment regimens dramatically extend the therapeutic options available to patients with HCV infection.
Vaniprevir is a rapidly reversible, noncovalent, competitive, selective inhibitor of the HCV nonstructural (NS)3/4A protease that is approved in Japan for the treatment of chronic HCV infection in combination with peginterferon + ribavirin.9, 10 In preclinical studies, vaniprevir had excellent liver exposure across various species, low‐nanomolar to sub‐nanomolar potency against genotype (GT)1 and 2 proteases, and potent activity in the HCV replicon system.11, 12 A substantial decrease in plasma HCV viral load (>5 logs) has been reported in an HCV‐infected chimpanzee model.13 In patients with HCV GT1 infection, vaniprevir was well tolerated and associated with a dose‐related (25 mg twice daily (b.i.d.) to 500 mg b.i.d.) decrease in HCV RNA of 1.8–4.6 log10 IU/mL following 1 week of monotherapy.14 Subsequent clinical trials have shown the combination of vaniprevir plus peginterferon and ribavirin to be safe and effective for treatment‐naive and treatment‐experienced patients with HCV GT1 infection.9, 10, 15, 16, 17 The aim of the present phase I study was to evaluate the safety, tolerability, and pharmacokinetics (PKs) of single and multiple doses of vaniprevir in healthy volunteers.
METHODS
These studies were carried out in accordance with the Declaration of Helsinki, current guidelines on Good Clinical Practices, and local ethical and legal requirements. All subjects provided voluntary written informed consent before trial entry.
Subjects
All subjects were healthy male volunteers aged 18–50 years with a body mass index (BMI) ≤30 kg/m2. All subjects were in good medical health based on medical history, physical examination, and laboratory safety tests, showed no clinically significant abnormality on electrocardiogram (ECG), and were nonsmokers. Subjects with clinically significant disease, including HCV and human immunodeficiency virus infection, or an estimated creatinine clearance of ≤80 mL/min were excluded. Subjects were also required to refrain from using any medications, including prescription and nonprescription drugs or herbal remedies, from ∼2 weeks before the initial dose in each panel of the study until its conclusion. Anticipated use of any prescription or nonprescription medication during the study was a criterion for exclusion.
STUDY DESIGN
Study 1
Protocol 001 was a double‐blind, placebo‐controlled, alternating‐panel, multiple‐period, single‐rising‐dose study. Two panels (panel A and panel B) of eight subjects alternately received single‐rising oral doses of vaniprevir (n = 6) or placebo (n = 2). Each panel consisted of five dosing periods: panel A subjects received vaniprevir single doses of 10, 40, 160, 350, and 825 mg or placebo, and panel B subjects received vaniprevir single doses of 20, 80, 240, and 550 mg or placebo. In period 5 of panel B, single‐dose vaniprevir (80 mg) was administered with a high‐fat meal (the dose of vaniprevir was identical in periods 2 and 5 of panel B, ensuring that the same subjects received the vaniprevir dose both with and without the high‐fat meal). A minimum washout interval of 48 h was observed between dose escalations to allow for PK sample collection and assessment of adverse events (AEs). There was also a minimum washout period of 3 days for each subject before receiving the next dose in the subsequent study period (except for subjects in panel B, period 5, who observed a ≥1‐week washout period from the previous dose). After each dose level, safety was reviewed; the decision to proceed to the next dose level was based on acceptable safety and tolerability.
Study 2
Protocol 002 was a double‐blind, randomized, placebo‐controlled, staggered incremental dose study consisting of six successive panels (panels A, B, C, D, E, and F). In each panel, eight subjects were randomized to receive vaniprevir (n = 6) or placebo (n = 2) in a blinded manner. Subjects in panels A, B, C, and F received a single dose of vaniprevir (100, 200, 400, or 800 mg, respectively) or placebo on days 1 and 14, and 2 daily doses of vaniprevir (100, 200, 400, or 800 mg b.i.d., respectively) or placebo on days 2–13. Subjects in panel D received a single dose of vaniprevir 1,000 mg or placebo followed by a 7‐day washout period. Then the subjects in panel D received single doses of vaniprevir 600 mg or placebo on days 1 and 14 and vaniprevir 600 mg b.i.d. or placebo on days 2–13. Subjects in panel E first received a single dose of vaniprevir 1,000 mg or placebo (period 1) followed by a ≥7‐day washout period, then a single dose of vaniprevir 1,300 mg or placebo (period 2) followed by a second ≥7‐day washout period, and finally a single dose of vaniprevir 1,600 mg or placebo (period 3). The administration of higher single doses in panel E was designed to provide experience with sufficiently high total daily exposures that would allow administration of 800 mg b.i.d. in panel F.
Assessments
Blood samples were obtained to assess the single‐dose and steady‐state plasma PKs of vaniprevir. Parameters evaluated included: area under the plasma concentration vs. time curve for 12 and 24 h postdose (AUC0–12h and AUC0–24h, respectively) as appropriate on day 1; AUC0–12h on day 14; maximum concentration of drug in the plasma (Cmax); plasma trough concentration (Ctrough); time to reach Cmax (tmax); apparent terminal elimination half‐life (t½); and accumulation ratio (AR). In addition, in study 2, AUC extrapolated to infinity (AUC0–∞) was calculated from concentration vs. time data collected following dosing on day 1, for comparison with AUC0–12h on day 14 of multiple dosing to determine predictability of multiple‐dose PKs from single‐dose PKs.
Urine samples for vaniprevir analysis were collected predose and up to 24 h postdose following administration of 160, 240, and 825‐mg single doses in study 1 (periods 3, 4, and 5). The safety and tolerability of vaniprevir was monitored by clinical assessment of AEs and by repeated measurements of vital signs, physical examinations, 12‐lead ECGs, and standard laboratory safety tests (hematology, chemistry, and urinalysis).
Analytical methods
Plasma and urine concentrations of vaniprevir were assayed at Merck & Co., Inc. (Kenilworth, NJ, USA; using high‐performance liquid chromatographic tandem mass spectrometry). The drug and internal standard were isolated from plasma using liquid–liquid extraction in the 96‐well format. The lower limit of quantitation (LLOQ) was 1 ng/mL and the linear calibration range was 1–1,000 ng/mL.
Data analysis
The primary objective for both studies was to establish the safety and tolerability of single and multiple dosing with vaniprevir. The secondary objectives were to determine the impact of a high‐fat meal on vaniprevir PKs relative to fasted administration (study 1) and whether at least one well‐tolerated, twice‐daily dose would result in a day 14 geometric mean (GM) Ctrough of >25 nM (study 2). A target Ctrough of 25 nM was selected based on preclinical and clinical data suggesting that a value of 35 nM was an optimal PK target, with a lower limit of 25 nM.
Safety assessments and clinical and laboratory AEs are summarized descriptively for placebo and each vaniprevir dose in both studies. Other safety measurements, such as laboratory tests, ECG parameters, and vital signs, were evaluated following multiple doses of vaniprevir. With the exception of C12h values, which were obtained using SAS, version 9.1 (SAS, Cary, NC), all PK parameters were calculated from the plasma‐concentration‐time data using the software WinNonlin (Pharsight, Mountain View, CA). All AUC parameters were calculated using the linear trapezoidal method for ascending concentrations and the log trapezoidal method for descending concentrations. For each subject, the apparent terminal t½ was calculated by regression of the terminal log‐linear portion of the plasma concentration–time profile.
Study 1 (Protocol 001)
The individual AUC0–∞, Cmax, and C12h values from both panels were natural‐log transformed and evaluated using linear mixed‐effects models containing treatment as a fixed effect and subject as a random effect. For each treatment, 95% confidence intervals (CIs) for the mean log concentration were calculated, and the CI limits then back‐transformed to construct the GMs and corresponding 95% CIs on the original scale. To evaluate the effect of food on vaniprevir PKs, geometric mean ratios (GMRs; fed/fasted) and corresponding 90% CIs were constructed for AUC0–∞ and Cmax from the aforementioned models. Although no formal testing was performed, appropriate summary statistics were provided for tmax and apparent terminal t½.
Study 2 (Protocol 002)
Individual AUC0–12h, Ctrough, and Cmax values (except for panel E) were natural log‐transformed and evaluated using a mixed‐effect model with fixed‐effects for dose, day, and dose‐by‐day interaction, and random effects for subject within dose. For AUC0–12h, a 90% CI for the mean between‐day difference (day 14/day 1) was calculated for each dose on the log‐scale based on the analysis of variance (ANOVA) model. The CIs were exponentiated to obtain the 90% CIs for the AR for each dose. For log‐AUC0–12h, log‐Ctrough, and log‐Cmax, a 95% CI for each dose level was calculated based on the ANOVA model. These CIs were exponentiated to obtain the 95% CIs for GM AUC0–12h, Ctrough, and Cmax. Day 1 AUC0–24h and AUC0–∞ were log‐transformed and evaluated by an ANOVA model, with dose as the single factor. The 95% CIs for day 1 GM AUC0–24h and AUC0–∞ were similarly constructed based on the ANOVA model. Descriptive statistics are provided for all PK parameters following single oral doses of vaniprevir (panels D and E), as well as tmax and apparent terminal t½ from all panels.
For the secondary hypothesis in study 2, the estimated SD for the natural log‐Ctrough was 0.22 nM. For a panel of six subjects, given a true GM Ctrough of 34 nM, there is ≥90% probability that the lower limit of the 90% CI of GM Ctrough exceeded 25 nM. Given a true GM Ctrough of 32 nM, there was ≥80% probability that the lower limit of the 90% CI of GM Ctrough exceeded 25 nM.
RESULTS
Study 1
Sixteen subjects were enrolled. All subjects were white, with a mean age of 30.7 years (range, 20–43 years) and a mean BMI of 23.7 kg/m2 (range, 19.6–27.6 k/m2). All 16 subjects completed the study and were included in the evaluation of PK and safety.
Safety
Single oral doses of vaniprevir up to 825 mg were generally well tolerated in healthy male subjects. Fifteen subjects reported a total of 72 AEs, of which 23 were considered related to treatment. The most common clinical AEs (regardless of relation to study treatment) were nasopharyngitis (n = 6), headache (n = 5), and influenza (n = 3). There was no apparent relationship between the frequency and intensity of AEs and vaniprevir dose, and no consistent, clinically relevant, treatment‐related, or dose‐related effects on ECGs, vital sign measurements, or laboratory safety tests. There were no serious AEs or laboratory AEs, no subject discontinued because of an AE, and there were no deaths.
Pharmacokinetics
For subjects receiving vaniprevir 10 mg, all vaniprevir plasma concentrations were below the assay LLOQ except for one sample. Similarly, at the 20‐mg dose, vaniprevir plasma concentrations were only detectable between 0.5 and 4 h postdose. Summary statistics and statistical comparisons were, therefore, not performed for the 10‐mg and 20‐mg doses. At higher doses, vaniprevir plasma AUC0–∞ and Cmax increased in a greater‐than‐dose‐proportional manner (Table 1). The GM apparent terminal t½ was ∼4–5 h independent of dose, and median tmax ranged from 1–3 h.
Table 1.
Vaniprevir pharmacokinetics following single oral doses in fed and fasted healthy male subjects
| Dose a (mg) | AUC0–∞ b (nM·h) n = 6 | Cmax b (nM) n = 6 | C12h b (nM) n = 6 | tmax c (h) n = 6 | Apparent terminal t½ d (h) n = 6 | |
|---|---|---|---|---|---|---|
| A | 40 | 43.24e (34.63–54.00) | 22.48 (15.48–32.65) | 1.35 (0.64–2.85) | 1.00 (1.00–1.50) | 3.9 (24.8) |
| 160 | 432.61 (354.35–528.14) | 162.71 (112.03–236.33) | 5.06 (3.77–6.80) | 1.50 (1.50–2.00) | 4.1 (33.6) | |
| 350 | 2,014.47 (1650.51–2458.70) | 857.09 (590.11–1,244.86) | 11.88 (8.84–15.96) | 2.00 (1.50–3.00) | 4.5 (22.3) | |
| 825 | 17,061.96 (13,975.65–20,829.83) | 6,524.18 (4,491.93–9,475.86) | 67.93 (50.54–91.29) | 2.00 (2.00–4.00) | 4.6 (32.7) | |
| B | 80 | 76.90 (62.27–94.95) | 33.24 (22.85–48.35) | 1.49 (0.98–2.28) | 2.00 (1.00–3.00) | 4.3 (23.2) |
| 240 | 600.32 (479.35–751.83) | 233.18 (160.54–338.69) | 6.95 (5.17–9.34) | 2.00 (1.50–3.00) | 4.4 (38.4) | |
| 550 | 4,006.30 (3279.05–4894.84) | 1,653.27 (1,138.24–2,401.36) | 24.19 (17.99–32.52) | 3.00 (2.00–3.00) | 5.0 (51.9) | |
| rMSEe | 0.144 | 0.396 | 0.308 |
AUC0–∞, area under the plasma concentration vs. time curve extrapolated to infinity; Cmax, maximum concentration; C12h, plasma concentration at 12 h postdose; rMSE, root mean square error; tmax, time taken to reach Cmax occurred; t½, elimination half‐life.
All doses administered after an 8‐h fast.
AUC0– ∞ and apparent terminal t½ values were not calculable for two subjects in the 40‐mg dose level, one subject in the 80‐mg fasted dose level, and two subjects in the 240‐mg dose level. Vaniprevir plasma concentrations were below the lower limit of quantitation for the 10‐mg and 20‐mg doses. bBack‐transformed least‐squares mean and confidence interval from mixed‐effects model performed on natural log‐transformed values. cMedian (minimum and maximum) reported for tmax. dGeometric mean and percent of geometric coefficient of variation reported for apparent terminal t½. erMSE: Square root of conditional mean squared error (residual error) from the linear mixed effects model. rMSE*100% approximates the within‐subject %CV on the raw scale.
Plasma vaniprevir concentrations were detectable up to 12 h following 80‐mg dosing in both fasted and fed states. The GMR (fed/fasted) and 90% CI were 1.22 (1.02–1.46) and 0.79 (0.54–1.17) for AUC0–∞ and Cmax, respectively (Table 2). Median tmax values were 2 h in fed and fasted patients receiving the 80‐mg dose. Under fed conditions, the apparent terminal t½ GM value for the 80‐mg dose was decreased by ∼2 h as compared with the 80‐mg administered fasted.
Table 2.
Comparison of vaniprevir (80 mg) in fasted and fed subjects (Protocol 001, panel B)
| Parametera | Vaniprevir, 80 mg fed | Vaniprevir, 80 mg fasted | Vaniprevir, 80 mg fed/fasted | |
|---|---|---|---|---|
| GM (95% CI) | GM (95% CI) | GMR (90% CI) | rMSEb | |
| AUC0–∞ c | 93.97 (75.13–117.55) | 76.90 (62.27–94.95) | 1.22 (1.02–1.46) | 0.144 |
| Cmax c | 26.39 (18.14–38.38) | 33.24 (22.85–48.35) | 0.79 (0.54–1.17) | 0.396 |
| tmax d | 2.00 (1.00–4.00) | 2.00 (1.00–3.00) | ||
| Apparent terminal t½ e (h) | 2.3 (32.6) | 4.3 (23.2) | ||
AUC, area under the curve; CI, confidence interval; Cmax, maximum concentration; GM, geometric least‐squares means; GMR, geometric least‐squares mean ratio (day 14/day 1); rMSE, root mean square error; tmax, time taken to reach Cmax occurred; t½, elimination half‐life.
rMSE*100% approximates the within‐subject %CV on the raw scale.
AUC0–∞ and apparent terminal t½ values were not calculable for two fed subjects and one fasted subject. brMSE: Square root of conditional mean squared error (residual error) from the linear mixed‐effects model. cBack‐transformed least‐squares mean and confidence interval from mixed‐effects model performed on natural‐log transformed values. dMedian (min, max) reported for tmax. eGeometric mean and percent of geometric coefficient of variation reported for apparent terminal t½.
Urine concentrations of vaniprevir were below LLOQ at all time points following the 160‐mg and 240‐mg doses. Approximately 0.2% of the vaniprevir dose was eliminated in urine as unchanged drug following the 825‐mg dose.
Study 2
Forty‐eight healthy male subjects were enrolled. The mean age of enrolled subjects was 37.1 years (range, 19–48 years), with a mean BMI of 24.8 kg/m2 (range, 20.2–30.2 kg/m2). Three subjects discontinued for reasons unrelated to the study drug (i.e., due to administrative reasons) and 45 subjects completed the study. All 48 subjects were included in the safety evaluation.
Safety
Forty‐two subjects reported a total of 105 AEs, all of which were of mild or moderate intensity. Twenty‐nine AEs were considered to be related to the study drug by the investigator. No patient discontinued treatment due to an AE and there were no serious AEs. No laboratory AE was reported. All AEs resolved with ongoing treatment, and there was no apparent relationship between the frequency or intensity of AEs and the dose of vaniprevir. The most common AEs in patients receiving single doses were headache (n = 6) and diarrhea (n = 3), and those in patients receiving multiple doses were diarrhea (n = 8), abdominal discomfort (n = 4), and nausea (n = 4). There were no consistent, clinically relevant, treatment‐related, or dose‐related effects on ECGs, vital sign measurements, or laboratory safety tests.
Pharmacokinetics
Accumulation of vaniprevir in the plasma occurred over the 14‐day dosing period in all subjects (Figure 1 and Table 3). The GM ARs for AUC0–12h and Cmax (day 14/day 1) ranged from 1.53–1.90 and from 1.41–1.92, respectively. On days 1 and 14, both AUC0–12h and Cmax seemed to increase in a greater‐than‐dose‐proportional manner at doses >100 mg. Trough values (C12h) also seemed to increase slightly in a greater‐than‐dose‐proportional manner on days 1 and 14. Steady‐state levels of vaniprevir were generally achieved on day 3 in panels A, B, and C (100, 200, and 400 mg, respectively), on day 8 in panel D (600 mg), and on day 4 in panel F (800 mg). The GM for day 14 C12h was >25 nM following the 600‐mg and 800‐mg b.i.d. dosing of vaniprevir.
Figure 1.
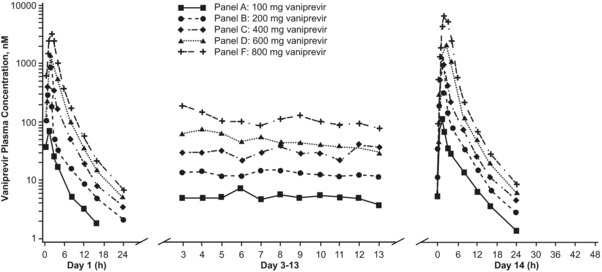
Mean plasma vaniprevir pharmacokinetic profiles following administration of multiple oral doses in healthy male subjects (n = 6 per dose level). Dosing was daily on days 1 and 14, and twice daily (i.e., every 12 h) on days 2 through 13. Serum concentrations were below level of assay quantification 24 h after the first dose on day 1 in patients receiving vaniprevir 100 mg.
Table 3.
Summary statistics of plasma vaniprevir pharmacokinetic parameters following multiple oral dosing in healthy male subjects (n = 6 for each panel)
| Panel | Dose (mg) | Day | AUC0–12h a (nM/h) | AUC0–24h a (nM/h) | AUC0–∞ a (nM/h) | Cmax a (nM) | C12h a (nM) | C24h a (nM) | tmax b (h) | Apparent t1/2 c (h) |
|---|---|---|---|---|---|---|---|---|---|---|
| A | 100d | 1 | 199.11 (132.23–299.81) | 215.81 (147.61–315.50) | 221.48 (147.67–332.18) | 8,782.07 (5,855.41–13,171.55) | 3.14 (2.18–4.51) | NA | 1.00 (0.50–2.00) | 4.6 (36.7) |
| 14 | 332.29 (220.68–500.34) | NA | NA | 120.92 (75.75–193.02) | 6.00 (4.17–8.61) | 2.01 (1.35–3.00) | 1.50 (0.50–2.03) | 5.1 (31.4) | ||
| GMR | 1.67 (1.35–2.06) | NA | NA | 1.41 (0.97–2.06) | 1.91 (1.61–2.28) | NA | NA | NA | ||
| B | 200d | 1 | 569.45 (378.18–857.45) | 614.88 (420.58–898.93) | 635.34 (423.61–952.90) | 295.41 (185.05–471.56) | 7.08 (4.93–10.18) | 2.45 (1.71–3.50) | 1.00 (1.00–2.00) | 6.5 (28.3) |
| 14 | 1,083.00 (719.24–1,630.74) | NA | NA | 498.99 (312.59–796.56) | 13.16 (9.16–18.90) | 3.20 (2.24–4.57) | 1.00 (1.00–2.00) | 4.4 (13.2) | ||
| GMR | 1.90 (1.54–2.35) | NA | NA | 1.69 (1.16–2.47) | 1.86 (1.56–2.21) | 1.31 (1.08–1.57) | NA | NA | ||
| C | 400d | 1 | 2,171.64 (1,442.23–3,269.97) | 2,262.90 (1,547.85–3,308.28) | 2,278.27 (1,519.03–3,417.00) | 871.18 (545.74–1,390.69) | 18.58 (12.93–26.69) | 3.03 (2.18–4.19) | 1.51 (1.50–2.00) | 4.1 (11.6) |
| 14 | 3,312.76 (2,200.06–4,988.21) | NA | NA | 1,484.32 (929.84–2,369.47) | 25.43 (17.70–36.53) | 4.36 (3.15–6.05) | 1.50 (1.00–2.00) | 3.9 (24.7) | ||
| GMR | 1.53 (1.24–1.88) | NA | NA | 1.70 (1.17–2.49) | 1.37 (1.15–1.63) | 1.44 (1.22–1.71) | NA | NA | ||
| D | 600d | 1 | 3,755.34 (2,493.99–5,654.63) | 3,912.96 (2,676.51–5,720.61) | 3,938.76 (2,626.15–5,907.44) | 1,363.86 (854.37–2,177.17) | 32.36 (22.53–46.49) | 4.95 (3.57–6.86) | 2.50 (2.00–3.00) | 4.0 (18.5) |
| 14 | 5,905.16 (3,921.72–8,891.74) | NA | NA | 2,019.46 (1,265.06–3,223.72) | 45.40 (31.61–65.22) | 5.80 (4.18–8.03) | 2.50 (1.00–4.00) | 3.8 (10.6) | ||
| GMR | 1.57 (1.28–1.94) | NA | NA | 1.48 (1.01–2.16) | 1.40 (1.18–1.67) | 1.17 (0.99–1.39) | NA | NA | ||
| F | 800d | 1 | 8,511.27 (5,652.48–12,815.92) | 8,755.13 (5,988.61–12,799.69) | 8,782.07 (5,855.41–13,171.55) | 2,909.88 (1,822.85–4,645.13) | 54.06 (37.63–77.66) | 6.19 (4.47–8.58) | 2.00 (2.00–3.00) | 3.4 (17.8) |
| 14 | 14,855.81 (9,866.00–22,369.26) | NA | NA | 5,583.31 (3,497.59–8,912.81) | 62.86 (43.76–90.30) | 7.64 (5.46–10.69) | 2.00 (2.00–3.00) | 5.4 (43.5) | ||
| GMR | 1.75 (1.42–2.15) | NA | NA | 1.92 (1.31–2.80) | 1.16 (0.98–1.38) | 1.23 (1.02–1.48) | NA | NA |
AUC, area under the curve; C12h, plasma concentration at 12 h postdose; C24h, plasma concentration at 24 h postdose; Cmax, maximum concentration; GM, geometric least‐squares means; GMR, geometric least‐squares mean ratio (day 14/day 1); NA, not applicable; tmax, time taken to reach Cmax occurred; t½, elimination half‐life.
Back‐transformed least‐squares mean and confidence interval from linear mixed effect model performed on natural log‐transformed values for the geometric mean (95% confidence interval) and geometric mean ratio (90% confidence interval). bMedian (range). cGeometric mean and percent of geometric coefficient of variation reported for t½. dDose was administered daily on days 1 and 14 and twice daily on days 2–13.
At higher single doses of vaniprevir (1,000 and 1,300 mg), there was appreciable variability (Figure 2 and Table 4). Substantially higher exposures were observed following administration of a single dose of vaniprevir 1,300 mg compared with lower single doses, confirming the greater‐than‐dose‐proportional increase in exposures seen at the lower doses evaluated in study 1. The observed GM AUC0–12h and Cmax values following single‐dose administration of vaniprevir 1,000 mg were between 12,518 and 16,505 nM/h and between 3,865 and 5,903 nM, respectively. Following single‐dose administration of vaniprevir 1,300 mg, the observed geometric AUC0–12h and Cmax were between 24,147 and 32,250 nM/h and between 7,579 and 9,199 nM, respectively.
Figure 2.
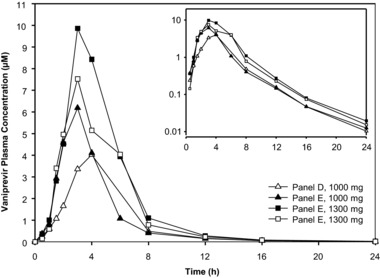
Mean plasma concentrations of vaniprevir following administration of single oral doses of vaniprevir.
Table 4.
Mean summary statistics of plasma vaniprevir pharmacokinetic parameters following administration of single oral doses (1,000 and 1,300 mg) in healthy male subjects
| Panel | Dose (mg) | AUC0–12h a (nM/h) | AUC0–∞ a (nM/h) | Cmax a (nM) | C12h a (nM) | C24h a (nM) | tmax b (h) | Apparent terminal t½ a (h) |
|---|---|---|---|---|---|---|---|---|
| D (period 1) | 1,000 | 12,517.93 (83.47) | 13,000.38 (83.05) | 3,865.39 (76.10) | 110.88 (105.65) | 8.85 (63.05) | 3.50 (3.00–4.00) | 3.1 (11.4) |
| E (period 1) | 1,000 | 16,505.14 (87.46) | 17,075.08 (86.01) | 5,903.18 (77.72) | 128.02 (57.51) | 10.48 (72.70) | 3.00 (2.00–4.02) | 3.3 (7.5) |
| D, E pooled | 1,000 | 14,373.94 (82.53) | 14,899.08 (81.61) | 4,776.83 (77.60) | 119.14 (78.10) | 9.63 (65.04) | 3.00 (2.00–4.02) | 3.2 (9.6) |
| E (period 2) | 1,300 | 32,250.61 (65.25) | 33,151.64 (65.05) | 9,198.88 (68.54) | 227.87 (67.46) | 16.38 (68.56) | 3.50 (3.00–6.00) | 2.9 (13.2) |
| E (period 3) | 1,300 | 24,146.87 (83.47) | 24,953.89 (82.85) | 7,578.83 (83.71) | 192.89 (79.74) | 12.52 (82.25) | 3.00 (3.00–6.00) | 3.0 (10.4) |
| E (periods 2 and 3) pooledc | 1,300 | 28,725.47 (69.66) | 29,590.91 (69.26) | 8,512.98 (70.23) | 213.17 (67.97) | 14.71 (70.72) | 3.00 (3.00–6.00) | 2.9 (11.7) |
AUC, area under the curve; C12h, plasma concentration at 12 h postdose; C24h, plasma concentration at 24 h postdose; Cmax, maximum concentration; tmax, time taken to reach Cmax occurred; t½, elimination half‐life.
Geometric mean and geometric coefficient of variation. bMedian (range). cData for repeat assessments to panel E (treatments E2 and E3) were pooled for overall pharmacokinetic summary following the 1,300‐mg (E2, E3) doses, respectively.
Vaniprevir had median tmax values ranging from 1.0–3.5 h following single and multiple doses (Tables 3 and 4). There was a trend toward increasing tmax with increasing dose. Following tmax, plasma vaniprevir concentrations declined in a biphasic manner with GM apparent terminal t½ values of 3.9–5.0 h and 3.4–6.5 h following single (Table 1) and b.i.d. dosing (Table 3), respectively. Across all single‐dose PK data in the current study, the geometric coefficients of variation for AUC0–∞ and Cmax ranged from 65–86% and from 69–84%, respectively.
DISCUSSION
Vaniprevir is a selective inhibitor of the HCV NS3/4A protease approved, in combination with peginterferon and ribavirin, in Japan for the treatment of treatment‐naive and treatment‐experienced patients with HCV infection.9, 10 Previous studies with this agent report potent in vitro activity in preclinical models11, 12 and antiviral activity when used in combination with peginterferon and ribavirin in patients with HCV infection.15, 16, 17 Data from the present studies support a twice‐daily dosing regimen with vaniprevir. Single‐dose administration of vaniprevir 80 mg with a high‐fat meal did not have a clinically meaningful effect on the plasma PKs of vaniprevir. Therefore, vaniprevir alone can be administered without regard to food. However, because vaniprevir is administered together with ribavirin and peginterferon, dosing instructions related to administration of peginterferon and ribavirin with food also apply to vaniprevir combination treatment.
Dose‐related increases in vaniprevir exposure occurred in a greater‐than‐dose‐proportional manner with both single and twice‐daily dosing. Maximum plasma concentrations were achieved within 1–3 h of dosing and plasma terminal t1/2 was 4–6 h at steady state. With multiple dosing, accumulation was observed with ratios of day 14/day 1 ranging between 1.5 and 1.9 for AUC and Cmax. Single‐dose administration does not predict steady‐state PKs following multiple‐dose administration, reflecting nonlinear PKs. Vaniprevir is a substrate of the CYP3A‐metabolizing enzyme and the hepatic organic anion transporting polypeptide‐1B1/3 uptake transporter. The nonlinear PKs of vaniprevir may be attributed to saturation of hepatic uptake and/or elimination.18 The longer time to achieve steady‐state relative to the half‐life also suggests saturable distribution and/or elimination of vaniprevir.
Safety assessments indicate that vaniprevir monotherapy was well tolerated in healthy volunteers, with mild‐to‐moderate headache and gastrointestinal AEs among the most commonly reported safety events. Increasing dose was not associated with an increase in AE frequency.
The PK profile observed in healthy subjects is consistent with that observed in Japanese HCV‐infected patients following co‐administration of vaniprevir 100, 300, or 600 mg b.i.d. with peginterferon and ribavirin. Similar to what was seen in healthy subjects, vaniprevir plasma PKs (i.e., AUC0–12 and Cmax) increased in a greater‐than‐dose‐proportional manner in Japanese HCV‐infected patients. However, higher exposures were observed in Japanese vs. non‐Japanese HCV‐infected patients. In Japanese patients, the mean steady‐state vaniprevir plasma exposure (i.e., AUC0–12) was 3.8, 25.3, and 78.5 μM/h following co‐administration of 100, 300, or 600 mg b.i.d. of vaniprevir with peginterferon and ribavirin, respectively.10, 19 Vaniprevir has low‐to‐moderate bioavailability; therefore, minor physiologic changes (e.g., body weight and liver size) affecting first‐pass processes (e.g., hepatic uptake) may be contributing to the moderate‐to‐high vaniprevir PK variability.20
The trends observed in the present PK data in healthy subjects are generally similar to those reported previously in patients with HCV infection, both when administered as a monotherapy14 and when used in combination with peginterferon and ribavirin.15 When administered as monotherapy in patients with HCV infection, greater‐than‐dose‐proportional increases in vaniprevir exposure were reported, with higher doses being associated with improved antiviral efficacy.14 Lawitz et al.14 reported that in patients with HCV GT1 infection, tmax ranged between 1 and 3 h, t1/2 was 4–9 h, and AUC0–12 and Cmax ARs ranged from 1.3–1.8. Similar to the present study 2, steady‐state levels were achieved by day 2 with twice‐daily dosing, whereas accumulation was not evident in patients receiving vaniprevir once daily (AUC0–12 and Cmax ratios of 0.88–1.2). Based on PK/pharmacodynamic correlation analysis in this monotherapy study, there was no particular association between decline in HCV RNA and any single vaniprevir PK parameter with the day 8 dose. AUC, Cmax, and Ctrough were each similarly predictive of virologic response. Vaniprevir PKs have also been shown to increase in a greater‐than‐dose‐proportional manner in patients with HCV infection receiving combination therapy with peginterferon and ribavirin.15 At vaniprevir doses of 300 and 600 mg b.i.d., AUC0–24 values were 5.15 and 23.36 μm·h, respectively, and Cmax and AUC ARs were 1.2–1.8. Collectively, these studies indicate that the trends in vaniprevir PKs are generally similar in healthy volunteers and in patients with HCV infection when administered as monotherapy or in combination with peginterferon and ribavirin.
In conclusion, the data yielded by this study support the administration of twice‐daily vaniprevir in combination with peginterferon and ribavirin for the treatment of patients with HCV infection. Based on the cumulative efficacy, safety, PK data, and data from the vaniprevir development program, a dose of 300 mg b.i.d. is approved for the treatment of chronic HCV infection in Japanese patients.9, 19
Acknowledgments
The authors are grateful to the study subjects who participated in the clinical trials and the clinical study site staff who facilitated them. Medical writing and editorial assistance were provided by Tim Ibbotson, PhD, of ApotheCom (Yardley, PA). This assistance was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
Author Contributions
L.C. and D.H.W. wrote the manuscript. L.C., J.B., J.W., and D.H.W. designed the research. J.d.H., M.D., and C.C. performed the research. D.H.W., L.C., J.d.H., M.D., C.C., J.M., W.G., D.P., Z.G., S.L.T., M.S.A., N.U., J.B., and J.W. analyzed the data.
Conflict of Interest
L.C., C.C., J.M., W.G., D.P., Z.G., S.L.T., M.S.A., N.U., J.B., J.W., and D.H.W. are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA or MSD at the time the study was conducted. J.d.H. received grants from Merck & Co., Inc., Kenilworth, NJ, USA to support the conduct of this study. M.D. declares no conflict of interest.
References
- 1. Poordad, F. et al Boceprevir for untreated chronic HCV genotype 1 infection. N. Engl. J. Med. 364, 1195–1206 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bacon, B.R. et al Boceprevir for previously treated chronic HCV genotype 1 infection. N. Engl. J. Med. 364, 1207–1217 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jacobson, I.M. et al Telaprevir for previously untreated chronic hepatitis C virus infection. N. Engl. J. Med. 364, 2405–2416 (2011). [DOI] [PubMed] [Google Scholar]
- 4. Zeuzem, S. et al Telaprevir for retreatment of HCV infection. N. Engl. J. Med. 364, 2417–2428 (2011). [DOI] [PubMed] [Google Scholar]
- 5. Afdhal, N. et al Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N. Engl. J. Med. 370, 1889–1898 (2014). [DOI] [PubMed] [Google Scholar]
- 6. Afdhal, N. et al Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. N. Engl. J. Med. 370, 1483–1493 (2014). [DOI] [PubMed] [Google Scholar]
- 7. Sulkowski, M.S. et al Ombitasvir, paritaprevir co‐dosed with ritonavir, dasabuvir, and ribavirin for hepatitis C in patients co‐infected with HIV‐1: a randomized trial. JAMA. 313, 1223–1231 (2015). [DOI] [PubMed] [Google Scholar]
- 8. Zeuzem, S. et al Grazoprevir‐elbasvir combination therapy for treatment‐naive cirrhotic and noncirrhotic patients with chronic hepatitis C virus genotype 1, 4, or 6 infection: a randomized trial. Ann. Intern. Med. 163, 1–13 (2015). [DOI] [PubMed] [Google Scholar]
- 9. Hayashi, N. et al Vaniprevir plus peginterferon alfa‐2b and ribavirin in treatment‐naive Japanese patients with hepatitis C virus genotype 1 infection: a randomized phase III study. J. Gastroenterol. 51, 390–403 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hayashi, N. , Mobashery N. & Izumi N. Vaniprevir plus peginterferon alfa‐2a and ribavirin in treatment‐experienced Japanese patients with hepatitis C virus genotype 1 infection: a randomized phase II study. J. Gastroenterol. 50, 238–248 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liverton, N.J. et al MK‐7009, a potent and selective inhibitor of hepatitis C virus NS3/4A protease. Antimicrob. Agents Chemother. 54, 305–311 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McCauley, J.A. , et al Discovery of vaniprevir (MK‐7009), a macrocyclic hepatitis C virus NS3/4a protease inhibitor. J. Med. Chem. 53, 2443–2463 (2010). [DOI] [PubMed] [Google Scholar]
- 13. Olsen, D.B. et al Sustained viral response in a hepatitis C virus‐infected chimpanzee via a combination of direct‐acting antiviral agents. Antimicrob. Agents Chemother. 55, 937–939 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lawitz, E. et al Characterization of vaniprevir, a hepatitis C virus NS3/4A protease inhibitor, in patients with HCV genotype 1 infection: safety, antiviral activity, resistance, and pharmacokinetics. Antiviral Res. 99, 214–220 (2013). [DOI] [PubMed] [Google Scholar]
- 15. Manns, M.P. et al Vaniprevir with pegylated interferon alpha‐2a and ribavirin in treatment‐naïve patients with chronic hepatitis C: a randomized phase II study. Hepatology 56, 884–893 (2012). [DOI] [PubMed] [Google Scholar]
- 16. Lawitz, E. , et al A phase 2B study of MK‐7009 (vaniprevir) in patients with genotype 1 HCV infection who have failed previous pegylated interferon and ribavirin treatment. J. Hepatol. 59, 11–17 (2013). [DOI] [PubMed] [Google Scholar]
- 17. Rodriguez‐Torres, M. et al Combination of vaniprevir with peginterferon and ribavirin significantly increases the rate of SVR in treatment‐experienced patients with chronic HCV genotype 1 infection and cirrhosis. Clin. Gastroenterol. Hepatol. 12, 1029–1037 (2014). [DOI] [PubMed] [Google Scholar]
- 18. Monteagudo, E. et al The metabolism and disposition of a potent inhibitor of hepatitis C virus NS3/4A protease. Xenobiotica. 40, 826–839 (2010). [DOI] [PubMed] [Google Scholar]
- 19. Hayashi, N. , et al Safety and efficacy of vaniprevir (MK‐7009) in combination with peg‐interferon a‐2a (peg‐IFN) / ribavirin (RBV) in genotype 1 treatment‐experienced HCV‐infected Japanese patients. Hepatology. 54(S1), 996A (2011). [Google Scholar]
- 20. Wright, D.H. et al Liver‐to‐plasma vaniprevir (MK‐7009) concentration ratios in HCV‐infected patients. Antivir. Ther. 20, 843–848 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]