

Abstract
Background
Chronic obstructive pulmonary disease (COPD) is a respiratory condition causing accumulation of mucus in the airways, cough, and breathlessness; the disease is progressive and is the fourth most common cause of death worldwide. Current treatment strategies for COPD are multi‐modal and aim to reduce morbidity and mortality and increase patients' quality of life by slowing disease progression and preventing exacerbations. Fixed‐dose combinations (FDCs) of a long‐acting beta2‐agonist (LABA) plus a long‐acting muscarinic antagonist (LAMA) delivered via a single inhaler are approved by regulatory authorities in the USA, Europe, and Japan for the treatment of COPD. Several LABA/LAMA FDCs are available and recent meta‐analyses have clarified their utility versus their mono‐components in COPD. Evaluation of the efficacy and safety of once‐daily LABA/LAMA FDCs versus placebo will facilitate the comparison of different FDCs in future network meta‐analyses.
Objectives
We assessed the evidence for once‐daily LABA/LAMA combinations (delivered in a single inhaler) versus placebo on clinically meaningful outcomes in patients with stable COPD.
Search methods
We identified trials from Cochrane Airways' Specialised Register (CASR) and also conducted a search of the US National Institutes of Health Ongoing Trials Register ClinicalTrials.gov (www.clinicaltrials.gov) and the World Health Organization International Clinical Trials Registry Platform (apps.who.int/trialsearch). We searched CASR and trial registries from their inception to 3 December 2018; we imposed no restriction on language of publication.
Selection criteria
We included parallel‐group and cross‐over randomised controlled trials (RCTs) comparing once‐daily LABA/LAMA FDC versus placebo. We included studies reported as full‐text, those published as abstract only, and unpublished data. We excluded very short‐term trials with a duration of less than 3 weeks. We included adults (≥ 40 years old) with a diagnosis of stable COPD. We included studies that allowed participants to continue using their ICS during the trial as long as the ICS was not part of the randomised treatment.
Data collection and analysis
Two review authors independently screened the search results to determine included studies, extracted data on prespecified outcomes of interest, and assessed the risk of bias of included studies; we resolved disagreements by discussion with a third review author. Where possible, we used a random‐effects model to meta‐analyse extracted data. We rated all outcomes using the GRADE (Grades of Recommendation, Assessment, Development and Evaluation) system and presented results in 'Summary of findings’ tables.
Main results
We identified and included 22 RCTs randomly assigning 8641 people with COPD to either once‐daily LABA/LAMA FDC (6252 participants) or placebo (3819 participants); nine studies had a cross‐over design. Studies had a duration of between three and 52 weeks (median 12 weeks). The mean age of participants across the included studies ranged from 59 to 65 years and in 21 of 22 studies, participants had GOLD stage II or III COPD. Concomitant inhaled corticosteroid (ICS) use was permitted in all of the included studies (where stated); across the included studies, between 28% to 58% of participants were using ICS at baseline. Six studies evaluated the once‐daily combination of IND/GLY (110/50 μg), seven studies evaluated TIO/OLO (2.5/5 or 5/5 μg), eight studies evaluated UMEC/VI (62.5/5, 125/25 or 500/25 μg) and one study evaluated ACD/FOR (200/6, 200/12 or 200/18 μg); all LABA/LAMA combinations were compared with placebo.
The risk of bias was generally considered to be low or unknown (insufficient detail provided), with only one study per domain considered to have a high risk of bias except for the domain 'other bias' which was determined to be at high risk of bias in four studies (in three studies, disease severity was greater at baseline in participants receiving LABA/LAMA compared with participants receiving placebo, which would be expected to shift the treatment effect in favour of placebo).
Compared to the placebo, the pooled results for the primary outcomes for the once‐daily LABA/LAMA arm were as follows: all‐cause mortality, OR 1.88 (95% CI 0.81 to 4.36, low‐certainty evidence); all‐cause serious adverse events (SAEs), OR 1.06 (95% CI 0.88 to 1.28, high‐certainty evidence); acute exacerbations of COPD (AECOPD), OR 0.53 (95% CI 0.36 to 0.78, moderate‐certainty evidence); adjusted St George's Respiratory Questionnaire (SGRQ) score, MD ‐4.08 (95% CI ‐4.80 to ‐3.36, high‐certainty evidence); proportion of SGRQ responders, OR 1.75 (95% CI 1.54 to 1.99). Compared with placebo, the pooled results for the secondary outcomes for the once‐daily LABA/LAMA arm were as follows: adjusted trough forced expiratory volume in one second (FEV1), MD 0.20 L (95% CI 0.19 to 0.21, moderate‐certainty evidence); adjusted peak FEV1, MD 0.31 L (95% CI 0.29 to 0.32, moderate‐certainty evidence); and all‐cause AEs, OR 0.95 (95% CI 0.86 to 1.04; high‐certainty evidence). No studies reported data for the 6‐minute walk test. The results were generally consistent across subgroups for different LABA/LAMA combinations and doses.
Authors' conclusions
Compared with placebo, once‐daily LABA/LAMA (either IND/GLY, UMEC/VI or TIO/OLO) via a combination inhaler is associated with a clinically significant improvement in lung function and health‐related quality of life in patients with mild‐to‐moderate COPD; UMEC/VI appears to reduce the rate of exacerbations in this population. These conclusions are supported by moderate or high certainty evidence based on studies with an observation period of up to one year.
Plain language summary
Once daily long‐acting beta2‐agonists and long‐acting muscarinic antagonists in a combined inhaler versus placebo for COPD
We wanted to know whether once‐daily treatment with a fixed‐dose combination of a long‐acting beta2 agonist (LABA) plus a long‐acting muscarinic antagonist (LAMA) delivered via a single inhaler is better than treatment with a dummy inhaler (placebo) for people with chronic obstructive pulmonary disease (COPD).
Background to the review
COPD is a disease of the lungs and is the fourth most common cause of death worldwide. People with COPD experience symptoms of cough, breathlessness and a build up of mucus, which become worse over time. Current treatments for COPD aim to manage these symptoms and improve the quality of life of people with the disease.
A combination of a LABA plus a LAMA taken once‐daily in a single inhaler (LABA/LAMA) has been shown to be more effective than taking each separately in individual inhalers. Several different combinations of inhaled LABA and LAMA are available (e.g. indacaterol/glycopyrronium, olodaterol/tiotropium, formoterol/aclidinium, and vilanterol/umeclidinium) and are used for the treatment of COPD. By gathering information from clinical trials that compare once‐daily LABA/LAMA with placebo in a dummy inhaler we will provide information to help future research decide which combination is best for treating people with COPD.
What did we find?
Twenty‐two studies (including 8641 people with COPD) compared once‐daily LABA/LAMA in a single inhaler with a dummy inhaler. People were allowed to continue to use their inhaled corticosteroids (ICS) during the studies; approximately a third to a half of people were using their ICS at the beginning of each study. The evidence presented in this review is current up to December 2018. The majority of people who took part in the studies had mild‐to‐moderate COPD and the average age of people in each study ranged from 59 to 65 years. Six studies evaluated the once‐daily combination of indacaterol/glycopyrronium, seven studies evaluated tiotropium/olodaterol, eight studies evaluated umeclidinium/vilanterol and one study evaluated aclidinium/formoterol.
People who took once‐daily LABA/LAMA using a single inhaler showed a greater improvement in quality of life than those taking placebo in a dummy inhaler; lung function was also improved in people taking once‐daily LABA/LAMA. People taking umeclidinium/vilanterol had fewer flare‐ups (exacerbations). There was no significant difference between groups (LABA/LAMA versus placebo) in the number of people who died, or in the number of people who experienced serious adverse events or any adverse event. The results were similar for the different LABA/LAMA combinations and doses that we evaluated.
The included studies were generally well designed and well reported. People in the studies and those performing the research did not know which treatment people were receiving, which ensures a fair evaluation of the treatments.
In three of the studies, people who were taking once‐daily LABA/LAMA had more severe COPD at the start of the study than people taking dummy inhalers; this could have reduced the treatment effect seen with LABA/LAMA in these studies so we can be confident that our findings do not overestimate the effect seen with once‐daily LABA/LAMA. One of the outcomes of interest (how far a person is able to walk in six minutes) was not reported by any of the included studies. Overall, we can be confident in the conclusions of this review.
Summary of findings
Summary of findings for the main comparison. Once‐daily LABA/LAMA in a combined inhaler compared with placebo in adults with COPD.
| Once daily LABA/LAMA in a combined inhaler compared with placebo in adults with COPD | ||||||
| Patient or population: Adults with COPD Setting: Clinical practice (primary care/secondary care/academic centres) Intervention: Once‐daily LABA/LAMA in a combined inhaler Comparison: Placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with once daily LABA/LAMA in a combined inhaler | |||||
| All‐cause mortality (3‐week to 1‐year follow‐up) | 1 per 1,000 | 2 per 1,000 (1 to 4) | OR 1.88 (0.81 to 4.36) | 8752 (18 RCTs) | ⊕⊕⊝⊝ LOW 1 ,2 | |
| Serious adverse events (3‐week to 1‐year follow‐up) |
47 per 1,000 | 50 per 1,000 (42 to 59) | OR 1.06 (0.88 to 1.28) | 10536 (22 RCTs) | ⊕⊕⊕⊕ HIGH | |
| Acute exacerbations of COPD (4‐week to 24‐week follow‐up) |
136 per 1,000 | 77 per 1,000 (53 to 109) | OR 0.53 (0.36 to 0.78) | 1127 (3 RCTs) | ⊕⊕⊕⊝ MODERATE 3 | Data limited to UMEC/VI versus placebo comparison |
| Difference vs placebo in adjusted SGRQ score (HRQoL) Scale 0‐100, lower on the scale is better. (12‐week to 1‐year follow‐up) |
Mean change from baseline in SGRQ score with placebo ranged from 6.39 lower to 0.12 higher | MD 4.08 points lower (4.8 lower to 3.36 lower) | ‐ | 4952 (8 RCTs) | ⊕⊕⊕⊕ HIGH | MD exceeded MCID (4 points). |
| Difference vs placebo in adjusted trough FEV1 at EOT (3‐week to 1‐year follow‐up) |
Mean change from baseline in trough FEV1 with placebo ranged from 0.08 L lower to 0.01 L higher | MD 0.20 L higher (0.19 higher to 0.21 higher) | ‐ | 6598 (13 RCTs) | ⊕⊕⊕⊝ MODERATE 4 | MD exceeded MCID. |
| Difference vs placebo in adjusted peak FEV1 (3‐week to 6‐month follow‐up) |
Mean change from baseline in peak FEV1 with placebo ranged from 0.04 to 0.1 L higher | MD 0.31 L higher (0.29 higher to 0.32 higher) | ‐ | 4188 (7 RCTs) | ⊕⊕⊕⊝ MODERATE 4 | |
| Adverse events (3‐week to 1‐year follow‐up) |
448 per 1,000 | 435 per 1,000 (411 to 458) | OR 0.95 (0.86 to 1.04) | 8235 (17 RCTs) | ⊕⊕⊕⊕ HIGH | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). COPD: chronic obstructive pulmonary disease; CI: confidence interval; EOT: end of treatment; FEV1: forced expiratory volume in 1 second; HRQoL: health‐related quality of life; LABA: long‐acting beta‐adrenoceptor agonist; LAMA: long‐acting muscarinic antagonist; MCID: minimum clinically important difference; MD: mean difference; OR: odds ratio; RR: risk ratio; SGRQ: St George's Respiratory Questionnaire; UMEC: umeclidinium; VI: vilanterol. | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded once for indirectness; duration of treatment varied widely: maximum duration 52 weeks, n = 3 studies duration < 6 weeks.
2 Downgraded once for imprecision; wide 95% confidence intervals due to low number of events – confidence intervals encompassed no effect, benefit, and risk.
3 Downgraded once for indirectness as all studies examined UMEC/VI.
4 Downgraded once for inconsistency; significant heterogeneity (overall I2 ≥ 68%), noting that effect sizes were similar between studies with the exception of ACLID/FORM.
Background
Description of the condition
Chronic obstructive pulmonary disease (COPD) is a progressive condition resulting from the complex interplay between environmental exposures (e.g. cigarette smoke) and genetic factors (Barnes 2015). The disease is characterised by a chronic limitation of airflow, which is not fully reversible, and intermittent exacerbations during which symptoms increase in severity. Symptoms include shortness of breath, increased sputum production and cough. The condition is diagnosed objectively by spirometric evaluation, with a post bronchodilator forced expiratory volume in one second/forced vital capacity (FEV1/FVC) < 0.70 confirming the presence of airflow limitation. COPD severity is graded by the extent of airflow limitation according to international guideline criteria (GOLD 2017).
COPD is the fourth most common cause of death worldwide (WHO 2015), and has an estimated prevalence of 6.4%; the burden on worldwide healthcare services is significant (CDC 2016; GOLD 2017).
Current treatment strategies are multi‐modal and aim to reduce morbidity and mortality and increase patients' quality of life by slowing disease progression and preventing exacerbations. Interventions include cessation of smoking and pulmonary rehabilitation, vaccination against influenza and pneumonia, and the use of inhaled corticosteroids (ICS) and bronchodilators (GOLD 2017). Supplemental oxygen is a life‐prolonging option in hypoxaemic patients. Although treatment is not curative, patients may occasionally be candidates for lung transplantation (GOLD 2017).
Description of the intervention
Long‐acting beta2‐agonists (LABA) and long‐acting anticholinergics (LAMA) are commonly used in patients with COPD as recommended by COPD guidelines (GOLD 2017; Wedzicha 2017). Each bronchodilator can be taken individually or in combination using either two separate inhalers or a single inhaler in a fixed‐dose combination (FDC; denoted herein by LABA/LAMA). Evidence suggests that combination of a LABA and tiotropium in individual inhalers offers benefits over the use of either component alone, in terms of lung function and quality of life (Farne 2015). The need for single‐inhaler fixed‐dose combinations arose for several reasons including the underwhelming efficacy of salmeterol and tiotropium administered via separate devices (Aaron 2007) and potential advantages in terms of convenience and adherence (Bangalore 2007). This review has synthesised the evidence for the safety and efficacy of once‐daily LABA/LAMA FDCs versus placebo in patients with COPD.
How the intervention might work
The co‐administration of LABA/LAMA in COPD has beneficial effects on lung function, dyspnoea scores, health‐related quality of life, and possibly in preventing acute exacerbations of COPD (AECOPD) (Calzetta 2016; Wedzicha 2014). Bronchodilation is thought to form the foundation of these benefits, but a reduction in hyperinflation, modulation of mucous production and clearance, and potentially anti‐inflammatory effects are theorised to contribute as well (Beeh 2016). In terms of bronchodilation, use of LABA and LAMA together is more effective compared to either agent alone (Singh 2014a; Van Noord 2005), but the nature of this interaction is not entirely clear, with in vitro and clinical studies suggesting that there is a synergistic rather than additive effect (Cazzola 2015). The mechanism of increased bronchodilation has mainly been attributed to the activation of presynaptic beta2‐receptors, which attenuates the release of junctional acetylcholine (Calzetta 2015). In addition, airway smooth muscle relaxation achieved by a LABA (via increased cyclic adenosine monophosphate) is amplified by the blockade of acetylcholine by inhibition of M3 muscarinic receptors (Cazzola 2010), and there is evidence to suggest that M2 receptors interact with adenyl cyclase as well (Beeh 2016).
Why it is important to do this review
Fixed‐dose combinations (FDCs) of a long‐acting beta2‐agonist (LABA) plus a long‐acting muscarinic antagonist (LAMA) delivered via a single inhaler are approved by regulatory authorities in the USA, Europe, and Japan for the treatment of COPD . The introduction of these inhalers follow guideline‐based recommendations to optimise inhaled bronchodilator use (Quaseem 2011; Vestbo 2013). Recent meta‐analyses have clarified the utility of LABA/LAMA combination inhalers compared to their mono‐components in COPD, particularly with respect to trough FEV1, transitional dyspnoea index (TDI), St. George’s Respiratory Questionnaire (SGRQ) and safety (Calzetta 2016; Calzetta 2017). They found statistically and clinically significant improvements in trough FEV1 for all fixed‐dose combinations (FDC) compared with their mono‐components. Though there were statistically significant improvements in TDI and SGRQ, these fell below previously established minimal clinically important differences (MCIDs), and thus the clinical meaning of this benefit is unclear. Side effects, including cardiac events, were no greater in those taking LABA/LAMA. There were no significant differences between different FDCs for the outcomes examined (Calzetta 2016; Calzetta 2017). Individual clinical trials have demonstrated a reduction in AECOPD with LABA/LAMA versus mono‐components and versus placebo (Bateman 2015; Wedzicha 2017). Unfortunately, the benefits of LABA/LAMA on AECOPD were not included in the meta‐analyses, and thus remain to be clarified. Evaluation of the efficacy and safety of once‐daily LABA/LAMA fixed‐dose combinations versus placebo will facilitate the comparison of different FDCs in future network meta‐analyses.
Objectives
To assess the effects of single‐inhaler LABA/LAMA combinations versus placebo on clinically meaningful outcomes in patients with stable COPD.
Methods
Criteria for considering studies for this review
Types of studies
We included parallel‐group and cross‐over randomised controlled trials (RCTs). We included studies reported as full‐text, those published as abstract only, and unpublished data. We excluded very short‐term trials (i.e. ≤ three weeks in duration).
Types of participants
We included adults (≥ 40 years old) with a diagnosis of stable COPD. We recorded study authors' definition of stable COPD. We did not exclude participants with comorbidities.
Types of interventions
We included trials comparing once‐daily LABA/LAMA in a single inhaler (i.e. fixed dose combination) versus placebo.
We included studies that allowed participants to continue using their ICS during the trial as long as the ICS was not part of the randomised treatment; if ICS was administered in combination with LABA prior to the trial, participants should be transitioned to the equivalent ICS monotherapy prior to study start. The effect of continued ICS use was planned to be examined by subgroup analysis (see Subgroup analysis and investigation of heterogeneity).
Types of outcome measures
Primary outcomes
All‐cause mortality.
Serious Adverse Events (SAE) of any cause.
Acute Exacerbations of COPD (AECOPD).
-
Respiratory Health‐related Quality of Life (HRQoL), as measured by the
St. George’s Respiratory Questionnaire (SGRQ).
Chronic Respiratory Questionnaire (CRQ).
Comments about primary outcomes
Serious adverse events
SAEs can include death, life‐threatening adverse reaction, hospitalisation or increased length of hospital stay, disability, and birth defects. We recorded each study's definition of an SAE if it varied from our definition.
Respiratory health‐related quality of life
CRQ and SGRQ are widely‐used, reliable and valid measures of patient‐reported health status in COPD (Guyatt 1987; Jones 1992). SGRQ scores three domains of health status (symptoms, patient activity, and disease impact), and reports scores ranging from zero (best) to 100 (worst). The Minimally Clinical Important Difference (MCID) is approximately four (Schunemann 2003). That is, a clinically meaningful change in health status is equal to a change of about four points on SGRQ. CRQ scores four domains (shortness of breath, fatigue, emotional function, and mastery), reports scores ranging from one (worst) to seven (best), and has an MCID of 0.5 (Schunemann 2005). While CRQ and SGRQ provide very similar information and are highly correlated, SGRQ is less responsive; it was shown to underestimate treatment effects when compared to CRQ in identical populations (Puhan 2006). Thus, pooling SGRQ data with CRQ data may spuriously suggest heterogeneity of treatment effect. Therefore, SGRQ and CRQ were considered as separate outcomes; this approach agrees with the recommendations of Puhan 2006, who suggest that mean differences for SGRQ and CRQ should be reported separately.
Acute exacerbations of COPD
We included AECOPD as a main outcome because exacerbations are consistently linked to mortality, morbidity, and costly hospitalisations. Since a consensus definition and standard reporting criteria do not exist for AECOPD (Cazzola 2008), we performed a meta‐analysis of AECOPD data only when study authors used one of the following definitions: increase in symptoms precipitating the use of antibiotics; increase in symptoms precipitating the use of systemic steroids; increase in symptoms precipitating emergency room visit; or hospitalisation. The MCID for AECOPD outcomes is not established: Calverley 2005 estimated an MCID of 20% to 25% using a crude anchor‐based approach, while Chapman 2013 used an expert consensus process to estimate an MCID of 11%.
Secondary outcomes
Trough (pre‐dose) Forced Expiratory Volume in One Second (FEV1).
Peak (post‐dose) FEV1.
Six‐minute walking test (6MWT).
Adverse effects.
Comments about secondary outcomes
Forced expiratory volume
FEV1 is the volume of air forcibly exhaled one second after maximum inhalation. FEV1 is often used for staging COPD (GOLD 2017): FEV1 is 20% lower than normal for patients with mild COPD and 70% lower than normal for patients with very severe COPD. FEV1 is also used to assess treatment effect. However, the MCID for FEV1 has not been quantitatively established (expert opinion proposes an MCID of 100 mL to 140 mL) (Cazzola 2008). Moreover, FEV1 is an intermediate endpoint, representing airflow as a surrogate for clinically important outcomes. Surrogate outcomes are not patient‐centred. Nevertheless, we included trough FEV1 because one meta‐analysis points to a modest correlation between increased trough FEV1 and improved SGRQ (Westwood 2011). For the purpose of this review we will consider the MCID for FEV1 to be 100 mL (Donohue 2005).
Six‐minute walking test
In the ECLIPSE study (a non‐interventional cohort study of treated COPD patients), one‐year change in 6MWT predicted death in the subsequent 12 months. The mean between‐group change between survivors and non‐survivors was 30 metres (95% CI 26 to 34). Using these results, Polkey 2013 proposed an MCID of about 30 metres.
Adverse effects
We analysed all‐cause adverse effects and serious adverse events reported in studies of LABA or LAMA.
Search methods for identification of studies
Electronic searches
We searched the Cochrane Airways Trials Register on 3 December 2018. The Cochrane Airways Trials Register is maintained by the Information Specialist for the Group and contains studies identified from several sources:
Monthly searches of the Cochrane Central Register of Controlled Trials (CENTRAL), through the Cochrane Register of Studies ‐ CRS Web;
Weekly searches of MEDLINE Ovid SP;
Weekly searches of Embase Ovid SP;
Monthly searches of PsycINFO Ovid SP;
Monthly searches of CINAHL EBSCO (Cumulative Index to Nursing and Allied Health Literature);
Monthly searches of AMED EBSCO (Allied and Complementary Medicine);
Handsearches of the proceedings of major respiratory conferences.
Studies contained in the Trials Register were identified through search strategies based on the scope of Cochrane Airways. Details of these strategies, as well as a list of handsearched conference proceedings, are in Appendix 1. See Appendix 2 for search terms used to identify studies for this review.
We searched the following trials registries on 3 December 2018:
US National Institutes of Health Ongoing Trials Register ClinicalTrials.gov (www.clinicaltrials.gov);
World Health Organization International Clinical Trials Registry Platform (apps.who.int/trialsearch).
We searched the Cochrane Airways Trials Register and additional sources from inception, with no restriction on language of publication.
Searching other resources
We checked reference lists of all primary studies and review articles for additional references. We searched relevant manufacturers' web sites for trial information.
We searched for errata or retractions from included studies published in full‐text on PubMed (www.ncbi.nlm.nih.gov/pubmed) and reported the date this was done within the review.
Data collection and analysis
Selection of studies
Two review authors (DE, UM, RW, or TH) independently screened each title and abstract for inclusion of all the potential studies we identified as a result of the search and coded them as 'retrieve' (eligible or potentially eligible/unclear) or 'do not retrieve'. We retrieved the full‐text study reports/publications that appeared eligible and two review authors (DE, UM, TH, or RW) independently screened each full‐text paper and identified studies for inclusion, or identified and recorded reasons for exclusion of the ineligible studies. We resolved any disagreement through discussion or, if required, we consulted a third person (DE, UM, RW, or TH). We identified and excluded duplicates and collated multiple reports of the same study so that each study rather than each report was the unit of interest in the review. We recorded the selection process in sufficient detail to complete a PRISMA flow diagram and Characteristics of excluded studies table.
Data extraction and management
We used a data collection form for study characteristics and outcome data which had been piloted on at least one study in the review. Two review authors (DE, UM, RW, or TH) extracted study characteristics from each included study. We extracted the following study characteristics.
Methods: study design, total duration of study, details of any 'run‐in' period, number of study centres and location, study setting, withdrawals, and date of study.
Participants: N, mean age, age range, gender, severity of condition, diagnostic criteria, baseline lung function, smoking history, inclusion criteria, and exclusion criteria.
Interventions: intervention, comparison, concomitant medications, and excluded medications.
Outcomes: primary and secondary outcomes specified and collected, and time points reported.
Notes: funding for trial, and notable conflicts of interest of trial authors.
Two review authors (DE, UM, RW, or TH) independently extracted outcome data from each included study. We noted in the Characteristics of included studies table if outcome data were not reported in a usable way. We resolved disagreements by consensus or by involving a third person (DE, UM, RW, or TH). One review author (DE) transferred data into the Review Manager file. We double‐checked that data have been entered correctly by comparing the data presented in the systematic review with the study reports. A second review author (RW) spot‐checked study characteristics for accuracy against the trial report.
Trials may report continuous outcomes as change scores (i.e. change from baseline) or final values. As per the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), we presented mean differences in change scores in one subgroup, mean differences in final values in another, and pooled both subgroups for an overall analysis.
Where multiple time points were reported for outcomes, we chose the time point that maximised length of follow‐up for the randomised treatment period.
Assessment of risk of bias in included studies
Two review authors (DE, KP, or FE) independently assessed the risk of bias for each study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We resolved any disagreements by discussion or by involving a third author (DE, KP, or FE). We assessed the risk of bias according to the following domains.
Random sequence generation.
Allocation concealment.
Blinding of participants and personnel.
Blinding of outcome assessment.
Incomplete outcome data.
Selective outcome reporting.
Other bias.
We graded each potential source of bias as high, low, or unclear and provided a quote from the study report together with a justification for our judgement in the 'Risk of bias' table. We summarised the risk of bias judgements across different studies for each of the domains listed. We considered blinding separately for different key outcomes, where necessary (e.g. for unblinded outcome assessment, risk of bias for all‐cause mortality may be very different than for a patient‐reported pain scale). Where information on risk of bias related to unpublished data or correspondence with a trialist, we noted this in the 'Risk of bias' table.
When considering treatment effects, we took into account the risk of bias for the studies that contributed to that outcome.
Assessment of bias in conducting the systematic review
We conducted the review according to this published protocol and reported any deviations from it in the Differences between protocol and review section of the systematic review.
Measures of treatment effect
We analysed dichotomous data as odds ratios and continuous data as mean differences or standardised mean differences. We entered data presented as a scale with a consistent direction of effect.
We performed meta‐analyses only where this was meaningful, i.e. if the treatments, participants, and the underlying clinical question were similar enough for pooling to make sense.
We narratively described skewed data reported as medians and interquartile ranges.
Where multiple trial arms were reported in a single trial, we included only the relevant arms. If two comparisons (e.g. drug A versus placebo and drug B versus placebo) were combined in the same meta‐analysis, we halved the control group to avoid double‐counting.
Unit of analysis issues
We analysed dichotomous data using participants as the unit of analysis (rather than events) to avoid counting the same participant more than once. Paired data from each participant in cross‐over trials were analysed using the Generic Inverse Variance method.
Dealing with missing data
We contacted investigators or study sponsors in order to obtain missing numerical outcome data where possible (e.g. when a study was identified as abstract only). That is, if study authors did not report true intention‐to‐treat (ITT) data, we attempted an available case analysis by including data for all participants for whom outcome data were collected (whether the participants completed or did not complete the trial). Please note that a case analysis is not a true ITT analysis, nor a per‐protocol analysis.
If we could not obtain missing data from study authors, we planned to:
compare our available case analysis with an imputed, true ITT analysis (see Sensitivity Analyses);
use an average standard deviation (SD) borrowed from other studies included in our meta‐analysis if the SD for a mean difference was unavailable (or incalculable);
use final values instead of the change‐from‐baseline values if the standard deviation for a change score was missing.
If the missing data were thought to introduce serious bias, we planned to explore the impact of including such studies in the overall assessment of results by a sensitivity analysis.
Assessment of heterogeneity
We used the I² statistic to measure heterogeneity among the trials in each analysis. If we identified substantial heterogeneity (i.e. I2 greater than 30%) we reported it and explored possible causes by prespecified subgroup analysis.
Assessment of reporting biases
If we were able to pool more than 10 trials, we planned to create and examine a funnel plot to explore possible small study and publication biases.
Data synthesis
We used a fixed‐effect model and performed a sensitivity analysis with a random‐effects model. Where study authors reported exacerbation rate, we meta‐analysed rate data when study authors accounted for duration of follow‐up and inter‐patient variability (Aaron 2008). The odds ratio was our primary summary statistic. Where possible, we also reported AECOPD as the percentage of participants experiencing at least one exacerbation. This way, AECOPD could be presented as a dichotomous outcome, and a patient‐based number needed to treat for an additional beneficial outcome (NNTB) could be reported. When possible, we also reported SGRQ and CRQ as dichotomous outcomes (i.e. participants who reached the MCID versus participants who did not).
Summary of findings table
We created a 'Summary of findings' table using the seven primary and secondary outcomes identified above; for health‐related quality of life, SGRQ was reported in the 'summary of findings' table. We used the five GRADE considerations (study limitations, consistency of effect, imprecision, indirectness, and publication bias) to assess the certainty of a body of evidence as it related to the studies which contributed data to the meta‐analyses for the prespecified outcomes. We used the methods and recommendations described in Section 8.5 and Chapter 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) using GRADEpro software. We justified all decisions to downgrade or upgrade the certainty of the evidence using footnotes and we made comments to aid reader's understanding of the review, where necessary.
Subgroup analysis and investigation of heterogeneity
We planned to carry out the following subgroup analyses:
participants with ICS use during the trial versus participants without ICS use during the trial;
different LABA/LAMA combinations (IND/GLY; UMEC/VI; TIO/OLO; ACM/FOR);
length of follow‐up (less than six months versus six months or longer);
baseline COPD severity (mild or moderate disease versus severe disease, according to GOLD criteria).
We used our primary outcomes in subgroup analyses.
We used the formal test for subgroup interactions in Review Manager.
Sensitivity analysis
We planned to carry out the following sensitivity analyses:
a comparison of available case analysis to true ITT analyses, where the ITT analyses were imputed with best‐case and worse‐case outcome data;
a comparison of results from fixed‐effect models with results from random‐effects models;
a comparison based on our 'risk of bias' assessments (i.e. exclusion of studies with a high risk of bias).
Results
Description of studies
The Characteristics of included studies tables presents details of the included studies; a summary table is also provided (Table 2). In the Characteristics of excluded studies table, we reported reasons for the exclusion of studies considered during review of full‐text articles.
1. Characteristics of included studies.
| Study | RCT Design | N rand. | Country/region | Interventiona (dose in μg) | Participant baseline characteristics | ||||
| Age, yearsb | Male (%)b | % pred. FEV1c | Smoking status | ICS use (%)b,d | |||||
| Bateman 2013 | Parallel | 709 | Worldwide | IND/GLY 110/50 | 64.0‐64.4 | 73‐76 | 55 | 40.1‐40.5 | 56.5‐57.8 |
| Beeh 2014 | Cross‐over | 85 | NS | IND/GLY 110/50 | 62.1 | 63 | 47 | 45 | 31 |
| Beeh 2015 | Cross‐over | 219 | Europe, N. America | TIO/OLO 2.5/5; 5/5 | 61.1 | 59 | 54 | 62.6 | 41.1 |
| Celli 2014 | Parallel | 678 | NS | UMEC/VI 125/25 | 62.2‐63.4 | 64‐66 | 48 | 50‐52 | 44‐50 |
| Dahl 2013 | Parallel | 339 | Europe, Canada, Asia and South Africa | IND/GLY 110/50 | 62.5‐62.9 | 76‐77 | 57.5 | 45.1‐45.3 | 38.9‐45.8 |
| Donohue 2013 | Parallel | 693 | Worldwide | UMEC/VI 62.5/25 | 62.2‐63.1 | 70‐74 | 47.5 | 49‐54 | 49‐51 |
| Feldman 2012 | Parallel | 51 | USA | UMEC/VI 500/25 | 58.7‐59.2 | 57‐78 | 50 | 57‐78 | NR |
| Larbig 2015 | Parallel | 811 | Worldwide | IND/GLY 110/50 | 64.6‐64.9 | 71‐77 | NR | NR | NR |
| Mahler 2014 | Cross‐over | 247 | Europe, N. America | IND/GLY 110/50 | 62.8 | 70 | 56 | 45.5 | 54.9 |
| Maltais 2014 | Parallel | 404 | Europe, N. America, S. America | TIO/OLO 2.5/5; 5/5 | 60.8‐63.1 | 65‐68 | 59 | NR | NR |
| Maltais 2014b | Cross‐over | 349 | Europe, N. America, S. Africa | UMEC/VI 62.5/25; 125/25 | 61.6 | 56 | 51 | 63.2 | 28.2 |
| Maltais 2014c | Cross‐over | 308 | Europe, N. America, Russia | UMEC/VI 62.5/25; 125/25 | 62.6 | 55 | 51 | 60.6 | 39.4 |
| NCT00626522 | Parallel | 425 | Europe, Australia, Russia | ACD/FOR 200/6; 200/12; 200/18 | 60.7‐63.9 | 75‐82 | NR | NR | NR |
| NCT02275052 | Cross‐over | 99 | USA | UMEC/VI 62.5/25 | 60.7 | 53 | NR | NR | NR |
| O'Donnell 2015a | Cross‐over | 586 | N. America, S. America, Oceania, Europe | TIO/OLO 2.5/5; 5/5 | 61.7 | 71 | 58 | 39.1 | NR |
| O'Donnell 2015b | Cross‐over | N, America, S. America, Russia, Europe | TIO/OLO 2.5/5; 5/5 | 61.7 | 71 | 58 | 39.1 | NR | |
| Siler 2016 | Parallel | 498 | Europe, USA, Russia | UMEC/VI 62.5/25 | 62.6‐64.1 | 58‐60 | 47.5 | 52‐55 | 45‐50 |
| Singh 2016a | Parallel | 608 | Europe, N. America, S. Africa | TIO/OLO 2.5/5; 5/5 | 64.7‐65.1 | 56‐62 | 55.5 | 43.1‐54.7 | 34.8‐41.9 |
| Singh 2016b | Parallel | 606 | Europe N. America, S. Africa, Oceania | TIO/OLO 2.5/5; 5/5 | 64.0‐65.2 | 58‐66 | 54.5 | 44.6‐47.0 | 35.1‐41.1 |
| Troosters 2016 | Parallel | 152 | Europe, Canada, Oceania | TIO/OLO 5/5 | 64.4‐65.0 | 63‐69 | NR | NR | NR |
| Watz 2016 | Cross‐over | 194 | Germany | IND/GLY 110/50 | 62.8 | 66 | 62 | 56.7 | NR |
| Zheng 2014 | Parallel | 580 | China, South‐east Asia | UMEC/VI 62.5/25; 125/5 | 63.7‐64.3 | 92‐94 | NR | 25‐34 | 51‐54 |
aPlacebo was the comparator in all studies. bRange across treatment arms reported where overall data were not reported. c % predicted FEV1 post‐bronchodilator. dConcomitant ICS use was permitted in all studies (not stated for Celli 2014, Larbig 2015; NCT00626522; Troosters 2016).
Abbreviations: ACD: aclidinium; FOR: formoterol; GLY: glycopyrronium; IND: indacaterol; NR: not reported; OLO: olodaterol; rand: randomised; RCT: randomised controlled trial; TIO: tiotropium; UMEC: umeclidinium; VI: vilanterol.
Results of the search
We identified 1114 records by performing electronic searches of bibliographic databases . Of a total of 1059 records (55 duplicates removed), we excluded 765 upon screening titles and abstracts. We examined full‐text articles of the remaining 294 records and excluded 220 records (reporting 96 studies; see Excluded studies). The remaining 74 records reported the findings of 23 studies, which we included in this review (studies included in quantitative analyses, n = 22; studies awaiting classification, n = 1). Figure 1 depicts the flow of information through the different stages of this systematic review.
1.
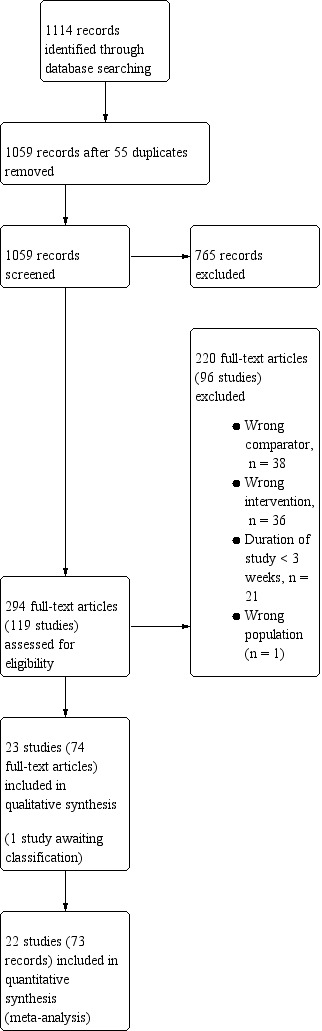
Study flow diagram.
Included studies
Of the 23 studies that met the inclusion criteria, there were twenty‐two included studies (Bateman 2013; Beeh 2014; Beeh 2015; Celli 2014; Dahl 2013; Donohue 2013; Feldman 2012; Larbig 2015; Mahler 2014; Maltais 2014b; Maltais 2014c; Maltais 2014; NCT00626522; NCT02275052; O'Donnell 2015a; O'Donnell 2015b; Siler 2016; Singh 2016a; Singh 2016b; Troosters 2016; Watz 2016; Zheng 2014) and one study awaiting classification (NCT02233543 2014). A majority of included studies were reported as full peer‐reviewed articles, with the exception of those reported as abstract only (Larbig 2015) or trial registry only (NCT00626522 and NCT02275052).
Methods
Of the included studies, 13 had a parallel‐group design and nine had a cross‐over design; all 22 studies were described as double‐blind (blinding of participants and staff occurred in 15 studies and was unclear in seven studies; blinding of outcome assessors occurred in six studies and was unclear in 16 studies). Studies had a randomly assigned treatment period ranging from three weeks to 52 weeks (mean 11 weeks; median 12 weeks; mode 12 weeks); a minority of studies had a duration of six months or longer (6 months, n = 4; 12 months, n = 2). All studies were multicentre studies; 19 of 22 studies were international, with the exception of trials performed solely in Germany (Watz 2016) or the USA (Feldman 2012; NCT02275052). Overall, there was good geographical coverage; the majority of studies (16/22) enrolled participants from both Europe and North America and studies also enrolled a proportion of participants from China and Asia (Bateman 2013; Dahl 2013; Donohue 2013; Larbig 2015; Siler 2016), Oceania (O'Donnell 2015a; O'Donnell 2015b; Singh 2016a; Singh 2016b; Troosters 2016), Russia (Maltais 2014c; NCT00626522; O'Donnell 2015a; O'Donnell 2015b; Siler 2016) and South Africa (Dahl 2013; Maltais 2014b; Singh 2016a; Singh 2016b). Study setting was poorly reported, but appeared to represent a mix of academic/clinical research centres and primary or secondary care units.
Participants
The twenty‐two included studies randomised a total of 8641 participants (Table 2). Baseline characteristics were generally consistent across studies. Inclusion criteria for the majority of studies (n = 21/22) specified either GOLD stage II/III, or criteria aligned with this disease severity (i.e. post‐bronchodilator FEV1 < 70% or 80%; post‐bronchodilator FVC/FEV1 < 70%; MRC dyspnoea score ≥ 2); Beeh 2015 permitted inclusion of participants with GOLD stage II to IV. The mean ages of participants across the relevant arms of all included studies ranged from 59 to 65 years; the proportion of current smokers generally ranged from 40% to 55% (n = 20; two outliers: 25% (Zheng 2014) and 78% (Feldman 2012)). In each trial, a majority of participants were male (range across studies 53% to 82%; one outlier, 92% to 94% (Zheng 2014)). Where reported, post‐bronchodilator percent predicted FEV1 ranged from 47% to 62% (median ˜58%); Zheng 2014 did not report % predicted FEV1, but pre‐bronchodilator FEV1 was 1.2 L to 1.3 L; Larbig 2015, NCT00626522, NCT02275052 and Troosters 2016 did not report baseline lung‐function (abstract or trial registry only). Concomitant inhaled corticosteroid (ICS) use was permitted in all of the included studies (where stated); across the included studies, between 28% to 58% of participants were using ICS at baseline.
Intervention
Of the 8641 randomised participants across the 22 studies, and accounting for the enrolment in multiple arms of cross‐over studies, a total of 6252 participants were randomised to receive once‐daily LABA/LAMA via a combined inhaler, and 3819 participants were randomised to receive placebo. In the subgroup of parallel‐group trials, 4124 participants were randomised to receive once‐daily LABA/LAMA via a combined inhaler and 2520 participants were randomised to receive placebo. Across the 22 studies, six studies evaluated the once‐daily combination of IND/GLY (110/50 μg), seven studies evaluated TIO/OLO (2.5/5 or 5/5 μg), eight studies evaluated UMEC/VI (62.5/5, 125/25 or 500/25 μg) and one study evaluated ACD/FOR (200/6, 200/12 or 200/18 μg); all LABA/LAMA combinations were compared with placebo. Where reported, concomitant treatment with ICS was permitted by all studies with various restrictions relating to prior use and stable dose for a prespecified time prior to study initiation; whether concomitant ICS was permitted was not reported for one study (Troosters 2016).
Outcomes
With the exception of the 6MWT (secondary outcome), all of the prespecified outcomes were reported by at least three of the included studies. All‐cause mortality was reported by 18 studies, SAEs, by all 22 studies, AECOPD by three studies, difference versus placebo in adjusted trough FEV1 by 13 studies, difference versus placebo in adjusted peak FEV1 by seven studies, difference versus placebo in adjusted SGRQ score by eight studies, and all‐cause AEs by 18 studies (Table 1). 6MWT was not reported by any of the included studies.
Excluded studies
Ninety‐six studies were excluded, primarily because either the intervention did not meet the inclusion criteria (i.e. the LAMA and LABA were not administered once‐daily in a fixed dose combination, or the combination was administered twice daily; n = 36 studies) or because the study did not include a placebo arm (n = 38). It was often difficult to ascertain from the abstract whether the LAMA and LABA were administered as a fixed‐dose combination and from the clinical trial record headers it was not always possible to identify whether a placebo group was included; this resulted in a high rate of exclusions at full‐text review stage. Other reasons for exclusion at this stage included 'duration < 3 weeks' (n = 21), 'wrong participant population' (healthy volunteers; n = 1).
Risk of bias in included studies
Please refer to the Characteristics of included studies tables for details on risk of bias and for supporting evidence for each study. Figure 2 provides a summary of 'risk of bias' judgements, presented by study and domain (sequence generation, allocation concealment, blinding, incomplete data, selective reporting and 'other'). Figure 3 depicts the risk of bias for each domain, presented as percentages across all included studies. Across 198 assessments (22 studies, nine risk of bias domains), 146 were considered to be at a low risk of bias, seven at a high risk of bias and 45 to have an unclear risk of bias.
2.
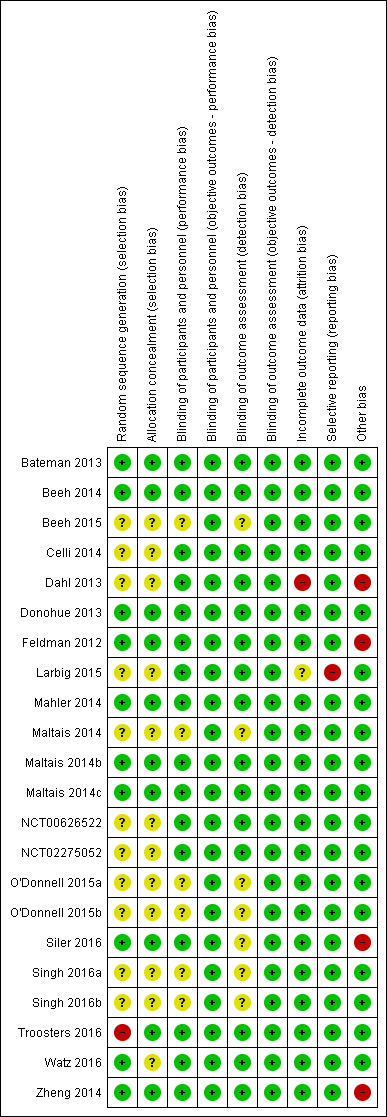
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
3.
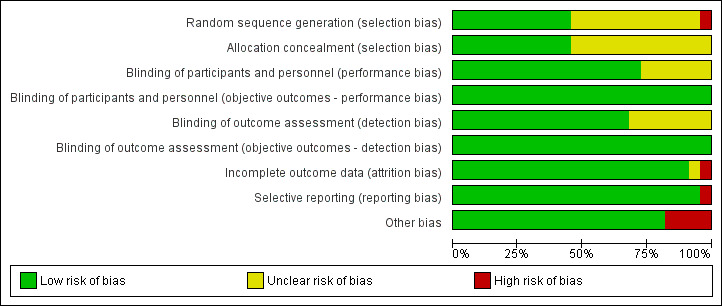
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
More than half of the included studies provided insufficient information regarding methods of random sequence generation (11 of 22 studies) and concealment of treatment allocation (12 of 22 studies) to allow a judgement on risk of bias; the risk of bias for these studies was rated as unclear. Ten studies employed adequate methods of random sequence generation and were considered to be at low risk of bias. (Bateman 2013; Beeh 2014; Donohue 2013; Feldman 2012; Mahler 2014; Maltais 2014b; Maltais 2014c; Siler 2016; Watz 2016; Zheng 2014) or adequate methods of allocation concealment (Bateman 2013; Beeh 2014; Donohue 2013; Feldman 2012; Mahler 2014; Maltais 2014b; Maltais 2014c; Siler 2016; Troosters 2016; Zheng 2014). Inadequate methods of random sequence generation (pseudo‐random number generator and block randomisation) were employed in one study (Troosters 2016), which was considered to be at high risk of bias.
Blinding
We considered the risk of performance and detection bias separately for objective and subjective outcomes. For objective outcomes (all‐cause mortality, SAEs, AECOPD, lung function and AEs) we considered that a lack of blinding would not result in a risk of detection or performance bias; therefore all studies were considered to be at low risk of bias with respect to these outcomes. The only subjective outcome relevant to this review was HRQoL based on assessment by SGRQ; sixteen studies were considered to be at a low risk of performance bias (Bateman 2013; Beeh 2014; Celli 2014; Dahl 2013; Donohue 2013; Feldman 2012; Larbig 2015; Mahler 2014; Maltais 2014b; Maltais 2014c; NCT00626522; NCT02275052; Siler 2016; Troosters 2016; Watz 2016; Zheng 2014); and the risk of performance bias was unclear for the remaining six studies (Beeh 2015; Maltais 2014; O'Donnell 2015a; O'Donnell 2015b; Singh 2016a; Singh 2016b). For HRQoL, the risk of detection bias was considered low for fifteen studies (Bateman 2013; Beeh 2014; Celli 2014; Dahl 2013; Donohue 2013; Feldman 2012; Larbig 2015; Maltais 2014b; Maltais 2014c; Mahler 2014; NCT00626522; NCT02275052; Watz 2016; Troosters 2016; Zheng 2014) and unclear in seven studies (Beeh 2015; Maltais 2014; O'Donnell 2015a; O'Donnell 2015b; Siler 2016; Singh 2016a; Singh 2016b).
Incomplete outcome data
We considered 20 of 22 studies to be at low risk of attrition bias on the basis of low and balanced rates of participant withdrawal, which were adequately documented in the trial reports. One study (Dahl 2013) was considered to be at high risk for attrition bias based on a greater than 20% rate of attrition in the placebo arm versus < 15% in the IND/GLY arm; insufficient information was reported by one study (Larbig 2015), resulting in a rating of unclear risk of attrition bias.
Selective reporting
We considered 21 of 22 studies to be at low risk of reporting bias. One study (Larbig 2015) was considered to be at high risk for reporting bias as the abstract (abstract only) did not report key prespecified outcomes (as reported on the trial registry site).
Other potential sources of bias
We considered there to be potential sources of bias present in four of the studies. In Dahl 2013, more participants in the QVA149 group had severe COPD versus those in the placebo group; however, this would likely skew treatment effect in favour of placebo. An imbalance in baseline characteristics in Feldman 2012 suggested that randomisation was not robust, although the limited sample size of the placebo group could also account for imbalance in baseline characteristics. In Siler 2016, a greater proportion of participants with GOLD category D were enrolled in the active treatment group, possibly favouring placebo and underestimation of the treatment effect. In Zheng 2014, a higher proportion of participants with GOLD Stage IV were enrolled in the UMEC/VI 62.5/25 µg group compared with placebo and could potentially skew the treatment effect in favour of placebo. These four studies were considered to be at high risk of 'other' bias; however, we noted that in three cases, the issue would tend to skew the results in favour or placebo, resulting in a potential underestimation of the treatment effect.
Effects of interventions
See: Table 1
Structure of the meta‐analysis
As per the protocol, we elected to perform a meta‐analysis only when interventions and outcomes were sufficiently similar for pooling of the data. We subgrouped the data in the forest plots according to the type and dose of LABA/LAMA combination. However, some comparisons (stated below) should be interpreted with caution because of the relatively small number of trials for each subgrouping, heterogeneity in study design (i.e. length, inclusion and exclusion criteria), and the low number of events for all‐cause mortality and SAEs.
Structure of the narrative synthesis
In the following sections, we present a narrative summary of study results according to the prespecified outcomes. We present primary outcomes (all‐cause mortality, SAEs, AECOPD, respiratory HRQoL) followed by secondary outcomes (trough FEV1, peak FEV1, 6MWT, AEs). For each outcome, we describe the overall effect of the intervention irrespective of LABA/LAMA type or dose, followed by the effect of the intervention in subgroups according to LABA/LAMA type and dose.
Primary outcomes
All‐cause mortality
Eighteen studies (8752 participants) reported all‐cause mortality, although the number of reported deaths was low. There was no significant difference in the number of deaths reported in participants receiving a once‐daily LABA/LAMA fixed‐dose combination compared with those receiving placebo (OR 1.88, 95% CI 0.81 to 4.36; I2 = 0%; Analysis 1.1). The overall certainty of the evidence for this outcome was rated as low, having been downgraded once for indirectness (duration of studies varied widely from six weeks to 52 weeks) and once for imprecision (wide confidence intervals due to a low number of events).
1.1. Analysis.
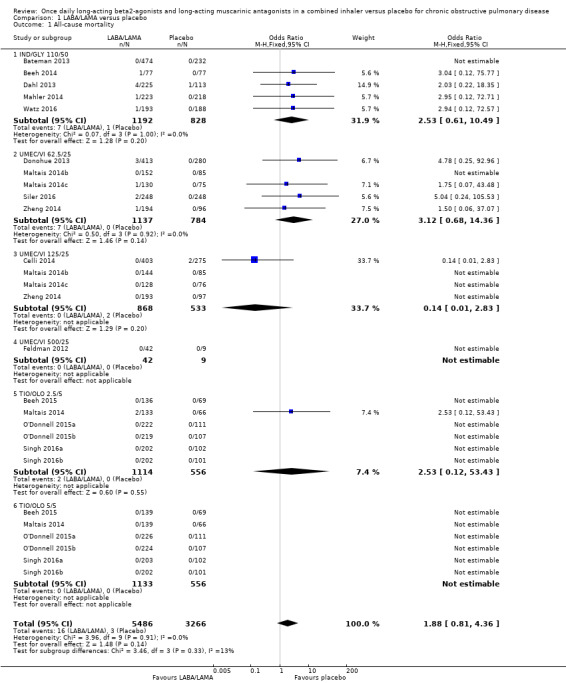
Comparison 1 LABA/LAMA versus placebo, Outcome 1 All‐cause mortality.
The results were generally consistent (i.e. overlapping CIs) across subgroups for different LABA/LAMA combinations and doses, with ORs ranging from 1.88 with UMEC/VI 500/25 μg to 3.12 with UMEC/VI 62.5/25 μg (Figure 4); the only exception was the UMEC/VI 125/25 μg subgroup with two deaths reported in the placebo arm of one of four studies and no other deaths reported in the remaining three studies, resulting in an OR of 0.14 (95% CI 0.01 to 2.83).
4.
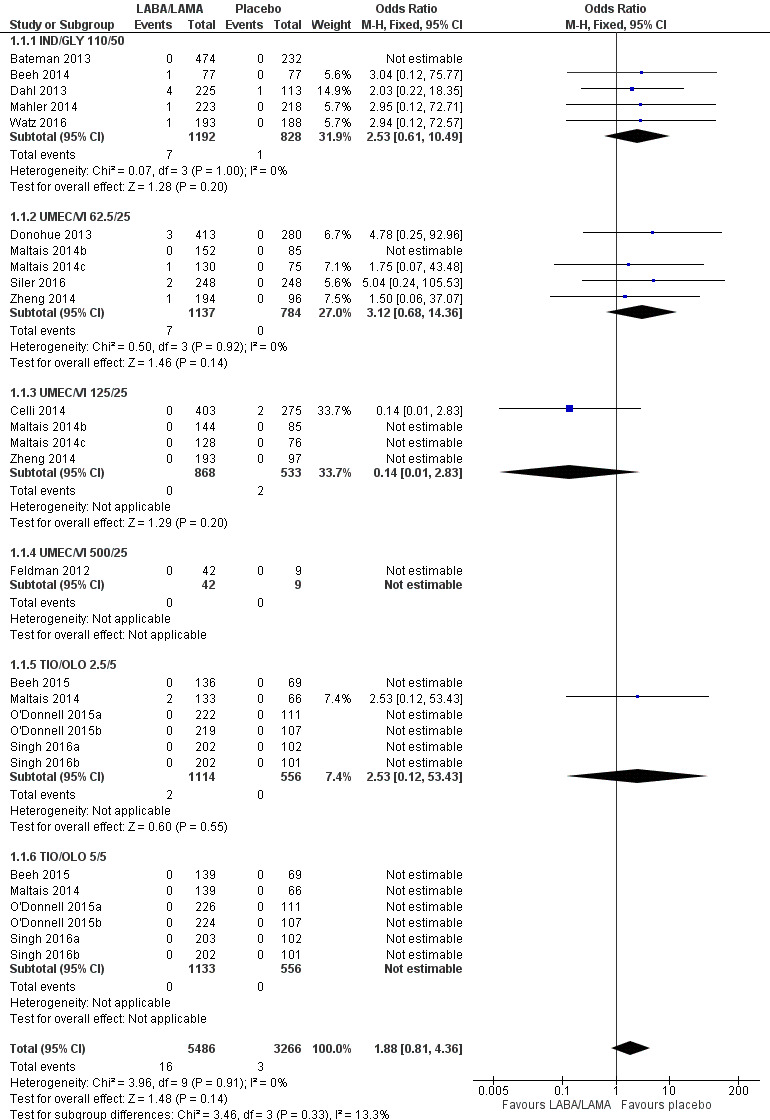
Forest plot of comparison: 1 LABA/LAMA versus placebo, outcome: 1.1 All‐cause mortality.
Serious adverse events
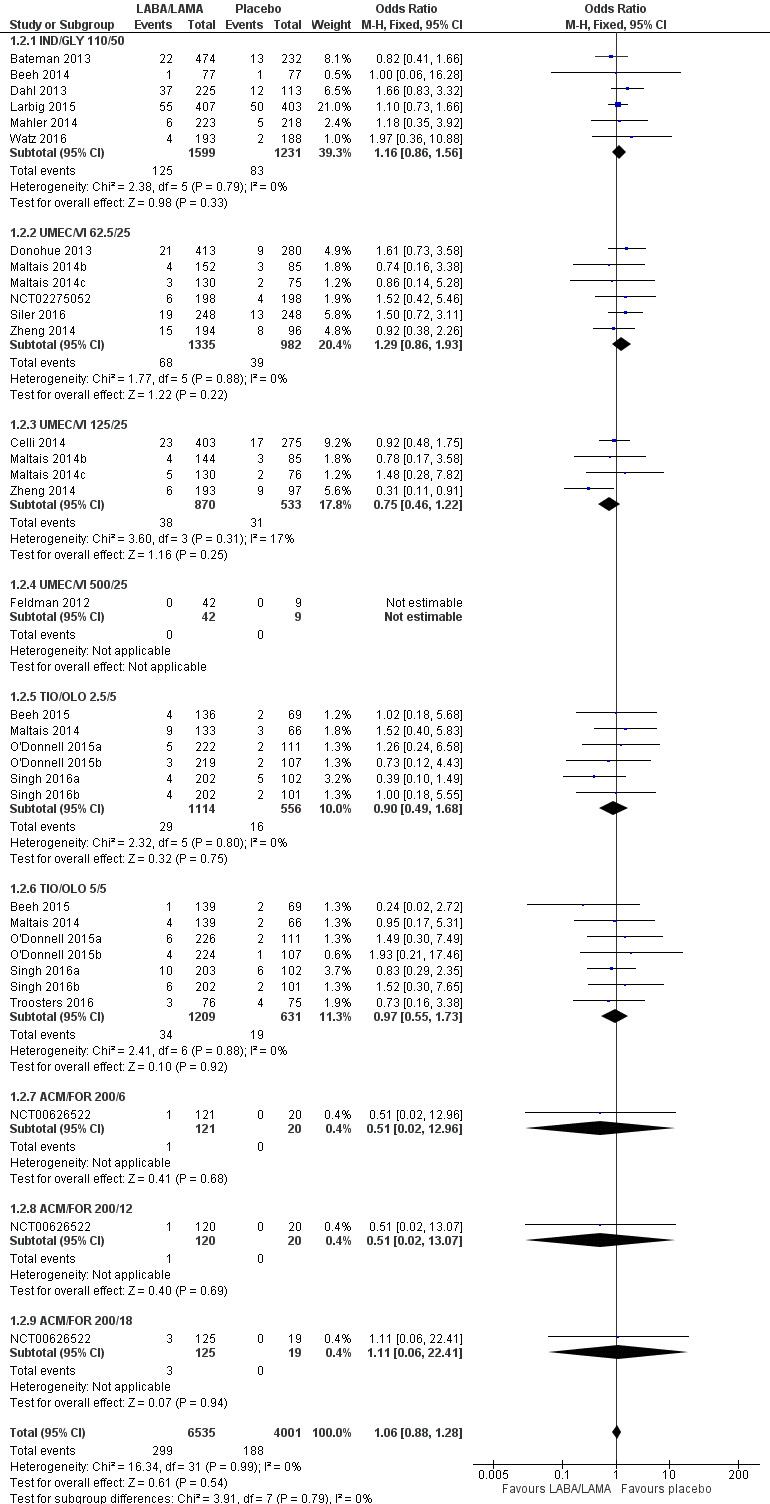
Twenty‐two studies (10,536 participants) reported the number of participants experiencing serious, but non‐fatal adverse events during the study period, for which there was no statistically significant difference (OR 1.06, 95% CI 0.88 to 1.28; I2 = 0%; Analysis 1.2). Compared with taking placebo, we estimated that taking once‐daily LABA/LAMA in a combined inhaler would result in three more people per 1000 experiencing a SAE, but the confidence intervals ranged from five fewer to nine more people per 1000. The overall certainty of the evidence for this outcome was rated as high.
1.2. Analysis.
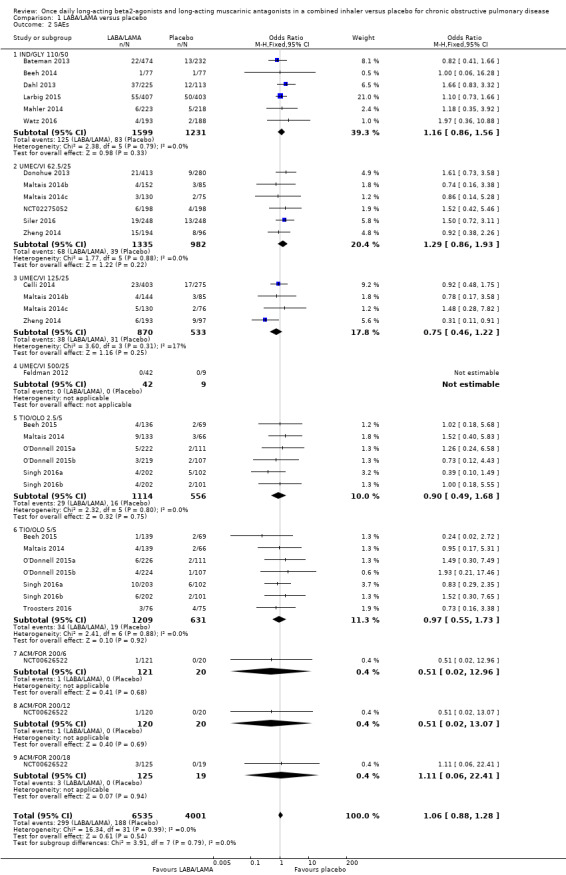
Comparison 1 LABA/LAMA versus placebo, Outcome 2 SAEs.
The results were generally consistent across subgroups for different LABA/LAMA combinations and doses, with ORs ranging from 0.75 with UMEC/VI 125/25 μg to 1.29 with UMEC/VI 62.5/25 μg (Figure 5). The only exceptions were the ACM/FOR 200/6 μg and 200/12 μg subgroups, where the ORs were 0.51 (95% CI 0.02 to 12.96) and 0.51 (95% CI 0.02 to 13.07), respectively; however, these results should be interpreted cautiously as they were based on a small sample size from a single study, resulting in wide confidence intervals.
5.
Forest plot of comparison: 1 LABA/LAMA versus placebo, outcome: 1.2 SAEs.
AECOPD
Three studies (1127 participants) reported the number of participants experiencing an AECOPD; all three studies examined UMEC/VI versus placebo. Significantly fewer people receiving once‐daily LABA/LAMA in a combined inhaler experienced an AECOPD compared with those receiving placebo (OR 0.53, 95% CI 0.36 to 0.78; I2 = 0%; Analysis 1.3; Figure 6). Compared with taking placebo, we estimated that taking once‐daily LABA/LAMA in a combined inhaler would result in 59 fewer people per 1000 experiencing an AECOPD, with the confidence intervals ranging from 27 to 83 fewer people per 1000. The overall certainty of the evidence for this outcome was rated as moderate, having been downgraded once for indirectness (all studies related to UMEC/VI). The results were consistent for two of three UMEC/VI doses examined, with ORs of 0.58 (95% CI 0.37 to 0.93) and 0.37 (0.17 to 0.78) for the 62.5/25 μg and 125/25 μg groups, respectively; the OR for the 500/25 μg dose was 1.68 (0.08 to 35.43) but was based on data from a small sample size (n = 51 participants).
1.3. Analysis.
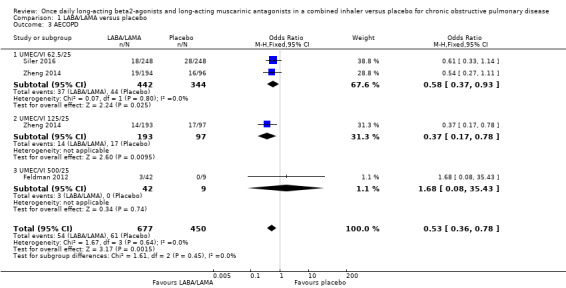
Comparison 1 LABA/LAMA versus placebo, Outcome 3 AECOPD.
6.
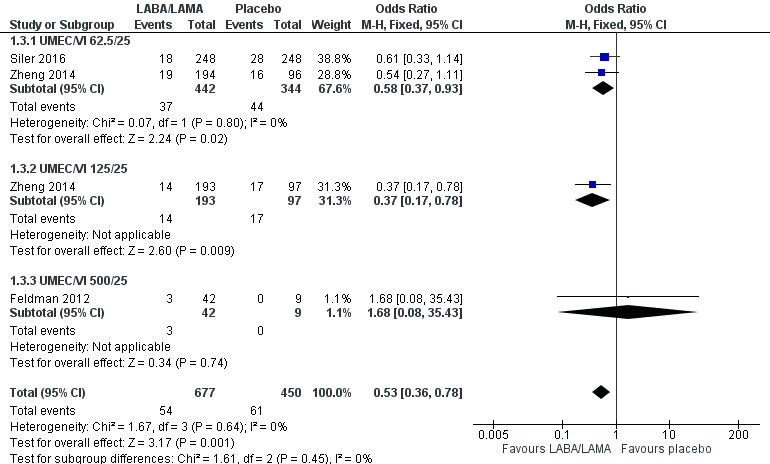
Forest plot of comparison: 1 LABA/LAMA versus placebo, outcome: 1.3 AECOPD.
Two studies (1371 participants), reported the time to first AECOPD; both studies examined UMEC/VI 125/25 μg versus placebo. The mean time to first AECOPD was statistically significantly longer in people receiving once‐daily LABA/LAMA in a combined inhaler compared with those receiving placebo (Hazard Ratio 0.44, 95% CI 0.31 to 0.63; Analysis 1.4).
1.4. Analysis.
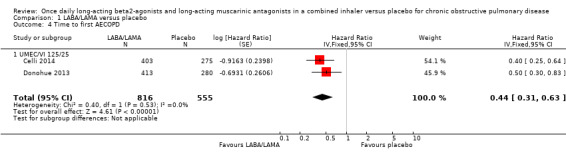
Comparison 1 LABA/LAMA versus placebo, Outcome 4 Time to first AECOPD.
Health‐related quality of life
SGRQ
Eight studies (4952 participants) reported health‐related quality of life as assessed using the SGRQ, at the end of treatment. A decrease in SGRQ represents an improvement in quality of life and the MCID is considered to be a change of four units (SGRQ‐C Manual). At the end of treatment in participants receiving once‐daily LABA/LAMA in a combined inhaler, the mean improvement versus placebo in adjusted SGRQ score was ‐4.08 (95% CI ‐4.80 to ‐3.36; Analysis 1.5), which was statistically significant and clinically relevant, exceeding the MCID. The overall certainty of the evidence for this outcome was rated as high.
1.5. Analysis.
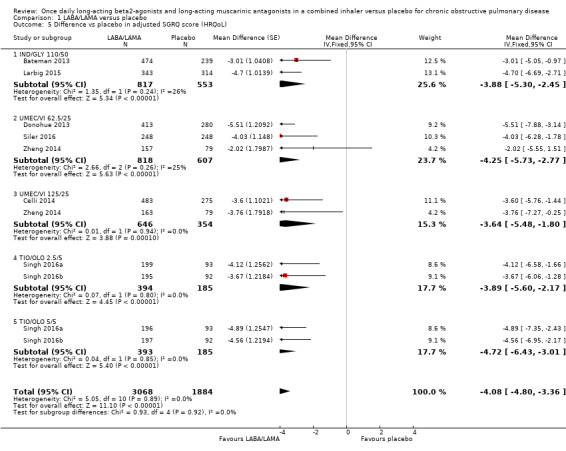
Comparison 1 LABA/LAMA versus placebo, Outcome 5 Difference vs placebo in adjusted SGRQ score (HRQoL).
The results were generally consistent across subgroups for different LABA/LAMA combinations and doses, with mean differences versus placebo ranging from ‐3.64 with UMEC/VI 125/25 μg to ‐4.72 with TIO/OLO 5/5 μg (Figure 7). The mean difference in SGRQ score versus placebo was statistically significant for all LABA/LAMA combinations and doses for which data were available (IND/GLY 110/50 μg; UMEC/VI 125/25 μg; UMEC/VI 62.5/25 μg; TIO/OLO 2.5/5 μg; TIO/OLO 5/5 μg); however, the MCID (4.00) was only exceeded with UMEC/VI 62.5/25 μg, TIO/OLO 2.5 μg and TIO/OLO 5/5 μg.
7.
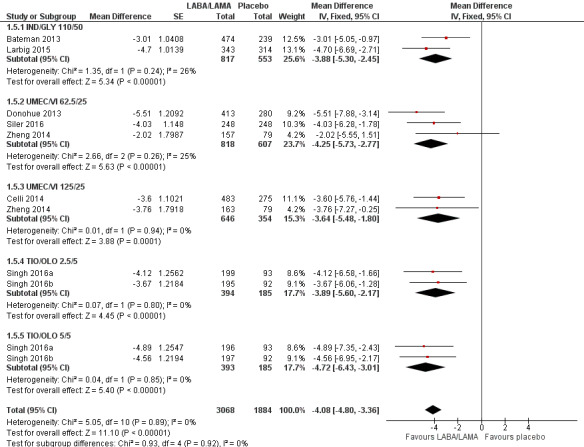
Forest plot of comparison: 1 LABA/LAMA versus placebo, outcome: 1.5 Difference vs placebo in adjusted SGRQ score (HRQoL).
Seven studies (4258 participants) reported SGRQ responder status (i.e. the proportion of participants who achieved a ≥ 4 point improvement from baseline in SGRQ total score) at the end of treatment. Compared with placebo, a greater proportion of participants receiving once‐daily LABA/LAMA were responders (OR 1.75, 95% CI 1.54 to 1.99; I2 = 0%; Analysis 1.6) and this difference was statistically significant. Compared with taking placebo, we estimated that taking a once‐daily LABA/LAMA in a combined inhaler would result in 138 more people per 1000 achieving a clinically meaningful improvement in quality of life, with the confidence intervals ranging from 106 to 170 more people per 1000. This finding is in agreement with the mean improvement in SGRQ total score for LABA/LAMA versus placebo, as reported above. The results were generally consistent across subgroups for different LABA/LAMA combinations and doses, with ORs versus placebo ranging from 1.70 with UMEC/VI 62.52/5 μg to 2.35 with TIO/OLO 5/5 μg; the exception was IND/GLY 110/50 μg for which the OR (95% CI) versus placebo was 1.35 (0.98 to 1.86), thus narrowly missing out on statistical significance. We note that the latter result was based on a single study, for which data were presented as percentages and extrapolated to participant numbers; given uncertainty around the precise raw data, this finding should be interpreted cautiously.
1.6. Analysis.
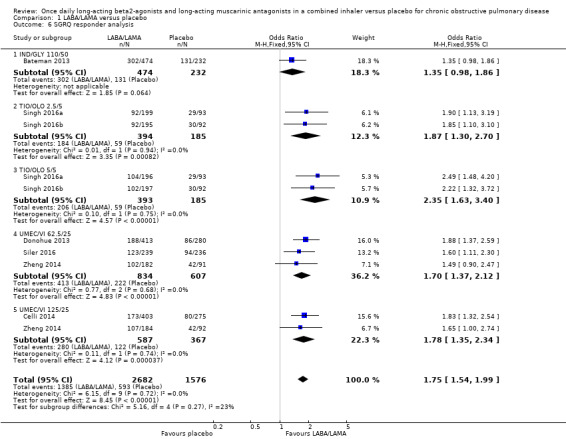
Comparison 1 LABA/LAMA versus placebo, Outcome 6 SGRQ responder analysis.
Secondary outcomes
Trough FEV1
Adjusted difference versus placebo in trough FEV1 at end of treatment
Thirteen studies (6598 participants) reported adjusted trough FEV1 at the end of treatment (i.e. change from baseline in FEV1). In participants receiving once‐daily LABA/LAMA in a combined inhaler, the mean difference versus placebo in adjusted trough FEV1 was 0.20 L (95% CI 0.19 to 0.21; Analysis 1.7), which was statistically significant and clinically relevant, exceeding the MCID of 100 mL (Donohue 2005). The overall certainty of the evidence for this outcome was rated as moderate, having been downgraded once for inconsistency (significant heterogeneity, I2= 71%), noting that heterogeneity was due to a different magnitude of treatment effect in a single study (NCT00626522; see below).
1.7. Analysis.
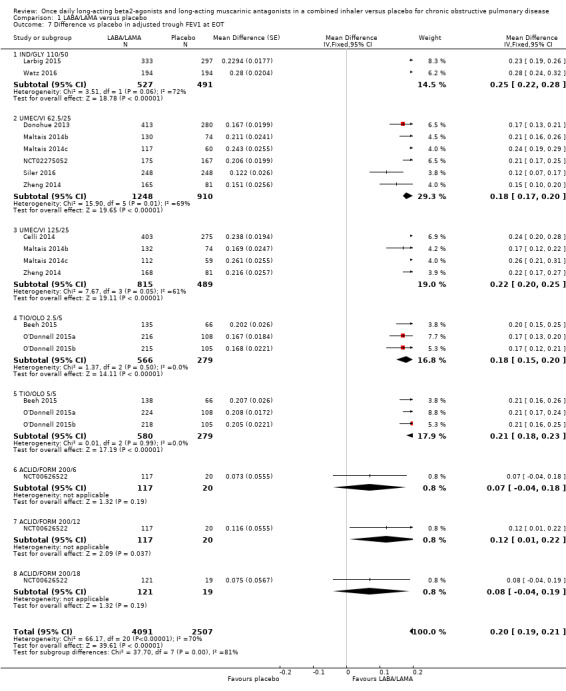
Comparison 1 LABA/LAMA versus placebo, Outcome 7 Difference vs placebo in adjusted trough FEV1 at EOT.
The results were generally consistent (i.e. overlapping CIs) across subgroups for different LABA/LAMA combinations and doses, with mean differences versus placebo ranging from 0.18 L with UMEC/VI 62.5/25 μg to 0.25 L with IND/GLY 110/50 μg; the exception was the results for ACLID/FORM, which were based on a single study (NCT00626522); mean differences were 0.07, 0.12 and 0.07 L for the 200/6, 200/12 and 200/18 μg subgroups, respectively. The MCID (0.1 L) was exceeded with IND/GLY 110/50 μg, UMEC/VI 62.5/25 μg, UMEC/VI 125/25 μg, TIO/OLO 2.5 μg, and TIO/OLO 5/5 μg.
Unadjusted difference versus placebo in trough FEV1 at end of treatment
Five studies (2330 participants) reported trough FEV1 at the end of treatment (i.e. not adjusted for baseline values). In participants receiving once‐daily LABA/LAMA in a combined inhaler, the mean difference versus placebo in trough FEV1 was 0.18 L (95% CI 0.16 to 0.20; Analysis 1.8), which was statistically significant and clinically relevant, exceeding the MCID of 100 mL (Donohue 2005). The overall certainty of the evidence for this outcome was rated as high.
1.8. Analysis.
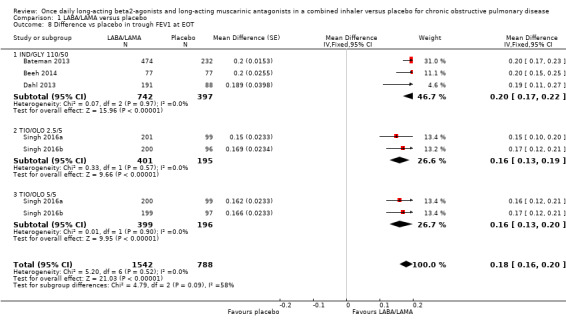
Comparison 1 LABA/LAMA versus placebo, Outcome 8 Difference vs placebo in trough FEV1 at EOT.
The results were consistent across subgroups for different LABA/LAMA combinations and doses, with mean differences versus placebo ranging from 0.16 L with TIO/OLO (2.5/5 and 5/5 μg doses) to 0.20 L with IND/GLY 110/50 μg. The MCID (0.1L) was exceeded with all LABA/LAMA combinations/doses for which data were available (IND/GLY 110/50 μg; TIO/OLO 2.5/5 μg; TIO/OLO 5/5 μg).
Pooled analyses for trough FEV1
When the adjusted and unadjusted data for trough FEV1 were pooled, there was no appreciable change in the overall mean difference (adjusted: MD 0.20 L, 95% CI 0.19 to 0.21; unadjusted: 0.18 L, 95% CI 0.16 to 0.20; pooled: MD 0.20 L, 95% CI 0.19 to 0.20 Analysis 1.9).
1.9. Analysis.
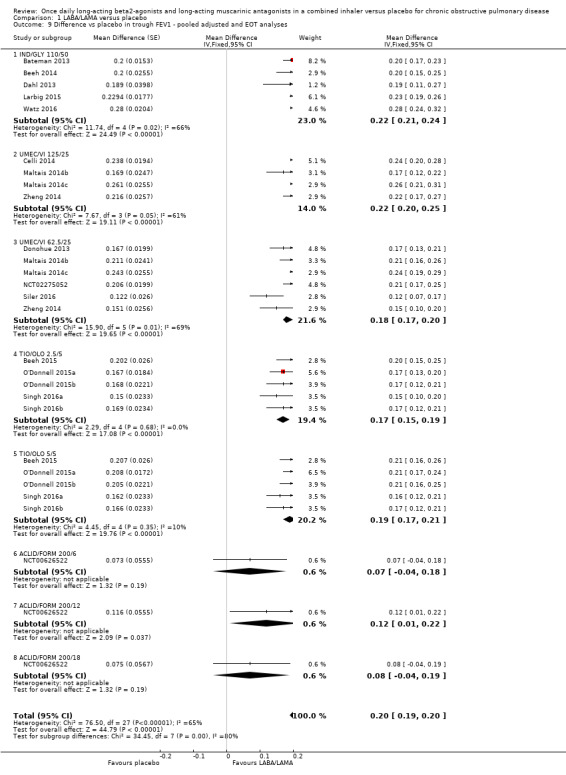
Comparison 1 LABA/LAMA versus placebo, Outcome 9 Difference vs placebo in trough FEV1 ‐ pooled adjusted and EOT analyses.
Adjusted peak FEV1
Seven studies (4188 participants) reported peak FEV1 at the end of treatment (i.e. peak FEV1 was explicitly specified, rather than 1‐hour FEV1, 2‐hour FEV1, etc). In participants receiving once‐daily LABA/LAMA in a combined inhaler, the mean difference versus placebo in peak FEV1 was 0.31 L (95% CI 0.29 to 0.32; Analysis 1.10), which was statistically significant. The overall certainty of the evidence for this outcome was rated as moderate, having been downgraded once for inconsistency (significant heterogeneity, I2= 68%).
1.10. Analysis.
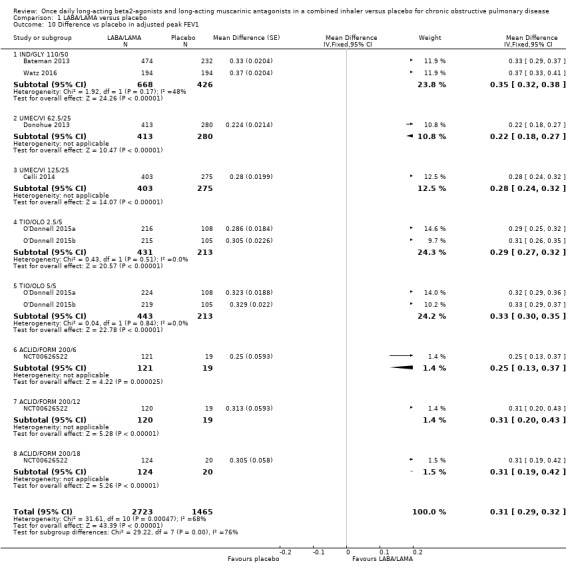
Comparison 1 LABA/LAMA versus placebo, Outcome 10 Difference vs placebo in adjusted peak FEV1.
The results were consistent across subgroups for different LABA/LAMA combinations and doses, with mean differences versus placebo ranging from 0.22 L with UMEC/VI 62.52/5 μg to 0.35 L with IND/GLY 110/50 μg.
6MWT
No studies reported data for this outcome.
Adverse events
Seventeen studies (8235 participants) reported the number of participants experiencing adverse events during the study period, for which there was no statistically significant difference (OR 0.95, 95% CI 0.86 to 1.04; I2 = 0%; Analysis 1.11). Compared with taking placebo, we estimated that taking once‐daily LABA/LAMA in a combined inhaler would result in 13 fewer people per 1000 experiencing a AE, with the confidence intervals ranging from 37 fewer to 10 more people per 1000. The overall certainty of the evidence for this outcome was rated as high.
1.11. Analysis.
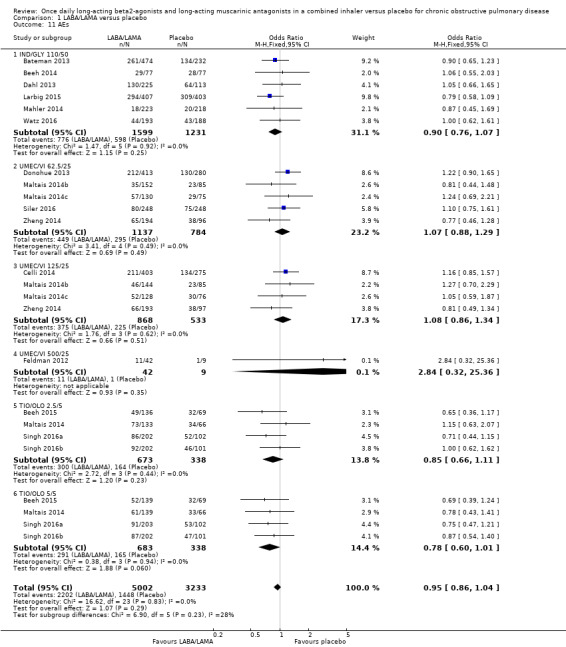
Comparison 1 LABA/LAMA versus placebo, Outcome 11 AEs.
The results were generally consistent across subgroups for different LABA/LAMA combinations and doses, with ORs ranging from 0.78 with TIO/OLO 5/5 μg to 1.08 with UMEC/VI 125/25 μg (Figure 5). The only exception was the UMEC/VI 500/25 μg subgroup (OR 2.84, 95% CI 0.32 to 25.36; participants = 51); however, these results should be interpreted cautiously as they were based on a small sample size from a single study, resulting in wide confidence intervals.
Subgroup analyses
Participants with ICS use during the trial versus participants without ICS use during the trial
All studies permitted the use of ICS during the trial, provided that participants had used ICS prior to the trial, and, in some cases, that the dose was stable prior to study initiation. Therefore, no subgroup analysis was performed.
Different LABA/LAMA combinations
The main analyses were split out by different LABA/LAMA combinations; please see the main results section above for a summary of different LABA/LAMA combinations.
Length of follow‐up (less than six months versus six months or longer)
Three studies had a duration of six months or longer (Bateman 2013; Dahl 2013; Larbig 2015) and all evaluated IND/GLY 110/50 versus placebo. This subanalysis was only relevant for three of the four primary outcomes as no studies evaluating IND/GLY contributed data to the meta‐analyses for AECOPD.
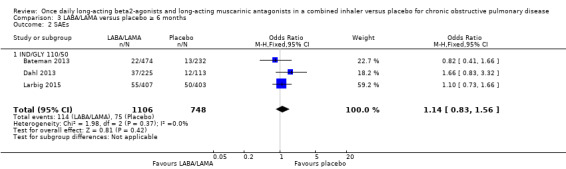
For all‐cause mortality, no significant difference between LABA/LAMA and placebo groups was identified, regardless of study duration (overall: < 6 months, OR 1.86, 95% CI 0.75 to 4.60 (25 studies); ≥ 6 months, OR 2.03, 95% CI 0.22 to 18.35 (2 studies); IND/GLY: < 6 months, OR 2.97, 95% CI 0.47 to 18.97 (3 studies); ≥ 6 months, OR 2.03, 95% CI 0.22 to 18.35 (2 studies)) (Analysis 2.1; Analysis 3.1).
2.1. Analysis.
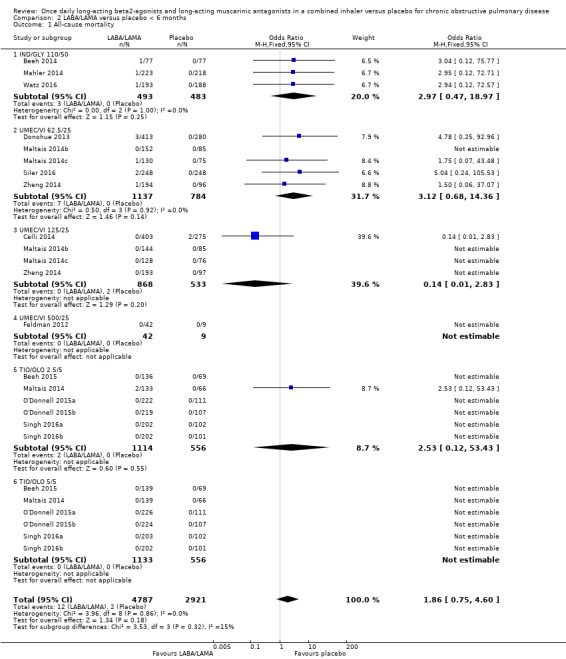
Comparison 2 LABA/LAMA versus placebo < 6 months, Outcome 1 All‐cause mortality.
3.1. Analysis.
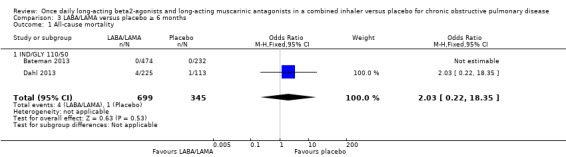
Comparison 3 LABA/LAMA versus placebo ≥ 6 months, Outcome 1 All‐cause mortality.
For SAEs, there was no statistically significant difference in the number of participants experiencing serious, but non‐fatal, adverse events during the study period, regardless of study duration (overall: < 6 months, OR 1.02, 95% CI 0.80 to 1.29 (19 studies); ≥ 6 months, OR 1.14, 95% CI 0.83 to 1.56 (3 studies); IND/GLY: < 6 months, OR 1.35, 95% CI 0.54 to 3.40 (3 studies); ≥ 6 months, OR 1.14, 95% CI 0.83 to 1.56 (3 studies)) (Analysis 2.2; Analysis 3.2).
2.2. Analysis.
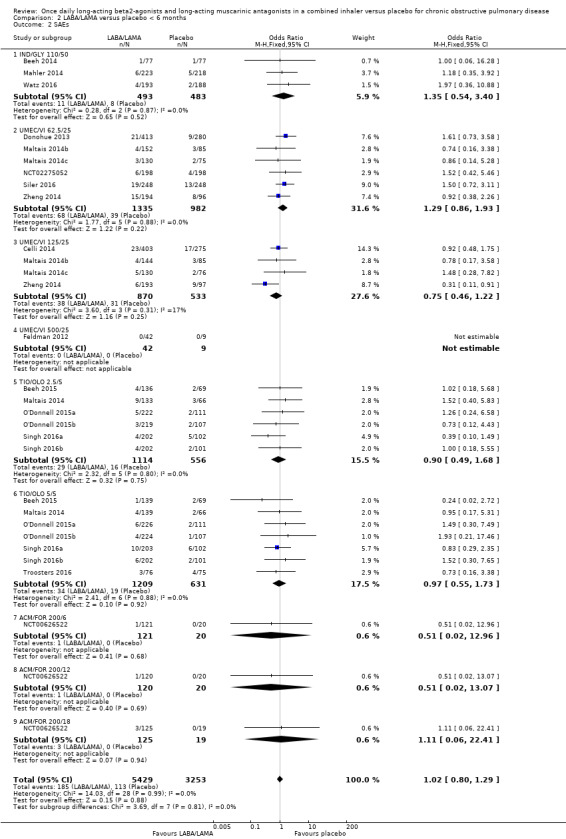
Comparison 2 LABA/LAMA versus placebo < 6 months, Outcome 2 SAEs.
3.2. Analysis.
Comparison 3 LABA/LAMA versus placebo ≥ 6 months, Outcome 2 SAEs.
For HRQoL, a statistically significant and clinically relevant improvement (i.e. exceeding MCID) in SGRQ score was observed with LABA/LAMA compared with placebo based on studies with a duration of < 6 months (MD ‐4.15, 95% CI ‐4.99 to ‐3.32; 9 studies). Three studies with a duration of ≥ 6 months reported SGRQ score and all evaluated IND/GLY 110/50 μg. As in the primary analyses, a statistically significant improvement was observed but did not exceed the MCID (MD ‐3.88, 95% CI ‐5.30 to ‐2.45; 2 studies).
For each outcome (all‐cause mortality, SAEs, and HRQoL), given the overlapping confidence intervals for the < 6‐month versus ≥ 6‐month comparison, we concluded that study duration had no statistically significant effect on the results.
Baseline COPD severity
All of the included studies that contributed data to the quantitative analyses enrolled a majority (> 97%) of participants with GOLD Stage II/III COPD. Therefore, subanalyses based on baseline disease severity were not performed.
Sensitivity analyses
The following sensitivity analyses were performed for the primary outcomes.
Available case analysis versus true ITT analysis
All included studies claimed to analyse the ITT population or 'full analysis set'; however, in the majority of studies it was not possible to determine whether missing values were imputed. Therefore, this sensitivity analysis was not performed.
Fixed‐ versus random‐effect models
The results were consistent regardless of choice of analysis model (fixed‐ versus random‐effects model) (Table 3).
2. Sensitivity analysis ‐ fixed‐ versus random‐effects model.
| Outcome | Fixed‐effect model | Random‐effects model |
| All‐cause mortality | OR 1.88, 95% CI 0.81 to 4.36 (P = 8752; S = 18) | OR 2.02, 95% CI 0.79 to 5.17 (P = 8752; S = 18) |
| SAEs | OR 1.06, 95% CI 0.88 to 1.28 (P = 10536; S = 22) | OR 1.05, 95% CI 0.87 to 1.27 (P = 10536; S = 22) |
| AECOPD | OR 0.53, 95% CI 0.36 to 0.78 (P = 1127; S = 3) | OR 0.52, 95% CI 0.35 to 0.78 (P = 1127; S = 3) |
| Adjusted SGRQ score (HRQoL) | MD ‐4.08, 95% CI ‐4.80 to ‐3.36 (P = 4952; S = 8) | MD ‐4.08, 95% CI ‐4.80 to ‐3.36 (P = 4952; S = 8) |
| Adjusted trough FEV1 at EOT | MD 0.19, 95% CI 0.20 to 0.21 (P = 6598; S = 13) | MD 0.20, 95% CI 0.18 to 0.21 (P = 6598; S = 13) |
| Adjusted peak FEV1 | MD 0.31, 95% CI 0.29 to 0.32 (P = 4188; S = 7) | MD 0.30, 95% CI 0.28 to 0.33 (P = 4188; S = 7) |
| AEs | OR 0.95, 95% CI 0.86 to 1.04 (P = 8235; S = 17) | OR 0.95, 95% CI 0.86 to 1.04 (P = 8235; S = 17) |
OR and MD are for LABA/LAMA relative to placebo. Note that studies and participants can be counted multiple times as analyses were split by dose groups (several dose groups were examined in a single trial).
CI: confidence interval; HRQoL: health‐related quality of life; OR: odds ratio; MD, mean difference; P: participants; S: studies; SGRQ: St George's Respiratory Questionnaire.
Risk of bias assessments
The results were consistent regardless of the inclusion of studies with a high risk of bias for one or more domains (i.e. any risk of bias versus low/unclear risk of bias) (Table 4).
3. Sensitivity analysis ‐ any risk of bias versus low/unclear risk of bias.
| Outcome | Any risk of bias | Low/unclear risk of bias |
| All‐cause mortality | OR 1.88, 95% CI 0.81 to 4.36 (P = 8752; S = 18) | OR 1.65, 95% CI 0.60 to 4.50 (P = 7287; S = 13) |
| SAEs | OR 1.06, 95% CI 0.88 to 1.28 (P = 10536; S = 22) | OR 1.09, 95% CI 0.86 to 1.40 (P = 8448; S = 17) |
| AECOPD | OR 0.53, 95% CI 0.36 to 0.78 (P = 1127; S = 3) | NA |
| Adjusted SGRQ score (HRQoL) | MD ‐4.08, 95% CI ‐4.80 to ‐3.36 (P = 4952; S = 8) | MD ‐4.12, 95% CI ‐4.99 to ‐3.24 (P = 3321; S = 3) |
| Adjusted trough FEV1 at EOT | MD 0.20, 95% CI 0.19 to 0.21 (P = 6598; S = 13) | MD 0.20, 95% CI 0.19 to 0.22 (P = 4977; S = 13) |
| Adjusted peak FEV1 | MD 0.31, 95% CI 0.29 to 0.32 (P = 4188; S = 7) | MD 0.31, 95% CI 0.29 to 0.32 (P = 4188; S = 7) |
| AEs | OR 0.95, 95% CI 0.86 to 1.04 (P = 8235; S = 17) | OR 0.97, 95% CI 0.86 to 1.08 (P = 5579; S = 10) |
OR and MD are for LABA/LAMA relative to placebo. Note that studies and participants can be counted multiple times as analyses were split by dose groups (several dose groups were examined in a single trial).
CI: confidence interval; HRQoL: health‐related quality of life; OR: odds ratio; MD: mean difference; NA: not applicable (no studies reported this outcome); P: participants; S: studies; SGRQ: St George's Respiratory Questionnaire.
Discussion
Summary of main results
We included twenty‐two studies (13 parallel‐group designs and nine cross‐over designs), which randomised a total of 8461 participants. All studies were RCTs that compared once‐daily LABA/LAMA via combination inhaler (n = 6252) with placebo (n = 3819). Most participants were adults with GOLD stage II/III COPD and between 28% to 58% of participants were using ICS at baseline. The duration of treatment ranged from three to 52 weeks (mean = 11 weeks; median = 12 weeks) and only three studies had a duration of six months or longer. All studies were performed at multiple centres and 19 of 22 studies were international. Most studies were well designed and considered to be at low risk of bias. Compared to placebo, once‐daily LABA/LAMA in a combined inhaler resulted in an improvement in HRQoL (measured using the SGRQ) and lung function and a decrease in AECOPD. Generally, the safety and tolerability of once‐daily LABA/LAMA appeared comparable to that observed in placebo‐treated participants, with similar rates of AEs and SAEs observed in each group. There was no significant difference in the number of deaths reported in participants receiving a once‐daily LABA/LAMA fixed‐dose combination (one per 1000) compared with those receiving placebo (2 per 1000); we assessed the certainty of the evidence to be low having been downgraded for imprecision and indirectness. Treatment effects were generally consistent across different LABA/LAMA combinations and doses. Improvements in HRQoL that statistically significantly exceeded the MCID were achieved with UMEC/VI 62.5/25 μg and TIO/OLO (2.5/5 and 5/5 μg) but not with IND/GLY 110/50 μg or UMEC/VI 125/25 μg. Improvements in lung function (trough FEV1 and peak FEV1) that statistically significantly exceeded the MCID were achieved with IND/GLY 110/50 μg, UMEC/VI (62.5/25 and 125/25 μg) and TIO/OLO (2.5/5 and 5/5 μg); these findings should be interpreted cautiously given the uncertainty around the MCID for FEV1 (see Types of outcome measures). A clinically significant improvement in peak FEV1, but not trough FEV1, was observed with ACLID/FORM, although the evidence for this combination was based only on a single study with a relatively small sample size. A statistically significant reduction in both the time to first AECOPD and rate of AECOPD was observed with UMEC/VI; data for these AECOPD outcomes were not available for other combinations.
Overall completeness and applicability of evidence
Demographics across the 8641 randomised participants were representative of patients with COPD (GOLD 2017). For example, participants had a mean age of around 60 to 65 years, were more often male and the majority either currently smoked or had a history of smoking. The inclusion criteria for 21 of 22 included studies specified either GOLD stage II/III, or criteria aligned with this disease severity; only one of the included studies permitted the enrolment of individuals with moderate‐to‐severe COPD. Therefore, the evidence synthesised herein is applicable to individuals with mild‐to‐moderate COPD. All prespecified outcomes, except for the 6MWT, were well reported across the 22 studies, although reporting of the number of participants experiencing AECOPD was based on only three studies and time to first AECOPD on only two studies; this was taken into account when evaluating the strength of the evidence for these outcomes. Six studies evaluated the once‐daily combination of IND/GLY (110/50 μg), seven studies evaluated TIO/OLO (2.5/5 or 5/5 μg), eight studies evaluated UMEC/VI (62.5/5 μg, 125/25 or 500/25 μg) and one study evaluated ACD/FOR (200/6, 200/12 or 200/18 μg). Subgrouping of studies by LABA/LAMA combination and dose had the effect of reducing the sample size for each comparison; in particular, only one study with a short duration examined the ACLID/FORM combination so we can be less certain of how the overall findings apply to the ACLID/FORM combination. The median study duration was 12 weeks; only three studies had a duration of six months or longer and all evaluated ING/GLY 110/50 μg. In these studies with a duration of six months or longer, the results of meta‐analyses for all‐cause mortality, SAEs, and HRQoL were consistent with those based on studies with a duration of less than six months.
Quality of the evidence
The certainty of the evidence was generally considered to be moderate or high with the exception of all‐cause mortality, which we considered to be low, having downgraded it once for indirectness and once for imprecision due to a low number of events. We considered the certainty of the evidence for SAEs, HRQoL, and AEs to be high. The certainty of the evidence for lung function (trough and peak FEV1) was considered to be moderate having been downgraded for inconsistency due to significant heterogeneity. The certainty of the evidence for AECOPD was considered moderate having been downgraded once for indirectness as the evidence related only to UMEC/VI. We could not rule out the possibility of publication bias for this outcome but were unable to demonstrate conclusively that publication bias existed, due to the low number of studies reporting this outcome (i.e. the validity of a funnel plot is limited when based on fewer than ten studies). Additionally, selective reporting for this outcome in studies of other LABA/LAMA combinations did not occur based on comparison of primary reports with trial registry entries.
Risk of bias in the included studies was generally considered to be low or was unclear due to the lack of necessary information provided in the study reports. Across 198 assessments (22 studies, nine domains each), over three‐quarters were considered to be at a low risk of bias, and only seven were considered to be at a high risk of bias. Risk of bias was considered unclear in the remaining 37 assessments. Four studies were considered to be at high risk for 'other' bias, in three cases, due to greater disease severity in the LABA/LAMA group compared with the placebo group; this problem would tend to skew the results in favour or placebo, resulting in a potential underestimation of the treatment effect. However, the results were robust to the removal of studies with any domain considered to be at high risk of bias and no downgrading of the strength of the evidence (by GRADE) was performed on the basis of risk of bias.
Potential biases in the review process
The review was conducted to the standards set by MECIR (MECIR 2018) and in accordance with the published protocol. In particular, two authors independently screened the search results, determined studies for inclusion, assessed the risk of bias, extracted the relevant data, and performed the GRADE assessment (i.e. all steps involving subjective decisions). There were several minor deviations from the protocol (see Differences between protocol and review). It is unlikely that any relevant studies were missed, as a skilled information specialist conducted the main electronic searches. Additionally, the main searches were supplemented by manual searches of reference lists of associated studies and reviews. Finally, this review has undergone editorial and peer review and thus considers the opinions of independent external experts. In summary, the review was conducted in a manner that should ensure that our conclusions fairly and accurately represent the results synthesised during the review process.
Agreements and disagreements with other studies or reviews
The majority of relevant systematic reviews compared LABA/LAMA FDCs with their mono‐components (Calzetta 2016; Calzetta 2017). However, our findings are consistent with those of a recent network meta‐analysis of LABA/LAMA versus their mono‐components and placebo (Oba 2016). For example, LABA/LAMA combinations demonstrated a mean improvement in trough FEV1 over placebo of 0.21 (95% CI 0.19, 0.23), 0.20 L (95% CI 0.17 to 0.23) and 0.24 L (95% CI 0.14 to 0.35) at three, six, and 12 months, respectively, agreeing with the 0.20 L reported herein. Clinically significant improvements in HRQoL were also seen with LABA/LAMA over placebo, with a mean change from baseline in SGRQ score of ‐4.6 (‐5.9, ‐3.3) at three months and ‐4.1 (‐5.9, ‐2.3) at six months, agreeing with the 4.08 point improvement reported herein. Furthermore and in agreement with our findings, no significant differences in mortality or total SAEs were observed between LABA/LAMA and placebo (Oba 2016).
Authors' conclusions
Implications for practice.
Compared with placebo, once‐daily LABA/LAMA (either IND/GLY, UMEC/VI or TIO/OLO) via a combination inhaler is associated with a clinically significant improvement in lung function and health‐related quality of life in patients with mild‐to‐moderate COPD; in addition, UMEC/VI appears to reduce the rate of exacerbations in this population. These conclusions are supported by moderate‐ or high‐certainty evidence from studies with an observation period of up to one year.
Implications for research.
Prespecified outcomes of interest for this review were generally well evaluated by the included studies, with the exception of the 6MWT, which was not evaluated by any of the studies. The 6MWT requires large sample sizes or large treatment effects to detect a statistically significant signal and thus may not be the most appropriate test for evaluating new interventions; alternative outcomes for assessing functional exercise capacity include the incremental shuttle walk test and the endurance shuttle walk test (Singh 2014b). Future research should focus on establishing the relative net clinical benefit (i.e. considering both efficacy and safety) for the different LABA/LAMA combinations; the findings of this review (relative to placebo) should facilitate this work.
Acknowledgements
Julia Walters was the Editor for this review and commented critically on the review.
We gratefully acknowledge Dr Emma Dennett for her role in refining our research question, and Mrs Elizabeth Stovold for developing the search strategy.
The Background and Methods section of the protocol were based on a standard template used by Cochrane Airways Group. Some of the content from an earlier Cochrane protocol has been used verbatim and modified after the original author team stepped down from the review (Sarai 2014).
This project was supported by the National Institute for Health Research (NIHR), via Cochrane Infrastructure funding to the Cochrane Airways Group. Dr Evans's post is funded by the Faculty of Health and Medicine, Lancaster University. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS, or the Department of Health.
Appendices
Appendix 1. Sources and search methods for the Cochrane Airways Trials Register
Electronic searches: core databases
| Database | Dates searched | Frequency of search |
| CENTRAL (via the Cochrane Register of Studies (CRS)) |
From inception | Monthly |
| MEDLINE (Ovid) | 1946 onwards | Weekly |
| EMBASE (Ovid) | 1974 onwards | Weekly |
| PsycINFO (Ovid) | 1967 onwards | Monthly |
| CINAHL (EBSCO) | 1937 onwards | Monthly |
| AMED (EBSCO) | From inception | Monthly |
Handsearches: core respiratory conference abstracts
| Conference | Years searched |
| American Academy of Allergy, Asthma and Immunology (AAAAI) | 2001 onwards |
| American Thoracic Society (ATS) | 2001 onwards |
| Asia Pacific Society of Respirology (APSR) | 2004 onwards |
| British Thoracic Society Winter Meeting (BTS) | 2000 onwards |
| Chest Meeting | 2003 onwards |
| European Respiratory Society (ERS) | 1992, 1994, 2000 onwards |
| International Primary Care Respiratory Group Congress (IPCRG) | 2002 onwards |
| Thoracic Society of Australia and New Zealand (TSANZ) | 1999 onwards |
MEDLINE search strategy used to identify trials for the Trials Register
COPD search
1. Lung Diseases, Obstructive/
2. exp Pulmonary Disease, Chronic Obstructive/
3. emphysema$.mp.
4. (chronic$ adj3 bronchiti$).mp.
5. (obstruct$ adj3 (pulmonary or lung$ or airway$ or airflow$ or bronch$ or respirat$)).mp.
6. COPD.mp.
7. COAD.mp.
8. COBD.mp.
9. AECB.mp.
10. or/1‐9
Filter to identify RCTs
1. exp "clinical trial [publication type]"/
2. (randomised or randomised).ab,.
3. placebo.ab,ti.
4. dt.fs.
5. randomly.ab,ti.
6. trial.ab,ti.
7. groups.ab,ti.
8. or/1‐7
9. Animals/
10. Humans/
11. 9 not (9 and 10)
12. 8 not 11
The MEDLINE strategy and RCT filter are adapted to identify trials in other electronic databases
Appendix 2. Search strategy to identify trials from the Cochrane Airways Trials Register
(Search platform: Cochrane Register of Studies ‐ CRS Web)
#1 MeSH DESCRIPTOR Pulmonary Disease, Chronic Obstructive Explode All
#2 MeSH DESCRIPTOR Bronchitis, Chronic
#3 (obstruct*) near3 (pulmonary or lung* or airway* or airflow* or bronch* or respirat*)
#4 COPD:MISC1
#5 (COPD OR COAD OR COBD):TI,AB,KW
#6 #1 OR #2 OR #3 OR #4 OR #5
#7 aclidinium*
#8 glycopyrronium*
#9 tiotropium*
#10 umeclidinium*
#11 #7 or #8 or #9 or #10
#12 vilanterol*
#13 indacaterol*
#14 olodaterol*
#15 formoterol*
#16 #12 or #13 or #14 or #15
#17 QVA149
#18 LAMA NEAR LABA
#19 (long‐acting or "long acting") AND (muscarinic* NEAR antagonist*)
#20 (#11 and #16)
#21 #17 or #18 or #19 or #20
#22 #6 and #21
[Note: in search line #4 MISC1 denotes the field in which the reference has been coded for condition, in this case, COPD]
Data and analyses
Comparison 1. LABA/LAMA versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 All‐cause mortality | 18 | 8752 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.88 [0.81, 4.36] |
| 1.1 IND/GLY 110/50 | 5 | 2020 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.53 [0.61, 10.49] |
| 1.2 UMEC/VI 62.5/25 | 5 | 1921 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.12 [0.68, 14.36] |
| 1.3 UMEC/VI 125/25 | 4 | 1401 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.01, 2.83] |
| 1.4 UMEC/VI 500/25 | 1 | 51 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 1.5 TIO/OLO 2.5/5 | 6 | 1670 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.53 [0.12, 53.43] |
| 1.6 TIO/OLO 5/5 | 6 | 1689 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2 SAEs | 22 | 10536 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.88, 1.28] |
| 2.1 IND/GLY 110/50 | 6 | 2830 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.86, 1.56] |
| 2.2 UMEC/VI 62.5/25 | 6 | 2317 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.29 [0.86, 1.93] |
| 2.3 UMEC/VI 125/25 | 4 | 1403 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.46, 1.22] |
| 2.4 UMEC/VI 500/25 | 1 | 51 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2.5 TIO/OLO 2.5/5 | 6 | 1670 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.49, 1.68] |
| 2.6 TIO/OLO 5/5 | 7 | 1840 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.55, 1.73] |
| 2.7 ACM/FOR 200/6 | 1 | 141 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.51 [0.02, 12.96] |
| 2.8 ACM/FOR 200/12 | 1 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.51 [0.02, 13.07] |
| 2.9 ACM/FOR 200/18 | 1 | 144 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.06, 22.41] |
| 3 AECOPD | 3 | 1127 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.53 [0.36, 0.78] |
| 3.1 UMEC/VI 62.5/25 | 2 | 786 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.37, 0.93] |
| 3.2 UMEC/VI 125/25 | 1 | 290 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.37 [0.17, 0.78] |
| 3.3 UMEC/VI 500/25 | 1 | 51 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.68 [0.08, 35.43] |
| 4 Time to first AECOPD | 2 | 1371 | Hazard Ratio (Fixed, 95% CI) | 0.44 [0.31, 0.63] |
| 4.1 UMEC/VI 125/25 | 2 | 1371 | Hazard Ratio (Fixed, 95% CI) | 0.44 [0.31, 0.63] |
| 5 Difference vs placebo in adjusted SGRQ score (HRQoL) | 8 | 4952 | Mean Difference (Fixed, 95% CI) | ‐4.08 [‐4.80, ‐3.36] |
| 5.1 IND/GLY 110/50 | 2 | 1370 | Mean Difference (Fixed, 95% CI) | ‐3.88 [‐5.30, ‐2.45] |
| 5.2 UMEC/VI 62.5/25 | 3 | 1425 | Mean Difference (Fixed, 95% CI) | ‐4.25 [‐5.73, ‐2.77] |
| 5.3 UMEC/VI 125/25 | 2 | 1000 | Mean Difference (Fixed, 95% CI) | ‐3.64 [‐5.48, ‐1.80] |
| 5.4 TIO/OLO 2.5/5 | 2 | 579 | Mean Difference (Fixed, 95% CI) | ‐3.89 [‐5.60, ‐2.17] |
| 5.5 TIO/OLO 5/5 | 2 | 578 | Mean Difference (Fixed, 95% CI) | ‐4.72 [‐6.43, ‐3.01] |
| 6 SGRQ responder analysis | 7 | 4258 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.75 [1.54, 1.99] |
| 6.1 IND/GLY 110/50 | 1 | 706 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.35 [0.98, 1.86] |
| 6.2 TIO/OLO 2.5/5 | 2 | 579 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.87 [1.30, 2.70] |
| 6.3 TIO/OLO 5/5 | 2 | 578 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.35 [1.63, 3.40] |
| 6.4 UMEC/VI 62.5/25 | 3 | 1441 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.70 [1.37, 2.12] |
| 6.5 UMEC/VI 125/25 | 2 | 954 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.78 [1.35, 2.34] |
| 7 Difference vs placebo in adjusted trough FEV1 at EOT | 13 | 6598 | Mean Difference (Fixed, 95% CI) | 0.20 [0.19, 0.21] |
| 7.1 IND/GLY 110/50 | 2 | 1018 | Mean Difference (Fixed, 95% CI) | 0.25 [0.22, 0.28] |
| 7.2 UMEC/VI 62.5/25 | 6 | 2158 | Mean Difference (Fixed, 95% CI) | 0.18 [0.17, 0.20] |
| 7.3 UMEC/VI 125/25 | 4 | 1304 | Mean Difference (Fixed, 95% CI) | 0.22 [0.20, 0.25] |
| 7.4 TIO/OLO 2.5/5 | 3 | 845 | Mean Difference (Fixed, 95% CI) | 0.18 [0.15, 0.20] |
| 7.5 TIO/OLO 5/5 | 3 | 859 | Mean Difference (Fixed, 95% CI) | 0.21 [0.18, 0.23] |
| 7.6 ACLID/FORM 200/6 | 1 | 137 | Mean Difference (Fixed, 95% CI) | 0.07 [‐0.04, 0.18] |
| 7.7 ACLID/FORM 200/12 | 1 | 137 | Mean Difference (Fixed, 95% CI) | 0.12 [0.01, 0.22] |
| 7.8 ACLID/FORM 200/18 | 1 | 140 | Mean Difference (Fixed, 95% CI) | 0.08 [‐0.04, 0.19] |
| 8 Difference vs placebo in trough FEV1 at EOT | 5 | 2330 | Mean Difference (Fixed, 95% CI) | 0.18 [0.16, 0.20] |
| 8.1 IND/GLY 110/50 | 3 | 1139 | Mean Difference (Fixed, 95% CI) | 0.20 [0.17, 0.22] |
| 8.2 TIO/OLO 2.5/5 | 2 | 596 | Mean Difference (Fixed, 95% CI) | 0.16 [0.13, 0.19] |
| 8.3 TIO/OLO 5/5 | 2 | 595 | Mean Difference (Fixed, 95% CI) | 0.16 [0.13, 0.20] |
| 9 Difference vs placebo in trough FEV1 ‐ pooled adjusted and EOT analyses | 18 | Mean Difference (Fixed, 95% CI) | 0.20 [0.19, 0.20] | |
| 9.1 IND/GLY 110/50 | 5 | Mean Difference (Fixed, 95% CI) | 0.22 [0.21, 0.24] | |
| 9.2 UMEC/VI 125/25 | 4 | Mean Difference (Fixed, 95% CI) | 0.22 [0.20, 0.25] | |
| 9.3 UMEC/VI 62.5/25 | 6 | Mean Difference (Fixed, 95% CI) | 0.18 [0.17, 0.20] | |
| 9.4 TIO/OLO 2.5/5 | 5 | Mean Difference (Fixed, 95% CI) | 0.17 [0.15, 0.19] | |
| 9.5 TIO/OLO 5/5 | 5 | Mean Difference (Fixed, 95% CI) | 0.19 [0.17, 0.21] | |
| 9.6 ACLID/FORM 200/6 | 1 | Mean Difference (Fixed, 95% CI) | 0.07 [‐0.04, 0.18] | |
| 9.7 ACLID/FORM 200/12 | 1 | Mean Difference (Fixed, 95% CI) | 0.12 [0.01, 0.22] | |
| 9.8 ACLID/FORM 200/18 | 1 | Mean Difference (Fixed, 95% CI) | 0.08 [‐0.04, 0.19] | |
| 10 Difference vs placebo in adjusted peak FEV1 | 7 | 4188 | Mean Difference (Fixed, 95% CI) | 0.31 [0.29, 0.32] |
| 10.1 IND/GLY 110/50 | 2 | 1094 | Mean Difference (Fixed, 95% CI) | 0.35 [0.32, 0.38] |
| 10.2 UMEC/VI 62.5/25 | 1 | 693 | Mean Difference (Fixed, 95% CI) | 0.22 [0.18, 0.27] |
| 10.3 UMEC/VI 125/25 | 1 | 678 | Mean Difference (Fixed, 95% CI) | 0.28 [0.24, 0.32] |
| 10.4 TIO/OLO 2.5/5 | 2 | 644 | Mean Difference (Fixed, 95% CI) | 0.29 [0.27, 0.32] |
| 10.5 TIO/OLO 5/5 | 2 | 656 | Mean Difference (Fixed, 95% CI) | 0.33 [0.30, 0.35] |
| 10.6 ACLID/FORM 200/6 | 1 | 140 | Mean Difference (Fixed, 95% CI) | 0.25 [0.13, 0.37] |
| 10.7 ACLID/FORM 200/12 | 1 | 139 | Mean Difference (Fixed, 95% CI) | 0.31 [0.20, 0.43] |
| 10.8 ACLID/FORM 200/18 | 1 | 144 | Mean Difference (Fixed, 95% CI) | 0.31 [0.19, 0.42] |
| 11 AEs | 17 | 8235 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.86, 1.04] |
| 11.1 IND/GLY 110/50 | 6 | 2830 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.76, 1.07] |
| 11.2 UMEC/VI 62.5/25 | 5 | 1921 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.88, 1.29] |
| 11.3 UMEC/VI 125/25 | 4 | 1401 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.86, 1.34] |
| 11.4 UMEC/VI 500/25 | 1 | 51 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.84 [0.32, 25.36] |
| 11.5 TIO/OLO 2.5/5 | 4 | 1011 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.66, 1.11] |
| 11.6 TIO/OLO 5/5 | 4 | 1021 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.60, 1.01] |
Comparison 2. LABA/LAMA versus placebo < 6 months.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 All‐cause mortality | 16 | 7708 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.86 [0.75, 4.60] |
| 1.1 IND/GLY 110/50 | 3 | 976 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.97 [0.47, 18.97] |
| 1.2 UMEC/VI 62.5/25 | 5 | 1921 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.12 [0.68, 14.36] |
| 1.3 UMEC/VI 125/25 | 4 | 1401 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.01, 2.83] |
| 1.4 UMEC/VI 500/25 | 1 | 51 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 1.5 TIO/OLO 2.5/5 | 6 | 1670 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.53 [0.12, 53.43] |
| 1.6 TIO/OLO 5/5 | 6 | 1689 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2 SAEs | 19 | 8682 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.80, 1.29] |
| 2.1 IND/GLY 110/50 | 3 | 976 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.35 [0.54, 3.40] |
| 2.2 UMEC/VI 62.5/25 | 6 | 2317 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.29 [0.86, 1.93] |
| 2.3 UMEC/VI 125/25 | 4 | 1403 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.46, 1.22] |
| 2.4 UMEC/VI 500/25 | 1 | 51 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2.5 TIO/OLO 2.5/5 | 6 | 1670 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.49, 1.68] |
| 2.6 TIO/OLO 5/5 | 7 | 1840 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.55, 1.73] |
| 2.7 ACM/FOR 200/6 | 1 | 141 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.51 [0.02, 12.96] |
| 2.8 ACM/FOR 200/12 | 1 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.51 [0.02, 13.07] |
| 2.9 ACM/FOR 200/18 | 1 | 144 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.06, 22.41] |
| 3 AECOPD | 3 | 1127 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.53 [0.36, 0.78] |
| 3.1 UMEC/VI 62.5/25 | 2 | 786 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.37, 0.93] |
| 3.2 UMEC/VI 125/25 | 1 | 290 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.37 [0.17, 0.78] |
| 3.3 UMEC/VI 500/25 | 1 | 51 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.68 [0.08, 35.43] |
| 4 Time to first AECOPD | 2 | Hazard Ratio (Fixed, 95% CI) | 0.44 [0.31, 0.63] | |
| 4.1 UMEC/VI 125/25 | 2 | Hazard Ratio (Fixed, 95% CI) | 0.44 [0.31, 0.63] | |
| 5 Difference vs placebo in adjusted SGRQ score (HRQoL) | 6 | Mean Difference (Fixed, 95% CI) | ‐4.15 [‐4.99, ‐3.32] | |
| 5.1 UMEC/VI 62.5/25 | 3 | Mean Difference (Fixed, 95% CI) | ‐4.25 [‐5.73, ‐2.77] | |
| 5.2 UMEC/VI 125/25 | 2 | Mean Difference (Fixed, 95% CI) | ‐3.64 [‐5.48, ‐1.80] | |
| 5.3 TIO/OLO 2.5/5 | 2 | Mean Difference (Fixed, 95% CI) | ‐3.89 [‐5.60, ‐2.17] | |
| 5.4 TIO/OLO 5/5 | 2 | Mean Difference (Fixed, 95% CI) | ‐4.72 [‐6.43, ‐3.01] | |
| 6 SGRQ responder analysis | 6 | 3552 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.84 [1.59, 2.12] |
| 6.1 TIO/OLO 2.5/5 | 2 | 579 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.87 [1.30, 2.70] |
| 6.2 TIO/OLO 5/5 | 2 | 578 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.35 [1.63, 3.40] |
| 6.3 UMEC/VI 62.5/25 | 3 | 1441 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.70 [1.37, 2.12] |
| 6.4 UMEC/VI 125/25 | 2 | 954 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.78 [1.35, 2.34] |
2.3. Analysis.
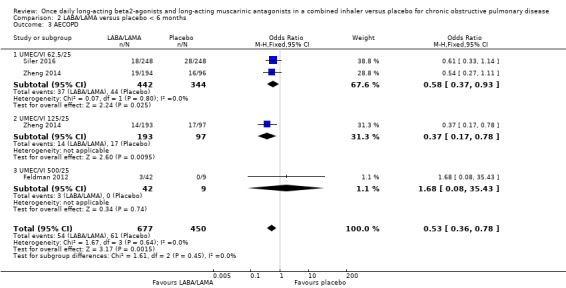
Comparison 2 LABA/LAMA versus placebo < 6 months, Outcome 3 AECOPD.
2.4. Analysis.
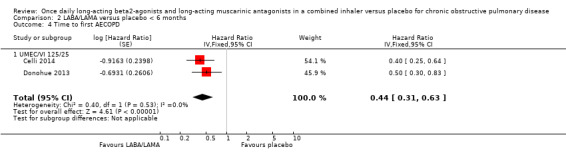
Comparison 2 LABA/LAMA versus placebo < 6 months, Outcome 4 Time to first AECOPD.
2.5. Analysis.
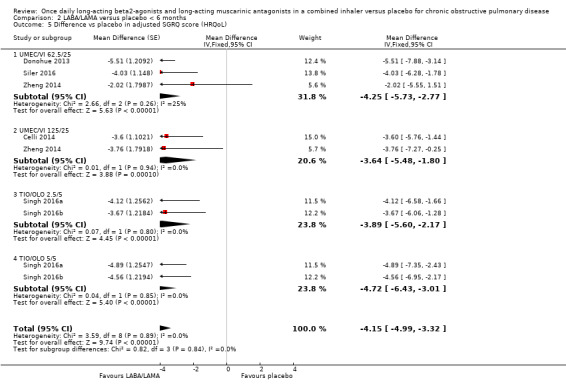
Comparison 2 LABA/LAMA versus placebo < 6 months, Outcome 5 Difference vs placebo in adjusted SGRQ score (HRQoL).
2.6. Analysis.
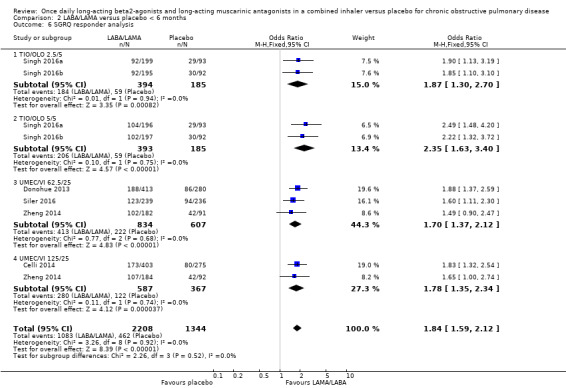
Comparison 2 LABA/LAMA versus placebo < 6 months, Outcome 6 SGRQ responder analysis.
Comparison 3. LABA/LAMA versus placebo ≥ 6 months.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 All‐cause mortality | 2 | 1044 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.03 [0.22, 18.35] |
| 1.1 IND/GLY 110/50 | 2 | 1044 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.03 [0.22, 18.35] |
| 2 SAEs | 3 | 1854 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.83, 1.56] |
| 2.1 IND/GLY 110/50 | 3 | 1854 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.83, 1.56] |
| 3 Difference vs placebo in adjusted SGRQ score (HRQoL) | 2 | Mean Difference (Fixed, 95% CI) | ‐3.88 [‐5.30, ‐2.45] | |
| 3.1 IND/GLY 110/50 | 2 | Mean Difference (Fixed, 95% CI) | ‐3.88 [‐5.30, ‐2.45] | |
| 4 SGRQ responder analysis | 1 | 706 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.35 [0.98, 1.86] |
| 4.1 IND/GLY 110/50 | 1 | 706 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.35 [0.98, 1.86] |
3.3. Analysis.

Comparison 3 LABA/LAMA versus placebo ≥ 6 months, Outcome 3 Difference vs placebo in adjusted SGRQ score (HRQoL).
3.4. Analysis.
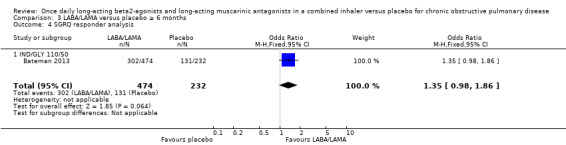
Comparison 3 LABA/LAMA versus placebo ≥ 6 months, Outcome 4 SGRQ responder analysis.
Comparison 4. Sensitivity analysis ‐ random‐effects model.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 All‐cause mortality | 18 | 8752 | Odds Ratio (M‐H, Random, 95% CI) | 2.02 [0.79, 5.17] |
| 1.1 IND/GLY 110/50 | 5 | 2020 | Odds Ratio (M‐H, Random, 95% CI) | 2.54 [0.61, 10.48] |
| 1.2 TIO/OLO 2.5/5 | 6 | 1670 | Odds Ratio (M‐H, Random, 95% CI) | 2.53 [0.12, 53.43] |
| 1.3 TIO/OLO 5/5 | 6 | 1689 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 1.4 UMEC/VI 62.5/25 | 5 | 1921 | Odds Ratio (M‐H, Random, 95% CI) | 2.92 [0.62, 13.79] |
| 1.5 UMEC/VI 125/25 | 4 | 1401 | Odds Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.83] |
| 1.6 UMEC/VI 500/25 | 1 | 51 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 2 SAEs | 22 | 10536 | Odds Ratio (M‐H, Random, 95% CI) | 1.05 [0.87, 1.27] |
| 2.1 IND/GLY 110/50 | 6 | 2830 | Odds Ratio (M‐H, Random, 95% CI) | 1.15 [0.85, 1.55] |
| 2.2 TIO/OLO 2.5/5 | 6 | 1670 | Odds Ratio (M‐H, Random, 95% CI) | 0.89 [0.47, 1.68] |
| 2.3 TIO/OLO 5/5 | 7 | 1840 | Odds Ratio (M‐H, Random, 95% CI) | 0.96 [0.53, 1.74] |
| 2.4 UMEC/VI 62.5/25 | 6 | 2317 | Odds Ratio (M‐H, Random, 95% CI) | 1.28 [0.85, 1.92] |
| 2.5 UMEC/VI 125/25 | 4 | 1403 | Odds Ratio (M‐H, Random, 95% CI) | 0.73 [0.41, 1.30] |
| 2.6 UMEC/VI 500/25 | 1 | 51 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 2.7 ACM/FOR 200/6 | 1 | 141 | Odds Ratio (M‐H, Random, 95% CI) | 0.51 [0.02, 12.96] |
| 2.8 ACM/FOR 200/12 | 1 | 140 | Odds Ratio (M‐H, Random, 95% CI) | 0.51 [0.02, 13.07] |
| 2.9 ACM/FOR 200/18 | 1 | 144 | Odds Ratio (M‐H, Random, 95% CI) | 1.11 [0.06, 22.41] |
| 3 AECOPD | 3 | 1127 | Odds Ratio (M‐H, Random, 95% CI) | 0.52 [0.35, 0.78] |
| 3.1 UMEC/VI 62.5/25 | 2 | 786 | Odds Ratio (M‐H, Random, 95% CI) | 0.58 [0.36, 0.93] |
| 3.2 UMEC/VI 125/25 | 1 | 290 | Odds Ratio (M‐H, Random, 95% CI) | 0.37 [0.17, 0.78] |
| 3.3 UMEC/VI 500/25 | 1 | 51 | Odds Ratio (M‐H, Random, 95% CI) | 1.68 [0.08, 35.43] |
| 4 Time to first AECOPD | 2 | Hazard Ratio (Random, 95% CI) | 0.44 [0.31, 0.63] | |
| 4.1 UMEC/VI 125/25 | 2 | Hazard Ratio (Random, 95% CI) | 0.44 [0.31, 0.63] | |
| 5 Difference vs placebo in adjusted SGRQ score (HRQoL) | 8 | Mean Difference (Random, 95% CI) | ‐4.08 [‐4.80, ‐3.36] | |
| 5.1 IND/GLY 110/50 | 2 | Mean Difference (Random, 95% CI) | ‐3.87 [‐5.53, ‐2.22] | |
| 5.2 UMEC/VI 125/25 | 2 | Mean Difference (Random, 95% CI) | ‐3.64 [‐5.48, ‐1.80] | |
| 5.3 UMEC/VI 62.5/25 | 3 | Mean Difference (Random, 95% CI) | ‐4.18 [‐5.92, ‐2.44] | |
| 5.4 TIO/OLO 2.5/5 | 2 | Mean Difference (Random, 95% CI) | ‐3.89 [‐5.60, ‐2.17] | |
| 5.5 TIO/OLO 5/5 | 2 | Mean Difference (Random, 95% CI) | ‐4.72 [‐6.43, ‐3.01] | |
| 6 SGRQ responder analysis | 7 | 4258 | Odds Ratio (M‐H, Random, 95% CI) | 1.74 [1.53, 1.99] |
| 6.1 IND/GLY 110/50 | 1 | 706 | Odds Ratio (M‐H, Random, 95% CI) | 1.35 [0.98, 1.86] |
| 6.2 TIO/OLO 2.5/5 | 2 | 579 | Odds Ratio (M‐H, Random, 95% CI) | 1.87 [1.30, 2.70] |
| 6.3 TIO/OLO 5/5 | 2 | 578 | Odds Ratio (M‐H, Random, 95% CI) | 2.35 [1.63, 3.40] |
| 6.4 UMEC/VI 62.5/25 | 3 | 1441 | Odds Ratio (M‐H, Random, 95% CI) | 1.70 [1.37, 2.12] |
| 6.5 UMEC/VI 125/25 | 2 | 954 | Odds Ratio (M‐H, Random, 95% CI) | 1.78 [1.35, 2.34] |
| 7 Difference vs placebo in adjusted trough FEV1 at EOT | 13 | Mean Difference (Random, 95% CI) | 0.20 [0.18, 0.21] | |
| 7.1 IND/GLY 110/50 | 2 | Mean Difference (Random, 95% CI) | 0.25 [0.20, 0.30] | |
| 7.2 UMEC/VI 125/25 | 4 | Mean Difference (Random, 95% CI) | 0.22 [0.18, 0.26] | |
| 7.3 UMEC/VI 62.5/25 | 6 | Mean Difference (Random, 95% CI) | 0.18 [0.15, 0.22] | |
| 7.4 TIO/OLO 2.5/5 | 3 | Mean Difference (Random, 95% CI) | 0.18 [0.15, 0.20] | |
| 7.5 TIO/OLO 5/5 | 3 | Mean Difference (Random, 95% CI) | 0.21 [0.18, 0.23] | |
| 7.6 ACLID/FORM 200/6 | 1 | Mean Difference (Random, 95% CI) | 0.07 [‐0.04, 0.18] | |
| 7.7 ACLID/FORM 200/12 | 1 | Mean Difference (Random, 95% CI) | 0.12 [0.01, 0.22] | |
| 7.8 ACLID/FORM 200/18 | 1 | Mean Difference (Random, 95% CI) | 0.08 [‐0.04, 0.19] | |
| 8 Difference vs placebo in trough FEV1 at EOT | 5 | Mean Difference (Random, 95% CI) | 0.18 [0.16, 0.20] | |
| 8.1 IND/GLY 110/50 | 3 | Mean Difference (Random, 95% CI) | 0.20 [0.17, 0.22] | |
| 8.2 TIO/OLO 2.5/5 | 2 | Mean Difference (Random, 95% CI) | 0.16 [0.13, 0.19] | |
| 8.3 TIO/OLO 5/5 | 2 | Mean Difference (Random, 95% CI) | 0.16 [0.13, 0.20] | |
| 9 Difference vs placebo in adjusted peak FEV1 | 7 | Mean Difference (Random, 95% CI) | 0.30 [0.28, 0.33] | |
| 9.1 IND/GLY 110/50 | 2 | Mean Difference (Random, 95% CI) | 0.35 [0.31, 0.39] | |
| 9.2 UMEC/VI 125/25 | 1 | Mean Difference (Random, 95% CI) | 0.28 [0.24, 0.32] | |
| 9.3 UMEC/VI 62.5/25 | 1 | Mean Difference (Random, 95% CI) | 0.22 [0.18, 0.27] | |
| 9.4 TIO/OLO 2.5/5 | 2 | Mean Difference (Random, 95% CI) | 0.29 [0.27, 0.32] | |
| 9.5 TIO/OLO 5/5 | 2 | Mean Difference (Random, 95% CI) | 0.33 [0.30, 0.35] | |
| 9.6 ACLID/FORM 200/6 | 1 | Mean Difference (Random, 95% CI) | 0.25 [0.13, 0.37] | |
| 9.7 ACLID/FORM 200/12 | 1 | Mean Difference (Random, 95% CI) | 0.31 [0.20, 0.43] | |
| 9.8 ACLID/FORM 200/18 | 1 | Mean Difference (Random, 95% CI) | 0.31 [0.19, 0.42] | |
| 10 AEs | 17 | 8235 | Odds Ratio (M‐H, Random, 95% CI) | 0.95 [0.86, 1.04] |
| 10.1 IND/GLY 110/50 | 6 | 2830 | Odds Ratio (M‐H, Random, 95% CI) | 0.90 [0.76, 1.07] |
| 10.2 TIO/OLO 2.5/5 | 4 | 1011 | Odds Ratio (M‐H, Random, 95% CI) | 0.85 [0.65, 1.11] |
| 10.3 TIO/OLO 5/5 | 4 | 1021 | Odds Ratio (M‐H, Random, 95% CI) | 0.78 [0.60, 1.01] |
| 10.4 UMEC/VI 62.5/25 | 5 | 1921 | Odds Ratio (M‐H, Random, 95% CI) | 1.07 [0.88, 1.29] |
| 10.5 UMEC/VI 125/25 | 4 | 1401 | Odds Ratio (M‐H, Random, 95% CI) | 1.08 [0.86, 1.34] |
| 10.6 UMEC/VI 500/25 | 1 | 51 | Odds Ratio (M‐H, Random, 95% CI) | 2.84 [0.32, 25.36] |
| 11 Difference vs placebo in trough FEV1 ‐ pooled adjusted and EOT analyses | 18 | Mean Difference (Random, 95% CI) | 0.20 [0.19, 0.21] | |
| 11.1 IND/GLY 110/50 | 5 | Mean Difference (Random, 95% CI) | 0.22 [0.19, 0.26] | |
| 11.2 UMEC/VI 125/25 | 4 | Mean Difference (Random, 95% CI) | 0.22 [0.18, 0.26] | |
| 11.3 UMEC/VI 62.5/25 | 6 | Mean Difference (Random, 95% CI) | 0.18 [0.15, 0.22] | |
| 11.4 TIO/OLO 2.5/5 | 5 | Mean Difference (Random, 95% CI) | 0.18 [0.15, 0.20] | |
| 11.5 TIO/OLO 5/5 | 5 | Mean Difference (Random, 95% CI) | 0.20 [0.18, 0.21] | |
| 11.6 ACLID/FORM 200/6 | 1 | Mean Difference (Random, 95% CI) | 0.07 [‐0.04, 0.18] | |
| 11.7 ACLID/FORM 200/12 | 1 | Mean Difference (Random, 95% CI) | 0.12 [0.01, 0.22] | |
| 11.8 ACLID/FORM 200/18 | 1 | Mean Difference (Random, 95% CI) | 0.08 [‐0.04, 0.19] |
4.1. Analysis.
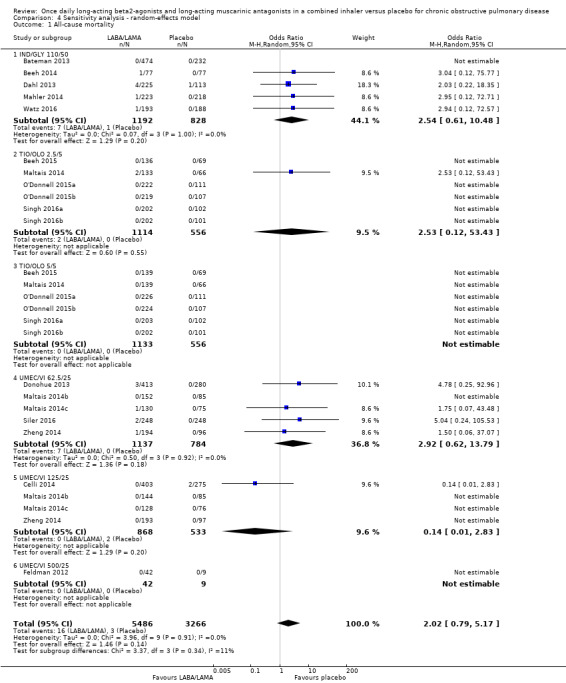
Comparison 4 Sensitivity analysis ‐ random‐effects model, Outcome 1 All‐cause mortality.
4.2. Analysis.
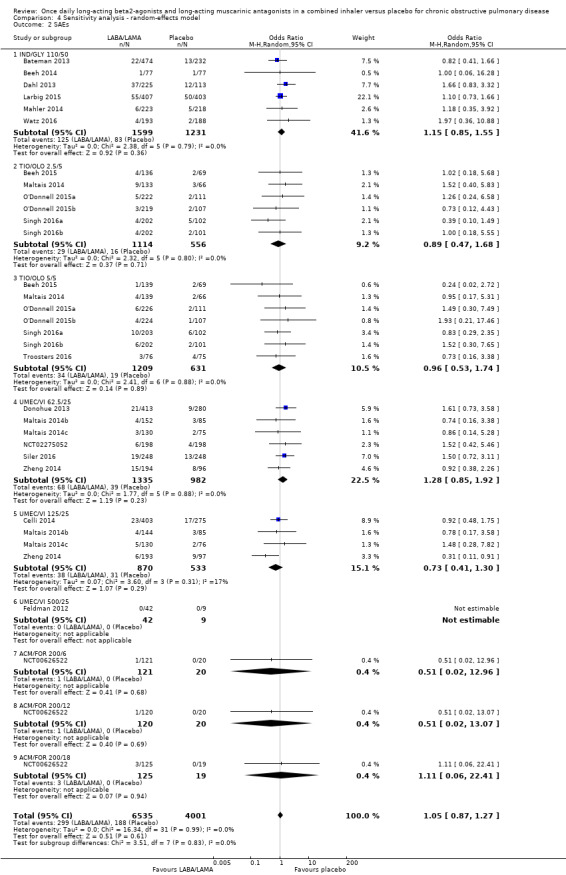
Comparison 4 Sensitivity analysis ‐ random‐effects model, Outcome 2 SAEs.
4.3. Analysis.
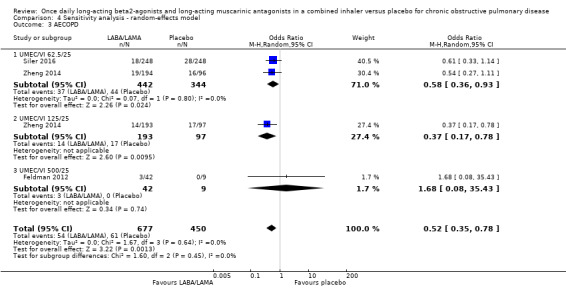
Comparison 4 Sensitivity analysis ‐ random‐effects model, Outcome 3 AECOPD.
4.4. Analysis.

Comparison 4 Sensitivity analysis ‐ random‐effects model, Outcome 4 Time to first AECOPD.
4.5. Analysis.
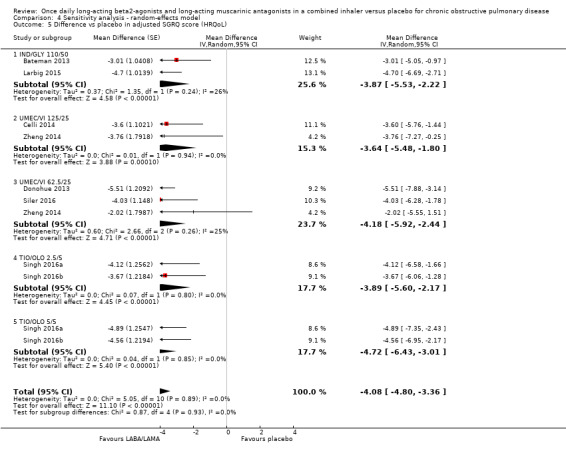
Comparison 4 Sensitivity analysis ‐ random‐effects model, Outcome 5 Difference vs placebo in adjusted SGRQ score (HRQoL).
4.6. Analysis.
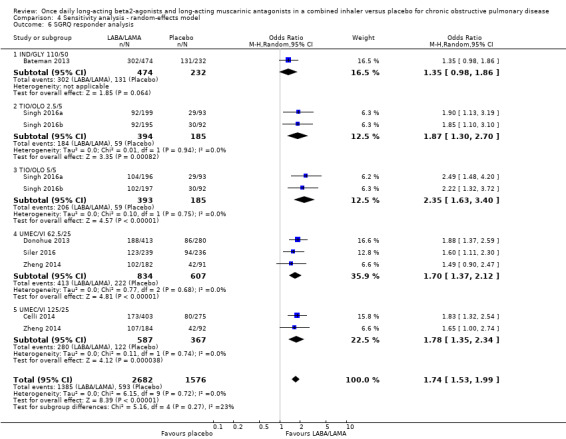
Comparison 4 Sensitivity analysis ‐ random‐effects model, Outcome 6 SGRQ responder analysis.
4.7. Analysis.
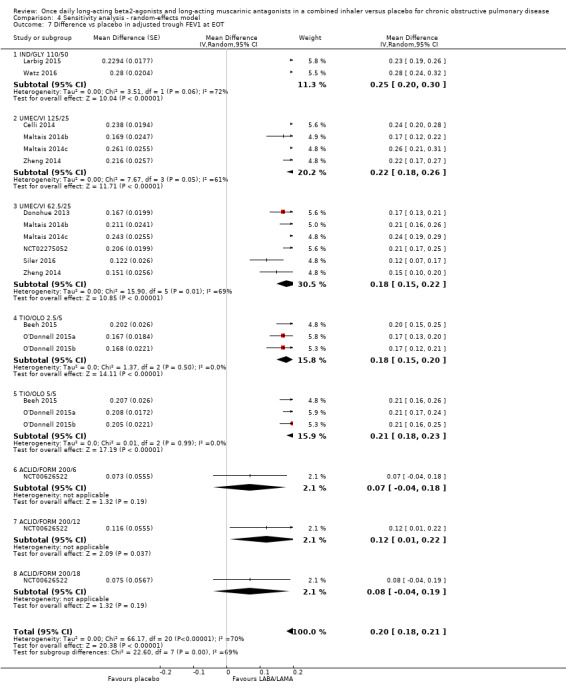
Comparison 4 Sensitivity analysis ‐ random‐effects model, Outcome 7 Difference vs placebo in adjusted trough FEV1 at EOT.
4.8. Analysis.
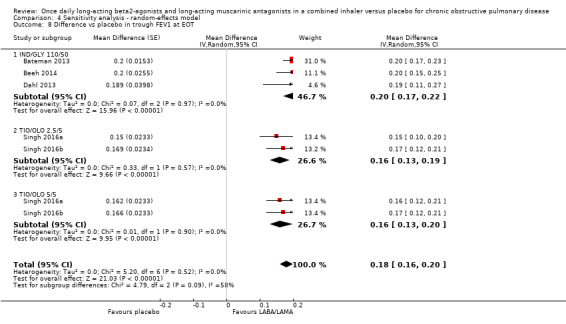
Comparison 4 Sensitivity analysis ‐ random‐effects model, Outcome 8 Difference vs placebo in trough FEV1 at EOT.
4.9. Analysis.
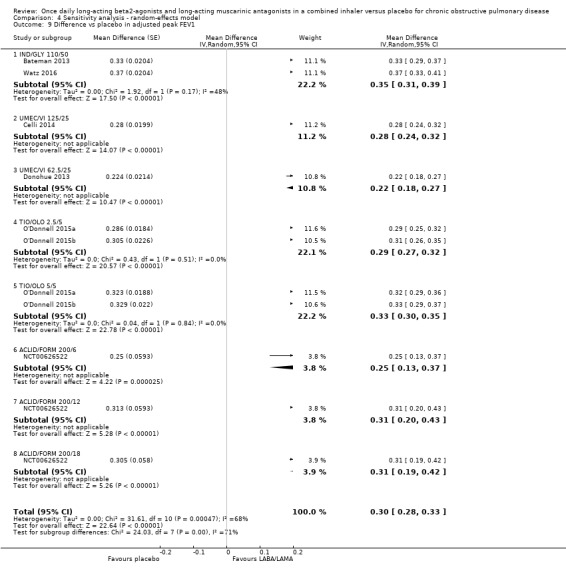
Comparison 4 Sensitivity analysis ‐ random‐effects model, Outcome 9 Difference vs placebo in adjusted peak FEV1.
4.10. Analysis.
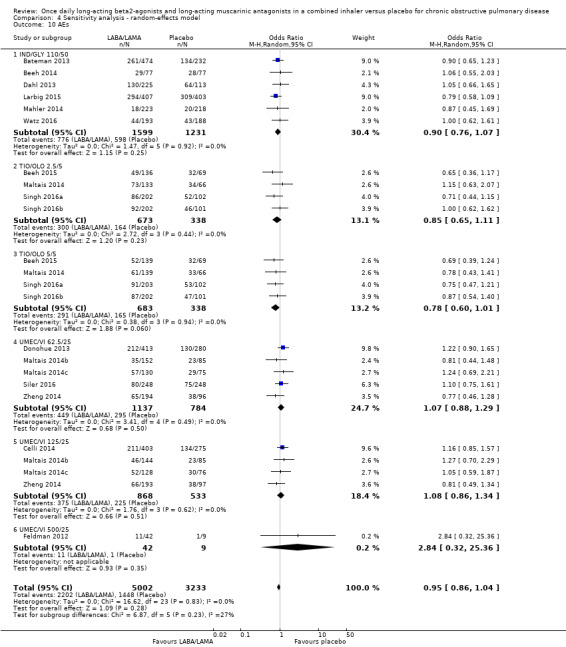
Comparison 4 Sensitivity analysis ‐ random‐effects model, Outcome 10 AEs.
4.11. Analysis.
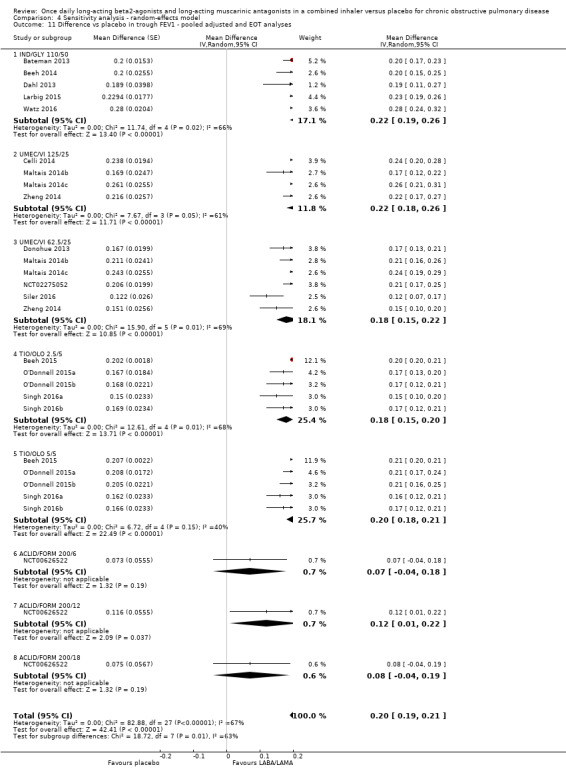
Comparison 4 Sensitivity analysis ‐ random‐effects model, Outcome 11 Difference vs placebo in trough FEV1 ‐ pooled adjusted and EOT analyses.
Comparison 5. Sensitivity analysis ‐ RoB.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 All‐cause mortality | 14 | 7287 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.65 [0.60, 4.50] |
| 1.1 IND/GLY 110/50 | 4 | 1682 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.97 [0.47, 18.97] |
| 1.2 TIO/OLO 2.5/5 | 6 | 1670 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.53 [0.12, 53.43] |
| 1.3 TIO/OLO 5/5 | 6 | 1689 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 1.4 UMEC/VI 62.5/25 | 3 | 1135 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.22 [0.38, 27.52] |
| 1.5 UMEC/VI 125/25 | 3 | 1111 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.01, 2.83] |
| 2 SAEs | 17 | 8448 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.86, 1.40] |
| 2.1 IND/GLY 110/50 | 5 | 2020 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.23 [0.79, 1.89] |
| 2.2 TIO/OLO 2.5/5 | 6 | 1670 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.49, 1.68] |
| 2.3 TIO/OLO 5/5 | 6 | 1689 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.54, 1.91] |
| 2.4 UMEC/VI 62.5/25 | 4 | 1531 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.34 [0.75, 2.40] |
| 2.5 UMEC/VI 125/25 | 3 | 1113 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.55, 1.66] |
| 2.6 ACM/FOR 200/6 | 1 | 141 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.51 [0.02, 12.96] |
| 2.7 ACM/FOR 200/12 | 1 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.51 [0.02, 13.07] |
| 2.8 ACM/FOR 200/18 | 1 | 144 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.06, 22.41] |
| 3 Time to first AECOPD | 2 | Hazard Ratio (Fixed, 95% CI) | 0.44 [0.31, 0.63] | |
| 3.1 UMEC/VI 125/25 | 2 | Hazard Ratio (Fixed, 95% CI) | 0.44 [0.31, 0.63] | |
| 4 Difference vs placebo in adjusted SGRQ score (HRQoL) | 5 | Mean Difference (Fixed, 95% CI) | ‐4.12 [‐4.99, ‐3.24] | |
| 4.1 IND/GLY 110/50 | 1 | Mean Difference (Fixed, 95% CI) | ‐3.01 [‐5.05, ‐0.97] | |
| 4.2 UMEC/VI 125/25 | 1 | Mean Difference (Fixed, 95% CI) | ‐3.60 [‐5.76, ‐1.44] | |
| 4.3 UMEC/VI 62.5/25 | 1 | Mean Difference (Fixed, 95% CI) | ‐5.51 [‐7.88, ‐3.14] | |
| 4.4 TIO/OLO 2.5/5 | 2 | Mean Difference (Fixed, 95% CI) | ‐3.89 [‐5.60, ‐2.17] | |
| 4.5 TIO/OLO 5/5 | 2 | Mean Difference (Fixed, 95% CI) | ‐4.72 [‐6.43, ‐3.01] | |
| 5 SGRQ responder analysis | 5 | 3234 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.81 [1.56, 2.10] |
| 5.1 IND/GLY 110/50 | 1 | 706 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.35 [0.98, 1.86] |
| 5.2 TIO/OLO 2.5/5 | 2 | 579 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.87 [1.30, 2.70] |
| 5.3 TIO/OLO 5/5 | 2 | 578 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.35 [1.63, 3.40] |
| 5.4 UMEC/VI 62.5/25 | 1 | 693 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.88 [1.37, 2.59] |
| 5.5 UMEC/VI 125/25 | 1 | 678 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.83 [1.32, 2.54] |
| 6 Difference vs placebo in adjusted trough FEV1 at EOT | 10 | Mean Difference (Fixed, 95% CI) | 0.20 [0.19, 0.22] | |
| 6.1 IND/GLY 110/50 | 1 | Mean Difference (Fixed, 95% CI) | 0.28 [0.24, 0.32] | |
| 6.2 UMEC/VI 125/25 | 3 | Mean Difference (Fixed, 95% CI) | 0.22 [0.20, 0.25] | |
| 6.3 UMEC/VI 62.5/25 | 4 | Mean Difference (Fixed, 95% CI) | 0.20 [0.18, 0.22] | |
| 6.4 TIO/OLO 2.5/5 | 3 | Mean Difference (Fixed, 95% CI) | 0.18 [0.15, 0.20] | |
| 6.5 TIO/OLO 5/5 | 3 | Mean Difference (Fixed, 95% CI) | 0.21 [0.18, 0.23] | |
| 6.6 ACLID/FORM 200/6 | 1 | Mean Difference (Fixed, 95% CI) | 0.07 [‐0.04, 0.18] | |
| 6.7 ACLID/FORM 200/12 | 1 | Mean Difference (Fixed, 95% CI) | 0.12 [0.01, 0.22] | |
| 6.8 ACLID/FORM 200/18 | 1 | Mean Difference (Fixed, 95% CI) | 0.08 [‐0.04, 0.19] | |
| 7 Difference vs placebo in trough FEV1 at EOT | 4 | Mean Difference (Fixed, 95% CI) | 0.18 [0.16, 0.20] | |
| 7.1 IND/GLY 110/50 | 2 | Mean Difference (Fixed, 95% CI) | 0.2 [0.17, 0.23] | |
| 7.2 TIO/OLO 2.5/5 | 2 | Mean Difference (Fixed, 95% CI) | 0.16 [0.13, 0.19] | |
| 7.3 TIO/OLO 5/5 | 2 | Mean Difference (Fixed, 95% CI) | 0.16 [0.13, 0.20] | |
| 8 Difference vs placebo in adjusted peak FEV1 | 7 | Mean Difference (Fixed, 95% CI) | 0.31 [0.29, 0.32] | |
| 8.1 IND/GLY 110/50 | 2 | Mean Difference (Fixed, 95% CI) | 0.35 [0.32, 0.38] | |
| 8.2 UMEC/VI 125/25 | 1 | Mean Difference (Fixed, 95% CI) | 0.28 [0.24, 0.32] | |
| 8.3 UMEC/VI 62.5/25 | 1 | Mean Difference (Fixed, 95% CI) | 0.22 [0.18, 0.27] | |
| 8.4 TIO/OLO 2.5/5 | 2 | Mean Difference (Fixed, 95% CI) | 0.29 [0.27, 0.32] | |
| 8.5 TIO/OLO 5/5 | 2 | Mean Difference (Fixed, 95% CI) | 0.33 [0.30, 0.35] | |
| 8.6 ACLID/FORM 200/6 | 1 | Mean Difference (Fixed, 95% CI) | 0.25 [0.13, 0.37] | |
| 8.7 ACLID/FORM 200/12 | 1 | Mean Difference (Fixed, 95% CI) | 0.31 [0.20, 0.43] | |
| 8.8 ACLID/FORM 200/18 | 1 | Mean Difference (Fixed, 95% CI) | 0.31 [0.19, 0.42] | |
| 9 AEs | 11 | 5579 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.86, 1.08] |
| 9.1 IND/GLY 110/50 | 3 | 1301 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.70, 1.19] |
| 9.2 TIO/OLO 2.5/5 | 4 | 1011 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.66, 1.11] |
| 9.3 TIO/OLO 5/5 | 4 | 1021 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.60, 1.01] |
| 9.4 UMEC/VI 62.5/25 | 3 | 1135 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.89, 1.46] |
| 9.5 UMEC/VI 125/25 | 3 | 1111 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.90, 1.48] |
| 10 Difference vs placebo in trough FEV1 ‐ pooled adjusted and EOT analyses | 14 | Mean Difference (Fixed, 95% CI) | 0.20 [0.20, 0.21] | |
| 10.1 IND/GLY 110/50 | 3 | Mean Difference (Fixed, 95% CI) | 0.22 [0.20, 0.25] | |
| 10.2 UMEC/VI 125/25 | 3 | Mean Difference (Fixed, 95% CI) | 0.22 [0.20, 0.25] | |
| 10.3 UMEC/VI 62.5/25 | 4 | Mean Difference (Fixed, 95% CI) | 0.20 [0.18, 0.22] | |
| 10.4 TIO/OLO 2.5/5 | 5 | Mean Difference (Fixed, 95% CI) | 0.20 [0.20, 0.20] | |
| 10.5 TIO/OLO 5/5 | 5 | Mean Difference (Fixed, 95% CI) | 0.21 [0.20, 0.21] | |
| 10.6 ACLID/FORM 200/6 | 1 | Mean Difference (Fixed, 95% CI) | 0.07 [‐0.04, 0.18] | |
| 10.7 ACLID/FORM 200/12 | 1 | Mean Difference (Fixed, 95% CI) | 0.12 [0.01, 0.22] | |
| 10.8 ACLID/FORM 200/18 | 1 | Mean Difference (Fixed, 95% CI) | 0.08 [‐0.04, 0.19] |
5.1. Analysis.
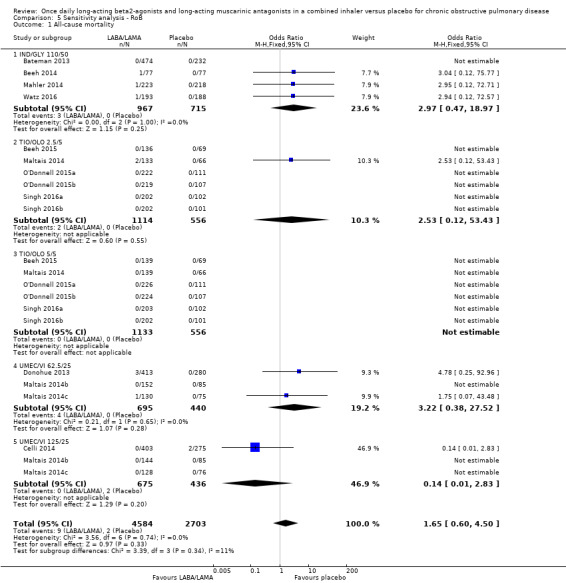
Comparison 5 Sensitivity analysis ‐ RoB, Outcome 1 All‐cause mortality.
5.2. Analysis.
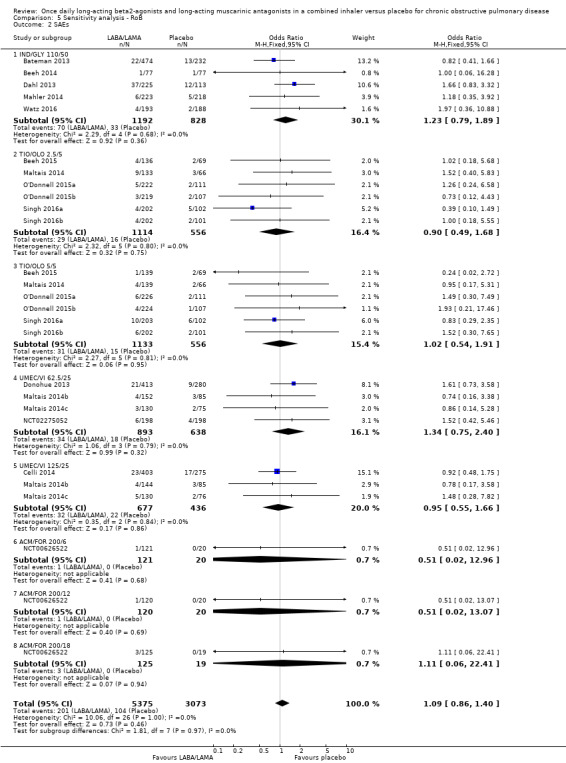
Comparison 5 Sensitivity analysis ‐ RoB, Outcome 2 SAEs.
5.3. Analysis.
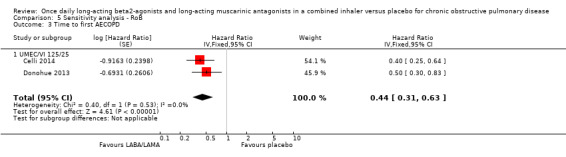
Comparison 5 Sensitivity analysis ‐ RoB, Outcome 3 Time to first AECOPD.
5.4. Analysis.
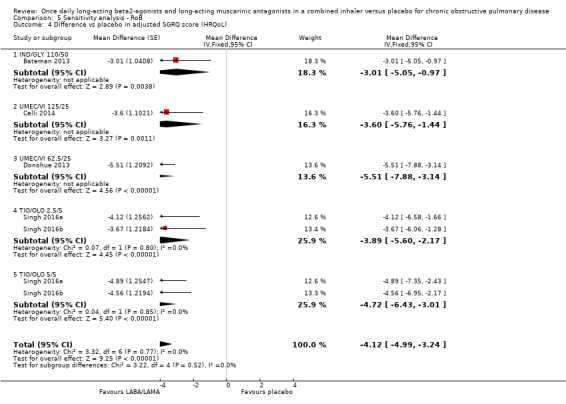
Comparison 5 Sensitivity analysis ‐ RoB, Outcome 4 Difference vs placebo in adjusted SGRQ score (HRQoL).
5.5. Analysis.
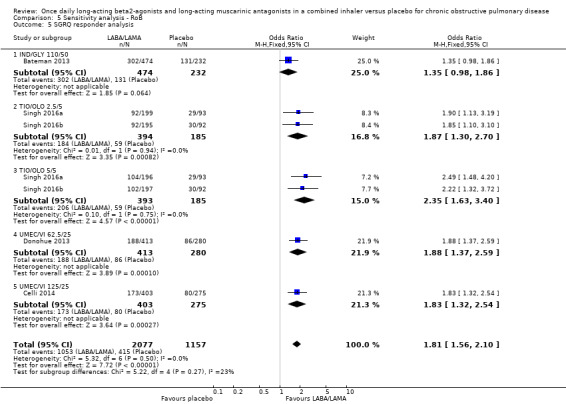
Comparison 5 Sensitivity analysis ‐ RoB, Outcome 5 SGRQ responder analysis.
5.6. Analysis.
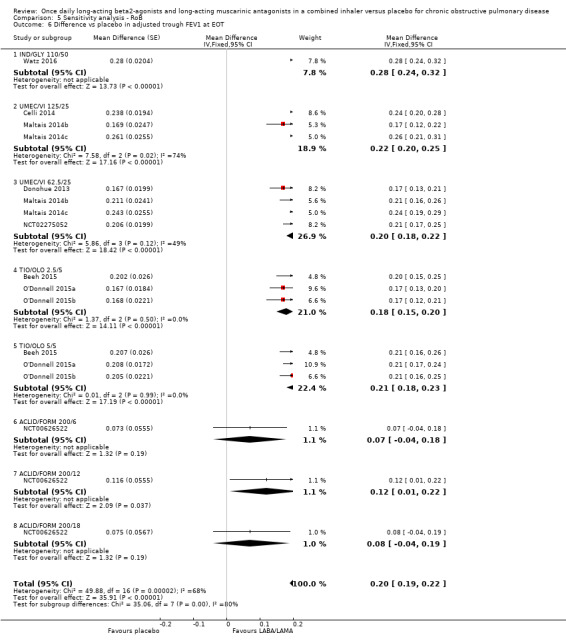
Comparison 5 Sensitivity analysis ‐ RoB, Outcome 6 Difference vs placebo in adjusted trough FEV1 at EOT.
5.7. Analysis.
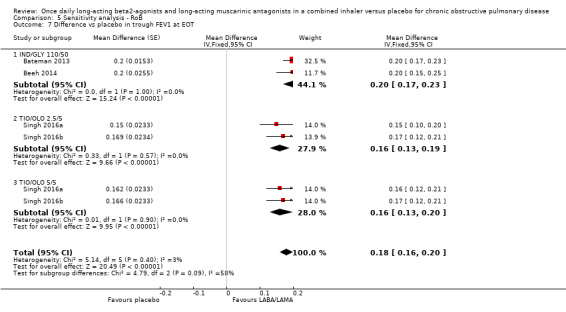
Comparison 5 Sensitivity analysis ‐ RoB, Outcome 7 Difference vs placebo in trough FEV1 at EOT.
5.8. Analysis.
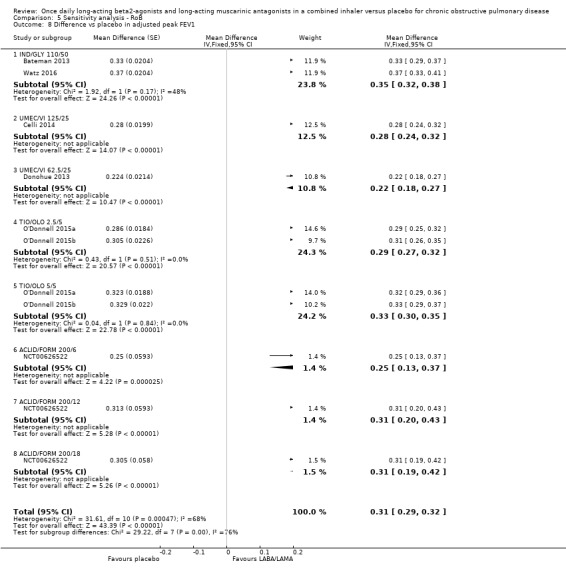
Comparison 5 Sensitivity analysis ‐ RoB, Outcome 8 Difference vs placebo in adjusted peak FEV1.
5.9. Analysis.
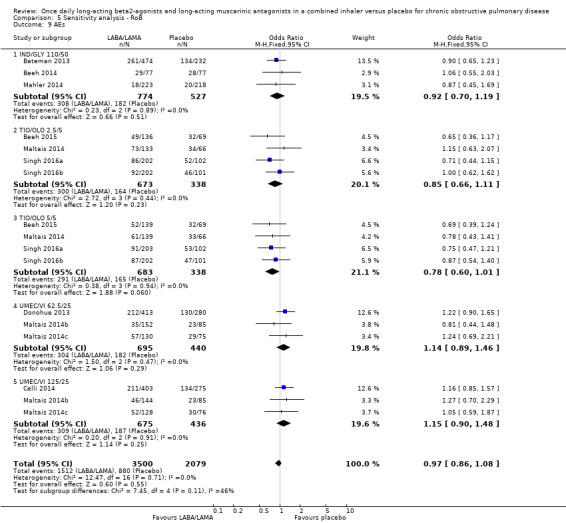
Comparison 5 Sensitivity analysis ‐ RoB, Outcome 9 AEs.
5.10. Analysis.
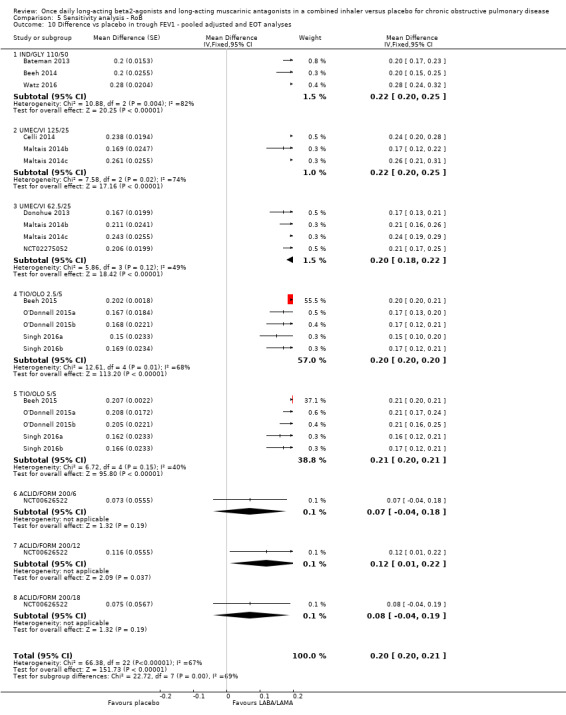
Comparison 5 Sensitivity analysis ‐ RoB, Outcome 10 Difference vs placebo in trough FEV1 ‐ pooled adjusted and EOT analyses.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bateman 2013.
| Methods |
Study ID and dates performed: NCT01202188 (SHINE); September 2010 to February 2012. Study design: Randomised, double‐blind, parallel‐group, placebo and active controlled study. Duration of study: Pre‐randomisation period (pre‐screening + run‐in): 3 weeks; treatment period: 26 weeks. Study setting, location, number of centres: 166 academic and clinical research centres in Europe, North America, South America, Asia (Philippines, Japan, India), Australia, China, Taiwan. and South Africa. Key inclusion criteria: Adults aged ≥ 40 years; signed consent; symptomatic moderate‐to‐severe stable COPD (GOLD 2008 Stage II or III); current or ex‐smokers (≥ 10 pack years); post‐BD FEV1 ≥ 30% and < 80% predicted normal AND post‐BD FEV1/FVC < 0.7 at visit 2 (day 14). Key exclusion criteria: Pregnant or women of child bearing potential; concomitant pulmonary disease; history of asthma; lung cancer or history of lung cancer; history of long QT syndrome; Type I or uncontrolled Type II diabetes; contraindication to or hypersensitive to anticholinergics, LABA, sympathomimetic amines, or lactose. Concomitant medications: Permitted: SSRI stable regimen ≥ 1 month prior to screening or during study; inactivated vaccine (not within 48 hours of study visit); ICS (constant doses and dose regimens of ≥ 1 month); H1 antagonists (constant doses and dose regimens). Excluded: long term O2 therapy. |
|
| Participants |
N randomised: IND/GLY 110/50 µg: 475; placebo: 234. N analysed: IND/GLY 110/50 µg: 474; placebo: 232. Mean age (SD), years: IND/GLY 110/50 µg: 64.0 (8.9); placebo: 64.4 (8.6). Gender male, n/N (%): IND/GLY 110/50 µg: 362/474 (76.4); placebo: 169/232 (72.8). Baseline lung function – mean (SD) post‐BD % predicted FEV1, %: IND/GLY 110/50 µg: 55.7 (13.2); placebo: 55.2 (12.7). Smoking status (current), n/N (%): IND/GLY 110/50 µg: 192/474 (40.5); placebo: 93/232 (40.1). |
|
| Interventions |
Intervention: Once‐daily QVA149 (IND/GLY 110/50 µg). Comparator: Once‐daily placebo. |
|
| Outcomes |
Prespecified outcomes:Primary: Trough FEV1 at week 26. Secondary: SGRQ score (week 26); SGRQ score week 12 and week 26; and number of participants with a MCID (4 units) improvement from baseline in SGRQ score (week 26); rate of moderate or severe COPD exacerbation; percentage of participants with ≥ 1 moderate or severe COPD exacerbation (26 weeks); AEs; SAEs. Reported outcomes: all prespecified outcomes reported. |
|
| Notes | Funding for trial; notable author COIs: The study was funded by Novartis Pharma AG. Authors were employed by Novartis or had received remuneration from Novartis for advisory boards/lectures. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Eligible participants were assigned a randomisation number via IRT system, linking the patient to a treatment arm and specific unique medication number for the study drug. The randomisation number was not communicated to the investigator contacting the IRT. |
| Allocation concealment (selection bias) | Low risk | See random sequence generation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinding of participants, investigator staff, personnel performing assessments and data analysts was maintained by ensuring randomisation data remained strictly confidential and inaccessible to anyone involved in the study until the time of unblinding. In addition, the identity of the treatments was concealed by the use of study drugs that were all identical in packaging, labelling, and schedule of administration, appearance, taste, and odour. |
| Blinding of participants and personnel (objective outcomes ‐ performance bias) Objective outcomes | Low risk | Knowledge of treatment allocation by participant or personnel would be unlikely to influence objective outcomes. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding of personnel performing assessments and data analysts was maintained by ensuring randomisation data remained strictly confidential and inaccessible to anyone involved in the study until the time of unblinding. In addition, the identity of the treatments was concealed by the use of study drugs that were all identical in packaging, labelling, and schedule of administration, appearance, taste, and odour. |
| Blinding of outcome assessment (objective outcomes ‐ detection bias) All outcomes | Low risk | Assessment of objective outcomes would be unlikely to be influenced by knowledge of treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition was low and consistent between treatment arms. |
| Selective reporting (reporting bias) | Low risk | Comparison of trial registration and published report information performed. Prespecified outcomes were well reported. |
| Other bias | Low risk | None identified. |
Beeh 2014.
| Methods |
Study ID and dates performed: NCT01294787 (BRIGHT); study dates not reported. Study design: Multicentre, randomised, double‐blind, double‐dummy, placebo‐controlled, three‐period cross‐over study. Duration of study: 18–25 day run‐in period; 3 x 3‐week treatment period, 21‐day washout period between treatments. Study setting, location, number of centres: Not reported. Key inclusion criteria: Participants ≥ 40 years of age; moderate‐to‐severe COPD (Stage II or III according to GOLD 2008 criteria); smoking history of ≥ 10 pack years (current or ex‐smokers); post‐bronchodilator FEV1 of ≥ 40% and < 70%. Key exclusion criteria: Pregnant women or nursing mothers; women of child‐bearing potential; contraindication for treatment with, or having a history of reactions/ hypersensitivity to any of the following inhaled drugs or drugs of a similar class: anticholinergic agents, long and short acting beta‐2 agonists, sympathomimetic amines, lactose or any of the other excipients; a history of long QT syndrome or whose QTc measured at screening (Fridericia method) is prolonged (> 450 ms for males and females) as confirmed by the central ECG assessor; a clinically significant abnormality on the screening ECG; Type I or uncontrolled Type II diabetes; Wmax value < 20 W (as determined by the incremental cycle endurance test) at visit 2); body mass index < 15 or > 40 kg/m2; contraindication to cardiopulmonary exercise testing; resting (5 min) oxygen SaO2 saturation on room air of < 85%; participants who do not maintain regular day/night, waking/sleeping cycles (e.g. night shift workers); participants whose endurance in the exercise test is limited by non‐respiratory conditions e.g. by neurologic, orthopaedic, or other disorders, narrow‐angle glaucoma, symptomatic prostatic hyperplasia or bladder‐neck obstruction or moderate to severe renal impairment or urinary retention; a history of malignancy of any organ system (including lung cancer); clinically relevant laboratory abnormality or a clinically significant condition. Concomitant medications: Short‐acting bronchodilators (salbutamol or albuterol) were provided for rescue use throughout the study but were not permitted within 6 hours of each visit. Prior ICS use permitted. |
|
| Participants |
Note: cross‐over study therefore participant data reported for whole cohort N randomised: 85 N analysed: 77 Mean age (SD), years: 62.1 (8.11) Gender ‐ male, n/N (%): 53/84 (63.1) Baseline lung function ‐ post‐bronchodilator % predicted FEV1: 46.5 (10.30) Smoking status, current smoker, n/N (%): 45 (53.6) |
|
| Interventions |
Intervention: Once‐daily IND/GLY (QVA149) 110/50 µg Comparator: Once‐daily placebo |
|
| Outcomes |
Prespecified outcomes:Primary: exercise tolerance comparison between QVA149 and placebo groups at 3 weeks. Secondary (all QVA149 vs placebo): dynamic inspiratory capacity at 3 weeks; trough 24‐hour post‐dose inspiratory capacity at 3 weeks; trough 24‐hour post‐dose FEV1 at 3 weeks; residual volume, slow vital capacity, specific airway conductance and functional residual capacity, each on day 1 and day 21, at 5 min and 15 min post‐dose as determined by body plethysmography; dynamic inspiratory capacity post‐dose pre‐exercise after three weeks of treatment; exertional dyspnoea (Borg CR10 Scale) at 3 weeks; leg discomfort (Borg CR10 Scale) during submaximal constant load cycle ergometry test after three weeks treatment; exercise endurance time during submaximal constant load cycle ergometry test cycle exercise test on day 1. Reported outcomes: prespecified outcomes well reported. |
|
| Notes | Funding for trial; notable author COIs: Novartis Pharma AG funded this study. All authors had relevant conflicts of interest relating to funding or employment provided by Novartis Pharma AG. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | IRT system was used to assign randomisation. |
| Allocation concealment (selection bias) | Low risk | IRT system was used to assign randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Personal communication from Dr Beeh (7 August 2018) confirmed that personnel and participants were blinded to treatment. |
| Blinding of participants and personnel (objective outcomes ‐ performance bias) Objective outcomes | Low risk | Personal communication from Dr Beeh (7 August 2018) confirmed that personnel and participants were blinded to treatment. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Personal communication from Dr Beeh (7 August 2018) confirmed that outcome assessors were blinded to treatment. |
| Blinding of outcome assessment (objective outcomes ‐ detection bias) All outcomes | Low risk | Personal communication from Dr Beeh (7 August 2018) confirmed that outcome assessors were blinded to treatment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropout rate was low (˜15%) and similar between QVA149 and placebo arms. |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes were generally well reported. |
| Other bias | Low risk | Safety results were analysed according to treatment received. No other issues identified. |
Beeh 2015.
| Methods |
Study ID and dates performed: NCT01559116; VIVACITO; dates not reported. Study design: Double‐blind, placebo‐controlled, multicentre, Phase III, incomplete cross‐over study. Duration of study: 38–42 weeks including 2–6 week run‐in period. Each treatment given for 6 weeks. Study setting, location, number of centres: 29 centres in seven countries (Belgium, Canada, Denmark, Germany, Hungary, The Netherlands, and the USA). Key inclusion criteria: A diagnosis of COPD; aged ≥ 40 years; smoking history of ≥ 10 pack‐years; relatively stable airway obstruction with a post‐bronchodilator FEV1 < 80% of predicted normal (in German sites only, FEV1 ≥ 30%) and FEV1/FVC < 70% of predicted normal. Key exclusion criteria: History of asthma or significant disease other than COPD; unstable or life‐threatening cardiac arrhythmia; hospitalisation for heart failure within the past year; history of myocardial infarction within 1 year of screening or a history of life‐threatening pulmonary obstruction. Concomitant medications: Participants could continue on inhaled corticosteroids during treatment periods (if taken as maintenance treatment at study entry) but not anticholinergics or LABAs. Short‐acting anticholinergics were permitted during screening and the washout periods, but had to be stopped 8 h before pulmonary function test at the first visit of the next treatment period. LAMAs and LABAs were not permitted during washout or screening periods. Open‐label salbutamol was provided to participants as rescue medication to be used at baseline and during screening, treatment, washout, and follow‐up periods. |
|
| Participants |
Note: incomplete cross‐over study therefore participant data reported for whole cohort N randomised: N = 219 N analysed: TIO/OLO 2.5/5 µg: n = 135 ; TIO/OLO 5/5 µg: n = 138 ; placebo: n = 130 Mean age (SD), years: 61.1 (7.7) Gender ‐ male, n/N (%): 129 (58.9%) Baseline lung function ‐ mean (SD) pre‐bronchodilator FEV1, L: 1.361 (0.471) Smoking status, current smoker, n/N (%): 137/219 (62.6) |
|
| Interventions |
Intervention: Once‐daily TIO/OLO 2.5/5 µg; once‐daily TIO/OLO 5/5 µg. Comparator: Once‐daily placebo. |
|
| Outcomes |
Prespecified outcomes: Primary: FEV1 AUC 0‐24 h response after 6 weeks treatment.Secondary (each after 6 weeks of treatment): FEV1 AUC 0‐12h response; FEV1 AUC 12‐24 h response; trough FEV1 response; peak (0‐3 h) FEV1 response; FVC AUC 0‐24 h response; FVC AUC 0‐12 h response; FVC AUC 12‐24h response; trough FVC response; peak (0‐3h) FVC response; safety. Reported outcomes: All prespecified outcomes were reported, plus functional residual capacity, residual volume, inspiratory capacity, and total lung capacity. |
|
| Notes | Funding for trial; notable author COIs: This study was funded by Boehringer Ingelheim Pharma GmbH & co. KG. Three of seven authors were employees of the company that funded the study; the remaining authors received no compensation in relation to development of the manuscript (no COIs provided) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information provided. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information provided (‘double‐blind’ stated but did not specify participant, personnel or outcome assessor). |
| Blinding of participants and personnel (objective outcomes ‐ performance bias) Objective outcomes | Low risk | Knowledge of treatment allocation by participant or personnel would be unlikely to influence objective outcomes. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information provided (‘double‐blind’ stated but did not specify participant, personnel or outcome assessor). |
| Blinding of outcome assessment (objective outcomes ‐ detection bias) All outcomes | Low risk | Assessment of objective outcomes would be unlikely to be influenced by knowledge of treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition was low and consistent between the groups, with the highest group dropout acknowledged as 5.8%. |
| Selective reporting (reporting bias) | Low risk | Comparison of trial registration and published report information performed. Prespecified outcomes were well reported. |
| Other bias | Low risk | None identified. |
Celli 2014.
| Methods |
Study ID and dates performed: NCT01313637; March 22, 2011 to April 19, 2012. Study design: Multicentre, randomised, placebo‐controlled, double‐blind, parallel‐group study. Duration of study: 24 weeks. Study setting, location, number of centres: 153 centres in 14 countries. Key inclusion criteria: Aged ≥ 40 years; history of COPD (ATS/ERS); current or former smoker with a history of ≥10 pack‐years; post‐albuterol (salbutamol) FEV1/FVC ratio < 0.70, FEV1 ≤ 70% predicted normal; a score of ≥ 2 on modified MRC dyspnoea scale at screening. Key exclusion criteria: Not reported. Concomitant medications: Not reported. |
|
| Participants |
N randomised: UMEC/VI 125/25 µg: n = 403 ; placebo: n = 275. N analysed: UMEC/VI 125/25 µg: 403/403 (100; ITT) ; placebo: 275/275 (100; ITT). Mean age (SD), years: UMEC/VI 125/25 µg: 63.4 (8.08); placebo: 62.2 (8.53). Gender ‐ male, n/N (%): UMEC/VI 125/25 µg: 264/403 (66); placebo: 175/275 (64). Baseline lung function ‐ mean (SD) post‐bronchodilator % predicted FEV1: UMEC/VI 125/25 µg: 47.7 (12.53); placebo: 47.6 (12.47). Smoking status, current smoker, n/N (%): UMEC/VI 125/25 µg: 200/403 (50); placebo: 143/275 (52). |
|
| Interventions |
Intervention: Once‐daily UMEC/VI 125/25 µg. Comparator: Once‐daily placebo. |
|
| Outcomes |
Prespecified outcomes: Primary: Change from baseline in trough FEV1 on day 169 (week 24). Secondary: Mean transition dyspnoea index focal score at day 168 (week 24); change from baseline in weighted mean 0‐6 hour FEV1 obtained post‐dose at day 168; safety. Other: change from baseline in the mean Shortness of Breath with Daily Activities score for week 24. Reported outcomes: All prespecified outcomes were reported, plus: the proportion of participants achieving an increase in FEV1 of ≥ 12% and ≥ 0.200 L above baseline at any time during 0–6 hours post‐dose on day 1; the proportion of participants achieving an increase of ≥ 0.100 L above baseline in trough FEV1, LSM peak FEV1, serial FEV1 and serial and trough FVC; SGRQ score and time to first COPD exacerbation. Serial FVC 0–24 h post‐dose was obtained in a subset of participants. |
|
| Notes | Funding for trial; notable author COIs: GSK funded the design/conduct of the study and manuscript development. All authors had received funding from, or were past or present employees, of GSK. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information provided. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Masking quadruple (participants, care provider, investigator, outcome assessor). |
| Blinding of participants and personnel (objective outcomes ‐ performance bias) Objective outcomes | Low risk | Knowledge of treatment allocation by participant or personnel would be unlikely to influence objective outcomes. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Masking quadruple (participants, care provider, investigator, outcome assessor). |
| Blinding of outcome assessment (objective outcomes ‐ detection bias) All outcomes | Low risk | Assessment of objective outcomes would be unlikely to be influenced by knowledge of treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Withdrawal rates were high and unbalanced between groups; however, ITT analysis performed. |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes (clinicaltrials.gov) were well reported. |
| Other bias | Low risk | None identified. |
Dahl 2013.
| Methods |
Study ID and dates performed: NCT01120717 (ENLIGHTEN); study dates not reported. Study design: Multicentre, randomised, double‐blind, parallel‐group, placebo‐controlled study. Duration of study: 55 weeks (7‐day pre‐screening; 14‐day run‐in period; 52‐week treatment period). Study setting, location, number of centres: Academic and clinical research centres in Europe, Canada, Asia (India, Korea) and South Africa. Key inclusion criteria: Aged ≥ 40 years of age; moderate‐to‐severe COPD (Stage II or III according to the GOLD 2008 criteria); smoking history of ≥ 10 pack‐years; post‐bronchodilator FEV1 of ≥ 30% and < 80% of the predicted normal and post‐bronchodilator FEV1/FVC < 0.70 at screening; total daily symptom score of ≥ 1 (obtained by adding the scores for the morning and evening symptoms (i.e. cough, wheezing, sputum production/colour, shortness of breath)) on 4 of the last 7 days prior to randomisation. Key exclusion criteria: COPD exacerbations that required treatment with antibiotics or oral steroids or hospitalisation in the 6 weeks prior to screening or between screening and randomisation; respiratory tract infection 4 weeks before or during screening; history of asthma; a clinically significant ECG abnormality. Concomitant medications: No participants received placebo treatment in isolation at any time during the study: placebo was included in participants’ established background COPD therapy (e.g. daily ICS). The short‐acting bronchodilator salbutamol (albuterol) was provided for rescue use throughout the study. participants were not permitted to use short‐acting (LAMAs, LABAs, theophylline) before the screening period (for at least 7 days for LAMAs and theophylline; 48 h for LABA and LABA/ICS combinations) or during the study. ICS use was maintained (i.e. participants taking combined LABA/ICS at screening were transitioned to the equivalent ICS monotherapy). |
|
| Participants |
N randomised: IND/GLY 110/50 µg: n = 226; placebo: n = 113. N analysed, n/N (%): IND/GLY 110/50 µg: 194/226 (85.8); placebo: 89/113 (78.8). Mean age (SD), years: IND/GLY 110/50 µg: 62.5 (8.81); placebo: 62.9 (8.14). Gender ‐ male, n/N (%): IND/GLY 110/50 µg: 174/225 (77.3); placebo: 86/113 (76.1). Baseline lung function ‐ post‐bronchodilator % predicted FEV1: IND/GLY 110/50 µg: 56.39 (13.27); placebo: 59.43 (12.50). Smoking status, current smoker, n/N (%): IND/GLY 110/50 µg: 102/225 (45.3); placebo: 51/113 (45.1). |
|
| Interventions |
Intervention: Once‐daily IND/GLY 110/50 µg. Comparator: Once‐daily placebo. |
|
| Outcomes |
Prespecified outcomes: Primary: Number of participants with AEs, SAEs or death. Secondary: Pre‐dose FEV1 at 52 weeks; number of participants with newly occurring or worsening clinically notable haematology values at any time point over the whole treatment period; number of participants with newly occurring or worsening clinically notable biochemistry values at any time point over the whole treatment period; number of participants with newly occurring or worsening clinically notable vital signs values at any time point over the whole treatment period; number of participants with notable change from baseline in Fridericia's QTc values at any time point over the whole treatment period. Reported outcomes: All prespecified outcomes (see above) reported. |
|
| Notes | Funding for trial; notable author COIs: The study was funded by Novartis Pharma AG. All authors had previously received funding/compensation from Novartis or were employees of Novartis. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information provided. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Masking quadruple (participants, care provider, investigator, outcome assessor). |
| Blinding of participants and personnel (objective outcomes ‐ performance bias) Objective outcomes | Low risk | Knowledge of treatment allocation by participant or personnel would be unlikely to influence objective outcomes. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Masking quadruple (participants, care provider, investigator, outcome assessor). |
| Blinding of outcome assessment (objective outcomes ‐ detection bias) All outcomes | Low risk | Assessment of objective outcomes would be unlikely to be influenced by knowledge of treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | High rates of attrition (> 20%) noted in placebo arm (versus < 15% in QVA149 arm). mITT analysis performed based on full analysis set (all participants randomised to treatment and who received at least one dose). |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes (clinicaltrials.gov) were well reported. |
| Other bias | High risk | More participants in the QVA149 group had severe COPD versus those in the placebo group (note: this would likely skew treatment effect in favour of placebo). |
Donohue 2013.
| Methods |
Study ID and dates performed: NCT01313650. 30 March 2011 to 5 April 2012. Study design: Randomised, double‐blind, placebo‐controlled, multicentre, parallel‐group study. Duration of study: 24 weeks. Study setting, location, number of centres: 163 centres in 13 countries. Key inclusion criteria: Current or former cigarette smokers; aged > 40 years; clinically established history of COPD characterised by airflow limitation that is not fully reversible (ATS/ERS criteria) and documented based on a smoking history of ≥ 10 pack years; post‐salbutamol FEV1/FVC ratio < 0.70; post‐salbutamol FEV1 of ≤ 70% of predicted normal values; dyspnoea score ≥ 2 on modified MRC dyspnoea scale. Key exclusion criteria: Current diagnosis of asthma or other known respiratory disorder; abnormal or clinically significant electrocardiogram or 24‐hour Holter ECG; clinically significant clinical laboratory finding. Concomitant medications: Permitted: Inhaled salbutamol as rescue medication; ICS at a stable dose of ≤ 1000 µg/day fluticasone propionate or equivalent from 30 days prior to screening onward. |
|
| Participants |
N randomised: UMEC/VI 62.5/25 µg: N = 413; placebo: N = 280. N analysed, n/N (%): UMEC/VI 62.5/25 µg: 413/413 (100); placebo: 280/280 (100). Mean age (SD), years: UMEC/VI 62.5/25 µg: 63.1 (8.71); placebo: 62.2 (9.04). Gender ‐ male, n/N (%): UMEC/VI 62.5/25 µg: 305/413 (74); placebo: 195/280 (70). Baseline lung function ‐ mean (SD) post‐bronchodilator % predicted FEV1: UMEC/VI 62.5/25 µg: 47.8 (13.19); placebo:46.7 (12.71). Smoking status, current smoker, n/N (%): UMEC/VI 62.5/25 µg: 203/413 (49); placebo: 150/280 (54). |
|
| Interventions |
Intervention: Once‐daily UMEC/VI 62.5/25 µg. Comparator: Once‐daily placebo. |
|
| Outcomes |
Prespecified outcomes: Primary endpoint: Pre‐dose trough FEV1 on day 169. Secondary lung function endpoints: Mean TDI focal score at day 168 (week 24); change from baseline in weighted mean 0‐6 hour FEV1 obtained post‐dose at day 168 baseline and day 168. Other outcomes: Change from baseline in the mean Shortness of Breath With Daily Activities (SOBDA) score for (baseline and week 24). Reported outcomes: Pre‐dose trough FEV1 on treatment day 169, defined as the mean of FEV1 values obtained 23 h and 24 h after dosing on day 168 (week 24 visit). Secondary and additional lung function endpoints: weighted mean FEV1 over 0–6 h post‐dose on day 168; trough and 0–6 h weighted mean FEV1 at other visits, serial FEV1 assessments, time to onset during 0–6 h post‐dose on day 1, proportion of participants achieving an increase in FEV1 of ≥ 12% and ≥ 0.2 L above baseline at any time during 0–6 h post‐dose on day 1, proportion of participants achieving an increase of ≥ 0.1 L above baseline in trough FEV1, peak FEV1 and serial and trough FVC. Serial FEV1 over 0–24 h post‐dose was obtained in a subset of participants to characterise changes in lung function over the dosing interval. |
|
| Notes | Funding for trial; notable author COIs: The design, concept and conduct of the study and development of the manuscript was funded by GSK. Both external authors had acted as consultants and received research grants from GSK. All other co‐authors were GSK employees. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Simple randomisation was performed centrally through a validated computerised system and an interactive voice response system was then used to communicate the randomisation to the study team. |
| Allocation concealment (selection bias) | Low risk | Participants were randomised using an automated, interactive voice response system. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Masking quadruple (participants, care provider, investigator, outcome assessor). |
| Blinding of participants and personnel (objective outcomes ‐ performance bias) Objective outcomes | Low risk | Knowledge of treatment allocation by participant or personnel would be unlikely to influence objective outcomes. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Masking quadruple (participants, care provider, investigator, outcome assessor). |
| Blinding of outcome assessment (objective outcomes ‐ detection bias) All outcomes | Low risk | Assessment of objective outcomes would be unlikely to be influenced by knowledge of treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition balanced across treatment groups; ITT analysis performed. |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes (clinicaltrials.gov) were well reported. |
| Other bias | Low risk | None identified. |
Feldman 2012.
| Methods |
Study ID and dates performed: NCT01039675; 14 January 2010 to 20 April 2010. Study design: Multicentre, randomised, placebo‐controlled, double‐blind, parallel‐group study. Duration of study: Run‐in period 5–8 days; 4‐week treatment period; 7‐day follow‐up. Study setting, location, number of centres: 4 centres in the USA. Key inclusion criteria: Males and females aged ≥ 40 years with an established clinical history of COPD under the ATS/ERS standards; a history of at least 10 pack‐years of cigarette smoking; a post‐albuterol/salbutamol FEV1/FVC ≤ 0.70 and a post‐albuterol/salbutamol FEV1 of ≤ 80% of predicted normal values calculated using NHANES III. Key exclusion criteria: Current diagnosis of asthma, alpha1‐antitrypsin deficiency; use of OCS, antibiotics, or had been hospitalised due to exacerbation of COPD or a lower respiratory tract infection within 3 months prior to screening; abnormal 12‐lead ECG that resulted in an active medical problem or had clinically significant abnormalities from 24‐h Holter ECG monitoring at screening; prior evidence of pathological QT waves on ECG at least 12 months prior to screening that were unchanged at screening were not exclusionary; use of ICS at a dose > 1000 µg/day of FP or equivalent within 30 days prior to screening or initiation or termination of ICS use within 30 days prior to screening; use of long‐term oxygen therapy; regular use of short‐acting bronchodilators, including nebulised therapy. Concomitant medications: Permitted concomitant medications: ICS ≤ 1000 µg/day of FP or equivalent) provided the dose remained constant for 30 days prior to the screening visit and throughout the study; antibiotics that were not strong inhibitors of cytochrome P450 3A4 for short‐term treatment (≤ 14 days) of acute non‐respiratory tract infections provided that the infection did not meet the criteria for a COPD exacerbation. Medications not permitted during study: systemic beta‐receptor antagonists (ophthalmic preparations were allowed); tricyclic antidepressants; monoamine oxidase inhibitors; anticonvulsants (such as barbiturates); and phenothiazines. |
|
| Participants |
N randomised: UMEC/VI 500/25 µg: N = 42; placebo: N = 9. N analysed, n/N (%): UMEC/VI 500/25 µg: 42/42 (100); placebo: 9/9 (100). Mean age (range), years: UMEC/VI 500/25 µg: 59.2 (40–83); placebo: 58.7 (42–69). Gender ‐ male, n/N (%): UMEC/VI 500/25 µg: 24/42 (57.1); placebo: 7/9 (78). Baseline lung function ‐ mean (SD) post‐bronchodilator % predicted FEV1: UMEC/VI 500/25 µg: 48.37 (15.376); placebo: 50.58 (15.609). Smoking status, current smoker, n/N (%): UMEC/VI 500/25 µg: 24/42 (57); placebo: 7/9 (78). |
|
| Interventions |
Intervention: Once‐daily UMEC/VI 500/25 µg. Comparator: Once‐daily placebo. |
|
| Outcomes |
Prespecified outcomes: Primary: Change from baseline in weighted mean pulse rate over 0 to 6 hours post‐dose at day 28 (baseline and day 28). Secondary: change from baseline in weighted mean pulse rate over 0 to 6 hours post‐dose at day 1 and day 14; change from baseline in maximum and minimum pulse rate 0 to 6 hours post‐dose on days 1, 14, and 28. Reported outcomes: Prespecified primary and secondary outcomes well reported; Other: weighted mean systolic and diastolic blood pressure 0–6 h post‐dose on days 1, 14, and 28; maximum systolic and minimum diastolic blood pressure on days 1, 14, and 28; 24‐h Holter ECG parameters at screening and day 28; maximum QTc with interval corrected by Fridericia’s method; (during 0–6 h post‐dose) on days 1, 14, and 28 (measured using 12‐lead ECG); changes in haematological and clinical chemistry parameters from baseline on days 14 and 29; incidence of AEs and SAEs throughout the 28‐day treatment period and follow‐up; incidence of COPD exacerbations; and plasma concentrations and derived PK Cmax, tmax, AUC for UMEC and VI parameters, and trough FEV1 on day 29. |
|
| Notes | Funding for trial; notable author COIs: The study was sponsored by GSK and administered by Greenville Pharmaceutical Research. All authors received funding from, or were employees of, GSK. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation schedule was generated by the sponsor using a validated computerised system (RandAll). |
| Allocation concealment (selection bias) | Low risk | Centralised system used. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and investigators were blinded to treatment allocation. |
| Blinding of participants and personnel (objective outcomes ‐ performance bias) Objective outcomes | Low risk | Knowledge of treatment allocation by participant or personnel would be unlikely to influence objective outcomes. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Participants for subjective outcomes were the outcome assessors; therefore, low risk of bias. |
| Blinding of outcome assessment (objective outcomes ‐ detection bias) All outcomes | Low risk | Assessment of objective outcomes would be unlikely to be influenced by knowledge of treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Differential withdrawal rates noted between UMEC/VI and placebo groups; however, ITT analysis performed. |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes (clinicaltrials.gov) were well reported. |
| Other bias | High risk | Imbalance in baseline characteristics suggested that randomisation was not robust; limited sample size of placebo group could also account for imbalance in baseline characteristics. |
Larbig 2015.
| Methods |
Study ID and dates performed: NCT01610037 (RADIATE); October 2012–February 2015; abstract only; additional info/data sourced from clinicaltrials.gov Study design: Multicentre, double‐blind (participant & investigator), parallel‐group, placebo‐ and active‐controlled study. Duration of study: 52 weeks. Study setting, location, number of centres: 116 locations ‐ international. Key inclusion criteria: Male and female adults aged ≥ 40 years; stable COPD according to GOLD strategy (GOLD 2011); airflow limitation indicated by a post‐bronchodilator FEV1 ≥ 30% and < 80% of the predicted normal, and a post‐bronchodilator FEV1/FVC < 0.70; current or ex‐smokers with a smoking history of at least 10 pack years; participants with an mMRC ≥ grade 2. Key exclusion criteria: History of long QT syndrome or prolonged QTc; COPD exacerbation that required treatment with antibiotics and/or systemic corticosteroids and/or hospitalisation in the 6 weeks prior to visit 1; type I or uncontrolled type II diabetes; history of asthma or have concomitant pulmonary disease; paroxysmal (e.g. intermittent) atrial fibrillation (only patients with persistent atrial fibrillation and controlled with a rate control strategy for at least six months could be eligible); clinically significant renal, cardiovascular, neurological, endocrine, immunological, psychiatric, gastrointestinal, hepatic, or haematological abnormalities which could interfere with the assessment of safety. Concomitant medications: Not reported. |
|
| Participants |
N randomised: IND/GLY 110/50 µg: N = 407; placebo: N = 404. N analysed, n/N (%): IND/GLY 110/50 µg: 407/407 (100); placebo: 403/404 (99.8). Mean age (SD), years: IND/GLY 110/50 µg: 64.6 (7.89); placebo: 64.9 (7.95). Gender ‐ male, n/N (%): IND/GLY 110/50 µg: 288/407; placebo: 310/404. Baseline lung function ‐ post‐bronchodilator % predicted FEV1: Not reported. Smoking status, current smoker, n/N (%): Not reported. |
|
| Interventions |
Intervention: Once‐daily IND/GLY 110/50 µg. Comparator: Once‐daily placebo. |
|
| Outcomes |
Prespecified outcomes: Primary: Number of participants with SAEs during study. Secondary: Percentage of participants with composite endpoint of all‐cause mortality, and serious cardio‐ and cerebrovascular events; post hoc analysis: percentage of participants with composite endpoint of cardiovascular death and MACE change from baseline in pre‐dose FEV1 (days 22, 43, 85, 183, 274, and 364); change from baseline at day 364 in health status as measured by SGRQ for COPD participants; change from baseline at week 52 in daily, morning, and evening symptom scores; change from baseline at week 52 in percentage of nights with no night‐time awakenings; change from baseline at week 52 in percentage of no daytime symptoms; change from baseline at week 52 in percentage of days able to perform usual daily activities; change from baseline in 1 hour post‐dose FVC measurements (days 1, 22, 43, 85, 183, 274, and 364); time to premature discontinuation; change from baseline in 1 hour post‐dose FEV1 measurements (days 1, 22, 43, 85, 183, 274, and 364). Reported outcomes: Prespecified outcome (see above) well reported. |
|
| Notes | Funding for trial; notable author COIs: The study was funded by Novartis Pharmaceuticals. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information provided (abstract only). |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided (abstract only). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Clinical trial registry states that 'participant and investigators' were blinded to treatment allocation. |
| Blinding of participants and personnel (objective outcomes ‐ performance bias) Objective outcomes | Low risk | Clinical trial registry states that 'participant and investigators' were blinded to treatment allocation. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Participants for subjective outcomes were the outcome assessors; therefore, low risk of bias. |
| Blinding of outcome assessment (objective outcomes ‐ detection bias) All outcomes | Low risk | Assessment of objective outcomes would be unlikely to be influenced by knowledge of treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information provided (abstract only). |
| Selective reporting (reporting bias) | High risk | Abstract did not report key prespecified outcomes. |
| Other bias | Low risk | None identified. |
Mahler 2014.
| Methods |
Study ID and dates performed: NCT01490125 (BLAZE); October 26, 2011 to August 29, 2012. Study design: Randomised, placebo‐controlled, multicentre, blinded, double‐dummy, three‐period cross‐over study. Duration of study: 22 weeks (2‐week screening period; 3 X 6‐week treatment periods with 2‐week washouts). Study setting, location, number of centres: 42 centres in 5 countries: Belgium, Canada, Germany, Spain, and the United Kingdom. “Data were collected in a clinical setting.” Key inclusion criteria: Aged ≥ 40 years; moderate‐to‐severe stable COPD (stage II or III according to the 2009 GOLD criteria); were either current smokers or ex‐smokers, with a smoking history of ≥ 10 pack‐years; post‐bronchodilator FEV1 of ≥ 30% and < 80% of predicted normal; post‐bronchodilator FEV1/FVC < 0.70 at screening (visit 2, day 14); modified MRC dyspnoea scale grade of ≥ 2 at visit 2. Key exclusion criteria: Participants were excluded if they: required long‐term oxygen therapy; had a COPD exacerbation (requiring antibiotics, systemic steroids, or hospitalisation) in the 6 weeks before screening, or between screening and randomisation; had a respiratory tract infection in the weeks before or during screening; had concomitant pulmonary disease or had undergone a lung lobectomy, volume reduction or transplantation; had asthma, eczema, known high IgE levels, blood eosinophil count > 600/mm3 at screening, or a known positive skin prick test in the previous 5 years; had allergic rhinitis and used an H1 antagonist or intra‐nasal corticosteroids; or if they had α‐1 antitrypsin deficiency. Concomitant medications: Participants were requested not to take short‐acting bronchodilators in the 6 hours prior to the start of each visit. |
|
| Participants |
Note: cross‐over study therefore participant data reported for whole cohort. N randomised: 247. N analysed: 218. Mean age (SD), years: 62.8 (8.2). Gender ‐ male, n/N (%): 173/246 (70.3). Baseline lung function ‐ mean (SD) post‐bronchodilator % predicted FEV1: 56.1 (12.3). Smoking status, current smoker, n/N (%): 112/246 (45.5). |
|
| Interventions |
Intervention: Once‐daily IND/GLY 110/50 µg. Comparator: Once‐daily placebo. |
|
| Outcomes |
Prespecified outcomes: Primary: Total Transient Dyspnea Index score after 6 weeks of treatment (QVA149 versus placebo). Secondary: Total Transient Dyspnea Index score after 6 weeks of treatment (QVA149 versus tiotropium); standardized FEV1 AUC 5min‐4h after first dose and 6 weeks of treatment (QVA149 vs placebo and tiotropium); standardised FVC AUC 5 min‐4 hrs after first dose and 6 weeks of treatment (QVA149 vs placebo and tiotropium); change from baseline in the Capacity of Daily Living during the Morning (CDLM) score averaged over 6 weeks of treatment; change from baseline in the mean daily number of puffs of rescue medication used over the 6 weeks of treatment; safety. Reported outcomes: All prespecified outcomes (see above) were reported. |
|
| Notes | Funding for trial; notable author COIs: The study was funded by Novartis Pharma AG. Author disclosures not available. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Investigators used an automated, interactive response technology. Randomisation numbers generated using this procedure ensured that treatment assignment was unbiased and concealed from participants and investigators. |
| Allocation concealment (selection bias) | Low risk | Investigators used an automated, interactive response technology. Randomisation numbers generated using this procedure ensured that treatment assignment was unbiased and concealed from participants and investigators. See supplemental section of Mahler 2014 for details |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinding of participants, investigator staff, personnel performing assessments and data analysts was maintained. Treatment allocation was blinded by the use of identical packaging, labelling, schedule of administration, appearance, taste, and colour. |
| Blinding of participants and personnel (objective outcomes ‐ performance bias) Objective outcomes | Low risk | Blinding of participants, investigator staff, personnel performing assessments and data analysts was maintained. Treatment allocation was blinded by the use of identical packaging, labelling, schedule of administration, appearance, taste, and colour. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding of participants, investigator staff, personnel performing assessments and data analysts was maintained. Treatment allocation was blinded by the use of identical packaging, labelling, schedule of administration, appearance, taste, and colour. |
| Blinding of outcome assessment (objective outcomes ‐ detection bias) All outcomes | Low risk | Blinding of participants, investigator staff, personnel performing assessments and data analysts was maintained. Treatment allocation was blinded by the use of identical packaging, labelling, schedule of administration, appearance, taste, and colour. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rate was relatively high at ˜23%; however, a modified ITT was used whereby attrition rate was effective ˜10‐15% across treatment groups. |
| Selective reporting (reporting bias) | Low risk | Key, prespecified primary and secondary endpoints were reported as per trial registry. Slight difference versus prespecified time points for FEV/FVC noted. |
| Other bias | Low risk | None identified. |
Maltais 2014.
| Methods |
Study ID and dates performed: NCT01525615 (TORRACTO); study dates not reported. Study design: Multicentre, multinational, randomised, double‐blind, placebo‐controlled, parallel‐group trial. Duration of study: 1‐week run‐in, 12‐week treatment period, 3‐week follow‐up. Study setting, location, number of centres: 58 centres in 10 countries. Key inclusion criteria: Aged 40–75 years; clinical diagnosis of COPD and stable airway obstruction; post‐bronchodilator FEV1/FVC < 70% and post‐bronchodilator FEV1 < 80% and ≥ 30% predicted normal; current or ex‐smokers with a smoking history of > 10 pack‐years. Key exclusion criteria: Significant disease other than COPD; a history of asthma, myocardial infarction in the previous year; unstable or life‐threatening cardiac arrhythmia, or hospitalisation for heart failure within the previous year; a recognised contraindication to exercise; participated in a pulmonary rehabilitation program within the 6 weeks prior to the screening visit; an exercise limitation other than leg fatigue or exertional dyspnoea (e.g. arthritis in the leg or morbid obesity). Concomitant medications: Participants continued with inhaled corticosteroids if taken at baseline. Open‐label salbutamol (albuterol) was provided as rescue medication throughout the study. |
|
| Participants |
N randomised: TIO/OLO 2.5/5 µg: N = 133; TIO/OLO 5/5 µg: N = 139; placebo: N = 132. N analysed, n/N: TIO/OLO 2.5/5 µg: 129/133 ; TIO/OLO 5/5 µg: n = 135/139; placebo: n = 121/132. Mean age (SD), years: TIO/OLO 2.5/5 µg: 61.9 (7.3); TIO/OLO 5/5 µg: 63.1 (7.5); placebo: 60.8 (7.6). Gender ‐ male, n/N (%): TIO/OLO 2.5/5 µg: 87 (65.4); TIO/OLO 5/5 µg: 95 (68.3); placebo: 87 (65.9). Baseline lung function ‐ mean (SD) post‐bronchodilator % predicted FEV1: Not reported. Smoking status, current smoker, n/N (%): Not reported. |
|
| Interventions |
Intervention: Once‐daily TIO/OLO 2.5/5 µg; once‐daily TIO/OLO 5/5 µg. Comparator: Once‐daily placebo. |
|
| Outcomes |
Prespecified outcomes: Primary: Adjusted mean endurance time during constant work rate cycle ergometry after 12 weeks. Secondary: Adjusted mean endurance time during endurance shuttle walk test after 12 weeks; adjusted mean inspiratory capacity at pre‐exercise after 12 weeks; adjusted mean endurance time during constant work rate cycle ergometry on day 1, after 6 weeks treatment; adjusted mean inspiratory capacity at pre‐exercise after 1 day and 6 weeks; adjusted mean slope of the intensity of breathing discomfort on day 1 and after weeks 6 and 12; adjusted mean 1‐hour, post‐dose FEV1 on day 1, and after 6 and 12 weeks. Reported outcomes: The majority of prespecified outcomes (see above) were reported although no data provided for FEV1‐related outcomes. |
|
| Notes | Funding for trial; notable author COIs: The study was sponsored by Boehringer Ingelheim Pharma GmbH & Co. KG. All authors except JBGI were either employees of BI or had received research funding/honoraria from BI. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information provided. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information provided. |
| Blinding of participants and personnel (objective outcomes ‐ performance bias) Objective outcomes | Low risk | Knowledge of treatment allocation by participant or personnel would be unlikely to influence objective outcomes. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information provided. |
| Blinding of outcome assessment (objective outcomes ‐ detection bias) All outcomes | Low risk | Assessment of objective outcomes would be unlikely to be influenced by knowledge of treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | According to clinicaltrials.gov results tab, noncompletion < 20% in each arm (in fact < 11%) and reasonably balanced across arms. |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes (clinicaltrials.gov) were well reported. |
| Other bias | Low risk | None identified. |
Maltais 2014b.
| Methods |
Study ID and dates performed: NCT01323660 (Study 417). Study design: Two multicentre, double‐blind, randomised cross‐over studies (incomplete treatment block). Duration of study: 12–21‐day run‐in period; two 12‐week treatment periods separated by a 14‐day washout period. Study setting, location, number of centres: 31 centres in 6 countries. Key inclusion criteria: Current or former smokers; ≤ 40 years of age; smoking history of ≥ 10 pack‐years; clinical diagnosis of moderate‐to‐severe stable COPD (post‐bronchodilator FEV1/FVC < 70% and FEV1 ≥ 35% and ≤ 70% predicted); score of ≥ 2 on the mMRC Dyspnoea Scale at visit 1; resting FRC ≥ 120% of predicted (to ensure participants were hyperinflated, as hyperinflation is associated with exercise intolerance). Key exclusion criteria: Comorbid conditions or current diagnosis of asthma. Concomitant medications: All participants were provided with salbutamol for use on an ‘as‐needed’ basis throughout the run‐in, washout, and treatment periods. Stable/regular doses of ICS were permitted. |
|
| Participants |
Note: cross‐over study therefore participant data reported for whole cohort. N randomised: N = 349. N analysed, n/N: 341/349. Mean age (SD), years: 61.6 (8.3). Gender ‐ male, n/N (%): 195/348 (56.0). Baseline lung function ‐ mean (SD) post‐bronchodilator % predicted FEV1: 51.3 (9.8). Smoking status, current smoker, n/N (%): 220/348 (63.2). |
|
| Interventions |
Intervention: Once‐daily UMEC/VI 62.5/25 µg; once‐daily UMEC/VI 125/25 µg. Comparator: Once‐daily placebo. |
|
| Outcomes |
Prespecified outcomes: Primary: Change from baseline in exercise endurance time post‐dose at week 12 of each treatment period; change from baseline in trough FEV1 at week 12 of each treatment period. Secondary: Change from baseline in inspiratory capacity (trough and 3 hours post‐dose) at week 12 of each treatment period; change from baseline in functional residual capacity (trough and 3 hours post‐dose) at week 12 of each treatment period; change from baseline in residual volume (trough and 3 hours post‐dose) at week 12 of each treatment period; change from baseline in 3 hours post‐dose FEV1 at week 12 of each treatment period. Reported outcomes: All prespecified outcomes (see above) plus safety were reported. |
|
| Notes | Funding for trial; notable author COIs: The studies were sponsored by GSK; all authors were employees of, or had received honoraria/research funding from, GSK. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Simple randomisation was performed through a validated computerised system, then communicated to the study team via an IVRS. |
| Allocation concealment (selection bias) | Low risk | Allocation of treatments was controlled by RAMOS, a telephone‐based IRVS. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Trial registry states that participants and investigators were blinded to treatment allocation. All six treatments options were administered in a ‘double‐blind fashion’ via the same model of inhaler. |
| Blinding of participants and personnel (objective outcomes ‐ performance bias) Objective outcomes | Low risk | Trial registry states that participants and investigators were blinded to treatment allocation. All six treatments options were administered in a ‘double‐blind fashion’ via the same model of inhaler. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Participants for subjective outcomes were the outcome assessors; therefore, low risk of bias. |
| Blinding of outcome assessment (objective outcomes ‐ detection bias) All outcomes | Low risk | Assessment of objective outcomes (including a co‐primary endpoint of exercise tolerance time) would be unlikely to be influenced by knowledge of treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low and comparable attrition in UME/VI and placebo arms. |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes (clinicaltrials.gov) were well reported. |
| Other bias | Low risk | None identified. |
Maltais 2014c.
| Methods |
Study ID and dates performed: NCT01328444 (Study 418). Study design: Two multicentre, double‐blind, randomised cross‐over studies (incomplete treatment block). Duration of study: 12–21‐day run‐in period; two 12‐week treatment periods separated by a 14‐day washout period. Study setting, location, number of centres: 31 centres in 6 countries. Key inclusion criteria: Current or former smokers; ≤ 40 years of age; smoking history of ≥ 10 pack‐years; clinical diagnosis of moderate‐to‐severe stable COPD (post‐bronchodilator FEV1/FVC < 70% and FEV1 ≥ 35% and ≤ 70% predicted); score of ≥ 2 on the mMRC Dyspnoea Scale at visit 1; resting FRC ≥ 120% of predicted (to ensure participants were hyperinflated, as hyperinflation is associated with exercise intolerance). Key exclusion criteria: Comorbid conditions or current diagnosis of asthma. Concomitant medications: All participants were provided with salbutamol for use on an ‘as‐needed’ basis throughout the run‐in, washout, and treatment periods. Stable/regular doses of ICS were permitted. |
|
| Participants |
Note: cross‐over study therefore participant data reported for whole cohort N randomised: N = 308. N analysed, n/N: 307/308. Mean age (SD), years: 62.6 (7.9). Gender ‐ male, n/N (%): 168/307 (54.7). Baseline lung function ‐ mean (SD) post‐bronchodilator % predicted FEV1: 51.3 (10.0). Smoking status, current smoker, n/N (%): 186/307 (60.6). |
|
| Interventions |
Intervention: Once‐daily UMEC/VI 62.5/25 µg; once‐daily UMEC/VI 125/25 µg. Comparator: Once‐daily placebo. |
|
| Outcomes |
Prespecified outcomes: Primary: Change from baseline in exercise endurance time post‐dose at week 12 of each treatment period; change from baseline in trough FEV1 at week 12 of each treatment period. Secondary: Change from baseline in inspiratory capacity (trough and 3 hours post‐dose) at week 12 of each treatment period; change from baseline in functional residual capacity (trough and 3 hours post‐dose) at week 12 of each treatment period; change from baseline in residual volume (trough and 3 hours post‐dose) at week 12 of each treatment period; change from baseline in 3 hours post‐dose FEV1 at week 12 of each treatment period. Reported outcomes: All prespecified outcomes (see above) plus safety were reported. |
|
| Notes | Funding for trial; notable author COIs: The studies were sponsored by GSK; all authors were employees of, or had received honoraria/research funding from, GSK. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Simple randomisation was performed through a validated computerised system, then communicated to the study team via an IVRS. |
| Allocation concealment (selection bias) | Low risk | Allocation of treatments was controlled by RAMOS, a telephone‐based IVRS. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Trial registry states that participants and investigators were blinded to treatment allocation. All six treatments options were administered in a ‘double‐blind fashion’ via the same model of inhaler. |
| Blinding of participants and personnel (objective outcomes ‐ performance bias) Objective outcomes | Low risk | Trial registry states that participants and investigators were blinded to treatment allocation. All six treatments options were administered in a ‘double‐blind fashion’ via the same model of inhaler. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Participants for subjective outcomes were the outcome assessors; therefore, low risk of bias. |
| Blinding of outcome assessment (objective outcomes ‐ detection bias) All outcomes | Low risk | Assessment of objective outcomes (including a co‐primary endpoint of exercise tolerance time) would be unlikely to be influenced by knowledge of treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low and comparable attrition in UME/VI and placebo arms. |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes (clinicaltrials.gov) were well reported. |
| Other bias | Low risk | None identified. |
NCT00626522.
| Methods |
Study ID and dates performed: NCT00626522; not published (source clinicaltrials.gov); study completed 2008. Study design: Randomised, 4‐week, placebo‐controlled, double‐blind, 6‐arm parallel‐group, dose‐finding trial. Duration of study: 4‐week treatment period. Study setting, location, number of centres: 11 sites in 4 countries. Key inclusion criteria: Aged 40–85 years; clinical diagnosis of stable moderate‐to‐severe COPD (GOLD 2006 stages II–III); current or ex‐cigarette smoker with a smoking history of ≥ 10 pack‐years; FEV1 at screening measured between 30‐45 minutes post‐inhalation of 400 μg of salbutamol was 30% ≤ FEV1 < 80% of the predicted normal value; FEV1/FVC at screening measured between 30‐ 45 minutes post inhalation of 400 μg of salbutamol was < 70%. Key exclusion criteria: History or current diagnosis of asthma, allergic rhinitis or atopy, or exercise‐induced bronchospasm; clinically significant respiratory conditions at the time of screening visit; hospitalisation due to COPD exacerbation within 3 months prior to screening; signs of COPD exacerbation or respiratory infection up to 6 weeks prior to screening visit; clinically significant cardiovascular conditions. Concomitant medications: Not reported. |
|
| Participants |
N randomised: ACL/FOR 200/6 µg: n = 121; ACL/FOR 200/12 µg: n = 120; ACL/FOR 200/18 µg: n = 125; placebo: n = 59. N analysed: ACL/FOR 200/6 µg: 121/121 (100); ACL/FOR 200/12 µg: 120/120 (100); ACL/FOR 200/18 µg: 125/125 (100); placebo: 59/59 (100). Mean age (SD), years: ACL/FOR 200/6 µg: 62.9 (9.0); ACL/FOR 200/12 µg: 63.6 (8.9); ACL/FOR 200/18 µg: 63.9 (8.1); placebo: 60.7 (7.8). Gender ‐ male, n/N: ACL/FOR 200/6 µg: 91/121; ACL/FOR 200/12 µg: 98/120; ACL/FOR 200/18 µg: 96/125; placebo: 44/59. Baseline lung function ‐ post‐bronchodilator % predicted FEV1: Not reported. Smoking status, current smoker, n/N (%): Not reported. |
|
| Interventions |
Intervention: Once‐daily ACL/FOR 200/6 µg; once‐daily ACL/FOR 200/12 µg; once‐daily ACL/FOR 200/18 µg. Comparator: Once‐daily placebo. |
|
| Outcomes |
Prespecified outcomes: Primary: Change from baseline in normalised FEV1 AUC for 0‐12 hr at week 4. Secondary: Change from baseline in trough FEV1 at week 4; change from baseline in peak FEV1 at week 4; change from baseline in normalised FEV1 AUC 0–3 hours at week 4; change from baseline in normalised FEV1 AUC 0–6 hours at week 4. Reported outcomes: All of the prespecified outcomes (see above) plus safety were reported on the clinicaltrials.gov site. |
|
| Notes | Funding for trial; notable author COIs: The study was sponsored by AstraZeneca. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information provided (trial registry only). |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided (trial registry only). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and investigators blinded. |
| Blinding of participants and personnel (objective outcomes ‐ performance bias) Objective outcomes | Low risk | Participants and investigators blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Participants for subjective outcomes were the outcome assessors; therefore, low risk of bias. |
| Blinding of outcome assessment (objective outcomes ‐ detection bias) All outcomes | Low risk | Assessment of objective outcomes would be unlikely to be influenced by knowledge of treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates were low and balanced between treatment groups. |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes (clinicaltrials.gov) were well reported. |
| Other bias | Low risk | None identified. |
NCT02275052.
| Methods |
Study ID and dates performed: NCT02275052 Study design: A multicenter, randomised, double‐blind, placebo‐controlled, 2‐period, complete block design cross‐over study. Duration of study: 2 x 12‐week treatment periods with washout of 12‐17 days. Run in period 12‐25 days. Total duration approximately 30 weeks including follow up. Study setting, location, number of centres: United States of America. Key inclusion criteria: Aged ≥ 40 years; diagnosis of COPD (ATS/ERS); current or former cigarette smokers with a history of cigarette smoking of ≥ 10 pack‐years; pre‐ and post‐albuterol FEV1/FVC < 0.70 and a post‐albuterol FEV1 of ≥ 30% and ≤ 70% of predicted normal value; dyspnoea score of ≥ 2 on the mMRC scale at visit 1; a resting FRC of ≥ 120% of predicted normal FRC at visit 1. Key exclusion criteria: Pregnancy; current diagnosis of asthma; other respiratory disorders; known alpha‐1 antitrypsin deficiency; active lung infections (such as tuberculosis), and lung cancer in remission for < 5 years. Concomitant medications: Permitted: All participants were provided with albuterol for use on an 'as needed' basis throughout the run‐in, washout, and study treatment periods while on investigational product. Use of the following medications according to the following defined time intervals prior to visit 1: Depot corticosteroids (12 weeks), systemic, oral or parenteral corticosteroids (Intra‐articular and epidural corticosteroid injections were permitted) (6 weeks), antibiotics (for lower respiratory tract infection and/or COPD exacerbation) (6 weeks), long‐acting beta agonist (LABA)/inhaled corticosteroid (ICS) combination products if LABA/ICS therapy was discontinued completely (30 days), LABA/ICS combination products only if discontinuing LABA therapy and switching to ICS monotherapy (dose of ICS that is switched to must not exceed 1000 µg of fluticasone propionate or equivalent) (48 hours for the salmeterol or formoterol component, 14 days for the vilanterol component), use of ICS at a dose > 1000 μg/day of fluticasone propionate or equivalent (use of ICS was permitted provided the dose did not exceed 1000 μg of fluticasone propionate or equivalent; ICS use not to be initiated or discontinued within 30 days prior to visit 1 except for participants on LABA/ICS therapy who may discontinue LABA/ICS therapy as indicated and switch to ICS monotherapy) (30 days), initiation or discontinuation of ICS use (30 days), PDE4 inhibitor (roflumilast) (14 days), Inhaled LABA: salmeterol, formoterol (48 hours); olodaterol, indacaterol (14 days), LAMA (tiotropium, aclidinium, glycopyrronium, umeclidinium) (7 days), LABA/LAMA combination products (whichever mono component had the longest washout), theophyllines (48 hours), oral beta2‐agonists (long‐acting (48 hours), short‐acting (12 hours), inhaled SABA (study provided prn albuterol was permitted during the study, except in the 4‐hour period prior to spirometry testing) (4 hours), inhaled short‐acting anticholinergics (permitted during the run‐in period between visits 1 and 4 and washout period between visits 7 and 9. Restricted/non‐permitted: Participants must discontinue use of short‐acting anticholinergics at least 4 hours before visit 4 and visit 9. Participants should not use short acting anticholinergics during the double‐blind treatment periods (4 hours), inhaled short‐acting anticholinergic/SABA combination products (4 hours), and any other investigational medication (30 days or within 5 drug half‐lives (whichever was longer)) |
|
| Participants |
Note: cross‐over study therefore participant data reported for whole cohort. N randomised: N = 99. N analysed, n/N: UMEC/VI 62.5/25 µg: 93/99; placebo: 90/99. Mean age (SD), years: 60.7 (9.47). Gender ‐ male, n/N (%): 104/198 (52.5). Baseline lung function ‐ post‐bronchodilator % predicted FEV1: Not reported. Smoking status, current smoker, n/N (%): Not reported. |
|
| Interventions |
Intervention: Once‐daily UMEC/VI 62.5/25 µg. Comparator: Once‐daily placebo. |
|
| Outcomes |
Prespecified outcomes: Primary outcome: change from baseline in exercise endurance time post‐dose at week 12 of each treatment period. Secondary outcome: change from baseline in trough FEV1 at week 12 of each treatment period; change from baseline in FRC 3 hours post‐dose at week 12 of each treatment period; change from baseline in IC 3 hours post‐dose at week 12 of each treatment period. Reported outcomes: All of the prespecified outcomes (see above) plus safety were reported on the clinicaltrials.gov site. |
|
| Notes | Funding for trial; notable author COIs: Study sponsored by GlaxoSmithKline. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information provided (trial registry only). |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided (trial registry only). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and investigators were blinded to treatment allocation. |
| Blinding of participants and personnel (objective outcomes ‐ performance bias) Objective outcomes | Low risk | Participants and investigators were blinded to treatment allocation. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Participants for subjective outcomes were the outcome assessors; therefore, low risk of bias. |
| Blinding of outcome assessment (objective outcomes ‐ detection bias) All outcomes | Low risk | Assessment of objective outcomes would be unlikely to be influenced by knowledge of treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rate < 20% and balanced between treatment arms. |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes (clinicaltrials.gov) were well reported. |
| Other bias | Low risk | None identified. |
O'Donnell 2015a.
| Methods |
Study ID and dates performed: NCT01533922 (MORACTO‐1); study dates not stated. Study design: Double‐blind, 6‐week incomplete cross‐over study. Duration of study: 38–40 weeks, including a 2–4 week run‐in period. Study setting, location, number of centres: 82 investigational sites in 13 countries. Key inclusion criteria: Aged 40–75 years; post‐bronchodilator (400 μg salbutamol) FEV1/FVC < 70%; post‐bronchodilator FEV1 ≥ 30% and < 80% of predicted normal (GOLD 2–3); current or ex‐smokers with a smoking history of > 10 pack‐years. Key exclusion criteria: Significant disease other than COPD; unstable or life‐threatening cardiac arrhythmia; hospitalisation for heart failure or myocardial infarction within the past year; regular use of daytime oxygen therapy for > 1 h per day; history of asthma and contraindications to exercise as per the ERS guidelines. Concomitant medications: Permitted: Participants continued with inhaled corticosteroids if taken at baseline; open‐label salbutamol (albuterol) was provided as rescue medication throughout the study. Restricted/not permitted: LABA or LAMA (other than study medication) during the baseline, treatment, and washout periods; short‐acting muscarinic antagonists during the treatment periods (permitted only during baseline and washout periods, with an 8‐h washout prior to assessments). |
|
| Participants |
Note: cross‐over study therefore participant data reported for whole cohort; baseline characteristics and participant flow reported for combined studies (MORACTO‐1 and MORACTO‐2). N randomised: N = 586. N analysed, n/N: TIO/OLO 2.5/5 µg: 424/442; TIO/OLO 5/5µg: 428/450; placebo: 413/438. Mean age (SD), years: 61.7 (7.7). Gender ‐ male, n/N (%): 417/586 (71.2). Baseline lung function ‐ mean (SD) post‐bronchodilator % predicted FEV1: 58 (13). Smoking status, current smoker, n/N (%): 229/586 (39.1). |
|
| Interventions |
Intervention: Once‐daily TIO/OLO 2.5/5 µg; once‐daily TIO/OLO 5/5 µg. Comparator: Once‐daily placebo. |
|
| Outcomes |
Prespecified outcomes: Primary: Inspiratory capacity at rest immediately before constant work rate cycle ergometry (assessed at 6 weeks); endurance time during constant work rate cycle ergometry. Secondary: Slope of the intensity of breathing discomfort during constant work rate cycle ergometry (assessed at 6 weeks); 1‐hour post‐dose FEV1 (assessed at 6 weeks). Reported outcomes: All prespecified outcomes (see above) plus FVC and safety and tolerability. |
|
| Notes | Funding for trial; notable author COIs: This work was funded by BI Pharma GmbH & Co. All authors were either employees of BI or had received research funding or honoraria from BI. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information provided. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information provided. |
| Blinding of participants and personnel (objective outcomes ‐ performance bias) Objective outcomes | Low risk | Knowledge of treatment allocation by participant or personnel would be unlikely to influence objective outcomes. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information provided. |
| Blinding of outcome assessment (objective outcomes ‐ detection bias) All outcomes | Low risk | Assessment of objective outcomes would be unlikely to be influenced by knowledge of treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar attrition with similar reasons for combined treatment arm and placebo arm; discontinuation rates < 5%. |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes (clinicaltrials.gov) were well reported. |
| Other bias | Low risk | None identified. |
O'Donnell 2015b.
| Methods |
Study ID and dates performed: NCT01533935 (MORACTO‐2); study dates not stated. Study design: Double‐blind, 6‐week incomplete cross‐over study. Duration of study: 38–40 weeks, including a 2–4 week run‐in period. Study setting, location, number of centres: 82 investigational sites in 13 countries. Key inclusion criteria: Aged 40–75 years; post‐bronchodilator (400 μg salbutamol) FEV1/FVC < 70%; post‐bronchodilator FEV1 ≥ 30% and < 80% of predicted normal (GOLD 2–3); current or ex‐smokers with a smoking history of > 10 pack‐years. Key exclusion criteria: Significant disease other than COPD; unstable or life‐threatening cardiac arrhythmia; hospitalisation for heart failure or myocardial infarction within the past year; regular use of daytime oxygen therapy for > 1 h per day; history of asthma and contraindications to exercise as per the ERS guidelines. Concomitant medications: Permitted: Participants continued with inhaled corticosteroids if taken at baseline; open‐label salbutamol (albuterol) was provided as rescue medication throughout the study. Restricted/not permitted: LABA or LAMA (other than study medication) during the baseline, treatment, and washout periods; short‐acting muscarinic antagonists during the treatment periods (permitted only during baseline and washout periods, with an 8‐h washout prior to assessments). |
|
| Participants |
Note: cross‐over study therefore participant data reported for whole cohort; baseline characteristics and participant flow reported for combined studies (MORACTO‐1 and MORACTO‐2). N randomised: N = 586. N analysed, n/N: TIO/OLO 2.5/5 µg: 424/442; TIO/OLO 5/5µg: 28/450; placebo: 413/438. Mean age (SD), years: 61.7 (7.7). Gender ‐ male, n/N (%): 417/586 (71.2). Baseline lung function ‐ mean (SD) post‐bronchodilator % predicted FEV1: 58 (13). Smoking status, current smoker, n/N (%): 229/586 (39.1). |
|
| Interventions |
Intervention: Once‐daily TIO/OLO 2.5/5 µg; once‐daily TIO/OLO 5/5µg. Comparator: Once‐daily placebo. |
|
| Outcomes |
Prespecified outcomes: Primary: Inspiratory capacity at rest immediately before constant work rate cycle ergometry (assessed at 6 weeks); endurance time during constant work rate cycle ergometry. Secondary: Slope of the intensity of breathing discomfort during constant work rate cycle ergometry (assessed at 6 weeks); 1‐hour post‐dose FEV1 (assessed at 6 weeks). Reported outcomes: All prespecified outcomes (see above) plus FVC and safety and tolerability. |
|
| Notes | Funding for trial; notable author COIs: This work was funded by BI Pharma GmbH & Co. All authors were either employees of BI or had received research funding or honoraria from BI. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information provided. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information provided. |
| Blinding of participants and personnel (objective outcomes ‐ performance bias) Objective outcomes | Low risk | Knowledge of treatment allocation by participant or personnel would be unlikely to influence objective outcomes. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information provided. |
| Blinding of outcome assessment (objective outcomes ‐ detection bias) All outcomes | Low risk | Assessment of objective outcomes would be unlikely to be influenced by knowledge of treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar attrition with similar reasons for combined treatment arm and placebo arm; discontinuation rates < 5%. |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes (clinicaltrials.gov) were well reported. |
| Other bias | Low risk | None identified. |
Siler 2016.
| Methods |
Study ID and dates performed: NCT02152605; September 2014 to March 2015. Study design: Multicentre, randomised, double‐blind, parallel‐group, placebo‐controlled study. Duration of study: 7–14 day run‐in period; 12‐week treatment period. Study setting, location, number of centres: 55 centres in Bulgaria, Germany, Hungary, Romania, Russian Federation, Ukraine, and US. Key inclusion criteria: ≥ 40 years of age; diagnosis of COPD; current or prior history of ≥ 10 pack‐years of cigarette smoking at screening; a pre‐ and post‐albuterol (salbutamol) FEV1/FVC < 0.70 and a post‐albuterol FEV1 ≤ 70% of predicted normal values at screening (based on NHANES III reference equations; a score ≥ 2 on the mMRC Dyspnoea Scale at screening. Key exclusion criteria: Current diagnosis of asthma or other known respiratory conditions (α1‐antitrypsin deficiency, active tuberculosis, lung cancer, bronchiectasis, sarcoidosis, lung fibrosis, pulmonary hypertension, interstitial lung diseases, or other active pulmonary diseases); hospitalisation for COPD or pneumonia within 12 weeks prior to visit 1; lung volume reduction surgery within the 12 months prior to visit 1; use of long‐term oxygen therapy (prescribed for > 12 h/day); severe hepatic impairment; any rapidly progressing disease or immediate life‐threatening illness (e.g. cancer); any condition that was likely to affect respiratory function (e.g. neurological condition); abnormal, clinically significant electrocardiogram finding at screening (atrial fibrillation with rapid ventricular rate > 120 bpm; sustained or nonsustained ventricular tachycardia; second‐degree heart block Mobitz type II, and third‐degree heart block (unless pacemaker or defibrillator had been inserted)). Concomitant medications: Use of study‐provided albuterol was permitted, except in the 4‐hour period prior to spirometry testing. Excluded medications prior to visit 1: depot corticosteroids; systemic, oral or parenteral corticosteroids; ICS/LABA combination products; ICS at a dose > 1000 µg/day of FP or equivalent; initiation or discontinuation of ICS use; PDE4 inhibitors; LAMAs, inhaled LABAs, LABA/LAMA combination products; theophyllines; oral beta2‐agonists; inhaled SABA or inhaled short‐acting anticholinergics, or any combination of the two. |
|
| Participants |
N randomised: UMEC/VI 62.5/25 µg: n = 249; placebo: n = 249. N analysed, n/N: UMEC/VI 62.5/25 µg: 248/249; placebo: 248/249. Mean age (SD), years: UMEC/VI 62.5/25 µg: 64.1 (8.70); placebo: 62.6 (8.23). Gender ‐ male, n/N (%): UMEC/VI 62.5/25 µg: 144/248 (58); placebo: 149/248 (60). Baseline lung function ‐ mean (SD) post‐bronchodilator % predicted FEV1: UMEC/VI 62.5/25 µg: 46.5 (12.81); placebo: 48.4 (14.06). Smoking status, current smoker, n/N (%): UMEC/VI 62.5/25 µg: 137/248 (55); placebo: 129/248 (52). |
|
| Interventions |
Intervention: Once‐daily UMEC/VI 62.5/25 µg. Comparator: Once‐daily placebo. |
|
| Outcomes |
Prespecified outcomes: Primary: Change from baseline in mean SGRQ total score at day 84. Secondary: Change from baseline in trough FEV1 at day 84; change from baseline in mean number of puffs of rescue medication per day used over weeks 1‐12. Reported outcomes: plus the proportion of SGRQ responders at days 28, 56, and 84; SGRQ total score at days 28 and 56 (SGRQ responders were defined as having a total score ≥ 4 units below baseline); percentage of rescue‐free days; trough FEV1 at days 28 and 56; trough FVC at days 28, 56, and 84; safety. |
|
| Notes | Funding for trial; notable author COIs: The study was funded by GSK. Lead author had received research funding from GSK. All other authors were employees of GSK. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Centralised IRVS system used for randomisation. |
| Allocation concealment (selection bias) | Low risk | Centralised IRVS system used for randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and investigators were blinded to treatment (see clinicaltrial.gov). |
| Blinding of participants and personnel (objective outcomes ‐ performance bias) Objective outcomes | Low risk | Participants and investigators were blinded to treatment (see clinicaltrial.gov). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information provided. |
| Blinding of outcome assessment (objective outcomes ‐ detection bias) All outcomes | Low risk | Assessment of objective outcomes would be unlikely to be influenced by knowledge of treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar loss in both arms (treatment and placebo) for similar reasons and loss < 10%. |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes (clinicaltrials.gov) were well reported. |
| Other bias | High risk | Greater proportion of participants with GOLD category D in the active treatment group; favoured placebo and underestimation of treatment effect. |
Singh 2016a.
| Methods |
Study ID and dates performed: NCT01964352 (OTEMTO 1). Study design: A multinational, double‐blind, parallel‐group, placebo‐controlled study. Duration of study: 2‐week run‐in period; 12‐week treatment period; 3‐week follow‐up. Study setting, location, number of centres: Not stated. Key inclusion criteria: Participants aged ≥ 40 years with moderate to severe COPD (GOLD 2–3); post‐bronchodilator FEV1 ≥ 30% and < 80% of predicted normal), FEV1/FVC < 70% predicted and a smoking history of > 10 pack‐years. Key exclusion criteria: A history of asthma, another significant disease, COPD exacerbation or symptoms of lower respiratory tract infection within the previous 3 months; unstable or life‐threatening cardiac arrhythmia, hospitalisation for heart failure within the past year; a history of myocardial infarction within 1 year of screening; a history of life‐threatening pulmonary obstruction. Concomitant medications: Participants were allowed to continue their ICS therapy (if they were on a stable dose for 6 weeks prior to screening). LAMAs or LABAs other than study medication were prohibited; short‐acting muscarinic antagonists were permitted only during the screening period. Open‐label salbutamol was provided as rescue medication for use throughout the study. |
|
| Participants |
N randomised: TIO/OLO 2.5/5 µg: n = 202; TIO/OLO 5/5 µg: n = 204; placebo: n = 204. N analysed, n/N (%): TIO/OLO 2.5/5 µg: 196/202 (97.0); TIO/OLO 5/5 µg: 195/204 (96.1); placebo: 178/204 (87.3). Mean age (SD), years: TIO/OLO 2.5/5 µg: 64.7 (8.2); TIO/OLO 5/5 µg: 64.7 (8.9); placebo: 65.1 (8.3). Gender ‐ male, n/N (%): TIO/OLO 2.5/5 µg: 116 (57.4); TIO/OLO 5/5 µg: 114 (56.2); placebo: 127 (62.3). Baseline lung function ‐ mean (SD) post‐bronchodilator % predicted FEV1: TIO/OLO 2.5/5 µg: 55.5 (13.7); TIO/OLO 5/5 µg: 54.9 (12.0); placebo: 56.3 (12.8). Smoking status, current smoker, n/N (%): TIO/OLO 2.5/5 µg: 98/202 (48.5); TIO/OLO 5/5 µg: 111/203 (54.7); placebo: 88/204 (43.1). |
|
| Interventions |
Intervention: Once‐daily TIO/OLO 2.5/5 µg; once‐daily TIO/OLO 5/5 µg. Comparator: Once‐daily placebo. |
|
| Outcomes |
Prespecified outcomes: Primary: FEV1 AUC 0‐3 h response at 12 weeks; trough FEV1 response at 12 weeks; SGRQ total score at 12 weeks. Secondary: trough FVC response (change from baseline) at 12 weeks; TDI focal score at 12 weeks; FVC AUC 0‐3 h response (change from baseline) at 12 weeks. Reported outcomes: All prespecified outcomes (see above) were reported. |
|
| Notes | Funding for trial; notable author COIs: This study was funded by Boehringer Ingelheim Pharma GmbH & Co. KG. Seven of nine authors were employees of, or had received funding/honoraria from, BI. Two authors disclosed no conflicts of interest. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information provided. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information provided. |
| Blinding of participants and personnel (objective outcomes ‐ performance bias) Objective outcomes | Low risk | Knowledge of treatment allocation by participant or personnel would be unlikely to influence objective outcomes. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information provided. |
| Blinding of outcome assessment (objective outcomes ‐ detection bias) All outcomes | Low risk | Assessment of objective outcomes would be unlikely to be influenced by knowledge of treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rate was < 15% in placebo and combined LABA/LAMA arms. |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes (clinicaltrials.gov) were well reported. |
| Other bias | Low risk | None identified. Note slightly higher rates of discontinuation in placebo arm ‐ likely accounted for due to the most severely ill participants dropping out. Potential positive placebo treatment effect not observed. |
Singh 2016b.
| Methods |
Study ID and dates performed: NCT02006732 (OTEMTO 2). Study design: One of two multinational, double‐blind, parallel‐group, placebo‐controlled studies. Duration of study: 2‐week run‐in period; 12‐week treatment period; 3‐week follow‐up. Study setting, location, number of centres: Not stated. Key inclusion criteria: Participants aged ≥ 40 years with moderate to severe COPD (GOLD 2–3); post‐bronchodilator FEV1 ≥ 30% and < 80% of predicted normal), FEV1/FVC < 70% predicted and a smoking history of > 10 pack‐years. Key exclusion criteria: A history of asthma, another significant disease, COPD exacerbation or symptoms of lower respiratory tract infection within the previous 3 months; unstable or life‐threatening cardiac arrhythmia, hospitalisation for heart failure within the past year; a history of myocardial infarction within 1 year of screening; a history of life‐threatening pulmonary obstruction. Concomitant medications: Participants were allowed to continue their ICS therapy (if they were on a stable dose for 6 weeks prior to screening). LAMAs or LABAs other than study medication were prohibited; short‐acting muscarinic antagonists were permitted only during the screening period. Open‐label salbutamol was provided as rescue medication for use throughout the study. |
|
| Participants |
N randomised: TIO/OLO 2.5/5 µg: n = 202; TIO/OLO 5/5 µg: n = 202; placebo: n = 202. N analysed, n/N: TIO/OLO 2.5/5 µg: 193/202; TIO/OLO 5/5 µg: 198/202; placebo: 182/202. Mean age (SD), years: TIO/OLO 2.5/5 µg: 64.4 (8.6); TIO/OLO 5/5 µg: 65.2 (8.5); placebo: 64.0 (8.3). Gender ‐ male, n/N (%): TIO/OLO 2.5/5 µg: 126 (62.4); TIO/OLO 5/5 µg: 133 (65.8); placebo: 117 (57.9). Baseline lung function ‐ mean (SD) post‐bronchodilator % predicted FEV1: TIO/OLO 2.5/5 µg: 54.5 (12.7); TIO/OLO 5/5 µg: 54.8 (12.8); placebo: 54.3 (13.4). Smoking status, current smoker, n/N (%): TIO/OLO 2.5/5 µg: 90/202 (44.6) ; TIO/OLO 5/5 µg: 92/202 (45.5); placebo: 95/202 (47.0). |
|
| Interventions |
Intervention: Once‐daily TIO/OLO 2.5/5 µg; once‐daily TIO/OLO 5/5 µg. Comparator: Once‐daily placebo. |
|
| Outcomes |
Prespecified outcomes: Primary: FEV1 AUC 0‐3 h response at 12 weeks; trough FEV1 response at 12 weeks; SGRQ total score at 12 weeks. Secondary: trough FVC response (change from baseline) at 12 weeks; TDI focal score at 12 weeks; FVC AUC 0‐3 h response (change from baseline) at 12 weeks. Reported outcomes: All prespecified outcomes (see above) were reported. |
|
| Notes | Funding for trial; notable author COIs: This study was funded by Boehringer Ingelheim Pharma GmbH & Co. KG. Seven of nine authors were employees of, or had received funding/honoraria from, BI. Two authors disclosed no conflicts of interest. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information provided. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information provided. |
| Blinding of participants and personnel (objective outcomes ‐ performance bias) Objective outcomes | Low risk | Knowledge of treatment allocation by participant or personnel would be unlikely to influence objective outcomes. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information provided. |
| Blinding of outcome assessment (objective outcomes ‐ detection bias) All outcomes | Low risk | Assessment of objective outcomes would be unlikely to be influenced by knowledge of treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rate was < 15% in placebo and combined LABA/LAMA arms. |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes (clinicaltrials.gov) were well reported. |
| Other bias | Low risk | None identified. Note slightly higher rates of discontinuation in placebo arm ‐ likely accounted for due to the most severely ill participants dropping out. Potential positive placebo treatment effect not observed. |
Troosters 2016.
| Methods |
Study ID and dates performed: NCT02085161; PHYSACTO; dates not reported. Study design: Randomised, partially double‐blinded, placebo‐controlled, parallel‐group trial. Duration of study: 19 weeks (4‐week run‐in; 12‐week treatment period; 3‐week follow‐up). Study setting, location, number of centres: 34 sites in Australia, New Zealand, USA, Canada, Europe (17 academic centres, 15 secondary care and 5 primary care centres). Key inclusion criteria: COPD; aged ≥ 40 years and ≤ 75 years; smoking history of > 10 pack‐years; post‐bronchodilator FEV1 ≥ 30% and < 80% of predicted normal (GOLD 2–3) and no acute exacerbations in the month prior to the study; post‐bronchodilator FEV1/FVC < 70%. Key exclusion criteria: Significant disease other than COPD; history of asthma; clinically relevant abnormal baseline haematology, blood chemistry or urinalysis; conditions excluding participants from exercise. Concomitant medications: Not reported. |
|
| Participants |
N randomised: TIO/OLO 5/5 µg: n = 76; placebo: n = 76. N analysed, n/N: TIO/OLO 5/5 µg: 72/76; placebo: 65/76. Mean age (SD), years: TIO/OLO 5/5 µg: 65.0 (6.9); placebo: 64.4 (6.6). Gender ‐ male, n/N (%): TIO/OLO 5/5 µg: 48/76 (63.2); placebo: 52/75 (69.3). Baseline lung function ‐ post‐bronchodilator % predicted FEV1: Not reported. Smoking status, current smoker, n/N (%): Not reported. |
|
| Interventions |
Intervention: Once‐daily TIO/OLO 5/5 µg. Comparator: Once‐daily placebo. |
|
| Outcomes |
Prespecified outcomes: Primary: Endurance time during endurance shuttle walk test (to symptom limitation) after 8 weeks. Secondary: average daily walking time measured by the activity monitor in the week prior to week 12; average daily walking intensity measured by the activity monitor in the week prior to 12 weeks of treatment; perceived difficulties as evaluated with functional performance inventory‐short form (FPI‐SF) total score at week 12; endurance time during endurance shuttle walk test (to symptom limitation) after 12 weeks; one‐hour, post‐dose FEV1 after 8 weeks of treatment; one‐hour, post‐dose FVC after 8 weeks of treatment; resting inspiratory capacity measured at 1.5 hours post‐dose after 8 weeks of treatment. Reported outcomes: Prespecified outcomes (clinicaltrials.gov) were well reported. |
|
| Notes | Funding for trial; notable author COIs: Study sponsored by BI. Authors had received funding or honoraria from BI, or were employees of BI. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | “Randomisation is performed using a pseudo‐random number generator and block randomisation is used to achieve balanced allocation”. |
| Allocation concealment (selection bias) | Low risk | Web‐based and telephone‐based response system used. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | 'Partially double‐blind' as it was not possible to blind participants and personnel to the receipt of exercise training or behavioural modifications. However; the groups of interest (TIO/OLO and placebo) received treatments in double‐blind fashion (participants and personnel were blinded). |
| Blinding of participants and personnel (objective outcomes ‐ performance bias) Objective outcomes | Low risk | Knowledge of treatment allocation by participant or personnel would be unlikely to influence objective outcomes. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Participants for subjective outcomes were the outcome assessors; therefore, low risk of bias. |
| Blinding of outcome assessment (objective outcomes ‐ detection bias) All outcomes | Low risk | Assessment of objective outcomes would be unlikely to be influenced by knowledge of treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rate was < 20% and the difference in attrition rates between relevant treatment groups was < 10% |
| Selective reporting (reporting bias) | Low risk | Data presented on clinicaltrials.gov website appeared in line with protocol. |
| Other bias | Low risk | None identified. |
Watz 2016.
| Methods |
Study ID and dates performed: ; NCT01996319 (MOVE); study dates not reported. Study design: Randomised, placebo‐controlled, double‐blind, multicentre, cross‐over study. Duration of study: Flexible run‐in period (duration dependent on COPD medication at baseline; two 21‐day treatment periods separated by a 14‐day washout. Study setting, location, number of centres: Multicentre, randomised, double‐blind, placebo‐controlled cross‐over study, conducted at 30 secondary care (pulmonology) practices in Germany. Key inclusion criteria: Stable COPD according to the current GOLD guidelines (GOLD 2013); current or ex‐smokers; smoking history of ≥ 10 pack years; airflow limitation indicated by a post‐bronchodilator FEV1 ≥ 40% and < 80% of the predicted normal, and a post‐bronchodilator FEV1/FVC < 0.70. Key exclusion criteria: Concomitant pulmonary disease, history of asthma, onset of respiratory symptoms prior to age 40 years, blood eosinophil count > 600/mm3 during run‐in, or a clinically significant abnormality that could interfere with the assessment of efficacy or safety of the study; COPD exacerbation in the 6 weeks prior to screening or during the run‐in period; respiratory tract infection within 4 weeks prior to screening or during the run‐in period. Concomitant medications: The following COPD medication was prohibited from the indicated time prior to visit 2 and for the duration of the study: LAMAs (7 days); LABAs (48 h; 7 days for indacaterol); xanthines and oral phosphodiesterase IV inhibitors (7 days). ICS were permitted, at a stable dose throughout the study (participants on a LABA/ICS combination were to be switched to the nearest equivalent dose of ICS monotherapy at least 48 h prior to visit 2). |
|
| Participants |
Note: cross‐over study therefore participant data reported for whole cohort. N randomised: N = 194. N analysed: N = 194. Mean age (SD), years: 62.8 (7.9). Gender ‐ male, n/N (%): 127/194 (65.5). Baseline lung function ‐ mean (SD), post‐bronchodilator % predicted FEV1: 61.6 (10.7). Smoking status, current smoker, n/N (%): 110/194 (56.7). |
|
| Interventions |
Intervention: Once‐daily IND/GLY 110/50 µg. Comparator: Once‐daily placebo. |
|
| Outcomes |
Prespecified outcomes: Primary: Change from baseline in peak IC (IND/GLY versus placebo); change from baseline in average physical activity level (IND/GLY versus placebo). Secondary: Average number of steps per day; change in the duration of at least moderate activity per day; change from baseline in peak IC; change from baseline in the trough IC; peak FEV1 at day 1; trough FEV1 comparison after 22 days. Reported outcomes: Prespecified outcomes (clinicaltrials.gov) were well reported. |
|
| Notes | Funding for trial; notable author COIs: This study was funded by Novartis Pharma. All authors were employees of Novartis, or received research funding/honoraria from Novartis. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The random number sequence was generated by the sponsor using a validated automated system. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants, staff, persons performing the assessments, sponsor, and data analysts were blinded to treatment. |
| Blinding of participants and personnel (objective outcomes ‐ performance bias) Objective outcomes | Low risk | Participants, staff, persons performing the assessments, sponsor, and data analysts were blinded to treatment. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Persons performing the assessments were blinded to treatment. |
| Blinding of outcome assessment (objective outcomes ‐ detection bias) All outcomes | Low risk | Persons performing the assessments were blinded to treatment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All randomised participants received at least one dose of allocated treatment and were included in the full analysis set. |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes (clinicaltrials.gov) were well reported. |
| Other bias | Low risk | Cross‐over design may not be appropriate for a study that examines physical activity end points, which require a change in lifestyle ‐ washout periods may be insufficient. However, physical activity end points were not included in the present meta‐analyses. |
Zheng 2014.
| Methods |
Study ID and dates performed: NCT01636713; dates not reported. Study design: Multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. Duration of study: 7–14 day run‐in; 24‐week treatment period. Study setting, location, number of centres: People’s Republic of China, Philippines, South Korea, Taiwan, Thailand. Key inclusion criteria: Aged ≥ 40 years at screening; established clinical history of COPD (ATS/ERS criteria); current or former smokers with a smoking history ≥ 10 pack‐years; post‐albuterol FEV1/FVC < 0.70 and a post‐albuterol FEV1 ≤ 70% of predicted normal values (NHANES III reference equations at visit 1); dyspnoea score of ≥ 2 on the mMRC Dyspnea Scale at screening. Key exclusion criteria: Current diagnosis of asthma or any other known respiratory disorder, including α1‐anti‐trypsin deficiency or active lung infection, e.g. tuberculosis, lung cancer, clinically significant bronchiectasis, pulmonary hypertension, sarcoidosis, or interstitial lung disease; previous history or current evidence of clinically significant or uncontrolled cardiovascular, neurological, psychiatric, renal, hepatic, immunological, endocrine, or haematological abnormalities. Concomitant medications: Permitted: Supplemental albuterol as rescue medication; ICS < 1000 µg/day of FP or equivalent; ICS not initiated or discontinued within 30 days prior to study entry. |
|
| Participants |
N randomised: UMEC/VI 62.5/25 µg: n = 194; UMEC/VI 125/25 µg: n = 193; placebo: n = 193. N analysed, n/N (100): UMEC/VI 62.5/25 µg: 194/194 (100); UMEC/VI 125/25 µg: 193/193 (100); placebo: 193/193 (100). Mean age (SD), years: UMEC/VI 62.5/25 µg: 64.0 (8.71); UMEC/VI 125/25 µg: 63.7 (8.26); placebo: 64.3 (8.78). Gender ‐ male, n (%): UMEC/VI 62.5/25 µg: 183 (94); UMEC/VI 125/25 µg: 182 (94); placebo: 177 (92). Baseline lung function ‐ mean (SD) post‐bronchodilator FEV1, L: UMEC/VI 62.5/25 µg: 1.131 (0.3965); UMEC/VI 125/25 µg: 1.195 (0.3889); placebo: 1.168 (0.3708). Smoking status, current smoker, n/N (%): UMEC/VI 62.5/25 µg: 56/194 (29); UMEC/VI 125/25 µg: 48/193 (25); placebo: 65/193 (34). |
|
| Interventions |
Intervention: Once‐daily UMEC/VI 62.5/25 µg; once‐daily UMEC/VI 125/25 µg. Comparator: Once‐daily placebo. |
|
| Outcomes |
Prespecified outcomes: Primary: Change from baseline in trough FEV1 on day 169 (week 24). Secondary: Transition Dyspnea Index (TDI) Focal Score at day 168 (week 24); change from baseline weighted mean 0‐6 hour FEV1 obtained post‐dose at day 1. Reported outcomes: Prespecified outcomes (clinicaltrials.gov) were well reported, plus trough FEV1 at other time points; serial FEV1 over 0–6 hours post‐dose at day 1; the proportion of participants achieving an increase in FEV1 of ≥ 12% and ≥ 0.200 L above baseline at any time 0–6 hours post‐dose on day 1; the proportion of participants achieving an increase of ≥ 0.100 L above baseline in trough FEV1; and trough and serial FVC and time to onset 0–6 hours post‐dose at day 1; TDI focal score recorded at other time points; proportion of TDI responders (a responder to TDI was defined as a participant who reported a TDI score of ≥ 1 unit); rescue‐albuterol use (percentage of rescue‐free days and puffs/day) and time to first COPD exacerbation (defined as an acute worsening of symptoms of COPD requiring the use of rescue albuterol or any treatment beyond study medication); safety. |
|
| Notes | Funding for trial; notable author COIs: GSK funded this study. Lead author has received lecture fees from GSK. 3 of 4 co‐authors are employees of GSK. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The randomisation schedule was generated by GSK using the validated computerised system RandAll version 2.5. |
| Allocation concealment (selection bias) | Low risk | Used the sponsors formal system for randomisation so although concealment not specifically stated, it seems likely that this was done. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and investigators were blinded to treatment allocation. |
| Blinding of participants and personnel (objective outcomes ‐ performance bias) Objective outcomes | Low risk | Participants and investigators were blinded to treatment allocation. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Participants for subjective outcomes were the outcome assessors; therefore, low risk of bias. |
| Blinding of outcome assessment (objective outcomes ‐ detection bias) All outcomes | Low risk | Assessment of objective outcomes would be unlikely to be influenced by knowledge of treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition based on completion rates < 20% across arms. Data reported for ITT population. |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes (clinicaltrials.gov) were well reported. |
| Other bias | High risk | Higher proportion of participants with GOLD Stage IV in the UMEC/VI 62.5/25 µg group compared with placebo; may skew the treatment effect in favour of placebo. Lower proportion of current smokers than comparable studies. |
AE: adverse event; AUC: area under the curve; ATS: American Thoracic Society; BD: bronchodilator; bpm: beats per minute; CDLM: capacity of daily living during the morning; COI: conflict of interest; COPD: chronic obstructive pulmonary disease; CR10: category ratio 10; ECG: echocardiogram; ERS: European Respiratory Society; FEV1: forced expiratory volume in 1 second; FP: fluticasone propionate; FPI‐SF: functional performance inventory‐short‐form; FRC: functional residual capacity; FVC: forced vital capacity; GOLD: Global Initiative for chronic obstructive pulmonary disease; H1: histamine 1; IC: inspiratory capacity; ICS: inhaled corticosteroid; IgE: immunoglobulin E; IND: indacaterol; IRT: interactive voice response system; ITT: intent to treat; IVRS: interactive voice response system; LABA: long‐acting beta‐adrenoceptor agonist; LAMA: long‐acting muscarinic antagonist; LSM: least squares mean; MACE: major adverse cardiovascular event; MCID: minimally clinically important difference; mITT: modified intent‐to‐treat; mMRC: modified Medical Research Council; MRC: Medical Research Council; N: number; O2: oxygen; OCS: oral corticosteroids; OLO: olodaterol; PDE4: phosphodiesterase 4; PK Cmax: pharmacokinetic maximum plasma concentration; prn: pro re nata (as needed); QT: Q‐T interval; RAMOS: registration and medical ordering system; SABA: short‐acting beta‐adrenoceptor agonist; SAE: serious adverse event; SaO2: oxygen saturation; SD: standard deviation; SGRQ: St George's Respiratory Questionnaire; SOBDA: shortness of breath with daily activities; SpO2: peripheral capillary oxygen saturation; SSRI: selective serotonin reuptake inhibitor; TDI: transition dyspnoea index; TIO: tiotropium; tmax:time to maximum plasma concentration; UMEC: umeclidinium; VI: vilanterol; W: Watt.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Aalbers 2015 | Wrong intervention (combination inhaler not used) |
| Asai 2013 | Wrong comparator (different LABA used in control arm) |
| Berton 2009 | Wrong intervention (LABA/LAMA not administered in combined inhaler) |
| Buhl 2011 INTENSITY | Wrong intervention (LABA/LAMA combination not evaluated) |
| Buhl 2015 TONADO | 2nd Round: Wrong comparator (no placebo) |
| D'Urzo 2014 AUGMENT | 2nd ROUND: Wrong intervention ‐ LAMA/LABA administered twice daily |
| Decramer 2014 | 2nd Round: Wrong comparator (no placebo) |
| Di Marco 2005 | Duration of treatment < 3 weeks |
| Donohue 2013b | 2nd ROUND: Wrong intervention ‐ LAMA/LABA administered twice daily |
| Donohue 2016 | Duration of treatment < 3 weeks |
| Donohue 2016b | 2nd Round: Wrong comparator (no placebo) |
| EUCTR2007‐003648‐31 | Duration of treatment < 3 weeks |
| EUCTR2007‐004435‐30 | Wrong intervention (combination inhaler not used) |
| EUCTR2009‐015901‐38 | Duration of treatment < 3 weeks |
| Evdokimov 2015 | 2nd Round: Wrong comparator (no placebo) |
| Evdokimov 2015b | 2nd Round: Wrong comparator (no placebo) |
| Ferguson 2015 | Wrong comparator (dose of indacaterol in the control group was greater than that in combination arm) |
| Fogarty 2014 | Duration of treatment < 3 weeks |
| Hanania 2016 | 2nd Round: Wrong comparator (no placebo) |
| Hoshino 2014 | Wrong intervention (combination inhaler not used) |
| Ichinose 2016 | No placebo group |
| Ichinose 2017 | 2nd Round: Wrong comparator (no placebo) |
| Imran 2015 | Duration of treatment < 3 weeks |
| Jones 2010 | Combined inhaler not used (LAMA plus LABA in individual inhalers) |
| Mahler 2015 | 2nd ROUND: Wrong intervention ‐ LAMA/LABA administered twice daily |
| Maltais 2010 | 2nd Round: Wrong comparator (no placebo) |
| NCT00308191 2006 | Wrong intervention (combination inhaler not used) |
| NCT00424528 2006 | Wrong intervention (combination inhaler not used) |
| NCT00696020 2008 | 2nd Round: Wrong comparator (no placebo) |
| NCT00720499 2008 | Wrong comparator (no placebo) |
| NCT00845728 2009 | Wrong intervention (LABA/LAMA combination not evaluated) |
| NCT00846586 2009 | Wrong intervention (combination inhaler not used) |
| NCT00877383 2009 | Wrong intervention (combination inhaler not used) |
| NCT01040689 2010 | Wrong intervention (LABA/LAMA combination not evaluated) |
| NCT01040728 2010 | Wrong intervention (LABA/LAMA combination not evaluated) |
| NCT01049360 2009 | Treatment duration < 3 weeks |
| NCT01437540 2011 | 2nd Round: Wrong comparator (no placebo) |
| NCT01476813 2012 | Wrong intervention (LABA/LAMA combination not evaluated) |
| NCT01491802 2012 | 2nd Round: Wrong comparator (no placebo) |
| NCT01529632 2012 | Wrong comparator (both individual LAMA and LABA received) |
| NCT01536262 2012 | 2nd Round: Wrong comparator (no placebo) |
| NCT01551888 2012 | Treatment duration < 3 weeks |
| NCT01574651 2012 | Wrong comparator (Indaceterol or glycopyrronium (i.e. FDC evaluated in intervention arm) not evaluated in comparator arm) |
| NCT01682863 2012 | Wrong comparator (indacaterol dose in control arm different than used in FDC in intervention arm) |
| NCT01697696 2012 | Wrong intervention (LABA/LAMA combination not evaluated) |
| NCT01703845 2012 | Wrong comparator (no LAMA or LABA alone, or placebo) |
| NCT01817764 2013 | Wrong comparator (no single agent, or placebo) |
| NCT01985334 2014 | Wrong comparator (individual LAMA or LABA could be used based on prior treatment) |
| NCT02030535 2014 | Wrong study design (single‐dose study) |
| NCT02059434 2013 | Treatment duration < 3 weeks (single ascending‐dose study) |
| NCT02196714 2014 | Wrong patient population (healthy volunteers) |
| NCT02231177 2008 | 2nd Round: Wrong comparator (no placebo) |
| NCT02296138 2015 | No placebo group |
| NCT02343458 2015 | LAMA/LABA administered twice daily |
| NCT02429765 2015 | LAMA/LABA administered twice daily |
| NCT02442206 2015 | Treatment duration < 3 weeks |
| NCT02465567 2015 | Wrong comparator (no single agent, or placebo) |
| NCT02487446 2015 | Wrong comparator (no single agent, or placebo) |
| NCT02487498 2015 | Wrong comparator (no single agent, or placebo) |
| NCT02579850 2015 | Wrong comparator (no single agent, or placebo) |
| NCT02643082 2015 | Treatment duration < 3 weeks |
| NCT02796677 2016 | LAMA/LABA administered twice daily |
| NCT02845752 2016 | Treatment duration < 3 weeks |
| NCT02937584 2016 | Wrong intervention (LABA/LAMA combination not evaluated) |
| NCT02988869 2016 | No placebo group |
| NCT03022097 2017 | LAMA/LABA administered twice daily |
| NCT03024346 2016 | Duration of treatment < 3 weeks |
| NCT03034915 2017 | No placebo group |
| Orevillo 2016 | Duration of treatment < 3 weeks |
| Rabe 2015 | 2nd ROUND: Wrong intervention ‐ LAMA/LABA administered twice daily |
| Reisner 2011 | Duration of treatment < 3 weeks |
| Reisner 2013 | Duration of treatment < 3 weeks |
| Reisner 2016 | 2nd ROUND: Wrong intervention ‐ LAMA/LABA administered twice daily |
| Reisner 2017 | Duration of treatment < 3 weeks |
| Reisner 2017b | 2nd Round: Wrong comparator (no placebo) |
| Sadigov 2014 | 2nd Round: Wrong comparator (no placebo) |
| Salomon 2017 | Wrong intervention (LABA/LAMA not administered in combined inhaler) |
| Setoguchi 2015 | 2nd Round: Wrong comparator (no placebo) |
| Singh 2014 | 2nd ROUND: Wrong intervention ‐ LAMA/LABA administered twice daily |
| Sliwinski 2010 | Wrong intervention (LABA/LAMA not administered in combined inhaler) |
| Tanaka 2015 | 2nd Round: Wrong comparator (no placebo) |
| Tashkin 2007 | Wrong intervention (LABA/LAMA not administered in combined inhaler) |
| Tashkin 2016 | Duration of treatment < 3 weeks |
| Ulubay 2005 | 2nd Round: Wrong comparator (no placebo) |
| Van de Maele 2010 | Duration of treatment < 3 weeks |
| Van Noord 2005 | Wrong intervention (LABA/LAMA not administered in combined inhaler) |
| Van Noord 2010 | Duration of treatment < 3 weeks |
| Velazquez‐Uncal 2016 | Wrong intervention (LABA/LAMA not evaluated) |
| Vincken 2013 | Wrong intervention (LABA/LAMA not administered in combined inhaler) |
| Vogelmeier 2008 | Wrong intervention (LABA/LAMA not administered in combined inhaler) |
| Vogelmeier 2013 | Wrong comparator (LABA/LAMA vs LABA steroids) |
| Watz 2017 | 2nd ROUND: Wrong intervention ‐ LAMA/LABA administered twice daily |
| Webb 2015 | 2nd Round: Wrong comparator (no placebo) |
| Wedzicha 2013 | 2nd Round: Wrong comparator (no placebo) |
| Yosuke 2014 | 2nd Round: Wrong comparator (no placebo) |
| ZuWallack 2014 | Wrong intervention (LABA/LAMA not administered in combined inhaler) |
FDC: LABA: LAMA:
to be completed
Characteristics of studies awaiting assessment [ordered by study ID]
NCT02233543 2014.
| Methods |
Study ID and dates performed: NCT02233543; Nov 2014 to June 2016; trial registry entry only (clinicaltrials.gov); no data submitted. Study design: Randomised, placebo‐controlled, cross‐over trial. Duration of study: 12 weeks (4 weeks per treatment, 2‐week washout). Study setting, location, number of centres: Not reported. Key inclusion criteria: Aged ≥ 40 years; clinical diagnosis of COPD (according to GOLD guidelines, updated 2014) with a post‐bronchodilator FEV1/FVC < 0.70; post‐bronchodilator FEV1 ≥ 30% and < 60% of the predicted normal value; resting daytime oxygen saturation levels measured by pulse oximetry of ≤ 95% SpO2; smoking history of at least 10 pack years (ten pack‐years are defined as 20 cigarettes a day for 10 years, or 10 cigarettes a day for 20 years). Key exclusion criteria: An exacerbation of COPD (treatment with oral or parenteral antibiotics and/or glucocorticosteroids and/or hospitalisation related to COPD) within 4 weeks prior to screening or during the run‐in period; diagnosed asthma; participants receiving regular long‐term oxygen therapy; ongoing/planned rehabilitation during the study period; three or more awakenings during the night leading to toilet visit or other reasons for exiting the bed during the last week prior to the screening visit due to non‐COPD reasons. Concomitant medications: Not reported. |
| Participants |
Note: cross‐over study therefore participant data reported for whole cohort; no data posted. N randomised: No data posted to trial registry site; no report available. N analysed: No data posted to trial registry site; no report available. Mean age (SD), years: No data posted to trial registry site; no report available. Gender ‐ male, n/N (%): No data posted to trial registry site; no report available. Baseline lung function ‐ post‐bronchodilator % predicted FEV1: No data posted to trial registry site; no report available. Smoking status, current smoker, n/N (%): No data posted to trial registry site; no report available. |
| Interventions |
Intervention: Once‐daily IND/GLY 85/43 μg. Comparator: Once‐daily placebo. |
| Outcomes |
Prespecified outcomes: Primary: Mean night‐time blood oxygenation at 4 weeks.Secondary: Time during the night spent below 90% in blood oxygen saturation at 4 weeks. Reported outcomes: No result posted. |
| Notes | Funding for trial; notable author COIs: Study sponsored by Novartis Pharmaceuticals. |
AE, adverse event; BD, bronchodilator; COI: conflict of interest; COPD: chronic obstructive pulmonary disease; FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity; GLY: glycopyrrolate; GOLD: Global Initiative for Chronic Obstructive Lung Disease; ICS: inhaled corticosteroid; IND: indacaterol; IRT, Interactive Response Technology; MCID, minimum clinically important difference; SAE, serious adverse event; SD, standard deviation; SGRQ, St George's Respiratory Questionaire; SpO2: oxygen saturation; SSRI, selective serotonin reuptake inhibitor.
Differences between protocol and review
The original protocol did not specify if studies with a cross‐over design should be included or excluded. We saw no reason to exclude studies with a cross‐over design, which we elected to include in the review; the relevant sentence in the methods has been revised to reflect this.
The protocol originally stated: "Specifically, we will report on the following prespecified events: any arrhythmia, acute coronary syndrome, cardiac mortality, hospitalisation due to cardiac event, and sudden deaths. Admittedly, the scope of our review of adverse effects will be narrow. A broader scope may be possible in a review dedicated to safety data." With the exception of sudden deaths, the prespecified events of interest were not typically reported in primary study reports.Therefore, we elected to perform a more standard analysis of safety by examining all‐cause adverse events and serious adverse events.
The protocol originally stated: "Where multiple time points were reported for dichotomous outcomes, we chose the time point that maximises length of follow‐up for the randomised treatment period. Where multiple time points are reported for continuous outcomes, we will pool results for three time points: six, twelve and 24 months." We elected to report data for dichotomous and continuous outcome using the time point that maximised the length of follow‐up for each study. Calzetta and colleagues performed a systematic review and meta‐analysis of fixed‐dose combination LABA/LAMA versus their mono‐components (Calzetta 2017). The authors reported that the overall superiority of LABA/LAMA fixed‐dose combinations over their mono‐components was greater after three months of treatment and diminished after six and 12 months of treatment, suggesting that continued improvements elicited by LABA/LAMA combinations can be expected during the initial three months of treatment, after which the benefits of dual bronchodilation remain stable. Based on the consistent treatment effects anticipated after three months, we took the view that it was of more interest to subgroup the studies by LABA/LAMA combination/dose rather than by study duration. Additional subgrouping by duration would also further reduce sample size.The original sentence in the protocol has been revised.
The protocol originally stated that "The rate ratio will be our primary summary statistic" for the analysis of AECOPD. However, we elected to report the odds ratio; the relevant sentence in the methods has been amended.
The protocol originally specified that CRQ will be reported in the 'summary of findings' table for the outcome HRQoL. We elected to report SGRQ because the majority of studies reported SGRQ and no studies reported CRQ; the relevant sentence in the methods has been revised.
We specified subgroups for different LABA/LAMA combinations and baseline COPD severity after publication of the protocol.
Contributions of authors
Dr Ho, Dr Maqsood, and Dr Evans updated and wrote the background section.
All authors contributed to the development of the review. See acknowledgement statement.
Sources of support
Internal sources
-
Lancaster University, UK.
David Evans received support in the form of a salary.
External sources
The authors declare that no such funding was received for this systematic review, Other.
Declarations of interest
U Maqsood: none known.
D Evans: provides freelance writing support to medical communications agencies.
T Ho: none known.
K Palmer: none known.
F Eccles: none known.
Ran Wang: none known.
M Munavvar: Received honorarium for lectures and to attend the European Respiratory Society meeting, in the past, from various companies, including Astra Zeneca, Chiesi, GSK, Almirall, Boehringer Ingelheim, and Pneumrx.
I Crossingham: none known.
New
References
References to studies included in this review
Bateman 2013 {published data only}
- Asai K, Hirata K, Hashimoto S, Fukuchi Y, Kitawaki T, Ikeda K, et al. Efficacy and safety of indacaterol/glycopyrronium in Japanese patients with COPD: pooled analysis of SHINE and ARISE. Respiratory Investigation 2016;54(6):428‐35. [2212‐5353] [DOI] [PubMed] [Google Scholar]
- Barnes N, Bateman E, Gallagher N, Green Y, Horton R, Henley M, et al. QVA149 once daily provides superior bronchodilation versus indacaterol, glycopyrronium, tiotropium and placebo: the SHINE study. Thorax 2012; Vol. 67:A147 [P192].
- Bateman E, Ferguson GT, Barnes N, Gallagher N, Green Y, Horton R, et al. Benefits of dual bronchodilation with QVA149 once daily versus placebo, indacaterol, NVA237 and tiotropium in patients with COPD: the SHINE study. European Respiratory Journal. 2012; Vol. 40:509s [2810].
- Bateman ED, Ferguson GT, Barnes N, Gallagher N, Green Y, Henley M, et al. Dual bronchodilation with QVA149 versus single bronchodilator therapy: the SHINE study. European Respiratory Journal 2013;42(6):1484‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman ED, Welte T, Hashimoto S, Gallagher N, Green Y, Horton R, et al. Dual bronchodilation with once‐daily QVA149 provides superior bronchodilation compared to its mono‐components and tiotropium in all subgroups of patients with COPD: the shine study. American Journal of Respiratory and Critical Care Medicine 2013;187:A4263. [Google Scholar]
- CQVA149A2303. A 26‐week treatment, multi‐center, randomized, double‐blind, parallel‐group, placebo and active controlled (open label) study to assess the efficacy, safety and tolerability of QVA149 (110/50 μg q.d.) in patients with moderate to severe chronic obstructive pulmonary disease (COPD). www.novctrd.com/ctrdWebApp/clinicaltrialrepository/ (first received 21 September 2010).
- Chapman K, Bateman E, Gallagher N, Hu H, Banerji D. Once‐daily QVA149 improves lung function, dyspnea, and health status independent of disease severity and prior medications: the shine study. Chest 2014;145:3. [Google Scholar]
- Chapman KR, Bateman ED, Gallagher N, Hu H, Banerji D. QVA149 once daily improves lung function, dyspnoea and health status independent of prior medications and disease severity: the SHINE study. Thorax 2013;68:A182 [P234]. [Google Scholar]
- Chapman KR, Bateman ED, Gallagher N, Hu H, Kim HJ, Banerji D. Once‐daily QVA149 improves lung function, dyspnea and health status regardless of disease severity and prior medications: the SHINE study. Respirology 2013;18:31 [OS100]. [1323‐7799] [Google Scholar]
- Frith P, Thompson P, Wark P, Lindstrom S, Bateman E. Benefits of dual bronchodilation with QVA149 once daily versus placebo, indacaterol, NVA237 and tiotropium in patients with COPD: the Shine study. Respirology 2013;18:20 [O041]. [1323‐7799] [Google Scholar]
- Hashimoto S, Ikeuchi H, Murata S, Kitawaki T, Ikeda K, Banerji D. Efficacy and safety of indacaterol/glycopyrronium in Japanese patients with COPD: a subgroup analysis from the SHINE study. International Journal of Chronic Obstructive Pulmonary Disease 2016;11:2543‐51. [1178‐2005] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01202188. A study to assess the efficacy, safety and tolerability of once‐daily (q.d.) QVA149 in patients with moderate to severe chronic obstructive pulmonary disease (COPD) (SHINE). clinicaltrials.gov/ct2/show/NCT01202188 (first received 15 September 2010).
- Wark P, Chapman K, Bateman E, Olsson P, Chen H, Banerji D, et al. Effect of QVA149 on lung function in the subgroup of patients with a history of exacerbations in the previous year and baseline ICS use from the shine study. Respirology 2015;20:81. [Google Scholar]
- Wark P, Tillmann H, Jones I, Fogel R, Banerji D. Qva149 provides significant symptomatic benefit to COPD patients as measured by a new symptom e‐diary: the SHINE study. Respirology 2016;21:128 [TP 065]. [Google Scholar]
- Welte T, Barnes N, Nagase T, Gallagher N, Green Y, Horton R, et al. QVA149 once daily is safe and well tolerated in patients with COPD: the SHINE Study. American Journal of Respiratory and Critical Care Medicine 2013;187:A1478. [Google Scholar]
- Welte T, Gallagher N, Green Y, Henley M, Banerji D. Dual bronchodilation with once‐daily QVA149 provides significantly improved clinical outcomes versus mono‐bronchodilator therapy: the SHINE study. American Journal of Respiratory and Critical Care Medicine 2013;187:A4272. [Google Scholar]
Beeh 2014 {published data only}
- Beeh KM, Korn S, Beier J, Jadayel D, Henley M, D'Andrea P, et al. Effect of QVA149 on lung volumes and exercise tolerance in COPD patients: the BRIGHT study. Respiratory Medicine 2014;108(4):584‐92. [DOI] [PubMed] [Google Scholar]
- Beeh KM, Korn S, Beier J, Jadayel D, Henley M, Tylek J, et al. QVA149 once daily improves exercise tolerance and lung function in patients with moderate to severe COPD: the BRIGHT study. Thorax 2012;67:A147 [P191]. [Google Scholar]
- Beeh KM, Korn S, Beier J, Jadayel D, Henley M, Tylek J, et al. QVA149 once daily reduces lung hyperinflation, improves lung function and volumes as measured by body plethysomography: the BRIGHT study. American Journal of Respiratory and Critical Care Medicine 2013;187:A4259. [Google Scholar]
Beeh 2015 {published data only}
- Beeh K, Westerman J, Kirsten A, Hebert J, Gronke L, Hamilton A, et al. The 24‐h lung‐function profile of once‐daily tiotropium and olodaterol fixed‐dose combination in chronic obstructive pulmonary disease. Pulmonary Pharmacology and Therapeutics 2015;32:53‐9. [1094‐5539] [DOI] [PubMed] [Google Scholar]
- Derom E, Westerman J, Groenke L, Hamilton A, Li C, Beeh K. The 24‐hour lung function profile of once‐daily tiotropium and olodaterol fixed‐dose combination compared with placebo and monotherapies in chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine 2014;69:A190‐1. [Google Scholar]
- NCT01559116. Characterization of 24‐hour lung function profiles of inhaled tiotropium + olodaterol fixed dose combination in patients suffering from chronic obstructive pulmonary disease. clinicaltrials.gov/ct2/show/NCT01559116 (first received 21 March 2012).
Celli 2014 {published data only}
- Celli B, Crater G, Kilbride S, Mehta R, Tabberer M, Kalberg C, et al. Once‐daily umeclidinium/vilanterol 125/25 mcg in COPD: a randomized, controlled study. Chest 2014;145(5):981‐91. [DOI] [PubMed] [Google Scholar]
- Celli BR, Crater G, Kilbride S, Mehta R, Tabberer M, Kalberg CJ, et al. A 24‐week randomized, double‐blind, placebo‐controlled study of the efficacy and safety of once‐daily umeclidinium/vilanterol 125/25 mcg in COPD. American Journal of Respiratory and Critical Care Medicine 2013;187:A2435. [Google Scholar]
- Donohue JF, Niewoehner D, Brooks J, O'Dell D, Church A. Safety and tolerability of once‐daily umeclidinium/vilanterol 125/25 mcg and umeclidinium 125 mcg in patients with chronic obstructive pulmonary disease: results from a 52‐week, randomized, double‐blind, placebo‐controlled study. Respiratory Research 2014;15:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01313637. A 24‐week evaluation of GSK573719/Vilanterol (125/25mcg) and components in COPD. clinicaltrials.gov/ct2/show/NCT01313637 (first received 14 March 2011).
Dahl 2013 {published data only}
- Dahl R, Chapman K, Rudolf M, Mehta R, Kho P, Alagappan V, et al. QVA149 administered once daily provides significant improvements in lung function over 1 year in patients with COPD: the ENLIGHTEN study. European Respiratory Journal 2012;40:529s [P2896]. [Google Scholar]
- Dahl R, Chapman K, Rudolf M, Mehta R, Kho P, Alagappan V, et al. QVA149 once daily provides significant improvements in lung function over 1 year in patients with COPD: the Enlighten study. Thorax 2012;67:A146 [P190]. [Google Scholar]
- Dahl R, Chapman KR, Rudolf M, Mehta R, Kho P, Alagappan VK, et al. Safety and efficacy of dual bronchodilation with QVA149 in COPD patients: the ENLIGHTEN study. Respiratory Medicine 2013;107(10):1558‐67. [DOI] [PubMed] [Google Scholar]
- Dahl R, Jadayel D, Alagappan V, Chen H, Banerji D. Once‐daily QVA149 provides the same efficacy as the free combination of its monocomponents indacaterol and glycopyrronium: the BEACON study. European Respiratory Society 23rd Annual Congress; 2013 Sep 7‐11; Barcelona. 2013; Vol. 42:694s [P3390].
- NCT01120717. A study to assess the long‐term safety of QVA149 (ENLIGHTEN). clinicaltrials.gov/ct2/show/NCT01120717 (first received 11 May 2010).
Donohue 2013 {published data only}
- Donohue JF, Maleki‐Yazdi MR, Kilbride S, Mehta R, Kalberg C, Church A. Efficacy and safety of once‐daily umeclidinium/vilanterol 62.5/25 mcg in COPD. Respiratory Medicine 2013;107(10):1538‐46. [DOI] [PubMed] [Google Scholar]
- NCT01313650. A 24‐week evaluation of GSK573719/Vilanterol (62.5/25mcg) and components in COPD (DB2113373). clinicaltrials.gov/ct2/show/NCT01313650 (first received 14 March 2011).
Feldman 2012 {published data only}
- Feldman G, Walker RR, Brooks J, Mehta R, Crater G. 28‐day safety and tolerability of umeclidinium in combination with vilanterol in COPD: a randomized placebo‐controlled trial. Pulmonary Pharmacology and Therapeutics 2012;25(6):465‐71. [DOI] [PubMed] [Google Scholar]
Larbig 2015 {published data only}
- Larbig M, Fowler‐taylor A, Maitra S, Schubert‐Tennigkeit A, Banerji D. Efficacy and safety of IND/GLY versus placebo and tiotropium in symptomatic patients with moderate‐to‐severe COPD: the 52‐week radiate study. Respirology 2015;20:44. [1323‐7799] [Google Scholar]
- NCT01610037. Comparison of long‐term safety of the combination product QVA149A against placebo and standard of care treatment in chronic obstructive pulmonary disease patients with moderate to severe airflow limitation. clinicaltrials.gov/ct2/show/NCT01610037 (first received 1 June 2012).
Mahler 2014 {published data only}
- D'Urzo A, Mahler D, Worth H, White T, Alagappan V, Chen H, et al. Patients with severe COPD show significant improvements in dyspnea and lung function with once‐daily QVA149: the blaze study. European Respiratory Society 23rd Annual Congress; 2013 Sep 7‐11; Barcelona. 2014; Vol. 145.
- D'Urzo A, Mahler DA, Decramer M, Worth H, White T, Alagappan VKT, et al. Superior lung function with once‐daily QVA149 translates into improvements in patient‐reported breathlessness compared with placebo and tiotropium in COPD patients: the BLAZE study. Thorax. 2013:A184 [P236].
- Mahler DA, Decramer M, D'Urzo A, Worth H, White T, Alagappan VKT, et al. Dual bronchodilation with QVA149 reduces patient‐reported dyspnoea in COPD: the BLAZE study. European Respiratory Journal 2014;43(6):1599‐609. [DOI] [PubMed] [Google Scholar]
- Mahler DA, Decramer M, D'Urzo AD, Worth H, White T, Alagappan V, et al. Superior lung function with once‐daily QVA149 translates into improvements in patient‐reported breathlessness compared with placebo and tiotropium in COPD patients: the BLAZE study. American Journal of Respiratory and Critical Care Medicine 2013;187:A6070. [Google Scholar]
Maltais 2014 {published data only}
- Maltais F, Galdiz Iturri JB, Kirsten A, Singh D, Hamilton A, Tetzlaff K, et al. Effects of 12 weeks of once‐daily tiotropium and olodaterol fixed‐dose combination on exercise endurance in patients with COPD. European Respiratory Journal 2014;44:P283. [Google Scholar]
- Maltais F, Galdiz Iturri JB, Kirsten A, Singh D, Hamilton A, Tetzlaff K, et al. Effects of 12 weeks of once‐daily tiotropium and olodaterol fixed‐dose combination on exercise endurance in patients with COPD. Thorax 2014;69:A186‐7. [Google Scholar]
- Maltais F, O'Donnell D, Gáldiz Iturri JB, Kirsten AM, Singh D, Hamilton A, et al. Effect of 12 weeks of once‐daily tiotropium/olodaterol on exercise endurance during constant work‐rate cycling and endurance shuttle walking in chronic obstructive pulmonary disease. Therapeutic Advances in Respiratory Disease 2018;12:1753465818755091. [DOI: 10.1177/1753465818755091] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01525615. A study to determine the effect of tiotropium + olodaterol fixed dose combination on exercise endurance time during constant work rate cycle ergometry test in COPD. clinicaltrials.gov/ct2/show/NCT01525615 (first received 3 February 2012).
Maltais 2014b {published data only}
- Maltais F, Singh S, Donald A, Church A, Crater G, Goh A, et al. Effects of a combination of vilanterol and umeclidinium on exercise endurance in subjects with COPD: two randomised clinical trials. European Respiratory Society 23rd Annual Congress; 2013 Sep 7‐11; Barcelona. Vol. 42:145s [P761].
- Maltais F, Singh S, Donald A, Crater G, Church A, Goh A. Effects of a combination of umeclidinium/vilanterol on exercise endurance in patients with chronic obstructive pulmonary disease: Two randomized, double‐blind clinical trials. Therapeutic Advances in Respiratory Disease 2016;10(3):Erratum in: Therapeutic Advances in Respiratory Disease 2014; 8: 169‐181; 289. [1753‐4666] [DOI] [PubMed] [Google Scholar]
- Maltais F, Singh S, Donald AC, Crater G, Church A, Goh AH, et al. Effects of a combination of umeclidinium/vilanterol on exercise endurance in patients with chronic obstructive pulmonary disease: two randomized, double‐blind clinical trials. Therapeutic Advances in Respiratory Disease 2014;8(6):169‐81. [1753‐4666] [DOI] [PubMed] [Google Scholar]
- Singh S, Maltais F, Tombs L, Fahy WA, Vahdati‐Bolouri M, Locantore N, et al. Relationship between exercise endurance and static hyperinflation in a post hoc analysis of two clinical trials in patients with COPD. International Journal of Chronic Obstructive Pulmonary Disease 2018;13:203‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Maltais 2014c {published data only}
- Maltais F, Singh S, Donald A, Church A, Crater G, Goh A, et al. Effects of a combination of vilanterol and umeclidinium on exercise endurance in subjects with COPD: two randomised clinical trials. European Respiratory Society 23rd Annual Congress; 2013 Sep 7‐11; Barcelona. 2013; Vol. 42:145s [P761].
- Maltais F, Singh S, Donald A, Crater G, Church A, Goh A. Effects of a combination of umeclidinium/vilanterol on exercise endurance in patients with chronic obstructive pulmonary disease: two randomized, double‐blind clinical trials. Therapeutic Advances in Respiratory Disease 2016;10(3):Erratum in: Therapeutic Advances in Respiratory Disease 2014; 8: 169‐181; 289. [1753‐4666] [DOI] [PubMed] [Google Scholar]
- Maltais F, Singh S, Donald AC, Crater G, Church A, Goh AH, et al. Effects of a combination of umeclidinium/vilanterol on exercise endurance in patients with chronic obstructive pulmonary disease: two randomized, double‐blind clinical trials. Therapeutic Advances in Respiratory Disease 2014;8(6):169‐81. [1753‐4666] [DOI] [PubMed] [Google Scholar]
- Singh S, Maltais F, Tombs L, Fahy WA, Vahdati‐Bolouri M, Locantore N, et al. Relationship between exercise endurance and static hyperinflation in a post hoc analysis of two clinical trials in patients with COPD. International Journal of Chronic Obstructive Pulmonary Disease 2018;13:203‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT00626522 {published data only}
- NCT00626522. Aclidinium/formoterol fixed combination dose finding study. clinicaltrials.gov/ct2/show/NCT00626522 (first received 29 February 2008).
NCT02275052 {published data only}
- NCT02275052. A randomized, double‐blind, placebo‐controlled evaluation of the effect of the combination of umeclidinium and vilanterol on exercise endurance time in subjects with COPD. clinicaltrials.gov/ct2/show/NCT02275052 (first received 27 October 2014).
O'Donnell 2015a {published data only}
- NCT01533922. Effect on exercise endurance and lung hyperinflation of tiotropium + olodaterol in COPD patients. clinicaltrials.gov/ct2/show/NCT01533922 (first received 16 February 2012).
- NCT01533935. Effect on exercise endurance and lung hyperinflation of tiotropium + olodaterol in COPD patients. clinicaltrials.gov/ct2/show/NCT01533935 (first received 16 February 2012).
- O'Donnell D, Casaburi R, Sousa D, Xue W, Frith P, Hamilton A, et al. Effects of 6 weeks' treatment with once‐daily tiotropium and olodaterol fixed‐dose combination on inspiratory capacity and exercise endurance in patients with COPD: the MORACTOTM studies. American Journal of Respiratory and Critical Care Medicine 2015;191:A3972. [Google Scholar]
- O'Donnell DE, Casaburi R, Frith P, Kirsten A, Sousa D, Hamilton A, et al. Effects of combined tiotropium/olodaterol on inspiratory capacity and exercise endurance in COPD. European Respiratory Journal 2017;49(4):1601348. [DOI: 10.1183/13993003.01348-2016] [DOI] [PMC free article] [PubMed] [Google Scholar]
O'Donnell 2015b {published data only}
- NCT01533922. Effect on exercise endurance and lung hyperinflation of tiotropium + olodaterol in COPD patients. clinicaltrials.gov/ct2/show/NCT01533922 (first received 16 February 2012).
- NCT01533935. Effect on exercise endurance and lung hyperinflation of tiotropium + olodaterol in COPD patients. clinicaltrials.gov/ct2/show/NCT01533935 (first received 16 February 2012).
- O'Donnell D, Casaburi R, Sousa D, Xue W, Frith P, Hamilton A, et al. Effects of 6 weeks' treatment with once‐daily tiotropium and olodaterol fixed‐dose combination on inspiratory capacity and exercise endurance in patients with COPD: the MORACTOTM studies. American Journal of Respiratory and Critical Care Medicine 2015;191:A3972. [Google Scholar]
- O'Donnell DE, Casaburi R, Frith P, Kirsten A, Sousa D, Hamilton A, et al. Effects of combined tiotropium/olodaterol on inspiratory capacity and exercise endurance in COPD. European Respiratory Journal 2017;49(4):pii 1601348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Siler 2016 {published data only}
- NCT02152605. A phase IIIb study to evaluate the efficacy of umeclidinium/vilanterol (UMEC/VI) in subjects with chronic obstructive pulmonary disease (COPD). clinicaltrials.gov/ct2/show/NCT02152605 (first received 2 June 2014).
- Siler TM, Donald AC, O'Dell D, Church A, Fahy WA. A randomized, parallel‐group study to evaluate the efficacy of umeclidinium/vilanterol 62.5/25 mug on health‐related quality of life in patients with COPD. International Journal of Chronic Obstructive Pulmonary Disease 2016;11(1):971‐9. [1178‐2005] [DOI] [PMC free article] [PubMed] [Google Scholar]
Singh 2016a {published data only}
- NCT01964352. Tiotropium + olodaterol fixed dose combination (FDC) in chronic obstructive pulmonary disease (OTEMTO 1). clinicaltrials.gov/ct2/show/NCT01964352 (first received 17 October 2013).
- NCT02006732. Tiotropium + olodaterol fixed dose combination (FDC) in chronic obstructive pulmonary disease (OTEMTO 2). clinicaltrials.gov/ct2/show/NCT02006732 (first received 10 December 2013).
- Singh D, Ferguson GT, Bolitschek J, Gronke L, Hallmann C, Bennett N, et al. Tiotropium + olodaterol fixed‐dose combination shows clinically meaningful improvements in quality of life versus placebo. European Respiratory Journal 2015;46:PA2958. [DOI: 10.1183/13993003.congress-2015.PA2958] [DOI] [PubMed] [Google Scholar]
- Singh D, Ferguson GT, Bolitschek J, Gronke L, Hallmann C, Bennett N, et al. Tiotropium + olodaterol shows clinically meaningful improvements in quality of life. Respiratory Medicine 2015;109(10):1312‐9. [DOI] [PubMed] [Google Scholar]
- Singh D, Ferguson GT, Bolitschek J, Grönke L, Hallmann C, Bennett N, et al. Tiotropium + olodaterol shows clinically meaningful improvements in quality of life versus placebo. Primary Care Respiratory Medicine 2016;26:21‐CR050. [DOI] [PubMed] [Google Scholar]
- Singh D, Gaga M, Schmidt O, Bjermer L, Gronke L, Voss F, et al. Effects of tiotropium + olodaterol versus tiotropium or placebo by COPD disease severity and previous treatment history in the OTEMTO(R) studies. Respiratory Research 2016;17(1):73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh D, Maleki‐Yazdi MR, Tombs L, Iqbal A, Fahy WA, Naya I. Prevention of clinically important deteriorations in COPD with umeclidinium/vilanterol. International Journal of Chronic Obstructive Pulmonary Disease 2016;11(1):1413‐24. [1178‐2005] [DOI] [PMC free article] [PubMed] [Google Scholar]
Singh 2016b {published data only}
- NCT01964352. Tiotropium + olodaterol fixed dose combination (FDC) in chronic obstructive pulmonary disease (OTEMTO 1). clinicaltrials.gov/ct2/show/NCT01964352 (first received 17 October 2013).
- NCT02006732. Tiotropium+olodaterol fixed dose combination (FDC) in chronic obstructive pulmonary disease (OTEMTO 2). clinicaltrials.gov/ct2/show/NCT02006732 (first received 10 December 2013).
- Singh D, Ferguson GT, Bolitschek J, Gronke L, Hallmann C, Bennett N, et al. Tiotropium + olodaterol fixed‐dose combination shows clinically meaningful improvements in quality of life versus placebo. European Respiratory Journal 2015;46:PA2958. [DOI: ] [DOI] [PubMed] [Google Scholar]
- Singh D, Ferguson GT, Bolitschek J, Gronke L, Hallmann C, Bennett N, et al. Tiotropium + olodaterol shows clinically meaningful improvements in quality of life. Respiratory Medicine 2015;109(10):1312‐9. [DOI] [PubMed] [Google Scholar]
- Singh D, Ferguson GT, Bolitschek J, Grönke L, Hallmann C, Bennett N, et al. Tiotropium + olodaterol shows clinically meaningful improvements in quality of life versus placebo. Primary Care Respiratory Medicine 2016;26(16022):21‐CR050. [ES:2055‐1010: IL:2055‐1010] [DOI] [PubMed] [Google Scholar]
- Singh D, Gaga M, Schmidt O, Bjermer L, Gronke L, Voss F, et al. Effects of tiotropium + olodaterol versus tiotropium or placebo by COPD disease severity and previous treatment history in the OTEMTO(R) studies. Respiratory Research 2016;17(1):73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh D, Maleki‐Yazdi MR, Tombs L, Iqbal A, Fahy WA, Naya I. Prevention of clinically important deteriorations in COPD with umeclidinium/vilanterol. International Journal of Chronic Obstructive Pulmonary Disease 2016;11(1):1413‐24. [1178‐2005] [DOI] [PMC free article] [PubMed] [Google Scholar]
Troosters 2016 {published data only}
- Bourbeau J, Lavoie KL, Sedeno M, Sousa D, Erzen D, Hamilton A, et al. Behaviour‐change intervention in a multicentre, randomised, placebo‐controlled COPD study: methodological considerations and implementation. BMJ Open 2016;6(4):e010109. [2044‐6055] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie K, Sedeno M, Li P, Troosters T, Hamilton A, Sousa D, et al. Effects of bronchodilator therapy and exercise with self‐management behaviour‐modification on psychological and cognitive outcomes in COPD. European Respiratory Journal 2017;50:OA4669. [Google Scholar]
- NCT02085161. To evaluate the effect of inhaled medication together with exercise and activity training on exercise capacity and daily activities in patients with chronic lung disease with obstruction of airways. clinicaltrials.gov/ct2/show/NCT02085161 (first received 12 March 2014).
- Troosters T, Bourbeau J, Maltais F, Leidy N, Erzen D, Sousa D, et al. Effect of 8 and 12 weeks' once‐daily tiotropium and olodaterol, alone and combined with exercise training, on exercise endurance during walking in patients with COPD. Pneumologie 2017;71(1):159‐60. [Google Scholar]
- Troosters T, Bourbeau J, Maltais F, Leidy N, Erzen D, Sousa D, et al. Enhancing exercise tolerance and physical activity in COPD with combined pharmacological and non‐pharmacological interventions: PHYSACTO randomised, placebo‐controlled study design. BMJ Open 2016;6(4):e010106. [2044‐6055] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troosters T, Maltais F, Leidy N, Lavoie KL, Sedeno M, Janssens W, et al. Effect of bronchodilation and exercise training with behavior modification on exercise tolerance and downstream effects on symptoms and physical activity in COPD. American Journal of Respiratory and Critical Care Medicine 2018;198:1021‐32. [DOI: 10.1164/rccm.201706-1288OC] [DOI] [PubMed] [Google Scholar]
Watz 2016 {published data only}
- Watz H, Mailaender C, Kirsten AM. Effects of indacaterol/glycopyrronium on lung function and physical activity in patients with moderate to severe COPD. Thorax 2015;70:A146‐7. [Google Scholar]
- Watz H, Mailander C, Baier M, Kirsten A. Effects of indacaterol/glycopyrronium (QVA149) on lung hyperinflation and physical activity in patients with moderate to severe COPD: a randomised, placebo‐controlled, crossover study (the MOVE Study). BMC Pulmonary Medicine 2016;16(1):95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zheng 2014 {published data only}
- Zheng J, Zhong N, Newlands A, Church A, Goh AH. Efficacy and safety of once‐daily inhaled umeclidinium/vilanterol in Asian patients with COPD: results from a randomized, placebo‐controlled study. International Journal of Chronic Obstructive Pulmonary Disease 2015;10(1):1753‐67. [1178‐2005] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng JP, Newlands AH, Church A, Goh AH. The efficacy and safety of inhaled umeclidinium bromide/vilanterol in Asian patients with chronic obstructive pulmonary disease. Respirology 2014;19:22. [Google Scholar]
References to studies excluded from this review
Aalbers 2015 {published data only}
- Aalbers R, Maleki‐Yazdi M, Hamilton A, Waitere‐Wijker S, Zhao Y, Amatto V, et al. Randomized, double‐blind, dose‐finding study for tiotropium when added to olodaterol, administered via the Respimat(R) inhaler in patients with chronic obstructive pulmonary disease. Advances in Therapy 2015;32(9):809‐22. [0741‐238X] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aalbers R, Reza Maleki‐Yazdi M, Hamilton A, Waitere‐Wijker S, Pivovarova A, Schmidt O, et al. Dose‐finding study for tiotropium and olodaterol when administered in combination via the Respimat inhaler in patients with COPD [Abstract]. European Respiratory Journal 2012;40:525s [P2882]. [Google Scholar]
- NCT01040403. Determining optimal free dose combination of tiotropium bromide and BI 1744 CL in chronic obstructive pulmonary disease (COPD). clinicaltrials.gov/ct2/show/NCT01040403 (first received 29 December 2009).
Asai 2013 {published data only}
- Asai K, Hirata K, Hashimoto S, Fukuchi Y, Kitawaki T, Ikeda K, et al. Efficacy and safety of indacaterol/glycopyrronium in Japanese patients with COPD: pooled analysis of SHINE and ARISE. Respiratory Investigation 2016;54(6):428‐35. [2212‐5353] [DOI] [PubMed] [Google Scholar]
- Asai K, Minakata Y, Hirata K, Fukuchi Y, Kitawaki T, Ikeda K, et al. QVA149 once‐daily is safe and well tolerated and improves lung function and health status in Japanese patients with COPD: the ARISE study. European Respiratory Society 23rdAnnual Congress; 2013 Sep 7‐11; Barcelona. 2013; Vol. 42:694s [P3392].
Berton 2009 {published data only}
- Berton D, Reis M, Siqueira A, Barroco A, Takara L, Bravo D. Comparative effects of formoterol monotherapy versus formoterol plus tiotropium on dynamic hyperinflation and exercise tolerance in COPD. European Respiratory Society 19th Annual Congress; 2009 Sep 12‐15; Vienna. 2009:[199].
- Berton DC, Chiappa GR, Takara LS, Figueiredo P, Reis M, Teixeira PJZ, et al. LABA‐LAMA combination is superior to LABA monotherapy in improving the cardiocirculatory responses to treadmill exercise in patients with moderate‐to‐severe COPD. European Respiratory Society 20th Annual Congress; 2010 Sep 18‐22; Barcelona. 2010:[E5411].
- Berton DC, Reis M, Siqueira AC, Barroco AC, Takara LS, Bravo DM, et al. Effects of tiotropium and formoterol on dynamic hyperinflation and exercise endurance in COPD. Respiratory Medicine 2010;104(9):1288‐96. [DOI] [PubMed] [Google Scholar]
Buhl 2011 INTENSITY {published data only}
- Buhl R, Dunn LJ, Disdier C, Lassen C, Amos C, Henley M, et al. Blinded 12‐week comparison of once‐daily indacaterol and tiotropium in COPD. European Respiratory Journal 2011;38(4):797‐803. [DOI] [PubMed] [Google Scholar]
- Dunn L, Buhl R, Lassen C, Henley M, Kramer B. Blinded 12‐week comparison of once‐daily indacaterol and tiotropium in COPD. Chest 2010;138(4):719A. [DOI] [PubMed] [Google Scholar]
- NCT00900731. A study to compare the lung effect of indacaterol and tiotropium in chronic obstructive pulmonary disease (COPD). clinicaltrials.gov/ct2/show/NCT00900731 (first received 13 May 2009).
Buhl 2015 TONADO {published data only}
- Abrahams R, Ferguson G, Clerisme‐Beaty E, Groenke L, Voss F, Korducki L, et al. Effect of tiotropium + olodaterol on the use of nighttime rescue medication in patients with COPD: results from four randomized, double‐blind studies. Chest 2015;148(4):731A. [Google Scholar]
- Bai C, Buhl R, Lee SH, Lee KH, Jons O, Bothner U, et al. Safety of once‐daily tiotropium plus olodaterol via the respimat in East Asian patients with chronic obstructive pulmonary disease (COPD). Respirology 2015;20:55. [1323‐7799] [Google Scholar]
- Bai C, Buhl R, Lee SH, Lee KH, Jons O, Zhao Y, et al. Lung function with tiotropium plus olodaterol in East Asian patients with chronic obstructive pulmonary disease (COPD). Respirology 2015;20:53. [Google Scholar]
- Buhl R, Abrahams R, Bjermer L, Derom E, Flezar M, Hebert J, et al. Pooled safety analysis of once‐daily tiotropium and olodaterol fixed‐dose combination via the respimat in patients with chronic obstructive pulmonary disease: two 1‐year studies. Chest 2014;146(4):48A. [Google Scholar]
- Buhl R, Abrahams R, Bjermer L, Derom E, Flezar M, Hebert J, et al. Safety of once‐daily tiotropium and olodaterol fixed‐dose combination via the respimat in chronic obstructive pulmonary disease in two 1‐year studies. Thorax 2014;69:A189‐90. [Google Scholar]
- Buhl R, Derom E, Ferguson G, Pizzichini E, Reid J, Watz H, et al. Once‐daily tiotropium and olodaterol fixed‐dose combination via the Respimat improves outcomes versus mono‐components in COPD in two 1‐year studies. Thorax 2014;69:A188‐9. [Google Scholar]
- Buhl R, Jons O, Bothner U, Tetzlaff K, Zhao Y, Ichinose M. Safety of once‐daily tiotropium plus olodaterol via the respimat in East Asian patients with chronic obstructive pulmonary disease (COPD). Respirology 2015;20:47. [1323‐7799] [Google Scholar]
- Buhl R, Jons O, Zhao Y, Ichinose M. Lung function with tiotropium plus olodaterol in East Asian patients with chronic obstructive pulmonary disease (COPD). Respirology 2015;20:51. [1323‐7799] [Google Scholar]
- Buhl R, Magder S, Bothner U, Tetzlaff K, Voss F, Loaiza L, et al. Long‐term general and cardiovascular safety of tiotropium/olodaterol in patients with moderate to very severe chronic obstructive pulmonary disease. Respiratory Medicine 2017;122:58‐66. [DOI] [PubMed] [Google Scholar]
- Buhl R, Maltais F, Abrahams R, Bjermer L, Derom E, Ferguson G, et al. Tiotropium and olodaterol fixed‐dose combination versus mono‐components in COPD (GOLD 2‐4). European Respiratory Journal 2015;45(4):969‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia M, Reid J, Buhl R, Abrahams R, Bjermer L, Derom E, et al. Safety of once‐daily tiotropium and olodaterol fixed‐dose combination via the respimat in chronic obstructive pulmonary disease in two 1‐year studies. Respirology 2015;20:77. [Google Scholar]
- Chia M, Reid J, Buhl R, Derom E, Ferguson G, Maltais F, et al. Once‐daily tiotropium and olodaterol fixed‐dose combination via the respimat improves outcomes vs mono‐components in COPD in two 1‐year studies. Respirology 2015;20:77. [1323‐7799] [Google Scholar]
- Derom E, Flezar M, Gronke L, Vob F, Buhl R. Effect of tiotropium + olodaterol therapy on COPD exacerbations in the TONADO studies. American Journal of Respiratory and Critical Care Medicine 2016;193:A6805. [Google Scholar]
- Ferguson G, Clerisme‐Beaty E, Groenke L, Voss F, Karpel J. Lung function response with tiotropium + olodaterol maintenance treatment in patients with COPD in the TONADO and OTEMTO studies: a subgroup analysis by age. Chest 2015;148:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson G, Clerisme‐Beaty E, Groenke L, Voss F, Korducki L, Bennett N, et al. Tiotropium + olodaterol provides improvements in SGRQ and dyspnea compared with monotherapy components in patients with COPD: results from four randomized, double‐blind studies. Chest 2015;148(4):726A. [Google Scholar]
- Ferguson GT, Flezar M, Korn S, Korducki L, Gronke L, Abrahams R, et al. Efficacy of tiotropium + olodaterol in patients with chronic obstructive pulmonary disease by initial disease severity and treatment intensity: a post hoc analysis. Advances in Therapy 2015;32(6):523‐36. [0741‐238X] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson GT, Karpel JP, Clerisme‐Beaty E, Gronke L, Voss F, Buhl R. Efficacy and safety of tiotropium + olodaterol maintenance treatment in patients with COPD in the TONADO(R) and OTEMTO(R) studies: a subgroup analysis by age. International Journal of Chronic Obstructive Pulmonary Disease 2016;11(1):2701‐10. [1178‐2005] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson GT, Pizzichini E, Flezar M, Gronke L, Korducki L. The fixed‐dose combination of tiotropium + olodaterol has a rapid onset of action in patients with COPD. European Respiratory Journal 2015;46:PA2957. [Google Scholar]
- Ichinose M, Takizawa A, Gronke L, Vos F, Fukuchi Y. Analysis of the efficacy and safety of tiotropium and olodaterol fixed‐dose combination via the respimat in patients with COPD by disease severity: Tonado trials. Respirology 2015;20:66. [Google Scholar]
- Ichinose M, Takizawa A, Hamilton A, Zhao Y, Fukuchi Y. Efficacy and safety of tiotropium and olodaterol fixed‐dose combination via the respimat in COPD patients: Japanese subgroup analysis of tonado 1 and 2. Respirology 2015;20:64. [Google Scholar]
- Ichinose M, Taniguchi H, Takizawa A, Gronke L, Loaiza L, Voss F, et al. The efficacy and safety of combined tiotropium and olodaterol via the Respimat((R)) inhaler in patients with COPD: results from the Japanese sub‐population of the Tonado((R)) studies. International Journal of Chronic Obstructive Pulmonary Disease 2016;11(1):2017‐27. [1178‐2005] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn S, Buhl R, Gronke L, Korducki L, Amato V, Ferguson G, et al. Analysis of the efficacy and safety of the combination of tiotropium + olodaterol in patients with COPD by previous usage of inhaled corticosteroids. Thorax 2015;70:A37. [Google Scholar]
- Maltais F, Groenke L, Korducki L, Buhl R. Efficacy of once‐daily tiotropium and olodaterol maintenance treatment for COPD: patient‐reported outcomes. Chest 2015;148(4):638A. [Google Scholar]
- NCT01431274. Tiotropium + olodaterol fixed dose combination (FDC) versus tiotropium and olodaterol in chronic obstructive pulmonary disease (COPD). clinicaltrials.gov/ct2/show/NCT01431274 (first received 9 September 2011).
- NCT01431287. Tiotropium + olodaterol fixed dose combination (FDC) versus tiotropium and olodaterol in chronic obstructive pulmonary disease (COPD). clinicaltrials.gov/ct2/show/NCT01431287 (first received 9 September 2011).
- Tashkin D, Ferguson G, Clerisme‐Beaty E, Vob F, Buhl R. Lung‐function response to tiotropium + olodaterol in patients with COPD with beta2‐agonist reversibility. American Journal of Respiratory and Critical Care Medicine 2016;193:A6808. [Google Scholar]
D'Urzo 2014 AUGMENT {published data only}
- D'Urzo A, Mergel V, Leselbaum A, Caracta C. Efficacy and safety of fixed‐dose combination aclidinium bromide/formoterol fumarate in patients with COPD: results from the AUGMENT COPD trial. Chest 2013;144(4):1025A. [Google Scholar]
- D'Urzo A, Rennard S, Kerwin E, Mergel V, He T, Leselbaum A, et al. One‐year efficacy of aclidinium/formoterol fixed‐dose combination in COPD patients: the AUGMENT COPD study. European Respiratory Journal 2014;44:P286. [Google Scholar]
- D'Urzo A, Rennard S, Mergel V, Garcia Gil E, Leselbaum A, Caracta C. The augment COPD trial: efficacy and safety of a fixed‐dose combination of aclidinium bromide and formoterol fumarate in COPD patients. Chest 2014;145(3):426A. [Google Scholar]
- D'Urzo AD, Rennard SI, Kerwin EM, He T, Leselbaum A, Caracta C. Efficacy of fixed‐dose combination aclidinium bromide/formoterol fumarate on bronchodilation over 1 year: AUGMENT COPD extension trial in patients with moderate to severe COPD. American Journal of Respiratory and Critical Care Medicine 2014;189:A6006. [Google Scholar]
- D'Urzo AD, Rennard SI, Kerwin EM, Mergel V, Leselbaum A, Caracta C. Twice‐daily aclidinium bromide/formoterol fumarate fixed‐dose combination: lung function improvements in the AUGMENT COPD trial in patients with moderate to severe COPD. American Journal of Respiratory and Critical Care Medicine 2014;189:A3776. [Google Scholar]
- D'Urzo AD, Rennard SI, Kerwin EM, Mergel V, Leselbaum AR, Caracta CF. Efficacy and safety of fixed‐dose combinations of aclidinium bromide/formoterol fumarate: the 24‐week, randomized, placebo‐controlled AUGMENT COPD study. Respiratory Research 2014;15(1):123‐41. [1465‐993X] [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Urzo AD, Singh D, Kerwin E, Lakkis H, Chuecos F, Miquel G. Efficacy and safety of aclidinium/formoterol fixed‐dose combination in patients with moderate to severe airflow obstruction, stratified by severity. European Respiratory Journal 2015;46:PA1499. [Google Scholar]
- NCT01437397. Efficacy, safety and tolerability of aclidinium bromide/formoterol fumarate compared with formoterol fumarate in patients with moderate to severe chronic obstructive pulmonary disease (COPD) (LAC). clinicaltrials.gov/ct2/show/NCT01437397 (first received 20 September 2011).
- NCT01572792. Efficacy, safety and tolerability of two fixed dose combinations of aclidinium bromide/formoterol fumarate, aclidinium bromide, formoterol fumarate and placebo for 28‐weeks treatment in patients with moderate to severe, stable chronic obstructive pulmonary disease (COPD). clinicaltrials.gov/ct2/show/NCT01572792 (first received 6 April 2012).
- Rennard SI, D'Urzo AD, Donohue JF, He T, Shrestha P, Leselbaum A, et al. Long‐term safety of fixed‐dose combination aclidinium bromide/formoterol fumarate in patients with moderate to severe COPD: the AUGMENT COPD extension trial. American Journal of Respiratory and Critical Care Medicine 2014;189:A6008. [Google Scholar]
- Rennard SI, D'Urzo AD, Jones PW, Mergel V, Leselbaum A, Caracta C. Fixed‐dose combination of aclidinium bromide/formoterol fumarate improve breathlessness and health status in COPD patients: AUGMENT COPD trial. American Journal of Respiratory and Critical Care Medicine 2014;189:A6007. [Google Scholar]
Decramer 2014 {published data only}
- Anzueto A, Decramer M, Kaelin T, Richard N, Tabberer M, Harris S, et al. The efficacy and safety of umeclidinium/vilanterol compared with tiotropium or vilanterol over 24 weeks in subjects with COPD. American Journal of Respiratory and Critical Care Medicine 2013;187:A4268. [Google Scholar]
- Decramer M, Anzueto A, Kerwin E, Kaelin T, Richard N, Crater G, et al. Efficacy and safety of umeclidinium plus vilanterol versus tiotropium, vilanterol, or umeclidinium monotherapies over 24 weeks in patients with chronic obstructive pulmonary disease: results from two multicentre, blinded, randomised controlled trials. Lancet Respiratory Medicine 2014;2(6):472‐86. [DOI] [PubMed] [Google Scholar]
- Decramer M, Anzueto A, Kerwin E, Richard N, Crater G, Tabberer M, et al. Efficacy and safety of umeclidinium/vilanterol compared with umeclidinium or tiotropium in COPD. European Respiratory Society 23rd Annual Congress; 2013 Sep 7‐11; Barcelona. 2013; Vol. 42:751s [P3640].
- NCT01316900. 24‐week trial comparing GSK573719/GW642444 with GW642444 and with tiotropium in chronic obstructive pulmonary disease. clinicaltrials.gov/ct2/show/NCT01316900 (first received 16 March 2011).
- NCT01316913. 24‐week trial comparing GSK573719/GW642444 with GSK573719 and with tiotropium in chronic obstructive pulmonary disease. clinicaltrials.gov/ct2/show/NCT01316913 (first received 16 March 2011).
Di Marco 2005 {published data only}
- Marco F, Verga M, Santus P, Cazzola M, Mondoni M, Belloi E, et al. Formoterol (Modulite®), tiotropium and their combination in patients with acute exacerbations of chronic bronchitis: preliminary data. European Respiratory Journal 2005;26:1905. [Google Scholar]
- Marcoa F, Verga M, Santus P, Morelli N, Cazzola M, Centanni S. Effect of formoterol, tiotropium, and their combination in patients with acute exacerbation of chronic obstructive pulmonary disease: a pilot study. Respiratory Medicine 2006;11:1925‐32. [DOI] [PubMed] [Google Scholar]
Donohue 2013b {published data only}
- Donohue J, Niewoehner D, Brooks J, O'Dell D, Church A. Long‐term safety and tolerability of umeclidinium/vilanterol and umeclidinium in COPD. European Respiratory Society 23rd Annual Congress; 2013 Sep 7‐11; Barcelona. 2013; Vol. 42:144s [P760].
Donohue 2016 {published data only}
- Donohue J, Singh D, Munzu C, Kilbride S, Church A. Magnitude of umeclidinium/vilanterol lung function effect depends on monotherapy responses: results from two randomised controlled trials. Respiratory Medicine 2016;112:65‐74. [DOI] [PubMed] [Google Scholar]
Donohue 2016b {published data only}
- Donohue JF, Soong W, Wu X, Shrestha P, Lei A. Long‐term safety of aclidinium bromide/formoterol fumarate fixed‐dose combination: results of a randomized 1‐year trial in patients with COPD. Respiratory Medicine 2016;116:41‐8. [DOI] [PubMed] [Google Scholar]
- Make B, Donohue J, Zhong X, Leselbaum A, Caracta C. Long‐term safety of a fixed‐dose combination of aclidinium bromide/formoterol fumarate in patients with stable moderate to severe COPD. Chest 2014;145(3):386A. [Google Scholar]
- Make B, Donohue JF, Soong W, Zhong X, Leselbaum A, Caracta C. Efficacy and tolerability of aclidinium bromide/formoterol fumarate fixed‐dose combination in patients with COPD: a 1‐year study. European Respiratory Journal 2014;44:P2413. [Google Scholar]
- Make BJ, Donohue JF, Soong W, Zhong X, Leselbaum A, Caracta C. Lung function and safety of aclidinium bromide/formoterol fumarate fixed‐dose combination: results of a 1‐year trial in patients with COPD. American Journal of Respiratory and Critical Care Medicine 2014;189:A6010. [Google Scholar]
EUCTR2007‐003648‐31 {published data only}
- EUCTR2007‐003648‐31. A phase IIa, randomised, multicentre, evaluator‐blinded, 4‐way crossover clinical trial to study the pharmacokinetics, safety, tolerability and effects on lung function of one day treatment of formoterol 12 µg qd delivered by 2 different dry powder inhalers (Aerolizer® and Almirall Inhaler), of the fixed dose combination formoterol 12 µg + aclidinium bromide 200 µg qd delivered by Almirall Inhaler, and of formoterol 12 µg bid delivered by Aerolizer®, in moderate to severe chronic obstructive pulmonary disease patients. www.clinicaltrialsregister.eu/ctr‐search/trial/2007‐003648‐31/DE (first received 21 February 2008).
EUCTR2007‐004435‐30 {published data only}
- EUCTR2007‐004435‐30. A randomised, 4‐week, placebo‐controlled, double‐blind, 6 arm parallel group, dose‐finding clinical trial, to assess the efficacy and safety of three different doses of formoterol (6, 12 & 18 µg) combined with the inhaled anticholinergic aclidinium bromide 200 µg, aclidinium bromide 200 µg monotherapy and formoterol 12 µg monotherapy all administered once daily by inhalation via Almirall inhaler in patients with stable moderate to severe Chronic Obstructive Pulmonary Disease. www.clinicaltrialsregister.eu/ctr‐search/search?query=eudract_number:2007‐004435‐30 (first received 14 April 2008).
EUCTR2009‐015901‐38 {published data only}
- EUCTR2009‐015901‐38. Efficacy, safety and tolerability of two fixed‐dose combinations of aclidinium bromide with two doses of formoterol fumarate compared with aclidinium bromide, formoterol fumarate and placebo all administered twice daily in stable, moderate to severe chronic obstructive pulmonary disease patients. apps.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2009‐015901‐38‐DE (first received 11 November 2009).
Evdokimov 2015 {published data only}
- Evdokimov V, Evdokimova A, Sheyanov M, Kozhina N. Clinical efficacy and safety of formoterol and tiotropium administration in patient with chronic heart failure due to coronary artery disease combined with chronic obstructive pulmonary disease. European Journal of Heart Failure 2015;17:68. [Google Scholar]
Evdokimov 2015b {published data only}
- Evdokimov V, Evdokimova A, Kovalenko E, Tebloev K. Prolonged use (18 months) of tiotropium and indacaterol in patient with chronic heart failure due to coronary artery disease combined with chronic obstructive pulmonary disease. European Journal of Heart Failure 2016;18:369. [1879‐0844] [Google Scholar]
- Evdokimov V, Evdokimova A, Kovalenko E, Tebloev K, Zolotova O. Clinical efficacy and safety of tiotropium and indacaterol administration in patient with chronic heart failure due to coronary artery disease combined with chronic obstructive pulmonary disease. European Heart Journal 2015;36:664. [Google Scholar]
- Evdokimov V, Evdokimova A, Tebloev K, Kovalenko E, Kozhina N, Zolotova O. Efficacy and safety of different types of bronchodilator therapy in patients with chronic heart failure due to coronary artery disease combined with chronic obstructive pulmonary disease. European Heart Journal 2014;35:385‐6. [Google Scholar]
- Evdokimov V, Evdokimova A, Tebloev K, Zolotova O. Clinical efficacy and safety of tiotropium and indacaterol administration in patient with chronic heart failure due to coronary artery disease combined with chronic obstructive pulmonary disease. European Journal of Heart Failure 2014;16:251. [1879‐0844] [Google Scholar]
Ferguson 2015 {published data only}
- Ferguson GT, Fowler‐Taylor A, Thach C, Wang Q, Schubert‐Tennigkeit AA, Banerji D. QVA149 is safe and well tolerated in patients with moderate‐to‐severe COPD: FLIGHT3, a 52 week safety study. American Journal of Respiratory and Critical Care Medicine 2015;191:A5762. [Google Scholar]
Fogarty 2014 {published data only}
- Fogarty C, Ortiz S. Pharmacokinetics, safety, and tolerability of aclidinium/formoterol fixed dose combination via pressair/genuair vs formoterol via foradil aerolizer in patients with moderate to severe COPD. Chest 2014;146(4 (2)):45A. [Google Scholar]
Hanania 2016 {published data only}
- Hanania NA, Tashkin DP, Kerwin E, Donohue J, Denenberg M, O'Donnell D, et al. Safety and efficacy of a novel LAMA/LABA co‐suspension technology glycopyrrolate/formoterol fixed‐dose combination delivered by MDI: results of a one‐year extension study in patients with COPD (PINNACLE‐3). American Journal of Respiratory and Critical Care Medicine 2016;193:A6791. [Google Scholar]
- Hanania NA, Tashkin DP, Kerwin EM, Donohue JF, Denenberg M, O'Donnell DE, et al. Long‐term safety and efficacy of glycopyrrolate/formoterol metered dose inhaler using novel Co‐Suspension Delivery Technology in patients with chronic obstructive pulmonary disease. Respiratory Medicine 2017;126:105‐15. [DOI] [PubMed] [Google Scholar]
Hoshino 2014 {published data only}
- Hoshino M. Quantitative computed tomography assessment of airway dimensions by combining tiotropium and indacaterol in patients with COPD. European Respiratory Journal 2014;44:4668. [Google Scholar]
- Hoshino M, Ohtawa J. Computed tomography assessment of airway dimensions with combined tiotropium and indacaterol therapy in COPD patients. Respirology 2014;19(3):403‐10. [DOI] [PubMed] [Google Scholar]
Ichinose 2016 {published data only}
- Ichinose M, Minakata Y, Motegi T, Ueki J, Seki T, Anzai T, et al. Study design of VESUTO®: efficacy of tiotropium/olodaterol on lung hyperinflation, exercise capacity, and physical activity in Japanese patients with chronic obstructive pulmonary disease. Advances in Therapy 2017;34(7):1622‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinose M, Minakata Y, Motegi T, Ueki J, Seki T, Anzai T, et al. Study design of vesuto; study to evaluate the efficacy of tiotropium + olodaterol vs tiotropium on lung hyperinflation, exercise capacity, and physical activity in Japanese COPD patients. Respirology 2016;21:179. [Google Scholar]
- NCT02629965. Comparing the efficacy of tiotropium + olodaterol fixed dose combination (FDC) over tiotropium in improvement of lung hyperinflation, exercise capacity and physical activity in Japanese COPD patients. clinicaltrials.gov/ct2/show/NCT02629965 (first received 15 December 2015).
Ichinose 2017 {published data only}
- Ichinose M, Minakata Y, Motegi T, Ueki J, Seki T, Anzai T, et al. Study design of VESUTO: efficacy of tiotropium/olodaterol on lung hyperinflation, exercise capacity, and physical activity in Japanese patients with chronic obstructive pulmonary disease. Advance Therapeutics 2017;4(7):1622‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Imran 2015 {published data only}
- Imran M, Chhabra S, Kotwani A. Combinations of long acting beta<inf>2</inf> agonists to tiotropium: a randomized, double‐blind, placebo‐controlled, active‐drug controlled, parallel design academic clinical trial in moderate COPD male patients. Archives of Pharmacy Practice 2015;6(2):19‐23. [Google Scholar]
Jones 2010 {published data only}
- Jones CDS. The Combined Effect of Tiotropium and Formoterol on the Functional Status of Patients with Moderate‐to‐Severe COPD [Dissertation]. Charleston: Proquest, Umi Dissertation Publishing, 2010. [978‐112‐405‐5602602] [Google Scholar]
Mahler 2015 {published data only}
- Ayers T, Fogel R, Banerji D, Maitra S, Schubert‐Tennigkeit AA. QVA149, twice daily, is well tolerated in patients with moderate‐to‐severe COPD and has a safety profile similar to placebo: FLIGHT1 and FLIGHT2 pooled analysis in the subgroup of patients from the USA. Chest 2015;148(4):725A. [Google Scholar]
- Fogel R, Ayers T, Banerji D, Maitra S, Schubert‐Tennigkeit AA. Cardiovascular safety of QVA149 in patients with moderate‐to‐severe COPD: pooled analysis of FLIGHT1 and FLIGHT2 clinical studies. Chest 2015;148(4):718A. [Google Scholar]
- Fowler TA, Fogel R, Banerji D, Maitra S. Improvement in lung function, dyspnea, and health status with QVA149 compared with placebo in the subgroup of patients with COPD from the USA: FLIGHT1 and FLIGHT2 pooled analysis. Chest. F.‐Y. Chao, Emory University, Atlanta, GA, United States, 2015; Vol. 148, issue 4:718A.
- Jones P, Larbig M, Maitra S, Fowler‐Taylor A, Banerji D. Analysis of improvement in SGRQ component scores with QVA149: pooled data from the FLIGHT1 and FLIGHT2 studies. Chest 2015;148(4):722A. [Google Scholar]
- Jones PW, Ayers T, Fowler‐Taylor A, Larbig M, Thach C, Maitra S, et al. QVA149 demonstrates superior improvements in health status, as measured by SGRQ, compared with placebo in patients with moderate‐to‐severe COPD: the FLIGHT2 study. American Journal of Respiratory and Critical Care Medicine 2015;191:A5747. [Google Scholar]
- Kerwin E, Ayers T, Banerji D, Maitra S, Patalano F. QVA149 significantly improves lung function and reduces rescue medication use compared with its monocomponents in COPD patients with moderate‐to‐severe airflow limitation: pooled analysis from the FLIGHT1 and FLIGHT2 studies. Chest 2015;148(4):729A. [Google Scholar]
- Kerwin E, Ayers T, Fowler‐Taylor A, Patalano F, Banerji D, Maitra S. QVA149 demonstrated significant improvement in lung function compared with placebo and its monocomponents: pooled analysis from the FLIGHT1 and FLIGHT2 studies. Chest 2015;148(4):741A. [Google Scholar]
- Kerwin EM, Ayers T, Fowler‐Taylor A, Larbig M, Thach C, Maitra S, et al. QVA149 provides superior improvement in lung function versus its monocomponents in patients with moderate‐to‐severe COPD: the FLIGHT2 study. American Journal of Respiratory and Critical Care Medicine 2015;191:A5741. [DOI] [PubMed] [Google Scholar]
- Kerwin EM, Fowler‐Taylor A, Ayers T, Thach C, Maitra S, Patalano F, et al. QVA149 improves lung function to a greater extent than monocomponents in patients with moderate‐to‐severe COPD: the FLIGHT1 study. American Journal of Respiratory and Critical Care Medicine 2015;191:A5743. [DOI] [PubMed] [Google Scholar]
- Mahler D, Larbig M, Patalano F, Maitra S, Banerji D. Dual bronchodilation with QVA149 improves dyspnea in patients with moderate‐to‐severe COPD: pooled analysis from the FLIGHT1 and FLIGHT2 studies. Chest 2015;148(4):730A. [Google Scholar]
- Mahler DA, Ayers T, Fowler‐Taylor A, Larbig M, Thach C, Maitra S, et al. QVA149 reduces dyspnea in patients with moderate‐to‐severe COPD as measured by the transition dyspnea index: the FLIGHT2 study. American Journal of Respiratory and Critical Care Medicine 2015;191:A5751. [Google Scholar]
- Mahler DA, D'Urzo A, Peckitt C, Lassen C, Kramer B, Filcek S, et al. Combining once‐daily bronchodilators In COPD: Indacaterol plus tiotropium versus tiotropium alone. American Journal of Respiratory and Critical Care Medicine 2011;183:A1591. [Google Scholar]
- Mahler DA, Kerwin E, Ayers T, Taylor AF, Maitra S, Thach C, et al. FLIGHT1 and FLIGHT2: efficacy and safety of QVA149 (indacaterol/glycopyrrolate) versus its monocomponents and placebo in patients with chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine 2015;192(9):1068‐79. [DOI] [PubMed] [Google Scholar]
- NCT01712516. A 12‐week treatment, multi‐center, randomized, double‐blind, parallel‐group, placebo and active controlled study to assess the efficacy, safety, and tolerability of indacaterol maleate glycopyrronium bromide in COPD patients with moderate to severe airflow limitation. clinicaltrials.gov/ct2/show/NCT01712516 (first received 23 October 2012).
- NCT01727141. A 12‐week treatment, multi‐center, randomized, double‐blind, parallel‐group, placebo and active controlled study to assess the efficacy, safety, and tolerability of indacaterol maleate / glycopyrronium bromide in COPD patients with moderate to severe airflow limitation. clinicaltrials.gov/ct2/show/NCT01727141 (first received 15 November 2012).
Maltais 2010 {published data only}
- Maltais F, Beck E, Webster D, Maleki‐Yazdi MR, Seibt JV, Arnoux A, et al. Four weeks once daily treatment with tiotropium + olodaterol (BI 1744) fixed dose combination compared with tiotropium in COPD patients. European Respiratory Journal 2010;36(Suppl 54):1014S. [Google Scholar]
NCT00308191 2006 {published data only}
- NCT00308191. A phase IIIB study to compare the efficacy and safety of concomitant treatment in patients with COPD. clinicaltrials.gov/ct2/show/NCT00308191 (first received 29 March 2006).
NCT00424528 2006 {published data only}
- NCT00424528. Efficacy safety study of Arformoterol/Tiotropium combination versus either therapy alone in chronic obstructive pulmonary disease (COPD). clinicaltrials.gov/ct2/show/NCT00424528 (first received 19 January 2006).
NCT00696020 2008 {published data only}
- NCT00696020. Combination of orally inhaled BI1744CL/tiotropium bromide in patients with chronic obstructive pulmonary disease (COPD). clinicaltrials.gov/ct2/show/NCT00696020 (first received 12 June 2008).
NCT00720499 2008 {published data only}
- NCT00720499. Efficacy and safety of 4 weeks of treatment with orally inhaled BI1744/tiotropium bromide in patients with chronic obstructive pulmonary disease (COPD). clinicaltrials.gov/ct2/show/NCT00720499 (first received 22 July 2008).
NCT00845728 2009 {published data only}
- NCT00845728. Exacerbation study (INVIGORATE). clinicaltrials.gov/ct2/show/NCT00845728 (first received 18 February 2009).
NCT00846586 2009 {published data only}
- NCT00846586. Efficacy and safety of indacaterol plus tiotropium versus tiotropium alone in patients with chronic obstructive pulmonary disease. clinicaltrials.gov/ct2/show/NCT00846586 (first received 18 February 2009).
NCT00877383 2009 {published data only}
- NCT00877383. Efficacy and safety of indacaterol plus tiotropium versus tiotropium alone in patients with chronic obstructive pulmonary disease. clinicaltrials.gov/ct2/show/NCT00877383 (first received 7 April 2009).
NCT01040689 2010 {published data only}
- NCT01040689. Characterization of 24 hour spirometry profiles of inhaled BI 1744 CL and inhaled tiotropium bromide in patients with chronic obstructive pulmonary disease. clinicaltrials.gov/ct2/show/NCT01040689 (first received 30 December 2009).
NCT01040728 2010 {published data only}
- NCT01040728. A randomized, double‐blind, 4‐way crossover study to evaluate the efficacy of 24 hour spirometry profiles of inhaled BI 1744 CL and inhaled tiotropium bromide in patients with chronic obstructive pulmonary disease. clinicaltrials.gov/ct2/show/NCT01040728 (first received 30 December 2009).
NCT01049360 2009 {published data only}
- NCT01049360. Efficacy and safety study of two fixed‐dose combinations of aclidinium bromide with formoterol fumarate compared with aclidinium bromide, formoterol fumarate and placebo. clinicaltrials.gov/ct2/show/NCT01049360 (first received 14 January 2010).
NCT01437540 2011 {published data only}
- NCT01437540. Safety and tolerability of aclidinium bromide/formoterol fumarate compared with formoterol fumarate in patients with moderate to severe chronic obstructive pulmonary disease (LAC). clinicaltrials.gov/ct2/show/NCT01437540 (first received 21 September 2011).
NCT01476813 2012 {published data only}
- NCT01476813. Randomized cross over study to assess efficacy and safety of BDP/FF and glycopyrrolate. clinicaltrials.gov/ct2/show/NCT01476813 (first received 22 November 2011).
NCT01491802 2012 {published data only}
- NCT01491802. Effect of a new combination bronchodilator on exercise in GOLD stage II moderate COPD. clinicaltrials.gov/ct2/show/NCT01491802 (first received 14 December 2011).
NCT01529632 2012 {published data only}
- NCT01529632. Comparison of safety and efficacy of the combination product QVA149A against the concurrent administration of the individual components, QAB149 and NVA237, in patients with chronic obstructive pulmonary disease (COPD). clinicaltrials.gov/ct2/show/NCT01529632 (first received 9 February 2012).
NCT01536262 2012 {published data only}
- NCT01536262. Japan long‐term safety for tiotropium plus olodaterol. clinicaltrials.gov/ct2/show/NCT01536262 (first received 22 February 2012).
NCT01551888 2012 {published data only}
- NCT01551888. Pharmacokinetic, safety and tolerability study of aclidinium/formoterol fixed dose combination and formoterol in patients with moderate to severe chronic obstructive pulmonary disease (COPD). clinicaltrials.gov/ct2/show/NCT01551888 (first received 13 March 2012).
NCT01574651 2012 {published data only}
- NCT01574651. The effect of QVA149 on health related quality of life in patients with chronic obstructive pulmonary disease (COPD) (QUANTIFY). clinicaltrials.gov/ct2/show/NCT01574651 (first received 10 April 2012).
NCT01682863 2012 {published data only}
- NCT01682863. A multi‐centre randomized double blind 52‐week study to assess the safety of QVA149 compared to QAB149 in patients with COPD who have moderate to severe airflow limitation. clinicaltrials.gov/ct2/show/NCT01682863 (first received 11 September 2012).
NCT01697696 2012 {published data only}
- NCT01697696. Long term safety study of NVA237 vs QAB149 in COPD patients. clinicaltrials.gov/ct2/show/NCT01697696 (first received 2 October 2012).
NCT01703845 2012 {published data only}
- NCT01703845. A study to characterize pharmacokinetics of tiotropium + olodaterol fixed‐dose combination in Japanese patients with COPD. clinicaltrials.gov/ct2/show/NCT01703845 (first received 11 October 2012).
NCT01817764 2013 {published data only}
- NCT01817764. A study to compare the efficacy and safety of umeclidinium/vilanterol and fluticasone propionate/salmeterol in subjects with chronic obstructive pulmonary disease (COPD). clinicaltrials.gov/ct2/show/NCT01817764 (first received 25 March 2013).
NCT01985334 2014 {published data only}
- NCT01985334. Study to evaluate the efficacy and safety of glycopyrronium or indacaterol maleate and glycopyrronium bromide fixed‐dose combination regarding symptoms and health status in patients with moderate COPD switching from treatment with any standard COPD regimen. clinicaltrials.gov/ct2/show/NCT01985334 (first received 15 November 2013).
NCT02030535 2014 {published data only}
- NCT02030535. Study to evaluate the effect on lung function and ECG when a combination of tiotropium plus olodaterol is administered to patients with COPD either from a single inhaler or each compound is administered after each other from two different inhalers. clinicaltrials.gov/ct2/show/NCT02030535 (first received 8 January 2014).
NCT02059434 2013 {published data only}
- NCT02059434. Two‐part pharmacokinetic and pharmacodynamic study of LAS190792 in patients with asthma and COPD. clinicaltrials.gov/ct2/show/NCT02059434 (first received 11 February 2014).
NCT02196714 2014 {published data only}
- NCT02196714. PK study of PT003 and PT001 in Japanese healthy subjects. clinicaltrials.gov/ct2/show/NCT02196714 (first received 22 July 2014).
NCT02231177 2008 {published data only}
- NCT02231177. Pharmacokinetics and safety of BI 1744 CL plus tiotropium bromide in chronic obstructive pulmonary disease (COPD). clinicaltrials.gov/ct2/show/NCT02231177 (first received 4 September 2014).
NCT02296138 2015 {published data only}
- NCT02296138. Comparing the efficacy of tiotropium + olodaterol (5/5 µg) fixed dose combination (FDC) over tiotropium 5µg in reducing moderate to severe exacerbations in patients with severe to very severe chronic obstructive pulmonary disease. clinicaltrials.gov/ct2/show/NCT02296138 (first received 20 November 2014).
NCT02343458 2015 {published data only}
- NCT02343458. Study to assess the efficacy and safety of PT003, PT005, and PT001 in subjects with moderate to very severe COPD. clinicaltrials.gov/ct2/show/NCT02343458 (first received 22 January 2015).
NCT02429765 2015 {published data only}
- NCT02429765. Effect of aclidinium/formoterol on nighttime lung function and morning symptoms in chronic obstructive pulmonary disease. clinicaltrials.gov/ct2/show/NCT02429765 (first received 29 April 2015).
NCT02442206 2015 {published data only}
- NCT02442206. Effect of QVA149 (indacaterol maleate/glycopyrronium bromide) on cardiac function in COPD patients (CLAIM). clinicaltrials.gov/ct2/show/NCT02442206 (first received 13 May 2015).
NCT02465567 2015 {published data only}
- NCT02465567. Study to assess the efficacy and safety of PT010 relative to PT003 and PT009 in subjects with moderate to very severe COPD (Ethos). clinicaltrials.gov/ct2/show/NCT02465567 (first received 8 June 2015).
NCT02487446 2015 {published data only}
- NCT02487446. Efficacy and safety study of QVA149 in COPD patients. clinicaltrials.gov/ct2/show/NCT02487446 (first received 1 July 2015).
NCT02487498 2015 {published data only}
- NCT02487498. Efficacy and safety study of indacaterol maleate/glycopyrronium bromide in chronic obstructive pulmonary disease (COPD) patients. clinicaltrials.gov/ct2/show/NCT02487498 (first received 1 July 2015).
NCT02579850 2015 {published data only}
- NCT02579850. 2‐arm parallel group study of fixed combination of CHF 5993 vs ultibro® in COPD patients (TRIBUTE). clinicaltrials.gov/ct2/show/NCT02579850 (first received 20 October 2015).
NCT02643082 2015 {published data only}
- NCT02643082. A study to assess the effects of PT003 and placebo MDI on specific image based parameters in subjects with moderate to severe COPD. clinicaltrials.gov/ct2/show/NCT02643082 (first received 30 December 2015).
NCT02796677 2016 {published data only}
- NCT02796677. AMPLIFY ‐ D6571C00001 Duaklir USA phase III study. clinicaltrials.gov/ct2/show/NCT02796677 (first received 13 June 2016).
NCT02845752 2016 {published data only}
- NCT02845752. The effect of STIOLTO ™ RESPIMAT® on fatigue in chronic obstructive pulmonary disease. clinicaltrials.gov/ct2/show/NCT02845752 (first received 27 July 2016).
NCT02937584 2016 {published data only}
- NCT02937584. A study to assess the effects of PT001 and PT005 MDI on specific image based parameters in subjects with moderate to severe COPD. clinicaltrials.gov/ct2/show/NCT02937584 (first received 18 October 2016).
NCT02988869 2016 {published data only}
- NCT02988869. Tiotropium/formoterol Via Discair® vs tiotropium monotherapy or tiotropium + formoterol free combination treatment. clinicaltrials.gov/ct2/show/NCT02988869 (first received 9 December 2016).
NCT03022097 2017 {published data only}
- NCT03022097. Study to patients with stable COPD to assess the efficacy and safety of aclidinium bromide/formoterol fumarate (DUAKLIR) (AVANT). clinicaltrials.gov/ct2/show/NCT03022097 (first received 16 January 2017).
NCT03024346 2016 {published data only}
- NCT03024346. A study to assess the effects of PT003 and Placebo MDI on specific image based parameters in subjects with moderate to severe COPD. https://clinicaltrials.gov/ct2/show/NCT02643082 (note: NCT03024346 is an obsolete identifier referring to NCT02643082) (first received 30 December 2015).
NCT03034915 2017 {published data only}
- NCT03034915. A 24‐week study to compare umeclidinium/vilanterol (UMEC/VI), UMEC and salmeterol in subjects with chronic obstructive pulmonary disease (COPD). clinicaltrials.gov/ct2/show/NCT03034915 (first received 27 January 2017).
Orevillo 2016 {published data only}
- Orevillo C, Miller J, DePetrillo P, Maes A, Siddiqui S, Reisner C. Pharmacokinetic and safety profile of the novel LAMA/LABA co‐suspension technology glycopyrrolate/formoterol fumarate fixed‐dose combination MDI, in Japanese healthy subjects. American Journal of Respiratory and Critical Care Medicine 2016;193:A6828. [Google Scholar]
Rabe 2015 {published data only}
- Martinez FJ, Rabe KF, Ferguson GT, Fabbri LM, Rennard S, Feldman GJ, et al. Efficacy and safety of glycopyrrolate/formoterol metered dose inhaler formulated using co‐suspension delivery technology in patients with COPD. Chest 2017;151(2):340‐57. [DOI] [PubMed] [Google Scholar]
- NCT01854658. Multi‐center study to assess the efficacy and safety of PT003, PT005, and PT001 in subjects with moderate to very severe COPD (PINNACLE 2). clinicaltrials.gov/ct2/show/NCT01854658 (first received 15 May 2013).
- Rabe K, Martinez F, Rodriguez‐Roisin R, Fabbri LM, Ferguson GT, Jones P, et al. PT003, a novel co‐suspension MDI glycopyrronium/formoterol fixed‐dose combination is superior to monocomponents in patients with COPD. European Respiratory Journal 2015;46:PA4363. [Google Scholar]
Reisner 2011 {published data only}
- Reisner C, Fogarty C, Spangenthal S, Dunn L, Kerwin E, Quinn D, et al. Novel combination of glycopyrrolate and formoterol MDI (GFF‐MDI) provides superior bronchodilation compared to its components administered alone, tiotropium DPI, and formoterol DPI in a randomized, double‐blind, placebo‐controlled phase 2b study In patients with COPD. American Journal of Respiratory and Critical Care Medicine 2011;183(1):A6435. [Google Scholar]
- Reisner C, Rennard SI, Fogarty C, Fischer T, Rose E, Fernandez C. Pearl therapeutics' combination LAMA/LABA MDI (GFF‐MDI, PT003) provides a significant benefit on home peak expiratory flow rate (PEFR) and reduces the need for rescue albuterol use compared to its components administered alone, Spiriva(R) Handihaler(R), and Foradil(R) Aerolizer(R) in a randomized, double‐blind, placebo‐controlled phase 2b study in patients with COPD. American Journal of Respiratory and Critical Care Medicine 2012;185:A2259. [Google Scholar]
Reisner 2013 {published data only}
- Reisner C, Orevillo CJ, Fernandez C, Rose E, Kollar C, Rutty D, et al. Pearl Therapuetics' combination LAMA/LABA (GFF MDI, PT003) provides comparable safety as measured by 24‐hour holter monitoring to its components (GP MDI, PT001 And FF MDI, PT005) and Foradil ® Aerolizer®. American Journal of Respiratory and Critical Care Medicine 2013;187:A1485. [Google Scholar]
Reisner 2016 {published data only}
- Reisner C, Gottschlich G, Fakih F, Koser A, Krainson J, Delacruz L, et al. 24‐Hour lung function profile of novel co‐suspension technology glycopyrrolate/formoterol metered dose inhaler versus placebo and spiriva respimat, in patients with moderate‐to‐very‐severe chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine 2016;193:A6823. [Google Scholar]
Reisner 2017 {published data only}
- Reisner C, Fabbri L, Kerwin E, Fogarty C, Spangenthal S, Rabe K, et al. A randomized, seven‐day study to assess the efficacy and safety of a glycopyrrolate/formoterol fumarate fixed‐dose combination metered dose inhaler using novel Co‐SuspensionTM Delivery Technology in patients with moderate‐to‐very severe chronic obstructive pulmonary disease. Respiratory Research 2017;18(1):8. [1465‐993X] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisner C, Fabbri LM, Kerwin EM, Fogarty C, Spangenthal S, Rabe KF, et al. A randomized, seven‐day study to assess the efficacy and safety of a glycopyrrolate/formoterol fumarate fixed‐dose combination metered dose inhaler using novel Co‐Suspension™ Delivery Technology in patients with moderate‐to‐very severe chronic obstructive pulmonary disease. Respiratory Research 2017;18(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisner C, Fernandez C, Orevillo C, Rose E, Kollar C. Pearl Therapeutics' combination LAMA/LABA MDI (GFF MDI, PT003) provides comparable metabolic and ECG safety to Spiriva(R) handihaler(R) and placebo In patients with COPD. American Journal of Respiratory and Critical Care Medicine 2012;185:A2925. [Google Scholar]
Reisner 2017b {published data only}
- NCT02347072. 24‐hour lung function in subjects with moderate to very severe COPD after treatment with PT003, open‐label Spiriva® Respimat® as an active control, and placebo. clinicaltrials.gov/ct2/show/NCT02347072 (first received 27 January 2015).
- NCT02347085. 24‐hour lung function in subjects with moderate to very severe COPD after treatment with PT003 and placebo MDI. clinicaltrials.gov/ct2/show/NCT02347085 (first received 27 January 2015).
- Reisner C, Gottschlich G, Fakih F, Koser A, Krainson J, Delacruz L, et al. 24‐h bronchodilation and inspiratory capacity improvements with glycopyrrolate/formoterol fumarate via co‐suspension delivery technology in COPD. Respiratory Research 2017;18(1):157. [DOI: 10.1186/s12931-017-0636-4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sadigov 2014 {published data only}
- Sadigov A, Akhundov S, Bagirov R. Analysis of chronic obstructive pulmonary disease exacerbations with the triple therapy compared with dual and single bronchodilator therapy: which treatment is better for patients with severe disease?. Chest 2014;145(3):425A. [Google Scholar]
Salomon 2017 {published data only}
- NCT01699685. Swiss studY for the treatment of COPD patients with the free combiNation of indacatERol and GlYcopyrroniumbromide (SYNERGY). clinicaltrials.gov/ct2/show/NCT01699685 (first received 4 October 2012).
- Salomon J, Stolz D, Domenighetti G, Frey JG, Turk AJ, Azzola A, et al. Indacaterol and glycopyrronium versus indacaterol on body plethysmography measurements in COPD ‐ a randomised controlled study. Respiratory Research 2017;18(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Setoguchi 2015 {published data only}
- Setoguchi Y. Indactaterol mono‐and combination therapy with inhaled tiotropium bromide in COPD: a 52‐week study. American Journal of Respiratory and Critical Care Medicine 2015;191:A5748. [Google Scholar]
- Setoguchi Y. The combination therapy with inhaled tiotropium bromide and indacaterol in COPD improves the peripheral airway function in static and dynamic status. European Respiratory Journal 2014;44:P896. [Google Scholar]
Singh 2014 {published data only}
- Bateman ED, Chapman KR, Singh D, D'Urzo AD, Molins E, Leselbaum A, et al. Aclidinium bromide and formoterol fumarate as a fixed‐dose combination in COPD: pooled analysis of symptoms and exacerbations from two six‐month, multicentre, randomised studies (ACLIFORM and AUGMENT). Respiratory Research 2015;16:92. [1465‐993X] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PW, Singh D, Bateman ED, Korn S, Serra C, Molins E, et al. The effect of aclidinium/formoterol fixed‐dose combination on COPD symptoms and health status in patients with COPD: results from the ACLIFORM/COPD study. American Journal of Respiratory and Critical Care Medicine 2014;189:A3764. [Google Scholar]
- Miravitlles M, Chapman KR, Chuecos F, Ribera A, Garcia Gil E. The efficacy of aclidinium/formoterol on lung function and symptoms in patients with COPD categorized by symptom status: a pooled analysis. International Journal of Chronic Obstructive Pulmonary Disease 2016;11(1):2041‐52. [1178‐2005] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miravitlles M, Garcia Gil E, Chuecos F, Lamarca R. Morning and nighttime symptoms in COPD and efficacy of aclidinium/formoterol in symptomatic vs asymptomatic patients. European Respiratory Journal 2015;46:OA1968. [Google Scholar]
- NCT01462942. Long‐term efficacy and safety of aclidinium bromide/formoterol fumarate fixed‐dose combination. clinicaltrials.gov/ct2/show/NCT01462942 (first received 1 November 2011).
- Singh D, Jones P, Bateman E, Korn S, Serra C, Molins E, et al. Evaluation of the efficacy and safety of two doses of aclidinium and formoterol in fixed‐dose combination in patients with COPD: the acliform study. Chest 2014;145(3):375A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh D, Jones PW, Bateman ED, Korn S, Serra C, Molins E, et al. Aclidinium/formoterol fixed‐dose combination improves bronchodilation and is well tolerated in patients with COPD: results from the ACLIFORM/COPD study. American Journal of Respiratory and Critical Care Medicine 2014;189:A5987. [Google Scholar]
- Singh D, Jones PW, Bateman ED, Korn S, Serra C, Molins E, et al. Efficacy and safety of aclidinium bromide/formoterol fumarate fixed‐dose combinations compared with individual components and placebo in patients with COPD (ACLIFORM‐COPD): a multicentre, randomised study. BMC Pulmonary Medicine 2014;14(1):178. [1471‐2466] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sliwinski 2010 {published data only}
- Sliwinski P, Perng DW, Chuchalin A, Jones PW. Efficacy and safety of once‐daily aclidinium bromide 200 muG in combination with formoterol in patients with COPD. Thorax 2010;65:A136 [P137]. [Google Scholar]
Tanaka 2015 {published data only}
- Tanaka Y, Hino M, Gemma A, Kobayashi Y, Katoh Y. 2nd interim report: Effect of tiotropium monotherapy versus tiotropium plus indacaterol in patients with chronic obstructive pulmonary disease ‐ single‐centre, randomized, prospective, real‐world study. Respirology 2015;20:37. [1323‐7799] [Google Scholar]
- Tanaka Y, Hino M, Naitoh T, Aoyama J, Kato Y, Kosaihira S, et al. 3rd interim report: Effect of tiotropium monotherapy versus tiotropium plus indacaterol in patients with chronic obstructive pulmonary disease ‐ single‐centre, randomised, prospective, real‐world study. Respirology 2016;21:176. [1323‐7799] [Google Scholar]
Tashkin 2007 {published data only}
- NCT00139932. Tiotropium bromide alone vs tiotropium bromide and formoterol fumarate in subjects with COPD (study P04272). clinicaltrials.gov/ct2/show/NCT00139932 (first received 31 August 2005).
- Tashkin D. Formoterol and tiotropium reduces rescue medication use more than tiotropium alone in patients with moderate COPD: findings from a 12 week randomized placebo controlled trial. American Thoracic Society International Conference; 2008 May 16‐21; Toronto. 2008:A647[#F5].
- Tashkin D, Pearle J, Iezzoni D, Varghese S. Formoterol and tiotropium compared with tiotropium alone for treatment of COPD. COPD 2009;6(1):17‐25. [1541‐2563] [DOI] [PubMed] [Google Scholar]
- Tashkin D, Pearle J, Varghese S. Improvement of lung function with coadministered formoterol and tiotropium, regardless of smoking status in patients with chronic obstructive pulmonary disease. Chest 2008;134(4):103002s. [Google Scholar]
- Tashkin D, Varghese S. Combined treatment with formoterol and tiotropium is more efficacious than treatment with tiotropium alone in patients with chronic obstructive pulmonary disease, regardless of smoking status, inhaled corticosteroid use, baseline severity, or gender. Pulmonary Pharmacology and Therapeutics 2011;24(1):147‐52. [1094‐5539] [DOI] [PubMed] [Google Scholar]
- Tashkin D, Varghese S. Formoterol treatment plus tiotropium results in greater improvements in lung function compared with tiotropium administered alone in patients with COPD. Journal of Allergy and Clinical Immunology 2007;119(1):S4 [13]. [Google Scholar]
- Tashkin D, Varghese S. The therapeutic effect of treatment with formoterol plus tiotropium was greater than the effect of treatment with tiotropium alone in COPD: findings from a 12‐week, multicenter, double‐blind, placebo‐controlled, trial. Chest 2007;132(4):529a. [Google Scholar]
Tashkin 2016 {published data only}
- Reisner C, Gotfried M, Denenberg MB, Fernandez C, Rose E, Fortner P, et al. Low doses of Pearl Therapeutics' LAMA/LABA Combination MDI (GFF MDI, PT003) provide superior bronchodilation compared to components and to open‐label spiriva handihaler in a randomized, double‐blind, placebo‐controlled phase IIb study in patients with COPD. American Journal of Respiratory and Critical Care Medicine 2013;187:A2434. [Google Scholar]
- Tashkin DP, Martinez FJ, Rodriguez‐Roisin R, Fogarty C, Gotfried M, Denenberg M, et al. A multicenter, randomized, double‐blind dose‐ranging study of glycopyrrolate/formoterol fumarate fixed‐dose combination metered dose inhaler compared to the monocomponents and open‐label tiotropium dry powder inhaler in patients with moderate‐to‐severe COPD. Respiratory Medicine 2016;120:16‐24. [DOI] [PubMed] [Google Scholar]
Ulubay 2005 {published data only}
- Ulubay G, Eyuboglu Oner F, Savas Bozbas S, Simsek A. Three regimens of inhaled bronchodilators for chronic obstructive pulmonary disease: comparison of pulmonary function and cardiopulmonary exercise test parameters. Turkish Respiratory Journal 2005;6(2):89‐94. [Google Scholar]
Van de Maele 2010 {published data only}
- Fabbri LM, Maele B, Lemmens B, Martin C, Horton R, Dolker M, et al. Cardiovascular safety of qva149, a novel combination of indacaterol and glycopyrronium, compared with indacaterol and placebo in patients with COPD. European Respiratory Society 19th Annual Congress; 2009 Sep 12‐15; Vienna. 2009:[P2027].
- Maele B, Fabbri LM, Martin C, Horton R, Dolker M, Overend T. Cardiovascular safety of QVA149, a combination of Indacaterol and NVA237, in COPD patients. COPD 2010;7:418‐27. [DOI] [PubMed] [Google Scholar]
Van Noord 2005 {published data only}
- Noord J, Aumann J, Janssens E, Mueller A, Cornelissen P. Relation between the acute response to salbutamol and long term FEV1 responses to tiotropium (TIO), formoterol (FORM) and its combination (T=F) in COPD patients. European Respiratory Journal 2003;22:153. [Google Scholar]
- Noord J, Qumann J, Janssens E, Mueller A, Cornelissen P, Aumann J, et al. Comparison of once daily tiotropium, twice daily formoterol and the free combination, once daily, in patients with COPD. American Thoracic Society 99th International Conference; 2003 May 16‐21; Seattle. 2003:B024 Poster 421.
- Noord JA, Aumann JL, Janssens E, Smeets JJ, Verhaert J, Disse B, et al. Comparison of tiotropium once daily, formoterol twice daily and both combined once daily in patients with COPD. European Respiratory Journal 2005;26(2):214‐22. [DOI] [PubMed] [Google Scholar]
Van Noord 2010 {published data only}
- Noord J, Buhl R, Laforce C, Martin C, Jones F, Dolker M, et al. QVA149 demonstrates superior bronchodilation compared with indacaterol or placebo in patients with chronic obstructive pulmonary disease. Thorax 2010;65(12):1086‐91. [DOI] [PubMed] [Google Scholar]
- Noord JA, Buhl R, LaForce C, Martin C, Jones F, Dolker M, et al. Qva149, a novel combination of indacaterol and glycopyrronium, demonstrates superior bronchodilation compared with indacaterol or placebo in patients with COPD. European Respiratory Society 19th Annual Congress; 2009 Sep 12‐15; Vienna. 2009:[E4347].
Velazquez‐Uncal 2016 {published data only}
- Velazquez‐Uncal M, Ramirez‐Venegas A, Sansores R, Perez‐Bautista O, Hernandez‐Zenteno R, Velazquez A, et al. Clinically significant impact of tiotropium and indacaterol on six‐minute walking test and inspiratory capacity in women with chronic obstructive pulmonary disease associated to biomass exposure. American Journal of Respiratory and Critical Care Medicine 2016;193:A3534. [Google Scholar]
Vincken 2013 {published data only}
- NCT01604278. Efficacy, safety and tolerability of the co‐administration of NVA237 plus indacaterol once daily versus indacaterol once daily in patients with moderate to severe chronic obstructive pulmonary disease (COPD) (GLOW6). clinicaltrials.gov/ct2/show/NCT01604278 (first received 23 May 2012).
- Vincken W, Aumann J, Chen H, Henley M, McBryan D, Goyal P. Efficacy and safety of coadministration of once‐daily indacaterol and glycopyrronium versus indacaterol alone in COPD patients: the GLOW6 study. International Journal of Chronic Obstructive Pulmonary Disease 2014;9:215‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincken W, Aumann J, Jack D, Chen H, Henley M, Goyal P. Once‐daily co‐administration of glycopyrronium and indacaterol via Breezhaler® device improves lung function and symptoms in patients with COPD versus indacaterol alone: the GLOW6 study. Thorax. 2013:A181 [P232].
Vogelmeier 2008 {published data only}
- Arievich H, Potena A, Fonay K, Vogelmeier CF, Overend T, Smith J, et al. Formoterol given either alone or together with tiotropium reduces the rate of exacerbations in stable COPD patients. European Respiratory Journal 2006;28:440s [P2514]. [Google Scholar]
- Vogelmeier C, Kardos P, Harari S, Gans S, Stenglein S, Thirlwell J. Formoterol mono‐ and combination therapy with tiotropium in patients with COPD: a 6‐month study. Respiratory Medicine 2008;102(11):1511‐20. [DOI] [PubMed] [Google Scholar]
- Vogelmeier CF, Harari SA, Fonay K, Beier J, Overend T, Till D, et al. Formoterol and tiotropium both improve lung function in stable COPD patients with some additional benefit when given together. European Respiratory Journal 2006;28:429s [P2506]. [Google Scholar]
Vogelmeier 2013 {published data only}
- Vogelmeier C, Bateman E, Pallante J, Bryant H, Alagappan V, D'Andrea P, et al. QVA149 once daily is safe and well tolerated in patients with COPD: the ILLUMINATE study. American Journal of Respiratory and Critical Care Medicine 2013;187:A1477. [Google Scholar]
- Vogelmeier CF, Bateman ED, Pallante J, Alagappan VK, D'Andrea P, Chen H, et al. Efficacy and safety of once‐daily QVA149 compared with twice‐daily salmeterol‐fluticasone in patients with chronic obstructive pulmonary disease (ILLUMINATE): a randomised, double‐blind, parallel group study. Lancet Respiratory Medicine 2013;1(1):51‐60. [DOI] [PubMed] [Google Scholar]
Watz 2017 {published data only}
- Aymerich JG, Watz H, Beeh KM, Paggiaro P, Moya M, Notari M, et al. Use of the Daily PROactive instrument to evaluate physical activity in patients with COPD: results from ACTIVATE. European Respiratory Journal 2017;50:PA4964. [Google Scholar]
- NCT02424344. Effect of aclidinium/formoterol on lung hyperinflation, exercise capacity and physical activity in moderate to severe COPD patients. clinicaltrials.gov/ct2/show/NCT02424344 (first received 23 April 2015).
- Watz H, Troosters T, Beeh KM, Garcia Aymerich J, Paggiaro P, Molins E, et al. ACTIVATE: effect of aclidinium/formoterol on physical activity in patients with COPD. European Respiratory Journal 2017;50:PA687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watz H, Troosters T, Beeh KM, Garcia‐Aymerich J, Paggiaro P, Molins E, et al. ACTIVATE: the effect of aclidinium/formoterol on hyperinflation, exercise capacity, and physical activity in patients with COPD. International Journal of Chronic Obstructive Pulmonary Disease 2017;12:2545‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
Webb 2015 {published data only}
- Webb KA, Ciavaglia CE, Preston M, O'Donnell DE. Effects of dual umeclidinium/vilanterol compared to umeclidinum on exertional dyspnea, respiratory mechanics and neural drive in patients with moderate COPD. American Journal of Respiratory and Critical Care Medicine 2015;191:A5735. [Google Scholar]
Wedzicha 2013 {published data only}
- CQVA149A2303. A 26‐week treatment, multi‐center, randomized, double‐blind, parallel‐group, placebo and active controlled (open label) study to assess the efficacy, safety and tolerability of QVA149 (110/50 μg q.d.) in patients with moderate to severe chronic obstructive pulmonary disease (COPD). www.novctrd.com/CtrdWeb/displaypdf.nov?trialresultid=7983 (first received 21 September 2010).
- Decramer M, Wedzicha JA, Ficker J, Fowler‐Taylor A, D'Andrea P, Arrasate C, et al. Once‐daily QVA149 improves health‐related quality of life in patients with severe to very severe COPD: the Spark study. American Journal of Respiratory and Critical Care Medicine 2013;187:A4257. [Google Scholar]
- Ficker JH, Wedzicha JA, Decramer M, Fowler‐Taylor A, D'Andrea P, Arrasate C, et al. QVA149 reduces COPD exacerbation rate in various subgroups of patients: the SPARK study. European Respiratory Society 23rd Annual Congress; 2013 Sep 7‐11; Barcelona. 2013; Vol. 42:3s [182].
- Mezzi K, Wedzicha J, Decramer M, Fowler‐Taylor A, Fogel R, Chen H, et al. QVA149 provides greater improvement in patient reported symptom scores compared to glycopyrronium and tiotropium in patients with severe to very severe COPD: the SPARK study [Abstract]. 7th International Primary Care Respiratory Group (IPCRG) World Conference; 2014 May 21‐24; Athens. 2014:PO005‐FRI.
- Meerhaeghe A, Vanhaeverbeek M. QVA149 versus glycopyrronium for COPD. Lancet Respiratory Medicine 2013;1(5):e22‐3. [2213‐2600] [DOI] [PubMed] [Google Scholar]
- Wedzicha J, Decramer M, Ficker J, Fowler Taylor A, D'Andrea P, Arrasate C, et al. Once‐daily glycopyrronium improves lung function and reduces exacerbations in severe‐to‐very severe COPD patients: The SPARK study [Abstract]. European Respiratory Society 23rd Annual Congress; 2013 Sep 7‐11; Barcelona. 2013; Vol. 42:3s [180].
- Wedzicha J, Decramer M, Ficker J, Fowler‐Taylor A, D'Andrea P, Arrasate C, et al. Dual bronchodilation with QVA149 reduces COPD exacerbations: the Spark study. American Journal of Respiratory and Critical Care Medicine 2013;187:A2428. [Google Scholar]
- Wedzicha J, Decramer M, Ficker J, Niewoehner D, Sandstrom T, Fowler‐Taylor A. Efficacy and safety of QVA149 versus glycopyrronium and tiotropium in severe to very severe COPD: the Spark study. American Journal of Respiratory and Critical Care Medicine 2013;187:A2429. [Google Scholar]
- Wedzicha J, Decramer M, Ficker J, Niewohner D, Sandstrom T, Fowler‐Taylor A, et al. Dual bronchodilation with once‐daily QVA149 reduces exacerbations, improves lung function and health status versus glycopyrronium and tiotropium in severe‐to‐very severe COPD patients: the SPARK study. European Respiratory Journal 2013;42:3s [181]. [Google Scholar]
- Wedzicha J, Decramer M, Ficker J, Sandstrom T, Taylor A, D'Andrea P, et al. Qva149 provides greater improvement in patient reported symptom scores compared to glycopyrronium and tiotropium in patients with severe to very severe COPD: the SPARK study. American Journal of Respiratory and Critical Care Medicine 2014;189:A3761. [Google Scholar]
- Wedzicha J, Ficker J, Fowler‐Taylor A, D'Andrea P, Arrasate C, Chen H, et al. Once‐daily QVA149 reduces exacerbations and improves health status in comparison with glycopyrronium and tiotropium in patients with severe‐to‐very severe COPD: the spark study. American College of Chest Physicians 2014;145(3):427A. [Google Scholar]
- Wedzicha J, Ficker J, Niewohner D, Sandstrom T, Fowler‐Taylor A, Banerji D. Dual bronchodilation with once‐daily QVA149 reduces exacerbations, improves lung function and health status versus glycopyrronium and tiotropium in severe‐to‐very severe COPD patients: the spark study. American College of Chest Physicians 2014;145(3):406A. [Google Scholar]
- Wedzicha JA, D'Urzo A, Mezzi K, Chen H, Banerji D, Fogel R. QVA149 showed significant improvements in lung function and health status and was well tolerated versus glycopyrronium and tiotropium in patients with severe COPD: the SPARK study. European Respiratory Journal 2014;44:1893. [Google Scholar]
- Wedzicha JA, Decramer M, Ficker JH, Niewoehner DE, Sandström T, Taylor AF, et al. Analysis of chronic obstructive pulmonary disease exacerbations with the dual bronchodilator QVA149 compared with glycopyrronium and tiotropium (SPARK): a randomised, double‐blind, parallel‐group study. Lancet Respiratory Medicine 2013;1(3):199‐209. [DOI] [PubMed] [Google Scholar]
- Wedzicha JA, Mezzi K, Fogel R, Banerji D. Baseline symptom scores and future risk of severe exacerbation: the SPARK study. European Respiratory Journal 2015;46(Suppl 59):PA1006. [Google Scholar]
Yosuke 2014 {published data only}
- Yosuke T, Mitsunori H, Naomi O, Hiroyuki T, Seiji K, Norihisa M, et al. Comparison of monotherapy with a long‐acting muscarinic antagonist versus combination therapy with a long‐acting muscarinic antagonist plus a long‐acting beta2‐agonist in patients with chronic obstructive pulmonary disease. Respirology 2014;19:122. [Google Scholar]
ZuWallack 2014 {published data only}
- NCT01694771. Co‐administration of olodaterol Respimat® and tiotropium Handihaler®. clinicaltrials.gov/ct2/show/NCT01694771 (first received 4 September 2014).
- NCT01696058. Co‐administration of olodaterol Respimat® and tiotropium Handihaler®. clinicaltrials.gov/ct2/show/NCT01696058 (first received 28 September 2012).
- Wallack RZ, Allen L, Hernandez G, Ting N, Abrahams R. Health status of patients with COPD receiving the combination of olodaterol respimat and tiotropium handihaler compared with tiotropium: results from two replicate, randomized, double‐blind studies. Chest 2014;146(4):46A. [Google Scholar]
- ZuWallack R, Allen L, Hernandez G, Ting N, Abrahams R. Efficacy and safety of combining olodaterol Respimat((R)) and tiotropium HandiHaler((R)) in patients with COPD: results of two randomized, double‐blind, active‐controlled studies. International Journal of Chronic Obstructive Pulmonary Disease 2014;9:1133‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies awaiting assessment
NCT02233543 2014 {published data only}
- NCT02233543. A multicenter, 4‐week crossover (total duration 12 weeks), placebo‐controlled, double‐blind study to determine the impact of QVA149 (indacaterol/glycopyrronium) 85/43 µg on nocturnal oxygen levels in chronic obstructive pulmonary disease (COPD). clinicaltrials.gov/ct2/show/NCT02233543 (first received 8 September 2014).
Additional references
Aaron 2007
- Aaron SD, Vandemheen KL, Fergusson D, Maltais F, Bourbeau J, Goldstein R, et al. Tiotropium in combination with placebo, salmeterol, or fluticasone‐salmeterol for treatment of chronic obstructive pulmonary disease: a randomized trial. Annals of Internal Medicine 2007;146(8):545‐55. [DOI] [PubMed] [Google Scholar]
Aaron 2008
- Aaron SD, Fergusson D, Marks GB, Suissa S, Vandemheen KL, Doucette S, et al. Counting, analysing and reporting exacerbations of COPD in randomised controlled trials. Thorax 2008;63(2):122‐8. [DOI] [PubMed] [Google Scholar]
Bangalore 2007
- Bangalore S, Kamalakkannan G, Parkar S, Messerli FH. Fixed‐dose combinations improve medication compliance: a meta‐analysis. American Journal of Medicine 2007;120(8):713‐9. [DOI] [PubMed] [Google Scholar]
Barnes 2015
- Barnes PJ, Burney PGJ, Silverman EK, Celli BR, Vestbo J, Wedzicha JA, et al. Chronic obstructive pulmonary disease. Nature Reviews Disease Primers 2015;122:15076. [DOI] [PubMed] [Google Scholar]
Bateman 2015
- Bateman ED, Chapman KR, Singh D, D’Urzo AD, Molins E, Leselbaum A, et al. Aclidinium bromide and formoterol fumarate as a fixed‐dose combination in COPD: pooled analysis of symptoms and exacerbations from two six‐month, multicentre, randomised studies (ACLIFORM and AUGMENT). Respiratory Research 2015;16(1):92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Beeh 2016
- Beeh KM, Burgel P‐R, Franssen FME, Lopez‐Campos JL, Loukides S, Hurst JR, et al. How do dual long‐acting bronchodilators prevent exacerbations of chronic obstructive pulmonary disease?. American Journal of Respiratory and Critical Care Medicine 2016;196(2):139‐49. [DOI] [PubMed] [Google Scholar]
Calverley 2005
- Calverley PMA. Minimal clinically important difference ‐ exacerbations of COPD. COPD 2005;2(1):143‐8. [DOI] [PubMed] [Google Scholar]
Calzetta 2015
- Calzetta L, Matera MG, Cazzola M. Pharmacological interaction between LABAs and LAMAs in the airways: optimizing synergy. European Journal of Pharmacology 2015;761(C):168–73. [DOI] [PubMed] [Google Scholar]
Calzetta 2016
- Calzetta L, Rogliani P, Matera MG, Cazzola M. A systematic review with meta‐analysis of dual bronchodilation with LAMA/LABA for the treatment of stable COPD. Chest 2016;149(5):1181‐96. [DOI] [PubMed] [Google Scholar]
Calzetta 2017
- Calzetta L, Rogliani P, Ora J, Puxeddu E, Cazzola M, Matera MG. LABA/LAMA combination in COPD: a meta‐analysis on the duration of treatment. European Respiratory Review 2017;26(143):160043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cazzola 2008
- Cazzola M, MacNee W, Martinez FJ, Rabe KF, Franciosi LG, Barnes PJ, et al. Outcomes for COPD pharmacological trials: from lung function to biomarkers. European Respiratory Journal 2008;31(2):416‐69. [DOI] [PubMed] [Google Scholar]
Cazzola 2010
- Cazzola M, Molimard M. The scientific rationale for combining long‐acting & beta2‐agonists and muscarinic antagonists in COPD. Pulmonary Pharmacology 2010;23(4):257–67. [DOI] [PubMed] [Google Scholar]
Cazzola 2015
- Cazzola M, Calzetta L, Ora J, Puxeddu E, Rogliani P, Matera MG. Searching for the synergistic effect between aclidinium and formoterol: from bench to bedside.. Respiratory Medicine 2015;109(10):1305‐11. [DOI] [PubMed] [Google Scholar]
CDC 2016
- Center for Disease Control. Chronic obstructive pulmonary disease among adults ‐ United States, 2016. www.cdc.gov/copd/index.html (accessed 1 Apr 2017).
Chapman 2013
- Chapman KR, Bergeron C, Bhutani M, Bourbeau J, Grossman RF, Hernandez P, et al. Do we know the minimal clinically important difference (MCID) for COPD exacerbations?. COPD 2013;10(2):243‐9. [DOI] [PubMed] [Google Scholar]
Donohue 2005
- Donohue JF. Minimal clinically important differences in COPD lung function. COPD 2005;2(1):111‐24. [DOI] [PubMed] [Google Scholar]
Farne 2015
- Farne HA, Cates CJ. Long‐acting beta2‐agonist in addition to tiotropium versus either tiotropium or long‐acting beta2‐agonist alone for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2015, Issue 10. [DOI: 10.1002/14651858.CD008989.pub3] [DOI] [PubMed] [Google Scholar]
GOLD 2017
- Global Initiative for Chronic Obstructive Lung Disease. From the global strategy for the diagnosis, management and prevention of COPD, global initiative for chronic obstructive lung disease (GOLD) 2018. goldcopd.org/gold‐reports/ (accessed prior to 28 September 2018).
Guyatt 1987
- Guyatt GH, Berman LB, Townsend M, Pugsley SO, Chambers LW. A measure of quality of life for clinical trials in chronic lung disease. Thorax 1987;42(10):773‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
Jones 1992
- Jones PW, Quirk FH, Baveystock CM, Littlejohns P. A self‐complete measure of health status for chronic airflow limitation. The St. George's Respiratory Questionnaire. American Review of Respiratory Disease 1992;145(6):1321‐7. [DOI] [PubMed] [Google Scholar]
MECIR 2018
- Higgins J, Lasserson T, Chandler J, Tovey D, Churchill R. Standards for the conduct and reporting of new Cochrane Intervention Reviews, reporting of protocols and the planning, conduct and reporting of updates (Version 1.05). community.cochrane.org/mecir‐manual (accessed prior to 28 September 2018).
Oba 2016
- Oba Y, Sarva ST, Dias S. Efficacy and safety of long‐acting β‐agonist/long‐acting muscarinic antagonist combinations in COPD: a network meta‐analysis. Thorax 2016;71(1):15‐25. [DOI] [PubMed] [Google Scholar]
Polkey 2013
- Polkey MI, Spruit M, Edwards LD, Watkins ML, Pinto‐Plata V, Vestbo JÃ, et al. Six‐minute‐walk test in chronic obstructive pulmonary disease: minimal clinically important difference for death or hospitalization. American Journal of Respiratory and Critical Care Medicine 2013;187(4):382‐6. [DOI] [PubMed] [Google Scholar]
Puhan 2006
- Puhan MA, Soesilo I, Guyatt GH, Schunemann HJ. Combining scores from different patient reported outcome measures in meta‐analyses: when is it justified?. Health and Quality of Life Outcomes 2006;4:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Quaseem 2011
- Qaseem A, Wilt TJ, Weinberger SE, Hanania NA, Criner G, Molen T, et al. Diagnosis and management of stable chronic obstructive pulmonary disease: a clinical practice guideline update from the American College of Physicians, American College of Chest Physicians, American Thoracic Society, and European Respiratory Society. Annals of Internal Medicine 2011;155:179‐91. [DOI] [PubMed] [Google Scholar]
Sarai 2014
- Sarai M, Sin D, FitzGerald JM, Aaron S. Long‐acting beta2‐agonists and long‐acting muscarinic antagonists in a combined inhaler versus either agent alone or placebo for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2014, Issue 9. [DOI: 10.1002/14651858.CD011282] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schunemann 2003
- Schunemann HJ, Griffith L, Jaeschke R, Goldstein R, Stubbing D, Guyatt GH. Evaluation of the minimal important difference for the feeling thermometer and the St. George's Respiratory Questionnaire in patients with chronic airflow obstruction. Journal of Clinical Epidemiology 2003;56(12):1170‐6. [DOI] [PubMed] [Google Scholar]
Schunemann 2005
- Schunemann HJ, Puhan M, Goldstein R, Jaeschke R, Guyatt GH. Measurement properties and interpretability of the chronic respiratory disease questionnaire (CRQ). COPD 2005;2(1):81‐9. [DOI] [PubMed] [Google Scholar]
SGRQ‐C Manual
- Jones P. St George’s Respiratory Questionnaire for COPD patients (SGRQ‐C). www.healthstatus.sgul.ac.uk/SGRQ_download/SGRQ‐C%20Manual%20March%202016.pdf (accessed 10 May 2018).
Singh 2014a
- Singh D, Jones PW, Bateman ED, Korn S, Serra C, Molins E, et al. Efficacy and safety of aclidinium bromide/formoterol fumarate fixed‐dose combinations compared with individual components and placebo in patients with COPD (ACLIFORM‐COPD): a multicentre, randomised study. BMC Pulmonary Medicine 2014;14:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Singh 2014b
- Singh SJ, Puhan MA, Andrianopoulos V, Hernandes NA, Mitchell KE, Hill CJ, et al. An official systematic review of the European Respiratory Society/American Thoracic Society: measurement properties of field walking tests in chronic respiratory disease. European Respiratory Journal 2014;44:1447‐78. [DOI] [PubMed] [Google Scholar]
Vestbo 2013
- Vestbo J, Hurd SS, Agustí AG, Jones PW, Vogelmeier C, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine 2013;187(4):347–65. [DOI] [PubMed] [Google Scholar]
Wedzicha 2014
- Wedzicha JA, Decramer M, Ficker JH, Niewoehner DE, Sandström T, Taylor AF, et al. Analysis of chronic obstructive pulmonary disease exacerbations with the dual bronchodilator QVA149 compared with glycopyrronium and tiotropium (SPARK): a randomised, double‐blind, parallel‐group study. Lancet Respiratory Medicine 2013;1(3):199–209. [DOI] [PubMed] [Google Scholar]
Wedzicha 2017
- Wedzicha JA, Calverley PMA, Albert RK, Anzueto A, Criner GJ, Hurst JR, et al. Prevention of COPD exacerbations: a European Respiratory Society/American Thoracic Society guideline. European Respiratory Journal 2017;50(3):1602265. [DOI] [PubMed] [Google Scholar]
Westwood 2011
- Westwood M, Bourbeau J, Jones PW, Cerulli A, Capkun‐Niggli G, Worthy G. Relationship between FEV1 change and patient‐reported outcomes in randomised trials of inhaled bronchodilators for stable COPD: a systematic review. Respiratory Research 2011;12:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 2015
- World Health Organization. Global health estimates ‐ year 2015. www.who.int/mediacentre/factsheets/fs310/en/ (accessed 1 April 2017).