

Abstract
Motivation: Many biological processes are guided by receptor interactions with linear ligands of variable length. One such receptor is the MHC class I molecule. The length preferences vary depending on the MHC allele, but are generally limited to peptides of length 8–11 amino acids. On this relatively simple system, we developed a sequence alignment method based on artificial neural networks that allows insertions and deletions in the alignment.
Results: We show that prediction methods based on alignments that include insertions and deletions have significantly higher performance than methods trained on peptides of single lengths. Also, we illustrate how the location of deletions can aid the interpretation of the modes of binding of the peptide-MHC, as in the case of long peptides bulging out of the MHC groove or protruding at either terminus. Finally, we demonstrate that the method can learn the length profile of different MHC molecules, and quantified the reduction of the experimental effort required to identify potential epitopes using our prediction algorithm.
Availability and implementation: The NetMHC-4.0 method for the prediction of peptide-MHC class I binding affinity using gapped sequence alignment is publicly available at: http://www.cbs.dtu.dk/services/NetMHC-4.0.
Contact: mniel@cbs.dtu.dk
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
A large number of biological processes are guided by receptor interactions with linear ligands (Gould et al., 2010). Proteins involved in interactions with a linear component include membrane receptors (e.g. the MHC molecules), enzymes (e.g. kinases and phosphatases) and carriers of peptide-recognition domains (e.g. SH3, PDZ, WW). Characterizing the specificity of such interactions is essential for our understanding of the underlying biological process and for the design of interventions aimed at altering or controlling the behavior of these processes. We have previously proposed an artificial neural network-based algorithm, NNAlign, for the identification of binding motifs in large-scale quantitative peptide datasets (Andreatta et al., 2011). However, this approach can only generate un-gapped sequence alignments and is therefore limited to the detection of motifs of a fixed length. In many cases, experimental data are derived using libraries of peptides that are longer than the basic receptor binding core. While still a linear problem, the number of residues directly involved in the interaction with the same receptor may generally vary for different ligands.
A prominent example of receptor that interacts with linear ligands of variable length is the MHC system. Major Histocompatibility Complex (MHC) class I molecules bind peptides derived from intracellular proteins and present them on the cell surface to CD8+ T cells. MHC molecules exist in numerous allelic variants with different physicochemical properties of their binding cleft. Consequently, the peptide repertoire recognized by different MHC molecules is very diverse, with marked allele-specific amino acid preferences. The major determinants of such amino acid preferences are found at the so-called anchor residues, generally corresponding to positions P2 and P9 of the minimum nine amino acid binding core (Rammensee et al., 1993). In some cases other positions play a crucial role: for instance HLA-B*08:01 has a strong preference for positively charged amino acids at P5, and HLA-A*01:01 for aspartic acid at P3 (Rapin et al., 2010). Constraints within peptides and intrapeptide contacts may also, to some extent, play a role in presentation and T-cell recognition (Theodossis et al., 2010).
MHC class I molecules exhibit preferences also with regard to the length of the peptides they can bind. Outside of a few exceptions where super-bulging peptides of up to 14 amino acids have been observed to bind to the MHC (Burrows et al., 2006), the closed conformation of the MHC class I binding cleft limits the length of bound peptides to 8–11 amino acids. Recent elution studies have attempted to quantify the length distribution of naturally processed and presented peptides (Bassani-Sternberg et al., 2015; Eichmann et al., 2014) and found that generally 9mer peptides were optimal. However, allele-specific preferences exist, for instance in the relatively high fraction of 8mer peptides found in complex with HLA-B*18:01 (Eichmann et al., 2014). Similarly, the murine allele H-2-Kb is known to have a comparable preference for 8 and 9mers (Deres et al., 1992) and HLA-B*44:03 a relative tendency toward longer peptides such as 10 and 11mers (Rist et al., 2013).
Considerable efforts have been dedicated to the development of accurate methods for the prediction of peptide binding to MHC molecules, applying many different approaches including similarity matrices (Kim et al., 2009), linear regression (Wang et al., 2015) and artificial neural networks (Hoof et al., 2009; Koch et al., 2013; Kuksa et al., 2015), among others. Of these methods, NetMHC (Nielsen et al., 2003) has been shown in several benchmark studies to be a state-of-the-art predictor for peptide–MHC binding affinity (Lundegaard et al., 2008; Peters et al., 2006). NetMHC was trained on MHC peptide binding data contained in the Immune Epitope Database (IEDB) (Vita et al., 2015). The IEDB has a large bias toward peptides of length nine (>72% of the data are for 9mers, whereas <3% of the data are for peptides of length 11). As the amount of available training data is crucial for the generation of accurate prediction models, the performance of data-driven predictors such as NetMHC will in general be limited for lengths different from nine. We have previously suggested a simple approximation approach that uses neural networks trained on 9mer data to extrapolate predictions for peptides of lengths other than nine (Lundegaard et al., 2008). This approximation was used in NetMHC to generate predictions for peptides of lengths 8, 10 and 11 for alleles with scarce binding affinity data. A more extreme approach has been taken for the development of the NetMHCpan method, which was trained only on 9mer peptides (Nielsen et al., 2007a). While these strategies have proven highly successful, they have the great limitation that they simply ignore all available data not conforming to the canonical 9mer peptide motif length.
In this article, we extend the NNAlign method to overcome this limitation and generate pan-length artificial neural networks trained on peptides of variable length. We demonstrate the performance of the method on a large set of MHC class I binding data, and show that it outperforms methods trained on single lengths and extrapolations from networks trained on 9mers only. Also, we addressed how the predicted location of deletions can aid the interpretation of the modes of binding of peptide-MHCs, as in the case of long peptides bulging out of the MHC groove or extending at either terminus. Finally, we analyzed to what degree the peptide length distribution of binders of the pan-length networks reflect the length preferences of different MHC class I alleles, and how such length preferences can potentially reduce the cost burden involved in rational epitope discovery.
2 Methods
2.1 Datasets
The prediction method for MHC class I affinity prediction was trained on a large set of quantitative peptide-MHC class I affinity measurements from the IEDB (Vita et al., 2015). We generated prediction models for all MHC class I molecules with at least 20 data points, of which at least four have IC50 affinity <500 nM, resulting in a set of 118 MHC class I (86 human, six murine, 26 primate) alleles. For several molecules, there were few or no measured data for certain peptide lengths. We introduced 100 random natural peptides for each of the lengths 8, 9, 10 and 11 as artificial negatives for each allele, to ensure the networks were exposed to a sufficiently diverse set of negative examples. Adding random data points with assumed weak affinity values was previously shown to have beneficial effects on ANN performance (Nielsen et al., 2007a). These random sequences were only used for training and were excluded from all evaluations.
As an external validation set, we extracted a set of 1540 ligands from SYFPEITHI (Rammensee et al., 1999) of length 8–11 that were not included in the training set, together with the complete sequence of the antigenic protein from which they were derived.
2.2 Neural network architecture
The method was implemented as a feed-forward artificial neural network ensemble with a single hidden layer as previously described (Nielsen and Lund, 2009). The amino acid sequence of training examples was encoded with 20 values for each position in the optimal nine amino acids binding core. We used both Blosum encoding, where these 20 values correspond to the BLOSUM matrix scores vector (Henikoff and Henikoff, 1992), and sparse encoding, where the 20 inputs are all set to a value of 0.05 except for the input corresponding to the observed amino acid which is set to 0.90. For peptides longer than nine amino acids, all possible consecutive deletions are applied to the primary sequence to reconcile the peptide to a core of nineamino acids. These include both deletions at the end terminals and internal consecutive deletions in all positions of the peptide. For peptides of length 8, the wildcard amino acid X (encoded as a vector of zeros) is inserted at each possible position to extend the peptide to a 9mer core. The sequence with the deletion or insertion that returns the highest predicted score with the current configuration of the neural network is taken as the optimal binding core (Fig. 1).
Fig. 1.
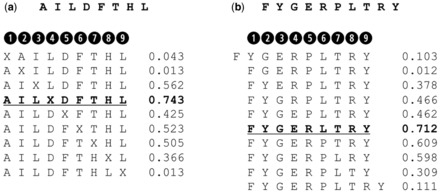
Examples of insertion and deletion applied to sequences of length different from nine. (a) Insertion: the wildcard amino acid X (encoded as a vector of zeros) is inserted in each possible position to complete the peptide to a 9mer core. The sequence with the insertion that returns the highest predicted score (in this case an insertion at P4) is taken as the optimal binding core. (b) Deletion: long peptides are reconciled to a 9mer amino acid core either by an extension at the terminals (first and last peptides in the example), or by deleting amino acids within the sequence. In this example a single deletion at P6 in the 10mer was found to be optimal
Other features of the training examples were encoded as input to the neural networks. They include the length of the deletion/insertion, the length and the composition of the peptide flanking regions in the case of a predicted extension at either the N- and C-terminus of the peptide with respect to the binding core. The length L of the peptide was encoded with four input neurons, corresponding to the four cases L ≤ 8, L = 9, L = 10, L ≥ 11. Encoding of the peptide length and eventual insertions/deletions enables the neural networks to learn the length preferences of a given MHC class I molecule.
The hidden layer of the networks consisted of five hidden neurons and the output layer of one neuron having as target value the binding affinity of the training example rescaled between 0 and 1 using the relationship 1 − log(aff)/log(50 ;000), where ‘aff’ is the IC50 affinity value in nM units (Nielsen et al., 2003).
2.3 Nested cross validation
In order to minimize the peptide overlap between training and testing data, the binding data for each molecule was partitioned into five subsets using a clustering approach as previously described (Nielsen et al. 2007b). Neural network training was performed using a nested cross-validation setup: three of the five subsets were used as training set and the fourth subset as a “stopping set”; network training was stopped when it reached the highest performance on this set, preventing over-fitting on the training set; all four combinations of the subsets were used to train and stop, resulting in an ensemble of four neural networks; these four networks were then applied on the fifth subset thus far excluded from the analysis as a test set; the process was repeated five times rotating the test subset to generate a complete cross-validated list of predictions. This setup ensures an unbiased evaluation of predictive performance, minimizing over-fitting on the training data.
2.4 Single length networks and L-mer approximation
Neural networks trained on all peptide lengths (allmer networks) were compared with the conventional approach of training individual networks for each peptide length. Where enough data was available (>20 data points and >3 binders), we trained length-specific networks using the same nested cross-validation strategy described above. In this case, the length of the binding core corresponds to the length of the peptides and no insertions/deletions are necessary.
A successful strategy employed in NetMHC-3.0 made use of an approximation algorithm (Lundegaard et al., 2008) to extrapolate predictions for peptide lengths different from nine. The L-mer approximation relies on networks trained only on 9mers, inserting/deleting amino acids at non-anchor positions in shorter/longer query peptides to conform the peptides to a series of 9mers and then averaging the predictions of the 9mer sequences. This approach was also considered for comparison to our approach trained on peptides of all lengths.
2.5 Validation on SYFPEITHI ligands
As an independent evaluation set, we extracted 1540 unique MHC class I ligands of length 8–11 from the SYFPEITHI database (Rammensee et al., 1999), excluding all peptide sequences found in the training set (SYF1 dataset). As previously discussed (Jørgensen et al., 2014; Trolle and Nielsen, 2014), SYFPEITHI contains a substantial number of sequences that do not match the canonical binding motif of the annotated MHC restriction element. We generated a filtered evaluation set (SYF2) by removing all peptide-MHCs with a predicted NetMHCpan rank >2%, resulting in a set of 1242 MHC ligands.
The source protein sequence of each validated ligand was scanned with a sliding window of 8–11 amino acids to generate all possible 8, 9, 10 and 11mers contained in the protein. These overlapping peptides were then ranked by the binding affinity predicted by our method, and for each protein we measured the relative rank of the validated ligand in the list of affinity predictions. The rank of the known ligand measures the fraction of peptides in the protein that would have to be tested before identifying the actual positive and can be used as a metric of predictive performance.
3 Results
3.1 Improved predictive performance by enrichment with peptides of different lengths
The extension of NNAlign implementing deletions and insertions was adapted to the MHC system and used to train the NetMHC-4.0 algorithm as described in the “Methods” section. For each MHC class I allele in the dataset, NetMHC-4.0 consists of an ensemble of neural networks trained on all peptides of lengths between 8 and 11. Networks trained on peptides of all lengths (allmer networks) showed significantly higher performance compared with networks trained on single lengths (Fig. 2, complete results in Supplementary Table S1). The improvement was particularly important for molecules where few data points were available for a given length, demonstrating how binding information can be learned across different peptide lengths. For example, there were only 61 affinity measurements for 8mer peptides to HLA-B*44:02 in the dataset, of which five had IC50 < 500 nM. A prediction method constructed on such small dataset performed close to random (PCC = 0.13). However, the dataset contains over 2000 measured 9- 10- and 11-mers for this molecule (including 429 binders); networks trained on this larger set of sequences of all lengths reach a PCC of 0.68 for the prediction of 8mers. Even on 9-mer peptides, where there are in most cases a fairly large number of measurements, including affinity data for other peptide lengths improved the predictive performance for 85 out of 118 alleles (p = 9 × 10−7, binomial test). The most extreme differences in PCC were observed when very limited data were available for all peptide lengths. For instance, the four alleles with highest gain in performance in favor of the allmer networks for the prediction of 9mers (second plot in Fig. 2) had an average of 86 data points (median 81), of which 31 were binders (median 22). Similarly, the four alleles with highest drop in performance had an average of 55 measured peptides (median 54), of which 29 binders (median 15). In contrast, the complete set of 118 MHC molecules contained an average number of data points per allele of 1 496 (median 807), of which on average 404 were measured binders (median 183).
Fig. 2.
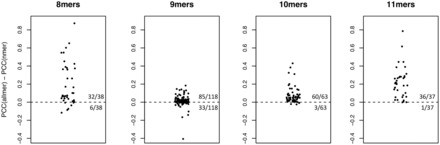
Difference in PCC (Pearson Correlation Coefficient) between networks trained on data for all peptide lengths (allmer) and networks trained on single lengths (nmer). Points above the baseline indicate alleles for which networks trained on all lengths give higher performance, and the deviation from the baseline shows the extent of the difference in terms of PCC. For 8mer peptides, allmer networks have higher performance in 32/38 alleles (p = 1 × 10−5), for 9mers in 85/118 alleles (p = 9 × 10−7), for 10mers in 60/63 alleles (p = 4 × 10−15), for 11mers in 36/37 alleles (p = 3 × 10−10)
The L-mer approximation algorithm (Lundegaard et al., 2008) relies on networks trained only on 9mers to extrapolate predictions for peptide lengths different from nine. For details on the L-mer approximation refer to the “Methods” section. Networks trained on all lengths gave higher PCC compared to the L-mer approximation with high significance for 10-mers (p = 8 × 10−12), and with lower but still significant P-values for 8-mers (P = 0.003) and 11-mers (P = 0.05) (see Fig. 3). A summary of the performance values of the networks trained on all peptide lengths compared with single lengths and the L-mer approximation is shown in Table 1.
Fig. 3.
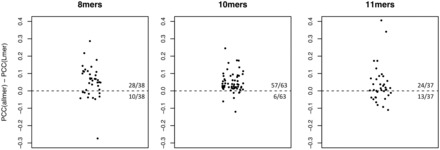
Difference in PCC between networks trained on data for all peptide lengths (allmer) and networks trained only on 9-mers with the L-mer approximation (Lmer). Points above the baseline indicate alleles for which networks trained on all lengths give higher performance, and the deviation from the baseline shows the extent of the difference in terms of PCC. For 8mer peptides, allmer networks have higher performance in 28/38 alleles (p = 0.003), for 10mers in 57/63 alleles (p = 8 × 10−12), for 11mers in 24/37 alleles (p = 0.05)
Table 1.
Summary of the performance (in PCC and AUC) of different prediction methods on the IEDB dataset
| PCC | AUC | |||||||
|---|---|---|---|---|---|---|---|---|
| Length | 8 | 9 | 10 | 11 | 8 | 9 | 10 | 11 |
| Alleles * | 38 | 118 | 63 | 37 | 38 | 118 | 63 | 37 |
| allmer | 0.717 | 0.717 | 0.744 | 0.706 | 0.895 | 0.884 | 0.882 | 0.888 |
| nmer | 0.524 | 0.702 | 0.672 | 0.488 | 0.775 | 0.875 | 0.845 | 0.775 |
| Lmer | 0.664 | 0.702 | 0.699 | 0.670 | 0.871 | 0.875 | 0.860 | 0.868 |
*For each length, only alleles with >20 data points and >3 binders are considered for validation.
allmer is the method trained on peptides of all lengths; nmer refers to networks trained only on peptides of length n; L-mer refers to networks trained on 9-mers and applied to peptides of different length using the L-mer approximation. Note that the L-mer approximation for 9mer reduces to the nmer method.
3.2 Learning the peptide length preferences of MHC molecules
In addition to the peptide primary sequence, the networks in the NetMHC-4.0 ensemble encode additional information including the length of the peptide and possible deletions/insertions with reference to the nine amino acid core. Depending on the amount of measured data points available to the networks for a given length, and the relative number of measured binders on the total number of data points, the neural networks can learn the length preferences of different MHC class I molecules.
In order to explore this aspect of network learning, we generated predictions for 400 000 random natural peptides (100 000 for each length between 8 and 11) for each allele, and analyzed the distribution of peptide lengths among the top 1% predicted peptides. An average profile over the 118 MHC class I molecules in the dataset shows a clear preference for 9mer peptides (54% of the top predicted binders), followed by 10mers (24%), 11mers (15%) and 8mers (6.5%). Such length distribution closely resembles the length profile of known ligands in the SYFPEITHI database (Rammensee et al., 1999), where about two-thirds of validated ligands are 9mers (Fig. 4a). In comparison, the L-mer approximation algorithm cannot account for peptide length preferences and returns a nearly flat profile of lengths.
Fig. 4.

Peptide length distributions predicted by NetMHC-4.0. (a) Length distribution for networks trained on peptides of all lengths and for the L-mer approximation networks, compared with the length distribution of ligands in the SYFPEITHI database. The allmer and L-mer profiles were calculated by running 400 000 random natural peptides through the predictors and calculating the relative number of peptides of different lengths among the top 1% predicted binders. (b) Predicted length distributions for selected alleles. For H-2-Kb the networks learn a preference for 8mers and 9mers, HLA-A*02:01 has a slight preference of 9mers over 10mers, HLA-B*07:02 favors 10mers and to a lesser extend 9mers, HLA-B*35:01 and HLA-C*04:01 have a strong preference for 9mer peptides
At the level of individual alleles, the length preferences learned by NetMHC-4.0 follow roughly the distribution of the data used to train the neural networks. For example, both for HLA-A*02:01 and HLA-B*07:02, ∼30% of the 9mer and 10mer peptides in the dataset are measured binders (IC50 < 500 nM), whereas very few 8mers and 11mers have high measured affinity for these molecules (see summary of the dataset in Supplementary Table S2). Among the 407 validated HLA-A*02:01 ligands in SYFPEITHI, 74% are 9mers, 13% 10mers, 7% 11mers, 3% 8mers and the remaining 3% are longer than 11. Similarly, a recent study of naturally presented peptides reported that 68% of 41 ligands restricted to HLA-A*02:01 were 9mers, 24% 10mers and only a handful had different length (Kowalewski et al., 2015). The murine allele H-2-Kb has a known preference for 8mers and 9mers (Moutaftsi et al., 2006) and a large number of measured binders in the training set have these lengths, a preference that reflects in the top predicted binders by the neural networks (Fig. 4b). Only 17 peptides are annotated as ligands to H-2-Kb in SYFPEITHI, and they all have length 8. For other alleles where very few or no affinity data are available for lengths other than 9 (e.g. HLA-B*35:01 and HLA-C*04:01), the predicted length profile has a marked preference for 9mer peptides. There are only two HLA-B*35:01 ligands in SYFPEITHI, both 9mers, and nearly all reported HLA-C*04:01 ligands are 9mers (50 out of 52), with the exception of one 8mer and one 10mer ligand.
3.3 Gapped alignment suggests modes of peptide-MHC binding
As described in the “Methods” section, insertions and deletions can reconcile sequences of different lengths to a common alignment core. In the context of peptide–MHC binding, the location of deletions may elucidate which positions of long peptides are involved in interactions with the dominant binding pockets of the MHC. Because bound peptides are deeply embedded in the MHC class I and the peptide-binding groove appears closed at both ends, peptides longer than nine amino acids are normally accommodated by bulging out from the middle of the groove. However, there is extensive evidence of an alternative mechanism of binding that involves protrusion of the peptide at either the N or C terminus rather than central bulging (Collins et al., 1994; Stryhn et al., 2000).
Based on the binding core predictions of NetMHC-4.0, we found that among the 10mers and 11mers in the IEDB dataset predicted to be binders (%rank ≤ 2), about 88% are expected to bulge out of the MHC pocket, 7% protrude from the C-terminal and 5% from the N-terminal. Importantly, the 12% of the data with such protruding non-canonical mode of binding cannot be accounted for by other prediction methods such as the L-mer approximation described above and NetMHCpan (Nielsen et al., 2007a).
In Figure 5, two three-dimensional structures of MHC molecules with a bound peptide from the Protein Data Bank (Rose et al., 2015) that illustrate these two mechanisms of binding are shown. The 10mer peptide MLLSVPLLLG bound to HLA-A*02:01 (PDB 2CLR) has its second (L) and ninth (L) residues in the two main pockets of the MHC molecule, with the extra Glycine (G) protruding at the C-terminus. NetMHC-4.0 correctly predicts the C-terminus extensions, placing the 9mer core at MLLSVPLLL with a deletion at the terminus (MLLSVPLLLG). In contrast, the 12mer peptide EECDSELEIKRY bound to HLA-B*44:03 (PDB 4JQX) shown in Figure 5b bulges out from the middle of the MHC binding groove and does not extend at any of the termini. The prediction of NetMHC-4.0 places a three-amino acid deletion after P5 (EECDSELEIKRY) with a minimal amino acid core of EECDSIKRY.
Fig. 5.

3D structures for two MHC class I molecules with bound peptides longer than 9 amino acids (PDB references 2CLR and 4JQX). (a) The 10mer peptide MLLSVPLLLG bound to HLA-A*02:01 extends at the C terminus with a glycine (G) amino acid. The residues at the anchor positions P2 (L) and P9 (L) are highlighted. (b) The 12mer EECDSELEIKRY bound to HLA-B*44:03 has anchors at its second (E) and last (Y) positions and bulges out from the middle of the MHC binding groove
3.4 Estimating the workload required to identify new epitopes
A typical bioinformatics-aided T cell epitope discovery study requires scanning protein sequences in search of potential epitopes, and select epitope candidates based on the predicted binding affinity to the MHC. A useful metric to assess the quality of an epitope predictor is the fraction of peptides in a given protein that would have to be tested before identifying the actual positive. On a set of 1242 proteins for which a known ligand is annotated in SYFPEITHI (SYF2 set), NetMHC-4.0 was applied to rank all peptides of length 8–11 that can be generated from the corresponding source protein, measuring the relative rank of the known ligand in the list of ordered predictions. Figure 6 shows that in 485 out of 1242 proteins (34%), the known ligand is the top predicted peptide by NetMHC-4.0; only one peptide per protein would have to be tested to identify the actual positive in these proteins. For 54% of the proteins the actual ligand is among the top three predicted peptides, and for 71% among the top seven peptides. In comparison, using networks trained only on 9-mer peptides and the L-mer approximation, the known ligand has the highest predicted binding affinity in only 25% of the cases, is within the top three predicted peptides in 46% of the proteins and within the top seven peptides in 63% of the proteins. In practical terms these numbers translate into at least a 25% reduction in the experimental effort and cost involved in rational epitope discovery based on peptide-binding prediction. On the unfiltered set SYF1 the curves converge more slowly toward 100% ligands identified, but the relative gain using NetMHC-4.0 is preserved (Supplementary Fig. S1).
Fig. 6.

Number of peptides per protein that should be tested to identify known ligands in the SYFPEITHI dataset. Antigenic proteins were digested into all possible peptides of length 8–11 as described in the text, which were then ranked by NetMHC-4.0 predicted affinity. The plot depicts the maximum number of peptides that would have to be tested for each protein before detecting the known ligand in the ranked list. The inset graph is a zoomed-out version of the curves of the main graph, showing eventual convergence to 100% identified ligands
4 Discussion
The machine-learning algorithm presented here stems from our previous work on sequence alignment based on artificial neural networks, namely the NNAlign method (Andreatta et al., 2011; Nielsen and Lund, 2009). By bringing together the training examples onto a common window of fixed length, the NNAlign training procedure effectively generates a multiple sequence alignment representing the minimal binding core of each peptide. The main innovation of the algorithm is the introduction of insertions and deletions into the NNAlign learning framework, essentially enabling the creation of gapped sequence alignments.
In the context of the MHC class I system, where the length of ligands is usually variable, insertions and deletions allow reconciling peptides of different lengths to a binding core of a common size. We demonstrated that models trained on all peptide lengths are superior to models made on individual lengths, especially for molecules with few measured experimental data of a given peptide length. These models also have higher performance than extrapolations from models constructed on 9mer peptides only.
Moreover, prediction of the core location can provide insight on the binding mode of linear peptides to their receptor, such as in the case of binders bulging out from the middle of the MHC groove or non-canonical binders protruding at the termini.
A further advantage of using a single model trained on peptide of all lengths is that the length preferences of the receptor can be learned by the method. We observed that for several MHC class I molecules the length distribution of the top predicted binders follows the known preferences for the different MHC alleles. If NetMHC-4.0 is used for a proteome scan in search for potential T cell epitopes, peptides of optimal length for the alleles of interest are therefore inherently prioritized. We quantified the experimental effort that can be saved by a bioinformatics-based selection of potential epitopes on a set of known ligands contained in the SYFPEITHI database, and found that on average the experimental effort and cost in identifying actual ligands could be reduced by at least 25% when using the prediction models trained with the pan-length algorithm.
The applications of the proposed machine-learning algorithm are not limited to the MHC class I system. The method is equally well suited to the identification of binding motifs in other peptide datasets characterized by a linear component, and we expect future applications will include MHC class II binding, pan-specific MHC class I and class II binding, and peptide interactions with PDZ, SH2 and SH3 domains.
The NetMHC-4.0 method for the prediction of peptide–MHC class I binding affinity is publicly available as a webserver at http://www.cbs.dtu.dk/services/NetMHC-4.0.
Supplementary Material
Acknowledgements
M.N. is a researcher at the Argentinean national research council (CONICET).
Funding
This work was supported by Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN272201200010C and from the Agencia Nacional de Promoción Científica y Tecnológica, Argentina (PICT-2012-0115).
Conflict of Interest: none declared.
References
- Andreatta M., et al. (2011) NNAlign: a web-based prediction method allowing non-expert end-user discovery of sequence motifs in quantitative peptide data. PLoS One, 6, e26781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani-Sternberg M., et al. (2015) Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol. Cell. Proteomics MCP, 14, 658–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows S.R., et al. (2006) Have we cut ourselves too short in mapping CTL epitopes?. Trends Immunol., 27, 11–16. [DOI] [PubMed] [Google Scholar]
- Collins E.J., et al. (1994) Three-dimensional structure of a peptide extending from one end of a class I MHC binding site. Nature, 371, 626–629. [DOI] [PubMed] [Google Scholar]
- Deres K., et al. (1992) Preferred size of peptides that bind to H-2 Kb is sequence dependent. Eur. J. Immunol., 22, 1603–1608. [DOI] [PubMed] [Google Scholar]
- Eichmann M., et al. (2014) Identification and characterisation of peptide binding motifs of six autoimmune disease-associated human leukocyte antigen-class I molecules including HLA-B*39:06. Tissue Antigens, 84, 378–388. [DOI] [PubMed] [Google Scholar]
- Gould C.M., et al. (2010) ELM: the status of the 2010 eukaryotic linear motif resource. Nucleic Acids Res., 38, D167–D180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S., Henikoff J.G. (1992) Amino acid substitution matrices from protein blocks. Proc. Natl Acad. Sci. U.S.A., 89, 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoof I., et al. (2009) NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics, 61, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen K.W., et al. (2014) NetMHCstab—predicting stability of peptide-MHC-I complexes; impacts for cytotoxic T lymphocyte epitope discovery. Immunology, 141, 18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y., et al. (2009) Derivation of an amino acid similarity matrix for peptide: MHC binding and its application as a Bayesian prior. BMC Bioinformatics, 10, 394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch C.P., et al. (2013) Scrutinizing MHC-I binding peptides and their limits of variation. PLoS Comput. Biol., 9, e1003088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalewski D.J., et al. (2015) HLA ligandome analysis identifies the underlying specificities of spontaneous antileukemia immune responses in chronic lymphocytic leukemia (CLL). Proc. Natl Acad. Sci. U.S.A., 112, E166–E175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuksa P.P., et al. (2015) High-order neural networks and kernel methods for peptide-MHC binding prediction. Bioinformatics, 31, 3600–3607. [DOI] [PubMed] [Google Scholar]
- Lundegaard C., et al. (2008) Accurate approximation method for prediction of class I MHC affinities for peptides of length 8, 10 and 11 using prediction tools trained on 9mers. Bioinformatics, 24, 1397–1398. [DOI] [PubMed] [Google Scholar]
- Moutaftsi M., et al. (2006) A consensus epitope prediction approach identifies the breadth of murine T(CD8+)-cell responses to vaccinia virus. Nat. Biotechnol., 24, 817–819. [DOI] [PubMed] [Google Scholar]
- Nielsen M., Lund O. (2009) NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinformatics, 10, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M., et al. (2003) Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. Publ. Protein Soc., 12, 1007–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M., et al. (2007a) NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS One, 2, e796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M., et al. (2007b) Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinformatics, 8, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters B., et al. (2006) A community resource benchmarking predictions of peptide binding to MHC-I molecules. PLoS Comput. Biol., 2, e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rammensee H., et al. (1999) SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics, 50, 213–219. [DOI] [PubMed] [Google Scholar]
- Rammensee H.G., et al. (1993) Peptides naturally presented by MHC class I molecules. Annu. Rev. Immunol., 11, 213–244. [DOI] [PubMed] [Google Scholar]
- Rapin N., et al. (2010) The MHC motif viewer: a visualization tool for MHC binding motifs. Curr. Protoc. Immunol. Ed. John E Coligan Al, Chapter 18, Unit 18.17. [DOI] [PubMed] [Google Scholar]
- Rist M.J., et al. (2013) HLA peptide length preferences control CD8 + T cell responses. J. Immunol, 191, 561–571. [DOI] [PubMed] [Google Scholar]
- Rose P.W., et al. (2015) The RCSB Protein Data Bank: views of structural biology for basic and applied research and education. Nucleic Acids Res., 43, D345–D356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stryhn A., et al. (2000) Longer peptide can be accommodated in the MHC class I binding site by a protrusion mechanism. Eur. J. Immunol., 30, 3089–3099. [DOI] [PubMed] [Google Scholar]
- Theodossis A., et al. (2010) Constraints within major histocompatibility complex class I restricted peptides: presentation and consequences for T-cell recognition. Proc. Natl Acad. Sci. U.S.A., 107, 5534–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trolle T., Nielsen M. (2014) NetTepi: an integrated method for the prediction of T cell epitopes. Immunogenetics, 66, 449–456. [DOI] [PubMed] [Google Scholar]
- Vita R., et al. (2015) The immune epitope database (IEDB) 3.0. Nucleic Acids Res., 43, D405–D412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., et al. (2015) Quantitative prediction of class I MHC/epitope binding affinity using QSAR modeling derived from amino acid structural information. Comb. Chem. High Throughput Screen., 18, 75–82. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.