

SUMMARY
NAD+ is a key metabolic redox cofactor that is regenerated from nicotinamide through the NAD+ salvage pathway. Here, we find that inhibiting the NAD+ salvage pathway depletes serine biosynthesis from glucose by impeding the NAD+-dependent protein, 3-phosphoglycerate dehydrogenase (PHGDH). Importantly, we find that PHGDHhigh breast cancer cell lines are exquisitely sensitive to inhibition of the NAD+ salvage pathway. Further, we find that PHGDH protein levels and those of the rate-limiting enzyme of NAD+ salvage, NAMPT, correlate in ER-negative, basal-like breast cancers. Although NAD+ salvage pathway inhibitors are actively being pursued in cancer treatment, their efficacy has been poor, and our findings suggest that they may be effective for PHGDH-dependent cancers.
Graphical Abstract
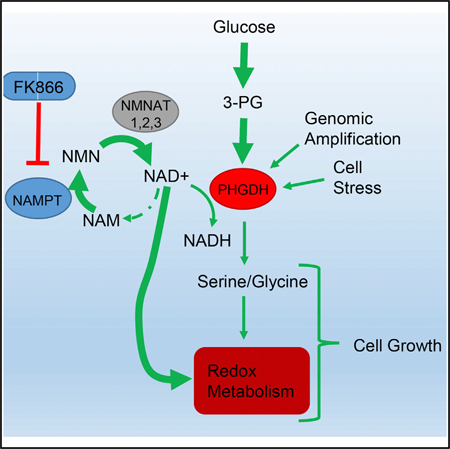
In Brief
Subsets of breast cancers depend on the serine biosynthesis enzyme PHGDH. Murphy et al. show that NAD+ used for PHGDH function requires the NAD+ salvage pathway and that PHGDH-dependent cancers are, thus, sensitive to NAD+ salvage inhibitors. Serine biosynthesis and NAD+ salvage pathway enzymes are also commonly co-expressed in breast cancers.
INTRODUCTION
Many critical metabolic reactions in cells are coupled to the redox co-factor NAD+ and are involved in neurodegenerative disease, cancer, and aging (Cantó et al., 2015). NAD+ levels are influenced both by its rate of utilization as an important biosynthetic substrate and by its regeneration (Chiarugi et al., 2012). Further, NAD+ can also be consumed as a substrate for the sirtuin lysine deacylases (SIRTs) (Haigis and Sinclair, 2010), poly-ADP ribose polymerases (PARPs) (Gupte et al., 2017), and cyclic ADP-ribose synthases (e.g., CD38) (Aksoy et al., 2006). Interestingly, NAD+ utilizing enzymes differ widely in abundance across cell types and physiological conditions, ultimately affecting how NAD+ is used. The use of NAD+ is also determined by subcellular compartmentalization in NAD+ pools, as has been observed across the SIRT families of proteins (Nikiforov et al., 2015).
Since NAD+ consumption removes it from redox pools, NAD+ must either be regenerated or synthesized continually. De novo synthesis occurs through the breakdown of tryptophan via the kynurenine pathway, which is active mostly in the brain, liver, and certain subpopulations of immune cells (Houtkooper et al., 2010). Alternatively, NAD+ regeneration occurs from nicotinamide through the NAD+ salvage pathway, which is favored in most cell types. In this pathway, the rate-limiting enzyme nicotinamide phosphoribosyl transferase (NAMPT) catalyzes the conversion of nicotinamide to nicotinamide mononucleotide (NMN), which is further converted to NAD+ by one of the three NMN adenylyl transferases (NMNATs; NMNAT1, −2, or −3) (Cantó et al., 2015). Pharmacological depletion of NAD+ is being explored broadly as a cancer treatment, leading to the development of drugs such as FK866/APO866 and epacadostat, inhibitors of salvage and de novo NAD+ biosynthesis, respectively (Hasmann and Schemainda, 2003; Hjarnaa et al., 1999). Recent work suggests that redox molecules such as NAD+ support stress responses in cancer cells by regulating amino acid metabolism that, in turn, supplies precursors for detoxifying reactive oxygen species (ROS) (Quirós et al., 2017). Indeed, 3-phospho-glycerate dehydrogenase (PHGDH), the first enzyme of the mammalian serine biosynthesis pathway (SBP), is NAD+ dependent. Moreover, certain breast cancers depend on genomically amplified PHGDH, which diverts glucose carbons away from glycolysis and into oxidative stress and biosynthetic pathways (Locasale et al., 2011; Possemato et al., 2011).
Although the SBP provides several precursors for glutathione, nucleotides, phospholipids, and porphyrins (Mattaini et al., 2016), the full benefit of amplified PHGDH to tumors is incompletely understood. The SBP is controlled by stress-related transcription factors, such as ATF4 (Ye et al., 2010), NRF2 (NFE2L2) (Mitsuishi et al., 2012), and p53 (Maddocks et al., 2016). Furthermore, stress-regulated NRF2 activation promotes the SBP in non-small-cell lung cancer (DeNicola et al., 2015), and high PHGDH levels are associated with aggressiveness and poor prognoses in lung adenocarcinomas (Zhang et al., 2017). Correspondingly, NAMPT (in the salvage pathway) is also induced by the stress response (Chiarugi et al., 2012), but coordination between global metabolic stress responses and the SBP has not been reported. Here, we investigate the proteomic changes during stress caused by depletion of NAD+ through complex I (CI) inhibition. These data and our laboratory’s previous stress-related findings (Sharif et al., 2016) further prompted an investigation into the requirement of NAD+ salvage for serine biosynthesis and growth of PHGDH-dependent breast cancers. We find that the NAD+ salvage pathway supports PHGDHhigh breast cancer cells and that they are exquisitely sensitive to NAMPT inhibition. We also find evidence for PHGDH and NAMPT co-expression in ER-negative, basal-like breast cancers. These findings suggest that the NAD+ salvage pathway is a potentially underappreciated target for the treatment of PHGDH-dependent cancers.
RESULTS
NAD+ Depletion Decreases Serine Biosynthetic Flux in PHGDH-Dependent Cells
To investigate the global proteome response to NAD+ depletion, we performed multiplexed, quantitative proteomics of SH-SY5Y cells treated with multiple CI inhibitors. A major observation from these datasets was the induction of SBP proteins by CI inhibition (Figure S1A; Data S1), supporting known roles of the SBP alongside other redox responses during cell stress. However, these NAD-depletion responses are particularly interesting, since PHGDH directly requires NAD+ redox to function. Furthermore, how NAD+ redox is supplied, or whether NAD+ regeneration co-comprises the stress response, in PHGDHhigh breast cancers is not established. To determine this, we first used mass spectrometry to measure the incorporation of fully 13C-labeled glucose into serine (Figure 1A) during NAD+ depletion of two major sources of NAD+ redox: NAD+ salvage and CI. To control for differences in glucose utilization and transaminase activity, we also measured glucose carbon incorporation into alanine, which is normally derived from transamination of pyruvate at the endpoint of glycolysis (Figure 1A). Western blotting confirmed a mild induction of PHGDH in MCF-7 and MDA MB 231 cells in response to CI inhibitors by the well-characterized NAMPT inhibitor FK866, but no effect on PHGDHhigh MDA MB 468 cells (Figure S1B). In comparison to CI inhibitors, FK866 almost completely ablated serine labeling in PHGDHhigh MDA MB 468 cells (Figure 1B), and it corresponded well to the degree of NAD+ depletion that we observed in these cells (Figure S1C). 13C-serine decreases were not significant, or occurred to a lesser degree, in PHGDHlow MDA MB 231 cells (Figure 1B). We observed no differences in alanine labeling (Figure 1C), and no serine labeling was observed in MCF-7 cells (data not shown).
Figure 1. Serine Biosynthesis Is Inhibited by NAD+ Depletion.

(A) Schematic of 13C-glucose incorporation into serine and alanine (from pyruvate).
(B) 13C-glucose incorporation into serine in cell lines treated with CI inhibitors (10 μM rotenone, 100 μM phenformin, 1 μM piericidin A, and 100 μM MPP+) or 10 μM NAD+ salvage inhibitor FK866.
(C) 13C-glucose incorporation into alanine following CI or NAD+ salvage inhibition, as in (B).
(D) Time course of 13C-glucose incorporation into serine, alanine, pyruvate, and 3-phosphoglycerate over 48 hr in MDA MB 468 and MDA MB 231 cells treated with DMSO (control), 10 μM FK866, or 10 μM FK866 and 100 μM NMN.
Error bars represent the mean ± SEM. *p < 0.05, t test. See also Figure S1 and Data S1.
Since NAD+ is a crucial cofactor for glycolysis, we also performed several experiments to determine whether decreased serine biosynthesis by FK866 results from decreased glycolytic supply to the SBP. First, we measured glucose uptake in MDA MB 468 cells using the glucose analog 2-NBDG and observed no effect of FK866 treatment on glucose uptake with or without the addition of 100 μM NMN, the product of NAMPT, for 48 hr (Figure S1D). In a time course experiment, we observed no temporal differences in alanine labeling in either MDA MB 231 or MDA MB 468 cells during FK866 treatment, suggesting that glycolytic flux remains intact (Figure 1D). In this same experiment, we observed a maximal amount of serine labeling at 24 hr, and this labeling was inhibited by 10 μM FK866 and rescued with 100 μM NMN. Strikingly, measurement of glucose incorporation into 3-phosphoglycerate and pyruvate also confirmed that branches of glycolysis most closely positioned to PHGDH were functioning normally in both cell lines (Figure 1D).
To address whether FK866 sensitivity resulted from PHGDHhigh cells having higher 12C pools, we also spiked in heavy amino-acid standards to PHGDHhigh MDA MB 468 cell and PHGDHlow MDA MB 231 cell extracts, thus determining absolute 12C- and 13C-serine amounts. This confirmed that PHGDHhigh cells are not more sensitive to FK866 because they have more or less 12C-serine or are depleted of serine faster than PHGDHlow cells (Figure S1E). Interestingly, although these main branches of glycolysis did not change, we observed a significantly decreased basal extracellular acidification rate (ECAR) (Figure S2A), oxygen consumption (Figure S2B), and lactate secretion (Figure S2C) by 10 μM FK866 treatment, and all were rescued with 100 μM NMN. As such, FK866 decreases lactate secretion, but unexpectedly, glycolytic reactions directly supplying PHGDH are not changed. Since the SBP combats ROS, we also determined whether the relationship between NAD+ inhibition and the SBP is a part of the mitochondrial stress or ROS response. We observed only small changes in mitochondrial superoxide production (Figure S2D) and ROS (Figure S2E) following 10 μM FK866 treatment, compared to the H2O2-posi-tive control. As such, we observed no correlation between ROS and SBP function. Together, these results show that the inhibition of the NAD+ salvage pathway disrupts serine biosynthesis in PHGDHhigh cells, independent of the main branch of glycolysis and ROS.
NAD+ Depletion Impedes the Growth of PHGDH-Dependent Cells
Multiple efforts are ongoing to target PHGDH directly, but modulating NAD+ levels in PHGDHhigh cancers has not been investigated. To determine the requirement for NAD+ salvage in PHGDH-dependent cells, we selected three PHGDHhigh (MDA MB 468, HCC70, and BT20) and three PHGDHlow (MCF10A, 48 hr. Western blotting confirmed high or low expression of PHGDH in the cell lines and unchanged levels due to FK866 treatment (with or without NMN) (Figure 2A). Mass spectrometry confirmed that FK866 treatment (10 μM) for 48 hr significantly decreased NAD+ levels in all cell lines, which was rescued by the addition of 100 μM NMN (Figure 2B). Accordingly, we next measured cell growth after treatment for 96 hr in either 0.01 or 10 μM FK866. We observed a markedly greater impairment of growth in PHGDHhigh cell lines than in PHGDHlow cell lines, which was rescued in all PHGDHhigh cell lines by the addition of 100 μM NMN (added every 24 hr) (Figure 2C).
Figure 2. PHGDH-Dependent Cells Are Sensitive to FK866.

(A) Western blotting analysis comparing PHGDH expression in PHGDHhigh and PHGDHlow cell lines, following treatment with 10 μM FK866 with or without 100 μM NMN. MCF-7 lysates are used to bridge between blots.
(B) Confirmed decreases in NAD+ levels by mass spectrometry in both PHGDHlow and PHGDHhigh cell lines treated for 48 hr with 10 μM FK866 and rescued with 100 μM NMN.
(C) Cell counts of PHGDHlow and PHGDHhigh breast cancer cell lines treated with two concentrations of FK866 (10 nM and 10 μM) for 96 hr with or without 100 μM NMN.
(D) 13C-glucose incorporation into serine and alanine in SBP-dependent cell lines treated with 10 μM FK866 and rescued with 100 μM NMN.
Error bars represent the mean ± SEM. *p < 0.05, t test. See also Figures S2, S3, and S4.
Finally, we observed significantly decreased serine labeling in all three PHGDHhigh cell lines treated with 10 μM FK866 for 48 hr, which was rescued by adding 100 μM NMN, with no effect on alanine labeling (Figure 2D). Importantly, by measuring NAD+ depletion over the time of FK866 treatment, we observed slower depletion in the MCF-7 cells but similar rates of decrease between MDA MB 231 and MDA MB 468 cells (Figure S3B), suggesting that FK866 susceptibility does not result from unequal NAD+ turnover rates. NAD+ depletion differences, particularly in MCF-7 cells, also do not appear to be due to their use of nicotinamide, since both MCF-7 and MDA MB 468 cells show a similar overall release (rather than consumption) of nicotinamide into the media during a time course of FK866 treatment (+/‒NMN) (Figure S3C). Furthermore, comparing these cell lines in low-dosage FK866 treatments (0 to 100 nM) showed that, when the degree of NAD+ depletion was equal across different dosages (Figure S3D), differential effects on cell growth remain apparent, particularly between MDA MB 231 (PHGDHlow) and MDA MB 468 (PHGDHhigh) cells (Figure S3E). Further supporting these data, these same dosages directly corresponded to decreases in 13C-glucose incorporation to 13C-serine in MDA MB 468 cells, with no change in 13C-alanine (Figure S3F).
We also investigated these effects in non-proliferative MCF10A cells (grown without epithelial growth factor) overexpressing either GFP (GFPOE) or PHGDH (PHGDHOE) (Figure S4A). Although non-proliferative MCF10A GFPOE cells were sensitive to FK866, we observed significantly more cell growth (after 96 hr) in PHGDHOE than in GFPOE MCF10A cells (Figure S4B). We also observed significantly greater cell growth rescue by NMN in PHGDHOE than in GFPOE cells treated with 10 μM FK866 (Figure S4B). Serine labeling in the non-proliferative model (GFPOE versus PHGDHOE) followed a similar trend as that in the cell growth experiments, and no effect was observed on alanine labeling by FK866 treatment (Figure S4C). These results suggest that PHGDHhigh and SBP-dependent, proliferative breast cancer cells are particularly sensitive to NAD+ salvage pathway inhibition.
NAMPT Produces NAD+ as a PHGDH Substrate in Breast Cancer Cells
To further support the data obtained through pharmacological NAD+ salvage inhibition, we performed short hairpin RNA (shRNA)-mediated knockdown (KD) of NAMPT, the rate-limiting enzyme of the NAD+ salvage pathway, in PHGDHlow (MCF-7) and PHGDHhigh (MDA MB 468) cells, using two separate shRNAs compared to a non-specific shRNA (Figure 3A). We observed only minor changes in PHGDH levels in NAMPT KD cells (in PHGDHlow MCF-7 cells only) grown with or without 100 μM NMN (Figure 3A). In both cell lines, NAMPT KD significantly decreased NAD+ levels after 48 hr, compared to the non-specific control, and the effect was rescued by adding 100 μM NMN (Figure 3B). Similar to the FK866 experiments, we observed significantly decreased serine labeling in NAMPT KD (PHGDHhigh) MDA MB 468 cells grown in 13C-glucose for 48 hr after puromycin selection, which was again rescued by adding 100 μM NMN (Figure 3C). Again, alanine labeling was unchanged in the NAMPT KD cells (Figure 3C). No 13C-serine labeling was detected in MCF-7 cells with or without 100 μM NMN (data not shown). We also observed significantly less cell growth (96 hr after puromycin selection) in NAMPT KD MDA MB 468 than in non-specific shRNA control cells, which was rescued by adding 100 μM NMN every 24 hr (Figure 3D). In NAMPT KD MCF-7 cells, the effect was less pronounced and not rescued by the presence of 100 μM NMN (Figure 3D). These findings, again, support the greater sensitivity of PHGDHhigh cells to NAD+ salvage pathway inhibition.
Figure 3. SBP Dependence Requires NAMPT.

(A) Analysis of NAMPT and PHGDH expression bywestern blotting in NAMPT KD MCF-7 and MDA MB 468 cells.
(B) NAD+ levels in NAMPT KD MCF-7 and MDA MB 468 cells grown in the presence or absence of 100 μM NMN (for 48 hr after puromycin selection).
(C) 13C-glucose incorporation into serine and alanine in NAMPT KD MDA MB 468 cells.
(D) Cell counts in NAMPT KD cells after 96 hr. ns, non-specific (non-targeting).
Error bars represent the mean ± SEM. *p < 0.05, t test.
To determine whether the rescue of cell growth by NMN is, indeed, SBP dependent, we performed shRNA-mediated KD of PHGDH in MDA MB 468 cells (Figure 4A). NAMPT levels were mostly unchanged in these cells grown with or without 100 μM NMN (Figure 4A). We observed significantly decreased 13C-glucose incorporation into serine in PHGDH KD cells, compared to control shRNA, which was not rescued by 100 μM NMN (Figure 4B). We observed no difference in alanine labeling following PHGDH KD (Figure 4B), nor did we observe any difference in glucose uptake based on 2-NBDG measurements in PHGDH KD cells with or without NMN (Figure S4D). We also determined cell growth 96 hr following puromycin selection and observed significant decreases, again, without any rescue by adding 100 μM NMN (Figure 4C). To determine whether these effects are specific to PHGDH, we also knocked down PSAT1 (which follows PHGDH in the SBP) expression in MDA MB 468 cells, which did not result in changes in the levels of NAMPT and PHGDH (Figure 4D). PSAT1 knockdown resulted in a more moderate effect on cell growth (Figure 4E) than PHGDH knockdown but had a similar effect on serine labeling that was, again, not rescued by 100 μM NMN (Figure 4F). PSAT1 knockdown did not alter glucose flux to alanine (Figure 4F). These data confirm that the rescue effects provided by NMN addition occur upstream of PHGDH as a source of NAD+ and not by either the rest of the pathway or a pleiotropic enhancement of metabolism. Based on these results and those of the NAMPT knockdown experiments, we propose a model of NAD+ salvage pathway-driven serine biosynthesis and suggest the requirement of NAMPT in support of SBP-dependent cancers (Figure 4G).
Figure 4. NMN Does Not Rescue PHGDH KD.

(A) Western blot for NAMPT and PHGDH expression in PHGDH KD MDA MB 468 cells (grown in the presence or absence of 100 μM NMN for 48 hr after puromycin selection).
(B) 13C-glucose incorporation into serine and alanine in PHGDH KD MDA MB 468 cells.
(C) Cell counts in PHGDH KD MDA MB 468 cells (grown in the presence or absence of 100 μM NMN for 96 hr after puromycin selection).
(D) Western blot for PSAT1, NAMPT, and PHGDH expression in PSAT1 KD MDA MB 468 cells (grown in the presence or absence of 100 μM NMN for 48 hr after puromycin selection).
(E) Cell counts in PSAT1 KD MDA MB 468 cells (grown in the presence or absence of 100 μM NMN for 96 hr after puromycin selection).
(F) 13C-glucose incorporation into serine and alanine in PSAT1 KD MDA MB 468 cells.
(G) A putative model of NAD+ salvage-supported serine biosynthesis. ns, non-specific (nontargeting) shRNA.
Error bars represent the mean ± SEM. *p < 0.05, t test.
Metabolomic Coordination between NAMPT and PHGDH Contributes to Redox Metabolites
Although PHGDH dependence is observed in subsets of cancers, the metabolic requirements it satisfies are not fully understood. Our experiments indicate that these requirements may be met only if cells also have an active NAD+ salvage pathway, which itself may have other PHGDH-independent roles. As such, we sought to examine whether the integrated metabolic benefits of coordinated utilization of both pathways perhaps supply key growth and oxidative stress response metabolites. The biosynthetic benefit of PHGDH flux is beyond the production of the amino acid itself, since serine is present at high amounts in most cell culture media, and exogenously supplied serine does not rescue PHGDH depletion (Possemato et al., 2011). Furthermore, PHGDHhigh MDA MB 468 cells are not more resistant to blocking serine flux by glucose withdrawal (Figure S5A), suggesting PHGDH supports growth through the SBP itself rather than obvious moonlighting functions.
The SBP also produces α-ketoglutarate as a by-product of using glutamate to transaminate 3-phosphohydroxypuruvate. This reaction is proposed to supply a significant amount of anapleurotic α-ketoglutarate to the tricarboxylic acid (TCA) cycle (Possemato et al., 2011). To first examine known metabolites met by NAD+ salvage-supported SBP, we tested whether FK866 affected the incorporation of fully labeled 13C-glutamine into α-ketoglutarate in PHGDHhigh MDA MB 468 cells (Figure 5A). We observed no change in fully labeled 13C-α-ketoglutarate following treatment with FK866 with or without the addition of NMN (Figure 5B), even though 13C-glutamate labeling was near complete (Figure 5C). We next determined whether FK866 affects α-ketoglutarate flux in MCF-7 and MDA MB 231 cells modified to overexpress PHGDH (versus GFP) (Figures 5D and 5E). Consistent with our model, we observed NAD+ levels to be slightly lower in the PHGDHOE MDA MB 231 cells and depleted further by 10 μM FK866 (Figure S5B). Increased incorporation of 13C-glucose into serine was, indeed, observed in PHGDHOE MDA MB 231 cells, which then decreased following 10 μM FK866 treatment and was rescued by adding 100 μM NMN (Figure 5F). PHGDH overexpression was not sufficient to initiate serine labeling in MCF-7 cells, perhaps due to their lower NAMPT levels (Figure 5D). Having confirmed the ability of PHGDH-overexpressing PHGDHlow/ NAMPThigh MDA MB 231 cells to increase serine labeling, we again performed 13C-glutamine labeling experiments and observed no decrease in 13C-α-ketoglutarate labeling (Figure 5G), which closely followed 13C-glutamate labeling patterns (Figure 5H). The decrease in 13C-glutamate in PHGDHOE cells might be explained by increased glutamate incorporation into glutathione downstream of serine biosynthesis. Together, these data suggest that the inhibition of NAD+ salvage fails to inhibit SBP-associated α-ketoglutarate synthesis.
Figure 5. Effect of NAD+ Salvage Inhibitors on the Incorporation of Glutamine Carbons into a-Ketoglutarate.

(A) Schematic of 13C-glutamine incorporation into glutamate and α-ketoglutarate.
(B and C) Shown are the fraction of fully labeled 13C-α-ketoglutarate (B) and C-glutamate (C) following 48 hr of FK866 treatment with or without 100 μM NMN.
(D) Western blot comparing PHGDH and NAMPTlevels in MCF-7, MDA MB 468, and MDA MB 231 cells.
(E) Western blot of MCF-7 and MDA MB 231 cells overexpressing GFP (GFPOE) or PHGDH (PHGDHOE) compared to MDA MB 468 cells.
(F) In PHGDHOE MDA MB 231 cells, 13C-glucose incorporation into serine following 10 μM FK866 treatment in the presence or absence of 100 μM NMN for 48 hr, compared to the GFPOE cells.
(G and H) In MDA MB 231 cells from (E), the fraction of fully labeled 13C-α-ketoglutarate (G) and 13 C-glutamate (H) following 48 hr FK866 treatment in the presence or absence of 100 μM NMN is shown.
Error bars represent the mean ± SEM. *p < 0.05, t test. See also Figure S5.
We next examined the global metabolic effects of coupling the NAD+ salvage pathway to the SBP. To perform this analysis, we used a hydrophilic interaction liquid chromatography-multiple reaction monitoring (HILIC-MRM) mass spectrometry method (Yuan et al., 2012), comparing three PHGDHlow and three PHGDHhigh breast cancer cell lines treated with 10 μM FK866 with or without 100 μM NMN. To further examine the PHGDHdriven metabolic effects, we also performed a global analysis of PHGDH KD in PHGDHhigh MDA MB 468 cells in the presence or absence of NMN. Although NAD+ salvage is required for serine biosynthesis in these cells, hierarchical clustering of metabolome data revealed distinct differences in the patterns of metabolites between FK866 and PHGDH KD experiments (Figure 6A). For example, we observed decreased overall kynurenine and glycerol-3-phosphate in FK866-treated cells, but these same metabolites were increased in PHGDH KD cells (Figure 6A). Interestingly, we also observed increases in nucleobases, including inosine, adenosine, and guanine, in PHGDH KD cells (Figure 6A). Metabolites that solely decreased in the PHGDH KD cells, but were not rescued in the presence of NMN, were numerous, including carnitine, acetylcarnitine, erythrose-4phosphate, DHAP, shikimate, and glycerophosphocholine (Figure 6A). These metabolites share common roles in protection from oxidative stress that are consistent with the known roles of serine biosynthesis and occur separately from the ones that are supported by the NAD+ salvage pathway.
Figure 6. Global Metabolite Profiling following NAD+ Salvage Inhibition and PHGDH KD.

(A) Hierarchically clustered heatmap of metabolites following FK866 treatment in PHGDHhigh and PHGDHlow cells (with or without 100 μM NMN) or PHGDH KD in MDA MB 468 cells (for both, mean log2 ratios compared to PBS-treated or ns controls).
(B) Relative taurine, UDP-glucose, and UDP-glucuronate levels across cell lines treated with 10 μM FK866, with or without 100 μM NMN, for 48 hr.
(C) Relative taurine, UDP-glucose, and UDP-glucuronate levels in PHGDH KD and ns shRNA control cells, with or without 100 μM NMN.
(D) A putative model of metabolic differences between PHGDHhigh and PHGDHlow cells highlighting the PHGDH-related and -unrelated effects of elevated NAMPT.
Error bars represent the mean ± SEM. *p < 0.05, t test. ns, non-specific (non-targeting) shRNA. See also Figure S6.
The most interesting cluster of metabolites from the dataset matched the profile of (1) decreased in the PHGDHhigh cells by 10 μM FK866 treatment and rescued by adding 100 μM NMN and (2) decreased by PHGDH KD but not rescued by adding 100 μM NMN (indicated by the red box in Figure 6A). This group represents metabolites that are likely supported through cooperation between NAD+ salvage and the SBP. Among the metabolites meeting these criteria was the sulfur amino acid taurine (Figures 6B and 6C), which is consistent with its biosynthesis from cysteine downstream of serine and the use of NAD+ as a cofactor in this process (Wu, 1982). These data and roles for taurine in counteracting oxidative stress (Marcinkiewicz and Kontny, 2014) suggest that it may comprise a part of the known stress-protective functions of the SBP. The remaining metabolites in the cluster are closely related uridine metabolites: uridine diphosphate (UDP)-glucose and UDP-glucuronate (Figures 6B and6C). Uridine metabolites are constituents of hexosamine biosynthesis and glycosylation, needed for cell-surface protein expression. To determine the role of NAD+ salvage in cell-surface protein levels, we performed a quantitative proteomic experiment with cell-surface glycoprotein biotinylation (Weekes et al., 2014), comparing cells treated for 24 hr in duplicate with PBS, 10 μM FK866, 100 μM NMN, 10 μM FK866 + 100 μM NMN, or 25 μM azaserine, an inhibitor of hexosamine biosynthesis (Figure S6A). In comparison to azaserine, we observed only minor effects of FK866 on cell-surface glycoproteins (Figure S6B) or proteins with predicted N- and O-linked glycosylation sites based on the UniProtKB database (Bairoch et al., 2008) (Figure S6C), suggesting a minimal effect of NAD+ salvage on uridine-metabolite-related changes in the expression of cell-surface proteins.
Altogether, these data indicate that NAD+ salvage-supported serine biosynthesis may support only a minor fraction of cellular α-ketoglutarate or cell-surface glycoproteins. Rather, we find that the pathway supports known redox roles for SBP-dependent metabolites such as taurine and suggests a model whereby coordination between NAD+ salvage and the SBP—through high levels of both PHGDH and NAMPT—contributes to several important redox metabolites (Figure 6D). This model is consistent with the already proposed role for the SBP (Mehrmohamadi et al., 2014) but adds coordinated metabolism happening through coupled NAMPT and PHGDH expression.
NAMPT and PHGDH Are Correlated in ER-Negative Breast Cancers
The requirement of adequate NAMPT levels for PHGDH function in breast cancer cell lines prompted us to investigate whether they are co-expressed in patient-derived breast tumors. Since proteins with coordinated functions are often more apparent at the protein than at the mRNA level (Lapek et al., 2017), we examined the relationship between NAMPT and PHGDH protein abundance, using a breast cancer proteomics dataset from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) containing a comprehensive proteomics analysis of 105 patient samples from The Cancer Genome Atlas (TCGA) (Mertins et al., 2016). Pearson correlation of the relative expression (normalized to a pooled sample) of all proteins in the CPTAC dataset revealed PHGDH to be among the top 25 most co-expressed proteins with NAMPT (Figure 7A). Other proteins included GLYATL2, PLOD3, CDC123, and others with varied functions (Figure 7A). On the other hand, the least co-expressed proteins included GATA3, EVL, and RABEP1, also with no immediately obvious functional similarities (Figure 7A). Besides PHGDH, we also observed significant correlation between NAMPT and PSAT1, the protein following PHGDH in the SBP that catalyzes the transamination of 3-PHP to 3-phosphoserine (Figure 7B). These proteins appear predominantly co-elevated in estrogen receptor (ER)-negative tumors (Figure 7B). Fitting with these data, previous reports indicate that PHGDH elevation is more frequent in ER-negative and basal-like tumor subsets (Possemato et al., 2011). Indeed, in the CPTAC dataset, we observed significantly higher relative PHGDH and NAMPT abundance in ER-negative tumors than in ER-positive tumors (Figure 7C). Although there were no significant differences in NAMPT or PHGDH between Her2‒and Her2+ cells (Figure S7A), there were several differences in NAMPT and PHGDH across the basal, Her2+, and luminal subtypes (Figure S7B). Fittingly, we also observed significantly higher NAMPT levels in tumors with above-median PHGDH expression, compared to those with below-median PHGDH expression, albeit in both ER-negative and ER-positive tumors (Figure S7C). A clear difference in subtype is exemplified in four of the most NAMPThigh patients: subtyped basal-like, progesterone receptor (PR) negative, and ER negative (Figure 7D).
Figure 7. Correlation of PHGDH and NAMPT Protein Levels within Breast Cancer Samples.

(A) 25 most co-expressed and least co-expressed proteins (by Pearson correlation) with NAMPT in the CPTAC dataset of protein expression across breast cancer samples (n = 105).
(B) Correlation among patients for PHGDH and PSAT with NAMPT in the CPTAC dataset.
(C) Relative protein abundance of PHGDH and NAMPT expression between ER-positive and ER-negative breast cancer samples in the CPTAC dataset.
(D) Top: relative abundance of PHGDH and PSAT1 in patients with the highest or lowest NAMPT expression in the dataset. Bottom: their corresponding tumor subtypes.
(E) Correlation among patients for PHGDH and ER- ER+ NAMPT in the METABRIC dataset.
(F) Relative transcript abundance (Z score) of PHGDH and NAMPT expression between ER-positive and ER-negative breast cancer samples in the CPTAC dataset.
See also Figure S7.
Finally, to support the observations from the CPTAC proteomics dataset, we assessed mRNA expression of NAMPT and PHGDH in 1,980 breast cancer patients, using the gene expression data from TCGA (METABRIC) (Pereira et al., 2016). Pearson correlation between NAMPT and PHGDH gene expression levels was significant, albeit more poorly correlated than at the proteome level (Figure 7E), but both proteins were significantly higher in ER-negative patients (Figure 7F). Such higher correlation at the protein level (but not by mRNA) is not surprising, since protein levels typically are more representative of function than transcript-level measurements (Lapek et al., 2017; Vogel and Marcotte, 2012). Taken together with our metabolomics data, these findings suggest that NAMPT expression is an unexplored vulnerability of SBP-dependent breast cancers. Importantly, NAMPThigh/PHGDHhigh cancers appear to be of the poorly tractable, ER-negative, PR-negative, and basal-like subsets. NAMPT inhibitors, currently only marginally successful for cancer therapy (Kennedy et al., 2016), might be more effective if strategically implemented to target PHGDHhigh tumors.
DISCUSSION
Our findings show that the inhibition of the NAD+ salvage pathway impairs serine biosynthesis and cell growth in SBPdependent breast cancers, and they highlight the pivotal role of NAD+ salvage-supported SBP in cell metabolism and biology. The long-appreciated role of NAD+ as a biosynthetic cofactor and substrate for important cancer proteins such as PARPs and SIRTs has already led to the development of several NAMPT inhibitors, including FK866 (used in this study, also called APO866) (Hasmann and Schemainda, 2003), GNE618 (Zheng et al., 2013), CHS-828 (Hjarnaa et al., 1999), and others (Adams et al., 2014; Fleischer et al., 2010). Unfortunately, in vitro studies have shown differential susceptibility among cancers to NAMPT inhibitors (Xiao et al., 2016), limiting their widespread clinical adoption. These limitations result from redundant NAD+-synthesizing metabolic pathways compensating for the NAMPTinhibition-mediated effects. For example, some cancers resist NAMPT inhibitors by activating alternative NAD+ synthesis routes such as the Preiss-Handler pathway (O’Brien et al., 2013; Piacente et al., 2017). Enhanced surface expression of CD73 also imparts resistance to NAMPT inhibition in some cells by enabling the utilization of extracellular NMN (Grozio et al., 2013). Such variations in NAD+ metabolism across different cancer subtypes may explain why clinical trials for NAMPT inhibitors have been largely unsuccessful (Holen et al., 2008). This cancertype specificity of NAD+ salvage inhibitors is further supported by findings showing that cell lines harboring mutations in the metabolic onco-protein IDH-1 are more susceptible to NAMPT inhibitors than their wild-type counterparts (Tateishi et al., 2015). Taken together with our findings that PHGDH-driven cancers are exquisitely sensitive to NAD+ salvage inhibition, improved clinical success for NAD+ salvage inhibitors may result if they are used for specific cancer subtypes. Accordingly, the co-expression of NAMPT and PHGDH in ER-negative breast cancers suggests that stratifying patients by PHGDH expression will improve the efficacy of NAMPT inhibitors in clinics. A better understanding of the full metabolic benefit of PHGDH is necessary, with particular respect to therapeutic limitations that may be encountered by NAD+ salvage support of serine production in immune cells.
Our global metabolomics findings suggest that the biosynthetic and redox precursors supplied by the SBP, while not fully understood, may be influenced, in part, through NAMPT-dependent actions arising from its co-expression with PHGDH. Since the main parts of the glycolysis pathway are not changed by NAMPT inhibition, our data suggest that NAD+ generated by NAMPT exists as a separate pool (than NAD+ for glycolysis) that is more closely linked to PHGDH. Better techniques to measure subcellular NAD+ pools are needed to assess this. Furthermore, the perceived effect on the SBP’s role in supplying α-ketoglutarate (Possemato et al., 2011) is inconsistent with our observations for α-ketoglutarate labeling in cells undergoing NAD+ salvage inhibition. Rather, we found evidence that NAMPT and PHGDH coupling sustains high levels of taurine and uracil metabolites, for which the requirement of NAD+ and serine metabolism have been established (Egger et al., 2011; Ueki and Stipanuk, 2007). Since total serine is not limited when the SBP is blocked, taurine and uracil metabolites might be beneficial bystander effects of elevated NAMPT in PHGDH-dependent cells. Stress response functions of implicated metabolites are consistent with known stress roles of NAMPT (Amelio et al., 2014; Wang et al., 2011). Interestingly, PHGDH has also been shown to be induced as a metabolic stress response (DeNicola et al., 2015; Quirós et al., 2017), and our data suggest that these scenarios would require support by a sufficiently active NAD+ salvage pathway. Although both NAMPT and PHGDH are regulated by the stress response (Cerna et al., 2012), the requirement for NAD+ salvage in cells with genomically amplified PHGDH levels is particularly important, since PHGDH inhibitors are only beginning to emerge and their efficacy is yet unproven.
In summary, we reveal metabolic coupling between the NAD+ salvage pathway and the SBP. We show that the SBP acquires NAD+ through the NAD+ salvage pathway for use as a redox substrate. Cells that depend on the SBP are exquisitely sensitive to NAD+ salvage inhibition, and although a full understanding of the metabolic benefit of the pathway coupling and subcellular compartmentalization should be pursued, our metabolomics data suggest that their expression is coupled to combat oxidative stress. We find that NAMPT and PHGDH expression are correlated in breast cancer tumors and are predominantly higher in ER-negative, basal-type breast cancers—the subtypes that are most difficult to treat. These data may highlight an approach for stratifying PHGDHhigh ER-negative, basal-like breast cancer patients for treatment with NAD+ salvage pathway inhibitors. Our findings may also be translatable to other PHGDH-dependent cancers such as those of the skin and lung (Mullarky et al., 2011). While the SBP was the main focus of this study, our proteomics dataset is a resourceful starting point for understanding the role of many other proteins involved in metabolic stress. The dataset is available through Mendeley Data (https://doi.org/10.17632/ptm7dzc25y.2). Furthermore, the SBP in other cell types, such as those of the brain and immune system, may also acquire NAD+ through the salvage pathway, and this relationship should be further explored for neurological and immune-related diseases.
STAR+METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by Dr. Shashi Gujar (shashi.gujar@dal.ca).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
MCF-7, MDA MB 231, and MDA MB 468 cells were maintained in DMEM (Invitrogen #11965) supplemented with 10% fetal bovine serum (Invitrogen #12484). MCF10A cells were maintained in DMEM/F12 (Invitrogen #11330), containing 5% horse serum (Invitrogen #16050), 2 ng/mL epithelial growth factor (EGF) (Peprotech #AF-100–15), 500 ng/mL hydrocortisone (Sigma #H-0888), 100 ng/mL cholera toxin (Sigma #C-8052), and 10 μg/mL insulin (Sigma #I-1882). For MCF10A cell growth experiments with GFP or PHGDH overexpression, EGF was excluded and DMEM or no glucose DMEM was used. BT20 cells were cultured in MEM (Invitrogen #11095) containing 10% fetal bovine serum (Invitrogen #12484) and 1X non-essential amino acids (Invitrogen #11140) and 1X sodium pyruvate (Invitrogen #11360). HCC70 cells were maintained in RPMI containing 10% fetal bovine serum (Invitrogen #12484). All cell lines were maintained in 1X antibiotic-antimycotic (Invitrogen #15240), grown at 37C and sub-cultured by dissociation with 0.5% trypsin-EDTA (Invitrogen #25300).
METHOD DETAILS
Quantitative Proteomics
Sample Preparation Following Treatment with Complex I Inhibitors
Cells were treated, in biological duplicate, for 48 hr with complex I inhibitors (as listed in the Key Resources Table) in 15 cm plates of cells at 50% confluence. Cells were rinsed with PBS and removed from plates by scraping into 2 mL of lysis buffer: 2% SDS, 150 mM NaCl, 50 mM Tris (pH 8.5), 5 mM DTT, and 1 tablet of protease inhibitor (Sigma #11836170001). Lysates were disrupted using an Omni homogenizer (Omni #TH115) with 3 cycles of 12 s, with cooling on ice between cycles. Samples were incubated at 56C for 30 min, cooled, then cysteines were alkylated using 14 mM iodoacetamide, followed by methanol-chloroform precipitation. Proteins were resolubilized in 1.5 mL of 8 M urea, 50 mM Tris, pH 8.8 and total protein content was measured using a BCA assay (Thermo Fisher #23227). An aliquot of 100 μg of protein was diluted to 1.5 M urea, digested for 2 hr with endoproteinase Lys-C (Wako #125–05061) at a ratio of 1:200 Lys-C:protein then overnight with trypsin (Promega #V5111) at a ratio of 1:100 trypsin:protein at 37°C. The pH of the digest was adjusted to < 3 using formic acid and peptides were desalted using 50 mg solid phase C18 extraction cartridges (Waters #WAT054955) then lyophilized. Dried peptides were resuspended in 100 μL of 100 mM HEPES, 30% acetonitrile and 10 μL of TMT10 reagents (Thermo Fisher #90110) pre-aliquoted at a concentration of 20 μg/mL in anhydrous acetonitrile. The reaction was quenched with 0.5% hydroxylamine (Sigma #159417), mixed equally, desalted using a 200 mg solid phase C18 extraction cartridge (Waters #WAT054945), and lyophilized. Peptides were fractionated using an Agilent 300-Extend, 4.6 mm × 250 mm, 5 μm particle size C18 column (Agilent #773995–902). A gradient of 5 to 40% acetonitrile (10 mM ammonium formate, pH 8) was applied at a flow rate of 800 μL/min using an Agilent 1100 pump. Fractions were collected every 0.38 min, beginning at 10 min; then, every 12th fraction was combined to a single sample to create 12 fractions, which were then desalted using homemade Stage-tips packed with Empore C18 extraction material (Sigma #66883-U) as previously described (Rappsilber et al., 2003), then lyophilized and subjected to LC-SPS-MS3.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Anti-PHGDH | Cell Signaling Technology | #66350;RRID:AB_2737030 |
| Rabbit Anti-NAMPT | Santa Cruz Biotechnology | #sc-67020;RRID;AB_2150246 |
| Mouse Anti-beta-Actin | Santa Cruz Biotechnology | #sc-47778;RRID:AB_2714189 |
| Rabbit Anti-PSAT1 | Thermo Fisher Scientific | #PA5–22124;RRID:AB_11153526 |
| Bacterial and Virus Strains | ||
| p-LJM1-EGFP | Addgene | #19319 |
| pDONR-PHGDH | hORFeome v8.1 | N/A |
| pLKO1.1 non-targeting shRNA | Dharmacon | #RHS6848 |
| pLKO1.1 NAMPT shRNA 1 | Dharmacon | #TRCN0000116177 |
| pLKO1.1 NAMPT shRNA 1 | Dharmacon | #TRCN0000116180 |
| pLKO1.1 PHGDH shRNA 1 | Dharmacon | #TRCN0000041626 |
| pLKO1.1 PHGDH shRNA 2 | Dharmacon | #TRCN0000041627 |
| pLKO1.1 PSAT1 shRNA 1 | Dharmacon | #TRCN0000035266 |
| pLKO1.1 PSAT1 shRNA 2 | Dharmacon | #TRCN0000035268 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Rotenone | Sigma | #R8775 |
| Phenformin HCl | Sigma | #P7045 |
| Piericidin A | Sigma | #P4368 |
| MPP+ iodide | Sigma | #D048 |
| FK866 | Sigma | #F8557 |
| Nicotinamide Mononucleotide | Sigma | #N3501 |
| Epithelial Growth Factor | Peprotech | #AF-100–15 |
| Complete-mini Protease Inhibitor | Sigma | #111836170001 |
| Endoproteinase Lys-C | Wako | #125–05061 |
| Sequencing Grade Trypsin | Promega | #V5111 |
| Empore C18 Extraction Material | Sigma | #66883-U |
| Magic C4 Resin | Michrom Bioresources | #PM5/641000/00 |
| Accucore 150A, 2.6 mm C18 Resin | Thermo Fisher | #16126–000 |
| 10-plex Tandem Mass Tags | Thermo Fisher | #90110 |
| Sodium Periodate | Sigma | #311448 |
| Aminoxy-biotin | Biotium | #90113 |
| Aniline | Sigma | #242384 |
| Streptavidin Agarose | Thermo Fisher | #20347 |
| Hydroxylamine | Sigma | #159417 |
| 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) amino]-2-deoxy-glucose | Thermo Fisher | #N13195 |
| MitoSOX Red | Thermo Fisher | #M36008 |
| H2-DCFDA | Thermo Fisher | #D399 |
| Polyethylenimine | Polysciences | #23966–1 |
| Polybrene | Sigma | #S2667 |
| Puromycin | Thermo Fisher | #A1113802 |
| Oligomycin | Sigma | #O4876 |
| Carbonyl cyanide 4-(triflutormethoxy)phenylhydrazone | Sigma | #C2920 |
| Antimycin A | Sigma | #A8674 |
| D-Glucose (U-13C6, 99%) | Cambridge Isotopes | #CLM-1396 |
| L-Glutamine (U-13C5, 95%) | Toronto Research Chemicals | #G597001 |
| Critical Commercial Assays | ||
| Bicinchoninic Acid Assay | Thermo Fisher | #23227 |
| Solid Phase Extraction Cartridges | Waters | #WAT054955 |
| Deposited Data | ||
| Raw CI mass spectrometry data and peptide identifications | This paper | PRIDE PXD007790 |
| Raw cell surface mass spectrometry data and peptide identifications | This paper | PRIDE PXD007791 |
| Experimental Models: Cell Lines | ||
| Human: MCF10A cells | ATCC | #CRL10317 |
| Human: MCF-7 cells | ATCC | #HTB-22 |
| Human: MDA MB 231 cells | ATCC | #HTB-26 |
| Human: MDA MB 468 cells | ATCC | #HTB-132 |
| Human: HCC70 cells | ATCC | #CRL-2315 |
| Human: BT20 cells | ATCC | #HTB-19 |
| Human: HEK293T cells | ATCC | #CRL-3216 |
| Oligonucleotides | ||
| Forward Primer: PHGDH amplification TTATGCTAGCGCCACCATG | This paper | N/A |
| GCTTTTGCAAATCTGCGGAAAGTGCTC | ||
| Reverse Primer: PHGDH amplification GCATTTCGAATTAGAAGTG | This paper | N/A |
| GAACTGGAAGGCTTCAGTCAC | ||
| Software and Algorithms | ||
| Sequest (version 28) | (Eng et al., 1994) | N/A |
| Multiquant | Sciex | N/A |
Cell Surface Biotinylation and Enrichment
As previously described (Weekes et al., 2012), cells (~ 1 3 107) were rinsed twice with PBS (pH 7.4), then cell surface proteins were biotinylated in the dark at 4°C for 2 hr with a biotinylation reaction mixture containing 1 mM sodium periodate (Sigma #311448), 100 μM aminoxybiotin (Biotium #90113), and 10 mM aniline (Sigma #242284) in ice-cold PBS (pH 6.7). The reaction was quenched with 1 mM glycerol, then cells were washed twice with PBS (pH 7.4) and were harvested by scraping in lysis buffer containing 1% Triton X-100, 150 mM NaCl, 5 mM iodoacetamide, 10 mM Tris-HCl pH 7.6 and 1 tablet of protease inhibitor (Sigma #11836170001) per 10 mL of lysis buffer. Lysates were centrifuged 3 times at 12,000 × g for 10 min. Biotinylated proteins were enriched by incubating with 150 μL of streptavidin-agarose resin (Thermo Fisher #20347) for 2 hr followed by washing four times (1.5 mL each) with lysis buffer, four times with PBS, 0.5% SDS (5 mL each), then incubated with 100 mM DTT (500 mL). Resin was further washed four times with 6 M urea, 100 mM Tris/HCl pH 8.5 (2 mL each), and three times with HPLC-grade water (2 mL each). All washes were performed on Polyprep columns (BioRad #731550). Proteins were digested on resin overnight with 100 mM HEPES, pH 8.5 (70 μL) containing 2 μg trypsin at 37°C. The next day, the peptide mixture was made up to 30% acetonitrile, labeled with TMT10 reagents (Thermo Fisher #90110), quenched with 0.5% hydroxylamine (Sigma #159417), mixed equally, and desalted using a 200 mg solid phase C18 extraction cartridge (Waters #WAT054945). Peptides were fractionated using an Onyx, 4.6 mm × 100 mm, monolithic C18 column (Phenomenex #CH0–7643). A gradient of 5 to 40% acetonitrile (10 mM ammonium formate, pH 8) was applied at a flow rate of 1 mL/min using an AKTA pure FPLC system (GE Healthcare). Fractions were collected every 0.4 min beginning at 10 min then combined to 6 fractions, which were lyophilized and desalted using home-made Stage-tips packed with Empore C18 extraction material (Sigma #66883-U) as previously described (Rappsilber et al., 2003), then lyophilized and subjected to LC-SPS-MS3.
LC-SPS-MS3
Each basic reverse phase fraction was resuspended in 0.1% formic acid and analyzed on a Thermo Fisher Orbitrap Fusion mass spectrometer with online chromatography using a Thermo Fisher EASY nLC. Peptides were separated on a 75 μm × 30 cm column packed with 0.5 cm of Magic C4 resin (Michrom Bioresources #PM5/64100/00) and 28 cm of C18 Accucore 120 resin (Thermo Fisher #16126–000). Peptides were separated at a flow rate of 300 nL/min using a gradient of 8%–26% acetonitrile (0.125% formic acid over 120 min followed by 10 min at 100% acetonitrile). Spectra were acquired using a synchronous precursor selection (SPS)-MS3 method on the mass spectrometer (McAlister et al., 2014). In this method, MS1 scans were acquired over 400–1400 m/z, 120,000 resolution, 2e5 AGC target, 100 msec maximum injection time. The 10 most abundant MS1 ions from charge states 2–6 were selected for fragmentation using an isolation window of 0.5 Th, CID activation at 35% energy, rapid scan rate, 4000 AGC target, 30 s dynamic exclusion, and 150 msec maximum injection time. MS3 scans were acquired using SPS of 10 isolation notches, 100–1000 m/z, 60 000 resolution, 5e4 AGC, HCD activation at 55% energy, 250 msec maximum injection time.
Peptide Identification and Protein Quantification
Mass spectrometry data files were converted to mzXML using a modified version of ReadW.exe. MS2 spectra were searched against the human Uniprot database (downloaded August, 2011) using Sequest (Ver28) (Eng et al., 1994) with TMT as a fixed modification (+229.162932) on lysine residues and peptide N-termini, and carbamidomethylation (15.99492) as a fixed modification on cysteine. The allowable precursor mass tolerance was 10 ppm and product ion mass tolerance was 1 Da. False positive rates were controlled using the target-decoy approach (Elias and Gygi, 2007) with a concatenated reversed database employing linear discriminant analysis to distinguish correct and incorrect peptide identifications based on XCorr, ΔCN, peptide length, and charge state. Peptides were grouped into proteins and their multiplied linear discriminant analysis probabilities were used to sort proteins and filter to a 1% maximum false discovery rate. The sum of all reporter ion summed signal to noise (S/N) values for peptides matching each protein was used for protein quantitation.
Global Metabolite and Flux Analysis
Metabolites were extracted from cells by scraping in cold (–20°C) 80% methanol. Samples were centrifuged at 13,000 × g for 5 min and a 25 μL aliquot of supernatant was added to 225 μL of hydrophilic interaction liquid chromatography (HILIC) loading buffer containing 95% acetonitrile, 2 mM ammonium hydroxide, and 2 mM ammonium acetate then centrifuged again at 13,000 × g for 5 min. Triplicate, 50 μL injections of the supernatant were loaded on an Acquity UPLC BEH Amide, 1.7 um particle size, 2.1 3 100 mm column (Waters #186004801). Multiple reaction monitoring (MRM) was performed using a Sciex 5500 QTRAP using a previously described acquisition method (Yuan et al., 2012). Peak heights for individual metabolites including NAD+ were extracted using Multiquant Software (Sciex). For absolute quantitation, 13C,15N-labeled amino acids (Cambridge Isotopes #MSK-A2–1.2) were spiked into cellular extracts to determine absolute measurements of serine and alanine flux. Absolute quantitation of the glucose labeled serine [M+3] to the 13C,15N-labeled amino acids were determined using a QExactive Orbitrap mass spectrometer operated using targeted selected ion monitoring (tSIM) scans (R = 70000) with an m/z window of 8 offset by 3.5 m/z. Extracted ion chromatograms of the [M], [M+3], and [M+4] masses in these windows were formed using a tolerance of 5 ppm implemented using Maven software. Determining fractional 13C-3-phosphoglyerate and 3C-pyruvate labeling were performed similarly. Data are presented as picomolar amounts per protein content based on protein determined in the same experiment by a BCA assay.
Glucose Uptake and ROS by Flow Cytometry
Glucose uptake was measured by incubating cells with a fluorescent glucose analog 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) amino]-2-deoxy-d-glucose (2-NBDG) (Thermo Fisher #N13195) at a concentration of 50 μM for 2 hr at 37C in growth media. The reaction was stopped by removing the treated media and washing the cells with PBS. Experiments were performed in triplicate. Mitochondrial superoxide was determined by detaching cells with 0.5% trypsin-EDTA and incubating in FACS buffer (PBS, pH 7.4 supplemented with 1% FBS and 1 mM EDTA) with 1 μM MitoSOX Red (Thermo Fisher #M36008) for 30 min at 37C. Reactive oxygen species were measured by detaching cells with 0.5% trypsin-EDTA and incubating in FACS buffer (PBS, pH 7.4 supplemented with 1% FBS and 1 mM EDTA) with 1 μM H2-DCFDA (Thermo Fisher #D399) for 30 min at 37C. For both H2-DCFDA and MitoSOX measurements, flow cytometry data were collected using a FACSCalibur flow cytometer (BD Bioscience) and analysis was performed using FCS Express (version 6). After live cell gating by FSC and SSC, mean fluorescence intensity of the channel associated with each dye was calculated (2-NBDG in channel FL1, MitoSOX in channel FL3, H2-DCFDA in channel FL1).
Lentiviral shRNA-mediated Knockdown
Lentiviral shRNA clones for PHGDH and NAMPT were from the RNAi consortium (TC-Hs1.0) as outlined in the Key Resources Table. Following bacterial propagation and plasmid isolation, transfection of HEK293T cells was performed to generate lentivirus using the PAX1 (packaging) and MD2G (envelope) plasmids with polyethylenimine (PEI) (Polysciences #23966–1) as a transfection reagent. Virus supernatant was used to infect MCF-7 or MDA MB 468 cells pretreated with 8 μg/mL polybrene (Sigma #S2667). After 48 hr, cells were subcultured into selection media containing 1 μg/mL puromycin (Thermo Fisher #A1113802) for 48 hr before plating out for experiments in the presence or absence of NMN.
Western Blotting
Cell lysis was performed in RIPA buffer (50 mM Tris, 150 mM NaCl, 0.1% SDS, 1% Triton, and 0.5% deoxycholate). Lysates were sonicated on ice for 3 cycles of 12 s with 20 s rests on ice, then cleared by centrifugation at 13,000 × g for 5 min. Total protein content was measured using a BCA assay (Thermo Fisher #23227). Extracts of 30 μg of protein were separated by SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted using the antibodies outlined in the Key Resources Table.
PHGDH Overexpression
The PHGDH pDONR plasmid was retrieved from the human ORFeome collection (version 8.1). PHGDH was amplified by PCR from the pDONR plasmid using the forward and reverse primers outlined in the Key Resources Table. The pLJM1-eGFP lentiviral expression plasmid (Addgene #19319) was digested with NheI/BstBI restriction enzymes to remove the eGFP fragment then the PHGDH PCR fragment was placed into the linearized pLJM1 by ligation. Transfection of HEK293T cells was performed to generate lentivirus using the PAX1 (packaging) and MD2G (envelope) plasmids with polyethylenimine (PEI) (Polysciences #23966–1) as a transfection reagent. Virus supernatant was used to infect MCF10A, MCF-7, or MDA MB 231 cells pretreated with 8 μg/mL polybrene (Sigma #S2667). After 48 hr, cells were subcultured into selection media containing 1 μg/mL puromycin (Thermo Fisher #A1113802) for 48 hr before plating experiments.
Bioenergetics Analysis
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured in XF assay media (unbuffered DMEM containing 10 mM glucose, 2 mM glutamine and 1 mM pyruvate) under basal conditions and in response to 1 μM oligomycin (Sigma #O4876), 1.5 mM carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (Sigma #C2920), 1 mM rotenone (Sigma #R8875) and 1 μM antimycin A (Sigma #A8674) starting with 1 3 105 cells on the XF24 extracellular flux analyzer (Seahorse Bioscience). Basal OCR was calculated by subtraction of the residual rate after antimycin-A treatment. Empty wells (no cells) were used as baselines for both ECAR and OCR.
QUANTIFICATION AND STATISTICAL ANALYSIS
Analysis of CPTAC and TCGA datasets
The protein level CPTAC ‘‘TCGA Breast Cancer’’ dataset was downloaded from the CPTAC data portal (https://cptac-data-portal.georgetown.edu/cptacPublic/). Protein relative abundance data values in this dataset were originally compared to an internal control sample and remain represented as such here. For all proteins, PAM50, ER, PR, and Her2 status were retrieved using TCGA barcodes retrieved from dataset metadata files. For NAMPT and PHGDH, protein abundance was matched to clinical data files based on the tumor TCGA barcodes. The transcript level ‘‘BRCA_METABRIC’’ TCGA dataset was downloaded from cBioPortal (http://www.cbioportal.org/study?id=brca_metabric#summary). Pairwise transcript z-scores for NAMPT and PHGDH were available for 817 tumors and used to correlate transcript abundance. ER status was assessed using the ‘‘ER_STATUS_BY_IHC’’ parameter from the dataset. All data manipulations for both proteome and transcriptome levels were performed using R. Correlations were performed using Pearson’s correlation coefficients and p values were calculated using the ‘‘Hmisc’’ package. Welch’s 2-sample t tests were performed to determine statistical significance between the ER, PR, and Her2 status, whereas ANOVAs followed by Tukey’s HSD tests were performed to determine statistical significance between PAM50 groups.
Student’s t tests and ANOVAs
Statistical analysis of metabolic flux, cell counts, NAD+ and all other data displayed in bar graph format was analyzed using a Student’s t test (unpaired,α= 0.05) or ANOVA (one way,α = 0.05) performed using Prism GraphPad.
DATA AND SOFTWARE AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (Vizcaíno et al., 2016) with the dataset identifiers PXD007790 (complex I proteomics) and PXD00791 (cell surface proteomics). These datasets are also made available by Mendeley Data (https://doi.org/10.17632/ptm7dzc25y.2).
Supplementary Material
Highlights.
The NAD+ salvage pathway is required for serine biosynthesis
PHGDHhigh cancer cells are highly sensitive to NAD+ salvage pathway inhibition
NAMPT and PHGDH correlate in ER-negative, basal-like breast cancer
ACKNOWLEDGMENTS
We gratefully acknowledge Dr. Jonathan Coloff (Harvard Medical School) and Dr. Paola Marcato (Dalhousie University) for providing editorial advice on the manuscript. We thank Dr. Alejandro Cohen (Dalhousie Proteomics Core Facility), Dr. Dev Pinto (National Research Council), and Ken Chisholm (National Research Council) for mass spectrometry assistance. This work was supported by grants from the Canadian Cancer Society Research Institute (CCSRI), Canadian Institutes of Health Research (CIHR), and the Terry Fox Research Institute (TFRI). J.P.M. and B.K. are supported through the Beatrice Hunter Cancer Research Institute (BHCRI). T.S. is supported by the CIHR. J.A.P. is funded by NIH/NIDDK grant K01 DK098285. S.G. is supported by the Dalhousie Medical Research Foundation (DMRF).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams DJ, Ito D, Rees MG, Seashore-Ludlow B, Puyang X, Ramos AH, Cheah JH, Clemons PA, Warmuth M, Zhu P, et al. (2014). NAMPT is the cellular target of STF-31-like small-molecule probes. ACS Chem. Biol 9, 2247–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksoy P, White TA, Thompson M, and Chini EN (2006). Regulation of intracellular levels of NAD: a novel role for CD38. Biochem. Biophys. Res. Commun 345, 1386–1392. [DOI] [PubMed] [Google Scholar]
- Amelio I, Cutruzzolá F, Antonov A, Agostini M, and Melino G (2014). Serine and glycine metabolism in cancer. Trends Biochem. Sci 39, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A, Apweiler R, Wu CH, and Barker WC; UniProt Consortium (2008). The universal protein resource (UniProt). Nucleic Acids Res 36, D190–D195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantó C, Menzies KJ, and Auwerx J (2015). NAD+ metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab 22, 31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerna D, Li H, Flaherty S, Takebe N, Coleman CN, and Yoo SS (2012). Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) activity by small molecule GMX1778 regulates reactive oxygen species (ROS)-mediated cytotoxicity in a p53- and nicotinic acid phosphoribosyltransferase1 (NAPRT1)-dependent manner. J. Biol. Chem 287, 22408–22417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarugi A, Dölle C, Felici R, and Ziegler M (2012). The NAD metabolome–a key determinant of cancer cell biology. Nat. Rev. Cancer 12, 741–752. [DOI] [PubMed] [Google Scholar]
- DeNicola GM, Chen P-H, Mullarky E, Sudderth JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE, et al. (2015). NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet 47, 1475–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger S, Chaikuad A, Kavanagh KL, Oppermann U, and Nidetzky B (2011). Structure and mechanism of human UDP-glucose 6-dehydrogenase. J. Biol. Chem 286, 23877–23887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, and Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Fleischer TC, Murphy BR, Flick JS, Terry-Lorenzo RT, Gao Z-H, Davis T, McKinnon R, Ostanin K, Willardsen JA, and Boniface JJ (2010). Chemical proteomics identifies Nampt as the target of CB30865, an orphan cytotoxic compound. Chem. Biol 17, 659–664. [DOI] [PubMed] [Google Scholar]
- Grozio A, Sociali G, Sturla L, Caffa I, Soncini D, Salis A, Raffaelli N, De Flora A, Nencioni A, and Bruzzone S (2013). CD73 protein as a source of extracellular precursors for sustained NAD+ biosynthesis in FK866-treated tumor cells. J. Biol. Chem 288, 25938–25949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupte R, Liu Z, and Kraus WL (2017). PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev 31, 101–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis MC, and Sinclair DA (2010). Mammalian sirtuins: biological insights and disease relevance. Annu. Rev. Pathol 5, 253–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasmann M, and Schemainda I (2003). FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res 63, 7436–7442. [PubMed] [Google Scholar]
- Hjarnaa P-JV, Jonsson E, Latini S, Dhar S, Larsson R, Bramm E, Skov T, and Binderup L (1999). CHS 828, a novel pyridyl cyanoguanidine with potent antitumor activity in vitro and in vivo. Cancer Res 59, 5751–5757. [PubMed] [Google Scholar]
- Holen K, Saltz LB, Hollywood E, Burk K, and Hanauske A-R (2008). The pharmacokinetics, toxicities, and biologic effects of FK866, a nicotinamide adenine dinucleotide biosynthesis inhibitor. Invest. New Drugs 26, 45–51. [DOI] [PubMed] [Google Scholar]
- Houtkooper RH, Cantó C, Wanders RJ, and Auwerx J (2010). The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr. Rev 31, 194–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy BE, Sharif T, Martell E, Dai C, Kim Y, Lee PWK, and Gujar SA (2016). NAD+ salvage pathway in cancer metabolism and therapy. Pharmacol. Res 114, 274–283. [DOI] [PubMed] [Google Scholar]
- Lapek JD Jr., Greninger P, Morris R, Amzallag A, Pruteanu-Malinici I, Benes CH,and Haas W(2017).Detectionofdysregulatedprotein-association networks by high-throughput proteomics predicts cancer vulnerabilities. Nat. Biotechnol 35, 983–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, et al. (2011). Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet 43, 869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddocks ODK, Labuschagne CF, Adams PD, and Vousden KH (2016). Serine metabolism supports the methionine cycle and DNA/RNA methylation through de novo ATP synthesis in cancer cells. Mol. Cell 61, 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinkiewicz J, and Kontny E (2014). Taurine and inflammatory diseases. Amino Acids 46, 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattaini KR, Sullivan MR, and Vander Heiden MG (2016). The importance of serine metabolism in cancer. J. Cell Biol 214, 249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wu€hr M, Huttlin EL, Erickson BK, Rad R, Haas W, and Gygi SP (2014). MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrmohamadi M, Liu X, Shestov AA, and Locasale JW (2014). Characterization of the usage of the serine metabolic network in human cancer. Cell Rep 9, 1507–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia F, et al. ; NCI CPTAC (2016). Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M, and Motohashi H (2012). Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22, 66–79. [DOI] [PubMed] [Google Scholar]
- Mullarky E, Mattaini KR, Vander Heiden MG, Cantley LC, and Locasale JW (2011). PHGDH amplification and altered glucose metabolism in human melanoma. Pigment Cell Melanoma Res 24, 1112–1115. [DOI] [PubMed] [Google Scholar]
- Nikiforov A, Kulikova V, and Ziegler M (2015). The human NAD metabolome: Functions, metabolism and compartmentalization. Crit. Rev. Biochem. Mol. Biol 50, 284–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien T, Oeh J, Xiao Y, Liang X, Vanderbilt A, Qin A, Yang L, Lee LB, Ly J, Cosino E, et al. (2013). Supplementation of nicotinic acid with NAMPT inhibitors results in loss of in vivo efficacy in NAPRT1-deficient tumor models. Neoplasia 15, 1314–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira B, Chin S-F, Rueda OM, Vollan H-KM, Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut S-J, et al. (2016). The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun 7, 11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piacente F, Caffa I, Ravera S, Sociali G, Passalacqua M, Vellone VG, Becherini P, Reverberi D, Monacelli F, Ballestrero A, et al. (2017). Nicotinic acid phosphoribosyltransferase regulates cancer cell metabolism, susceptibility to NAMPT inhibitors and DNA repair. Cancer Res 77, 3857–3869. [DOI] [PubMed] [Google Scholar]
- Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo H-K, Jang HG, Jha AK, et al. (2011). Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirós PM, Prado MA, Zamboni N, D’Amico D, Williams RW, Finley D, Gygi SP, and Auwerx J (2017). Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol 216, 2027–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Ishihama Y, and Mann M (2003). Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem 75, 663–670. [DOI] [PubMed] [Google Scholar]
- Sharif T, Ahn D-G, Liu R-Z, Pringle E, Martell E, Dai C, Nunokawa A, Kwak M, Clements D, Murphy JP, et al. (2016). The NAD+ salvage pathway modulates cancer cell viability via p73. Cell Death Differ 23, 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebel F, Lelic N, Wiederschain D, Bedel O, Deng G, Zhang B, et al. (2015). Extreme vulnerability of IDH1 mutant cancers to NAD+ depletion. Cancer Cell 28, 773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueki I, and Stipanuk MH (2007). Enzymes of the taurine biosynthetic pathway are expressed in rat mammary gland. J. Nutr 137, 1887–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, et al. (2016). 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44 (D1), D447–D456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C, and Marcotte EM (2012). Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet 13, 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Hasan MK, Alvarado E, Yuan H, Wu H, and Chen WY (2011). NAMPT overexpression in prostate cancer and its contribution to tumor cell survival and stress response. Oncogene 30, 907–921. [DOI] [PubMed] [Google Scholar]
- Weekes MP, Antrobus R, Talbot S, Hör S, Simecek N, Smith DL, Bloor S, Randow F, and Lehner PJ (2012). Proteomic plasma membrane profiling reveals an essential role for gp96 in the cell surface expression of LDLR family members, including the LDL receptor and LRP6. J. Proteome Res 11, 1475–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weekes MP, Tomasec P, Huttlin EL, Fielding CA, Nusinow D, Stanton RJ, Wang ECY, Aicheler R, Murrell I, Wilkinson GWG, et al. (2014). Quantitative temporal viromics: an approach to investigate host-pathogen interaction. Cell 157, 1460–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JY (1982). Purification and characterization of cysteic acid and cysteine sulfinic acid decarboxylase and L-glutamate decarboxylase from bovine brain. Proc. Natl. Acad. Sci. USA 79, 4270–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Kwong M, Daemen A, Belvin M, Liang X, Hatzivassiliou G, and O’Brien T (2016). Metabolic response to NAD depletion across cell lines is highly variable. PLoS ONE 11, e0164166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Kumanova M, Hart LS, Sloane K, Zhang H, De Panis DN, Bobrovnikova-Marjon E, Diehl JA, Ron D, and Koumenis C (2010). The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J 29, 2082–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Breitkopf SB, Yang X, and Asara JM (2012). A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat. Protoc 7, 872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Zheng A, Hydbring P, Ambroise G, Ouchida AT, Goiny M, Vakifahmetoglu-Norberg H, and Norberg E (2017). PHGDH defines a metabolic subtype in lung adenocarcinomas with poor prognosis. Cell Rep 19, 2289–2303. [DOI] [PubMed] [Google Scholar]
- Zheng X, Bair KW, Bauer P, Baumeister T, Bowman KK, Buckmelter AJ, Caligiuri M, Clodfelter KH, Feng Y, Han B, et al. (2013). Identification of amides derived from 1H-pyrazolo[3,4-b]pyridine-5-carboxylic acid as potent inhibitors of human nicotinamide phosphoribosyltransferase (NAMPT). Bioorg. Med. Chem. Lett 23, 5488–5497. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (Vizcaíno et al., 2016) with the dataset identifiers PXD007790 (complex I proteomics) and PXD00791 (cell surface proteomics). These datasets are also made available by Mendeley Data (https://doi.org/10.17632/ptm7dzc25y.2).