

Abstract
Inheritance of a coding variant of the protein tyrosine phosphatase non-receptor type 22 (PTPN22) gene is associated with increased susceptibility to autoimmunity and infection. Efforts to elucidate the mechanisms whereby the PTPN22-C1858T variant modulates disease risk revealed that PTPN22 exerts signaling function in multiple biochemical pathways and cell types. Capable of both enzymatic activity and adaptor functions, PTPN22 modulates signaling through both antigen and innate immune receptors. PTPN22 plays roles in lymphocyte development and activation, establishment of tolerance, and innate immune cell-mediated host defense and immunoregulation. The disease-associated PTPN22-R620W variant protein is likely involved in multiple stages of the pathogenesis of autoimmunity. Establishment of a tolerant B cell repertoire is disrupted by PTPN22-R620W action during immature B cell selection, and PTPN22-R620W alters mature T cell responsiveness. However, after autoimmune attack has initiated tissue injury, PTPN22-R620W may foster inflammation through modulating the balance of myeloid cell-produced cytokines.
Keywords: PTPN22, Tyrosine phosphatase, Autoimmunity, Type 1 diabetes, Rheumatoid arthritis, Signaling
Introduction
In the past decade, knowledge of the complete human genome sequence and subsequent construction of dense single nucleotide polymorphism maps has permitted definitive identification of numerous susceptibility alleles for polygenic autoimmune diseases. The burden of polygenic disease attributable to any single polymorphism or allele is typically small. However, detailed knowledge of “risk” genes has set the stage for functional studies with potential to define molecular pathways important for pathogenesis of these often poorly-understood conditions. The hope of using genetic data to improve mechanistic understanding of disease has therefore focused major funding and attention on what in many cases were obscure and poorly-characterized molecules. Resulting investigations have provided exquisitely detailed characterization of the biochemistry, cellular biology, and whole-organism physiology attendant to a number of disease “risk” alleles. Protein tyrosine phosphatase non-receptor type 22 (PTPN22) is one such gene.
In the 12 years between the report of Ptpn22 molecular cloning and the initial observations of PTPN22 association with multiple human autoimmune diseases, a total of 12 papers concerning the molecule appeared. In the 9-plus years since the first reports of disease association, 650 additional PTPN22-focused manuscripts were catalogued by Pubmed. The vast peer-reviewed literature on PTPN22 dooms the notion of comprehensive review to failure. We attempt a summary of key observations that underpin current functional models for the gene and for its autoimmunity-associated variant.
Section 1. Immunobiology of PTPN22
1.A. Molecular Biology of PTPN22
1.A.1. Cloning and historical aspects
Molecular cloning of Ptpn22 was reported in 1992. Matthew Thomas’ laboratory derived Ptpn22 cDNA from mouse spleen during a screen for novel genes encoding conserved tyrosine phosphatase domains(1). The new molecule exhibited almost exclusive hematopoietic-specific expression. In addition to a N-terminal canonical phosphatase domain, the 802-residue predicted protein product contained five C-terminal proline, glutamic/aspartic acid, serine, threonine-containing PEST sequences(1). The molecule’s structural features prompted the name PEST-domain Enriched Phosphatase (“Pep”). An initial clue to its biochemical significance came in 1996 when the Veillette group identified Ptpn22 in a screen for proteins interacting with the SH3 domain of C-terminal Src kinase (Csk), a T cell receptor signaling regulator(2). Several years later, Chaim Roifman’s laboratory reported the cloning of an 807-amino acid Ptpn22 human homologue(3). The newly identified human phosphatase displayed near-exclusive expression in thymus and spleen, inspiring the name Lymphoid phosphatase (“Lyp”).
1.A.2. Classification and structural aspects
PTPN22 and Ptpn22 are class I protein tyrosine phosphatases, and their catalytic domains are highly homologous to the catalytic domains of other classical tyrosine-specific PTPs with known immune system functions, including CD45, SHP-1 and TC-PTP (for reviews of PTP biology, see(4, 5)). Two additional PTPs --PTP-PEST and BDP1 (also called PTP20 or PTP-HCSF), encoded by the PTPN12 and PTPN18 genes respectively-- display proline-rich motifs in their C-terminal domains. Together with PTPN22/Ptpn22, these enzymes comprise the “proline-rich” subclass of Class I PTPs(4). PTP-PEST and BDP1 are expressed at high levels in immune cells(6, 7). PTP-PEST also interacts with the SH3 domain of Csk(8); however, its expression patterns and immunological functions do not overlap with those of PTPN22(6). The in vivo function of BDP1 remains to be elucidated.
The catalytic domains at the N-termini of PTPN22 and Ptpn22 share almost complete identity (Fig. 1). The PTPN22 catalytic domain has been crystallized(9, 10), and the structural data corroborate functional data from mutational analyses. The ability to dephosphorylate tyrosine is critically dependent on PTPN22-C227, which acts as a nucleophilic acceptor of a phosphate moiety, and on D195, which facilitates hydrolysis of the phosphate-enzyme intermediate (Fig. 1). Substitutions at PTPN22 C227 and D195 inactivate the enzyme, and may enhance its capacity to trap phosphorylated substrates(11). The Cysteine-based mechanism of catalysis makes Class I PTPs like PTPN22 and Ptpn22 prone to regulation by oxidation (for review, see(12)). One PTPN22 crystal structure analysis showed a disulfide bond between residues C227 and C129 in the backbone of the catalytic domain(10), suggesting how PTPN22 enzymatic activity could be regulated by reversible oxidation of the catalytic domain(12).
Fig. 1. Schematic of the structure of PTPN22 and of its interaction with Csk and TRAF3.
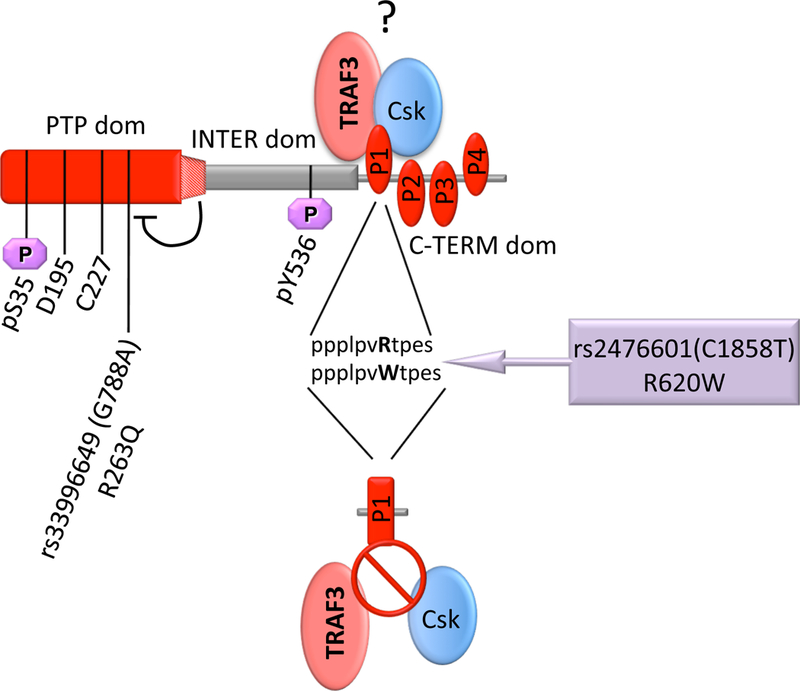
Three major domains of PTPN22 [N-terminal/PTP domain (aa 1–300), Interdomain (aa 301–600), and C-terminal domain (aa 601–807)] with high conservation (>90%) between human PTPN22 and murine Ptpn22 are indicated in red. Residues D195 and C227 are critical for catalytic function(11). PTPN22 forms a high stoichiometry complex with Csk (depicted in light blue) through interaction between the P1 motif in the C-terminal domain of PTPN22 and the SH3 domain in the N-terminus of Csk. PTPN22 also interacts directly with TRAF3 (depicted in light red) in myeloid cells, but it is unclear whether a ternary PTPN22-Csk-TRAF3 complex is formed (question mark). PTPN22 phosphorylation sites at S35(9)and Y536(27)- are shown in purple. A conserved region of the interdomain that constitutively inhibits the phosphatase activity is localized between amino acids 300 and 320(13). R263Q variation in the PTP domain causes decreased phosphatase activity(155), while the R620W variation in the P1 motif of the C-terminal domain decreases binding affinity between Csk and PTPN22(35, 42), and between TRAF3 and PTPN22(58).
C-terminal to the catalytic domain, PTPN22 contains an approximately 300 amino acid region termed the “interdomain”. Compared to the nearly identical catalytic domains, the interdomains show reduced conservation between human and mouse (52% identity, 65% conservation)(Fig. 1). Mutational analysis revealed that the interdomain of PTPN22 may regulate the catalytic activity(13). A motif between PTPN22 residues 300 and 320 forms an intramolecular interaction with the catalytic domain and inhibits enzymatic activity(13).
The C-terminal domains of PTPN22/Ptpn22 include the P1 motif, a highly conserved proline-rich region that interacts with the Csk SH3 domain(2). Three additional proline-rich motifs called P2, P3 and P4 are recognizable(3)(Fig. 1). Although their evolutionarily conserved sequences suggest functional roles for the P2-P4 motifs, no specific interactors for these domains have been identified. PEST motifs like those found in the C termini of PTPN22/Ptpn22 are frequently found in proteins exhibiting rapid turnover. However, pulse-chase studies showed that the resting cell half-life of Ptpn22 is greater than 5 hours(14), suggesting that Ptpn22 is not rapidly degraded.
1.A.3. Isoforms and subcellular localization
Several transcripts encoding variant PTPN22 isoforms have been described. An alternatively spliced isoform that encodes a putative, truncated protein called Lyp2 has been detected in T cells(3). The predicted 691 amino acid sequence of Lyp2 is identical to that of a truncated full-length PTPN22, except that Lyp2 harbors a unique 7 aa C-terminal tail. According to one report, Lyp2 is the most abundant isoform in resting peripheral blood human T cells; TCR stimulation inhibits Lyp2 expression while upregulating full-length PTPN22 (“Lyp1”)(3). An increased ratio of full-length PTPN22/Lyp2 transcripts was found in peripheral blood mononuclear cells (PBMC) from patients carrying the PTPN22-associated diagnosis of Rheumatoid Arthritis (RA)(15). While Lyp2 protein amounts were not assessed in this report, the data suggest that the expression of full-length PTPN22 and Lyp2 mRNAs might be independently regulated. Another isoform found in human PBMC, termed Lyp3, encodes a putative protein bearing a 28 aa deletion between the P1 and P3 domains(16). Biochemical investigations are further advanced for PTPN22.6, an isoform cloned from human CD4+ T cells(17). PTPN22.6 protein lacks a portion of the catalytic domain, and harbors a unique 8 aa C-terminal sequence(17). Low levels of an endogenous PTPN22.6 protein can be detected in cell lines. When co-expressed with full-length PTPN22 in Jurkat T cells, PTPN22.6 behaves as a dominant-negative in TCR signaling (see section 2.B). Positive correlation between PTPN22.6 mRNA levels and disease activity was found in a cohort of RA patients(17).
No murine Ptpn22 isoforms homologous to the above-described human transcripts have been reported. Taken together, published data suggest that variants of PTPN22 are transcriptionally regulated, that PTPN22 isoforms may have utility as biomarkers of disease activity, and that they have potential to modulate cellular function. However, details of the expression and regulation of non full-length PTPN22 isoforms at the protein level remain incompletely known.
1.A.4. Substrates of PTPN22 and Ptpn22
Recognition that PTPN22 and Ptpn22 encode tyrosine phosphatases that are highly expressed in T cells led to searches for their substrates among TCR signaling proteins (Fig. 2). The Veillette group reported that Ptpn22-D195A, a “substrate-trapping” mutant, could precipitate FynT (T-cell specific isoform of Fyn), TCR-CD3ζ, and Zap-70 from lysates of transfected COS-1 cells(18). Tryptic peptide mapping revealed that Ptpn22 can selectively dephosphorylate FynT at Y420(18). Y420 represents a highly conserved tyrosine in Src family kinase catalytic domains that promotes kinase activity upon phosphorylation. Later use of a substrate-trapping PTPN22-D195A/C227S mutant, expressed in pervanadate-treated Jurkat T cells, led to identification of Zap-70, CD3ε, TCR-CD3ζ, and the Src family kinase Lck as primary substrates of PTPN22(11).
Fig. 2. Summary of the putative substrates of PTPN22 in T cells.
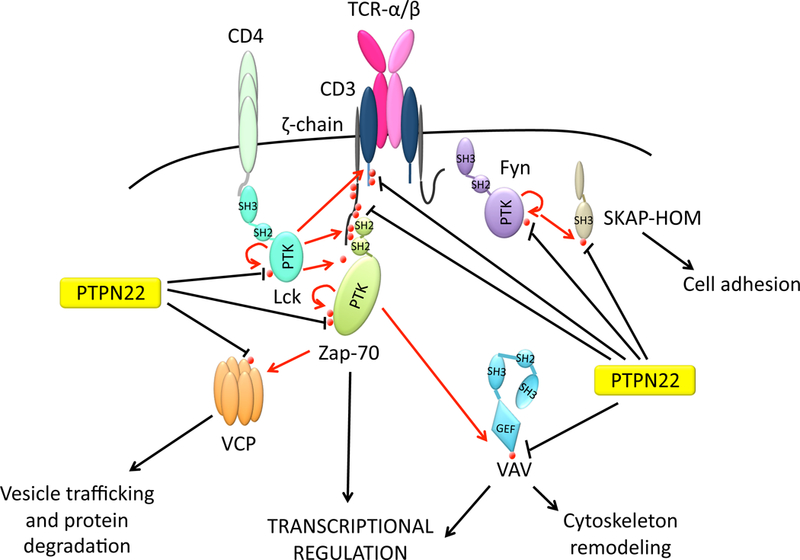
Established substrates include Y420 and Y394 in activation loops of Src family kinases Fyn(18) and Lck(36] respectively, and the autoactivatory Y493 of Zap-70){Wu, 2006 #35). Putative PTPN22 substrates that undergo tyrosine phosphorylation upon TCR engagement include i) TCR-CD3ζ and CD3ε, ii) the guanine nucleotide exchange factor Vav, iii) the ATPase valosin containing protein (VCP)(11). Y75 of Src-kinase associated protein homolog (SKAP-HOM) is a recently-described T cell substrate for PTPN22(31).
In vitro, PTPN22 can efficiently dephosphorylate Lck Y394 (homologous to Y420 of FynT) and Zap-70 Y493, residues that reflect activation of the respective kinases when phosphorylated(11). Observations of increased Lck phosphorylation in TCR-stimulated Ptpn22-deficient T cells(19, 20) or in human Jurkat T cells treated with chemical PTPN22 inhibitors(21), provided further evidence that Lck is a physiological substrate for PTPN22/Ptpn22. TCR-CD3ζ is also efficiently dephosphorylated in vitro by recombinant PTPN22(11)(Fig. 2), and displays increased phosphorylation in primary human T cells treated with a PTPN22 inhibitor(21), suggesting that it too serves as a bona fide substrate(11). However, it is unknown which of the several tandem phospho-tyrosine residues of TCR-CD3ζ and/or of CD3ε(22) are targeted by PTPN22/Ptpn22. In general, the proven capacity for PTPN22/Ptpn22 to dephosphorylate TCR-associated signaling mediators--especially Lck, Fyn and Zap-70 at their autoactivation sites-is consistent with their established roles of as potent suppressors of TCR signaling(22)(see section 1.B.1).
Proteins downstream of membrane-proximal TCR signaling molecules also likely serve as PTPN22 substrates (Fig. 2). The guanine nucleotide exchange factor (GEF) Vav and the ATPase VCP/p97 are also reported to be trapped by PTPN22-D195A/C227S in T cell lysates(11). These molecules are phosphorylated after TCR stimulation and play important roles in cytoskeletal rearrangement, autophagy, protein degradation, and endosomal sorting(23–25).
Additional putative substrates of PTPN22 awaiting further verification include c-Casitas B-lineage Lymphoma (c-Cbl), Bcr-Abl, and Src-kinase Associated Protein of 55kDa Homolog (SKAP-HOM). C-Cbl is an E3-ubiquitin ligase that undergoes tyrosine phosphorylation during TCR signaling(26). C-Cbl and PTPN22 co-precipitate from Jurkat T cells and PTPN22 can dephosphorylate c-Cbl upon co-expression in COS-1 cells(3). However, interpretation of co-precipitation experiments supporting a possible enzyme-substrate relationship for PTPN22 and c-Cbl is complicated by the facts that PTPN22 overlaps in molecular weight with c-Cbl, and that PTPN22 is itself tyrosine phosphorylated in Jurkat T cells(27, 28). Tyrosine phosphorylation of the oncogenic fusion protein Bcr-Abl is depressed in lysates of a chronic myeloid leukemia (CML) cell line in which PTPN22 is overexpressed(29). Further, phosphorylation of downstream Bcr-Abl substrates in these cells is impaired in a PTPN22-dependent manner. Substrate trapping experiments will be helpful to further assess functional interaction between PTPN22 and either c-Cbl or Bcr-Abl.
SKAP-HOM, an adaptor protein thought to regulate lymphocyte adhesion(30), is the only putative PTPN22 substrate identified using a substrate profiling approach(31). The peptide YGEEpYDDLY was identified as a preferred substrate of PTPN22 through inverse alanine scanning of a library. The peptide displays high similarity to sequence surrounding Y75 of SKAP-HOM(31). Although both trapping and in vitro peptide dephosphorylation studies suggest that SKAP-HOM can serve as a PTPN22 substrate(31), direct evidence that PTPN22 controls SKAP-HOM Y75 phosphorylation under physiologic conditions is lacking.
Identification of PTPN22 substrates among signaling proteins in membrane-proximal and cytoplasmic regions suggests that PTPN22 might reside in these compartments. However, studies addressing the subcellular localization of PTPN22 and Ptpn22 have yielded conflicting results. An initial report showed that Ptpn22 is entirely localized in the nucleus when expressed in HeLa cells(14). The P4 domain of Ptpn22 has been identified as a possible nuclear localization sequence(14). In contrast, others observed that overexpressed epitope- or GFP-tagged PTPN22 displays exclusive cytosolic localization, albeit with perinuclear enrichment(3, 29). A third group showed that overexpressed epitope-tagged Ptpn22 localizes to both the nucleus and peri-membrane cytosol in Jurkat T cells(32). The Ptpn22 C-terminus is necessary for the observed peri-membrane enrichment, while deletion of either the C-terminus or of the N-terminal catalytic domain abolishes Ptpn22 low-level nuclear localization(32). During cellular fractionation studies in Jurkat T cells, significant amounts of endogenous PTPN22 are found in the nuclear fraction (N. Bottini, unpublished data). Overall, the relative paucity of definitive evidence regarding subcellular localization of endogenous PTPN22/Ptpn22 continues to stem from unavailability of detection reagents useful in immunofluorescence studies. The preponderance of current evidence suggests that large fractions of PTPN22/Ptpn22 reside in the cytosol; possibly, smaller amounts are localized in the nucleus.
1.B. Regulation of T cell biology by PTPN22
1.B.1. Regulation of T cell signaling by PTPN22 and Ptpn22
Experiments using genetic or pharmacologic manipulation of PTPN22/Ptpn22 provide strong evidence that these phosphatases serve as negative regulators of signaling through the TCR. Thymocytes and effector/memory T cells from Ptpn22−/− mice display increased Lck phosphorylation on Y394 and Ca2+ mobilization after TCR triggering with anti-CD3 and anti-CD28(19, 20, 33). Ptpn22−/− mice also exhibit enhanced proliferation and expansion of effector/memory T cells in vivo and in vitro, as well as enhanced primary and secondary T cell dependent Ig response to antigen(19, 20). Human peripheral T cells or Jurkat T cells subjected to PTPN22 knockdown, or exposed to PTPN22 phosphatase inhibitors, display increased phosphorylation of TCR-CD3ζ(21), Zap-70(21), and Slp-76(34), and augmented activation of a NFAT-AP1-responsive reporter after TCR stimulation(21, 35). Conversely, overexpression of PTPN22 in Jurkat T cells or primary human T cells leads to reduction of anti-CD3-induced TCR-CD3ζ, Lck, and Zap-70 phosphorylation, and inhibited activation of an NFAT-AP1-responsive reporter(27, 36, 37). Together, these data indicate that PTPN22/Ptpn22 act as brakes on TCR signaling.
The inhibitory action of PTPN22/Ptpn22 on TCR signaling depends upon enzymatic activity. This function is exerted mainly through dephosphorylation of Lck and Zap-70, although dephosphorylation of TCR-CD3ζ or Vav might be relevant as well (Fig. 2). Interestingly, PTPN22/Ptpn22 pharmacological inhibition does not enhance phosphorylation of Lck Y394, TCR-CD3ζ, or Zap-70 in unstimulated mouse thymocytes(34), Jurkat T cells(21, 34), or primary human T cells(21). Moreover, Lck phosphorylation is not enhanced in resting naïve Ptpn22−/− T cells, suggesting that Ptpn22 is dispensable for Lck Y394 basal phosphorylation. Since enzymatically-active PTPN22 can be immunoprecipitated from resting Jurkat T cells(27), the apparent absence of basal PTPN22/Ptpn22 effects on Lck phosphorylation cannot be explained by simple restriction of phosphatase activity. One possibility is that PTPN22/Ptpn22 specifically inhibit high phosphorylation levels of target proteins under stimulation conditions, while basal phosphorylation levels are maintained by redundant phosphatases that serve to avoid inadvertent triggering of the TCR. Alternatively, PTPN22/Ptpn22 might be spatially sequestered away from substrates, specifically in unstimulated T cells.
Two non-mutually exclusive hypotheses can be advanced to explain why early TCR signaling is not enhanced in naïve Ptpn22−/− T cells. First, Ptpn22 expression may be lower in naïve T cells. Ptpn22 is subject to NFAT-dependent transcriptional induction after TCR stimulation(38), which correlates with higher levels of Ptpn22 in effector/memory compared to naïve T cells(39). In cells where scant protein resides, Ptpn22 may be expected to exert little effect on signaling. Second, different substrates might be critically targeted by PTPN22/Ptpn22, depending on the differentiation status of the T cell (naïve vs. effector phenotype). Interestingly, a recent report suggests that tyrosine phosphorylation of Lck is unchanged after TCR stimulation in primary human T cells, raising the possibility that PTPN22/Ptpn22 may modulate TCR signaling through functions other than dephosphorylation of Lck(40).
1.B.2. The PTPN22-Csk complex regulates PTPN22 function
In T cells, PTPN22/Ptpn22 bind the Csk tyrosine kinase(2, 3, 18). Csk phosphorylates the inhibitory tyrosine in the C-terminus of Src family kinases, and behaves as a strong negative regulator of TCR signaling(41). 25–50% of Ptpn22/PTPN22 is in complex with 5–6% of Csk in resting mouse T cells and primary human T cells(2, 21). Overexpression studies comparing wild-type (WT) Ptpn22 with a mutant lacking the required Csk-binding P1 domain, as well as co-expression experiments with Ptpn22 and Csk in Jurkat T cells, suggested that physical interaction between Ptpn22 and Csk enables synergistic inhibition of signaling between the kinase and the phosphatase(18, 36)(see “synergism model”, Fig. 5). Inactivation of Lck requires both dephosphorylation of Y394 (important for autoactivation) and phosphorylation of inhibitory Y505 (maintains inactive conformation)(40). Thus, it was proposed that complementary action of PTPN22 dephosphorylation of Lck Y394, coupled with Csk phosphorylation of Y505, facilitates complete inactivation of Lck (and other Src kinases subject to auto-inhibition, including Fyn)(36). However, the molecular mechanism behind PTPN22/Csk synergism remains hypothetical.
Figure 5. Proposed models for gain-of-function phenotype of disease-predisposing variant PTPN22-R620W in TCR signaling.
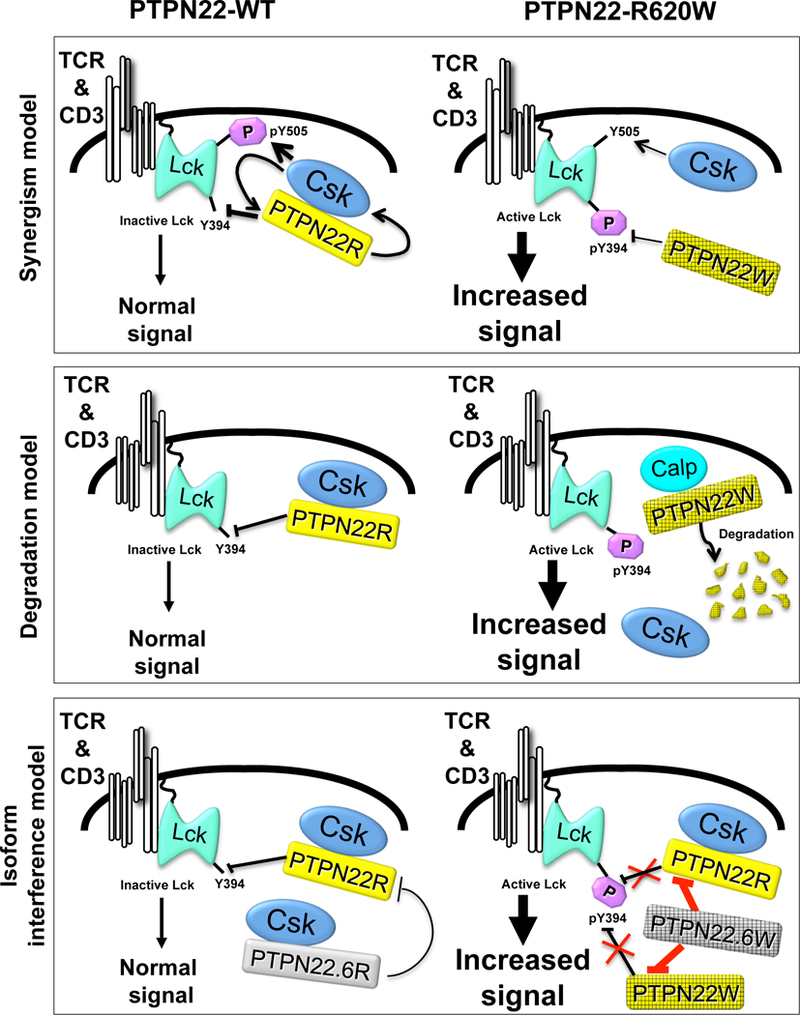
(A) In the lipid rafts model(21) PTPN22-WT is sequestered by non- raft- associated Csk and has limited accessibility to substrate Y394 within the activation loop of raft-localized Lck. The PTPN22-R620W variant loses capacity to bind Csk, and displays increased localization in lipid rafts containing proximal TCR signaling machinery. Lck activity is repressed by decreased phosphorylation of Y394, and downstream TCR signals are reduced. (B) In the phosphorylation model(27), PTPN22-WT undergoes basal and TCR-induced Csk-dependent phosphorylation on Y536 which inhibits PTPN22 phosphatase activity. The current model postulates that cross-activation of Lck molecules triggers phosphorylation of PTPN22-Y536, which facilitates optimal transduction of downstream signals in a positive feedback loop. The PTPN22-R620W variant loses capacity to bind Csk and displays decreased phosphorylation and increased phosphatase activity; thus, Lck Y394 phosphorylation is inhibited and downstream TCR signals are reduced.
Discovery that PTPN22-R620W, the autoimmunity-predisposing variant, shows impaired ability to interact with Csk(35, 42) stimulated increasing interest in the PTPN22/Csk complex. Zikherman et al. found that co-expressed PTPN22-R620W or Ptpn22-R619W variants, which are incapable of full interaction with Csk, inhibit TCR signaling less effectively(39). These data supported the idea of association-dependent synergistic function. Other reports, however, challenged the notion that Csk and PTPN22 cooperatively inhibit TCR function. Fiorillo et al. reported that formation of a complex with inactive Csk does not affect intrinsic enzymatic activity of PTPN22. Rather, active Csk promotes Lck-mediated phosphorylation of PTPN22 Y536 in the interdomain. Phosphorylation at Y536 inhibits PTPN22 phosphatase activity, which in turn results in enhanced TCR signaling(27). Moreover, an overexpressed PTPN22-Y536F mutant that is resistant to Csk-mediated phosphorylation behaves as a gain-of-function (i.e. more effective inhibitor) in TCR signaling. These findings support a model wherein PTPN22 interaction with Csk inhibits the activity of PTPN22 by promoting phosphorylation of PTPN22 at an inhibitory residue(27)(Fig 4; see section 2B). PTPN22-Y536 has been independently confirmed as a major Lck phosphorylation site (28). Vang et al. recently reported another potential function of Csk-PTPN22 interaction: membrane subregion localization of PTPN22(21). This group found that the Csk binding-defective PTPN22-R620W variant of PTPN22 displays significantly increased segregation into lipid rafts, which correlates with reduced TCR signaling (see section 2.B).
Figure 4. Proposed models for loss-of-function phenotype of disease-predisposing variant PTPN22-R620W in TCR signaling.
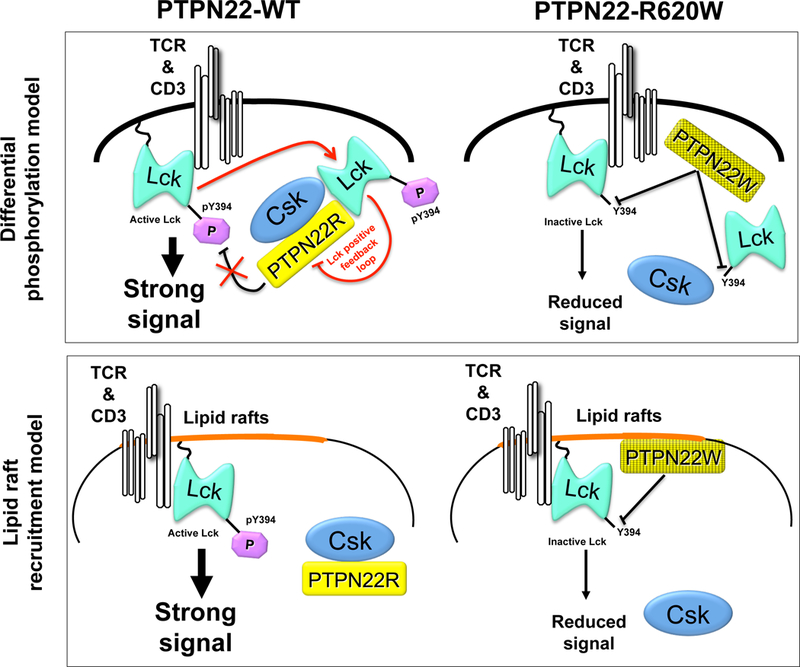
(A) In the initial model(18, 36, 39) the interaction between PTPN22-WT and Csk leads to a synergistic reciprocal potentiation of negative regulation of TCR signaling. The synergism is disrupted by the R620W variation that results in increased phosphorylation of Y394 and decreased phosphorylation of Y505. Interaction with Csk might enhance the function of PTPN22/Ptpn22, however evidence of a reciprocal action of PTPN22/Ptpn22 on Csk is lacking. (B) In the isoform model(17), PTPN22.6-WT, a catalytically-inactive isoform binds to Csk and acts as a dominant negative isoform by inhibiting the action of active PTPN22-WT. The autoimmune-predisposing SNP leads to the appearance of a variant PTPN22.6-R620W isoform. PTPN22.6-R620W displays reduced binding to Csk. However, PTPN22.6-R620W causes enhanced TCR signaling through dominant interference with the negative regulatory effects of PTPN22. As a consequence, Lck may undergo increased phosphorylation of Y394, and downstream TCR signals are enhanced. It is still unclear whether PTPN22.6-R620W inhibits the action of PTPN22-WT and PTPN22-R620W to the same extent. (C) In the degradation model(47), the half-life of PTPN22 is controlled by calpain binding which directs PTPN22 to proteasomal degradation. Csk competes with calpain for binding to PTPN22; thus PTPN22-R620W undergoes increased binding to calpain and proteasomal degradation. Lck is activated by increased phosphorylation of Y394, and downstream TCR signals are enhanced.
Conflicting data concerning dynamic regulation of the PTPN22/Csk complex have been reported. Compelling and independent reports find variously that the stoichiometry of the complex is unaffected(27), increased(21), or decreased(28) after TCR triggering in Jurkat T cells or in primary human T cells. Future studies of the Csk-PTPN22 complex will likely address whether additional effector molecules are recruited and required for function, and whether the stoichiometry of the complex is conditional.
Possible Csk-independent regulation of PTPN22/Ptpn22 function has been suggested. Protein Kinase C-mediated phosphorylation of PTPN22 S35 in the catalytic domain is associated with inhibited phosphatase activity(9). However, multiple, as-yet unknown regulatory mechanisms likely exist and await further biochemical characterization.
1.B.3. Role of PTPN22 in T cell development and differentiation
Establishment of an immunocompetent and self-tolerant T cell repertoire depends upon interactions between MHC-bearing thymic stroma and TCR-expressing immature thymocytes. Current models hold that TCR signal intensity (affinity) is a dominant determinant of both positive and negative thymocyte selection(43). Since Ptpn22 is expressed in developing T cells, and functions as a TCR signaling regulator (see section 1.B.1), potential roles for Ptpn22 in thymocyte selection have been carefully examined (see Table 1).
Table 1:
Immune Phenotypes reported for Ptpn22 deficiency
| Immune cell development |
Tolerance/Autoimmunity | Host defense |
|---|---|---|
|
T cells: ↑ positive selection of CD4+ T cells(19) ↑ memory T cells with age(19) Treg: ↑ Treg fraction, thymus and/or periphery(20, 33, 45) ↑ immunosuppressive capacity(20) ↑ integrin-mediated adhesion(20) ↑ IL-10(20) |
Multigenic systemic autoimmunity(39): ↑ lymph node cellularity ↑ lymphocyte activation ↑ autoantibodies EAE(33): ↓ severity ↑ Treg: Teff ratio Type 1 Diabetes: ↓Incidence in NOD(45) Inflammatory arthritis: ↓TLR-mediated disease suppression(58) Irritant Colitis: ↑ severity, colon mucosal damage(58) |
LCMV response(58): ↓ serum type 1 Interferon ↓ DC activation ↓ CD8+ T cell expansion and effector function EMCV response(58): ↑ mortality ↑ viral titers |
Hasegawa et al. found that Ptpn22 selectively restrains thymocyte positive selection(19). Ptpn22-/- mice, either “polyclonal” or TCR transgenic, display increased fractions and numbers of CD4 single positive (CD4 SP) cells. Although CD8 SP thymocyte numbers are not altered in Ptpn22-/- non-transgenic “polyclonal” animals, they are increased in Ptpn22-/- H-Y TCR transgenic thymi, and both CD4+ and CD8+ T cell numbers are increased in the Ptpn22-/- periphery. Zikherman et al. described subtle TCR hyper-responsiveness and evidence of increased TCR signal strength among Ptpn22-/- immature thymocytes(39), suggesting that altered antigen receptor thresholds could contribute to the observed enhancement of positive selection.
Negative selection occurs normally in the absence of Ptpn22. Neither deletion of autoreactive thymocytes in H-Y males, nor thymocyte apoptosis in response to injected anti-CD3, is affected by Ptpn22 deficiency(19). However, high efficiency of negative selection in these models could make Ptpn22-dependent augmentation of clonal deletion --which might be predicted with loss of the TCR signaling inhibitory function of Ptpn22 --difficult to detect.
Ptpn22 regulates homeostasis of previously-activated peripheral T cells. Ptpn22-/- mice contain increased numbers of peripheral effector/memory CD62LlowCD44high CD4+ and CD8+ T cells(19, 20). Notably, while naïve T cell proliferation after TCR stimulation is unaffected by loss of Ptpn22, TCR signaling, expansion, and function of effector/memory Ptpn22-/- T cells is enhanced. Adoptively-transferred Ptpn22-/- T cells proliferate more vigorously than controls (19, 20), demonstrating that the Ptpn22-/- phenotype of increased T cell expansion and function is T cell-intrinsic. Interestingly, loss of Ptpn22 does not affect response of effector/memory T cells to IL-2 or IL-15 in vitro, nor does it affect activation-induced T cell death or effector population contraction after antigen challenge in vivo(19). These data suggest that accumulation of memory T cells in Ptpn22-/- mice is not due to impaired apoptosis or removal mechanisms, but could result from increased tonic TCR signaling.
1.B.4. Role of PTPN22 in Treg biology
T regulatory cells (Treg) comprise a subset of CD4+ T lymphocytes that promote peripheral tolerance, and that frequently express the transcription factor FoxP3(44). The Ptpn22 promoter is among those targeted by FoxP3(38), suggesting that Ptpn22 might modulate development or function of Treg (see Table 1). A FoxP3+ T cell hybridoma shows reduced upregulation of Ptpn22 expression after TCR engagement, which has been interpreted as evidence that FoxP3 inhibits the expression of Ptpn22(38). Two groups independently reported increased size of the peripheral CD25+Foxp3+CD4+ T cell compartment in Ptpn22-/- mice(20, 33). The reports differ in important ways: Maine et al. found increased fractions and numbers of Treg and Treg precursors in the Ptpn22-/- thymus and periphery(33); Brownlie et al. found increased Treg numbers only in the periphery of Ptpn22-/- mice, associated with enhanced expansion of inducible Tregs(20). One study reported increased Ptpn22-/- cell-intrinsic Treg suppressive capacity, as measured by the ability to suppress effector T cell proliferation and attendant colitis in Rag-/- mice(20); the other found no alteration in Treg suppressive function(33). Brownlie et al. also found that Treg from Ptpn22-/- mice display an activated phenotype, but unexpectedly do not exhibit increased TCR signaling. The authors propose that Ptpn22 deficiency results in increased Treg expression of LFA-1 through control of Rap1 phosphorylation. The data imply that Ptpn22 could modulate integrin-dependent T cell migration and adhesion.
The Kissler laboratory examined Treg function in NOD mice engineered to undergo inducible knock-down of Ptpn22 in T cells(45). Ptpn22 knockdown was associated with expanded numbers of FoxP3+ peripheral T cells, but with normal thymic Treg numbers, and unchanged Treg suppressive capacity in vitro(45). The mechanism of action and physiological relevance of Ptpn22 in Treg biology await further investigation. However, at a minimum, the data collectively suggest that neither Treg quantity nor Treg suppressive activity is reduced by loss of Ptpn22 function.
1.C. Regulation of B cell biology by PTPN22
Ptpn22 expression is observed, and can be dynamically-regulated, in primary murine B cells(7, 39). In vitro CpG stimulation augments(46), while LPS stimulation suppresses(7), Ptpn22 mRNA or protein levels. As it does in T cells, Ptpn22 associates constitutively with Csk in a B cell line(2).
Surprisingly, few cell-intrinsic roles for Ptpn22 in B cell ontogeny, cellular activation, and B cell-dependent immune responses have been established. Hasegawa et al. reported no differences in total peripheral B cell numbers in Ptpn22-/- mice(19). Likewise, Zikherman et al. found no effect of Ptpn22 deficiency on cellularity of follicular mature, T1, T2, marginal zone B cell compartments, nor on plasma cell numbers(39). Although modest increases in selected Ig subtype levels are found in Ptpn22-/- mice(19), total Ig levels and levels of autoantibodies (ANA, anti-DNA) are not different from controls(19, 39). Increased frequency of B cell-rich germinal centers in lymphoid tissues is observed in Ptpn22-/- animals. However disinhibited T cell function in Ptpn22-/- mice likely accounts for the observed alteration in germinal center and immunoglobulin levels(19).
Ptpn22 also appears dispensable for BCR signaling. BCR-induced calcium mobilization, ERK phosphorylation, global tyrosine phosphorylation, CD69 upregulation, and proliferation in Ptpn22-/- primary B cells show no differences compared to wild type(19, 39, 46). Taken together, these data support the view that Ptpn22 is dispensable for B lymphocyte development and signaling. Notably, these negative findings contrast sharply with recent reports of significantly altered B cell development and function observed in humans or mice carrying an autoimmunity-associated variant (PTPN22-R620W) or its engineered homologue (Ptpn22-R619W)(46–51) (see Table 2 and section 2.C).
Table 2:
Phenotypes reported for disease-associated PTPN22-R620W or Ptpn22 R619W in knock-in and or BAC transgenic models
| Immune cell development |
Immune cell function | Tolerance/Autoimmunity |
|---|---|---|
|
T cells: • ↓ Pep619W protein stability in T lineage cells(47) • No defect in 619W stability in T cells(46) B cells: • Pep619W expression in B cells sufficient for: ↑ numbers of T1, germinal center, and “ABC” B cells(46) • ↑GC area(47) Myeloid cells: • ↑number and activation of CD11c+ cells in spleen(47) |
T cells: • ↑ proximal TCR signaling(46, 47) • ↑ TCR-induced proliferation(46) B cells: • ↑ BCR-induced Syk, PLCg2 phosphorylation(46) • ↑ BCR-driven proliferation(47) • Apoptosis resistance(46) Myeloid cells: • ↓ TLR4-induced type 1 IFN production(58) • ↑ DC costimulation capacity(47) |
Inflammatory arthritis: • ↓ TLR-mediated suppression of IL-1, leukocyte infiltrates, and synovitis(58) Diabetes: • ↑ anti-insulin antibodies; ↑ sensitivity to streptozocin-induced diabetes(46) Systemic autoimmunity: • ↑ ANA, anti-dsDNA antibodies; lymphoid tissue infiltrates(46) |
PTPN22 may play a role in malignant B cell signaling. High levels of PTPN22 are found in chronic lymphocytic leukemia (CLL) blasts(52). Knockdown of PTPN22 or inhibition of its enzymatic function promotes BCR-triggered apoptosis in CLL blasts, suggesting that PTPN22 may protect malignant cells from antigen receptor-induced death(52). Altered PTPN22 levels may both inhibit and promote different pathways downstream of the BCR in CLL, as PTPN22 knockdown leads to hyperactive Lyn but strongly decreased activation of Akt (52).
1.D. Regulation of myeloid cell biology by PTPN22
Myeloid cells, including dendritic cells (DC) and macrophages, exert innate immune functions, including pro-inflammatory cytokine secretion, initiation of host defense programs, and antigen presentation/co-stimulation. Myeloid cell pattern recognition receptors (PRR) can translate recognition of microbial substances into engagement of these functions(53).
Ptpn22 is expressed in myeloid cells. High basal levels are observed in splenic dendritic cells and macrophages(7, 54); Ptpn22 protein has also been reported in myeloid-derived hippocampal glial cells(55). Ptpn22 expression is suppressed after DC stimulation with Toll-like receptor (TLR) agonists(7, 56), but is induced in RAW 264.7 cells after LPS treatment(55). Monocytes stimulated with type 2 interferon exhibit increased PTPN22 expression(57). Ptpn22 appears dispensable for development of surface phenotype-defined populations of blood monocytes, CD11b+ splenocytes, and subsets of dendritic cells in lymphoid organs(19, 58).
Ptpn22 functions as a selective promoter of (PRR) signaling. Bone marrow-derived macrophages (BMM) from Ptpn22-/- mice display impaired induction of type 1 IFNs, after TLR4 agonist stimulation(58). Similar reduction of TLR4 agonist-stimulated upregulation of type 1 IFN is observed in Ptpn22-/- DC, and in human and murine myeloid cells subjected to PTPN22/Ptpn22 knock-down. In addition, Ptpn22 is required for efficient type 1 IFN induction after myeloid cell stimulation with ligands for TLR3, 7, and 9, and for intracellular RNA receptors (Fig. 3). Moreover, Ptpn22-/- mice display reduced capacity to upregulate systemic type 1 IFN after exposure to TLR4 ligand in vivo.
Figure 3. TLR and RLR signaling in myeloid cells require Ptpn22 for efficient TRAF3 autopolyubiqutination.
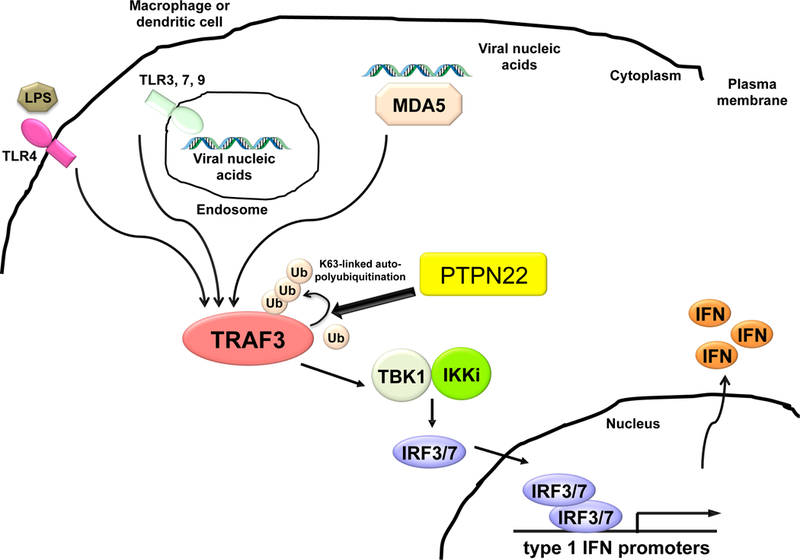
Type 1 interferon-inducing Pattern Recognition receptors (TLR3, 4, 7, 9; MDA-5) engage microbial products in extracellular milieu, endosomes, or cytoplasm of dendritic cells or macrophages(53). Pattern recognition receptor ligation induces lysine-63-linked autoubiquitination of TRAF3. Ptpn22 associates with TRAF3 and promotes TRAF3 K63-linked polyubiquitination in a phosphatase-independent manner (Wang et al, 2013). TRAF3 ubiquitination results in activation of serine-threonine kinases TBK1 and IKKε. Substrates for TBK1/IKKε include transcription factors IRF3 and IRF7; upon phosphorylation, the latter translocate to the nucleus and activate type 1 interferon transcriptional upregulation.
Ptpn22 function appears to be restricted to those PRR signaling pathways that lead to type 1 IFN induction by myeloid cells. Strikingly, Ptpn22-/- cells and mice display no defects in TLR4 agonist-induced upregulation of Tnf, Il1b, or Il6, either in vitro or in vivo(58). Thus, Ptpn22 is dispensable for TLR4-engaged signaling that controls NF-κB-dependent transactivation of many pro-inflammatory cytokine genes(53). Commensurate with the idea that Ptpn22 selectively promotes “interferogenic” TLR signals, TLR4-induced IRF3 phosphorylation is decreased, while NF-κB pathway activation is intact, in Ptpn22-/- macrophages(58).
A search for PTPN22/Ptpn22 binding partners among proteins implicated in interferogenic PRR signaling(53) revealed that PTPN22/Ptpn22 constitutively associate with TNF receptor associated factor 3 (TRAF3) in myeloid cells(58). TRAF3 is an E3 ubiquitin ligase and a key mediator of TLR-induced type 1 IFN production(59). After PRR stimulation, K63-linked autoubiquitination of TRAF3 is required for upregulation of type 1 IFN(60). In Ptpn22-/- or Ptpn22 knockdown myeloid cells, TLR-stimulated K63-linked polyubiquitination of TRAF3 is reduced(58), suggesting that PTPN22/Ptpn22 occupy a biochemical niche upstream of type 1 IFN promoters (Fig. 3). Interestingly, PTPN22 enzymatic activity is dispensable, both for PTPN22 association with TRAF3, and for promotion of TLR4-stimulated type 1 IFN(58). Whether the Ptpn22-TRAF3 interaction is required for TRAF3 ubiquitin ligase activity, and whether additional effectors contribute to positive regulation of TRAF3 signaling by Ptpn22, remain unknown.
Ptpn22 may promote myeloid cell functions unrelated to interferogenic signaling. Ptpn22 is a potential modulator of LPS-induced synthesis of anandamide, a lipid in the endocannabinoid pathway(55). Another report documented diminished phosphorylation of Signal Transducer and Activator of Transcription-1 (STAT1) and STAT3 in THP-1 cells subjected to PTPN22 knockdown and stimulated with Interferon-γ(57). PTPN22 appears in immunoprecipitates of SOCS1, an inhibitor of IFN-γ signaling(61). PTPN22 knock-down results in increased SOCS1 activation, suggesting that PTPN22 could promote IFN-γ signaling through repressing SOCS1.
Interestingly, Ptpn22-/- macrophages also show reduced STAT1 phosphorylation after TLR stimulation(58). However, the Ptpn22-dependent defect in STAT1 activation does not map to interferon signaling, since treatment of Ptpn22-/- cells with type 1 IFN induces normal STAT1 phosphorylation(58). Whether Ptpn22-/- primary cells mimic the phenotype of PTPN22-knockdown THP-1 cells in IFN-γ signaling, and whether the mechanisms by which Ptpn22 regulates PRR signaling (e.g. promoting TRAF3 ubiquitination) also hold for cytokine receptors, remain important questions.
PTPN22 might also regulate signaling in myeloid leukemia. PTPN22 is highly expressed in chronic myelogenous leukemia (CML) cell lines, and PTPN22 levels in CML positively correlate with resistance to Imatinib(62). PTPN22 overexpression in CML cells inhibits signaling downstream of Bcr-Abl(29), although whether PTPN22 affects blast survival in CML akin to its function in CLL is not known.
1.E. Role of Ptpn22 in tolerance and immunoregulation
While Ptpn22-/- mice do not develop spontaneous autoimmunity(19), deletion or knock-down of Ptpn22 can either increase or reduce the severity of disease in models of autoimmunity (see Table 1). Increased severity of disease is observed when Ptpn22 deficiency is combined with the “wedge” E613R mutation of CD45, which promotes autoimmunity mainly through promotion of BCR signaling(39). The phenotype of the Ptpn22-/- CD45-E613R mice is likely due to enhanced T cell help, given observations that a) Ptpn22 deficiency is associated with T cell hyper-responsiveness, b) Ptpn22 is dispensable for BCR signaling (see section 1.C), and c) the Treg compartment is not compromised by loss of Ptpn22 (see section 1.B.4). On the other hand, Ptpn22-/- mice display reduced severity of experimental autoimmune encephalitis (EAE). The protective phenotype is associated with increased Ptpn22-/- Treg numbers(33). However, variant human PTPN22 may regulate differentiation of Th17, CD4+ T cells that are pathogenic in EAE (see section 2.B). Thus, reduced Th17 numbers and/or function could also contribute to altered EAE severity observed in Ptpn22-/- mice.
The role of Ptpn22 in regulation of T1 diabetes (T1D) in the NOD background has been examined. In one study, NOD mice transgenic for Ptpn22 silencing RNA display transgene dose-dependent protection from diabetes(45). Surprisingly, a separate analysis of NOD mice overexpressing wild-type Ptpn22 in T cells also show reduced disease(63). In NOD mice with silenced Ptpn22, expanded peripheral Tregs are observed(45); in Ptpn22 overexpressing-mice, Treg are normal, but function of diabetogenic Teffectors is suppressed(63). Thus, differential regulation of Treg and Teffector compartments in the presence of either overabundant or Ptpn22 silencing might account for the paradoxical disease incidence results. Aberrant dephosphorylation of TCR signaling activators in T cells overexpressing wild-type Ptpn22 might also account for some discrepancies.
Ptpn22 may modulate expression of autoimmune diseases by promoting type 1 IFN-driven suppression of inflammation. Myeloid cell-generated type 1 IFNs can exert immunoregulatory function through antagonism of pro-inflammatory cytokines interleukin-1 (IL-1β) and TNFα(64, 65). Ptpn22 is required for optimal TLR-driven, myeloid-cell dependent amelioration of synovial inflammation in an IL-1β-dependent arthritis model(58). More severe disease in poly(I:C)-treated, arthritic Ptpn22-/- mice is associated with reduced synovial type 1 IFN-dependent genes, but elevated IL-1β mRNA(58). In a type 1 IFN-sensitive model of irritant colitis, Ptpn22-/- mice display more severe mucosal injury, associated with diminished type 1 IFN-driven mRNA, but augmented Tnf and Il1b in colon(58). Moreover, TLR9 agonist treatment does not protect Ptpn22-/- mice from irritant colonic injury, as it does control animals.
Taken together, the data suggest that altered autoimmune or inflammatory responses observed in Ptpn22-/- mice might reflect Ptpn22 roles in both T cell mediated peripheral tolerance and myeloid-dependent immunoregulatory processes.
1.F. Role of Ptpn22 in immune response to infections
Since Ptpn22 regulates induction of type 1 IFN, and since type 1 IFN is important for protective immune responses to diverse infections(53), a role for Ptpn22 in host defense was hypothesized. Indeed, Ptpn22-/- mice display impaired anti-viral immunity(58). After lymphochoriomeningitis virus (LCMV) exposure, systemic type 1 IFN induction, DC activation, and expansion of virus-specific CD8+ T cells are all impaired in Ptpn22-/- mice. Further, Ptpn22-/- mice infected with encephalomyocarditis virus (EMCV) show increased mortality and viral persistence. Together, these findings indicate that Ptpn22 serves as an anti-viral host defense gene, and are commensurate with Ptpn22-dependent promotion of signaling through virus-detecting TLR and intracellular RNA sensors such as MDA5(66, 67). Dissection of the relative contributions of Ptpn22 action in lymphocytes or myeloid cells to observed phenotypes in host defense, autoimmunity, and inflammation will require studies in animals lacking Ptpn22 only in selected immune cell types.
Section 2. PTPN22 in human disease
2.A. Genetic Association between PTPN22 and human disease
2.A.1. Association between PTPN22-C1858T and human autoimmunity
A missense single nucleotide polymorphism in PTPN22 exon 14 (PTPN22-C1858T) was initially identified by candidate gene approach in 2004 and reported to increase predisposition to type 1 diabetes (T1D), rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) (35, 42, 68). PTPN22-C1858T leads to an R-to-W substitution within the P1 motif (position 620) that diminishes strength of the interaction between Csk and Ptpn22. Subsequent genome-wide association (GWA) studies also identified association between PTPN22-C1858T and increased risk of RA, T1D(69), SLE(70) and vitiligo(71). These findings spurred a vast amount of genetic investigation. More than 500 publications addressing the genetic association between PTPN22-C1858T and autoimmune disease have appeared since 2006. Here we will attempt a summary the data, with a focus on the most replicated findings.
PTPN22-C1858T prevalence varies significantly in human populations. The SNP is present at the highest frequency (minor allele frequency >10%) in Caucasians of Northern European descent. The frequency is lower in Western Europeans (7%−8%), and even lower in Southern Europeans, including Italians and Sardinians (<5%)(72). PTPN22-C1858T is rare (<1%) in African, Middle Eastern, Amerindian, and Asian populations. The cause of these intriguing interpopulation frequency differences is unknown; evolutionary genetics analyses suggest that they are related to the ability of PTPN22-C1858T to confer survival advantage, perhaps against selected infections (see section 2.A.2)(73).
The association between PTPN22-C1858T and disease predisposition is well established for T1D(42, 69, 74, 75), RA(35, 69), SLE(68, 70), juvenile idiopathic arthritis (JIA)(76, 77), vitiligo(71, 78), Graves’ disease(74, 79), Hashimoto’s thyroiditis(80), Addison’s disease(79), myasthenia gravis (MG)(81–83), idiopathic thrombocytopenic purpura (ITP)(84), and alopecia areata(85). Several of these associations have been confirmed in multiple populations. Importantly, PTPN22-C1858T explains a T1D locus identified through genetic linkage analysis(86). Carriage of the PTPN22-C1858T variant increases the risk of disease 1.3–2 times per allele, depending on the study and the disease, with the strongest effects being observed in T1D, RA, and vitiligo. A weaker effect (OR for carriage of one PTPN22-C1858T allele ~ 1.16) has been established in systemic sclerosis(72, 87, 88). PTPN22-C1858T also predisposes to co-occurrence of major autoimmune diseases, including T1D and thyroid disease(89), or RA and T1D(90).
Other diseases are conclusively not associated with PTPN22-C1858T. The most established are celiac disease(91), multiple sclerosis (MS)(92), ulcerative colitis (UC)(93), and psoriasis(80). There is also evidence for no association with ankylosing spondylitis(94), aplastic anemia(95), chronic urticaria(96), pemphigus(97), primary biliary cirrhosis(98), primary sclerosing cholangitis(76), or uveitis(99). A wide range of genetic effects has been reported for PTPN22-C1858T in vasculitides. Large vessel vasculitides (giant-cell arteritis(100) and Takayasu’s disease(101)) are not associated with PTPN22-C1858T. Among ANCA-associated vasculitides, PTPN22-C1858T increases risk of granulomatosis with polyangiitis according to some reports(102), but a large genome-wide association study challenged this result(103). Micropolyangiitis and Churg-Strauss vasculitis show no association(104). PTPN22-C1858T is not associated with Henoch-Schönlein purpura(105); however it shows a significant protective effect in Behcet’s disease(106). Notably, a small but robust protective effect for PTPN22-C1858T is also observed in Crohn’s disease (CD)(107).
Reports about the association of PTPN22-C1858T with other diseases-including Sjögren’s syndrome, psoriatic arthritis, myositis, achalasia, Meniere’s disease, atherosclerosis and endometriosis- have been less conclusive and are awaiting further confirmation.
PTPN22-C1858T has been associated with particular clinical subsets of autoimmune disease patients. PTPN22-C1858T associates much more strongly with the subset of RA patients who are seropositive for autoantibodies (either RF+(108, 109) or anti-CCP+(108, 110, 111)). Robust studies in European populations showed that a three-way interaction between PTPN22, the “shared epitope” in HLA-DRB1 (the strongest genetic risk factor for RA in Caucasians) and smoking confers risk of anti-CCP+ RA in Caucasians(110, 112, 113). The association with seropositivity is particularly strong for anti-CCP+ patients who also have antibodies against citrullinated alpha-enolase}(113). One group has not found association between PTPN22-C1858T and the seropositive RA subset(114). Studies of the association of PTPN22-C1858T with radiographic progression of RA have yielded mixed results(108, 111).
Several groups described association between PTPN22-C1858T and the presence of autoantibodies in T1D(115–117), but others could not confirm these results(118). Some studies suggest that PTPN22-C1858T predisposes to increased risk of progression from autoantibody-positive status to pancreatic islet destruction have found no significant effect(119), while others find that PTPN22-C1858T does not affect progression risk(117). PTPN22-C1858T might also associate with worse metabolic control at T1D diagnosis(116).
2.A.2. Association between PTPN22-C1858T and infectious diseases
Carriage of PTPN22-C1858T has been linked with differential susceptibility to several infectious diseases. PTPN22-C1858T carriage is associated with resistance to active pulmonary M. tuberculosis(120, 121). Two studies did not confirm association between tuberculosis (TB) resistance and PTPN22-C1858T carriage(122, 123), albeit in populations (Brazilian-Amazonian, Iranian) with low overall PTPN22-C1858T frequency. However, meta-analysis concluded that PTPN22-C1858T does play a protective role in TB infection(123). In a sample of allogeneic hematopoietic stem cell transplant recipients, donor carriage of PTPN22-C1858T reduced the risk of post-transplantation bacterial infections(124).
Remarkably, PTPN22-C1858T also can increase susceptibility to infection. PTPN22-C1858T carriage predisposes to invasive infections with the Gram-positive bacterium S. pneumoniae(125) and to leprosy(126). PTPN22-C1858T is also associated with a higher incidence of bacterial pulmonary infections among patients with chronic mucocutaneous candidiasis (CMC), a condition in which PTPN22-C1858T carriage is enriched(127). Notably, although Ptpn22-/- mice show defective type 1 IFN-dependent anti-viral responses((58); see section 1.F), no link between PTPN22-C1858T carriage and susceptibility to particular viruses has yet been reported. In fact, one group found lack of association between PTPN22-C1858T carriage and hepatitis C infection(128).
Taken together, the data support the view that PTPN22-C1858T differentially modifies susceptibility to selected bacterial infections. The observations that PTPN22-C1858T confers resistance to TB, yet increases susceptibility to Gram-positive bacteria, produce a paradox. The apparent inconsistency might be explained by findings that PTPN22-C1858T behaves as a loss of function variant in PRR signaling to type 1 IFN promoters(58). Type 1 IFN induced via TLR signals can drive protective responses to Gram -positive pathogens, including S. pneumoniae(129). Conversely, during TB infection, type 1 IFN exerts anti-inflammatory and pro-bacterial effects, mediated in part through suppression of IL-1β(130, 131). These observations suggest how PTPN22-C1858T carriage could simultaneously engender enhanced immunity and selective immunodeficiency.
2.A.3. Association between PTPN22-C1858T and leukemias
PTPN22-C1858T is linked to both lymphoid and myeloid leukemias. Hebbring et al. reported increased frequency of PTPN22-C1858T carriage in chronic lymphocytic leukemia (CLL) cohorts(132, 133). PTPN22-R620W variant-specific effects on CLL physiology remain to be described. However, PTPN22 can suppress some subsets of BCR signals while augmenting others in CLL blasts ((52); see section 1.C), supporting the idea that the role of PTPN22–620W in BCR signaling may not be easily grasped through simple “gain-of-function” or “loss-of-function” models. Guillem et al. reported that chronic myeloid leukemia (CML) patients carrying PTPN22-C1858T show increased risk of failed or inadequate response to treatment(134). Although evidence of differential PTPN22-C1858T signaling function in CML is lacking, oncogenic fusion protein signaling in myeloid cells can be regulated by PTPN22 ((29); see section 1.D).
2.A.4. Genetic landscape of PTPN22-C1858T in human diseases
Several studies addressed whether additional PTPN22 SNPs that do not co-segregate with PTPN22-C1858T (in other words, non PTPN22-C1858T-carrying haplotypes) also associate with autoimmune disease. Haplotypic analyses have been carried out in case-control or family-based cohorts of Caucasian RA(135–138) or T1D(139–141) patients. The unanimous conclusion of all studies is that the PTPN22-C1858T-bearing haplotype explains most or all of the association between PTPN22 and risk for these syndromes. One report(135) suggested that non C1858T-carrying PTPN22 haplotypes associate with RA in North-Americans; however, this observation has not been replicated(137). It is possible that some non C1858T-carrying haplotypes could tag causal variants located in additional autoimmunity susceptibility genes close to PTPN22. For example, studies of psoriasis showed that although PTPN22-C1858T does not associate with disease, variations in linkage disequilibrium (LD) blocks spanning PTPN22 and the nearby locus RSBN1 display a positive association signal(142, 143).
The hypothesis that the PTPN22-C1858T SNP is not causal, but simply marks co-segregating causal SNP(s) within the PTPN22 gene, has been tested. Observations in multiple populations strongly suggested that the PTPN22-C1858T variant is casual, because haplotypes identical at all other SNPs but lacking C1858T did not display association with RA(135–137), T1D(139, 141) or Graves’ disease(144).
SNP rs2488457, specifying G-1123C in the PTPN22 promoter, segregates in almost complete LD with C1858T(145, 146), prompting interest in a possible causal role of G-1123C. However, later studies of T1D and JIA (141, 147) concluded that G-1123C does not have an effect independent of PTPN22-C1858T. Although it is unlikely that G-1123C has a major effect in the presence of PTPN22-C1858T, it might still have an etiologic role in selected populations or diseases. For example, in Asian populations, where carriage of PTPN22-C1858T is almost absent, some data suggest association between G-1123C and disease risk(145, 148). Notably, other groups report no such association in Asian cohorts(149, 150). Moreover, the G-1123C SNP might increase risk of chronic urticaria(96) and ulcerative colitis(151), conditions that are not associated with PTPN22-C1858T.
Several additional SNPs in the PTPN22 gene that were seemingly irrelevant in Caucasian populations are significantly associated with disease risk in Asian populations(149, 152). Thus, further studies of the functional effect of these SNPs in the absence of C1858T on PTPN22 expression and/or splicing are warranted.
As more detailed genetic analyses are carried out, covering an increasing range of PTPN22-associated diseases, some variations in the genetic landscape of PTPN22 among diseases might also emerge. For example, a recent GWAS on JIA found that SNP rs6679677, located near PTPN22 between RSBN1 and PHTF1, but in strong LD with PTPN22-C1858T, represents the strongest non-HLA association signal genome-wide(153).
2.A.5. Additional disease-relevant missense polymorphisms and mutations of PTPN22
The G788A variant is a rare (<7% across different populations) SNP that leads to an R263Q substitution in the PTPN22 catalytic domain (154). PTPN22-G788A displays a weakly protective effect against SLE(155), RA(156) and UC(93) in large multi-population cohorts. G788A segregates with different haplotypes than those containing PTPN22-C1858T, and thus independently influences disease susceptibility(155, 157). R263Q leads to reduction of PTPN22 catalytic activity, and thus can be considered a true “loss-of-function” variant(155, 158). No association was found with CD(93) or SSc(157). Interestingly, PTPN22-G788A might substantially increase the risk of pulmonary tuberculosis(120).
Rare (<1% minor allele frequency) autoimmune disease-associated PTPN22 variants have been identified by PTPN22 locus resequencing. The Concannon group described a PTPN22-G2250C variant which is overtransmitted to T1D patients in families independently of PTPN22-C1858T(139). Another rare variant PTPN22-C1108A (H370N) has been identified through resequencing in CD patients(159). C1108A is notable because it modestly increases risk of disease(159), whereas the more common PTPN22-C1858T variation has a protective effect in CD(107). The H370N substitution maps to the PTPN22 interdomain; however its functional effect remains unknown.
2.B. Functional genetics of PTPN22-R620W in T cells
In line with established roles for PTPN22/Ptpn22 as regulators of signaling through the TCR, most work aimed at unraveling the functional effect of the PTPN22-R620W variant has been focused on T cells. Studies carried out in primary human T cells and in overexpression systems have not reached unequivocal conclusions. One early study found that peripheral T cells from T1D patient PTPN22-C1858T carriers display reduced IL-2 production after CD3/CD28 stimulation(37). These data led to initial classification of PTPN22-R620W as a gain-of-function inhibitor of TCR signaling(37). The Buckner group subsequently reported findings of reduced TCR-induced calcium mobilization, more marked in memory than in naive T cells, among homozygous PTPN22-C1858T patient carriers(49). Although TCR-stimulated IL-10 was reduced significantly among PTPN22-C1858T carriers, production of IL-2, IFNγ, and TNFα, and proliferation of CD4+CD25- T cells were not different from controls(49). The authors also observed a significant increase in the fraction of memory T cells in PTPN22-C1858T carriers(49). A small study of CD4+ cells from juvenile Finnish T1D patients also found decreased TCR-induced calcium mobilization and proliferation of PTPN22-C1858T carrier T cells. Interestingly, no PTPN22-C1858T-dependent anomalies were found in healthy or non-diabetic, autoantibody-positive subjects(160).
Indirect support for a PTPN22-R620W gain of function model in T cell activation came from studies of vasculitis(161) and myasthenia gravis-associated thymomas(83), wherein reduced basal levels of ERK activation and of IL-2 production are observed in PTPN22-C1858T carrier patient PBMC and thymomas, respectively. Interpretation of these findings is complicated because purified T cells were not studied. Further, a separate report on myasthenia gravis found increased IL-2 production by stimulated PTPN22-C1858T carrier patient PBMC, in line with a opposing “loss of function” model for PTPN22-R620W.
More support for the latter model came from Zhang et al., who studied peripheral CD4+ T cells from PTPN22-C1858T homozygotes and non-carrier subjects, both healthy donors and RA patients(47). Carriers display increased anti-CD3/CD28-induced ERK phosphorylation and cell proliferation in this report.
More recently, a comprehensive functional assessment of peripheral blood T cells from healthy PTPN22-C1858T carriers(162) revealed a complex pattern of TCR signaling alterations. PTPN22-C1858T carrier CD4+ T cells show reductions in TCR-CD3ζ phosphorylation and in phosphoPKA-substrate reactivity early after anti-CD3/CD28 treatment, but clear increases in Erk, Slp-76-Y128, Akt-S473, and IKKα phosphorylation 15 minutes later(162). The latter findings of increased TCR-induced 2nd messenger activation agree with those of Zikherman and other proponents of a loss-of-function model for PTPN22-R620W in TCR signaling(39, 47). Ex vivo cytokine production patterns by differentiated effector T cells or by restimulated memory cells suggest Th1 skewing, with increased production of IL-2, IFNγ, and TNFα, but reduced production of IL-17, in PTPN22-C1858T carriers(162).
Isolated CD4+ Treg from carriers show the same signaling alterations observed in bulk CD4+ T cells. However, PTPN22-C1858T homozygous Treg show reduced ability to suppress IFNγ production by Teff(162). Thus, the TCR signaling phenotype in human PTPN22-C1858T carriers generally aligns with that of Ptpn22 deficiency(19), but the impaired Treg suppressive capacity is the opposite of that observed in Ptpn22-/- mice(20) (see section 1.B.4).
Overall, the data from human genotyped cells need to be interpreted with caution, since several of the studies assessed small numbers of subjects, and/or focused on narrow definitions of T cell signaling and function. Further, differences between studies in subject genetic background, cell type or differentiation state, PTPN22-C1858T allele number, and autoimmune diagnoses reduce capacity to draw general conclusions about genotype-dependent alterations in signaling. The ultimate effect of PTPN22-C1858T might differ among T cell subsets, perhaps in a manner dependent upon PTPN22 expression. It is clear that the data collected so far are difficult to reconcile with simple “gain-of-function” or “loss-of-function” models. Perhaps they are best interpreted as representing a “change in function”, a TCR signaling model suggested by recent mechanistic studies of the PTPN22-R620W variant in mice(46)(see below).
Mechanistic work on the effect of the PTPN22-R620W variation in TCR signaling was initially carried out by comparing Jurkat T cells or primary human T cells bearing overexpressed variants. Initial experiments showed that overexpressed PTPN22-R620W was more efficient than PTPN22-WT at inhibiting TCR-induced calcium mobilization, phosphorylation of Lck, TCR-CD3ζ and Zap-70; and induction of an NFAT-AP1-sensitive reporter(37). These data provided support to the “gain-of-function” theory. Consistent with an increased ability to inhibit TCR signaling, overexpressed PTPN22-R620W displays a small but significant increase in its phosphatase activity in vitro(27, 37). One scenario (Fig. 4, “differential phosphorylation model”) posits that the increased phosphatase activity of PTPN22-R620W is due in part to decreased Csk-dependent phosphorylation of PTPN22 on Y536, a residue that when phosphorylated by Lck reportedly inhibits PTPN22 phosphatase activity(27). Gain-of-function inhibition of TCR signaling by overexpressed PTPN22-R620W was subsequently replicated by another group(17), and increased phosphatase activity of PTPN22-R620W vs. PTPN22-WT was also observed in a study of genotyped human PBMC(161).
These results were challenged by Zikherman et al. who found that if PTPN22 is overexpressed in Jurkat T cells together with Csk, the PTPN22–620W variant behaves as a loss of function(39) (see “synergism model” in Fig. 5). These authors reasoned that Ptpn22 needs to be overexpressed together with Csk in order to avoid alterations of the phosphatase/kinase complex stoichiometry. However, due to the asymmetric stoichiometry of the endogenous interaction (50% of PTPN22 in complex with 5% Csk(18, 21)) overexpression of Csk also has potential to alter the physiology of the PTPN22/Csk complex. Thus, further studies in overexpression systems should include experimental assessment of the stoichiometry of the complex.
Vang et al. recently proposed an additional model to explain the “gain-of-function” phenotype of PTPN22-R620W in TCR signaling(21). In their working model (“lipid raft recruitment” model, Fig. 4), Csk normally sequesters PTPN22 away from lipid rafts, but Csk interacts more weakly with PTPN22-R620W, freeing up more phosphatase for recruitment to lipid rafts(21). Although this model is supported by some elegant chemical biology data(21), it relies on data produced in overexpression systems. Notably, another group observed no compartmentalization of Ptpn22 into lipid rafts in mouse T cells(163). Thus, refinement of the molecular mechanism whereby PTPN22 is recruited to lipid rafts in human T cells will be critical in order to increase confidence in this interesting model. In general, pitfalls of overexpression studies include the difficulty of achieving comparable expression levels, and the need to work in a narrow window of low overexpression, where excessive inhibition of TCR signaling by the ectopically expressed phosphatase is avoided.
Zhang et al. were the first to model the functional effects of the PTPN22-R620W variant in a physiological system. They reported studies of animals bearing knocked-in Ptpn22-R619W, a Ptpn22 mutant that is homologous to PTPN22-R620W (see Table 2). Mice harboring the Ptpn22-R619W variant exhibit a hyperactive T cell phenotype(47). Similar to aging Ptpn22-deficient mice(19), Ptpn22-R619W-expressing mice exhibit increased numbers of memory CD4+ T cells in peripheral lymphoid organs. Ptpn22-R619W-bearing thymocytes show augmented proliferative responses and increased tyrosine phosphorylation of Ptpn22 substrates Lck Y394 and Zap-70 after TCR stimulation, consistent with loss of TCR signal inhibitory function by Ptpn22-R619W.
Zhang et al. further reported reduced expression and stability of both Ptpn22-R619W and PTPN22-R620W variants, associated with increased ubiquitination and calpain- and proteasome-mediated degradation, suggesting a mechanism whereby the R-to-W substitution engenders loss of Ptpn22 function(47)(Fig. 5; “degradation” model). Notably, the reduced protein stability model has been vigorously challenged by Dai et al., who reported on independently-generated Ptpn22-R619W knock-in mice(46). They found that the R-to-W substitution in Ptpn22 does not alter protein stability, half-life, or calpain-1-mediated protein degradation(46). The increased PTPN22-R620W degradation phenotype reported by Zhang et al. remains controversial, since other groups failed to confirm altered PTPN22-R620W protein stability(21, 58, 162). Nevertheless, the report by Zhang et al. clearly showed that the immunological phenotype of PepR619W mice is hard to reconcile with simple “gain-of-function” inhibition of TCR signaling.
Another hypothesis to account for observations of PTPN22-R620W-associated TCR hyperresponsiveness proposes dominant interference by a PTPN22–620W-containing PTPN22.6 isoform (see section 1.A.3)(17). In the “isoform interference” model (Fig. 5), the inducible PTPN22.6 isoform acts as a dominant-negative, impairing the capacity of full-length PTPN22 to repress TCR signaling and yielding T cell hyperreactivity. Evidence for this model comes from T cell lines or CD4+ T cells in which overexpression of PTPN22-R620W leads to inhibition of NFAT-AP1 activation (17, 37). Co-overexpression of the dominant-negative PTPN22.6–620W results in an opposite phenotype of increased TCR signaling(17), suggesting that the R620W substitution confers upon PTPN22.6 an ability to inhibit full length PTPN22 function(17). The authors speculate that the expression ratio of PTPN22.6W/full-length PTPN22 may determine the net effect of PTPN22 on TCR signaling. Cells with high PTPN22.6W/full-length PTPN22 ratios will likely exhibit TCR signaling upmodulation, whereas those with low ratios will display relatively attenuated TCR signaling.
The “isoform interference” model is attractive because it has potential to reconcile “gain-of-function” and “loss-of-function” theories of PTN22–620W action in TCR signaling. However, limitations of the supporting dataset include the fact that functional assessments were carried out using overexpression systems, and the genotypes of transfected primary CD4+ T cell donors were not reported(17). It thus remains unclear whether PTPN22-WT and PTPN22-R620W are equally sensitive to the inhibitory action of PTPN22.6-W620.
Another functional model for PTPN22–620W in TCR signaling, termed here “change in function”, has been recently proposed by Dai et al., based on the immunophenotyping of an independently generated Ptpn22-R619W knock-in mouse ((46); see Table 2). Many T cell-associated phenotypes in Ptpn22-R619W mice recapitulate those observed by Zhang et al. and in Ptpn22-/- animals. Memory/effector CD4+ T cells accumulate in aging Ptpn22-R619W mice. Ptpn22-R619W CD4+ effector T cells proliferate more robustly and produce greater amounts of IL-2 after stimulation. Ptpn22-R619W thymocytes exhibit enhanced positive selection, associated with enhanced proximal antigen receptor signaling(46). However, there are some distinctions between Ptpn22-R619W and Ptpn22-/- T cell compartments. Naïve Ptpn22-R619W CD4+ T cells show augmented proximal TCR signaling, while Ptpn22-/- cells do not. Ptpn22-R619W mice display numerous autoantibodies not observed in Ptpn22-/- mice; it is likely that expansion of autoreactive B cell clones is dependent upon help from activated T cells. Ptpn22-/- Treg display enhanced suppressive function; Ptpn22-R619W Treg do not. Finally, the spectrum of TCR-induced tyrosine-phosphorylated molecules is expanded in Ptpn22-R619W effector T cells compared to Ptpn22-/- T cells, suggesting that the R-to-W substitution confers altered enzymatic specificity(46). Together, these differences support a model of altered function, rather than complete loss of function, exerted by the Ptpn22-R619W molecule in TCR signaling.
2.D. Functional genetics of PTPN22-R620W in B cells
B cell antigen receptor (BCR) signaling is required for tolerance-inducing processes, including clonal deletion, receptor editing, and anergy(164). These processes serve to censor developing B lymphocytes and limit the tissue-damaging potential of the autoreactive repertoire. The majority of PTPN22-C1858T-associated autoimmune syndromes feature circulating autoantibodies, which by definition represent disrupted B cell tolerance. Since PTPN22-R620W-expressing T cells display altered proximal TCR signaling, these observations gave impetus to the hypothesis that PTPN22-R620W might also differentially regulate BCR signaling, leading to dysfunction in tolerance induction and contributing to clinical autoimmunity (see Table 2).
Conflicting evidence concerning the role of PTPN22–620W in BCR signaling has come from studies in primary human cells and in animal models. CD19+ B cells from healthy PTPN22-C1858T carriers exhibit diminished proliferation after IgM/IgG stimulation(51). Reduced proliferation capacity correlates with decreased BCR-dependent tyrosine phosphorylation, both globally and on proximal BCR signaling effectors Syk and PLCγ−2. Further, Ca2+ mobilization after BCR engagement on PTPN22-C1858T carrier memory B cells is diminished(49). Treatment with a specific PTPN22 inhibitor(9) normalizes BCR-induced tyrosine phosphorylation in PTPN22-C1858T carrier B cells(51), suggesting that increased enzymatic activity associated with the PTPN22–620W variant contributes to the observed impairment in BCR signaling. Overall, data from primary human cells suggest that PTPN22–620W represents a phosphatase activity “gain-of-function” in BCR signaling. The “gain of function” model is indirectly supported by findings that a polymorphism in the gene encoding PTPN22 binding partner CSK is associated both with increased risk of the PTPN22-C1858T-associated syndrome SLE, and with augmented BCR signaling(165).
Animal studies have challenged the BCR gain of function model for PTPN22–620W. Both Dai et al. and Zhang et al. described B cell function in mice harbouring the knocked-in murine Ptpn22-R619W(46, 47). Both groups reported slightly increased proliferation of anti-IgM stimulated peripheral B cells harbouring Ptpn22-R619W. Dai et al. also found modestly enhanced tyrosine phosphorylation of Syk, PLCγ−2, and other signalling intermediates after BCR stimulation(46). Thus, Ptpn22-R619W in mice, and PTPN22–620W in humans both appear to perturb BCR signaling, but in opposite directions. Potential explanations for the discrepancy: a) murine and human PTPN22/Ptpn22 proteins may exhibit species-specific behaviors in the context of the autoimmunity-associated substitution; b) impaired BCR signaling in human PTPN22-C1858T carrier memory B cells may represent an adaptation to chronic receptor hyperresponsiveness.
B cell development and homeostasis are clearly affected by the presence of PTPN22–620W. PTPN22-C1858T carriers exhibit increased frequency of circulating transitional and anergic B cells(48), strongly suggesting increased passage of autoreactive BCR-bearing cells through developmental checkpoints(166). Indeed, increased frequency of polyreactive B cells in healthy PTPN22-C1858T carrier subjects has been documented using painstaking single-cell antibody cloning techniques(50). PTPN22-C1858T healthy carrier B cells bearing potentially-autoreactive BCR appear at increased frequency in both transitional/new emigrant and in mature, naïve compartments. Findings of increased autoreactivity in both populations strongly suggest that PTPN22–620W interferes with autoreactive cell removal at both central and peripheral “checkpoints”.
Ptpn22-R619W knock-in mice also display altered B cell homeostasis(46). Ptpn22-R619W-expressing mice display increased fractions of autoreactive B cells within the marginal zone compartment, and contain increasing numbers of splenic transitional B cells, germinal center B cells, and Age-related B Cells (“ABC”; capable of secreting autoantibodies(167)) as they age. Together, the data from human blood and animal tissues strongly suggest that PTPN22–620W expression can promote development of a B cell repertoire with enhanced autoreactive potential.
Interestingly, many patients with PTPN22-C1858T-associated diseases including T1D, SLE, and RA display defects in central and peripheral B cell tolerance(48, 50, 168, 169). Defects in B cell censoring can occur regardless of PTPN22 genotype, however, and are independent of disease activity and the presence or absence of active inflammation. These observations suggest that multiple autoimmune susceptibility alleles with potential to regulate BCR signaling thresholds(170) could influence B cell tolerance through the same checkpoints regulated by PTPN22–620W.
Altered B cell homeostasis in Ptpn22-R619W mice correlates with development of frank autoimmunity when the mice are bred on a mixed C57Bl/6:129 background. Lymphocyte infiltration of multiple tissues accompanies increased serum reactivity with numerous autoantigens, including dsDNA and insulin(46). Importantly, mice in which expression of Ptpn22-R619W is restricted to B cells also display autoimmune phenomena, including anti-dsDNA production and glomerulopathy. These findings indicate that B cell-intrinsic Ptpn22-R619W is sufficient to break B cell tolerance and provoke tissue injury, given a permissive genetic milieu.
2.C. Functional genetics of PTPN22-R620W in myeloid cells
Evidence that PTPN22–620R and PTPN22–620W differentially regulate myeloid cell function comes from studies in primary human cells and from animal models. Healthy human PTPN22-C1858T carrier PBMC or cultured DC exhibit significantly reduced upregulation of type 1 IFN, and of type 1 IFN-inducible genes after LPS (TLR4) stimulation(58). In contrast, LPS-induced expression of proinflammatory cytokines including TNFα and IL-6 is comparable in PTPN22-C1858T carrier and non-carrier cells. Since selective impairment of TLR-induced type 1 IFN is also observed in Ptpn22 deficiency, the primary human myeloid cell findings suggest that PTPN22–620W behaves as a reduced function variant in TLR signaling (Fig. 6). Observations that PTPN22-WT, but not PTPN22-R620W, promotes K63-linked autopolyubiquitination of TRAF3(58) provide a candidate mechanism for how PTPN22-R620W could engender defects in TLR signaling leading to upregulation of type 1 IFN.
Figure 6. Expression of disease-predisposing variant PTPN22-R620W is associated with diminished TLR signaling.
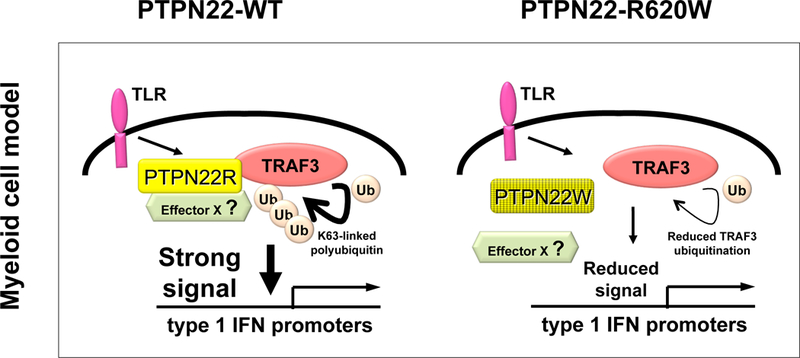
PTPN22-WT (but not PTPN22-R620W), can promote K63-linked autopolyubiquitination of TRAF3 and signaling leading to upregulation of type 1 IFN (left panel). Candidate mechanisms whereby PTPN22-R620W could result in reduced TRAF3 ubiquitination include partial reduction in direct PTPN22 and TRAF3 interaction(58), or diminished recruitment of an as-yet unidentified effector (Effector X) to the complex (R hand panel). Enzymatic activity of PTPN22 is dispensable for promotion of TLR-induced TRAF3 ubiquitination.
Studies in mice made transgenic for human PTPN22 corroborate findings in human primary myeloid cells. After LPS stimulation, BMDC from mice lacking endogenous Ptpn22, but transgenic for human PTPN22-WT, exhibit increased Ifnb1 expression and increased expression of activation markers compared to BMDC from non-transgenic Ptpn22-/- mice(58). In contrast, LPS stimulation of Ptpn22-/-, PTPN22–620W-expressing BMDC does not augment Ifnb1 expression, activation marker expression above the diminished levels observed in stimulated Ptpn22-/- cells. Thus, human PTPN22-WT, but not -R620W, can promote TLR-driven type 1 IFN upregulation and type 1 IFN-dependent myeloid cell activation. These data suggest a mechanism whereby PTPN22-R620W carriage could contribute to expression of autoimmunity: defective myeloid cell capacity for type 1 IFN-driven suppression of inflammation. Expression of human PTPN22-WT, but not of R620W, restores the defective capacity of Ptpn22-/- mice (see section 1.D) to suppress arthritis in response to type 1 IFN-inducing poly(I:C) injection(58, 171). In addition to suffering persistent arthritis despite ameliorating poly(I:C) treatment, PTPN22-R620W-expressing Ptpn22-/- mice exhibit reduced systemic type 1 IFN levels, and increased synovial leukocyte infiltration and IL-1 levels. Together, these data support the notion that PTPN22-R620W exhibits loss of function in promoting type 1 IFN-driven immunoregulation(172), and suggest a mechanism whereby PTPN22-R620W carriage could enable a pro-inflammatory milieu in inflamed tissues.
In contrast to data obtained in human cells and mice transgenic for human PTPN22 variants(58), mice bearing Ptpn22-R619W, the PTPN22-R620W orthologue, exhibit a hyperactive myeloid cell phenotype(47). Ptpn22-R619W-bearing DC manifest enhanced capacity to activate T cells and show increased basal and LPS-induced activation marker expression in vitro. These data suggest an antigen presenting cell-based model(173) for how PTPN22-R620W expression could set the stage for inappropriate T cell activation and autoimmunity. The extent to which PTPN22-R620W and Ptpn22-R619W-expressing mouse models can individually shed further light on how the myeloid compartment contributes to modulation of human disease susceptibility by PTPN22-R620W will be clarified by comparative analysis of in vivo myeloid cell functions including interferogenic TLR signaling, T cell costimulation, and host defense in these models.
Section 3. Mechanisms of PTPN22 action in autoimmune disease susceptibility
Elucidation of the cellular and molecular biology of PTPN22 has permitted construction of increasingly sophisticated models for its function in disease. Evidence continues to accumulate that PTPN22-C1858T exerts altered function in select immune cell developmental processes, in signaling, and in host defense responses.
How does PTPN22-C1858T selectively increase susceptibility to some autoimmune syndromes? The explanation could relate to the immune compartments that dominate in the expression of individual diseases. PTPN22-W620-associated major disease states (RA, T1D, thyroid disease, SLE) each feature prominent dysregulation of humoral responses, including production of autoantibodies. In contrast, in conditions such as inflammatory bowel disease (IBD), MS, or Behcet’s disease, where PTPN22-W620 does not increase disease risk, dysregulated B cells are thought to be less important drivers of pathogenesis. IBD and Behcet’s indeed exemplify syndromes in which PTPN22-C1858T likely plays a protective role. Reduced myeloid cell activation capacity encoded by PTPN22-C1858T could play out as a restraining influence on the development of autoinflammatory syndromes in which such cells play important roles. In MS, altered T cell signaling conferred by PTPN22-C1858T could skew autoreactive T cells away from a pathogenic Th17 differentiation fate, and towards a more benign Th1 fate(162), thus accounting for the lack of risk-enhancing effect.
Here, we briefly sketch out hypotheses that address potential causal links between the PTPN22-C1858T genotype and distinct organ-specific and systemic autoimmune phenotypes. Note that proposed models do not attempt to answer a related question arching over much PTPN22 research: how can a single amino acid substitution (PTPN22-R620W) predispose to diverse organ-specific and systemic autoimmune diseases? Available data do not allow a simple answer to this query. Since each PTPN22-C1858T-associated syndrome represents a complex interaction between numerous host genes and mostly-unknown environmental triggers (e.g. infections), the sources of individual clinical phenotypes likely reside in specific combinations of these factors.
3.A. Rheumatoid arthritis
Rheumatoid arthritis features disinhibition of autoreactive CD4+ T cells, dysregulated B cell homeostasis, and synovium-focused chronic inflammation driven by pro-inflammatory cytokines(174). One model holds that PTPN22-R620W contributes to disease initiation by interfering with BCR-driven censoring of B cell clones bearing receptors with affinity for altered self peptides such as citrullinated α-enolase((113); Fig 7A). After an unknown environmental stimulus, humoral autoimmunity develops, perhaps enabled by augmented T cell help provided by hyper-responsive PTPN22-R620W-bearing CD4+ T cells (Fig 7B). Later in disease, PTPN22-R620W contributes to an imbalance of myeloid cell-produced cytokines within the inflamed synovium. Diminished capacity for production of anti-inflammatory type 1 IFN by PTPN22-R620W-bearing myeloid cells leads to increased prevalence and pro-inflammatory function of IL-1β and TNFα (Fig. 7C). Design of specific PTPN22-R620W-targeted therapies for RA must take into account that PTPN22-R620W-engendered proclivity to humoral autoimmunity is likely dependent upon phosphatase enzymatic function, whereas PTPN22-R620W influence upon type 1 IFN-mediated immunoregulation is not.
Figure 7. Autoimmunity-associated variant PTPN22-R620W may contribute to disease predisposition through differential regulation of B cell repertoire development, T cell signaling, and TLR-driven type 1 IFN production.
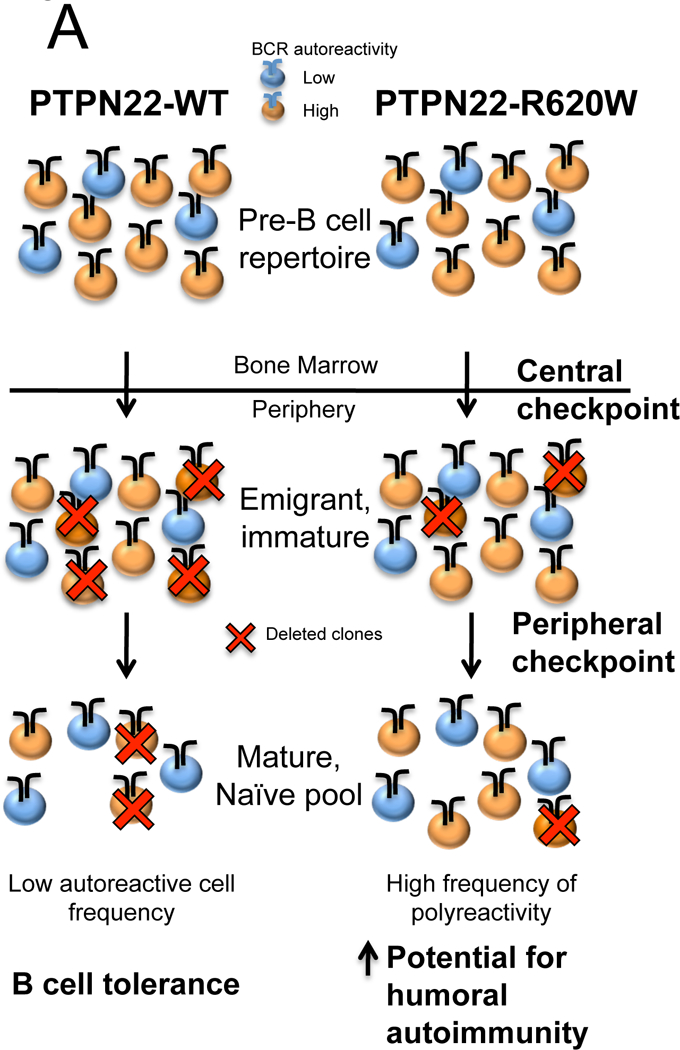
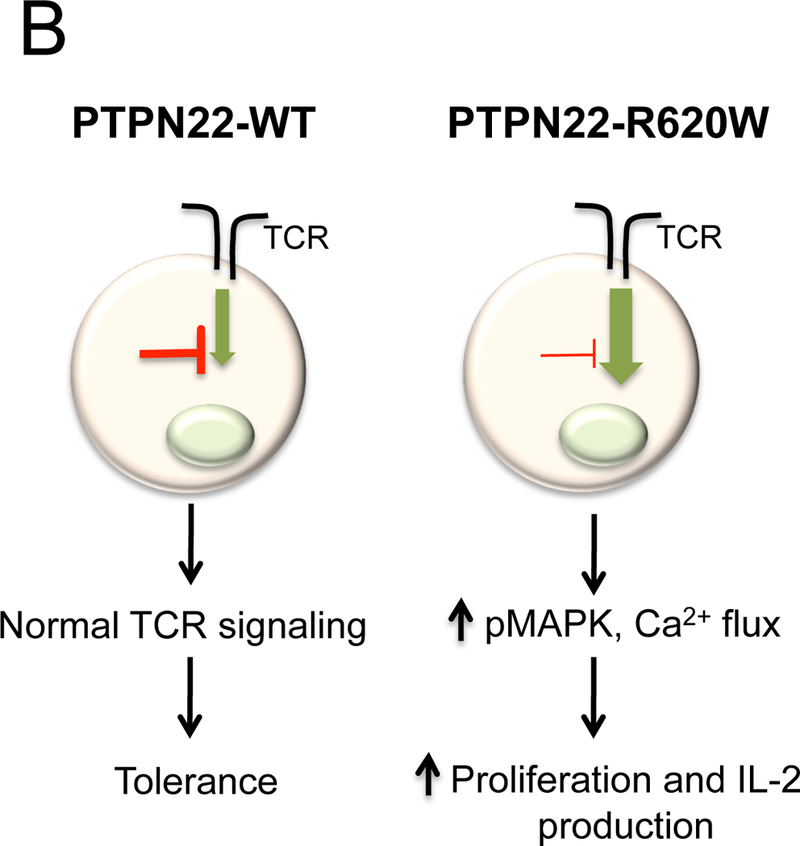
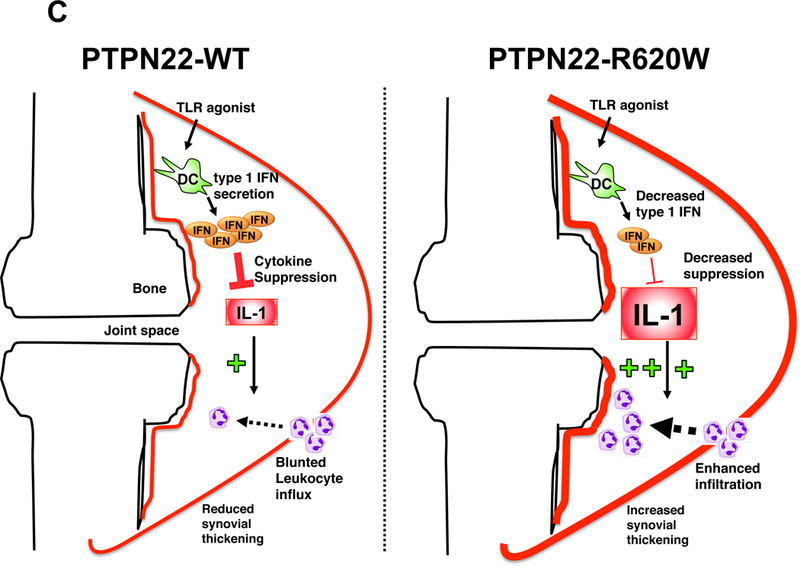
(A) Selective removal of B lymphocytes bearing high-affinity self-reactive B cell antigen receptors occurs at distinct developmental checkpoints in the bone marrow and in the periphery. In PTPN22-R620W carriers, increased numbers of polyreactive B cells escape BCR-mediated deletion as cells progress through both central and peripheral checkpoints. Thus, the PTPN22-R620W carrier naïve B cell repertoire harbors a greater tendency toward autoantibody production(50). (B) Diminished negative regulation of proximal TCR signaling in the presence of PTPN22-R620W (or murine orthologue Ptpn22-R619W) results in augmented 2nd messenger activity and enhanced capacity for cellular activation after TCR stimulus(39, 46, 47). (C) TLR signals drive type 1 IFN which can suppress activity of pro-inflammatory cytokines such as IL-1 in inflammatory arthritis(171). After treatment with TLR agonist, arthritic PTPN22-R620W-bearing animals display reduced synovial type 1 IFN, but increased IL-1, leukocyte infiltration, and joint swelling compared to PTPN22-WT-bearing animals(58).
3.B. Type 1 Diabetes
The subset of “autoimmune” T1D associated with PTPN22-C1858T may reflect infections that either directly destroy functioning islets, or that prime maladaptive lymphocyte responses. Viral infections are major risk factors for T1D(175); type 1 IFN-dependent responses are critical for islet cell defense against viruses(176), and pancreaticotropic viruses cause diabetes when type 1 IFN-promoting signaling proteins are deleted(67). Ptpn22-/- mice exhibit reduced clearance of a potentially-diabetogenic virus, and PTPN22-W620 exhibits loss of function in TLR signaling to type 1 IFN promoters(58). Together, these considerations prompt the hypothesis that PTPN22-C1858T predisposes to T1D by increasing host vulnerability to islet-damaging viral infections. PTPN22-C1858T -associated TCR hypersensitivity and heightened B cell autoreactivity also likely play essential roles in islet cell destruction. Virus infection-induced release of normally-sequestered islet cell antigens could result in recognition by autoreactive lymphocytes, which then mount a destructive autoimmune attack on the endocrine pancreas.
3.C. Systemic lupus erythematosus
SLE represents a collection of clinically-heterogenous syndromes featuring loss of lymphocyte self-tolerance, circulating autoantibodies reactive with DNA and nucleic acid-interacting proteins, and cytokine-mediated inflammatory tissue injury(177). As with RA, PTPN22-C1858T likely influences SLE pathogenesis through both humoral and innate immune processes. Defective censoring of autoreactive B lymphocytes in PTPN22-C1858T carriers (Fig. 7A) could contribute to polyclonal autoreactivity and autoantibody formation observed in SLE. SLE is frequently viewed as a disorder of overabundant type 1 IFN(178); thus, observations of SLE-associated PTPN22-R620W behaving as a loss of function variant in type 1 IFN production(58) appear paradoxical at one level. However, it is notable that inflammatory lesions in SLE target tissues (kidneys and joints) are characterized by high levels of the same pro-inflammatory cytokines (e.g. IL-1β)(179, 180) that are potently suppressed by type 1 IFN (Fig. 7C), and that are associated with tissue destruction in RA. Through modulation of cytokine balance in tissues subjected to autoantibody-mediated injury, PTPN22-R620W may therefore predispose to aggravated inflammation.
b). Summary Points
PTPN22 and Ptpn22 encode hematopoietic-specific, intracellular tyrosine phosphatases that regulate signaling through antigen and pattern recognition receptors.
C1858T, a coding polymorphism in PTPN22, causes elevated risk for major human autoimmune syndromes, including Type 1 Diabetes and Rheumatoid Arthritis, through altered function of a variant PTPN22-R620W protein.
Ptpn22 restrains T cell receptor signals, but promotes pattern recognition receptor signaling in myeloid cells; enzymatic activity is required for the former, but is dispensable for the latter.
PTPN22/Ptpn22 form a complex with the kinase Csk, dependent upon interaction between a proline-rich (P1) motif of the phosphatase, and upon the Csk SH3 domain.
Association with Csk is important for regulation of PTPN22/Ptpn22 function. The autoimmune-predisposing R620W variation within the P1 motif reduces the ability of PTPN22 to interact with Csk.
In myeloid cells, Ptpn22 is selectively required for efficient pattern recognition receptor signal-induced K63-linked ubiquitination of TRAF3, and for subsequent upregulation of type 1 interferon.
Animals harboring PTPN22 “risk” variants or homologues display impaired B cell tolerance, enhanced TCR signaling, and/or impaired type 1 Interferon upregulation in myeloid cells.
Humans carrying risk variant PTPN22-C1858T display enhanced B lymphocyte autoreactivity, dysregulated T cell receptor signaling, and reduced capacity for TLR-induced type 1 Interferon production.
c). Future Issues
Associations between candidate PTPN22 promoter polymorphisms and disease-risk modulation require verification in high-prevalence populations.
Correlation between PTPN22 genotype and exposure to infections (e.g. Coxsackie virus) implicated in pathogenesis of Type 1 diabetes will be sought.
Correlations between PTPN22 genotype and the balance of type 1 Interferons and pro-inflammatory cytokines in inflamed human tissues will be sought.
Conditional alleles of Ptpn22 or “risk” PTPN22 variants will help map PTPN22-related immune developmental, host defense, and autoimmune phenotypes to T cell, B cell, or myeloid compartments.
The significance of PTPN22 interaction with binding partner Csk in the modulation of B cell receptor and Toll-like receptor signaling should be probed.
Substrate mapping and protein association studies in B cells and myeloid cells will illumine molecular mechanisms of PTPN22–620W action.
Therapeutic trials of recently-described PTPN22/Ptpn22 phosphatase inhibitors will suggest dependence of autoimmunity associated with “risk” variant homologue Ptpn22–619W upon enzymatic activity of the Ptpn22 protein.
Acknowledgments:
We apologize in advance to all the investigators whose work could not be appropriately cited owing to space limitations. We extend special thanks to our many collaborators for thoughtful discussions. We are especially grateful to Dr. Bryce Binstadt for critical reading of the manuscript. Funding sources include grants from NIH R01AR057781, the Rheumatology Research Foundation Within Our Reach Campaign, the American Diabetes Association, the Alliance for Lupus Research, and Lupus Foundation of Minnesota (to E.J.P.), and NIH R01AI070544 (to N.B.).
A). Acronyms and Definitions
- Protein tyrosine phosphatase non-receptor type 22: PTPN22
human
- Ptpn22
murine
- Lymphoid phosphatase (Lyp)
human PTPN22 protein
- PEST-enriched phosphatase (Pep)
mouse Ptpn22 protein
- PTPN22-C1858T
PTPN22 disease-associated variant allele-DNA
- PTPN22-R620W
PTPN22 disease-associated variant-protein
- Ptpn22-R619W
murine homologue to PTPN22-R620W, bearing engineered substitution
- Substrate trapping
mutant phosphatase bearing a substitution (e.g. C227S or D195A or both together) in the catalytic domain that binds to, but does not dephosphorylate, substrate
- C-terminal Src kinase (Csk)
kinase that phosphorylates Src family kinases on the C-terminal inhibitory tyrosine residue
- Single nucleotide polymorphism (SNP)
variation of the genomic sequence at the single nucleotide level. Important SNPs and rare variants in PTPN22 include: rs2476601: C1858T (SNP, missense, exon 14, R620W) rs33996649: G788A (SNP, missense, exon 10, R263Q) rs2488457: G-1123C (SNP, promoter) rs72650671: C1108A (rare variant, missense, exon 13, H370N)
- Linkage disequilibrium
non-random association of variants at different loci.
- Haplotype
combination of variants at different loci on the same chromosome
- TRAF3
Tumor Necrosis Factor Receptor Associated Factor 3
- K63-linked ubiquitination
covalent attachment of ubiquitin monomers to ubiquitin residue Lysine63; does not target protein for degradation
- Pattern Recognition Receptors (PRR)
comprise transmembrane Toll-like receptors and cytoplasmic RIG-I-like receptors that recognize microbial nucleic acids and products
Contributor Information
Nunzio Bottini, Erik J. Peterson, M.D., University of Minnesota, 2101 6th St. SE, 2-112 WMBB, Minneapolis, MN 55455, Office 612-625-5634, Fax 612-625-2199, peter899@umn.edu.
Erik J. Peterson, Nunzio Bottini, M.D., Ph.D., La Jolla Institute for Allergy and Immunology, 9420 Athena Circle, La Jolla, CA 92037, Office: 858-752-6815, Fax: 858-752-6985, nunzio@liai.org
REFERENCES
- 1.Matthews RJ, Bowne DB, Flores E, Thomas ML. 1992. Characterization of hematopoietic intracellular protein tyrosine phosphatases: description of a phosphatase containing an SH2 domain and another enriched in proline-, glutamic acid-, serine-, and threonine-rich sequences. Mol Cell Biol 12: 2396–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cloutier JF, Veillette A. 1996. Association of inhibitory tyrosine protein kinase p50csk with protein tyrosine phosphatase PEP in T cells and other hemopoietic cells. EMBO J 15: 4909–18 [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen S, Dadi H, Shaoul E, Sharfe N, Roifman CM. 1999. Cloning and characterization of a lymphoid-specific, inducible human protein tyrosine phosphatase, Lyp. Blood 93: 2013–24 [PubMed] [Google Scholar]
- 4.Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. 2004. Protein tyrosine phosphatases in the human genome. Cell 117: 699–711 [DOI] [PubMed] [Google Scholar]
- 5.Tonks NK. 2013. Protein tyrosine phosphatases--from housekeeping enzymes to master regulators of signal transduction. Febs J 280: 346–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Veillette A, Rhee I, Souza CM, Davidson D. 2009. PEST family phosphatases in immunity, autoimmunity, and autoinflammatory disorders. Immunol Rev 228: 312–24 [DOI] [PubMed] [Google Scholar]
- 7.Arimura Y, Yagi J. 2010. Comprehensive expression profiles of genes for protein tyrosine phosphatases in immune cells. Science signaling 3: rs1. [DOI] [PubMed] [Google Scholar]
- 8.Davidson D, Cloutier JF, Gregorieff A, Veillette A. 1997. Inhibitory tyrosine protein kinase p50csk is associated with protein-tyrosine phosphatase PTP-PEST in hemopoietic and non-hemopoietic cells. J Biol Chem 272: 23455–62 [DOI] [PubMed] [Google Scholar]
- 9.Yu X, Sun JP, He Y, Guo X, Liu S, Zhou B, Hudmon A, Zhang ZY. 2007. Structure, inhibitor, and regulatory mechanism of Lyp, a lymphoid-specific tyrosine phosphatase implicated in autoimmune diseases. Proc Natl Acad Sci U S A 104: 19767–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsai SJ, Sen U, Zhao L, Greenleaf WB, Dasgupta J, Fiorillo E, Orru V, Bottini N, Chen XS. 2009. Crystal structure of the human lymphoid tyrosine phosphatase catalytic domain: insights into redox regulation. Biochemistry 48: 4838–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu J, Katrekar A, Honigberg LA, Smith AM, Conn MT, Tang J, Jeffery D, Mortara K, Sampang J, Williams SR, Buggy J, Clark JM. 2006. Identification of substrates of human protein-tyrosine phosphatase PTPN22. J Biol Chem 281: 11002–10 [DOI] [PubMed] [Google Scholar]
- 12.den Hertog J, Groen A, van der Wijk T. 2005. Redox regulation of protein-tyrosine phosphatases. Arch Biochem Biophys 434: 11–5 [DOI] [PubMed] [Google Scholar]
- 13.Liu Y, Stanford SM, Jog SP, Fiorillo E, Orru V, Comai L, Bottini N. 2009. Regulation of lymphoid tyrosine phosphatase activity: inhibition of the catalytic domain by the proximal interdomain. Biochemistry 48: 7525–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flores E, Roy G, Patel D, Shaw A, Thomas ML. 1994. Nuclear localization of the PEP protein tyrosine phosphatase. Mol Cell Biol 14: 4938–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ronninger M, Guo Y, Shchetynsky K, Hill A, Khademi M, Olsson T, Reddy PS, Seddighzadeh M, Clark JD, Lin LL, O’Toole M, Padyukov L. 2012. The balance of expression of PTPN22 splice forms is significantly different in rheumatoid arthritis patients compared with controls. Genome Med 4: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang S, Dong H, Han J, Ho WT, Fu X, Zhao ZJ. 2010. Identification of a variant form of tyrosine phosphatase LYP. BMC Mol Biol 11: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang HH, Tai TS, Lu B, Iannaccone C, Cernadas M, Weinblatt M, Shadick N, Miaw SC, Ho IC. 2012. PTPN22.6, a dominant negative isoform of PTPN22 and potential biomarker of rheumatoid arthritis. PLoS One 7: e33067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cloutier JF, Veillette A. 1999. Cooperative inhibition of T-cell antigen receptor signaling by a complex between a kinase and a phosphatase. J Exp Med 189: 111–21 Ptpn22 forms a complex with Csk and formation of the complex regulates phosphatase function in T cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hasegawa K, Martin F, Huang G, Tumas D, Diehl L, Chan AC. 2004. PEST domain-enriched tyrosine phosphatase (PEP) regulation of effector/memory T cells. Science 303: 685–9 Ptpn22 deficiency results in augmented TCR signaling and thymocyte positive selection; no B cell-intrinsic phenotype. [DOI] [PubMed] [Google Scholar]
- 20.Brownlie RJ, Miosge LA, Vassilakos D, Svensson LM, Cope A, Zamoyska R. 2012. Lack of the phosphatase PTPN22 increases adhesion of murine regulatory T cells to improve their immunosuppressive function. Sci Signal 5: ra87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vang T, Liu WH, Delacroix L, Wu S, Vasile S, Dahl R, Yang L, Musumeci L, Francis D, Landskron J, Tasken K, Tremblay ML, Lie BA, Page R, Mustelin T, Rahmouni S, Rickert RC, Tautz L. 2012. LYP inhibits T-cell activation when dissociated from CSK. Nat Chem Biol 8: 437–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kane LP, Lin J, Weiss A. 2000. Signal transduction by the TCR for antigen. Curr Opin Immunol 12: 242–9 [DOI] [PubMed] [Google Scholar]
- 23.Tybulewicz VL. 2005. Vav-family proteins in T-cell signalling. Curr Opin Immunol 17: 267–74 [DOI] [PubMed] [Google Scholar]
- 24.Egerton M, Ashe OR, Chen D, Druker BJ, Burgess WH, Samelson LE. 1992. VCP, the mammalian homolog of cdc48, is tyrosine phosphorylated in response to T cell antigen receptor activation. EMBO J 11: 3533–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meyer H, Bug M, Bremer S. 2012. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol 14: 117–23 [DOI] [PubMed] [Google Scholar]
- 26.Duan L, Reddi AL, Ghosh A, Dimri M, Band H. 2004. The Cbl family and other ubiquitin ligases: destructive forces in control of antigen receptor signaling. Immunity 21: 7–17 [DOI] [PubMed] [Google Scholar]
- 27.Fiorillo E, Orru V, Stanford SM, Liu Y, Salek M, Rapini N, Schenone AD, Saccucci P, Delogu LG, Angelini F, Manca Bitti ML, Schmedt C, Chan AC, Acuto O, Bottini N. 2010. Autoimmune-associated PTPN22 R620W variation reduces phosphorylation of lymphoid phosphatase on an inhibitory tyrosine residue. J Biol Chem 285: 26506–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de la Puerta ML, Trinidad AG, Rodriguez Mdel C, de Pereda JM, Sanchez Crespo M, Bayon Y, Alonso A. 2013. The autoimmunity risk variant LYP-W620 cooperates with CSK in the regulation of TCR signaling. PLoS One 8: e54569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chien W, Tidow N, Williamson EA, Shih LY, Krug U, Kettenbach A, Fermin AC, Roifman CM, Koeffler HP. 2003. Characterization of a myeloid tyrosine phosphatase, Lyp, and its role in the Bcr-Abl signal transduction pathway. J Biol Chem 278: 27413–20 [DOI] [PubMed] [Google Scholar]
- 30.Konigsberger S, Peckl-Schmid D, Zaborsky N, Patzak I, Kiefer F, Achatz G. 2010. HPK1 associates with SKAP-HOM to negatively regulate Rap1-mediated B-lymphocyte adhesion. PLoS One 5: e12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu X, Chen M, Zhang S, Yu ZH, Sun JP, Wang L, Liu S, Imasaki T, Takagi Y, Zhang ZY. 2011. Substrate specificity of lymphoid-specific tyrosine phosphatase (Lyp) and identification of Src kinase-associated protein of 55 kDa homolog (SKAP-HOM) as a Lyp substrate. J Biol Chem 286: 30526–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gjorloff-Wingren A, Saxena M, Han S, Wang X, Alonso A, Renedo M, Oh P, Williams S, Schnitzer J, Mustelin T. 2000. Subcellular localization of intracellular protein tyrosine phosphatases in T cells. Eur J Immunol 30: 2412–21 [DOI] [PubMed] [Google Scholar]
- 33.Maine CJ, Hamilton-Williams EE, Cheung J, Stanford SM, Bottini N, Wicker LS, Sherman LA. 2012. PTPN22 alters the development of regulatory T cells in the thymus. J Immunol 188: 5267–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stanford SM, Krishnamurthy D, Falk MD, Messina R, Debnath B, Li S, Liu T, Kazemi R, Dahl R, He Y, Yu X, Chan AC, Zhang ZY, Barrios AM, Woods VL Jr., Neamati N, Bottini N 2011. Discovery of a novel series of inhibitors of lymphoid tyrosine phosphatase with activity in human T cells. J Med Chem 54: 1640–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, Ardlie KG, Huang Q, Smith AM, Spoerke JM, Conn MT, Chang M, Chang SY, Saiki RK, Catanese JJ, Leong DU, Garcia VE, McAllister LB, Jeffery DA, Lee AT, Batliwalla F, Remmers E, Criswell LA, Seldin MF, Kastner DL, Amos CI, Sninsky JJ, Gregersen PK. 2004. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet 75: 330–7 PTPN22-C1858T carriage associates with increased risk of rheumatoid arthritis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gjorloff-Wingren A, Saxena M, Williams S, Hammi D, Mustelin T. 1999. Characterization of TCR-induced receptor-proximal signaling events negatively regulated by the protein tyrosine phosphatase PEP. Eur J Immunol 29: 3845–54 [DOI] [PubMed] [Google Scholar]
- 37.Vang T, Congia M, Macis MD, Musumeci L, Orru V, Zavattari P, Nika K, Tautz L, Tasken K, Cucca F, Mustelin T, Bottini N. 2005. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet 37: 1317–9 PTPN22-C1858T carrier T1D patients display hyporesponsive T cells, suggesting PTPN22-W620 “gain of function”. [DOI] [PubMed] [Google Scholar]
- 38.Marson A, Kretschmer K, Frampton GM, Jacobsen ES, Polansky JK, MacIsaac KD, Levine SS, Fraenkel E, von Boehmer H, Young RA. 2007. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature 445: 931–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zikherman J, Hermiston M, Steiner D, Hasegawa K, Chan A, Weiss A. 2009. PTPN22 deficiency cooperates with the CD45 E613R allele to break tolerance on a non-autoimmune background. J Immunol 182: 4093–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nika K, Soldani C, Salek M, Paster W, Gray A, Etzensperger R, Fugger L, Polzella P, Cerundolo V, Dushek O, Hofer T, Viola A, Acuto O. 2010. Constitutively active Lck kinase in T cells drives antigen receptor signal transduction. Immunity 32: 766–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmedt C, Saijo K, Niidome T, Kuhn R, Aizawa S, Tarakhovsky A. 1998. Csk controls antigen receptor-mediated development and selection of T-lineage cells. Nature 394: 901–4 [DOI] [PubMed] [Google Scholar]
- 42.Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, MacMurray J, Meloni GF, Lucarelli P, Pellecchia M, Eisenbarth GS, Comings D, Mustelin T. 2004. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet 36: 337–8 PTPN22-C1858T carriage associates with increased risk of type 1 diabetes; variant protein shows defective Csk binding. [DOI] [PubMed] [Google Scholar]
- 43.Moran AE, Hogquist KA. 2012. T-cell receptor affinity in thymic development. Immunology 135: 261–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu LF, Rudensky A. 2009. Molecular orchestration of differentiation and function of regulatory T cells. Genes Dev 23: 1270–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng P, Kissler S. 2013. PTPN22 silencing in the NOD model indicates the type 1 diabetes-associated allele is not a loss-of-function variant. Diabetes 62: 896–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dai X, James RG, Habib T, Singh S, Jackson S, Khim S, Moon RT, Liggitt D, Wolf-Yadlin A, Buckner JH, Rawlings DJ. 2013. A disease-associated PTPN22 variant promotes systemic autoimmunity in murine models. J Clin Invest 123: 2024–36 Ptpn22-R619W knock-in mice show autoantibodies and leukocyte infiltrates, enhanced TCR signaling; no Ptpn22 stability defect. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang J, Zahir N, Jiang Q, Miliotis H, Heyraud S, Meng X, Dong B, Xie G, Qiu F, Hao Z, McCulloch CA, Keystone EC, Peterson AC, Siminovitch KA. 2011. The autoimmune disease-associated PTPN22 variant promotes calpain-mediated Lyp/Pep degradation associated with lymphocyte and dendritic cell hyperresponsiveness. Nat Genet 43: 902–7 Ptpn22-R619W knock-in mice display hyper-responsive T cells; variant protein shows reduced stability. [DOI] [PubMed] [Google Scholar]
- 48.Habib T, Funk A, Rieck M, Brahmandam A, Dai X, Panigrahi AK, Luning Prak ET, Meyer-Bahlburg A, Sanda S, Greenbaum C, Rawlings DJ, Buckner JH. 2012. Altered B cell homeostasis is associated with type I diabetes and carriers of the PTPN22 allelic variant. J Immunol 188: 487–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rieck M, Arechiga A, Onengut-Gumuscu S, Greenbaum C, Concannon P, Buckner JH. 2007. Genetic variation in PTPN22 corresponds to altered function of T and B lymphocytes. J Immunol 179: 4704–10 PTPN22-C1858T carriers display altered BCR signaling. [DOI] [PubMed] [Google Scholar]
- 50.Menard L, Saadoun D, Isnardi I, Ng YS, Meyers G, Massad C, Price C, Abraham C, Motaghedi R, Buckner JH, Gregersen PK, Meffre E. 2011. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. J Clin Invest 121: 3635–44 Healthy PTPN22-C1858T carriers show defects at central and peripheral B cell tolerance checkpoints. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arechiga AF, Habib T, He Y, Zhang X, Zhang ZY, Funk A, Buckner JH. 2009. Cutting edge: the PTPN22 allelic variant associated with autoimmunity impairs B cell signaling. J Immunol 182: 3343–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Negro R, Gobessi S, Longo PG, He Y, Zhang ZY, Laurenti L, Efremov DG. 2012. Overexpression of the autoimmunity-associated phosphatase PTPN22 promotes survival of antigen-stimulated CLL cells by selectively activating AKT. Blood 119: 6278–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kawai T, Akira S. 2011. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34: 637–50 [DOI] [PubMed] [Google Scholar]
- 54.Heng TS, Painter MW. 2008. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 9: 1091–4 [DOI] [PubMed] [Google Scholar]
- 55.Liu J, Wang L, Harvey-White J, Osei-Hyiaman D, Razdan R, Gong Q, Chan AC, Zhou Z, Huang BX, Kim HY, Kunos G. 2006. A biosynthetic pathway for anandamide. Proc Natl Acad Sci U S A 103: 13345–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chevrier N, Mertins P, Artyomov MN, Shalek AK, Iannacone M, Ciaccio MF, Gat-Viks I, Tonti E, DeGrace MM, Clauser KR, Garber M, Eisenhaure TM, Yosef N, Robinson J, Sutton A, Andersen MS, Root DE, von Andrian U, Jones RB, Park H, Carr SA, Regev A, Amit I, Hacohen N. 2011. Systematic discovery of TLR signaling components delineates viral-sensing circuits. Cell 147: 853–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Spalinger MR, Lang S, Weber A, Frei P, Fried M, Rogler G, Scharl M. 2013. Loss of Protein Tyrosine Phosphatase Nonreceptor Type 22 Regulates Interferon-gamma-Induced Signaling in Human Monocytes. Gastroenterology 144: 978–88 [DOI] [PubMed] [Google Scholar]
- 58.Wang Y, Shaked I, Stanford SM, Binstadt B, Balfour HH Jr., Chan A, Ley K, Bottini N, Peterson E 2013. Ptpn22 potentiates Toll-like receptor-driven, type 1 interferon-dependent immunity. Immunity in press, 2013 Ptpn22 deficiency and PTPN22-R620W expression associate with impaired TLR signaling, host defense, and inflammation suppression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hacker H, Tseng PH, Karin M. 2011. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat Rev Immunol 11: 457–68 [DOI] [PubMed] [Google Scholar]
- 60.Tseng PH, Matsuzawa A, Zhang W, Mino T, Vignali DA, Karin M. 2010. Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat Immunol 11: 70–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoshimura A, Suzuki M, Sakaguchi R, Hanada T, Yasukawa H. 2012. SOCS, Inflammation, and Autoimmunity. Front Immunol 3: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Villuendas R, Steegmann JL, Pollan M, Tracey L, Granda A, Fernandez-Ruiz E, Casado LF, Martinez J, Martinez P, Lombardia L, Villalon L, Odriozola J, Piris MA. 2006. Identification of genes involved in imatinib resistance in CML: a gene-expression profiling approach. Leukemia 20: 1047–54 [DOI] [PubMed] [Google Scholar]
- 63.Yeh LT, Miaw SC, Lin MH, Chou FC, Shieh SJ, Chuang YP, Lin SH, Chang DM, Sytwu HK. 2013. Different Modulation of Ptpn22 in Effector and Regulatory T Cells Leads to Attenuation of Autoimmune Diabetes in Transgenic Nonobese Diabetic Mice. J Immunol 191: 594–607 [DOI] [PubMed] [Google Scholar]
- 64.Corr M, Boyle DL, Ronacher LM, Lew BR, van Baarsen LG, Tak PP, Firestein GS. 2011. Interleukin 1 receptor antagonist mediates the beneficial effects of systemic interferon beta in mice: implications for rheumatoid arthritis. Ann Rheum Dis 70: 858–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cantaert T, Baeten D, Tak PP, van Baarsen LG. 2010. Type I IFN and TNFalpha cross-regulation in immune-mediated inflammatory disease: basic concepts and clinical relevance. Arthritis Res Ther 12: 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, Diamond MS, Colonna M. 2006. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci U S A 103: 8459–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McCartney SA, Vermi W, Lonardi S, Rossini C, Otero K, Calderon B, Gilfillan S, Diamond MS, Unanue ER, Colonna M. 2011. RNA sensor-induced type I IFN prevents diabetes caused by a beta cell-tropic virus in mice. J Clin Invest 121: 1497–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kyogoku C, Langefeld CD, Ortmann WA, Lee A, Selby S, Carlton VE, Chang M, Ramos P, Baechler EC, Batliwalla FM, Novitzke J, Williams AH, Gillett C, Rodine P, Graham RR, Ardlie KG, Gaffney PM, Moser KL, Petri M, Begovich AB, Gregersen PK, Behrens TW. 2004. Genetic association of the R620W polymorphism of protein tyrosine phosphatase PTPN22 with human SLE. Am J Hum Genet 75: 504–7 PTPN22-C1858T carriage associates with increased risk of SLE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.2007. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447: 661–78 Genome-wide, case-control study shows genome-wide signal for PTPN22-C1858T in T1D, RA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, Tsao BP, Vyse TJ, Langefeld CD, Nath SK, Guthridge JM, Cobb BL, Mirel DB, Marion MC, Williams AH, Divers J, Wang W, Frank SG, Namjou B, Gabriel SB, Lee AT, Gregersen PK, Behrens TW, Taylor KE, Fernando M, Zidovetzki R, Gaffney PM, Edberg JC, Rioux JD, Ojwang JO, James JA, Merrill JT, Gilkeson GS, Seldin MF, Yin H, Baechler EC, Li QZ, Wakeland EK, Bruner GR, Kaufman KM, Kelly JA. 2008. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 40: 204–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jin Y, Birlea SA, Fain PR, Gowan K, Riccardi SL, Holland PJ, Mailloux CM, Sufit AJ, Hutton SM, Amadi-Myers A, Bennett DC, Wallace MR, McCormack WT, Kemp EH, Gawkrodger DJ, Weetman AP, Picardo M, Leone G, Taieb A, Jouary T, Ezzedine K, van Geel N, Lambert J, Overbeck A, Spritz RA. 2010. Variant of TYR and autoimmunity susceptibility loci in generalized vitiligo. N Engl J Med 362: 1686–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zheng J, Ibrahim S, Petersen F, Yu X. 2012. Meta-analysis reveals an association of PTPN22 C1858T with autoimmune diseases, which depends on the localization of the affected tissue. Genes Immun 13: 641–52 [DOI] [PubMed] [Google Scholar]
- 73.McPartland JM, Norris RW, Kilpatrick CW. 2007. Tempo and mode in the endocannaboinoid system. J Mol Evol 65: 267–76 [DOI] [PubMed] [Google Scholar]
- 74.Smyth D, Cooper JD, Collins JE, Heward JM, Franklyn JA, Howson JM, Vella A, Nutland S, Rance HE, Maier L, Barratt BJ, Guja C, Ionescu-Tirgoviste C, Savage DA, Dunger DB, Widmer B, Strachan DP, Ring SM, Walker N, Clayton DG, Twells RC, Gough SC, Todd JA. 2004. Replication of an association between the lymphoid tyrosine phosphatase locus (LYP/PTPN22) with type 1 diabetes, and evidence for its role as a general autoimmunity locus. Diabetes 53: 3020–3 [DOI] [PubMed] [Google Scholar]
- 75.Onengut-Gumuscu S, Ewens KG, Spielman RS, Concannon P. 2004. A functional polymorphism (1858C/T) in the PTPN22 gene is linked and associated with type I diabetes in multiplex families. Genes Immun 5: 678–80 [DOI] [PubMed] [Google Scholar]
- 76.Viken MK, Amundsen SS, Kvien TK, Boberg KM, Gilboe IM, Lilleby V, Sollid LM, Forre OT, Thorsby E, Smerdel A, Lie BA. 2005. Association analysis of the 1858C>T polymorphism in the PTPN22 gene in juvenile idiopathic arthritis and other autoimmune diseases. Genes Immun 6: 271–3 [DOI] [PubMed] [Google Scholar]
- 77.Hinks A, Barton A, John S, Bruce I, Hawkins C, Griffiths CE, Donn R, Thomson W, Silman A, Worthington J. 2005. Association between the PTPN22 gene and rheumatoid arthritis and juvenile idiopathic arthritis in a UK population: further support that PTPN22 is an autoimmunity gene. Arthritis Rheum 52: 1694–9 [DOI] [PubMed] [Google Scholar]
- 78.Canton I, Akhtar S, Gavalas NG, Gawkrodger DJ, Blomhoff A, Watson PF, Weetman AP, Kemp EH. 2005. A single-nucleotide polymorphism in the gene encoding lymphoid protein tyrosine phosphatase (PTPN22) confers susceptibility to generalised vitiligo. Genes Immun 6: 584–7 [DOI] [PubMed] [Google Scholar]
- 79.Velaga MR, Wilson V, Jennings CE, Owen CJ, Herington S, Donaldson PT, Ball SG, James RA, Quinton R, Perros P, Pearce SH. 2004. The codon 620 tryptophan allele of the lymphoid tyrosine phosphatase (LYP) gene is a major determinant of Graves’ disease. J Clin Endocrinol Metab 89: 5862–5 [DOI] [PubMed] [Google Scholar]
- 80.Criswell LA, Pfeiffer KA, Lum RF, Gonzales B, Novitzke J, Kern M, Moser KL, Begovich AB, Carlton VE, Li W, Lee AT, Ortmann W, Behrens TW, Gregersen PK. 2005. Analysis of families in the multiple autoimmune disease genetics consortium (MADGC) collection: the PTPN22 620W allele associates with multiple autoimmune phenotypes. Am J Hum Genet 76: 561–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vandiedonck C, Capdevielle C, Giraud M, Krumeich S, Jais JP, Eymard B, Tranchant C, Gajdos P, Garchon HJ. 2006. Association of the PTPN22*R620W polymorphism with autoimmune myasthenia gravis. Ann Neurol 59: 404–7 [DOI] [PubMed] [Google Scholar]
- 82.Lefvert AK, Zhao Y, Ramanujam R, Yu S, Pirskanen R, Hammarstrom L. 2008. PTPN22 R620W promotes production of anti-AChR autoantibodies and IL-2 in myasthenia gravis. J Neuroimmunol 197: 110–3 [DOI] [PubMed] [Google Scholar]
- 83.Chuang WY, Strobel P, Belharazem D, Rieckmann P, Toyka KV, Nix W, Schalke B, Gold R, Kiefer R, Klinker E, Opitz A, Inoue M, Kuo TT, Muller-Hermelink HK, Marx A. 2009. The PTPN22(gain-of-function)+1858T(+) genotypes correlate with low IL-2 expression in thymomas and predispose to myasthenia gravis. Genes Immun 10: 667–72 [DOI] [PubMed] [Google Scholar]
- 84.D’Silva KJ, Zamora MB, Gerlach J, Schwartz KA. 2010. Increased representation of the PTPN22 mutation in patients with immune thrombocytopenia. J Thromb Haemost 8: 2076–8 [DOI] [PubMed] [Google Scholar]
- 85.Kemp EH, McDonagh AJ, Wengraf DA, Messenger AG, Gawkrodger DJ, Cork MJ, Tazi-Ahnini R. 2006. The non-synonymous C1858T substitution in the PTPN22 gene is associated with susceptibility to the severe forms of alopecia areata. Hum Immunol 67: 535–9 [DOI] [PubMed] [Google Scholar]
- 86.Smyth DJ, Cooper JD, Howson JM, Walker NM, Plagnol V, Stevens H, Clayton DG, Todd JA. 2008. PTPN22 Trp620 explains the association of chromosome 1p13 with type 1 diabetes and shows a statistical interaction with HLA class II genotypes. Diabetes 57: 1730–7 [DOI] [PubMed] [Google Scholar]
- 87.Gourh P, Tan FK, Assassi S, Ahn CW, McNearney TA, Fischbach M, Arnett FC, Mayes MD. 2006. Association of the PTPN22 R620W polymorphism with anti-topoisomerase I- and anticentromere antibody-positive systemic sclerosis. Arthritis Rheum 54: 3945–53 [DOI] [PubMed] [Google Scholar]
- 88.Dieude P, Guedj M, Wipff J, Avouac J, Hachulla E, Diot E, Granel B, Sibilia J, Cabane J, Meyer O, Mouthon L, Kahan A, Boileau C, Allanore Y. 2008. The PTPN22 620W allele confers susceptibility to systemic sclerosis: findings of a large case-control study of European Caucasians and a meta-analysis. Arthritis Rheum 58: 2183–8 [DOI] [PubMed] [Google Scholar]
- 89.Dultz G, Matheis N, Dittmar M, Rohrig B, Bender K, Kahaly GJ. 2009. The protein tyrosine phosphatase non-receptor type 22 C1858T polymorphism is a joint susceptibility locus for immunthyroiditis and autoimmune diabetes. Thyroid 19: 143–8 [DOI] [PubMed] [Google Scholar]
- 90.Liao KP, Gunnarsson M, Kallberg H, Ding B, Plenge RM, Padyukov L, Karlson EW, Klareskog L, Askling J, Alfredsson L. 2009. Specific association of type 1 diabetes mellitus with anti-cyclic citrullinated peptide-positive rheumatoid arthritis. Arthritis Rheum 60: 653–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rueda B, Nunez C, Orozco G, Lopez-Nevot MA, de la Concha EG, Martin J, Urcelay E. 2005. C1858T functional variant of PTPN22 gene is not associated with celiac disease genetic predisposition. Hum Immunol 66: 848–52 [DOI] [PubMed] [Google Scholar]
- 92.Begovich AB, Caillier SJ, Alexander HC, Penko JM, Hauser SL, Barcellos LF, Oksenberg JR. 2005. The R620W polymorphism of the protein tyrosine phosphatase PTPN22 is not associated with multiple sclerosis. Am J Hum Genet 76: 184–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Diaz-Gallo LM, Espino-Paisan L, Fransen K, Gomez-Garcia M, van Sommeren S, Cardena C, Rodrigo L, Mendoza JL, Taxonera C, Nieto A, Alcain G, Cueto I, Lopez-Nevot MA, Bottini N, Barclay ML, Crusius JB, van Bodegraven AA, Wijmenga C, Ponsioen CY, Gearry RB, Roberts RL, Weersma RK, Urcelay E, Merriman TR, Alizadeh BZ, Martin J. 2011. Differential association of two PTPN22 coding variants with Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis 17: 2287–94 [DOI] [PubMed] [Google Scholar]
- 94.Orozco G, Garcia-Porrua C, Lopez-Nevot MA, Raya E, Gonzalez-Gay MA, Martin J. 2006. Lack of association between ankylosing spondylitis and a functional polymorphism of PTPN22 proposed as a general susceptibility marker for autoimmunity. Ann Rheum Dis 65: 687–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Graf SA, Calado RT, Young NS. 2007. PTPN22 620W allele is not associated with aplastic anemia. Am J Hematol 82: 291–2 [DOI] [PubMed] [Google Scholar]
- 96.Brzoza Z, Grzeszczak W, Trautsolt W, Moczulski D. 2011. Protein tyrosine phosphatase-22 (PTPN-22) polymorphism in the pathogenesis of chronic urticaria. Allergy 66: 1392–3 [DOI] [PubMed] [Google Scholar]
- 97.Sachdev A, Bhanusali DG, Patterson KC, Zamora MB, Ghuman A, Gerlach JA, Sinha AA. 2011. PTPN22 1858T is not a risk factor for North American pemphigus vulgaris. Exp Dermatol 20: 514–9 [DOI] [PubMed] [Google Scholar]
- 98.Milkiewicz P, Pache I, Buwaneswaran H, Liu X, Coltescu C, Heathcote EJ, Siminovitch KA. 2006. The PTPN22 1858T variant is not associated with primary biliary cirrhosis. Tissue Antigens 67: 434–7 [DOI] [PubMed] [Google Scholar]
- 99.Cenit MC, Marquez A, Cordero-Coma M, Fonollosa A, Llorenc V, Artaraz J, Diaz Valle D, Blanco R, Canal J, Salom D, Garcia Serrano JL, de Ramon E, del Rio Jose M, Gorrono-Echebarria MB, Martin-Villa JM, Molins B, Ortego-Centeno N, Martin J. 2013. Lack of association between the protein tyrosine phosphatase non-receptor type 22 R263Q and R620W functional genetic variants and endogenous non-anterior uveitis. Mol Vis 19: 638–43 [PMC free article] [PubMed] [Google Scholar]
- 100.Gonzalez-Gay MA, Oliver J, Orozco G, Garcia-Porrua C, Lopez-Nevot MA, Martin J. 2005. Lack of association of a functional single nucleotide polymorphism of PTPN22, encoding lymphoid protein phosphatase, with susceptibility to biopsy-proven giant cell arteritis. J Rheumatol 32: 1510–2 [PubMed] [Google Scholar]
- 101.Sahin N, Aksu K, Kamali S, Bicakcigil M, Ozbalkan Z, Fresko I, Ozer H, Akar S, Onat AM, Cobankara V, Kiraz S, Ozturk MA, Tunc E, Yucel E, Ates A, Keser G, Inanc M, Direskeneli H, Saruhan-Direskeneli G. 2008. PTPN22 gene polymorphism in Takayasu’s arteritis. Rheumatology 47: 634–5 [DOI] [PubMed] [Google Scholar]
- 102.Jagiello P, Aries P, Arning L, Wagenleiter SE, Csernok E, Hellmich B, Gross WL, Epplen JT. 2005. The PTPN22 620W allele is a risk factor for Wegener’s granulomatosis. Arthritis Rheum 52: 4039–43 [DOI] [PubMed] [Google Scholar]
- 103.Lyons PA, Rayner TF, Trivedi S, Holle JU, Watts RA, Jayne DR, Baslund B, Brenchley P, Bruchfeld A, Chaudhry AN, Cohen Tervaert JW, Deloukas P, Feighery C, Gross WL, Guillevin L, Gunnarsson I, Harper L, Hruskova Z, Little MA, Martorana D, Neumann T, Ohlsson S, Padmanabhan S, Pusey CD, Salama AD, Sanders JS, Savage CO, Segelmark M, Stegeman CA, Tesar V, Vaglio A, Wieczorek S, Wilde B, Zwerina J, Rees AJ, Clayton DG, Smith KG. 2012. Genetically distinct subsets within ANCA-associated vasculitis. N Engl J Med 367: 214–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Martorana D, Maritati F, Malerba G, Bonatti F, Alberici F, Oliva E, Sebastio P, Manenti L, Brugnano R, Catanoso MG, Fraticelli P, Guida G, Gregorini G, Possenti S, Moroni G, Leoni A, Pavone L, Pesci A, Sinico RA, Di Toma L, D’Amico M, Tumiati B, D’Ippolito R, Buzio C, Neri TM, Vaglio A. 2012. PTPN22 R620W polymorphism in the ANCA-associated vasculitides. Rheumatology 51: 805–12 [DOI] [PubMed] [Google Scholar]
- 105.Orozco G, Miranda-Filloy JA, Martin J, Gonzalez-Gay MA. 2007. Lack of association of a functional single nucleotide polymorphism of PTPN22, encoding lymphoid protein phosphatase, with susceptibility to Henoch-Schonlein purpura. Clin Exp Rheum 25: 750–3 [PubMed] [Google Scholar]
- 106.Baranathan V, Stanford MR, Vaughan RW, Kondeatis E, Graham E, Fortune F, Madanat W, Kanawati C, Ghabra M, Murray PI, Wallace GR. 2007. The association of the PTPN22 620W polymorphism with Behcet’s disease. Ann Rheum Dis 66: 1531–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, Bitton A, Dassopoulos T, Datta LW, Green T, Griffiths AM, Kistner EO, Murtha MT, Regueiro MD, Rotter JI, Schumm LP, Steinhart AH, Targan SR, Xavier RJ, Libioulle C, Sandor C, Lathrop M, Belaiche J, Dewit O, Gut I, Heath S, Laukens D, Mni M, Rutgeerts P, Van Gossum A, Zelenika D, Franchimont D, Hugot JP, de Vos M, Vermeire S, Louis E, Cardon LR, Anderson CA, Drummond H, Nimmo E, Ahmad T, Prescott NJ, Onnie CM, Fisher SA, Marchini J, Ghori J, Bumpstead S, Gwilliam R, Tremelling M, Deloukas P, Mansfield J, Jewell D, Satsangi J, Mathew CG, Parkes M, Georges M, Daly MJ. 2008. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet 40: 955–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lie BA, Viken MK, Odegard S, van der Heijde D, Landewe R, Uhlig T, Kvien TK. 2007. Associations between the PTPN22 1858C->T polymorphism and radiographic joint destruction in patients with rheumatoid arthritis: results from a 10-year longitudinal study. Ann Rheum Dis 66: 1604–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Song GG, Bae SC, Kim JH, Lee YH. 2013. The PTPN22 C1858T polymorphism and rheumatoid arthritis: a meta-analysis. Rheum Int Epub 1–31-2013 [DOI] [PubMed] [Google Scholar]
- 110.Morgan AW, Thomson W, Martin SG, Carter AM, Erlich HA, Barton A, Hocking L, Reid DM, Harrison P, Wordsworth P, Steer S, Worthington J, Emery P, Wilson AG, Barrett JH. 2009. Reevaluation of the interaction between HLA-DRB1 shared epitope alleles, PTPN22, and smoking in determining susceptibility to autoantibody-positive and autoantibody-negative rheumatoid arthritis in a large UK Caucasian population. Arthritis Rheum 60: 2565–76 [DOI] [PubMed] [Google Scholar]
- 111.van Nies JA, Knevel R, Daha N, van der Linden MP, Gregersen PK, Kern M, le Cessie S, Houwing-Duistermaat JJ, Huizinga TW, Toes RE, van der Helm-van Mil AH. 2010. The PTPN22 susceptibility risk variant is not associated with the rate of joint destruction in anti-citrullinated protein antibody-positive rheumatoid arthritis. Ann Rheum Dis 69: 1730–1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kallberg H, Padyukov L, Plenge RM, Ronnelid J, Gregersen PK, van der Helm-van Mil AH, Toes RE, Huizinga TW, Klareskog L, Alfredsson L 2007. Gene-gene and gene-environment interactions involving HLA-DRB1, PTPN22, and smoking in two subsets of rheumatoid arthritis. Am J Hum Genet 80: 867–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mahdi H, Fisher BA, Kallberg H, Plant D, Malmstrom V, Ronnelid J, Charles P, Ding B, Alfredsson L, Padyukov L, Symmons DP, Venables PJ, Klareskog L, Lundberg K. 2009. Specific interaction between genotype, smoking and autoimmunity to citrullinated alpha-enolase in the etiology of rheumatoid arthritis. Nat Genet 41: 1319–24 [DOI] [PubMed] [Google Scholar]
- 114.Pierer M, Kaltenhauser S, Arnold S, Wahle M, Baerwald C, Hantzschel H, Wagner U. 2006. Association of PTPN22 1858 single-nucleotide polymorphism with rheumatoid arthritis in a German cohort: higher frequency of the risk allele in male compared to female patients. Arthritis Res Ther 8: R75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chelala C, Duchatelet S, Joffret ML, Bergholdt R, Dubois-Laforgue D, Ghandil P, Pociot F, Caillat-Zucman S, Timsit J, Julier C. 2007. PTPN22 R620W functional variant in type 1 diabetes and autoimmunity related traits. Diabetes 56: 522–6 [DOI] [PubMed] [Google Scholar]
- 116.Petrone A, Suraci C, Capizzi M, Giaccari A, Bosi E, Tiberti C, Cossu E, Pozzilli P, Falorni A, Buzzetti R. 2008. The protein tyrosine phosphatase nonreceptor 22 (PTPN22) is associated with high GAD antibody titer in latent autoimmune diabetes in adults: Non Insulin Requiring Autoimmune Diabetes (NIRAD) Study 3. Diabetes care 31: 534–8 [DOI] [PubMed] [Google Scholar]
- 117.Steck AK, Zhang W, Bugawan TL, Barriga KJ, Blair A, Erlich HA, Eisenbarth GS, Norris JM, Rewers MJ. 2009. Do non-HLA genes influence development of persistent islet autoimmunity and type 1 diabetes in children with high-risk HLA-DR,DQ genotypes? Diabetes 58: 1028–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kordonouri O, Hartmann R, Badenhoop K, Kahles H, Ilonen J. 2010. PTPN22 1858T allele is associated with younger age at onset of type 1 diabetes and is not related to subsequent thyroid autoimmunity. Hum Immunol 71: 731–2 [DOI] [PubMed] [Google Scholar]
- 119.Lempainen J, Hermann R, Veijola R, Simell O, Knip M, Ilonen J. 2012. Effect of the PTPN22 and INS risk genotypes on the progression to clinical type 1 diabetes after the initiation of beta-cell autoimmunity. Diabetes 61: 963–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lamsyah H, Rueda B, Baassi L, Elaouad R, Bottini N, Sadki K, Martin J. 2009. Association of PTPN22 gene functional variants with development of pulmonary tuberculosis in Moroccan population. Tissue Antigens 74: 228–32 [DOI] [PubMed] [Google Scholar]
- 121.Gomez LM, Anaya JM, Martin J. 2005. Genetic influence of PTPN22 R620W polymorphism in tuberculosis. Hum Immunol 66: 1242–7 [DOI] [PubMed] [Google Scholar]
- 122.Kouhpayeh HR, Hashemi M, Hashemi SA, Moazeni-Roodi A, Naderi M, Sharifi-Mood B, Taheri M, Mohammadi M, Ghavami S. 2012. R620W functional polymorphism of protein tyrosine phosphatase non-receptor type 22 is not associated with pulmonary tuberculosis in Zahedan, southeast Iran. Genet Mol Res 11: 1075–81 [DOI] [PubMed] [Google Scholar]
- 123.Boechat AL, Ogusku MM, Sadahiro A, Dos Santos MC. 2013. Association between the PTPN22 1858C/T gene polymorphism and tuberculosis resistance. Infect Genet Evol 16: 310–3 [DOI] [PubMed] [Google Scholar]
- 124.Azarian M, Busson M, Rocha V, Ribaud P, Peffault de Latour R, Bleux H, Lepage V, Charron D, Toubert A, Socie G, Loiseau P. 2008. The PTPN22 R620W polymorphism is associated with severe bacterial infections after human leukocyte antigen geno-identical haematopoietic stem-cell transplantations. Transplantation 85: 1859–62 [DOI] [PubMed] [Google Scholar]
- 125.Chapman SJ, Khor CC, Vannberg FO, Maskell NA, Davies CW, Hedley EL, Segal S, Moore CE, Knox K, Day NP, Gillespie SH, Crook DW, Davies RJ, Hill AV. 2006. PTPN22 and invasive bacterial disease. Nat Genet 38: 499–500 [DOI] [PubMed] [Google Scholar]
- 126.Rani R, Singh A, Israni N, Sharma P, Kar HK. 2009. The role of polymorphic protein tyrosine phosphatase non-receptor type 22 in leprosy. J Invest Dermatol 129: 2726–8 [DOI] [PubMed] [Google Scholar]
- 127.Nahum A, Bates A, Sharfe N, Roifman CM. 2008. Association of the lymphoid protein tyrosine phosphatase, R620W variant, with chronic mucocutaneous candidiasis. J Allergy Clin Immunol 122: 1220–2 [DOI] [PubMed] [Google Scholar]
- 128.Montes-Cano MA, Garcia-Lozano JR, Aguilar-Reina J, Romero-Gomez M, Barroso N, Nunez-Roldan A, Martin J, Gonzalez-Escribano MF. 2008. PTPN22 C1858T polymorphism and the outcome of hepatitis C virus infection. Viral Immunol 21: 491–4 [DOI] [PubMed] [Google Scholar]
- 129.Koppe U, Suttorp N, Opitz B. 2012. Recognition of Streptococcus pneumoniae by the innate immune system. Cell Microbiol 14: 460–6 [DOI] [PubMed] [Google Scholar]
- 130.Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, Oland S, Gordon S, Sher A. 2011. Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity 35: 1023–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Novikov A, Cardone M, Thompson R, Shenderov K, Kirschman KD, Mayer-Barber KD, Myers TG, Rabin RL, Trinchieri G, Sher A, Feng CG. 2011. Mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL-1beta production in human macrophages. J Immunol 187: 2540–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hebbring SJ, Slager SL, Epperla N, Mazza JJ, Ye Z, Zhou Z, Achenbach SJ, Vasco DA, Call TG, Rabe KG, Kay NE, Caporaso NE, Lanasa MC, Camp NJ, Strom SS, Goldin LR, Cerhan JR, Brilliant MH, Schrodi SJ. 2013. Genetic evidence of PTPN22 effects on chronic lymphocytic leukemia. Blood 121: 237–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Efremov DG. 2013. Genetic evidence of PTPN22 effects on chronic lymphocytic leukemia--author’s response to Hebbring. Blood 121: 238–9 [DOI] [PubMed] [Google Scholar]
- 134.Guillem V, Amat P, Cervantes F, Alvarez-Larran A, Cervera J, Maffioli M, Bellosillo B, Collado M, Marugan I, Martinez-Ruiz F, Hernandez-Boluda JC. 2012. Functional polymorphisms in SOCS1 and PTPN22 genes correlate with the response to imatinib treatment in newly diagnosed chronic-phase chronic myeloid leukemia. Leuk Res 36: 174–81 [DOI] [PubMed] [Google Scholar]
- 135.Carlton VE, Hu X, Chokkalingam AP, Schrodi SJ, Brandon R, Alexander HC, Chang M, Catanese JJ, Leong DU, Ardlie KG, Kastner DL, Seldin MF, Criswell LA, Gregersen PK, Beasley E, Thomson G, Amos CI, Begovich AB. 2005. PTPN22 genetic variation: evidence for multiple variants associated with rheumatoid arthritis. Am J Hum Genet 77: 567–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wesoly J, Hu X, Thabet MM, Chang M, Uh H, Allaart CF, Toes RE, Houwing-Duistermaat JJ, Begovich AB, Huizinga TW. 2007. The 620W allele is the PTPN22 genetic variant conferring susceptibility to RA in a Dutch population. Rheumatology 46: 617–21 [DOI] [PubMed] [Google Scholar]
- 137.Hinks A, Eyre S, Barton A, Thomson W, Worthington J. 2007. Investigation of genetic variation across the protein tyrosine phosphatase gene in patients with rheumatoid arthritis in the UK. Ann Rheum Dis 66: 683–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wan Taib WR, Smyth DJ, Merriman ME, Dalbeth N, Gow PJ, Harrison AA, Highton J, Jones PB, Stamp L, Steer S, Todd JA, Merriman TR. 2010. The PTPN22 locus and rheumatoid arthritis: no evidence for an effect on risk independent of Arg620Trp. PLoS One 5: e13544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Onengut-Gumuscu S, Buckner JH, Concannon P. 2006. A haplotype-based analysis of the PTPN22 locus in type 1 diabetes. Diabetes 55: 2883–9 [DOI] [PubMed] [Google Scholar]
- 140.Steck AK, Baschal EE, Jasinski JM, Boehm BO, Bottini N, Concannon P, Julier C, Morahan G, Noble JA, Polychronakos C, She JX, Eisenbarth GS. 2009. rs2476601 T allele (R620W) defines high-risk PTPN22 type I diabetes-associated haplotypes with preliminary evidence for an additional protective haplotype. Genes Immun 10 Suppl 1: S21–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zoledziewska M, Perra C, Orru V, Moi L, Frongia P, Congia M, Bottini N, Cucca F. 2008. Further evidence of a primary, causal association of the PTPN22 620W variant with type 1 diabetes. Diabetes 57: 229–34 [DOI] [PubMed] [Google Scholar]
- 142.Huffmeier U, Reis A, Steffens M, Lascorz J, Bohm B, Lohmann J, Wendler J, Traupe H, Kuster W, Wienker TF, Burkhardt H. 2006. Male restricted genetic association of variant R620W in PTPN22 with psoriatic arthritis. J Invest Dermatol 126: 932–5 [DOI] [PubMed] [Google Scholar]
- 143.Li Y, Liao W, Chang M, Schrodi SJ, Bui N, Catanese JJ, Poon A, Matsunami N, Callis-Duffin KP, Leppert MF, Bowcock AM, Kwok PY, Krueger GG, Begovich AB. 2009. Further genetic evidence for three psoriasis-risk genes: ADAM33, CDKAL1, and PTPN22. J Invest Dermatol 129: 629–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Heward JM, Brand OJ, Barrett JC, Carr-Smith JD, Franklyn JA, Gough SC. 2007. Association of PTPN22 haplotypes with Graves’ disease. J Clin Endocrinol Metab 92: 685–90 [DOI] [PubMed] [Google Scholar]
- 145.Kawasaki E, Awata T, Ikegami H, Kobayashi T, Maruyama T, Nakanishi K, Shimada A, Uga M, Kurihara S, Kawabata Y, Tanaka S, Kanazawa Y, Lee I, Eguchi K. 2006. Systematic search for single nucleotide polymorphisms in a lymphoid tyrosine phosphatase gene (PTPN22): association between a promoter polymorphism and type 1 diabetes in Asian populations. Am J Med Genet A 140: 586–93 [DOI] [PubMed] [Google Scholar]
- 146.Viken MK, Olsson M, Flam ST, Forre O, Kvien TK, Thorsby E, Lie BA. 2007. The PTPN22 promoter polymorphism −1123G>C association cannot be distinguished from the 1858C>T association in a Norwegian rheumatoid arthritis material. Tissue Antigens 70: 190–7 [DOI] [PubMed] [Google Scholar]
- 147.Cinek O, Hradsky O, Ahmedov G, Slavcev A, Kolouskova S, Kulich M, Sumnik Z. 2007. No independent role of the −1123 G>C and+2740 A>G variants in the association of PTPN22 with type 1 diabetes and juvenile idiopathic arthritis in two Caucasian populations. Diabetes Res Clin Pract 76: 297–303 [DOI] [PubMed] [Google Scholar]
- 148.Liu F, Liu J, Zheng TS, Li Q, Wang C, Pan XP, Lu H, Zhao YW. 2012. The −1123G>C variant of PTPN22 gene promoter is associated with latent autoimmune diabetes in adult Chinese Hans. Cell Biochem Biophys 62: 273–9 [DOI] [PubMed] [Google Scholar]
- 149.Ichimura M, Kaku H, Fukutani T, Koga H, Mukai T, Miyake I, Yamada K, Koda Y, Hiromatsu Y. 2008. Associations of protein tyrosine phosphatase nonreceptor 22 (PTPN22) gene polymorphisms with susceptibility to Graves’ disease in a Japanese population. Thyroid 18: 625–30 [DOI] [PubMed] [Google Scholar]
- 150.Taniyama M, Maruyama T, Tozaki T, Nakano Y, Ban Y. 2010. Association of PTPN22 haplotypes with type 1 diabetes in the Japanese population. Hum Immunol 71: 795–8 [DOI] [PubMed] [Google Scholar]
- 151.Chen Z, Zhang H, Xia B, Wang P, Jiang T, Song M, Wu J. 2013. Association of PTPN22 gene (rs2488457) polymorphism with ulcerative colitis and high levels of PTPN22 mRNA in ulcerative colitis. Int J Colorectal Dis Epub 3–3-2013 [DOI] [PubMed] [Google Scholar]
- 152.Gu LQ, Zhu W, Zhao SX, Zhao L, Zhang MJ, Cui B, Song HD, Ning G, Zhao YJ. 2010. Clinical associations of the genetic variants of CTLA-4, Tg, TSHR, PTPN22, PTPN12 and FCRL3 in patients with Graves’ disease. Clin Endocrinol (Oxf) 72: 248–55 [DOI] [PubMed] [Google Scholar]
- 153.Hinks A, Cobb J, Marion MC, Prahalad S, Sudman M, Bowes J, Martin P, Comeau ME, Sajuthi S, Andrews R, Brown M, Chen WM, Concannon P, Deloukas P, Edkins S, Eyre S, Gaffney PM, Guthery SL, Guthridge JM, Hunt SE, James JA, Keddache M, Moser KL, Nigrovic PA, Onengut-Gumuscu S, Onslow ML, Rose CD, Rich SS, Steel KJ, Wakeland EK, Wallace CA, Wedderburn LR, Woo P, Bohnsack JF, Haas JP, Glass DN, Langefeld CD, Thomson W, Thompson SD. 2013. Dense genotyping of immune-related disease regions identifies 14 new susceptibility loci for juvenile idiopathic arthritis. Nat Genet 45: 664–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Skinningsrud B, Husebye ES, Gervin K, Lovas K, Blomhoff A, Wolff AB, Kemp EH, Egeland T, Undlien DE. 2008. Mutation screening of PTPN22: association of the 1858T-allele with Addison’s disease. Eur J Hum Genet 16: 977–82 [DOI] [PubMed] [Google Scholar]
- 155.Orru V, Tsai SJ, Rueda B, Fiorillo E, Stanford SM, Dasgupta J, Hartiala J, Zhao L, Ortego-Centeno N, D’Alfonso S, Arnett FC, Wu H, Gonzalez-Gay MA, Tsao BP, Pons-Estel B, Alarcon-Riquelme ME, He Y, Zhang ZY, Allayee H, Chen XS, Martin J, Bottini N. 2009. A loss-of-function variant of PTPN22 is associated with reduced risk of systemic lupus erythematosus. Hum Mol Genet 18: 569–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Rodriguez-Rodriguez L, Taib WR, Topless R, Steer S, Gonzalez-Escribano MF, Balsa A, Pascual-Salcedo D, Gonzalez-Gay MA, Raya E, Fernandez-Gutierrez B, Gonzalez-Alvaro I, Bottini N, Witte T, Viken MK, Coenen MJ, van Riel PL, Franke B, den Heijer M, Radstake TR, Wordsworth P, Lie BA, Merriman TR, Martin J. 2011. The PTPN22 R263Q polymorphism is a risk factor for rheumatoid arthritis in Caucasian case-control samples. Arthritis Rheum 63: 365–72 [DOI] [PubMed] [Google Scholar]
- 157.Diaz-Gallo LM, Gourh P, Broen J, Simeon C, Fonollosa V, Ortego-Centeno N, Agarwal S, Vonk MC, Coenen M, Riemekasten G, Hunzelmann N, Hesselstrand R, Tan FK, Reveille JD, Assassi S, Garcia-Hernandez FJ, Carreira P, Camps MT, Fernandez-Nebro A, de la Pena PG, Nearney T, Hilda D, Gonzalez-Gay MA, Airo P, Beretta L, Scorza R, Herrick A, Worthington J, Pros A, Gomez-Gracia I, Trapiella L, Espinosa G, Castellvi I, Witte T, de Keyser F, Vanthuyne M, Mayes MD, Radstake TR, Arnett FC, Martin J, Rueda B. 2011. Analysis of the influence of PTPN22 gene polymorphisms in systemic sclerosis. Ann Rheum Dis 70: 454–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Liu J, Chen M, Li R, Yang F, Shi X, Zhu L, Wang HM, Yao W, Liu Q, Meng FG, Sun JP, Pang Q, Yu X. 2012. Biochemical and functional studies of lymphoid-specific tyrosine phosphatase (Lyp) variants S201F and R266W. PLoS One 7: e43631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, Zhang CK, Boucher G, Ripke S, Ellinghaus D, Burtt N, Fennell T, Kirby A, Latiano A, Goyette P, Green T, Halfvarson J, Haritunians T, Korn JM, Kuruvilla F, Lagace C, Neale B, Lo KS, Schumm P, Torkvist L, Dubinsky MC, Brant SR, Silverberg MS, Duerr RH, Altshuler D, Gabriel S, Lettre G, Franke A, D’Amato M, McGovern DP, Cho JH, Rioux JD, Xavier RJ, Daly MJ. 2011. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet 43: 1066–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Aarnisalo J, Treszl A, Svec P, Marttila J, Oling V, Simell O, Knip M, Korner A, Madacsy L, Vasarhelyi B, Ilonen J, Hermann R. 2008. Reduced CD4(+)T cell activation in children with type 1 diabetes carrying the PTPN22/Lyp 620Trp variant. J Autoimmun 31: 13–21 [DOI] [PubMed] [Google Scholar]
- 161.Cao Y, Yang J, Colby K, Hogan SL, Hu Y, Jennette CE, Berg EA, Zhang Y, Jennette JC, Falk RJ, Preston GA. 2012. High basal activity of the PTPN22 gain-of-function variant blunts leukocyte responsiveness negatively affecting IL-10 production in ANCA vasculitis. PLoS One 7: e42783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Vang T, Landskron J, Viken MK, Oberprieler N, Torgersen KM, Mustelin T, Tasken K, Tautz L, Rickert RC, Lie BA. 2013. The autoimmune-predisposing variant of lymphoid tyrosine phosphatase favors T helper 1 responses. Hum Immunol 74: 574–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Davidson D, Bakinowski M, Thomas ML, Horejsi V, Veillette A. 2003. Phosphorylation-dependent regulation of T-cell activation by PAG/Cbp, a lipid raft-associated transmembrane adaptor. Mol Cell Biol 23: 2017–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Meffre E 2011. The establishment of early B cell tolerance in humans: lessons from primary immunodeficiency diseases. Ann N Y Acad Sci 1246: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Manjarrez-Orduno N, Marasco E, Chung SA, Katz MS, Kiridly JF, Simpfendorfer KR, Freudenberg J, Ballard DH, Nashi E, Hopkins TJ, Cunninghame Graham DS, Lee AT, Coenen MJ, Franke B, Swinkels DW, Graham RR, Kimberly RP, Gaffney PM, Vyse TJ, Behrens TW, Criswell LA, Diamond B, Gregersen PK. 2012. CSK regulatory polymorphism is associated with systemic lupus erythematosus and influences B-cell signaling and activation. Nat Genet 44: 1227–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Duty JA, Szodoray P, Zheng NY, Koelsch KA, Zhang Q, Swiatkowski M, Mathias M, Garman L, Helms C, Nakken B, Smith K, Farris AD, Wilson PC. 2009. Functional anergy in a subpopulation of naive B cells from healthy humans that express autoreactive immunoglobulin receptors. J Exp Med 206: 139–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, Marrack P. 2011. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c(+) B-cell population is important for the development of autoimmunity. Blood 118: 1305–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Menard L, Samuels J, Ng YS, Meffre E. 2011. Inflammation-independent defective early B cell tolerance checkpoints in rheumatoid arthritis. Arthritis Rheum 63: 1237–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yurasov S, Wardemann H, Hammersen J, Tsuiji M, Meffre E, Pascual V, Nussenzweig MC. 2005. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med 201: 703–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Gregersen PK, Olsson LM. 2009. Recent advances in the genetics of autoimmune disease. Annu Rev Immunol 27: 363–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Yarilina A, DiCarlo E, Ivashkiv LB. 2007. Suppression of the effector phase of inflammatory arthritis by double-stranded RNA is mediated by type I IFNs. J Immunol 178: 2204–11 [DOI] [PubMed] [Google Scholar]
- 172.Gonzalez-Navajas JM, Lee J, David M, Raz E. 2012. Immunomodulatory functions of type I interferons. Nat Rev Immunol 12: 125–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Mayer CT, Berod L, Sparwasser T. 2012. Layers of dendritic cell-mediated T cell tolerance, their regulation and the prevention of autoimmunity. Front Immunol 3: 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Imboden JB. 2009. The immunopathogenesis of rheumatoid arthritis. Annu Rev Pathol 4: 417–34 [DOI] [PubMed] [Google Scholar]
- 175.Boettler T, von Herrath M. 2011. Protection against or triggering of Type 1 diabetes? Different roles for viral infections. Expert Rev Clin Immunol 7: 45–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Swiecki M, Colonna M. 2011. Type I interferons: diversity of sources, production pathways and effects on immune responses. Curr Opin Virol 1: 463–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Choi J, Kim ST, Craft J. 2012. The pathogenesis of systemic lupus erythematosus-an update. Curr Opin Immunol 24: 651–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Niewold TB. 2011. Interferon alpha as a primary pathogenic factor in human lupus. J Interferon Cytokine Res 31: 887–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Postal M, Appenzeller S. 2011. The role of Tumor Necrosis Factor-alpha (TNF-alpha) in the pathogenesis of systemic lupus erythematosus. Cytokine 56: 537–43 [DOI] [PubMed] [Google Scholar]
- 180.Ostendorf B, Iking-Konert C, Kurz K, Jung G, Sander O, Schneider M. 2005. Preliminary results of safety and efficacy of the interleukin 1 receptor antagonist anakinra in patients with severe lupus arthritis. Ann Rheum Dis 64: 630–3 [DOI] [PMC free article] [PubMed] [Google Scholar]