

Abstract
Cardiac fibrosis is the most important mechanism contributing to cardiac remodeling after myocardial infarction (MI). VPO1 is a heme enzyme that uses hydrogen peroxide (H2O2) to produce hypochlorous acid (HOCl). Our previous study has demonstrated that VPO1 regulates myocardial ischemic reperfusion and renal fibrosis. We investigated the role of VPO1 in cardiac fibrosis after MI. The results showed that VPO1 expression was robustly upregulated in the failing human heart with ischemic cardiomyopathy and in a murine model of MI accompanied by severe cardiac fibrosis. Most importantly, knockdown of VPO1 by tail vein injection of VPO1 siRNA significantly reduced cardiac fibrosis and improved cardiac function and survival rate. In VPO1 knockdown mouse model and cardiac fibroblasts cultured with TGF-β1, VPO1 contributes to cardiac fibroblasts differentiation, migration, collagen I synthesis and proliferation. Mechanistically, the fibrotic effects following MI of VPO1 manifested partially through HOCl formation to activate Smad2/3 and ERK1/2. Thus, we conclude that VPO1 is a crucial regulator of cardiac fibrosis after MI by mediating HOCl/Smad2/3 and ERK1/2 signaling pathways, implying a promising therapeutic target in ischemic cardiomyopathy.
Keywords: Cardiac fibrosis, Myocardial infarction, Vascular peroxidase1, Cardiac remodeling, Cardiac fibroblasts
Graphical abstract
During periods of cardiac stress, VPO1 is activated by elevated levels of TGF-β1 and promotes cardiac fibroblasts differentiation, migration, proliferation and collagen I synthesis by regulating Smad2/3 and ERK1/2 pathways, ultimately leading to cardiac fibrosis and dysfunction.
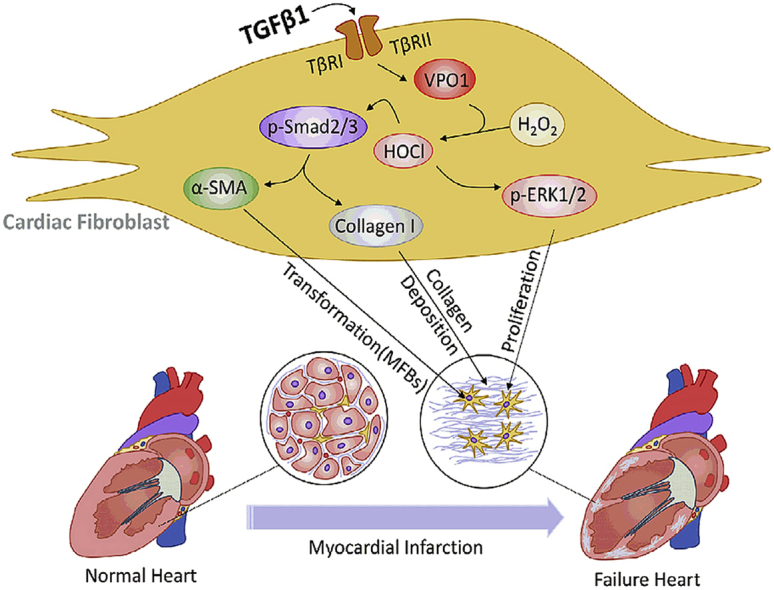
Highlights
-
•
VPO1 is increased in failing human hearts of ischemic cardiomyopathy (ICM) patients, murine model of myocardial infarction (MI), and cardiac fibroblasts cultured with transforming growth factor-β1 (TGF-β1).
-
•
VPO1 regulates cardiac fibrosis and cardiac function during cardiac remodeling after MI.
-
•
VPO1 activates HOCl/Smad2/3 and HOCl/ERK1/2 signaling pathways to regulate cardiac fibroblasts differentiation, collagen I synthesis and proliferation.
Abbreviations
- MI
Myocardial Infarction
- VPO1
Vascular peroxidase 1
- MPO
Myeloperoxidase
- siRNA
Small interference RNA
- TGF-β1
Transforming growth factor-β1
- HOCl
Hypochlorous acid
- H2O2
Hydrogen peroxide
- ICM
Ischemic cardiomyopathy
- EF
Ejection fraction
- FS
Fractional shortening
- LVIDd
Left ventricular internal dimension at end-diastole
- LVIDs
Left ventricular internal dimension at systole
- Smad2/3
Mothers against decapentaplegic homolog 2/3
- ERK1/2
Extracellular regulated protein kinases 1/2
- α-SMA
Alpha smooth muscle actin
- ECM
Extracellular matrix
- PXDN
Peroxidasin
1. Introduction
Myocardial infarction (MI) is characterized by high rates of morbidity and mortality [1]. A large number of patients experience the adverse cardiac remodeling process following MI [2]. Cardiac fibrosis is an important pathological process contributing to the pathogenesis of cardiac remodeling after MI, which is a transition from an early inflammatory phase to fibrotic granulation and maturation stage of cardiac remodeling [3].
Myeloperoxidase (MPO), released by activated neutrophils and monocytes, is a well-studied member of the peroxidase family [4]. MPO deficient (MPO−/-) mice have improved cardiac function and decreased risk of ventricular arrhythmias as well as ventricular fibrosis following MI [5]. The effects of MPO are attributed to its regulation of cardiac fibroblasts differentiation. It has been previously reported that MPO plasma levels are increased 3–5 days after MI before subsequently declining [5]. Therefore, although MPO derived from neutrophils and monocytes has been verified to impair normal cardiac function after MI, it primarily participates in the inflammatory phase of cardiac fibrosis and may have a minimal effect on fibrotic granulation and the maturation of cardiac fibrosis.
Peroxidasin (PXDN), a newly identified heme-containing peroxidase, has been renamed vascular peroxidase1 (VPO1) as it is primarily expressed in the cardiovascular system [6]. Recent reports suggest that VPO1 plays a critical signaling role in mediating the development and progression of cardiovascular disease [[7], [8], [9]]. Specifically, VPO1 promotes extracellular matrix (ECM) formation in fibrotic kidney and collagen IV crosslinking by catalyzing the formation of sulfilimine chemical bonds [10,11]. We previously reported that VPO1 aggravates myocardial injury in an ischemic reperfusion mouse model by activating the JNK/p38 MAPK pathway [12]. Based on previous study that VPO1 is related to fibrosis and myocardial ischemia, we hypothesized that VPO1 may regulate the formation of granulation and maturation stages of cardiac fibrosis after MI.
Here, we investigated for the first time, the functional effects of VPO1 on cardiac fibrosis after MI. Using a combination of in vivo and in vitro studies, we studied the mechanistic role of VPO1 in cardiac fibroblasts differentiation, migration, collagen I synthesis and proliferation. Moreover, our findings suggest that inhibition of VPO1 in vivo may represent a potential therapeutic strategy against cardiac fibrosis after MI.
2. Methods
Refer to the supplement methods for supplementary material.
2.1. Human studies
This study of human heart specimens followed the ethical guidelines of Central South University. Failing hearts were obtained from five patients with ischemic cardiomyopathy (ICM) who underwent heart transplantation. Normal hearts were obtained from five healthy donors who were declared brain dead.
2.2. Animal studies and mouse model of MI
All animal experiments were approved by the institutional Animal Care and Use Committee regulations at Central South University. Male C57BL/6J wild-type (WT) mice ranging from 6 to 8 weeks in age were used for this study. MI was induced in mice as previously described [13]. Briefly, mice were anesthetized by intraperitoneal injection of pentobarbital sodium (60 mg/kg). Then, MI was induced by permanent ligation of the left coronary artery with a 8-0 suture, while the sham mice underwent the same procedure without ligation. Subsequently, mice were sacrificed at 7 days, 14 days or 28 days after MI.
2.3. Histology and immunochemistry
Human and mouse heart tissues were fixed overnight in 4% paraformaldehyde. Samples were embedded in paraffin wax and sliced into 4 μm sections. Masson trichrome stain (Sigma-Aldrich, USA) was used according to the manufacture's protocol. Infarct size, expressed as a percentage, was calculated by dividing the sum of infarct areas from all sections by the sum of LV areas from all sections (including those without infarct scar) and multiplying by 100 [14]. Infarct expansion index was calculated using the following formula: expansion index = (LV cavity area/total heart area) × (uninfarcted septal thickness/infarcted LV free wall thickness) [15]. Rabbit anti-VPO1 (1:200; ABS1675, Millipore, USA), anti-3-chlorotyrosine (1:100; HP5002, Hycult Biotech, Netherlands), anti-collagen I (1:200; ab3471, Abcam, UK), anti-ki67 (1:200, ab16667, Abcam, UK), and mouse anti-α-SMA antibody (1:200; ab7817, Abcam, UK) were used as primary antibodies for immunochemical staining, followed by secondary antibodies (1:100; ZSGB-BIO, China) and DAB reagents. Quantification of fibrosis, VPO1, α-SMA and collagen I in heart tissues was determined by Imag-Pro-Plus 6.0 software.
2.4. Immunofluorescence staining
Frozen heart sections (8 μm) were incubated with primary antibody against α–SMA (1:200, Abcam, UK) and VPO1 (1:200; Millipore, USA) overnight at 4 °C. Subsequently, secondary antibodies were incubated at room temperature for an hour. Nuclei were stained with DAPI (1:1000; Sigma-Aldrich, USA).
2.5. Isolation and culture of primary mice cardiac fibroblasts
Cardiac fibroblasts were isolated from one to three-day-old C57BL/6 mice as described previously [13]. Left ventricles were finely cut into pieces and digested with trypsin/EDTA (Gibco, USA) and collagenase II (Sigma-Aldrich, USA) at 37 °C. Cells suspensions were centrifuged, re-suspended and plated for 2 h. Adherent cells were cardiac fibroblasts that positively expressed vimentin. Cardiac fibroblasts were cultured with a supplemented DMEM solution, containing 15% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin and incubated at 37 °C in humidified chamber. Passages 1–3 were used for our experiments.
2.6. Western blot
Lysates of cultured cardiac fibroblasts and tissues lysates of infarcted hearts were extracted using a lysis buffer (Beyotime, Shanghai, China) with a protease inhibitor cocktail (Roche, Germany). Protein blotting was performed with a modified standard protocol using primary antibodies to VPO1 (1:1000, ABS1675, Millipore, USA), α-SMA (1:1000; ab7817, Abcam, UK), collagen I (1:1000; ab3471, Abcam, UK), smad2/3 (#8685), P-smad2/3 (#9510), ERK1/2 (#4695), P-ERK1/2 (#4370) (1:1000; Cell Signaling Technology, USA), 3-chlorotyrosine (1:500; HP5002, Hycult Biotech, Netherlands), GAPDH (1:5000, G8795, Sigma-Aldrich, USA) overnight at 4 °C. PVDF membranes were incubated with HRP--conjugated secondary antibodies and bands were visualized by a gel documentation system (Bio-Rad, USA).
2.7. Statistical analysis
Data were presented as the mean ± SEM. N indicates the number of independent experiments performed at different cell passages, the number of mice, or human patients examined. For statistical comparisons, we evaluated whether the data were normally distributed using Kolmogorov-Smirnov test. T-test or ANOVA test was used in data with normal distribution; otherwise, non-parametric test was used. Statistical significance of differences between groups was measured using unpaired Student's t-test. Differences among ≥3 groups were compared using one-way ANOVA with Tukey Kramer test. Kaplan-Meier survival curves were analyzed using the log-rank test. Statistical analysis was performed using Graph Prism 6 or SPSS version. 22.
3. Results
3.1. VPO1 expression is upregulated in fibrotic human heart
To investigate a potential role of VPO1 in cardiac fibrosis, VPO1 protein expression was compared between normal hearts and failing hearts. The clinical information and echocardiography parameters for ICM patients and healthy donors are presented in Supplementary Table 1. Immunochemistry indicated that VPO1 was significantly increased in failing human hearts compared to the normal hearts (Fig. 1A left panel). However, MPO, another important member of the peroxidase family, was not significant elevated in failing human hearts (Fig. 1A middle panel). 3-chlorotyrosine (3-Cl Tyr) is recognized as a specific marker of VPO1 and MPO-generated HOCl that has reacted with tyrosine residues [16]. Our results showed that the failing human hearts exhibited increased 3-Cl Tyr levels consistent with the elevated levels of VPO1 expression (Fig. 1A right panel).
Fig. 1.

VPO1 expression is upregulated in fibrotic human heart. (A) Representative images of immunochemical staining and quantification of VPO1, MPO, 3-Cl Tyr in human heart tissues obtained from control subjects and patients with ischemic cardiomyopathy (ICM).Scale bar: 20 μm (B) Representative images of immunochemical staining and quantification of fibrotic area, Collagen I and α-SMA in human heart tissues obtained from control subjects and patients with ICM. Scale bar: 20 μm. (C) Immunofluorescence images labeling VPO1 (red), α-SMA (green) and DAPI (blue) in the left ventricular section of normal and failing hearts with ICM. Scale bar: 50 μm (D) Immunofluorescence images labeling VPO1 (red), Collagen I (green) and DAPI (blue) in the left ventricular section of normal and failing hearts with ICM. Scale bar: 50 μm. In A and B, Quantitative analysis of protein levels indicated by positive staining area/total area was determined by image pro plus. Data are presented as mean ± SEM. n = 5, *P < 0.05, **P < 0.01. NS: no significance. (Student's t-test). The difference of fibrotic area was measured by using unpaired test. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
In failing hearts, up-regulation of VPO1 and 3-Cl Tyr expression was accompanied by severe fibrosis. Of note, the left ventricle of failing human hearts tissues had an abundance of collagen evident by the heavy Masson trichrome staining (Fig. 1B left panel). Collagen I accounts for approximately 80% of total collagen in the myocardial interstitium and is crucial to maintain structure and integrity of heart [17]. In our study, the expression of collagen I in the failing heart was significantly upregulated compared with the normal heart (Fig. 1B middle panel). α-SMA, a marker of differentiation of fibroblast into myofibroblast, was also elevated in the failing human heart tissues (Fig. 1B right panel). Furthermore, α-SMA was co-stained with vimentin as shown in Fig. S1, which suggested that α-SMA was expressed in vimentin-positive cardiac fibroblasts. Additionally, immunofluorescence results revealed that elevated expression of VPO1 and α-SMA or Collagen I were co-localized in fibrotic areas of the failing heart (Fig. 1C&D). Collectively, these results indicate a strong correlation between VPO1 expression and cardiac fibrosis in the failing human heart with ICM.
3.2. VPO1 expression is upregulated during cardiac fibrosis after MI
To characterize the potential mechanistic link between VPO1 and cardiac fibrosis following cardiac injury in vivo, a murine model of MI was applied. Masson trichrome staining showed a significant increase in fibrosis with the extended period of MI (Fig. 2A). Consistent with our results in the failing human heart tissues, immunofluorescence staining revealed upregulation of VPO1 expression and co-localization of VPO1 and α-SMA in the fibrotic area of mice with MI (Fig. 2A). As evident by the western blot, VPO1 expression was gradually increased in parallel with the up-regulation of α-SMA and collagen I at 14 days and 28 days after MI as shown by western blot (Fig. 2B). To identify the specific cell type that expressed VPO1, we performed immunofluorescence staining in the fibrotic murine heart. VPO1 expressed in vimentin-positive fibroblasts was higher than that in CD31-positive endothelial cells, cTnI-positive cardiomyocytes, and α-SMA-positive vascular smooth muscle cells in the fibrotic area of infarcted heart after 28 days of ischemia (Fig. S2). Interestingly, MPO expression was increased > 2-fold after 7 days of MI compared with the sham group, but 28 days after MI was similar to the sham mice as measured by western blot and immunofluorescence staining (Fig. 2A&B). Consistent with the increase in VPO1 expression, the 3-Cl Tyr expression level was significantly increased after 7 days and 14 days of MI, and slightly decreased but remained significantly elevated after 28 days of MI compared with the sham group (Fig. 2C, upper). These findings suggest that VPO1 may play a more central role in the fibrotic phase of cardiac fibrosis after MI than MPO.
Fig. 2.

VPO1 expression is upregulated during cardiac fibrosis after myocardial infarction (MI). (A) Representative images of masson trichrome staining and immunofluorescence staining of MPO (red), VPO1(red), α-SMA (green) and DAPI (blue) in the infarct zone of mouse hearts in sham group and 7 days, 14 days, 28days after MI. Scale bar, 50 μm. (B) Levels of VPO1, MPO, α-SMA and Collagen I in the sham and MI mouse hearts as determined by western blot and quantified by Image Lab software. n = 6. (C) Immunochemical staining of Ki67 (a proliferation marker) and 3-Cl Tyr in sham and post MI at 7 days, 14 days, 28 days. Quantification of Ki67 positive cells and 3-Cl Tyr staining level in sham and subjected MI mouse. n = 6. Scale bar: 50 μm (upper), 20 μm (bottom). Data are presented as mean ± SEM. In B, Student's t-test in comparison with Sham group. *P < 0.05 vs Sham, **P < 0.01 vs Sham, NS: no significance vs Sham. In C, One-way ANOVA test was used. *P < 0.05, **P < 0.01. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Proliferation of cardiac fibroblasts is another vital mechanism of cardiac fibrosis [18]. In this study, we found that ki67 (a marker of proliferation) positive cells were gradually increased in the fibrotic area following MI (Fig. 2C, bottom). Taken together, these results indicate that VPO1 mediates the development of cardiac fibrosis by regulating cardiac fibroblasts differentiation, collagen I synthesis and proliferation.
3.3. Knockdown of VPO1 attenuates cardiac fibrosis and improves cardiac function in vivo
To further illuminate the important role of VPO1 in cardiac fibrosis and function, VPO1 small interfering RNA (si-VPO1) was used to achieve inhibition of VPO1 expression in mice prior to MI. Mice received a tail vein injection of either si-VPO1 or negative control small interfering RNA (si-NC). After initial small interfering RNA (siRNA) injection three times a week, mice underwent permanent left artery ligation, and subsequently injected with additional siRNA three times per week until sacrificed at 28 days after experimental MI (Fig. 3A). The efficiency of VPO1 knockdown was assessed by western blot. We found that VPO1 expression was significantly inhibited by si-VPO1 injection (Fig. 3B). Moreover, we detected VPO1 expression in different tissues. VPO1 was highly expressed in heart and lung, and significantly inhibited by injection with si-VPO1 after 7 days, 14 days and 28 days (Fig. S3). Mice treated with si-VPO1 had an increased survival proportion (14/19, 73.6%) compared to the si-NC treated mice (13/24, 54.2%) (Fig. 3C). To evaluate the critical role of VPO1 in cardiac function, echocardiography was performed. Depletion of VPO1 significantly improved cardiac function compared to si-NC-injected mice 28 days following MI. As shown in Fig. 3D–H, left ventricular ejection fraction (EF), fraction shortening (FS) were upregulated, while left ventricular internal dimension at end-diastole (LVIDd) and left ventricular internal dimension at systole (LVIDs) were downregulated compared to the si-NC-injected mice after 28 days of MI (Supplementary Table 2). While no difference in cardiac function between si-NC sham and si-VPO1 sham group was observed (Fig. 3D–H).
Fig. 3.

Knockdown of VPO1 attenuates cardiac fibrosis and improves cardiac function in vivo. (A) Experimental procedure for injecting mice with siRNA. (B) The efficiency of VPO1 knockdown by siRNA was assessed by western blot and quantified with Image Lab software. n = 6 per group. (C) Kaplan-Meier survival curves of mice injected with si-NC and si-VPO1 28days after MI. (D) Representative M-mode images of sham and MI mice injected with si-NC and si-VPO1. (E–H) Ejection traction (EF), fractional shortening (FS), left ventricular internal dimension at end-diastole (LVIDd) and left ventricular internal dimension at systole (LVIDs) were analyzed by echocardiography. n = 7 in sham group, n = 8 in MI group. (I) Representative images of Masson trichrome at various magnifications. n = 6. Upper scale bar:1 mm, bottom scale bar: 100 μm. Quantification of the total fibrotic area using Image Pro Plus. (J) Quantification of infarct area size (infarct area/total area) of left ventricle. (K) Quantification of infarct expansion index ((LV cavity area/total heart area) ✖ (uninfarcted septal thickness/infarcted LV free wall thickness)). n = 6. Data are presented as mean ± SEM. In B, Student's t-test in comparison with si-NC group. In C, the difference between the two groups was tested by the Log-Rank test. In E-H, One-way ANOVA test was used to detect the difference. In I, J and K, Student's t-test in comparison with si-NC MI group. *P < 0.05, **P < 0.01, ***P < 0.001.
Although infarct area of left ventricular was not different between the si-NC MI group and the si-VPO1 MI group (Fig. 3J), the si-VPO1 MI group showed less overall collagen deposition than the si-NC MI group, as demonstrated by Masson trichrome staining (Fig. 3I). As shown in Fig. 3K, we found that the infarct expansion index was significantly reduced with injection of si-VPO1 compared with the si-NC MI group. These findings suggest that knockdown of VPO1 ameliorates cardiac fibrosis and cardiac dysfunction induced by MI and thus improves the survival rate post-MI.
3.4. VPO1 induces cardiac fibroblasts differentiation and proliferation in vivo
The differentiation of fibroblasts into myofibroblasts is a critical process in the development and progression of cardiac fibrosis [19]. To determine whether VPO1 is necessary for cardiac fibroblasts differentiation, markers of differentiation were analyzed in mice with VPO1 knockdown after MI. VPO1 and α-SMA expression of heart tissues was upregulated in si-NC treated mice 28 days after MI compared to the sham group. However, the induction of VPO1 and α-SMA by MI was blocked by treatment with si-VPO1 (Fig. 4A). Myofibroblasts are the major source of collagen during pathological matrix remodeling [20]. As expected, we observed that collagen I and matrix metalloproteinase 9 (MMP9) were elevated after 28 days after MI. And si-VPO1 succeeded to inhibit the collagen I synthesis and MMP9 expression (Fig. 4A, Fig. S4A).
Fig. 4.

VPO1 induces cardiac fibroblasts differentiation and proliferation in vivo. (A) VPO1, α-SMA and Collagen I protein levels in heart tissues of mice injected with siRNA were assessed by western blot and quantified by Image Lab. n = 6. (B). Representative images of immunostaining of Ki67 staining in mouse hearts subjected to MI and injected with si-NC and si-VPO1. Quantification of Ki67 positive cells in each group. n = 6. Data are presented as mean ± SEM. One-way ANOVA test was used for A-B. *P < 0.05, **P < 0.01.
Proliferation of cardiac fibroblasts is required for the pathogenesis of cardiac fibrosis. A significant increase in ki67 positive cells was observed in si-NC mice hearts following MI. The induction of ki67 positive cells by MI was attenuated in mice treated with si-VPO1 (Fig. 4B). Collectively, our results suggest that VPO1 may play an important role in the differentiation and proliferation of cardiac fibroblast in vivo.
3.5. VPO1 regulates cardiac fibroblasts differentiation, migration and collagen I synthesis
Cardiac fibroblasts were positive for vimentin and harvested from one to three-day-old C57BL/6 mice (Fig. S5A). Numerous studies suggest that transforming growth factor-β1 (TGF-β1) is the most important cytokine for cardiac fibroblast differentiation [21,22]. In this study, by immunochemistry and western blot, TGF-β1 levels were increased in MI-induced mice and remained significantly elevated at 28 days after MI (Figs. S6A–C). Therefore, in the present study, TGF-β1 was used to study the role of VPO1 in fibrosis in vitro. TGF-β1 up-regulated VPO1 protein expression in both a concentration- and time-dependent manner, which peaked at 24 h and 10 ng/ml of stimulation (Figs. S7A–D). Additionally, we observed a similar induction in α-SMA and collagen I expression.
We next sought to determine whether VPO1 is a key regulator of the fibrotic response of cardiac fibroblasts in response to TGF-β1. Cultured primary cardiac fibroblasts were transfected with si-VPO1 and the efficiency of knockdown was confirmed by western blot (Fig. 5A). We examined the effects of VPO1 knockdown on cardiac fibroblasts differentiation, migration, collagen I synthesis. Western blot and immunofluorescence staining revealed that TGF-β1 induced α-SMA expression in cultured cardiac fibroblasts and this effect was attenuated by VPO1 depletion (Fig. 5A&B). Myofibroblasts are the primary source of collagen during pathological fibrotic response. Our data revealed that VPO1 knockdown not only inhibited cardiac fibroblasts differentiation, but also reduced collagen I synthesis. Next, we assessed cell migration using a wound healing assay. Cardiac fibroblasts transfected with si-NC migrated into the wound upon TGF--β1 stimulation whereas cells with VPO1 depletion migrated significantly less (Fig. 5C). Thus, together these data suggested that VPO1 contributes to cardiac fibroblasts differentiation, migration and collagen I synthesis.
Fig. 5.

VPO1 regulates cardiac fibroblasts differentiation, migration and collagen I synthesis. (A) Protein levels of VPO1, α-SMA and Collagen I in cultured cardiac fibroblasts transfected with si-NC or si-VPO1 were measured by western blot and quantified by immunoblots. n = 4. (B) Immunofluorescence staining and quantification of VPO1 (red), α-SMA (green) and DAPI (blue) in cultured cardiac fibroblasts transfected with si-NC, si-VPO1 treated with or without TGF-β1 for 24 h. Scale bar: 50 μm n = 4. (C) Representative images of scratch assay of cardiac fibroblasts transfected with si-NC or si-VPO1 and treated with or without TGF-β1 for 24 h and statistical analysis of the migratory distance (μm) after 24 h. n = 4. Scale bar:100 μm. (D–E) Western blot and quantification of the phosphorylation of Smad2/3 in MI mice injected with si-NC and si-VPO1(D), or (E) cultured cardiac fibroblasts transfected with siRNA and incubated with or without TGF-β1. n = 6. Data are presented as mean ± SEM. One-way ANOVA test was used for A-E. *P < 0.05, **P < 0.01. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Several studies suggested that activation of the TGF-β1/Smad2/3 pathway is essential for cardiac fibroblasts differentiation, migration, wound contraction and deposition of extracellular matrix proteins during the formation of pathological cardiac fibrosis [[23], [24], [25]]. Therefore, we investigated the effects of VPO1 on the TGF-β1/Smad2/3 pathway in our murine MI model. Western blot revealed that the phosphorylation levels of Smad2/3 was increased significantly in the hearts of si-NC group with MI and that VPO1 knockdown blocked this increase in phosphorylation (Fig. 5D). In vitro experiments further support the finding that VPO1 regulates phosphorylation of Smad2/3. As shown Fig. 5E, si-VPO1 alleviated TGF-β1-induced phosphorylation of Smad2/3 in cultured cardiac fibroblasts.
3.6. VPO1 mediates cardiac fibroblasts proliferation
BrdU and CCK-8 were used to measure cardiac fibroblasts proliferation in vitro. Consistent with our findings in vivo, TGF-β1 stimulated cardiac fibroblasts proliferation and differentiation. Fibroblasts proliferation was significantly inhibited by si-VPO1. (Fig. 6A–B).
Fig. 6.

VPO1 mediates cardiac fibroblasts proliferation. (A) Representative immunofluorescence images of BrdU (red), α-SMA (green) and DAPI (blue) staining in cultured cardiac fibroblasts transfected with siRNA and treated with or without TGF-β1. Scale bar: 50 μm. Quantification of BrdU positive and α-SMA staining cells. n = 4. (B) CCK-8 proliferation assay of primary cardiac fibroblasts transfected with siRNA and exposed to TGF-β1for 24 h n = 7. (C–E) Western blot and quantification of Smad2/3 phosphorylation in MI mice injected with si-NC and si-VPO1 (C), or cultured cardiac fibroblasts transfected with siRNA and incubated with or without TGF-β1 (D). n = 6. Data are presented as mean ± SEM. In A-D, one-way ANOVA test was used. *P < 0.05, **P < 0.01, ***P < 0.001. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
The TGF-β1/Smad2/3 pathway is believed to have an anti-proliferative effect on cardiac fibroblasts [26]. Previous studies have reported that ERK1/2 plays an important signaling role in mediating cardiac fibroblasts proliferation [25,27]. We have previous reported that HOCl promotes activation of ERK1/2 in various pathological process [7,8]. In this study, ERK1/2 was activated following MI in the si-NC MI group and phosphorylation was blocked in mice of VPO1 knockdown (Fig. 6C). Consistent with our findings in vivo, cardiac fibroblasts that transfected with si-VPO1 and incubated with TGF-β1 exhibited reduced activation of ERK1/2 compared with si-NC (Fig. 6D).
3.7. VPO1-derived HOCl activates Smad2/3 to regulate cardiac fibroblasts differentiation, and collagen I synthesis and ERK1/2 to regulate cardiac fibroblasts proliferation
We previously reported that VPO1 is predominantly expressed in cardiovascular tissues and is the main peroxidase responsible for HOCl production in the cardiovascular system [6]. In this study, we assessed the effect of VPO1 knockdown expression on HOCl production in heart tissues. As shown Fig. 7A, western blot revealed that 3-Cl Tyr levels were increased in the si-NC with MI group compared to the si-VPO1 group. In cultured cardiac fibroblasts, we also found that VPO1 depletion significantly suppressed 3-Cl Tyr expression induced by TGF-β1 (Fig. 7B).
Fig. 7.

VPO1-derived HOCl activates Smad2/3 to regulate cardiac fibroblasts differentiation, and collagen I synthesis and ERK1/2 to regulate cardiac fibroblasts proliferation. (A–B) 3-Cl Tyr levels in MI mice injected with si-NC and si-VPO1(A), or cultured cardiac fibroblasts transfected with siRNA and incubated with or without TGF-β1(B) were measured by western blot. (C) 1 × 10(6) cardiac fibroblasts were seeded in 6 cm plates and incubated with HOCl (100 μmoL/L, diluted in PBS solution) for 2 h, and then changed to normal DMEM and incubated for 24 h before harvesting. α-SMA, Collagen I, Smad2/3 and ERK1/2 were measured by western blot and quantified by Image Lab. n = 4. (D) Cultured cardiac fibroblasts were stimulated with HOCl, then incubated with U0126 (10 μmoL/l). Proliferation was assessed by CCK-8 assay. n = 6. (E) Cultured cardiac fibroblasts were stimulated with HOCl, then incubated with SIS3 (10 μmoL/l). Cardiac fibroblasts were co-stained with α-SMA (red) and phalloidin (green). (F) The expression of α-SMA and collagen I was measured by western blot. n = 4. Data are presented as mean ± SEM. In C, Student's t-test was used in comparison with control group. In D and F, One-way ANOVA test was used. *P < 0.05, **P < 0.01, ***P < 0.001. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
To investigate the role of HOCl in the pathogenesis of cardiac fibrosis, 1 × 10(6) cardiac fibroblasts were seeded in 6 cm plates and incubated with HOCl (100 μmoL/L, diluted in PBS solution) for 2 h, and then changed to normal DMEM and incubated for 24 h before harvesting. We found that HOCl treatment increased α-SMA and collagen I expression, the phosphorylation of Smad2/3 and ERK1/2, and cardiac fibroblast proliferation (Fig. 7C&D). Next, the cultured cardiac fibroblasts were infected with SIS3 (1 μmoL/L, a specific inhibitor of Smad3) or U0126 (10 μmoL/L, a specific inhibitor of ERK1/2). SIS3 inhibited the induction of α-SMA and collagen I expression by HOCl treatment (Fig. 7F). Additionally, U0126 reduced cardiac fibroblast proliferation induced by HOCl treatment (Fig. 7D). To further clarify the role of HOCl on cardiac fibroblast differentiation, cardiac fibroblasts were co-stained with α-SMA and stress-fibers (phalloidin). We found that HOCl treatment induced α-SMA expression incorporating with phalloidin, which could be inhibited by SIS3 (Fig. 7E). These results suggested that VPO1 regulates the pathogenesis of cardiac fibrosis through catalyzing the HOCl formation, which contributes to cardiac fibroblast differentiation via activation of Smad2/3 and cardiac fibroblast proliferation via activation of ERK1/2.
4. Discussion
Cardiac fibrosis is the most crucial mechanism of the pathological cardiac remodeling following MI [[28], [29], [30]]. The progress of cardiac fibrosis after MI is a cascade reaction, initiated by inflammatory infiltration (inflammatory phase at 3–5 day in mice), followed by a granulation and maturation stage in which cardiac fibroblasts differentiate, proliferate, migrate and deposit collagen over the subsequent days [3,31]. MPO, an enzyme released by activated neutrophils and macrophages, is a well-studied member of peroxidase family [32]. It has been reported that MPO generated HOCl regulates cardiac remodeling after MI [33]. Likewise, VPO1, a newly discovered member of the peroxidase family, is highly expressed in cardiovascular system that also catalyzes the reaction of H2O2 with halide ions to produce HOCl [6]. VPO1 has been verified to regulates kidney fibrosis [11]. Using a mouse ischemia reperfusion model, our previous study showed that VPO1 induced cardiomyocyte apoptosis, while its inhibitor ameliorated this effect [12]. Additionally, miR-29b, a protective factor against cardiac fibrosis, has been found to target VPO1 [34,35]. Herein, we report that VPO1 expression was pathologically increased in fibrotic human hearts; contrarily, MPO expression did not increase. In a murine model of MI, VPO1 expression was increased gradually over the duration of ischemia and with the aggravation of cardiac fibrosis in the MI mouse model.
To study the role of VPO1 in cardiac fibrosis, VPO1 global knockdown mice were generated by tail vein injection of si-VPO1. Here, we observed that inhibition of VPO1 expression significantly attenuated the adverse cardiac fibrosis and improved cardiac function, resulting in significantly improved survival rates at 28 days after MI, suggesting that VPO1 could be identified as a key regulator of cardiac remodeling and fibrosis after MI. Short-term depletion of VPO1 attenuated cardiac fibrosis development following MI, implying that VPO1 may be a potential therapeutic target.
Previously, several groups have found that cardiac fibroblasts differentiation is the major pathological mechanism of cardiac fibrosis [19,20,36]. In murine model of MI, VPO1 expression was significantly upregulated in the infarct zone, along with the elevation of α-SMA and collagen I expression. Notably, the inhibition of VPO1 expression suppressed cardiac fibroblasts differentiation and collagen I deposition. In addition, we report that VPO1 knockdown reduced differentiation and migration induced by TGF-β1 treatment in cardiac fibroblasts. It has been previously reported that TGF-β1/Smad2/3 signaling mediates cardiac fibroblasts differentiation in cardiac fibrosis after MI. Smad3-null mice and cardiac fibroblasts exhibited impaired differentiation, migration potential and collagen synthesis [24,37,38]. In this study, VPO1 depletion attenuated the phosphorylation of Smad2/3 in MI model and cardiac fibroblasts treated with TGF-β1 respectively.
Of the various key fibrogenic pathways contributing to the progression of cardiac fibrosis, proliferation of cardiac fibroblasts is a primary mechanism [39,40]. We found that fibrotic mouse hearts exhibited an increased number of Ki67-positive cells 28 days after MI. Remarkably, VPO1 depletion prevented the increase number of Ki67-positive cells in mouse model of cardiac fibrosis after MI. Consistent with our in vivo findings, VPO1 knockdown in cultured cardiac fibroblasts significantly inhibited the proliferation stimulated by TGF-β1. Smad3 has been found to have anti-proliferative effects on cardiac fibroblasts [26]. TGFβ1 also signals through a non-canonical pathway, such as ERK1/2 and Akt [27]. Studies have reported that ERK1/2 phosphorylation facilitates cardiac fibroblast proliferation in an angiotensin II-induced cardiac fibrosis mouse model, which is dependent on PKC and calcium [41]. Here, ERK1/2 phosphorylation was upregulated in mouse model of cardiac fibrosis following MI. ERK1/2 phosphorylation was suppressed by the inhibition of VPO1 expression. Similarly, in vitro VPO1 knockdown also inhibited expression P-ERK1/2 in vitro.
MPO-generated HOCl produced by leukocytes increases left ventricular dilation and aggravates cardiac function following MI [33]. MPO-deficient (MPO−/-) mice exhibited improved cardiac function following MI and decreased susceptibility to ventricular arrhythmias in the MI mouse model. Significantly, cardiac fibroblasts differentiation was inhibited in theses mice and as a result fibrotic tissue formation was attenuated [6]. It has been verified, however, that MPO expression level peaked only at 5–7 days after MI, and subsequently dropped in the absence of an inflammatory reaction [5]. Consistent with these reports, we observed that MPO expression was increased substantially at 7 days following MI and subsequently decreased to levels that similar to that of the sham group. Additionally, heart tissues from ICM patients exhibited no significant elevation of MPO expression. However, VPO1 expression was gradually upregulated over the duration of MI, especially 28 days after MI. Interestingly, 3-Cl Tyr level were significantly increased at 7 days after MI compared to the sham group, and remained elevated at 28 days after MI. The elevated level of 3-Cl Tyr may be due to the high level of VPO1 expression. The primary function of VPO1 entails the utilization of H2O2 to produce HOCl. As expected, VPO1 depletion reduced HOCl production in vivo and in vitro. Therefore, it is reasonable to speculate that VPO1, but not MPO, may be the primary regulator of the granulation and maturation stage of cardiac fibrosis. The effect of VPO1 on the granulation and maturation phases of cardiac fibrosis is mediated by HOCl formation. HOCl treatment significantly induced differentiation and proliferation in cardiac fibroblasts. Furthermore, Smad2/3 and ERK1/2 were also activated in cultured cardiac fibroblasts by HOCl treatment. Inhibitors of Smad2/3 and ERK1/2 significantly reversed the response of cardiac fibroblasts to HOCl treatment. Collectively, these results suggested that VPO1 mediates cardiac fibrosis after MI by regulating HOCl/Smad2/3 and HOCl/ERK1/2 signaling pathways.
Certainly, this study bears some limitations. Although we have investigated the role of VPO1 in cardiac fibrosis in vivo and in vitro, it would be more reliable if cardiac fibroblasts specific knockout and overexpression mice were used in our study. In addition, whether VPO1 could be a prognostic marker in MI patients remains unknown. Thus, the clinical value of VPO1 as a biomarker for cardiac fibrosis needs to be further studied.
5. Conclusions
We propose VPO1 is a novel regulator of cardiac fibrosis following MI. Mechanistically, VPO1-mediated HOCl promotes fibrotic remodeling, including cardiac fibroblast differentiation, migration and proliferation through activation of Smad2/3 and ERK1/2. Therefore, targeting VPO1 may represent a potential therapeutic strategy for the prevention of cardiac fibrosis after MI.
Declarations of interest
None.
Acknowledgments
The authors extend thanks to Guiping Zhang (Guangzhou Medical University) for his work in echocardiography. We are grateful for the technical assistance provided by Quan Sun, Xueting Qiu, Zhengshi Zhou, Kai Xie (All from Central South University).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2019.101151.
Contributor Information
Ruizheng Shi, Email: xyshiruizheng@csu.edu.cn.
Guogang Zhang, Email: zhangguogang@csu.edu.cn.
Funding
This work was supported by grants from the National Nature Science Foundation of China (No81570453 to S.R.Z); the National Nature Science Foundation of China (No. 81670267 to Z.G.G.) and the National Basic Research Program of China (973 Program) (No.2014CB542402); the Fundamental Research Funds for the Central Research Funds for the Central Universities of Central South University (NO2018zzts255 to L.Z.Y).
Disclosures
None.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.McManus D.D., Gore J., Yarzebski J., Spencer F., Lessard D., Goldberg R.J. Recent trends in the incidence, treatment, and outcomes of patients with STEMI and NSTEMI. Am. J. Med. 2011;124:40–47. doi: 10.1016/j.amjmed.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braunwald E. The war against heart failure: the Lancet lecture. Lancet. 2015;385:812–824. doi: 10.1016/S0140-6736(14)61889-4. [DOI] [PubMed] [Google Scholar]
- 3.van Nieuwenhoven F.A., Turner N.A. The role of cardiac fibroblasts in the transition from inflammation to fibrosis following myocardial infarction. Vasc. Pharmacol. 2013;58:182–188. doi: 10.1016/j.vph.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 4.Prokopowicz Z., Marcinkiewicz J., Katz D.R., Chain B.M. Neutrophil myeloperoxidase: soldier and statesman. Arch. Immunol. Ther. Exp (Warsz). 2012;60:43–54. doi: 10.1007/s00005-011-0156-8. [DOI] [PubMed] [Google Scholar]
- 5.Mollenhauer M., Friedrichs K., Lange M., Gesenberg J., Remane L., Kerkenpass C., Krause J., Schneider J., Ravekes T., Maass M., Halbach M., Peinkofer G., Saric T., Mehrkens D., Adam M., Deuschl F.G., Lau D., Geertz B., Manchanda K., Eschenhagen T., Kubala L., Rudolph T.K., Wu Y., Tang W.H.W., Hazen S.L., Baldus S., Klinke A., Rudolph V. Myeloperoxidase mediates postischemic arrhythmogenic ventricular remodeling. Circ. Res. 2017;121:56–70. doi: 10.1161/CIRCRESAHA.117.310870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng G., Salerno J.C., Cao Z., Pagano P.J., Lambeth J.D. Identification and characterization of VPO1, a new animal heme-containing peroxidase. Free Radic. Biol. Med. 2008;45:1682–1694. doi: 10.1016/j.freeradbiomed.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang Y., Xu Q., Peng H., Liu Z., Yang T., Yu Z., Cheng G., Li X., Zhang G., Shi R. The role of vascular peroxidase 1 in ox-LDL-induced vascular smooth muscle cell calcification. Atherosclerosis. 2015;243:357–363. doi: 10.1016/j.atherosclerosis.2015.08.047. [DOI] [PubMed] [Google Scholar]
- 8.Yang W., Liu Z., Xu Q., Peng H., Chen L., Huang X., Yang T., Yu Z., Cheng G., Zhang G., Shi R. Involvement of vascular peroxidase 1 in angiotensin II-induced hypertrophy of H9c2 cells. J. Am. Soc. Hypertens. 2017;11:519–529 e1. doi: 10.1016/j.jash.2016.08.002. [DOI] [PubMed] [Google Scholar]
- 9.Peng H., Chen L., Huang X., Yang T., Yu Z., Cheng G., Zhang G., Shi R. Vascular peroxidase 1 up regulation by angiotensin II attenuates nitric oxide production through increasing asymmetrical dimethylarginine in HUVECs. J. Am. Soc. Hypertens. 2016;10:741–751 e3. doi: 10.1016/j.jash.2016.06.036. [DOI] [PubMed] [Google Scholar]
- 10.Bhave G., Cummings C.F., Vanacore R.M., Kumagai-Cresse C., Ero-Tolliver I.A., Rafi M., Kang J.S., Pedchenko V., Fessler L.I., Fessler J.H., Hudson B.G. Peroxidasin forms sulfilimine chemical bonds using hypohalous acids in tissue genesis. Nat. Chem. Biol. 2012;8:784–790. doi: 10.1038/nchembio.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peterfi Z., Donko A., Orient A., Sum A., Prokai A., Molnar B., Vereb Z., Rajnavolgyi E., Kovacs K.J., Muller V., Szabo A.J., Geiszt M. Peroxidasin is secreted and incorporated into the extracellular matrix of myofibroblasts and fibrotic kidney. Am. J. Pathol. 2009;175:725–735. doi: 10.2353/ajpath.2009.080693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y.S., He L., Liu B., Li N.S., Luo X.J., Hu C.P., Ma Q.L., Zhang G.G., Li Y.J., Peng J. A novel pathway of NADPH oxidase/vascular peroxidase 1 in mediating oxidative injury following ischemia-reperfusion. Basic Res. Cardiol. 2012;107:266. doi: 10.1007/s00395-012-0266-4. [DOI] [PubMed] [Google Scholar]
- 13.Tao L., Bei Y., Chen P., Lei Z., Fu S., Zhang H., Xu J., Che L., Chen X., Sluijter J.P., Das S., Cretoiu D., Xu B., Zhong J., Xiao J., Li X. Crucial role of miR-433 in regulating cardiac fibrosis. Theranostics. 2016;6:2068–2083. doi: 10.7150/thno.15007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takagawa J., Zhang Y., Wong M.L., Sievers R.E., Kapasi N.K., Wang Y., Yeghiazarians Y., Lee R.J., Grossman W., Springer M.L. Myocardial infarct size measurement in the mouse chronic infarction model: comparison of area- and length-based approaches. J. Appl. Physiol. (1985) 2007;102:2104–2111. doi: 10.1152/japplphysiol.00033.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sutton N.R., Hayasaki T., Hyman M.C., Anyanwu A.C., Liao H., Petrovic-Djergovic D., Badri L., Baek A.E., Walker N., Fukase K., Kanthi Y., Visovatti S.H., Horste E.L., Ray J.J., Goonewardena S.N., Pinsky D.J. Ectonucleotidase CD39-driven control of postinfarction myocardial repair and rupture. JCI Insight. 2017;2 doi: 10.1172/jci.insight.89504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winterbourn C.C., Kettle A.J. Biomarkers of myeloperoxidase-derived hypochlorous acid. Free Radic. Biol. Med. 2000;29:403–409. doi: 10.1016/s0891-5849(00)00204-5. [DOI] [PubMed] [Google Scholar]
- 17.Bashey R.I., Martinez-Hernandez A., Jimenez S.A. Isolation, characterization, and localization of cardiac collagen type VI. Associations with other extracellular matrix components. Circ. Res. 1992;70:1006–1017. doi: 10.1161/01.res.70.5.1006. [DOI] [PubMed] [Google Scholar]
- 18.Zhang W., Liu H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12:9–18. doi: 10.1038/sj.cr.7290105. [DOI] [PubMed] [Google Scholar]
- 19.Chistiakov D.A., Orekhov A.N., Bobryshev Y.V. The role of cardiac fibroblasts in post-myocardial heart tissue repair. Exp. Mol. Pathol. 2016;101:231–240. doi: 10.1016/j.yexmp.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 20.van den Borne S.W., Diez J., Blankesteijn W.M., Verjans J., Hofstra L., Narula J. Myocardial remodeling after infarction: the role of myofibroblasts. Nat. Rev. Cardiol. 2010;7:30–37. doi: 10.1038/nrcardio.2009.199. [DOI] [PubMed] [Google Scholar]
- 21.Leask A. TGFbeta, cardiac fibroblasts, and the fibrotic response. Cardiovasc. Res. 2007;74:207–212. doi: 10.1016/j.cardiores.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 22.Leask A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ. Res. 2010;106:1675–1680. doi: 10.1161/CIRCRESAHA.110.217737. [DOI] [PubMed] [Google Scholar]
- 23.Bujak M., Ren G., Kweon H.J., Dobaczewski M., Reddy A., Taffet G., Wang X.F., Frangogiannis N.G. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation. 2007;116:2127–2138. doi: 10.1161/CIRCULATIONAHA.107.704197. [DOI] [PubMed] [Google Scholar]
- 24.Verrecchia F., Chu M.L., Mauviel A. Identification of novel TGF-beta/Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J. Biol. Chem. 2001;276:17058–17062. doi: 10.1074/jbc.M100754200. [DOI] [PubMed] [Google Scholar]
- 25.Khalil H., Kanisicak O., Prasad V., Correll R.N., Fu X., Schips T., Vagnozzi R.J., Liu R., Huynh T., Lee S.J., Karch J., Molkentin J.D. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Invest. 2017;127:3770–3783. doi: 10.1172/JCI94753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dobaczewski M., Bujak M., Li N., Gonzalez-Quesada C., Mendoza L.H., Wang X.F., Frangogiannis N.G. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ. Res. 2010;107:418–428. doi: 10.1161/CIRCRESAHA.109.216101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chung C.C., Kao Y.H., Liou J.P., Chen Y.J. Curcumin suppress cardiac fibroblasts activities by regulating proliferation, migration, and the extracellular matrix. Acta Cardiol. Sin. 2014;30:474–482. [PMC free article] [PubMed] [Google Scholar]
- 28.Shinde A.V., Frangogiannis N.G. Fibroblasts in myocardial infarction: a role in inflammation and repair. J. Mol. Cell. Cardiol. 2014;70:74–82. doi: 10.1016/j.yjmcc.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krenning G., Zeisberg E.M., Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J. Cell. Physiol. 2010;225:631–637. doi: 10.1002/jcp.22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weber K.T., Sun Y., Bhattacharya S.K., Ahokas R.A., Gerling I.C. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat. Rev. Cardiol. 2013;10:15–26. doi: 10.1038/nrcardio.2012.158. [DOI] [PubMed] [Google Scholar]
- 31.Talman V., Ruskoaho H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 2016;365:563–581. doi: 10.1007/s00441-016-2431-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma Y., Yabluchanskiy A., Lindsey M.L. Neutrophil roles in left ventricular remodeling following myocardial infarction. Fibrogenesis Tissue Repair. 2013;6:11. doi: 10.1186/1755-1536-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vasilyev N., Williams T., Brennan M.L., Unzek S., Zhou X., Heinecke J.W., Spitz D.R., Topol E.J., Hazen S.L., Penn M.S. Myeloperoxidase-generated oxidants modulate left ventricular remodeling but not infarct size after myocardial infarction. Circulation. 2005;112:2812–2820. doi: 10.1161/CIRCULATIONAHA.105.542340. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y., Huang X.R., Wei L.H., Chung A.C., Yu C.M., Lan H.Y. miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosis by targeting TGF-beta/Smad3 signaling. Mol. Ther. 2014;22:974–985. doi: 10.1038/mt.2014.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oliveira L.H., Schiavinato J.L., Fraguas M.S., Lucena-Araujo A.R., Haddad R., Araujo A.G., Dalmazzo L.F., Rego E.M., Covas D.T., Zago M.A., Panepucci R.A. Potential roles of microRNA-29a in the molecular pathophysiology of T-cell acute lymphoblastic leukemia. Cancer Sci. 2015;106:1264–1277. doi: 10.1111/cas.12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Porter K.E., Turner N.A. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol. Ther. 2009;123:255–278. doi: 10.1016/j.pharmthera.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 37.Flanders K.C. Smad3 as a mediator of the fibrotic response. Int. J. Exp. Pathol. 2004;85:47–64. doi: 10.1111/j.0959-9673.2004.00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Le A.V., Cho J.Y., Miller M., McElwain S., Golgotiu K., Broide D.H. Inhibition of allergen-induced airway remodeling in Smad 3-deficient mice. J. Immunol. 2007;178:7310–7316. doi: 10.4049/jimmunol.178.11.7310. [DOI] [PubMed] [Google Scholar]
- 39.Virag J.I., Murry C.E. Myofibroblast and endothelial cell proliferation during murine myocardial infarct repair. Am. J. Pathol. 2003;163:2433–2440. doi: 10.1016/S0002-9440(10)63598-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frangogiannis N.G., Perrard J.L., Mendoza L.H., Burns A.R., Lindsey M.L., Ballantyne C.M., Michael L.H., Smith C.W., Entman M.L. Stem cell factor induction is associated with mast cell accumulation after canine myocardial ischemia and reperfusion. Circulation. 1998;98:687–698. doi: 10.1161/01.cir.98.7.687. [DOI] [PubMed] [Google Scholar]
- 41.Hu J., Wang X., Wei S.M., Tang Y.H., Zhou Q., Huang C.X. Activin A stimulates the proliferation and differentiation of cardiac fibroblasts via the ERK1/2 and p38-MAPK pathways. Eur. J. Pharmacol. 2016;789:319–327. doi: 10.1016/j.ejphar.2016.07.053. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.