

Abstract
The poly(ADP-ribose) polymerase (PARP) family of enzymes plays a crucial role in cellular and molecular processes including DNA damage detection and repair, and transcription; indeed, PARP inhibitors are under clinical evaluation as chemotherapeutic adjuncts given their capacity to impede genomic DNA repair in tumor cells. Conversely, overactivation of PARP can lead to NAD+ depletion, mitochondrial energy failure, and cell death. Since PARP activation facilitates genomic but impedes mitochondrial DNA repair, non-selective PARP inhibitors are likely to have opposing effects in these cellular compartments. Herein, we describe the synthesis and evaluation of the mitochondria-targeting PARP inhibitor, XJB-veliparib. Attachment of the hemigramicidin S pentapeptide isostere for mitochondrial targeting using a flexible linker at the primary amide site of veliparib did not disrupt PARP affinity or inhibition. XJB-veliparib was effective at low nanomolar concentrations (10–100 nM) and more potent than veliparib in protection from oxygen glucose deprivation (OGD) in primary cortical neurons. Both XJB-veliparib and veliparib (10 nM) preserved mitochondrial NAD+ after OGD; however, only XJB-veliparib prevented release of NAD+ into cytosol. XJB-veliparib (10 nM) appeared to inhibit poly(ADP-ribose) polymer formation in mitochondria and preserve mitochondrial cytoarchitecture after OGD in primary cortical neurons. After 10 nM exposure, XJB-veliparib was detected by LC-MS in mitochondria-but not nuclear-enriched fractions in neurons and was observed in mitoplasts stripped of the outer mitochondrial membrane obtained from HT22 cells. XJB-veliparib was also effective at preventing glutamate-induced HT22 cell death at micromolar concentrations. Importantly, in HT22 cells exposed to H2O2 to produce DNA damage, XJB-veliparib (10 µM) had no effect on nuclear DNA repair, in contrast to veliparib (10 µM) where DNA repair was retarded. XJB-veliparib and analogous mitochondria-targeting PARP inhibitors warrant further evaluation in vitro and in vivo, particularly in conditions where PARP overactivation leads to mitochondrial energy failure and maintenance of genomic DNA integrity is desirable, e.g. ischemia, oxidative stress, and radiation exposure.
Keywords: ABT-888, Hypoxia-ischemia, mtPARP, PARP, Veliparib, XJB-veliparib
Grahical Abstract
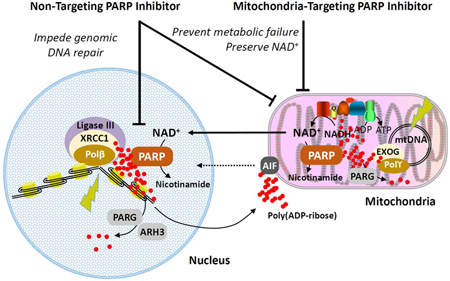
Proposed mechanisms of action for mitochondria-targeting PARP inhibitors. Abbreviations: AIF, apoptosis-inducing factor; ARH3, ADP-ribosylhydrolase 3; EXOG, exo/endonuclease G; mtDNA, mitochondrial DNA; PARG, poly(ADP-ribose) glycohydrolase; PARP, poly(ADP-ribose) polymerase; Polβ, DNA polymerase β; Polγ, DNA polymerase γ; XRRC1, X-ray repair cross-complementing protein 1. 127×79mm (300 × 300 DPI)
INTRODUCTION
Poly(ADP-ribosyl)ation (PARylation) plays a crucial role in cellular and molecular processes including DNA damage detection and repair, transcription, and the maintenance of genomic integrity.1 The currently identified 17 members of the poly(ADP-ribose) polymerase (PARP) family induce the cleavage of NAD+ into nicotinamide and ADP-ribose moieties and mediate their polymerization on target proteins, with links to cellular redox homeostasis, inflammatory, and metabolic networks.2, 3 PARP1 binds to nuclear DNA single-strand break (SSBs) sites and recruits repair proteins to the DNA, subsequently dissociating itself from the SSB. The most potent small-molecule inhibitors, rather than just off-setting PARP1’s enzymatic activity, trap it at the site of DNA damage and stabilize PARP–DNA complexes, ultimately causing DNA double-strand breaks that require more complex repair mechanisms.4
DNA damage induced by irradiation or oxidative stress leads to overactivation of PARP1 and induces depletion of cellular NAD+ and ATP levels, leading to cell dysfunction and necrotic cell death. The primary location of NAD+ is in mitochondria, where it is utilized for oxidative phosphorylation. Furthermore, mitochondrial DNA (mtDNA) is constantly being exposed to damaging species such as reactive oxygen and nitrogen species and is efficiently repaired through at least a subset of the mechanisms involved in nuclear DNA repair, including regulation by PARP1.5 However, opposing roles for PARP1 have been observed in genomic versus mtDNA repair with PARP1 facilitating DNA repair in the nucleus and impeding DNA repair in mitochondria.6 Overactivation of PARP1 and NAD+ depletion has been linked to the pathogenesis of central nervous system (CNS) disorders, including ischemia, traumatic brain injury (TBI), neuroinflammation, and neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases, which have a pronounced mitochondrial component.1, 7 Preserving NAD+ levels may reverse mitochondrial dysfunction and prevent energy failure.8 If suitable PARP inhibitors could be targeted exclusively to the mitochondria, they might reduce NAD+ depletion-associated energy failure while allowing repair of damaged DNA in the nucleus and facilitating mtDNA repair.6
We have previously reported evidence for the localization of PARP in mitochondria and mitochondria-specific PARylation by immunocytochemistry, electron microscopy, and by western blot analysis of purified mitochondrial fractions.9, 10 Furthermore, we demonstrated that the inhibition of mtPARP preserved mitochondrial transmembrane potential and reduced neuronal cell death triggered by oxidative stress or excitotoxicity.9 However, to the best of our knowledge, the hypothesis that mitochondria-targeted PARP-inhibitors would be therapeutically effective in neurodegeneration and other CNS and non-CNS conditions associated with energy failure, and a desire to maintain genomic integrity, has not yet been addressed.
While there are broader therapeutic implications for synthetic PARP modulators, research and development of poly(ADP-ribose) polymerase-1 (PARP1) inhibitors in cancer research has intensified since the regulatory approval of olaparib and rucaparib for patients with BRCA1/2 mutant ovarian cancer, and niraparib for recurrent gynecologic cancers (Figure 1).11–13 Veliparib (1, 2-[(S)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide, ABT-888) is a promising PARP1 inhibitor that has entered clinical phase I/II/III trials for several forms of cancer, including breast cancer and solid tumor neoplasm, either as a single agent or as a combination with chemotherapeutics. A phase III trial is currently ongoing to assess the efficacy, safety and tolerability of veliparib in patients with previously untreated ovarian cancer.14 The (S)- and (R)-enantiomers of veliparib have been shown to have identical biological activity,15 and relative to other FDA approved PARP1 inhibitors veliparib seems to be the least potent in terms of trapping PARP at the site of DNA damage.16
Figure 1.
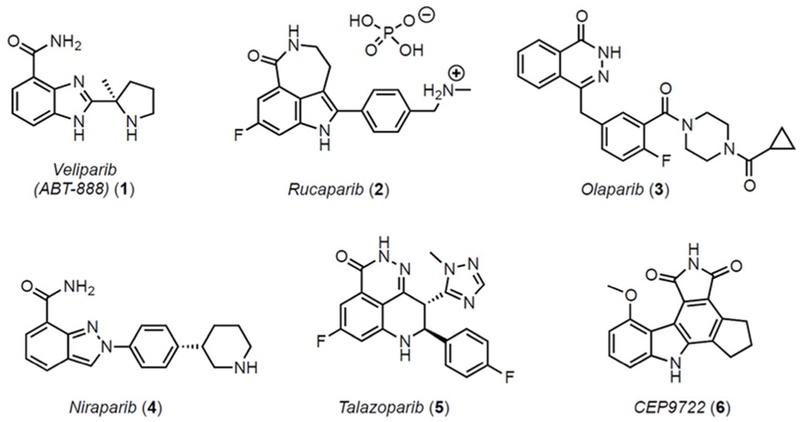
PARP inhibitors in clinical use or in current trials.
The XJB pentapeptide isostere is a mitochondrial targeting unit17–20 derived from the cyclopeptide antibiotic gramicidin S (GS). The XJB moiety has been used successfully to deliver various payloads to mitochondria, including nitroxides,19 a nitric oxide synthase (NOS) antagonist (AMT),21 and a derivative of the natural anticancer product β-lapachone.22 In line with these previous studies, we used XJB to deliver the payload veliparib to mitochondria. Here we show that XJB-veliparib is enriched in mitochondria, and that the compound potently inhibits PARP-mediated cell death in multiple models of cytotoxicity. XJB-veliparib is particularly effective in preventing cell death from hypoxia-ischemia in neurons, warranting further evaluation and development of this class of mitochondria-targeting PARP inhibitors for neurological diseases.
RESULTS AND DISCUSSION
Mitochondria-targeting Veliparib
The chemical synthesis of the mitochondria-targeting XJB-veliparib hybrid molecule is shown in Scheme 1 and fully described in the Methods. Since (S)- and (R)-enantiomers of veliparib have shown identical biological activity,15 we utilized racemic veliparib for the preparation of the XJB-linked hybrid molecule. Furthermore, we hypothesized that the attachment of a flexible linker at the primary amide would be unlikely to disrupt veliparib’s high PARP affinity since, while energetically not as favored, the amide could adopt a cis-configuration and engage Gly429 in PARP with two hydrogen bonds, or, in the trans-amide configuration, a hydrophopic alkyl linker chain could reinforce the single remaining amide carbonyl hydrogen bond to Gly429.23 We selected the XJB pentapeptide isostere as the mitochondrial targeting sequence (Figure 2).17–20 The alkene peptide isostere segment in XJB is a surrogate of the leucyl-D-phenylalanine dipeptide in the bacterial membrane-associating antibiotic gramicidin S (GS), and its side chain-protected ornithylvalylproline tripeptide subunit is taken directly from GS. The D-Phe-Pro sequence is based on the reverse turn inducing sequence of GS and folds into a type II′ β-turn structure that buries several polar amide groups inside the molecule and thus may facilitate membrane transport.24–27 This moiety has previously been used in combination with a nitroxide payload to generate XJB-5–131, a reactive oxygen species (ROS) scavenger that validated the targeting design and was found to be ca. 600-fold enriched in mitochondria over the cytosol.19 XJB-5–131 has shown in vivo efficacy in rodent models of Huntington’s disease (HD),28–30 TBI,31 ischemia-reperfusion injury,32, 33 and hemorrhagic shock.34 In the radiation protector XJB-AMT, a nitric oxide synthase (NOS) antagonist (AMT) was conjugated to the targeting sequence, with the goal to counteract the activation of mitochondrial NOS by ionizing radiation, which can lead to inhibition of the respiratory chain, a burst of superoxide and peroxynitrite, and cellular damage.21 XJB-Lapachone introduced a derivative of the natural anticancer compound β-lapachone into mitochondria and triggered extensive cellular vacuolization and autophagy, as well as stimulating ROS generation in mitochondria.22
Scheme 1.
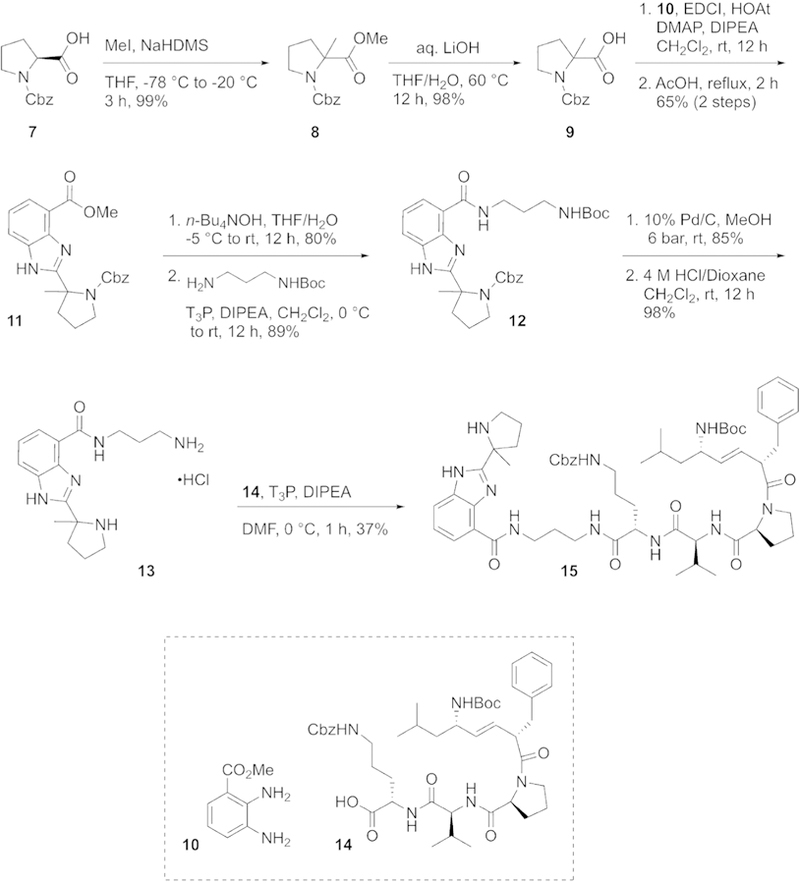
Synthesis of XJB-Veliparib (refer to Methods for chemical transformations).
Figure 2.
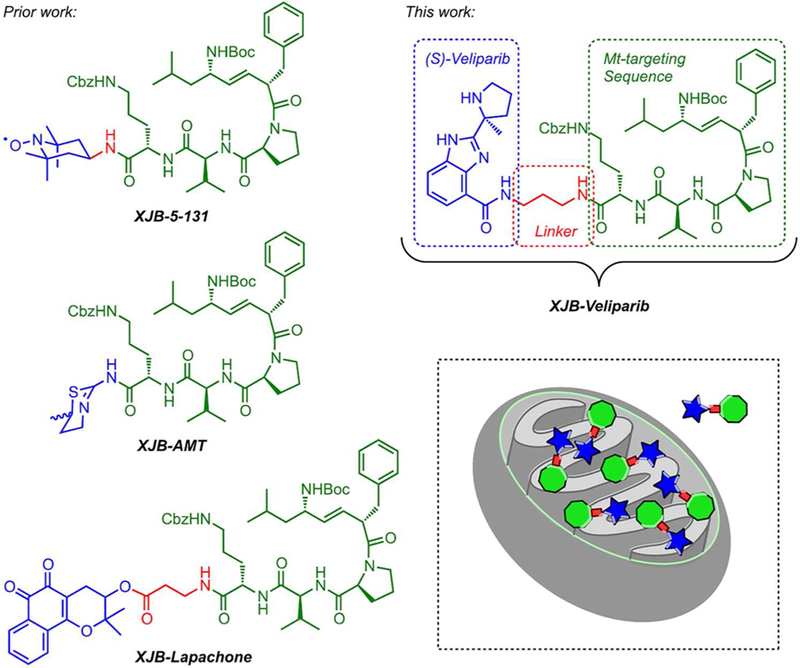
Structures of mitochondrially targeted 4-amino-TEMPO (XJB-5–131), lapachone (XJB-lapachone) and veliparib (XJB-veliparib). The therapeutically active payload is in shown blue, the linker region is in red, and the XJB mitochondrial targeting moiety is in green.
XJB-veliparib and Veliparib PARP1 Enzyme Inhibition ex vivo
To determine whether the linkage to the mitochondria-targeting moiety on 15 affected PARP1 inhibition, we tested the capacity of veliparib and XJB-veliparib to inhibit active PARP1 enzyme (10 U) ex vivo. PARP1 inhibition was similar between untargeted veliparib and the XJB-veliparib conjugate 15 (Figure 3; experiments performed in triplicate), and is consistent with the reported Ki of veliparib of 5.2 nmol/L.35
Figure 3.
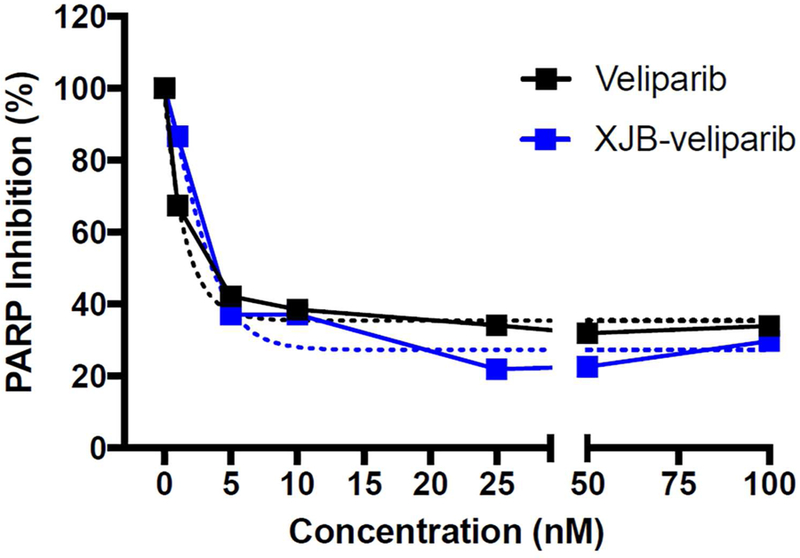
Capacity of XJB-veliparib (15) to inhibit PARP1 enzyme. Varying concentrations of XJB-veliparib or veliparib (0–100 nM) were applied to active PARP1 enzyme (10 U) ex vivo (performed in triplicate).
XJB-veliparib and Veliparib Cytotoxicity in Primary Cortical Neurons
To investigate the biological properties and potential cytotoxicity of XJB-veliparib, we exposed rat primary cortical neurons to varying concentrations of XJB-veliparib and veliparib. Cytotoxicity was assessed by lactate dehydrogenase (LDH) release from dying neurons at 24 h. Both XJB-veliparib and veliparib showed a concentration-dependent cytotoxicity profile; however, XJB-veliparib was significantly less toxic compared with unconjugated veliparib (Figure 4; n = 6/group; mean ± standard deviation [SD]; *P < 0.05). Neurotoxicity defined as >10% cell death was seen with veliparib at 1 µM concentration, vs. a 10 µM concentration required for XJB-veliparib. Significant cytotoxicity has previously been reported when leukemia cells are exposed to micromolar concentrations of veliparib.36 Cytotoxicity produced by PARP inhibitors in clinical use, including veliparib, have been associated with inability to efficiently repair DNA damage and genomic instability.37 However, recent evidence further points to trapping of PARP1 enzyme itself in double-strand DNA (dsDNA) breaks by PARP1 inhibitors.16 Trapped PARP-dsDNA complexes retain catalytic activity and enhance genotoxicity and lethality of chemotherapeutic agents. Relevant here, veliparib concentrations of > 100 µM are tumoricidal in most cancer cell lines.38 Importantly, mitochondria-targeting PARP inhibitors would avoid formation of trapped PARP-dsDNA complexes, and accordingly may have lower toxicity and a therapeutic advantage where prevention of cell death is desired.
Figure 4.
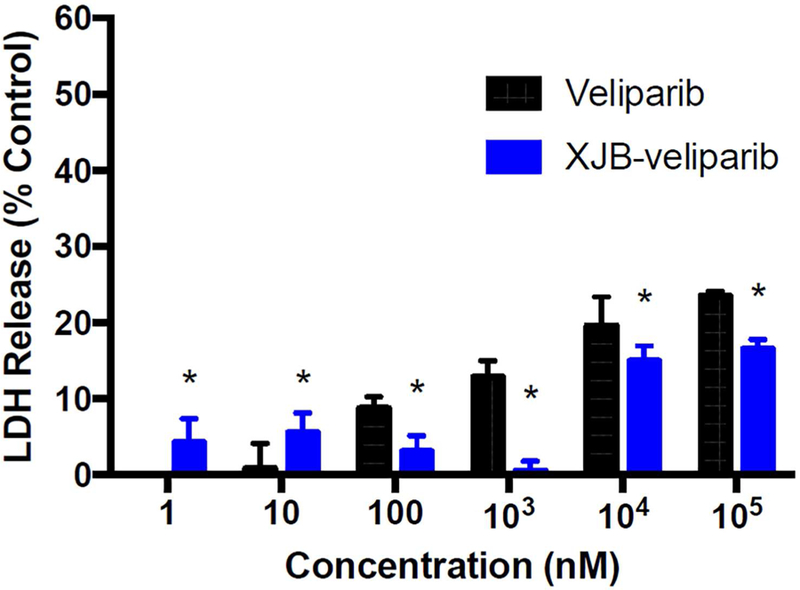
Cytotoxicity studies. Rat primary cortical neurons (DIV 10) were exposed to varying concentrations of XJB-veliparib or veliparib (0–100 µM) for 24 h. Cytotoxicity was determined by LDH release measured at 24 h (n = 6/group; mean ± SD).
Effect of XJB-veliparib and Veliparib after Oxygen-glucose Deprivation in Primary Cortical Neurons
To determine whether mitochondria-targeting XJB-veliparib can promote neuronal survival in ischemia-like conditions in vitro, we subjected primary cortical neurons at 12 DIV to oxygen-glucose deprivation (OGD).33 Cultured neurons were exposed to a hypoxic and glucose-depleted environment for 2 h, followed by normal culture conditions for 24 h to mimic ischemia/reperfusion injury. As shown in Figure 5a, treatment with XJB-veliparib significantly attenuated OGD-induced cell death at low nanomolar concentrations. Specifically, 10 nM XJB-veliparib reduced cell death by 65% (LDH release 12.1 ± 3.1% vs. 34.2 ± 4.0%, 10 nM XJB-veliparib vs. vehicle; n = 30/group; mean ± SD; *P < 0.05).
Figure 5.
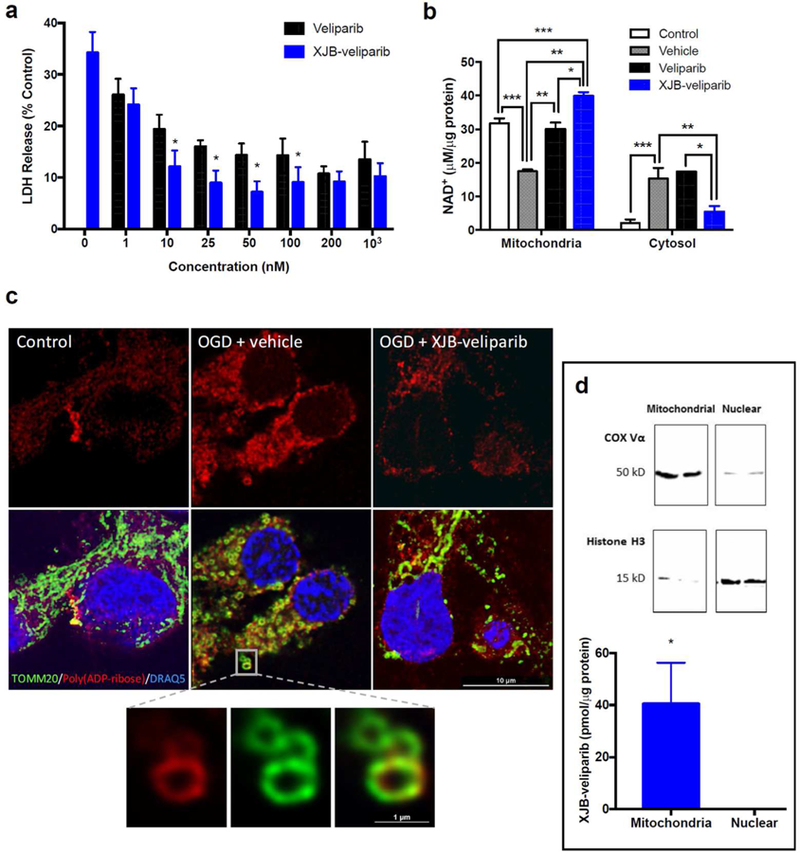
Ischemia-reperfusion injury (2 h of OGD) in rat primary cortical neurons treated with XJB-veliparib or veliparib.
(a) Cell death was determined by LDH release at 24 h. Drug concentrations of 1 nM −1 µM were protective against OGD, and XJB-veliparib was more potent than veliparib at concentrations of 10–100 nM (*P < 0.05 vs. veliparib; n = 12–30/group; mean ± SD).
(b) Mitochondrial and cytosolic NAD+ concentration in primary cortical neurons 24 h after OGD. XJB-veliparib or veliparib (each 10 nM) administered before OGD preserved mitochondrial NAD+ concentration (*P < 0.05 vs. veliparib; **P < 0.05 vs. vehicle; ***P < 0.05 vs. control; n = 6/group; mean ± SD).
(c) STED images showing mitochondrial morphology and PAR polymers 24 h after OGD. Mitochondria were labelled with anti-TOMM20 antibody (green), PAR polymers were labelled with anti-PAR antibody (red), and nuclei were labelled with DRAQ5 (blue). XJB-veliparib (10 nM) was administered before OGD and immunohistochemistry was performed 24 h after OGD. PAR co-localization in mitochondria after OGD is shown in higher magnification.
(d) Mitochondrial enrichment of XJB-veliparib. Rat primary cortical neurons (DIV 10) were exposed to 10 nM XJB-veliparib for 24 h. Cells were then harvested and mitochondrial and nuclear-enriched subfractions were obtained. XJB-veliparib was detected in mitochondria-but not nuclear-enriched fractions (3 independent experiments). Western blot showing mitochondrial and nuclear enrichment using antibodies against COX Vα and histone H3, respectively.
Treatment with concentrations >100 nM of either veliparib or XJB-veliparib conjugate provided no additional protection. Both XJB-veliparib and non-targeting veliparib appear more protective against OGD compared with other published PARP1 inhibitors, where protection is observed in micromolar ranges.39, 40 In vivo, PARP1 inhibition is highly effective at reducing neuronal death caused by ischemia-reperfusion injury.41
To determine whether XJB-veliparib and veliparib related neuroprotection was associated with preservation of intracellular NAD+, NAD+ concentrations in mitochondria-and cytosol-enriched fractions were measured 24 h after OGD in primary cortical neurons. Ten nM XJB-veliparib or non-targeting veliparib preserved mitochondrial NAD+ stores after OGD (Fig. 5b; n = 6/group; mean ± SD; *P < 0.05 vs. vehicle). However, XJB-veliparib, but not veliparib, also prevented efflux of NAD+ from the mitochondria to the cytosol after OGD (**P < 0.05 vs. vehicle and veliparib). These results are consistent with preservation of mitochondrial NAD+ as one of the mechanisms behind PARP-mediated neuroprotection after OGD.
To evaluate the impact of XJB-veliparib on mitochondrial structure after OGD, we used stimulation emission depletion (STED) microscopy and immunohistochemistry. Primary rat cortical neurons were treated with 10 nM XJB-veliparib or vehicle and then exposed to OGD for 2 h. Vehicle treated neurons showed increased PARP activation at 1 h, assessed by immunofluorescence staining with anti-PAR antibody, and swollen, circular mitochondria (labelled with translocase of outer mitochondrial membrane (OMM) 20 [TOMM20]), consistent with fission vs. coiling and compaction compared with control (no ischemia) neurons (Figure 5c; performed in triplicate). In contrast, PAR immunoreactivity was reduced in neurons treated with XJB-veliparib (vs. vehicle) and mitochondrial architecture appeared partially preserved. The relative increase in PAR immunoreactivity observed in mitochondria vs. nuclei in neurons after OGD may be explainable by more effective PAR metabolism by poly(ADP-ribose) glycohydrolase in cell nuclei vs. ADP-ribosylhydrolase 3 in mitochondria.42
To verify mitochondrial enrichment at a therapeutic dose, rat primary cortical neurons were exposed to 10 nM XJB-veliparib for 24 h. Mitochondria- and nuclear-enriched subcellular fractions were obtained as described and verified by western blot using antibodies against the cytochrome oxidase subunit Vα (COX Vα) and histone H3, respectively (Figure 5d).9 Liquid chromatography-mass spectrometry (LC-MS) analysis of XJB-veliparib showed a concentration of 40.5 ± 15.8 pmol/10 µg protein in mitochondria-enriched fractions (Figure 5d; mean ± SD; n = 3 independent experiments). XJB-veliparib was undetectable in nuclear-enriched fractions.
Detection of XJB-veliparib in Mitoplasts
To verify that XJB-veliparib detected in mitochondria-enriched fractions shown in Figure 5d was not simply associated with the OMM, we isolated mitoplasts stripped of the OMM obtained from HT22 cells exposed to 10 nM XJB-veliparib for 45 min. Mitochondria were isolated using differential centrifugation following a standardized protocol (ThermoFisher #89874). The OMM was stripped from the inner mitochondrial membrane (IMM)/matrix using digitonin (0.15 – 1.5 mg/mL). The purity of the mitoplast containing fractions was verified by the absence of translocase of the outer mitochondrial membrane 40 (TOM40) and presence of manganese superoxide dismutase (MnSOD) observed on western blot. LC-MS analysis of XJB-veliparib showed a concentration of 62.6 ± 16.3 pM in mitoplast fractions (Figure 6; mean ± SD; n = 8 samples from 3 independent experiments). For comparison, the concentration of XJB-veliparib in whole mitochondria was 85.1 ± 5.3 pM in HT22 cells after a 45 min exposure to 10 nM XJB-veliparib (n = 3).
Figure 6.
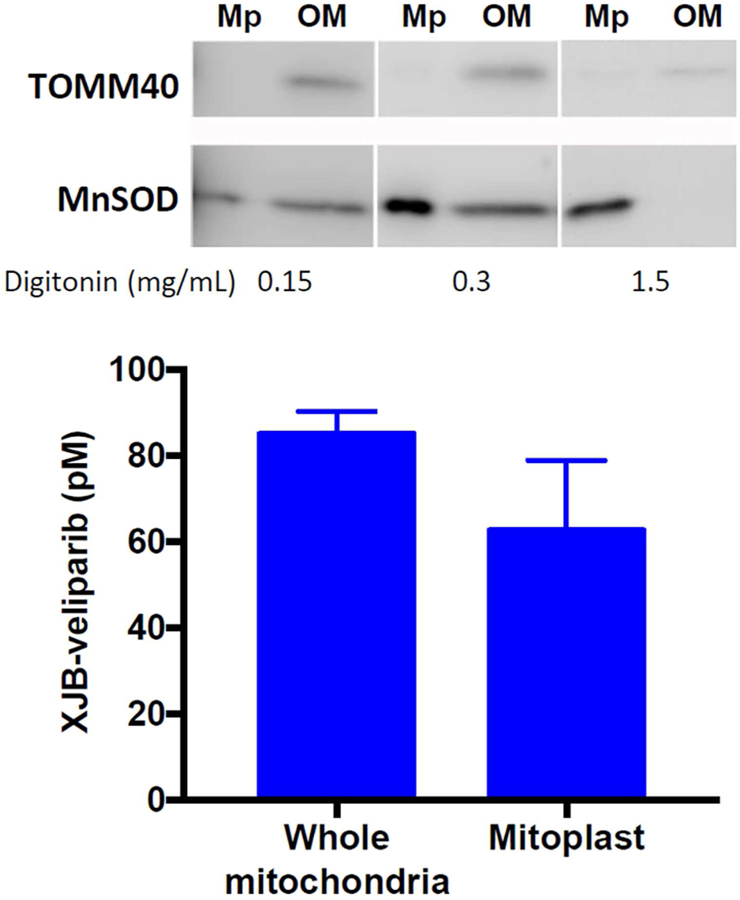
XJB-veliparib concentrates in mitoplasts obtained from HT22 cells exposed to 10 nM XJB-veliparib for 45 min. The OMM was digested from IMM and matrix containing mitoplasts using digitonin (0.15 – 1.5 mg/mL). The purity of the mitoplast containing fractions was verified by the absence of TOM40 and presence of MnSOD observed on western blot (mean ± SD; n = 8 samples from 3 independent experiments). For comparison, the concentration of XJB-veliparib in whole mitochondria from HT22 cells exposed to 10 nM XJB-veliparib for 45 min is shown (n = 3). Mp, mitoplast; OM, outer mitochondrial membrane.
Effect of XJB-veliparib and Veliparib after Excitotoxic Injury in Primary Cortical Neurons and Hippocampal Neuronal Cells
Previous studies have shown an important role for nuclear PARP1 activation in inhibiting excitotoxic neuronal death in vitro.43 In addition, PARP1 inhibition in vivo is effective at reducing N-methyl-D-aspartate (NMDA) but not non-NMDA excitotoxicity.44 To determine whether XJB-Veliparib was also effective in attenuating excitotoxic cell death in vitro, primary cortical neurons were exposed to 10 µM L-glutamate and 10 µM glycine with varying concentrations of XJB-Veliparib or naked veliparib for 24 h as previously described.9 PARP inhibition reduced cell death after glutamate/glycine exposure in a concentration-dependent manner (Fig. 7a; n = 6/group; mean ± SD; *P < 0.05 vs. vehicle). Differing from in vitro ischemia/reperfusion, veliparib was more potent than XJB-veliparib in reducing LDH release. The underlying basis for the differential effectiveness of targeting versus non-targeting PARP inhibitors in models of OGD versus excitotoxicity is beyond the scope of the present study. However, one possible explanation is that since glutamate stimulates DNA repair via activation of transcription factors including cyclic AMP response element binding protein and upregulation of apurinic endonuclease 1,45 any impact of non-targeting PARP inhibitors on nuclear DNA repair and formation of trapped PARP-dsDNA complexes may be mitigated.
Figure 7.
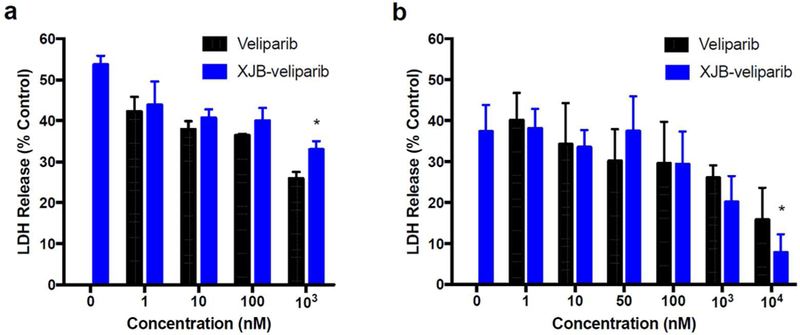
Excitiotoxicity in rat primary cortical neurons or HT22 cells treated with XJB-veliparib or veliparib.
(a) Glutamate-glycine excitotoxicity in primary cortical neurons. Neurons were exposed to 10 µM L-glutamate and 10 µM glycine, with 1–100 µM of XJB-veliparib or veliparib for 24 h (*P < 0.05 vs. veliparib, n = 6/group; mean ± SD).
(b) Glutamate excitotoxicity in an immortalized hippocampal neuronal HT22 cell line. XJB-veliparib and veliparib were both effective at inhibiting excitotoxic cell death, with XJB-veliparib slightly more effective at higher doses (*P < 0.05 vs. naked veliparib; n = 5/group; mean ± SD).
In addition to rat primary cortical neuron cultures, we also determined the effect of veliparib and XJB-veliparib in a stable mouse hippocampal neuronal cell line HT22, in which ferroptotic cell death is induced by high concentrations of glutamate.46 HT22 cells grown to confluence were exposed to 5 mM glutamate and various concentrations of veliparib or XJB-veliparib with LDH release measured at 24 h. Both veliparib and XJB-veliparib similarly inhibit glutamate-induced ferroptosis in HT22 cells, with XJB-veliparib slightly more effective than veliparib at higher (10 µM) concentrations (Fig. 7b; n = 5/group; mean ± SD; *P < 0.05 vs. vehicle).
Effect of XJB-veliparib and Veliparib on DNA Repair in Hippocampal Neuronal Cells Exposed to H2O2
To determine whether XJB-veliparib and/or veliparib impacted DNA repair, we evaluated DNA damage serially in HT22 cells exposed to 200 µM H2O2 using the comet tail intensity assay as previously described.47 Exposure to H2O2 produced DNA damage with evidence of DNA repair over 30 min (Fig. 8). Veliparib, but not XJB-veliparib (both 10 µM), impeded DNA repair (Fig. 8; *P < 0.05 vs. veliparib; **P < 0.05 vs. vehicle; n = 52–60 cells/group; mean ± SEM). These data suggest that mitochondria-targeting PARP inhibitors at a dose that protects HT22 cells from glutamate toxicity (Fig. 7b) do not impact repair of damaged DNA.
Figure 8.
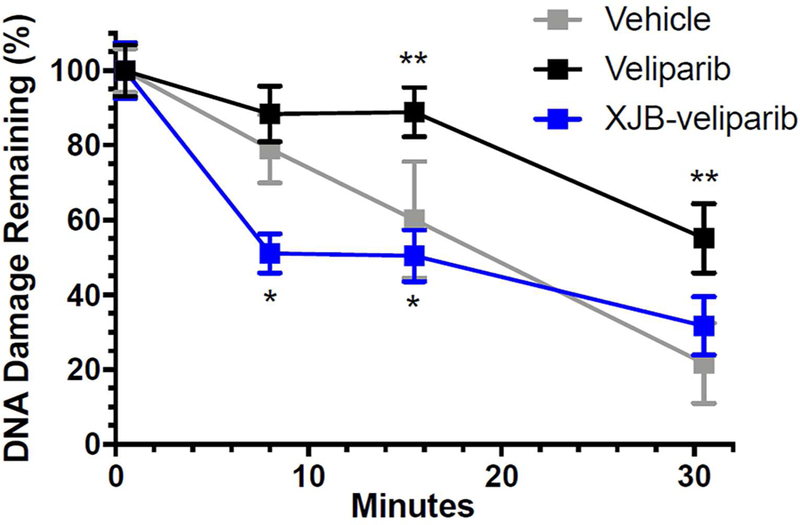
DNA damage and repair in HT22 cells exposed to H2O2 treated with XJB-veliparib (10 µM) or veliparib (10 µM). HT22 cells were exposed to 200 µM H2O2 for 30 min. Tail moments were determined for each condition using the comet tail intensity assay and expressed as percent DNA damage remaining. Exposure to H2O2 produced DNA damage with evidence of DNA repair over 30 min. Veliparib, but not XJB-veliparib, impeded DNA repair (*P < 0.05 vs. veliparib; **P < 0.05 vs. vehicle; n = 52–60 cells/group; mean ± SEM).
Role for Mitochondria-targeting PARP Inhibitors
An undisputed role for mitochondrial PARP, and in particular mitochondrial PARP1, in health and disease is controversial;48 however, at least a partial role for mitochondrial PARP in mtDNA repair and cellular metabolism is becoming more apparent.5 Several early studies have shown the presence of ADP-ribosylation in mitochondria from testes,49 liver,50, 51 and brain.52 More recently, inhibition of mitochondrial PARP1 using genetic approaches was shown to increase the efficiency of mtDNA base excision repair via interaction with exo/endonuclease G and DNA polymerase γ;6 as opposed to inhibition of nuclear PARP1 which decreases the efficiency of genomic DNA repair.53 Here we show that mitochondria-targeting XJB-veliparib has no effect on nuclear DNA repair after H2O2 exposure in neuronal cells, in contrast to non-targeting veliparib (Fig. 8).
PARP1 inhibition has been shown to increase mitochondrial metabolism via silent mating type information regulation 2 homolog 1 (SIRT1),54 and directly preserve mitochondrial function and prevent cell death via maintenance of NAD+ stores.55 Here we show that mitochondria-targeting XJB-veliparib is very effective at preserving mitochondrial NAD+ levels and preventing efflux of NAD+ into cytosol in neurons after OGD (Fig. 5b). We have previously reported PARylation of the electron transport chain proteins F1F0 ATPase, cytochrome c oxidase, and cytochrome c reductase, and preservation of respiration in isolated brain mitochondria under conditions of nitrosative stress with PARP inhibition or addition of poly(ADP-ribose) glycohydrolase.10 In addition, we have shown that mitochondrial PARP inhibition using PARP1 gene deletion or relatively selective PARP1 inhibitors protects primary cortical neurons and fibroblasts from oxidative stress and excitotoxicity.9
Powerful neuroprotection using non-mitochondria targetinggene deletion has been demonstrated in pre-clinical models of stroke,41 TBI,56, 57 and neurodegenerative disease.42, 58 In these and other CNS diseases, an important role for mitochondrial energy failure is implicated. XJB-veliparib (and similar compounds) that concentrate in mitochondria would be expected to directly inhibit mitochondrial PARP, reduce NAD+ depletion, prevent energy failure, and reduce cell death while having minimal impact on genomic DNA repair and integrity (please refer to Abstract Graphic). Furthermore, XJB-veliparib (and similar compounds) may also enhance mtDNA repair6 when used to treat these diseases. Mitochondria-targeting PARP inhibitors may also reduce PARylation of apoptosis-inducing factor (AIF), potentially mitigating AIF release from mitochondria, translocation of AIF into nuclei, and parthanatos-related cell death.59
Conclusions
Mitochondria-targeting PARP inhibitors may have unique and important advantages over PARP inhibitors that are non-selective to cellular compartments, including directly preserving mitochondrial function and viability and preventing mitochondrial energy failure. Moreover, mitochondria-targeting PARP inhibitors could have clinical efficacy in diseases where the pathophysiology includes mitochondrial dysfunction and/or energy failure, but efficient nuclear DNA repair is desired—such as ischemia-reperfusion injury, radiation toxicity, and other diseases involving oxidative stress. As PARP inhibitors are being reconsidered for many clinical indications,60 further development and in vitro and in vivo evaluation of mitochondria-targeting PARP inhibitors appears warranted.
METHODS
Synthesis of XJB-veliparib
For the synthesis of mitochondrially targeted veliparib, XJB-Veliparib, N-Cbz-L-proline methyl ester (7) was treated with NaHMDS and MeI, followed by saponification, to give the acid 9 in 98% yield over 2 steps (Scheme 1). One-pot EDCl coupling to methyl 2,3-diaminobenzoate (10) and acid-catalyzed cyclization provided the benzimidazole 11 in good overall yield. Hydrolysis of the methyl ester followed by acylation with the N-Boc-1,3-diaminopropane spacer group afforded 12. Removal of the Cbz-group via hydrogenation followed by Boc-deprotection provided 13, which was coupled to the mitochondrial targeting sequence 1461 to afford the desired XJB-Veliparib conjugate 15.
General Experimental Conditions. All moisture- and air-sensitive reactions were performed in oven dried glassware under a positive pressure of argon. All reagents and solvents were used as received unless otherwise specified. THF and Et2O were distilled over sodium/benzophenone ketyl; CH2Cl2 was distilled over CaH2, MeCN and DMF were dried over molecular sieves. Reactions were monitored by TLC analysis (pre-coated silica gel 60 F254 plates, 250 µm layer thickness) and visualization was accomplished with a 254/280 nm UV light and/or by staining with KMnO4 solution (1.5 g KMnO4 and 1.5 g K2CO3 in 100 mL of a 0.1% NaOH solution), a ninhydrin solution (2 g ninhydrin in 100 mL EtOH), a PMA solution (5 g phosphomolybdic acid in 100 mL EtOH), or a p-anisaldehyde solution (2.5 mL p-anisaldehyde, 2 mL AcOH and 3.5 mL conc. H2SO4 in 100 mL EtOH). Flash chromatography was performed on silica gel (40–63 µm). Melting points were determined on a Mel-Temp II capillary melting point apparatus fitted with a Fluke 51 II digital thermometer. Infrared spectra were recorded on an ATR spectrometer. NMR spectra were recorded on 300, 400, 500 or 700 MHz instruments. Chemical shifts were reported in parts per million (ppm) and referenced to residual solvent. 1H NMR spectra are tabulated as follows: chemical shift, multiplicity (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constant(s), number of protons. 13C NMR spectra were obtained using a proton-decoupled pulse sequence and are tabulated by observed peak. LC-MS analyses were performed on a Shimadzu UFLC instrument equipped with an Applied Biosystem MDS SCIEX API 2000 mass spectrometer (ESI), under the following conditions: column: Varian Polaris C18-A (100 × 4.6 mm, 5µm) equilibrated at 40 °C; buffer A: 0.1% aqueous AcOH, buffer B: 0.1% AcOH in MeCN; 30 min gradient: 5% buffer B in buffer A for 1 min, then 5 to 95% buffer B in buffer A over 13 min, then 95% buffer B in buffer A for 4 min, then 95–5% buffer B in buffer A over 7 min, then 5% buffer B in buffer A for 5 min; flow rate: 0.2 mL/min; detection: TIC and/or UV λ = 254/280 nm. Intracellular distribution and HRMS data were obtained on a Thermo Scientific Exactive Orbitrap LC-MS (ESI positive ion mode) coupled to a Thermo Scientific Accela HPLC system using a 3.5 µM Water XTerra C18 column (2.1 × 50 mm; 10 min gradient elution with MeCN/H2O/MeOH containing 0.1% formic acid at a flow rate of 500 µL/min from 3:92:5 at 0–0.5 min to 93:2:5 at 4.0 min, back to 3:92:5 from 6.0 to 7.5 min).
1-Benzyl 2-methyl 2-methylpyrrolidine-1,2-dicarboxylate (8).
To a −78 °C solution of N-Cbz-L-proline 7 (0.43 mL, 1.9 mmol) and iodomethane (0.24 mL, 3.8 mmol) in THF (3.5 mL) was added dropwise NaHMDS (1 M in THF, 3.8 mL, 3.8 mmol). The resulting mixture was warmed to −20 °C, stirred for 3 h, quenched with H2O, acidified with 2N HCl and extracted with EtOAc (3 x). The combined organic layers were washed with brine (1x), dried over MgSO4, filtered and concentrated to dryness. The residue was purified by chromatography on SiO2 (30% EtOAc/hexanes) to give 8 as a pale yellow oil (0.52 g, 99%). Spectral data of the mixture of rotamers are in accordance with literature values.54 1H NMR (300 MHz, CDCl3) δ 1.54 (s, 1.5 H), 1.61 (s, 1.5 H), 1.86–1.99 (m, 3 H), 2.13–2.24 (m, 1 H), 3.46 (s, 1.5 H), 3.56–3.69 (m, 2 H), 3.71 (s, 1.5 H), 4.99–5.23 (m, 2 H), 7.24–7.40 (m, 5 H).
1-((Benzyloxy)carbonyl)-2-methylpyrrolidine-2-carboxylic acid (9).
Methyl ester 8 (1.53 g, 5.52 mmol) was dissolved in THF (10.5 mL) and H2O (5.4 mL) and treated with a solution of LiOH (265 mg, 11.0 mmol) in H2O (5.4 mL). MeOH (1.5 mL) was then added and the resulting homogenous solution heated to 60 °C overnight. The organic solvents were removed and the aqueous layer acidified to pH 2 using 2 N HCl and extracted with EtOAc (3x). The combined organic layers were washed with water (1x), dried (MgSO4) filtered and concentrated to dryness to give 9 as a colorless solid (1.42 g, 98%). Spectral data of the mixture of rotamers are in accordance with literature values.54 1H NMR (500 MHz, DMSO-d6) δ 1.44 (s, 1.5 H), 1.45 (s, 1.5 H), 1.80–1.96 (m, 3 H), 2.02–2.19 (m, 1 H), 3.43–3.53 (m, 2 H), 4.94–5.11 (m, 2 H), 7.24–7.41 (m, 5 H), 12.51 (br s, 1 H).
Methyl 2-(1-((benzyloxy)carbonyl)-2-methylpyrrolidin-2-yl)-1H-benzo[d]imidazole-4-carboxylate (11).
To a solution of 9 (200 mg, 0.760 mmol) and methyl diaminobenzoate 10 (189 mg, 1.14 mmol) in CH2Cl2 (15 mL) was added DIPEA (0.13 mL, 0.76 mmol) followed by addition of EDCI (218 mg, 1.14 mmol), HOAt (155 mg, 1.14 mmol) and DMAP (9.3 mg, 0.076 mmol), and the resulting solution was stirred at room temperature overnight. The reaction mixture was quenched with sat. aqueous NH4Cl solution and extracted with CH2Cl2 (2x). The combined organic layers were washed with brine (1x), dried (MgSO4), filtered and concentrated to give the product as a brown oil. The crude residue was redissolved in AcOH (5 mL) and heated at reflux for 2 h. The solvent was evaporated, the crude residue poured onto sat. aqueous NaHCO3 and extracted with EtOAc (3x). The combined organic layers were washed with brine (1x), dried (MgSO4) and concentrated. The crude residue was purified via chromatography on SiO2 (30–90% EtOAc/hexanes) to give 11 as a pale yellow oil (198 mg, 65% over 2 steps): HRMS (ESI+) m/z calcd for C22H24N3O4 [M+H] 394.1761, found 394.1758.
2-(1-((Benzyloxy)carbonyl)-2-methylpyrrolidin-2-yl)-1H-benzo[d]imidazole-4-carboxylic acid.
Methyl ester 11 (280 mg, 0.712 mmol) was dissolved in THF/H2O (14:1, 21 mL) and cooled to −5 °C. A 40% n-Bu4NOH (aqueous) solutionq (4.6 mL, 7.1 mmol) was added slowly and the reaction mixture was stirred for 30 min at −5 °C and at room temperature over night. The solution was acidified with aqueous AcOH and extracted with EtOAc (3x). The combined organic layers were washed with brine, dried (MgSO4), filtered and concentrated under reduced pressure. The crude residue was purified by chromatography on SiO2 (0–10% MeOH/CH2Cl2) to give 2-(1-((benzyloxy)carbonyl)-2-methylpyrrolidin-2-yl)-1H-benzo[d]imidazole-4-carboxylic acid as a pale-yellow foam (215 mg, 80%): 1H NMR (400 MHz, DMSO-d6) δ 7.86 (br s, 1 H), 7.78 (d, J = 7.5 Hz, 1 H), 7.41 – 7.36 (m, 4 H), 7.33 – 7.30 (m, 1 H), 7.30 – 7.25 (m, 1H), 7.06 (t, J = 7.4 Hz, 0.5 H), 6.91 (t, J = 7.0 Hz, 1H), 6.70 (br s, 1 H), 5.15 – 5.09 (m, 0.5 H), 5.07 – 5.04 (m, 1H), 3.87 – 3.54 (m, 3 H), 2.76 (br s, 1 H), 1.92 (s, 3 H), 1.87 (br s, 2 H), 1.86 (br s, 1 H); 13C-NMR (700 MHz, DMSO-d6) δ 167.5, 160.5, 159.8, 154.4, 154.1, 137.4, 136.8, 128.9, 128.2, 127.9, 127.7, 127.1, 124.4, 124.3, 121.7, 66.3, 66.2, 62.9, 55.4, 48.8, 48.3, 42.7, 40.5, 24.4, 24.2, 22.9, 22.4; IR (ATR, neat) 1682.8, 1407.6, 1351.5, 1255.8, 746.4 cm−1; Mp 102.3 – 104.6 °C; HRMS (ESI+) m/z calcd for C21H22N3O4 [M+H] 380.1605, found 380.1610.
Benzyl 2-(4-((3-((TERT-butoxycarbonyl)amino)propyl)carbamoyl)-1H-benzo[d]imidazol-2-yl)-2-methylpyrrolidine-1-carboxylate (12).
To a solution of N-Boc-1,3-propanediamine (0.28 mL, 1.6 mmol) and 2-(1-((benzyloxy)carbonyl)-2-methylpyrrolidin-2-yl)-1H-benzo[d]imidazole-4-carboxylic acid (342 mg, 1.05 mmol) in CH2Cl2 (12 mL) at 0 °C was added DIPEA (0.46 mL, 2.6 mmol), followed by the dropwise addition of T3P (50 wt% in EtOAc, 0.92 mL, 1.6 mmol). The resulting mixture was stirred at room temperature overnight, washed with 5% aqueous Na2CO3 and brine, dried (MgSO4), filtered and concentrated. The crude residue was purified via chromatography on SiO2 (100% CH2Cl2 to 10% MeOH/CH2Cl2) to give 12 as a pale-yellow foam (503 mg, 89%): 1H NMR (600 MHz, DMSO-d6) δ 12.76 (br s, 0.5 H), 12.69 (br s, 0.5 H), 9.93 (br s, 1H), 7.86 – 7.80 (m, 1 H), 7.64 (d, J = 7.7 Hz, 0.5 H), 7.60 (d, J = 7.7 Hz, 0.5 H), 7.39 – 7.34 (m, 2 H), 7.33 – 7.27 (m, 2 H), 6.94 – 6.81 (m, 2 H), 6.73 – 6.65 (m, 1 H), 5.09 – 4.97 (m, 1 H), 4.94 – 4.79 (m, 1 H), 3.86 – 3.77 (m, 1 H), 3.70 – 3.60 (m, 1 H), 3.47 – 3.37 (m, 2 H), 3.11 – 3.00 (m, 2 H), 2.28 – 2.10 (m, 2 H), 2.02 – 1.94 (m, 2 H), 1.91 (br s, 1.5 H), 1.89 (br s, 1.5 H), 1.73 – 1.65 (m, 3 H), 1.38 (s, 9 H); 13C-NMR (600 MHz, DMSO-d6) δ 165.1, 160.4, 160.2, 156.1, 154.1, 153.7, 140.7, 140.6, 137.5, 136.7, 135.3, 135.2, 128.8, 128.2, 127.8, 127.7, 127.0, 122.5, 122.4, 122.3, 122.2, 122.1, 115.2, 115.1, 77.9, 66.2, 62.6, 62.2, 55.4, 49.0, 48.2, 43.4, 42.1, 38.2, 36.9, 30.3, 30.3, 28.7, 24.4, 23.3, 23.0, 22.5 (mixture of rotamers); Mp 112.7 – 116.2 °C; HRMS (ESI+) m/z calcd for C29H38N5O5 [M+H] 536.2867, found 536.2867.
tert-Butyl (3-(2-(2-methylpyrrolidin-2-yl)-1H-benzo[d]imidazole-4-carboxamido)propyl)carbamate.
A mixture of 12 (500 mg, 0.933 mmol) and 10% Pd/C (99 18 mg, 0.093 mmol) in MeOH (5 mL) in a high-pressure hydrogenator was purged with argon and flushed with hydrogen (3x). Thepressure was set to 6 bar and the mixture was stirred at this pressure at room temperature overnight. The solution was filtered through a plug of Celite® and washed with CH2Cl2. The filtrate was concentrated and the crude residue was purified via chromatography on SiO2 (100% CH2Cl2 to 10% MeOH/CH2Cl2) to give tert-butyl (3-(2-(2-methylpyrrolidin-2-yl)-1H-benzo[d]imidazole-4-carboxamido)propyl)carbamate as a white foam (375 mg, 85%): 1H NMR (600 MHz, DMSO-d6) δ 12.50 (br s, 2 H), 9.94 (br s, 1 H), 7.78 (d, J = 7.5 Hz, 1 H), 7.62 (d, J = 7.5 Hz, 1 H), 7.25 (t, J = 7.7 Hz, 1 H), 6.92 – 6.87 (m, 1 H), 3.45 – 3.38 (m, 2 H), 3.12 – 3.04 (m, 3H), 2.93 – 2.86 (m, 1 H), 2.45 – 2.38 (m, 1 H), 1.89 – 1.80 (m, 2 H), 1.72 – 1.64 (m, 3 H), 1.59 (br s, 3 H), 1.38 (s, 9 H); 13C-NMR (600 MHz, DMSO-d6) δ 165.3, 156.2, 122.1, 121.8, 79.7, 79.4, 79.2, 77.9, 62.6, 60.2, 55.4, 49.1, 46.4, 38.1, 36.8, 30.4, 28.7, 27.6, 25.7, 21.3, 14.6; IR (ATR, neat) 3269, 2973, 1694, 1645, 1612, 1523, 1406, 1365, 1245, 1166, 1046, 988, 758 cm−1; Mp 79.8 – 83.2 °C; HRMS (ESI+) m/z calcd for C21H32N5O3 [M+H] 402.2500, found 402.2499.
N-(3-Aminopropyl)-2-(2-methylpyrrolidin-2-yl)-1H-benzo[d]imidazole-4-carboxamide hydrochloride (13).
To a solution of tert-butyl (3-(2-(2-methylpyrrolidin-2-yl)-1H-benzo[d]imidazole-4-carboxamido)propyl)carbamate (32.8 mg, 0.0817 mmol) in CH2Cl2 (1 mL) was added 4 M HCl in dioxane (0.2 mL, 0.8 mmol). The resulting mixture was stirred at room temperature for 2 h. The product was filtered off, washed with hexanes, and the resulting white solid (27.1 mg, 98%) was used in the next step without further purification: HRMS (ESI+) m/z calcd for C16H24N5O [M+H] 302.1975, found 302.1974.
tert-Butyl ((4S,7S,E)-7-benzyl-2-methyl-8-((2S)-2-(((2S)-3-methyl-1-(((8S)-15-(2-(2-methylpyrrolidin-2-yl)-1H-benzo[d]imidazol-4-yl)-3,9,15-trioxo-1-phenyl-2-oxa-4,10,14-triazapentadecan-8-yl)amino)-1-oxobutan-2-yl)carbamoyl)pyrrolidin-1-yl)-8-oxooct-5-en-4-yl)carbamate (“XJB-Veliparib”, 15).
To a solution of XJB-acid 14 (36.9 mg, 0.0458 mmol) and 13 (23.2 mg, 0.0687 mmol) in DMF (0.92 mL, 0.05M) at 0 °C was added DIPEA (40 µL, 0.23 mmol), followed by the dropwise addition of T3P (50% in DMF, 35 µL, 0.060 mmol). The reaction mixture was stirred at 0 °C for 1 h, quenched with aqueous NH4Cl, and extracted with CH2Cl2 (3x). The combined organic layers were washed with 5% aqueous LiCl solution (1x), dried (MgSO4), filtered and concentrated to give the crude product as a pale yellow oil that was purified by chromatography on SiO2 (100% CH2Cl2 to 15% MeOH/CH2Cl2) to afford 15 as a colorless solid (18.5 mg, 37%): 1H NMR (400 MHz, DMSO-d6, 100 °C) δ 9.32 (br s, 1 H), 7.88 (d, J = 7.44 Hz, 1 H), 7.72 (d, J = 7.96 Hz, 1 H), 7.69 – 7.59 (m, 1 H), 7.47 – 7.38 (m, 1 H), 7.37 – 7.31 (m, 5 H), 7.30 – 7.26 ( m, 1 H), 7.25 – 7.12 (m, 5 H), 6.79 (br s, 1 H), 6.12 (d, J = 7.72 Hz, 1 H), 5.52 – 5.42 (m, 2 H), 5.02 (s, 2 H), 4.44 – 4.37 (m, 1 H), 4.30 – 4.23 (m, 1 H), 4.18 – 4.09 (m, 1 H), 3.93 – 3.84 (m, 1 H), 3.53 – 3.35 (m, 6 H), 3.29 – 3.21 (m, 3 H), 3.10 – 3.03 (m, 4 H), 2.72 – 2.00 (m, 3 H), 1.98 – 1.88 (m, 2 H), 1.84 (br s, 3 H), 1.80 – 1.71 (m, 5 H), 1.66 – 1.57 (m, 1 H), 1.55 – 1.43 (m, 3 H), 1.38 (s, 9 H), 1.30 – 1.25 (m, 4 H), 0.90 – 0.78 (m, 12 H); 13C NMR (700 MHz, DMSO-d6) δ 172.7, 172.5, 172.2, 171.9, 171.8, 171.7, 171.4, 171.3, 171.2, 164.8, 156.6, 155.3, 155.2, 139.7, 139.5, 137.7, 135.0, 134.6, 129.6, 128.8, 128.4, 128.3, 128.2, 127.8, 126.4, 126.2, 122.8, 78.0, 77.8, 65.6, 64.9, 64.6, 59.7, 59.6, 59.3, 58.2, 53.1, 53.0, 50.7, 50.5, 49.3, 48.8, 47.2, 47.1, 45.5, 38.9, 38.2, 36.5, 36.4, 35.2, 32.4, 30.9, 30.7, 30.2, 29.8, 28.7, 26.5, 25.0, 24.7, 24.4, 24.3, 23.8, 23.0, 22.8; Mp 147.2 – 152.6 °C; IR (ATR, CH2Cl2) 3291.7, 2955.8, 1647.1, 1528.8, 1439.1, 1365.7, 1248.0, 1167.2, 1028.3, 758.2, 698.1 cm−1; HRMS (ESI) m/z calcd for C60H85N10O9 1089.6496, found 1089.6495.
PARP1 Activity
The capacity to inhibit PARP1 was determined using a commercial assay (Trevigen, Gaithersburg, MD) as per manufacturer’s direction. Various concentrations of XJB-veliparib or veliparib were added to histone-coated wells containing active PARP1 enzyme and NAD+ in surplus.
Cell Cultures
Primary cortical neuron-enriched cultures were prepared from 16–17 day old Sprague-Dawley rat embryos as described.9 Dissociated cell suspensions were filtered through a 70 µm nylon cell strainer and seeded in 96-well plates (5×104 cells/well) or on poly-D-lysine coated glass coverslips, and maintained in Neurobasal medium with B27 supplements (Life Technologies, Carlsbad, CA). Experiments were performed 12 days in vitro (DIV).
HT22 cells were cultured at 37°C in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen Inc., Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, San Jose, CA) and 1% penicillin-streptomycin (ATCC, Manassas, VA) in an atmosphere containing 5% CO2. Cells were cultured for 24 to 48 h before use.
Separation of Subcellular Proteins
Separation of cytosol-, nuclear-, and mitochondria-enriched protein fractions was performed using differential centrifugations as previously described.62 Samples were homogenized in lysis buffer then centrifuged at 1025 g for 15 min at 4°C to separate nuclei. Pellets containing nuclei were resuspended in buffer and centrifuged at 16,000 g at 4°C for 20 min with these supernatants containing nuclear-enriched fractions. Supernatants after separation of nuclei containing mitochondria and cytosolic proteins were then centrifuged at 20,000 g for 50 min at 4°C to separate cytosol-enriched protein fractions. The pellets were lysed, sonicated until frothy, and re-suspended in lysis buffer then centrifuged at 16,000 g for 25 min at 4°C. These mitochondria-enriched pellets were then washed in 5 µM CaCl and centrifuged at 1,600 g for 10 min. Western blots were performed using antibodies against cytochrome c oxidase (BD Biosciences, San Jose, CA) and histone H3 (Abcam, Cambridge, MA) to verify purity of subcellular fractions.
Isolation of Mitoplasts
Mitochondria from HT22 cells (25 × 106 cells/mL) were isolated using differential centrifugation following a standardized protocol using a commercial isolation kit (#89874; ThermoFisher Scientific, San Jose, CA). A low spin speed (3,000 g) was used to collect only heavy mitochondria for OMM removal. Mitochondria were resuspended in 325 µL mitochondrial isolation reagent C, then 100 µL of suspension was centrifuged at 3,000 g. The pellet was then digested with 0.15 – 1.5 mg/mL freshly dissolved digitonin in 125 µL volume. The samples were agitated at 750 rpm for 15 min at 4ºC, then digestion was stopped by adding an equal volume of reagent C. Suspensions were then centrifuged at 10,000 g for 10 min at 4ºC with the pellet containing mitoplasts (IMM and matrix) and the supernatant containing OMM and protein from the intermembrane space. The purity of the mitoplast containing fractions was verified by the absence of TOM40 and presence of MnSOD on western blot.
Quantification of XJB-veliparib
The mitochondrial fraction (50 µL) or nuclear fraction (50 µL), treated with XJB-veliparib was added to a 5:1 ratio of CH2Cl2:MeOH (950 µL) and vortexed (30 sec). Water (150 µL) was added and the solution was vortexed (15 sec) and set aside to equilibrate at room temperature (30 min). The resulting suspension was placed in an Eppendorf Centrifuge 5702 (4400 rpm, 20 °C) for 12 min. The organic layer was extracted and filtered through a 0.45 µm filter for analysis.
XJB-veliparib was quantified on a Thermo Scientific Exactive Orbitrap LC-MS (ESI positive ion mode) coupled to a Thermo Scientific Accela HPLC system using a 3.5 µM Water XTerra C18 column (2.1 × 50 mm; 20 min gradient elution with MeCN/H2O containing 0.1% formic acid at a flow rate of 500 µL/min from 5:95 at 0–1.0 min to 95:5 at 12.0 min, back to 5:95 from 16.0 to 16.1 min). Calibration curves for XJB-veliparib were run in duplicate from 102 nM to 5.7 nM. Samples (10 µL) were injected in triplicate and Thermo Xcalibur software was used to determine the concentration of XJB-veliparib in mitochondrial and nuclear fractions (n = 3). The concentration was reported as pM concentration of XJB-veliparib per 10 µg of protein with corresponding standard deviation values.
Oxygen-glucose Deprivation
To model ischemia-reperfusion in vitro, culture medium was replaced with a pre-equilibrated low glucose (0.5 mM) medium. Neurons were transferred into a sealed hypoxic chamber (Coy Laboratory Products Inc., Grass Lake, MI) set to an atmosphere of 95% N2 with 5% CO2 at 37∘C for 2 h. After OGD neurons were removed from the chamber and returned to the incubator.
Excitotoxicity
To model excitotoxicity in primary cortical neurons, cells were exposed to 10 µM L-glutamate with 10 µM glycine. Neuronal cells from HT22 cell line were exposed to 5 mM L-glutamate.
Assessment of Cell Death
Cell death was quantified by measuring lactate dehydrogenase (LDH) released into supernatant using a colorometric assay.9 LDH values were normalized to 100% cell death caused by 0.5% Triton X-100 exposure. Data are reported as the percentage of dead cells relative to total cells and presented as mean ± standard deviation (SD).
Measurement of NAD+
NAD+ levels were quantified in subcellular fractions by the enzymatic cycling method using alcohol dehydrogenase as described.9 A standard curve was generated using known concentrations of NAD+ and levels were calculated.
STED Imaging
Neurons grown on poly-D-lysine coated glass coverslips were fixed in 2% paraformaldehyde and permeabilized with TritonX-100. Coverslips were then incubated in a 1:200 dilution of mouse monoclonal antibody against PAR (SA216, ENzo Life Sciences, Inc., Farmingdale, NY) and an antibody against TOMM20 (Abcam, Cambridge, MA) followed by incubation in the appropriate secondary antibodies. STED imaging was performed using a Leica TCS SP8 super resolution STED microscope with a pulsed white light laser and AOBS detection system was used (Leica Microsystems, Wetzlar, Germany). Images were collected using the 775 nm STED laser line with 30% 3D STED using the Leica STED WHITE oil objective lens (HC PL APO 100x/1.40 OIL) with a 200 Hz scan speed and 2× line averaging. Pixel size was set to 45 nm/pixel, step size was set to 160 µm, and pinhole was set at 132.8 µm (0.875 AU). TOMM20 was visualized using Alexa Fluor 555, exciting at 553 nm and detecting between 558–599 nm and temporally gate between 0.83–4.33 nm. PAR was visualized with Alexa Fluor 594, exciting at 598 nm and detecting between 603–666 nm and temporally gated between 0.3–6.0 nsec. DRAQ5 (ThermoFisher Scientific, Waltham, MA) used to label cell nuclei was excited at 662 and detected between 667 to 780 nm and temporally gated between 0.3–6.0 nsec. Channels were collected between stacks, sequentially.
COMET Tail Assay
HT22 cells were incubated for 60 min at 37°C in growth media (DMEM) with vehicle (DMSO, 0.1%), 10 µM XJB-veliparib, or 10 µM veliparib. Cells were then washed with PBS and exposed to 200 µM H2O2 in the presence of vehicle, XJB-veliparib, or veliparib for 30 min on ice. Cells were rewashed to remove H2O2, and media containing vehicle, XJB-veliparib, or veliparib was replaced for specified times before collection via rubber policeman. Cells were washed in 4°C PBS, counted, resuspended in DMEM (10% DMSO, 40% FBS), and frozen (−80°C) for batch analysis. The comet tail intensity assay was performed as previously described.47
Acknowledgements
We thank D. Franicola for technical assistance with the comet assay. HT22 cells were a generous gift from D. Schubert (The Salk Institute, La Jolla, CA).
Funding Sources
NIH grants U19 AI068021 (HB, ME, JG, PW) and R01 NS084604 (HB, RC, PW).
Footnotes
Disclosures
United States Patent Application No. 62/407,639 and International Patent Application PCT/US2017/056503 have been filed for Mitochondrially Targeted PARP Inhibitors.
References
- (1).Curtin NJ, and Szabo C (2013) Therapeutic applications of PARP inhibitors: anticancer therapy and beyond, Mol Aspects Med 34, 1217–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bai P (2015) Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance, Mol Cell 58, 947–958. [DOI] [PubMed] [Google Scholar]
- (3).Jubin T, Kadam A, Jariwala M, Bhatt S, Sutariya S, Gani AR, Gautam S, and Begum R (2016) The PARP family: insights into functional aspects of poly (ADP-ribose) polymerase-1 in cell growth and survival, Cell Prolif 49, 421–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Shen Y, Aoyagi-Scharber M, and Wang B (2015) Trapping Poly(ADP-Ribose) Polymerase, J Pharmacol Exp Ther 353, 446–457. [DOI] [PubMed] [Google Scholar]
- (5).Brunyanszki A, Szczesny B, Virag L, and Szabo C (2016) Mitochondrial poly(ADP-ribose) polymerase: The Wizard of Oz at work, Free Radic Biol Med 100, 257–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Szczesny B, Brunyanszki A, Olah G, Mitra S, and Szabo C (2014) Opposing roles of mitochondrial and nuclear PARP1 in the regulation of mitochondrial and nuclear DNA integrity: implications for the regulation of mitochondrial function, Nucleic Acids Res 42, 13161–13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Martire S, Mosca L, and d’Erme M (2015) PARP-1 involvement in neurodegeneration: A focus on Alzheimer’s and Parkinson’s diseases, Mech Ageing Dev 146–148, 53–64. [DOI] [PubMed] [Google Scholar]
- (8).Guarente L (2014) Linking DNA damage, NAD(+)/SIRT1, and aging, Cell Metab 20, 706–707. [DOI] [PubMed] [Google Scholar]
- (9).Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SC, Graham SH, Carcillo JA, Szabo C, and Clark RSB (2003) Intra-mitochondrial poly-ADP-ribosylation contributes to NAD+ depletion and cell death induced by oxidative stress, J Biol Chem 278, 18426–18433. [DOI] [PubMed] [Google Scholar]
- (10).Lai Y, Chen Y, Watkins SC, Nathaniel PD, Guo F, Kochanek PM, Jenkins LW, Szabo C, and Clark RS (2008) Identification of poly-ADP-ribosylated mitochondrial proteins after traumatic brain injury, J Neurochem 104, 1700–1711. [DOI] [PubMed] [Google Scholar]
- (11).Wang YQ, Wang PY, Wang YT, Yang GF, Zhang A, and Miao ZH (2016) An Update on Poly(ADP-ribose)polymerase-1 (PARP-1) Inhibitors: Opportunities and Challenges in Cancer Therapy, J Med Chem 59, 9575–9598. [DOI] [PubMed] [Google Scholar]
- (12).Brown JS, Kaye SB, and Yap TA (2016) PARP inhibitors: the race is on, Br J cancer 114, 713–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Sisay M, and Edessa D (2017) PARP inhibitors as potential therapeutic agents for various cancers: focus on niraparib and its first global approval for maintenance therapy of gynecologic cancers, Gynecol Oncol Res Pract 4, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wagner LM (2015) Profile of veliparib and its potential in the treatment of solid tumors, Onco Targets Ther 8, 1931–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Penning TD, Zhu GD, Gandhi VB, Gong J, Liu X, Shi Y, Klinghofer V, Johnson EF, Donawho CK, Frost DJ, Bontcheva-Diaz V, Bouska JJ, Osterling DJ, Olson AM, Marsh KC, Luo Y, and Giranda VL (2009) Discovery of the Poly(ADP-ribose) polymerase (PARP) inhibitor 2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide (ABT-888) for the treatment of cancer, J Med Chem 52, 514–523. [DOI] [PubMed] [Google Scholar]
- (16).Pommier Y, O’Connor MJ, and de Bono J (2016) Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action, Sci Transl Med 8, 362ps317. [DOI] [PubMed] [Google Scholar]
- (17).Frantz MC, and Wipf P (2010) Mitochondria as a target in treatment, Environ Mol Mutagen 51, 462–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Hoye AT, Davoren JE, Wipf P, Fink MP, and Kagan VE (2008) Targeting mitochondria, Acc Chem Res 41, 87–97. [DOI] [PubMed] [Google Scholar]
- (19).Wipf P, Xiao J, Jiang J, Belikova NA, Tyurin VA, Fink MP, and Kagan VE (2005) Mitochondrial targeting of selective electron scavengers: synthesis and biological analysis of hemigramicidin-TEMPO conjugates, J Am Chem Soc 127, 12460–12461. [DOI] [PubMed] [Google Scholar]
- (20).Yamada Y, and Harashima H (2008) Mitochondrial drug delivery systems for macromolecule and their therapeutic application to mitochondrial diseases, Adv Drug Deliv Rev 60, 1439–1462. [DOI] [PubMed] [Google Scholar]
- (21).Kanai A, Zabbarova I, Amoscato A, Epperly M, Xiao J, and Wipf P (2007) Mitochondrial targeting of radioprotectants using peptidyl conjugates, Org Biomol Chem 5, 307–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ma J, Lim C, Sacher JR, Van Houten B, Qian W, and Wipf P (2015) Mitochondrial targeted beta-lapachone induces mitochondrial dysfunction and catastrophic vacuolization in cancer cells, Bioorg Med Chem Lett 25, 4828–4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Karlberg T, Hammarstrom M, Schutz P, Svensson L, and Schuler H (2010) Crystal structure of the catalytic domain of human PARP2 in complex with PARP inhibitor ABT-888, Biochemistry 49, 1056–1058. [DOI] [PubMed] [Google Scholar]
- (24).R, T. B., Jeffrey A, Siahaan TJ, Gangwar S, and Pauletti GM. (1997) Improvement of oral peptide bioavailability: Peptidomimetics and prodrug strategies, Adv Drug Deliv Rev 27, 235–256. [DOI] [PubMed] [Google Scholar]
- (25).Wipf P, Xiao J, and Stephenson CR (2009) Peptide-Like Molecules (PLMs): A Journey from Peptide Bond Isosteres to Gramicidin S Mimetics and Mitochondrial Targeting Agents, Chimia 63, 764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Xiao J, Weisblum B, and Wipf P (2005) Electrostatic versus steric effects in peptidomimicry: synthesis and secondary structure analysis of gramicidin S analogues with (E)-alkene peptide isosteres, J Am Chem Soc 127, 5742–5743. [DOI] [PubMed] [Google Scholar]
- (27).Xiao J, Weisblum B, and Wipf P (2006) Trisubstituted (E)-alkene dipeptide isosteres as beta-turn promoters in the gramicidin S cyclodecapeptide scaffold, Org Lett 8, 4731–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Polyzos A, Holt A, Brown C, Cosme C, Wipf P, Gomez-Marin A, Castro MR, Ayala-Pena S, and McMurray CT (2016) Mitochondrial targeting of XJB-5–131 attenuates or improves pathophysiology in HdhQ150 animals with well-developed disease phenotypes, Hum Mol Genet 25, 1792–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Xun Z, Rivera-Sanchez S, Ayala-Pena S, Lim J, Budworth H, Skoda EM, Robbins PD, Niedernhofer LJ, Wipf P, and McMurray CT (2012) Targeting of XJB-5–131 to Mitochondria Suppresses Oxidative DNA Damage and Motor Decline in a Mouse Model of Huntington’s Disease, Cell Rep 2, 1137–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Polyzos AA, Wood NI, Williams P, Wipf P, Morton AJ, and McMurray CT (2018) XJB-5–131-mediated improvement in physiology and behaviour of the R6/2 mouse model of Huntington’s disease is age- and sex-dependent, PLoS One 13, e0194580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ji J, Kline AE, Amoscato A, Samhan-Arias AK, Sparvero LJ, Tyurin VA, Tyurina YY, Fink B, Manole MD, Puccio AM, Okonkwo DO, Cheng JP, Alexander H, Clark RS, Kochanek PM, Wipf P, Kagan VE, and Bayir H (2012) Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury, Nat Neurosci 15, 1407–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Escobales N, Nunez RE, Jang S, Parodi-Rullan R, Ayala-Pena S, Sacher JR, Skoda EM, Wipf P, Frontera W, and Javadov S (2014) Mitochondria-targeted ROS scavenger improves post-ischemic recovery of cardiac function and attenuates mitochondrial abnormalities in aged rats, J Mol Cell Cardiol 77, 136–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Ji J, Baart S, Vikulina AS, Clark RS, Anthonymuthu TS, Tyurin VA, Du L, St Croix CM, Tyurina YY, Lewis J, Skoda EM, Kline AE, Kochanek PM, Wipf P, Kagan VE, and Bayir H (2015) Deciphering of mitochondrial cardiolipin oxidative signaling in cerebral ischemia-reperfusion, J Cereb Blood Flow Metab 35, 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Macias CA, Chiao JW, Xiao J, Arora DS, Tyurina YY, Delude RL, Wipf P, Kagan VE, and Fink MP (2007) Treatment with a novel hemigramicidin-TEMPO conjugate prolongs survival in a rat model of lethal hemorrhagic shock, Ann Surg 245, 305–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, Bontcheva-Diaz VD, Cox BF, DeWeese TL, Dillehay LE, Ferguson DC, Ghoreishi-Haack NS, Grimm DR, Guan R, Han EK, Holley-Shanks RR, Hristov B, Idler KB, Jarvis K, Johnson EF, Kleinberg LR, Klinghofer V, Lasko LM, Liu X, Marsh KC, McGonigal TP, Meulbroek JA, Olson AM, Palma JP, Rodriguez LE, Shi Y, Stavropoulos JA, Tsurutani AC, Zhu GD, Rosenberg SH, Giranda VL, and Frost DJ (2007) ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models, Clin Cancer Res 13, 2728–2737. [DOI] [PubMed] [Google Scholar]
- (36).Horton TM, Jenkins G, Pati D, Zhang L, Dolan ME, Ribes-Zamora A, Bertuch AA, Blaney SM, Delaney SL, Hegde M, and Berg SL (2009) Poly(ADP-ribose) polymerase inhibitor ABT-888 potentiates the cytotoxic activity of temozolomide in leukemia cells: influence of mismatch repair status and O6-methylguanine-DNA methyltransferase activity, Mol Cancer Ther 8, 2232–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ito S, Murphy CG, Doubrovina E, Jasin M, and Moynahan ME (2016) PARP Inhibitors in Clinical Use Induce Genomic Instability in Normal Human Cells, PLoS One 11, e0159341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, and Pommier Y (2012) Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors, Cancer Res 72, 5588–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Chiarugi A, Meli E, Calvani M, Picca R, Baronti R, Camaioni E, Costantino G, Marinozzi M, Pellegrini-Giampietro DE, Pellicciari R, and Moroni F (2003) Novel isoquinolinone-derived inhibitors of poly(ADP-ribose) polymerase-1: pharmacological characterization and neuroprotective effects in an in vitro model of cerebral ischemia, J Pharmacol Exp Ther 305, 943–949. [DOI] [PubMed] [Google Scholar]
- (40).Moroni F, Meli E, Peruginelli F, Chiarugi A, Cozzi A, Picca R, Romagnoli P, Pellicciari R, and Pellegrini-Giampietro DE (2001) Poly(ADP-ribose) polymerase inhibitors attenuate necrotic but not apoptotic neuronal death in experimental models of cerebral ischemia, Cell Death Differ 8, 921–932. [DOI] [PubMed] [Google Scholar]
- (41).Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, and Dawson VL (1997) Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia, Nat Med 3, 1089–1095. [DOI] [PubMed] [Google Scholar]
- (42).Narne P, Pandey V, Simhadri PK, and Phanithi PB (2017) Poly(ADP-ribose)polymerase-1 hyperactivation in neurodegenerative diseases: The death knell tolls for neurons, Semin Cell Dev Biol 63, 154–166. [DOI] [PubMed] [Google Scholar]
- (43).Zhang J, Dawson VL, Dawson TM, and Snyder SH (1994) Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity, Science 263, 687–689. [DOI] [PubMed] [Google Scholar]
- (44).Mandir AS, Poitras MF, Berliner AR, Herring WJ, Guastella DB, Feldman A, Poirier GG, Wang ZQ, Dawson TM, and Dawson VL (2000) NMDA but not non-NMDA excitotoxicity is mediated by Poly(ADP-ribose) polymerase, J Neurosci 20, 8005–8011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Yang JL, Sykora P, Wilson DM 3rd, Mattson MP, and Bohr VA (2011) The excitatory neurotransmitter glutamate stimulates DNA repair to increase neuronal resiliency, Mech Ageing Dev 132, 405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Liu Y, Wang W, Li Y, Xiao Y, Cheng J, and Jia J (2015) The 5-Lipoxygenase Inhibitor Zileuton Confers Neuroprotection against Glutamate Oxidative Damage by Inhibiting Ferroptosis, Biol Pharm Bull 38, 1234–1239. [DOI] [PubMed] [Google Scholar]
- (47).Berhane H, Epperly MW, Goff J, Kalash R, Cao S, Franicola D, Zhang X, Shields D, Houghton F, Wang H, Wipf P, Parmar K, and Greenberger JS (2014) Radiologic differences between bone marrow stromal and hematopoietic progenitor cell lines from Fanconi Anemia (Fancd2(−/−)) mice, Radiat Res 181, 76–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Hong SJ, Dawson TM, and Dawson VL (2004) Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling, Trends Pharmacol Sci 25, 259–264. [DOI] [PubMed] [Google Scholar]
- (49).Burzio LO, Saez L, and Cornejo R (1981) Poly (ADP-ribose) synthetase activity in rat testis mitochondria, Biochem Biophys Res Commun 103, 369–375. [DOI] [PubMed] [Google Scholar]
- (50).Kun E, Zimber PH, Chang AC, Puschendorf B, and Grunicke H (1975) Macromolecular enzymatic product of NAD+ in liver mitochondria, Proc Natl Acad Sci U S A 72, 1436–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Richter C, Winterhalter KH, Baumhuter S, Lotscher HR, and Moser B (1983) ADP-ribosylation in inner membrane of rat liver mitochondria, Proc Natl Acad Sci U S A 80, 3188–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Masmoudi A, Islam F, and Mandel P (1988) ADP-ribosylation of highly purified rat brain mitochondria, J Neurochem 51, 188–193. [DOI] [PubMed] [Google Scholar]
- (53).Audebert M, Salles B, and Calsou P (2004) Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining, J Biol Chem 279, 55117–55126. [DOI] [PubMed] [Google Scholar]
- (54).Bai P, Canto C, Oudart H, Brunyanszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper RH, Schoonjans K, Schreiber V, Sauve AA, Menissier-de Murcia J, and Auwerx J (2011) PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation, Cell Metab 13, 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Ying W, Alano CC, Garnier P, and Swanson RA (2005) NAD+ as a metabolic link between DNA damage and cell death, J Neurosci Res 79, 216–223. [DOI] [PubMed] [Google Scholar]
- (56).Satchell MA, Zhang X, Kochanek PM, Dixon CE, Jenkins LW, Melick J, Szabo C, and Clark RS (2003) A dual role for poly-ADP-ribosylation in spatial memory acquisition after traumatic brain injury in mice involving NAD+ depletion and ribosylation of 14–3-3gamma, J Neurochem 85, 697–708. [DOI] [PubMed] [Google Scholar]
- (57).Whalen MJ, Clark RS, Dixon CE, Robichaud P, Marion DW, Vagni V, Graham SH, Virag L, Hasko G, Stachlewitz R, Szabo C, and Kochanek PM (1999) Reduction of cognitive and motor deficits after traumatic brain injury in mice deficient in poly(ADP-ribose) polymerase, J Cereb Blood FlowMetab 19, 835–842. [DOI] [PubMed] [Google Scholar]
- (58).Fan J, Dawson TM, and Dawson VL (2017) Cell Death Mechanisms of Neurodegeneration, Adv Neurobiol 15, 403–425. [DOI] [PubMed] [Google Scholar]
- (59).Wang Y, Kim NS, Haince JF, Kang HC, David KK, Andrabi SA, Poirier GG, Dawson VL, and Dawson TM (2011) Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos), Sci Signal 4, ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Berger NA, Besson VC, Boulares AH, Burkle A, Chiarugi A, Clark RS, Curtin NJ, Cuzzocrea S, Dawson TM, Dawson VL, Hasko G, Liaudet L, Moroni F, Pacher P, Radermacher P, Salzman AL, Snyder SH, Soriano FG, Strosznajder RP, Sumegi B, Swanson RA, and Szabo C (2017) Opportunities for the repurposing of PARP inhibitors for the therapy of non-oncological diseases, Br J Pharmacol [DOI] [PMC free article] [PubMed]
- (61).Skoda EM, Davis GC, and Wipf P (2012) Allylic Amines as Key Building Blocks in the Synthesis of (E)-Alkene Peptide Isosteres, Org Process Res Dev 16, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Zhang X, Chen J, Graham SH, Du L, Kochanek PM, Draviam R, Guo F, Nathaniel PD, Szabo C, Watkins SC, and Clark RS (2002) Intranuclear localization of apoptosis-inducing factor (AIF) and large scale DNA fragmentation after traumatic brain injury in rats and in neuronal cultures exposed to peroxynitrite, J Neurochem 82, 181–191. [DOI] [PubMed] [Google Scholar]