

Summary:
The reaction of di(2-pyridyl) carbonate with a variety of alcohols including hindered secondary, tertiary and protected glycols afforded the corresponding mixed carbonate which was efficiently transformed into various carbamates in high yield under mild conditions.
The alkoxycarbonylation of amines is an important reaction which is used frequently in the synthesis of pharmacologically important organic molecules.1 In recent years, several alkoxycarbonylation methods have appeared in the literature.2 However, the scope of existing methodologies are limited by the need for specialized reagents, poor reactivity towards sterically hindered alcohols and operational complexity due to use of highly toxic phosgene. In connection with work aimed at synthesizing biologically active compounds for probing enzyme active-sites, we required a general and synthetically reliable method for the synthesis of various carbamate derivatives. In this letter, we describe the utility of di(2-pyridyl) carbonate3 as an efficient, high-yielding and convenient alkoxycarbonylation reagent for amines that overcomes many of the limitations of existing methodologies.
Di(2-pyridyl) carbonate4 (DPC) 1 was readily prepared from commercially available 2-hydroxypyridine and triphosgene in methylene chloride in the presence of triethylamine. The product was isolated by extractive workup with aqueous sodium bicarbonate followed by the crystallization of the crude residue from ether and petroleum ether (mp=78°C, 81%). DPC is quite stable and can be stored at room temperature for several months without any noticeable decomposition.
(+)-Menthol 2 was reacted with 1.5 equiv. of DPC and 1.5 equiv. of triethylamine in methylene chloride at 23°C for 12 h to provide the mixed carbonate 3 after work-up with aqueous sodium bicarbonate solution. The carbonate 3 was fairly stable and was purified by silica gel chromatography to obtain analytically pure 3. The identity of structure 3 was confirmed by 1H-NMR spectrum and mass spectral analysis. Reaction of mixed carbonate 3 with 1.2 equiv. of protected riboseamine 45 in methylene chloride afforded the corresponding pure carbamate 6 after silica gel chromatography (81%). Furthermore, reaction of carbonate 3 with 1.2 equiv. of amine 5 under the same reaction conditions resulted in carbamate 7 (70%).
As evidenced in table I, these reaction conditions were shown to be suitable for functionally diverse primary and secondary alcohols6 (entry 1-8). In the case of entry 9 and 10, the reaction of tertiary alcohol7 17 with DPC in the presence of various amine bases afforded only a low yield (<10%) of the desired mixed carbonate. However, treatment of 17 with potassium hydride (1.5 equivalents at 0°C to 23°C for 1 h) and DPC (1.5 equivalents at 23°C for 4 h) in dry tetrahydrofuran, followed by workup with saturated ammonium chloride furnished the corresponding mixed carbonate.9 Reaction of the amines 4 and 5 (1.2 equivalents) with the mixed carbonate of 17 in methylene chloride provided the corresponding carbamates 18 and 19 in good yield.
Table I:
Synthesis of carbamates with DPCa
| Entry | Alcohols | Amines | Carbamates | Method | Yields(%)b |
|---|---|---|---|---|---|
| 1. | 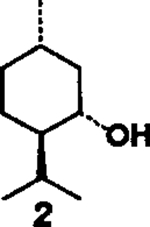 |
4 | 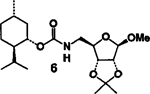 |
A | 81 |
| 2. | 2 | 5 | 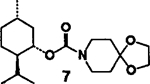 |
A | 70 |
| 3. | 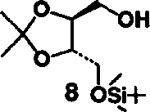 |
4 | 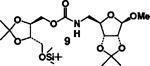 |
A | 79 |
| 4. | 8 | 5 | 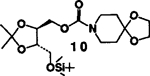 |
A | 87 |
| 5. | 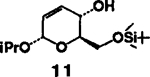 |
4 | 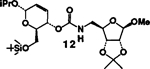 |
A | 77 |
| 6. | 11 | 5 | 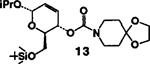 |
A | 83 |
| 7. | 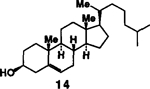 |
4. | 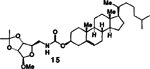 |
A | 81 |
| 8. | 14 | 5 | 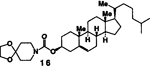 |
A | 83 |
| 9. | 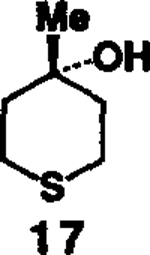 |
4 | 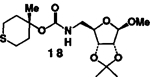 |
B | 72 |
| 10. | 17 | 5 | 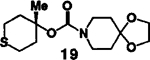 |
B | 68 |
All reactions were carried out under N2
Yield of pure products after silica gel chromatography (0.5–4.0 mmol scale)
In conclusion, the results presented in this paper considerably extend the scope and utility of di(2-pyridyl) carbonate in the synthesis of a variety of functionalized carbamates. Application of this methodology in the synthesis of various biologically active carbamates is currently under investigation. The following procedures are illustrative.10
Preparation of Di(2-pyridyl) carbonate 1:
To a stirred solution of triphosgene (10 mmol, 3 g) and 2-hydroxypyridine (60 mmol, 5.7 g) in CH2Cl2 (500 ml) at 0°C was added Et3N (75 mmol, 10.5 ml) dropwise over 15 min. The mixture was stirred at 23°C for 5 h and the solvent was evaporated under reduced pressure. The residue was dissolved in EtOAc (500 ml), washed with sat’d NaHCO3 (300 ml), brine (200 ml) and dried over Na2SO4. Removal of the solvent and subsequent recrystallization of the redidue from ether and petroleum ether afforded di(2-pyridyl) carbonate (5.2 g, 81% yield) as a white solid: mp 76–78°C.
Preparation of Carbamate 6 (method A):
To a stirred solution of (+)-menthol 2 (1.0 mmol, 0.16 g) in CH2Cl2 (5 ml) at 23°C were added DPC (1.5 mmol, 0.32 g) and Et3N ( 1.5 mmol, 0.2 ml). The contents were stirred for 12 h and then diluted with CH2Cl2 (25 ml). The mixture was washed with sat’d NaHCO3 (10 ml), brine (10 ml) and dried over Na2SO4. The solvent was evaporated and the residue was used directly for the next reaction.
The residue was dissolved in CH2Cl2 (2 ml) and added to a stirred solution of amine 4 (1.2 mmol, 0.24 g) in CH2Cl2 (5 ml). The mixture was stirred for 12 h and then diluted with CH2Cl2 (20 ml). The resulting mixture was washed with 10% aqueous citric acid (10 ml), sat’d NaHCO3 (10 ml) and dried over Na2SO4. Removal of the solvent, followed by chromatography over silica gel (25% EtOAc-hexanes) afforded carbamate 6 (0.28 g, 81%) as a white solid: mp 68–70°C; IR (neat) 1702 cm−1; 1H NMR (CDCl3) δ 5.2 (br s, 1H), 5.0 (s, 1H), 4.6 (m, 3H), 4.3 (t,1H, J=6.1 Hz), 3.35 (s, 3H), 3.31 (m, 2H,), 1.85–2.1 (m, 2H), 1.0–1.7 (m, 7H), 1.48 (s, 3H), 1.32 (s, 3H), 0.9 (d, 6H, J=7.0 Hz), 0.82 (d, 3H, J=6.9 Hz).
Preparation of Carbamate 19 (method B):
To a stirred suspension KH (1.2 mmol, 0.05 g, pre-washed) in THF (2 ml) at 0°C was added a solution of alcohol 17 (0.83 mmol, 0.11 g) in THF (2 ml) dropwise over 5 min. The mixture was stirred (0°C to 23°C) for 1 h and DPC (1.2 mmol, 0.27 g) was added. The mixture was stirred for 4 h and the reaction was cautiously quenched with sat’d NH4Cl solution. The solvent was removed under reduced pressure, and the residue was diluted with sat’d NaHCO3 (5 ml) and extracted with EtOAc (2×10 ml). The combined extracts were dried over Na2SO4 and concentrated. The residue was then dissolved in CH2Cl2 (2 ml) and added to a stirred solution of amine 5 (1.0 mmol, 0.14 g) in CH2Cl2 (3 ml). After stirring for 12 h (23°C), the reaction mixture was worked-up and purified according to above procedure to afford 19 (0.17 g, 68%) as a white solid: mp 76–78°C; IR (neat) 1695 cm−1; 1H NMR (CDCl3) δ 3.98 (s, 4H), 3.52 (t, 4H, J=5.8 Hz), 2.85 (m, 2H), 2.35–2.61 (m, 4H), 1.7 (m, 6H), 1.51 (s, 3H).
Scheme 1.

Acknowledgement:
The authors thank Dr. Wayne J. Thompson for helpful discussions and acknowledge the encouragement and support of Dr. Joel R. Huff and Dr. Paul S. Anderson.
References and Notes:
- 1.(a) Matassa VG, Maduskuie TP, Shapiro HS, Hesp B, Snyder DW, Aharony D, Krell RD and Keith RA; J. Med. Chem, 1990,33, 1781; [DOI] [PubMed] [Google Scholar]; (b) Firestone RA, Pisano JM, Falck JR, McPhaul MM and Krieger M; J. Med. Chem, l984,22,1037. [DOI] [PubMed] [Google Scholar]
- 2.(a) Eckert H and Forster B; Anaew Chem Int Ed, 1987, 26, 894; [Google Scholar]; (b) Denarie M, Grenouillat D, Malfroot T, Senet J-P, Sennyey G and Wolf P; Tetrahedron Letters, 1987, 28,5823; [Google Scholar]; (c) Pedersen U; Weyl Houben, Methoden der Organischen Chemie 1983, E4, 144; [Google Scholar]; (d) Takeda K, Tsuboyama K, Yamaguchi K, Ogura H; j. Org. Chem, 1985,50, 273; [Google Scholar]; (e) Shute RE and Rich DH; synthesis, 1987, 346. [DOI] [PubMed] [Google Scholar]
- 3.Kim S, Lee JI and Ko YK; Tetrahedron Letters, 1984, 25, 4943. [Google Scholar]
- 4.This reagent has been known in the literature by a variety of names:; (a) O,O’-Carbonylbis(α-hydroxypyridine); Cramer F, CA 64:6625d; [Google Scholar]; (b) O,O’-Carbonyldihydroxypyridine; Kampe W, Angew Chem,1963, 75(13), 641; [Google Scholar]; (c) Di(2-pyridyl) carbonate; Kampe W, Chem. Ber,1965, 98(4) 1031. [Google Scholar]
- 5.(a) Levene PA and Stiller ET; J. Biol. Chem, 1934, 106, 421; [Google Scholar]; (b) Leonard NJ and Carraway KL; J Het. Chem, 1966,3,485. [Google Scholar]
- 6. (a).Alcohol 11 was prepared by Ferrier rearrangement (ref. 8) of tri-O-acetyl-D-glucal with isopropanol, hydrolysis of the resulting acetates with aqueous methanol and triethylamine, followed by selective protection of primary alcohol as TBDMS ether (63%, overall yield):; for alcohol 8, see: lida H, Yamazaki N and Kibayashi C; J. Org. Chem, 1987,52, 3337.; [Google Scholar]
- 7.Alcohol 17 was obtained by reaction of methylmagnesium bromide and tetrahydrothiopyran-4 one in dry tetrahydrofuran at 23°C for 12h (51% isolated yield).
- 8.Ferrier RJ and Prasad NJ; J. Chem. Sot. (C), 1969,570. [Google Scholar]
- 9.Mixed carbonate often undergoes rearrangement to the corresponding alkoxycarbonyl 2-pyridone derivative (5-10%), which imposes no complication to subsequent reaction.
- 10.All new compounds gave satisfactory spectroscopic and analytical results.