

Abstract
Sirtuin inhibitors have attracted much interest due to the involvement of sirtuins in various biological processes. Several SIRT2-selective inhibitors have been developed, and some exhibit anticancer activities. To facilitate the choice of inhibitors in future studies and the development of better inhibitors, we directly compared several reported SIRT2-selective inhibitors: AGK2, SirReal2, Tenovin-6, and TM. In vitro, TM is the most potent and selective inhibitor, and only TM could inhibit the demyristoylation activity of SIRT2. SirReal2, Tenovin-6, and TM all showed cytotoxicity in cancer cell lines, with Tenovin-6 being the most potent, but only TM showed cancer cell-specific toxicity. All four compounds inhibited the anchorage-independent growth of HCT116 cells, but the effect of TM was most significantly affected by SIRT2 overexpression, suggesting that the anticancer effect of TM depends more on SIRT2 inhibition. These results not only provide useful guidance about choosing the right SIRT2 inhibitor in future studies, but also suggest general practices that should be followed for small molecule inhibitor development activities.
Keywords: Inhibitors, Sirtuins, SIRT2, Cytotoxicity, Cancer
Graphical Abstract
Direct comparison of SIRT2 inhibitors identified substrate-dependent inhibition of enzymes and highlight good general practices for small molecule inhibitor development.
The nicotinamide adenine dinucleotide (NAD)-dependent protein lysine deacylases, or sirtuins, have attracted a lot of attention as potential targets to treat cancer and various other diseases such as neurodegeneration.[1] There are seven mammalian sirtuins, SIRT1-SIRT7, which have been shown to play important roles in vivo by regulating processes such as DNA repair, transcription, cell cycle, and metabolism.[2]
The physiological function of sirtuins is a result of their enzymatic activity on different substrate proteins. Sirtuins can remove various acyl modifications from lysine residues on histone and non-histone proteins. SIRT1, SIRT2 and SIRT3 can efficiently deacetylate proteins, while SIRT5 preferentially hydrolyzes succinyl and malonyl groups, and SIRT1, SIRT2, SIRT3, SIRT6, and SIRT7 can all hydrolyze long-chain fatty acyl groups. [3]
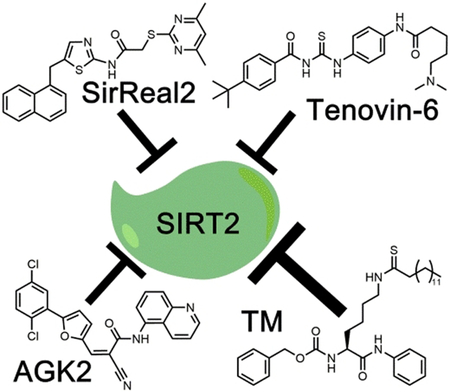
Among the mammalian seven sirtuins, inhibition of SIRT2 has been reported to have beneficial effects in cancer and neurodegenerative diseases.[1-2] SIRT2 plays an important role in regulating the cell cycle, oxidative stress response, metabolism, apoptosis, autophagy, differentiation, and aging.[1-2, 4] Initial studies presented conflicting results that suggested SIRT2 has both tumor suppressing and promoting roles.[5] More recently, it has been shown that SIRT2 depletion, or inhibition, has anticancer effects.[5a, 6] However, the full potential of inhibiting SIRT2 as a therapeutic option requires the development of potent and selective SIRT2 inhibitors. In the past decade, several SIRT2 inhibitors have been reported. Several of them, AGK2, SirReal2, Tenovin-6, and a thiomyristoyl lysine compound TM (Figure 1) are commercially available.
Figure 1.

Structures of SIRT2 inhibitors AGK2, SirReal2, Tenovin-6, and TM.
AGK2 was identified from a screening of 200 compounds as a potent SIRT2 inhibitor that could rescue α-synuclein-mediated toxicity.[7] It has also been reported to exhibit an anticancer effect in a few cancer cells.[8] SirReal2 was also identified through a compound screening that was aimed at identifying more potent and selective sirtuin inhibitors.[9] However, there have not been any studies reported regarding its effect on cancer cells. Tenovin-6 was identified when screening compounds that could activate the tumor suppressor p53.[10] Tenovin-6 has been shown to effectively inhibit SIRT1 and SIRT2 in cells, and shows promising anticancer activity. Recently, our lab developed TM, a mechanism-based selective SIRT2 inhibitor. TM exhibits broad anticancer effects and promotes the degradation of the c-Myc oncoprotein in several cancer cell lines.[5a] TM exhibits broad anticancer effects and promotes the degradation of the c-Myc oncoprotein in several cancer cell lines. Given the availability of different SIRT2 inhibitors, we reasoned that a direct comparison would be useful to help users choose the right compounds for the proper applications. This is especially true because in most studies, only in vitro IC50 values (concentrations that lead to 50% of SIRT2 inhibition) were reported, which is known to be dependent on the exact experimental conditions used. Thus, it is still ambiguous as to which inhibitor is the most potent, selective, and exhibits the best anticancer effect. Furthermore, it is also unclear which enzymatic activity (deacetylation or defatty-acylation) these inhibitors are potent against. Here, we directly compare the potency and selectivity of these compounds to help identify which inhibitor should be used to study the function of SIRT2, and explore its therapeutic potential.
The SIRT2 inhibition potency and selectivity of the four inhibitors was determined using a high-performance liquid chromatography (HPLC)-based assay. In this assay, we first incubated the enzyme with the inhibitors for 15 min and then added the substrates to initiate the reactions. The activity of the enzymes was detected using two different substrates, histone H3 lysine 9 (H3K9) acetyl (H3K9Ac) and myristoyl (H3K9Myr) peptides. We determined the IC50 values for inhibiting the deacetylation activity of SIRT1–3 and the demyristoylation activity of SIRT2 and SIRT6 (Table 1). We found TM was the most potent and selective SIRT2 inhibitor in vitro among all the tested compounds. TM was able to inhibit both the deacetylation (IC50 0.038 μM) and demyristoylation (IC50 0.049 μM) activity of SIRT2. Furthermore, TM is the most selective inhibitor as it inhibited SIRT2 activity 650-fold more efficiently than it could inhibit SIRT1, and it essentially did not inhibit SIRT3 or SIRT6.
Table 1.
In vitro IC50 values (μM, average and standard deviation from three independent experiments)
| AGK2 | SirReal2 | Tenovin-6 | TM | |
|---|---|---|---|---|
| SIRT1 H3K9Ac | 42 ± 4 | 82 ± 14 | 26 ± 10 | 26 ± 15 |
| SIRT2 H3K9Ac | 8 ± 5 | 0.23 ± 0.08 | 9 ± 7 | 0.04 ± 0.02 |
| SIRT2 H3K9Myr | >100 | >100 | >200 | 0.05 ± 0.03 |
| SIRT3 H3K9Ac | >50 | >50 | >50 | >50 |
| SIRT6 H3K9Myr | >100 | >200 | >200 | >200 |
| Selectivity[a] | 5 | 357 | 3 | 650 |
(IC50 for SIRT1 on H3K9Ac) / (IC50 for SIRT2 on H3K9Ac)
SirReal2 inhibits the deacetylation activity of SIRT2 with a relatively low IC50 value of 0.23 μM, however, consistent with a recent report it was unable to inhibit the demyristoylation activity of SIRT2 at the highest concentration tested.[11] AGK2 and Tenovin-6 inhibited both SIRT1 and SIRT2 deacetylation with similar IC50 values, suggesting that these two inhibitors are not very selective. Like SirReal2, these compounds did not inhibit the demyristoylation activity of SIRT2 at the highest concentration tested.
Because competitive inhibitors do not require pre-incubating the enzyme and inhibitor, while the mechanism-based inhibitor TM requires pre-incubation to reach maximum SIRT2 inhibition, we also determined the IC50 values for the inhibitors without pre-incubation (Supplemental Table 1). As expected, the IC50 values for the competitive inhibitors with and without pre-incubation were comparable. For TM, the IC50 values for inhibiting SIRT2 deacetylation were similar with and without pre-incubation, but TM did not inhibit SIRT2 demyristoylation activity without pre-incubation.
In the absence of pre-incubation, TM needs to compete with H3K9Ac or H3K9Myr for binding to SIRT2. H3K9Myr likely binds much stronger than TM because of the extra hydrogen bonding provided by the peptide backbone, making TM ineffective at inhibiting demyristoylation. However, TM likely binds stronger than H3K9Ac to SIRT2 because the thiomyristoyl group contributes a lot to the binding free energy because of the strong hydrophobic interaction, rendering TM an efficient inhibitor for the deacetylation activity of SIRT2. This is consistent with the fact that SIRT2 has much higher Km values, which are upper estimates of Kd values, for acetyl peptides than for myristoyl peptides.[3a] Thus, TM, being a thiomyristoyl lysine compound, binds more tightly to SIRT2 than the acetyl lysine peptide substrate. Hence, it can effectively compete with the acetyl peptide substrate in the reaction mixture even without pre-incubation. On the other hand, TM could not compete with the myristoyl peptide substrate because the myristoyl peptide backbone provides additional hydrogen bonding interactions with SIRT2, making it an even more tight binder than TM. This explains why without pre-incubation, SIRT2 is not inhibited by TM if myristoyl peptide is used as a substrate.
However, with pre-incubation, TM will first form an ADP-ribosyl covalent intermediate, which binds to SIRT2 much more tightly than TM due to binding interactions from both the ADP-ribose and TM. This covalent intermediate can efficiently prevent both the H3K9Ac and H3K9Myr peptides from binding to SIRT2, rendering TM an effective inhibitor for both deacetylation and demyristoylation under pre-incubation conditions. In vivo and in cells, there is no real way for pre-incubation, as the substrate either is or is not present. However, cells are dynamic and protein localization is constantly changing. Therefore, it is possible that small molecule inhibitors such as TM may encounter SIRT2 prior to its interaction with substrates allowing it to potentially inhibit the defatty-acylation of certain substrates. Thus, the in-cell potency of SIRT2 inhibition by TM may be between with pre-incubation and without preincubation. For other inhibitors that are not mechanism-based, the inhibitors are competitive with the H3K9Ac or H3K9Myr peptides. Because H3K9Myr binds SIRT2 more tightly than H3K9Ac, the inhibitors cannot effectively compete with H3K9Myr, making them ineffective at inhibiting the demyristoylation activity of SIRT2.
To further study the selectivity of these SIRT2 inhibitors, we next examined their effects on the acetylation levels of known SIRT1 and SIRT2 substrates. The tumor suppressor p53 is a well-established SIRT1 deacetylation target.[12] To determine if the compounds can efficiently inhibit SIRT1 in cells, we looked at the acetylation levels of p53 in MCF-7 cells after treating the cells with the inhibitors at 25 mM. Only Tenovin-6 increased p53 acetylation (Figure 2A), which is consistent with a previous report showing that Tenovin-6 can increase Ac-p53 levels through SIRT1 inhibition.[10] These results suggest that only Tenovin-6 inhibits the deacetylation activity of SIRT1 in cells.
Figure 2.

Evaluation of the inhibition of SIRT1 and SIRT2 by different inhibitors in cells. (A) Detection of acetyl p53 (Ac-p53) levels in MCF-7 cells after 6-hour treatment with indicated inhibitors and 200 nM of TSA, an HDAC inhibitor. (B) Detection of acetyl α-tubulin (Ac-α-tubulin) levels by immunofluorescence in MCF-7 cells after 6-hour treatment with indicated inhibitors.
To examine the inhibition of SIRT2 in cells, we detected acetylation levels on α-tubulin, a well-established SIRT2 substrate, using immunofluorescence after inhibitor treatment.[4e] As expected, all the compounds increased the acetylation of α-tubulin in MCF-7 cells at 25 μM (Figure 2B). Thus, it appears that all the compounds can inhibit SIRT2’s tubulin deacetylation activity.
Tenovin-6, with an in vitro IC50 value of 9 μM for SIRT2 deacetylation, was able to inhibit tubulin deacetylation in cells at 25 μM. TM and SirReal2, with much lower in vitro IC50 values for SIRT2 deacetylation, still require tens of μM concentrations to inhibit tubulin deacetylation. Differences in cellular uptake and solubility of the inhibitors may lead to this observation. The in vitro and in cell inhibition of SIRT1 is also consistent with this explanation. For example, both TM and Tenovin-6 inhibit SIRT1 in vitro with an IC50 values of ~26 μM, but in cells at 25 μM, only Tenovin-6 was able to inhibit SIRT1 (measured by p53 deacetylation). Thus, TM is the most potent and selective in vitro SIRT2 inhibitor and the SIRT2 selectivity is maintained in cells, while Tenovin-6 is not very selective for SIRT2, but it may have better solubility and cellular uptake.
To evaluate the anticancer effect of these four inhibitors, we first looked at cytotoxicity of these compounds in several breast cancer cell lines (MDA-MB-468, MDA-MB-231, MCF-7, SK-BR-3) and two normal mammary epithelial cell lines (MCF10A and HME-1 cells). To evaluate the cytotoxicity, we looked at the GI50 value, or the small molecule inhibitor concentration which inhibits 50% of cell growth. As shown in Figure 3, Tenovin-6 was the most potent compound, exhibiting GI50 values of a few μM (Figure 3, Supplemental Table 2, Supplemental Figure 1). The anticancer effect of SirReal2 had not previously been studied, it was interesting to note that it had a modest effect on several of the cell lines tested, including the normal mammary epithelial cell lines. TM was not as potent as Tenovin-6, but was more potent than SirReal2 (Figure 3). More interestingly, TM was more potent in the cancer cell lines than in the normal mammary epithelial cell lines (GI50 >50 μM in MCF10A and HME-1 cells), suggesting it selectively targets cancer cells. AGK2 had very weak effects on the cell proliferation of the cell lines tested, with all the GI50 values >50 μM.
Figure 3.

Heat map of GI50 (μM) values of different SIRT2 inhibitors in various breast cancer and normal breast cell lines. The values represent the average of three independent experiments done in duplicate.
We next determined the GI50 values of these inhibitors in various other cancer cell lines, including colon cancer (HCT116, SW948, and HT29), lung cancer (A549 and H520), leukemia (K562), cervical cancer (HeLa), and pancreatic cancer (ASPC1 and CFPAC1) cell lines (Figure 4, Supplemental Table 3, and Figure S2). Like the results obtained with breast cancer cell lines, Tenovin-6 was the most potent compound in almost every cell line tested. TM was the second most potent compound, followed by SirReal2 in almost all of the cell lines tested, while AGK2 was in general the least potent compound. As AGK2 efficiently inhibits the deacetylation of α-tubulin, but it is not particularly toxic to cancer cells, our data suggests that inhibiting tubulin deacetylation does not contribute to the anticancer activity of these inhibitors.
Figure 4.

Heat map of GI50 values (μM) of different SIRT2 inhibitors in various cancer cell lines. The values represent an average of three independent experiments done in duplicate.
Cancer cells have the ability to grow on soft agar without attaching to extracellular matrix, while normal cells cannot. Thus, a soft agar anchorage-independent growth assay is typically used to examine the transformed phenotype of cancer cells. We therefore examined the effect of SIRT2 inhibitors on anchorage-independent growth. Because this assay is more labor-intensive and time consuming, we limited our study to one cancer cell line, HCT116. Interestingly, we saw a different activity trend from the cytotoxicity assay (Table 2, Supplemental Figure S4). Tenovin-6 is still the most potent compound with a GI50 value of 2.1 μM. TM is the second most potent compound with a GI50 value of 13.5 μM. SirReal2 was not very active in this assay, with a GI50 value of 55.8 μM. In contrast, AGK2, which was not active in the cytotoxicity assay, showed much better activity in the soft agar assay with an GI50 value of 24.1 μM.
Table 2.
The GI50 (μM) values of different SIRT2 inhibitors on the anchorage-independent growth of HCT116 cells with and without SIRT2 overexpression.
| AGK2 | SirReal2 | Tenovin-6 | TM | |
|---|---|---|---|---|
| Control | 24.1 | 55.8 | 2.1 | 13.5 |
| SIRT2 expression | 27.6 | 58.1 | 2.3 | 24.2 |
| Fold Change[a] | 1.2 | 1 | 1.1 | 1.8 |
(GI50 for SIRT2 expression) / (GI50 for Control)
Results were obtained from all individual samples from at least three replicates, standard deviation are presented in supplemental information.
To study if the effect on anchorage-independent growth is a result of SIRT2 inhibition, we obtained the GI50 values of these inhibitors on the anchorage-independent growth of HCT116 cells with SIRT2 overexpression and compare to that on HCT116 cells without SIRT2 overexpression (Table 2, Supplemental Figure S4). SIRT2 overexpression increased the GI50 value of TM by 1.8-fold, suggesting that the suppressive effect on anchorage-independent growth is dependent on SIRT2 inhibition. In contrast, SIRT2 overexpression had no or very little effect on the GI50 values of SirReal2, Tenovin-6, or AGK2, suggesting possible off-target effects. This is consistent with a recent report suggesting that Tenovin-6 impairs autophagy independent of sirtuins.[13] We also examined the effect of SIRT2 overexpression on the cytotoxicity of these inhibitors in MDA-MB-468 and HCT116 cells and the results were similar to that on anchorage-independent growth (Supplemental Figure S5, S6).
In summary, we have compared four established SIRT2 inhibitors: AGK2, SirReal2, Tenovin-6, and TM. This study will help people interested in using SIRT2 inhibitors to choose the proper compounds to elucidate the function of SIRT2 and to explore the therapeutic potential of inhibiting SIRT2. TM, while not the most potent inhibitor against cancer cells, is the most potent and selective SIRT2 inhibitor and its anticancer activity is dependent on SIRT2 inhibition. We suggest future studies to understand the physiological function of SIRT2 should use TM before better SIRT2 inhibitors are developed.
Our study also highlights a previously under-appreciated point regarding small molecule inhibitors for enzymes, which is the substrate or activity-dependent inhibition of enzymes. SIRT2 has been reported to have both deacetylation and demyristoylation activities. Interestingly, most of the inhibitors tested here only inhibited the deacetylation activity, but not the demyristoylation activity. TM is the only compound that can inhibit both activities, but the inhibition of demyristoylation is less potent. We think this is likely because myristoyl peptides have much higher binding affinities (reflected by the much lower Km values) compared to acetyl peptides. Therefore, it is easier for the inhibitors to displace the acetyl peptide than to displace the myristoyl peptide from SIRT2 active site. It may be beneficial to inhibit the demyristoylation activity of SIRT2 in order to achieve anticancer activity, given the recent report that lysine fatty acylated KRas-4a is less effective at promoting anchorage-independent growth compared to the deacylated KRas-4a.[14] Furthermore, our study underlies the importance of careful comparative and validation studies for the development of small molecule inhibitors. As we embrace the power of small molecule inhibitors to probe biology and treat human diseases, we must be careful as off-target effects are very common. Small molecule inhibitor development should be accompanied by detailed validation studies (such as target knockdown or overexpression) to make sure that the biological effects observed is due to target engagement. Similarly, because IC50 values are dependent on the experimental conditions (e.g. enzyme, substrate concentrations and specific activity of different batches of enzymes) used, it will be particularly informative if direct comparisons are performed. We therefore strongly recommend the chemical biology community to adopt these practices.
Supplementary Material
Acknowledgements
The work was supported in part by HHMI, Cornell University, and NIH/NIGMS grant R01GM086703. Imaging data was acquired through the Cornell University Biotechnology Resource Center, with NYSTEM (CO29155) and NIH (S10OD018516) funding for the shared Zeiss LSM880 confocal/ multiphoton microscope. This work made use of the Cornell University NMR Facility, which is supported, in part, by the NSF under Award Number CHE-1531632.
Footnotes
Experimental Section
All experimental details can be found in the supplemental information.
References:
- [1].Chopra V, Quinti L, Kim J, Vollor L, Narayanan KL, Edgerly C, Cipicchio PM, Lauver MA, Choi SH, Silverman RB, Ferrante RJ, Hersch S, Kazantsev AG, Cell Rep 2012, 2, 1492–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Haigis MC, Sinclair DA, Annu Rev Pathol 2010, 5, 253–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a Teng YB, Jing H, Aramsangtienchai P, He B, Khan S, Hu J, Lin H, Hao Q, Sci Rep 2015, 5, 8529; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C, Du J, Kim R, Ge E, Mostoslavsky R, Hang HC, Hao Q, Lin H, Nature 2013, 496, 110–113; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Du Y. Z. Jintang, Su Xiaoyang, Yu Jiu Jiu, Khan Saba, Jiang Hong, Kim Jungwoo, Woo Jimin, Kim Jun Huyn, Choi Brian Hyun, He Bin, Chen Wei, Zhang Sheng, Cerione Richard A., Auwerx Johan, Hao Quan, Lin Hening, Science 2011, 334, 806–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S, Wang D, Feng J, Yu L, Zhu WG, Nat Cell Biol 2010, 12, 665–675; [DOI] [PubMed] [Google Scholar]; b Wang YP, Zhou LS, Zhao YZ, Wang SW, Chen LL, Liu LX, Ling ZQ, Hu FJ, Sun YP, Zhang JY, Yang C, Yang Y, Xiong Y, Guan KL, Ye D, EMBO J 2014, 33, 1304–1320; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wang F, Nguyen M, Qin FX, Tong Q, Aging Cell 2007, 6, 505–514; [DOI] [PubMed] [Google Scholar]; d North BJ, Rosenberg MA, Jeganathan KB, Hafner AV, Michan S, Dai J, Baker DJ, Cen Y, Wu LE, Sauve AA, van Deursen JM, Rosenzweig A, Sinclair DA, EMBO J 2014, 33, 1438–1453; [DOI] [PMC free article] [PubMed] [Google Scholar]; e North BJ, Marshall BL, Borra MT, Denu JM, Verdin E, Mol Cell 2003, 11, 437–444; [DOI] [PubMed] [Google Scholar]; f Li Y, Matsumori H, Nakayama Y, Osaki M, Kojima H, Kurimasa A, Ito H, Mori S, Katoh M, Oshimura M, Inoue T, Genes Cells 2011, 16, 34–45; [DOI] [PubMed] [Google Scholar]; g Jing E, Gesta S, Kahn CR, Cell Metab 2007, 6, 105–114; [DOI] [PMC free article] [PubMed] [Google Scholar]; h Jiang W, Wang S, Xiao M, Lin Y, Zhou L, Lei Q, Xiong Y, Guan KL, Zhao S, Mol Cell 2011, 43, 33–44; [DOI] [PMC free article] [PubMed] [Google Scholar]; i Inoue T, Nakayama Y, Li Y, Matsumori H, Takahashi H, Kojima H, Wanibuchi H, Katoh M, Oshimura M, FEBS J 2014, 281, 2623–2637; [DOI] [PubMed] [Google Scholar]; j Dryden SC, Nahhas FA, Nowak JE, Goustin AS, Tainsky MA, Mol Cell Biol 2003, 23, 3173–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a Jing H, Hu J, He B, Negron Abril YL, Stupinski J, Weiser K, Carbonaro M, Chiang YL, Southard T, Giannakakou P, Weiss RS, Lin H, Cancer Cell 2016, 29, 767–768; [DOI] [PubMed] [Google Scholar]; b Zhao D, Zou SW, Liu Y, Zhou X, Mo Y, Wang P, Xu YH, Dong B, Xiong Y, Lei QY, Guan KL, Cancer Cell 2013, 23, 464–476; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Serrano L, Martinez-Redondo P, Marazuela-Duque A, Vazquez BN, Dooley SJ, Voigt P, Beck DB, Kane-Goldsmith N, Tong Q, Rabanal RM, Fondevila D, Munoz P, Kruger M, Tischfield JA, Vaquero A, Genes Dev 2013, 27, 639–653; [DOI] [PMC free article] [PubMed] [Google Scholar]; d Liu PY, Xu N, Malyukova A, Scarlett CJ, Sun YT, Zhang XD, Ling D, Su SP, Nelson C, Chang DK, Koach J, Tee AE, Haber M, Norris MD, Toon C, Rooman I, Xue C, Cheung BB, Kumar S, Marshall GM, Biankin AV, Liu T, Cell Death Differ 2013, 20, 503–514; [DOI] [PMC free article] [PubMed] [Google Scholar]; e Kim HS, Vassilopoulos A, Wang RH, Lahusen T, Xiao Z, Xu X, Li C, Veenstra TD, Li B, Yu H, Ji J, Wang XW, Park SH, Cha YI, Gius D, Deng CX, Cancer Cell 2011, 20, 487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mellini P, Itoh Y, Tsumoto H, Li Y, Suzuki M, Tokuda N, Kakizawa T, Miura Y, Takeuchi J, Lahtela-Kakkonen M, Suzuki T, Chem Sci 2017, 8, 6400–6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tiago Fleming Outeiro EK, Altmann Stephen M., Kufareva Irina, Strathearn Katherine E., Amore Allison M., Volk Catherine B., Maxwell Michele M., Rochet Jean-Christophe, McLean Pamela J., Young Anne B., Abagyan Ruben,, B. T. H. Feany Mel B., Kazantsev Aleksey G., Science 2007, 317, 516–519. [DOI] [PubMed] [Google Scholar]
- [8].Rotili D, Tarantino D, Nebbioso A, Paolini C, Huidobro C, Lara E, Mellini P, Lenoci A, Pezzi R, Botta G, Lahtela-Kakkonen M, Poso A, Steinkuhler C, Gallinari P, De Maria R, Fraga M, Esteller M, Altucci L, Mai A, J Med Chem 2012, 55, 10937–10947. [DOI] [PubMed] [Google Scholar]
- [9].Rumpf T, Schiedel M, Karaman B, Roessler C, North BJ, Lehotzky A, Olah J, Ladwein KI, Schmidtkunz K, Gajer M, Pannek M, Steegborn C, Sinclair DA, Gerhardt S, Ovadi J, Schutkowski M, Sippl W, Einsle O, Jung M, Nat Commun 2015, 6, 6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, McCarthy A, Appleyard V, Murray KE, Baker L, Thompson A, Mathers J, Holland SJ, Stark MJ, Pass G, Woods J, Lane DP, Westwood NJ, Cancer Cell 2008, 13, 454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kudo N, Ito A, Arata M, Nakata A, Yoshida M, Philos Trans R Soc Lond B Biol Sci 2018, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA, Cell 2001, 107, 149–159. [DOI] [PubMed] [Google Scholar]
- [13].Yuan H, Tan B, Gao SJ, Cell Death Dis 2017, 8, e2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jing H, Zhang X, Wisner SA, Chen X, Spiegelman NA, Linder ME, Lin H, Elife 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.