

SUMMARY
Little is known about how variation in sequence composition alters transcription factor occupancy to precisely recruit large transcription complexes. A key model for understanding how transcription complexes are targeted is the Drosophila dosage compensation system in which the male-specific lethal (MSL) transcription complex specifically identifies and regulates the male X chromosome. The chromatin-linked adaptor for MSL proteins (CLAMP) zinc-finger protein targets MSL to the X chromosome but also binds to GA-rich sequence elements throughout the genome. Furthermore, the GAGA-associated factor (GAF) transcription factor also recognizes GA-rich sequences but does not associate with the MSL complex. Here, we demonstrate that MSL complex recruitment sites are optimal CLAMP targets. Specificity for CLAMP binding versus GAF binding is driven by variability in sequence composition within similar GA-rich motifs. Therefore, variation within seemingly similar cis elements drives the context-specific targeting of a large transcription complex.
Graphical Abstract
In Brief
Kaye et al. investigate two transcription factors, GAF and CLAMP, that target similar GA-rich sequences yet have distinct occupancy. They identify specific features that distinguish binding of each factor and reveal that CLAMP and GAF both function in recruitment of the MSL complex that promotes X chromosome dosage compensation.
INTRODUCTION
Precise regulation of gene expression is essential for cell viability and requires the activity of large transcription complexes. Transcription factors that bind to DNA and recruit these large complexes are therefore critical regulators of diverse cellular processes. Often, multiple transcription factors are able to recognize very similar cis elements. DNA sequence changes such as SNPs within the genome can alter transcription factor binding sites such that the binding of one transcription factor is favored compared with another factor with similar binding sites (Barrera et al., 2016; Inukai et al., 2017). Differential occupancy of transcription factors with similar binding sequence preferences then drives the recruitment of different large transcription complexes to perform specific gene-regulatory functions. Currently, little is understood about the mechanisms by which sequence variation within similar cis elements drives the recruitment of different large transcription complexes to regulate gene expression.
Dosage compensation is a model for understanding how transcription complexes are specifically targeted to generate domains of coordinated gene expression because all of the genes along the length of the X chromosome are targeted for compensation. In mammals, dosage compensation is achieved by identifying and increasing transcription of most X-linked genes in both sexes (Deng et al., 2011, 2013), followed by a second step: the random inactivation of one of the two female X chromosomes (Lyon, 1961). Alternatively, dosage compensation is accomplished through a single step in D. melanogaster, in which transcription is increased 2-fold on the single male X chromosome while the female X chromosomes remain unaffected (Hamada et al., 2005). In all species, X chromosome identification is the critical first step in dosage compensation but remains poorly understood.
Therefore, we use D. melanogaster dosage compensation as a model system to study this first step of X chromosome identification. The master regulator of dosage compensation in D. melanogaster is the male-specific lethal (MSL) complex, a ribonucleoprotein complex assembled only in males (Belote and Lucchesi, 1980). The MSL complex is specifically targeted to the single male X chromosome, where it deposits the histone 4 lysine 16 acetylation (H4K16ac) mark, which promotes transcriptional elongation (Larschan et al., 2011). The MSL complex is enriched on the X chromosome within 1.5 kb regions at the 3′ ends of genes called chromatin entry sites (CESs) (Alekseyenko et al., 2008; Straub et al., 2008). These CESs contain one or more 21 bp GA-rich DNA motifs called MSL recognition elements (MREs) (Alekseyenko et al., 2008).
Localization of MSL complex to MREs is dependent on an additional co-factor, chromatin-linked adaptor for MSL proteins (CLAMP) (Soruco et al., 2013). CLAMP was identified in a genome-wide RNAi screen to identify proteins required for MSL complex recruitment that were missed by MSL screens because they are essential in both sexes (Larschan et al., 2012). CLAMP binds directly to GA-rich MRE sequences in vitro and in vivo (Soruco et al., 2013) and was found in a chromatin immunoprecipitation (ChIP) mass spectrometry experiment identifying proteins that physically interact with MSL complex (Wang et al., 2013a). Moreover, we recently determined that CLAMP globally enhances chromatin accessibility on the X chromosome using both MSL-dependent and MSL-independent mechanisms (Urban et al., 2017a).
Despite performing a critical role in MSL complex recruitment, immunostaining and ChIP sequencing (ChIP-seq) analysis reveals that CLAMP is not an X-specific protein and that only 3% of its binding sites overlap with MSL complex (Soruco et al., 2013). CLAMP is also not sex specific: a decrease in CLAMP protein levels by either RNAi or mutation is lethal to both males and females by the pupal stage of development (Soruco et al., 2013; Urban et al., 2017b). CLAMP is a highly conserved protein across insect species, and it has a more ancient, non-sex-specific function in histone gene regulation (Rieder et al., 2017) that was co-opted for male-specific dosage compensation through the evolutionarily recent enrichment of clustered degenerate GA-repeat cis-element sequences on the X chromosome (Ellison and Bachtrog, 2013; Kuzu et al., 2016). Because the role of transcription factors in gene regulation often depends on their occupancy relationship with co-factors, we hypothesized that the presence or absence of a specific combination of co-factors promotes the context-specific function of CLAMP on the male X chromosome compared with autosomes.
We previously determined that CLAMP occupies long, clustered GA-repeat motifs that are enriched on the X chromosome, especially within CESs (Kuzu et al., 2016). CLAMP, however, is not the only protein capable of binding to GA-rich sequences. In fact, prior to the identification of CLAMP, a different well-studied transcription factor encoded by the Trithorax-like (Trl) gene was known to bind directly to GA-rich sequences, earning the name GAGA-associated factor (GAF) (Farkas et al., 1994). Despite sharing similar binding sites, there are key differences between GAF and CLAMP. The CLAMP DNA binding domain has six C2H2 zinc fingers that are sufficient for interaction with its binding sites (Kuzu et al., 2016), as well as a currently uncharacterized N-terminal domain. GAF has three well-studied domains, including an N-terminal BTB protein-protein interaction domain (Benyajati et al., 1997; Wilkins and Lis, 1999; Zollman et al., 1994), a single C2H2 zinc-finger DNA binding domain, and a C-terminal glutamine (Gln, Q) rich domain known to allow multimerization (Wilkins and Lis, 1999).
Functional analysis determined hypomorphic mutations that decrease GAF function have MSL phenotypes (Greenberg et al., 2004). However, GAF was not essential for recruiting MSL complex to the X chromosome at the resolution of polytene chromosome staining, because only a single MSL complex polytene band was lost after depletion of GAF (Greenberg et al., 2004). In contrast to CLAMP, GAF was not identified as a protein associating with MSL complex (Wang et al., 2013a). Therefore, the function of GAF in dosage compensation and MSL recruitment leading to male-specific lethality remained unknown.
There are several lines of evidence leading us to hypothesize that the relationship between CLAMP and GAF promotes MSL complex targeting. First, we recently determined that GAF and CLAMP are components of the same insulator protein complex that physically interacts with CES (Kaye et al., 2017). Second, both GAF and CLAMP can recruit NURF301, a chromatin-remodeling enzyme shown to promote chromatin accessibility at CESs (Lomaev et al., 2017; Tsukiyama et al., 1994; Bai et al., 2007; Urban et al., 2017a). Third, CLAMP and GAF are the only two known direct GA-binding transcription factors in Drosophila that both regulate chromatin (Urban et al., 2017a; Fuda et al., 2015). Fourth, CESs, which are the optimal MSL complex recruitment sites, are enriched for clustered GA-rich motifs (Kuzu et al., 2016). Finally, CLAMP null mutant males die earlier during development than females (Urban et al., 2017b), and GAF hypomorphic alleles confer MSL phenotypes (Greenberg et al., 2004). Therefore, we hypothesized that the relative occupancy of CLAMP and GAF differs at CESs compared with other regions in the genome to promote accurate targeting of MSL complex.
Using a combination of genetic, genomic, machine learning, and biochemical approaches, we determined the mechanism by which CLAMP reduces GAF occupancy at CESs that recruit MSL complex. We demonstrated that GAF and CLAMP can directly compete for binding sites and that specific X-enriched GA-rich cis elements favor CLAMP binding because of increased variability of their sequence composition. For instance, GAF exhibits strong binding preference for a contiguous GAGAG pentamer, a sequence often absent at the CLAMP binding sites, which are still GA-rich. Surprisingly, GAF promotes accurate targeting of MSL complex binding to CESs despite not directly interacting with MSL complex or stably localizing to most CESs. CLAMP and GAF function synergistically to alter chromatin, providing a likely mechanism by which GAF promotes MSL complex targeting. Overall, we provide insight into how variability of sequence composition within similar cis elements drives differential occupancy of transcription factors to promote accurate targeting of a large transcription complex.
RESULTS
MSL Complex Binding Sites Have High CLAMP Occupancy and Low GAF Occupancy
It has been previously observed that CLAMP co-localizes with MSL on the X chromosome and is required for targeting of the MSL complex to CESs (Soruco et al., 2013). In contrast, although MSL phenotypes have been observed for combinations of GAF (Trl) and MSL mutant alleles, polytene immunostaining of chromosomes in surviving larval stage animals revealed that only one MSL recruitment site was lost (Greenberg et al., 2004). In order to define the relationship among CLAMP, GAF, and MSL complex, we first examined their relative localization on wild-type male polytene chromosomes (Figure 1A). We found that MSL complex co-localizes with CLAMP, as previously reported (Soruco et al., 2013). In contrast, we observed less co-localization between GAF and MSL complex. Together, these data qualitatively demonstrate that CLAMP and GAF exhibit different occupancy patterns compared with each other and with MSL complex.
Figure 1. MSL Complex Binding Sites Have High CLAMP Occupancy and Low GAF Occupancy.
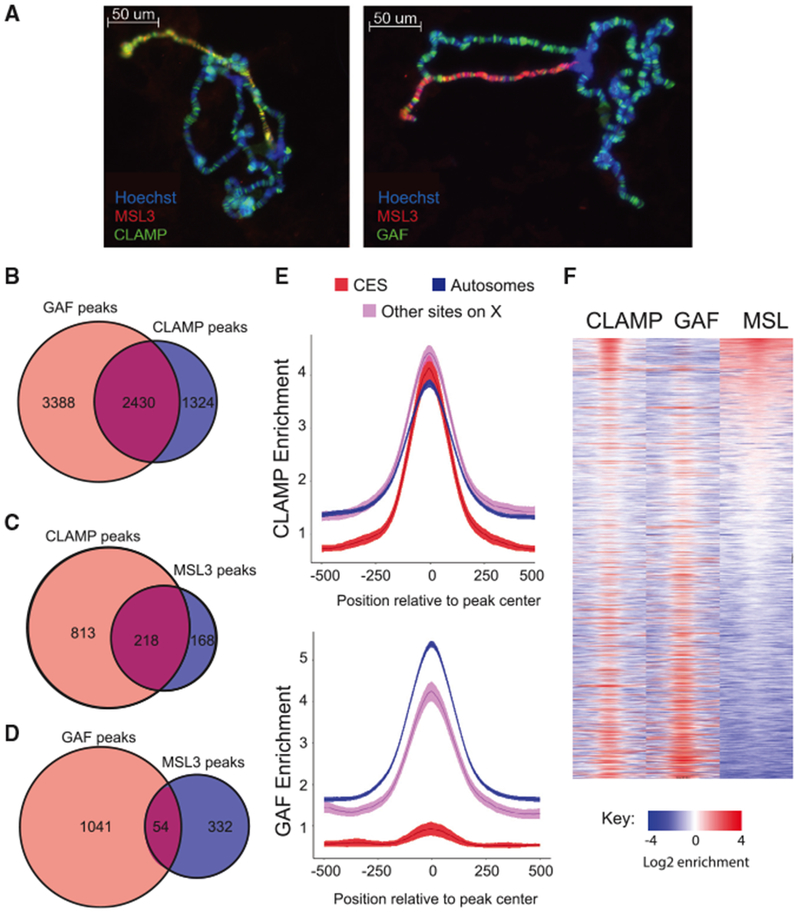
(A) Immunostaining of wild-type male Drosophila salivary gland polytene chromosomes. DNA is visualized with Hoechst staining (blue), while proteins are detected with Alexa Fluor secondary antibodies to identify MSL3 in red and CLAMP (left) or GAF (right) in green. Overlap in localization of CLAMP or GAF with MSL3 is observed in yellow.
(B) Venn diagram showing the number of GAF (light red) and CLAMP (blue) peaks and shared peaks where both GAF and CLAMP are present (purple).
(C) Venn diagrams showing the number of CLAMP (light red) or MSL3 (blue) peaks and peaks where both MSL3 and CLAMP are present (purple) on the X chromosome.
(D) Venn diagram showing the number of GAF (light red) or MSL3 (blue) peaks and peaks where both MSL3 and GAF are present (purple) on the X chromosome.
(E) Fold enrichment profiles of CLAMP (top) and GAF (bottom) from ChIP-seq data. Averages were plotted for peaks on autosomes (blue), CESs (red), and other sites on the X chromosome (pink), with the 95% confidence interval represented by shading around the line.
(F) Heatmap of 1 kb regions centered on CLAMP and/or GAF peaks on the X chromosome. White regions indicate background enrichment, while red and blue represent above (red) and below (blue) background enrichment, respectively (see key). Sites are rank-ordered by MSL3 enrichment.
To map the occupancy of CLAMP and GAF at higher resolution than possible with polytene chromosomes, we performed ChIP-seq on all three factors in male Drosophila S2 cells. Four replicates were performed for each immunoprecipitation condition, and we defined reproducibility of ChIP-seq peaks across replicates to assess data quality and filter out the replicates showing low levels of reproducibility (Figures S1A–S1C; Table S1). After peak calling (see Supplemental Experimental Procedures), we determined the extent of GAF and CLAMP peak overlap (Figure 1B). There were more total GAF peaks (5,818) than CLAMP peaks (3,754). Also, there was a greater percentage of unique GAF peaks compared with total GAF peaks (58%) than the percentage of unique CLAMP peaks (35%) out of total CLAMP peaks. We further analyzed peak overlap by separating peaks on the X chromosome from autosomal peaks (Figures S1D and S1E). On the X chromosome, CLAMP had a modestly reduced percentage of peaks overlapping with GAF (56%) compared with autosomes (68%). Next, we measured overlap between X-linked GAF and CLAMP peaks and MSL complex binding sites (Figures 1C and 1D). Consistent with polytene staining, we found that CLAMP and MSL complex occupancy on the X chromosome overlaps at 21% of CLAMP peaks (218 shared peaks), which is 4 times greater than the 5% of GAF peaks that are co-occupied by MSL complex (54 shared peaks). Therefore, CLAMP and MSL complex occupancy overlap more frequently than GAF and MSL complex occupancy.
However, peak-counting analysis does not account for differences in peak magnitude. Therefore, we plotted enrichment profiles over peaks at CESs compared with other genomic binding sites on the X chromosome or on autosomes (Figure 1E; shading around center line indicates 95% confidence intervals). CLAMP binds to CESs at levels similar to CLAMP peaks not located within CESs (Figure 1E, top), with a slight preference for X chromosome sites compared with autosomal sites. These data are consistent with our previous finding that the number of total sites and the density and clustering of these sites enhance CLAMP occupancy on the X chromosome rather than increased CLAMP occupancy at individual binding sites in both males and females (Kuzu et al., 2016).
In contrast, GAF occupancy differs dramatically between subclasses of sites (Figure 1E, bottom). CESs are significantly less occupied by GAF compared with other sites on X and sites on autosomes. To define how GAF and CLAMP binding occupancy compares with MSL complex occupancy, we next rank-ordered all sites on the X chromosome that are occupied by CLAMP, GAF, or MSL complex by MSL complex occupancy levels (Figure 1F). The sites most enriched for MSL complex occupancy (Figure 1F, top rows) were depleted for GAF, suggesting an inverse relationship between GAF and MSL occupancy levels. Therefore, we tested for an inverse correlation between GAF and MSL complex occupancies and obtained a Pearson’s correlation coefficient (r) equal to −0.39 (Figure S1C). Interestingly, CLAMP and GAF co-occupy sites at the bottom of the MSL complex occupancy rank-ordered heatmap, suggesting that their co-occupancy is inversely correlated with MSL complex occupancy (Figure 1F).
CLAMP and GAF Compete for the Same Binding Sites In Vitro
Although GAF and CLAMP both contain C2H2 zinc-finger DNA binding domains, CLAMP has six tandem zinc fingers and a predicted N-terminal zinc finger, whereas GAF has only a single zinc finger (Figure 2A). We performed predictions of prion-like domains and calculated the glutamine (Q) sequence composition in both GA-binding proteins to reveal more about their biochemical properties (http://plaac.wi.mit.edu). We determined that GAF contains one long disordered domain, which is consistent with reports that it can aggregate into prion-like structures (Michelitsch and Weissman, 2000; Tariq et al., 2013) (Figure 2A). In contrast, CLAMP has two shorter, central disordered prion-like domains (Figure 2A). Therefore, both proteins feature zinc-finger DNA binding domains and Q-rich stretches leading to predicted unstructured domains. Notably, the DNA binding domain is longer in CLAMP, while GAF has a longer unstructured domain. In fact, GAF has two different isoforms (519 versus 581 aa), and the main difference is additional polyQ stretches (Benyajati et al., 1997; Wilkins and Lis, 1999).
Figure 2. GAF and CLAMP Compete for the Same Binding Sites In Vitro.
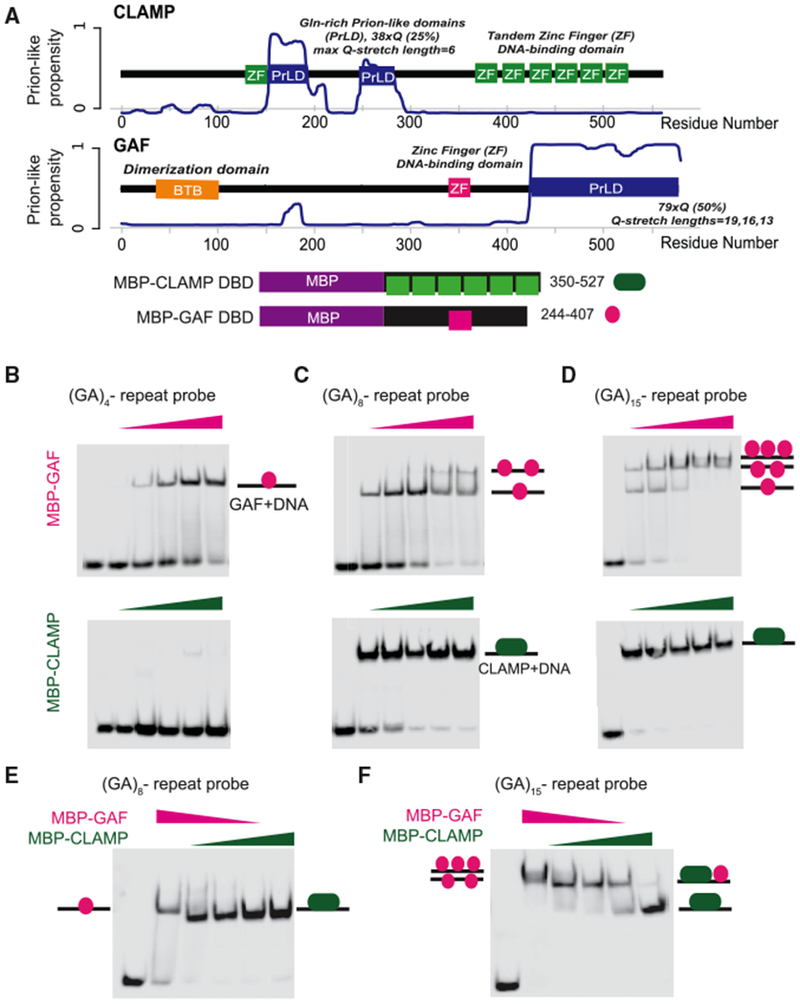
(A) Plot of CLAMP and GAF prion-like (PrL) propensity over the length of the proteins. Amino acid residue numbers are indicated as the x axis. PrL domains (i.e., resemblance to Q/N-rich yeast prion sequences) are the result of long glutamine (Gln, Q) stretches, which are also noted. Other known and predicted domains are displayed, with green boxes as CLAMP DBD zinc fingers (ZFs), pink as the single GAF ZF, and orange for the GAF BTB dimerization domain. Schematic representations of MBP-tagged fusion constructs cloned and expressed for binding assays are also shown. Amino acid residue numbers to the right correspond to regions of the full-length proteins used to make fusion proteins.
(B) EMSA using a biotin-labeled DNA probe containing an 8 bp (GA)4 repeat and GAF (top) or CLAMP (bottom) DBD MBP-fusion proteins. Shifts of protein-bound DNA are indicated by lines with pink colored circles (GAF) or elongated green ovals (CLAMP).
(C) EMSA as in (B), using DNA containing a 16 bp (GA)8-repeat probe. DNA shifted by multiple proteins (i.e., two GAF proteins) indicated by two or three circles.
(D) EMSA as in (B) and (C), now using DNA containing a 30 bp (GA)15-repeat probe.
(E) Competition EMSA using the 16 bp GA8 repeat probe sequence. The first lane is a DNA-only control, the second lane is GAF only, lanes 3–5 are competition with both proteins in the reaction, and the final lane is CLAMP only. Shifts of protein-bound DNA are indicated by arrows.
(F) Competition EMSA as in (E), now using the 30 bp GA15-repeat probe. Shifts of protein-bound DNA are indicated by schematics, and as above, DNA shifted by multiple proteins is indicated by double or triple circles.
Because CLAMP and GAF have different in vivo occupancy patterns (Figure 1F), we asked whether they directly compete with each other for GA-rich sequences using an electrophoretic mobility shift assay (EMSA). Therefore, we expressed and purified MBP fusions with the GAF and CLAMP DNA binding domains (Figure 2A). Next, we tested binding of each protein individually to 60 bp probes containing different numbers of GA-repeat sequences (Figures 2B–2D). GAF can bind to probes containing a shorter 8 bp (GA)4 repeat in addition to longer sequences, consistent with its smaller DNA binding domain. Interestingly, as GAF protein concentration was increased, additional shifted species were observed for the probes containing longer GA stretches, suggesting that multiple GAF-DNA binding domain fusion proteins may be interacting with the same probe (Figures 2C and 2D). Consistent with previous results (Kuzu et al., 2016), we found that CLAMP bound well to probes that contained longer 16 or 30 bp of GA-repeat sequence (Figures 2C and 2D) but not to a probe containing a shorter 8 bp (GA)4 repeat (Figure 2B). In contrast to GAF, we did not observe multiple shifted species for CLAMP. Therefore, CLAMP requires a longer GA-rich binding sequence for interaction with its DNA binding sites than GAF.
We next performed competition EMSAs to test whether both GA-binding proteins compete for the same DNA binding sequence and whether changing relative protein concentration or GA-repeat length can alter binding. CLAMP outcompetes GAF for binding to a 16 bp (GA)8 repeat probe, and GAF was unable to compete even at a 3:1 GAF-to-CLAMP stoichiometric ratio (Figure 2E). Interestingly, we observed an intermediate shift between the separate CLAMP and GAF binding signals for the 30 bp (GA)15 repeat (Figure 2F). Because this shift did not match that observed for either protein alone, it is possible that it represents DNA bound by both GAF and CLAMP in tandem because of the long 30 bp GA-repeat sequence that could interact with both proteins. Overall, these results demonstrate that CLAMP and GAF can compete with each other for binding to GA repeat-containing DNA sequences and that their relative occupancy can be altered by changing the number of continuous GA repeats.
Variation of Sequence Composition within GA-Rich cis Elements Favors CLAMP or GAF Binding In Vitro and In Vivo
After determining that competition between GAF and CLAMP can occur in vitro, we wanted to more specifically define the sequence composition that favors direct binding of one protein or the other using a genomic-context protein binding microarray (gcPBM) that we had previously used to measure CLAMP occupancy (Kuzu et al., 2016). Protein binding microarrays (PBMs) detect GST-tagged protein binding to double-stranded DNA probes on a microarray using a fluorescently conjugated anti-GST secondary antibody (Berger et al., 2006). Therefore, we produced a GST-GAF DNA binding domain fusion and assessed its binding to our gcPBMs that contain GA-rich probes that are either bound or unbound by CLAMP in vivo and control probes that contain in vivo sequences that are not GA rich (Kuzu et al., 2016). Next, we plotted CLAMP and GAF signal intensity Z scores to compare binding of each protein to the same sequence (see Supplemental Experimental Procedures). Each point represents a probe sequence, and sequences are color coded on the basis of their GA-repeat content. Consistent with our EMSA experiments (Figure 2), we observed that GAF can bind to sequences with shorter GA repeats that are not strongly bound by CLAMP (Figure 3A). Furthermore, sequences bound by both GAF and CLAMP have longer GA-repeat sequences.
Figure 3. Variation of Sequence Composition within GA-Rich Elements Favors CLAMP or GAF Binding In Vitro and In Vivo.
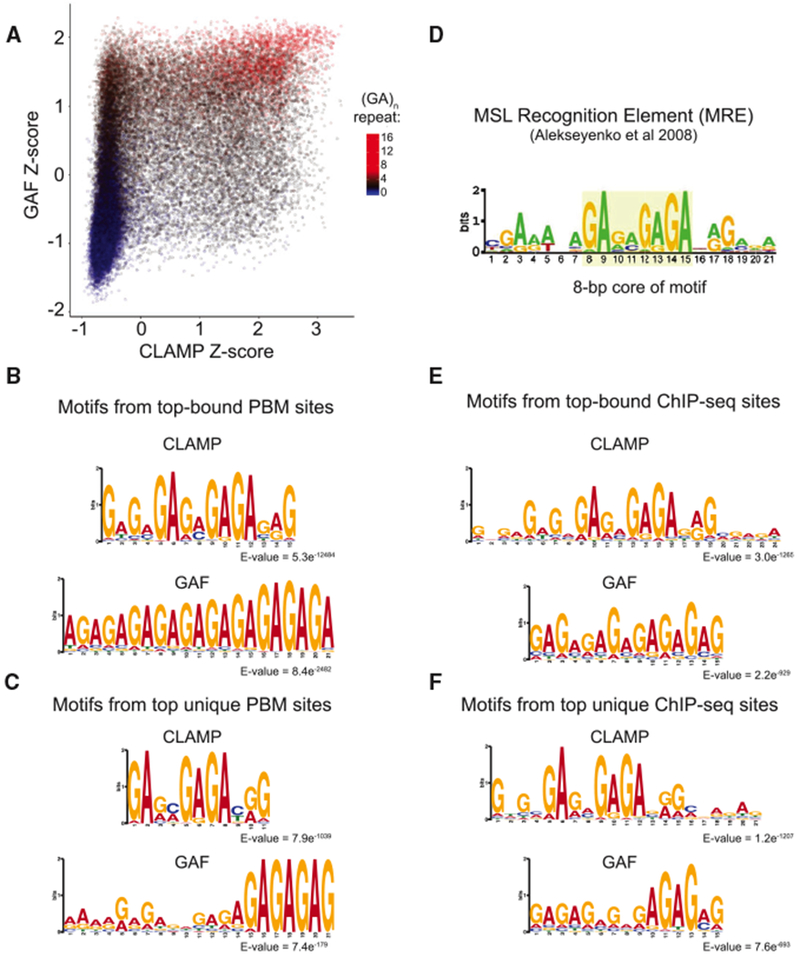
(A) Scatterplot of log10 signal intensity Z scores from the CLAMP and GAF PBM experiments. The number of consecutive GAs in each probe is indicated on the color scale.
(B) Top: CLAMP MEME motif logo for PBM data using sites with Z scores ≥ 2 (n = 2,657). Bottom: GAF MEME motif logo for PBM data using sites with Z scores ≥ 2 (n = 2,218).
(C) Top: CLAMP MEME motif logo for PBM data using sites with CLAMP Z scores ≥ 3 and GAF Z score < 0 (n = 268). Bottom: GAF MEME motif logo for PBM data using sites with GAF Z scores ≥ 3, CLAMP Z score < 0 (n = 156).
(D) MSL recognition element (MRE) motif previously determined from ChIP-seq (Alekseyenko et al., 2008).
(E) Top: sequence motif from MEME-ChIP using a 500 bp region centered on CLAMP peak summits under control RNAi conditions. Bottom: GAF sequence motif from MEME-ChIP using a 500 bp region centered on GAF peak summits under control RNAi conditions.
(F) Top: sequence motifs from MEME-ChIP using a 500 bp region centered on CLAMP peak summits that do not overlap GAF peaks under control RNAi conditions. Bottom: sequence motifs from MEME-ChIP using a 500 bp region centered on GAF peak summits that do not overlap CLAMP peaks under control RNAi conditions.
Next, we refined our comparison of in vitro binding specificities for GAF and CLAMP to extract sequence preference information for CLAMP and GAF. We determined position weight matrices (PWMs) for the strongest CLAMP binding sites and the strongest GAF binding sites (Figure 3B) (see Experimental Procedures). Although the strongest CLAMP binding motifs contain variability within GA stretches, the strongest GAF motifs show a uniform GA-rich motif with at least 5 bp of contiguous GA-repeat sequence. Therefore, although CLAMP and GAF both bind to GA-rich motifs, the specific sequence composition of their motifs differs: (1) unlike GAF, CLAMP can bind to sequences that contain non-contiguous GA repeats, similar to the previously reported MRE (Figure 3D), and (2) unlike CLAMP, GAF can bind to shorter sequences as long as they are composed of contiguous GA repeats (see below for a more detailed analysis of these features).
We next compared in vitro binding motifs from PBMs with in vivo binding motifs from ChIP-seq to determine whether differences in specificity between GAF and CLAMP in vitro were also observed in vivo. We first compared the PBM CLAMP binding motif with the in vivo CLAMP binding motif from ChIP-seq using MEME for motif identification (Machanick and Bailey, 2011) (Figures 3B and 3E). Consistent with our previous reports (Kuzu et al., 2016), the CLAMP binding motif in vitro (Figure 3B) and in vivo (Figure 3E) includes a core 8 bp region with a high degree of conservation of the first two “GA” nucleotides within the repeat followed by two less conserved “GA” or “GC” nucleotides and four additional conserved “GAGA” nucleotides.
We previously demonstrated that both the 8 bp core and additional flanking sequences are required for CLAMP occupancy (Kuzu et al., 2016). In contrast, the GAF binding motif is shorter but requires more highly conserved stretches of GA repeats because the minimal motif contains 5 highly conserved contiguous GA-base pairs (GAGAG), consistent with a previously described GAF-DNA crystal structure (Omichinski et al., 1997). We next determined the motifs from sites bound by only CLAMP or GAF, excluding sites bound by both proteins (Figures 3C and 3F). Although both motifs are still GA rich, the sequences that are recognized only by GAF have either one or two 5 or 6 bp contiguous GA consensus sequences. In contrast, sites that bind only to CLAMP but not GAF rarely have the 5 bp GAF consensus. Instead, CLAMP occupies its binding sites even if there are different bases within the GA repeat that are less conserved, especially at the third and fourth positions within the core 8 bp region. MREs (Figure 3D) that are known to recruit MSL complex (Alekseyenko et al., 2008) exhibit similar variability within the GA-rich motif at the third and fourth positions within the core 8 bp region. Overall, our data are consistent with a model in which CLAMP but not GAF recognizes MREs because of variation in sequence composition within GA-rich motifs.
Factors beyond Sequence Motifs Are Predictive of CLAMP and GAF Binding
To quantify the extent to which the differences between the CLAMP and GAF motifs and additional factors beyond DNA sequence influence relative occupancy of CLAMP and GAF, we performed computational analysis to identify the features that distinguish CLAMP from GAF binding sites in vivo. First, we sought to evaluate the predictive power of the in vivo CLAMP and GAF motifs (Figures 3E and 3F) in the absence of other genomic features. Therefore, we measured how well the sequences under GAF or CLAMP peaks (±214 from center of peak) match the GAF and CLAMP PWMs that were derived from these sequences (Figure 4A). Using this initial approach, we were able to distinguish GAF from CLAMP peaks with a 78.3% success rate for GAF peaks and a 62.8% success rate for CLAMP peaks. Although the success rate of this simple classification on the basis of PWMs derived from ChIP-seq is higher than expected by a random chance (50% success rate), this analysis showed that factors other than those captured by motifs PWMs substantially influence protein binding.
Figure 4. Factors beyond Sequence Motifs Are Predictive of CLAMP and GAF Binding.
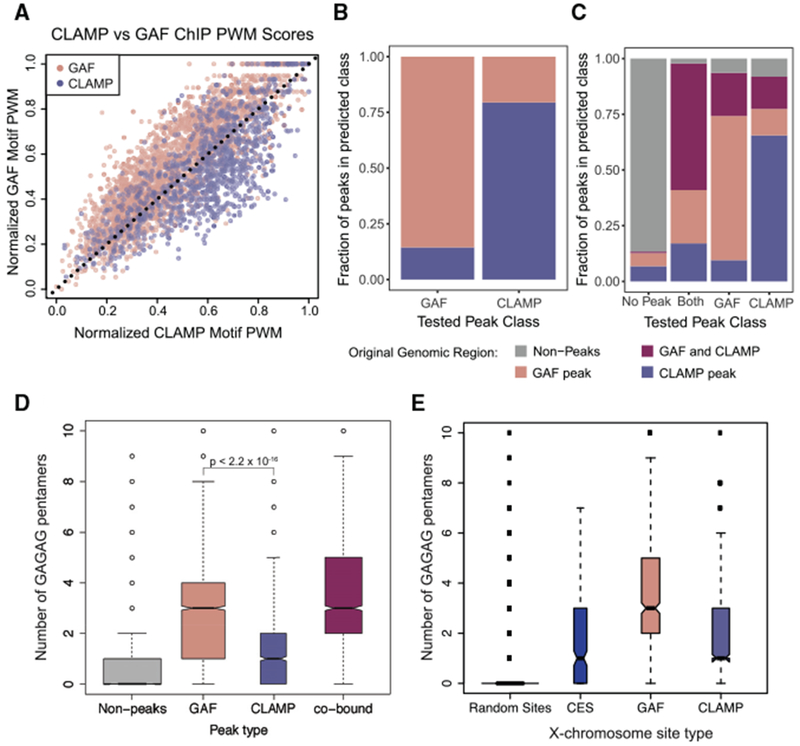
(A) The position weight matrices (PWMs) of GAF and CLAMP unique motifs (Figure 3F) were normalized to the same scale and applied to non-co-bound GAF peaks and CLAMP peaks (Figure 1B). GAF motif scores for each peak are plotted on the y axis and CLAMP motif scores on the x axis, and a dotted line from (0,0) to (1,1) was drawn.
(B) The machine learning algorithm XGBoost (Chen and Guestrin, 2016) was used to develop a classifier model for using GAF-only and CLAMP-only peaks with features including PWMs (Figures 3C and 3F), chromatin states from the nine-state Drosophila genome model (Kharchenko et al., 2010), and the distance (in base pairs) to the nearest TSS. The x axis indicates peak category in the set of peaks used to test the model, and the y axis indicates the predictions from the classifier model.
(C) The machine learning algorithm XGBoost was used to develop a classifier model for using GAF-only, CLAMP-only, and GAF and CLAMP overlapping peaks along with randomly sampled regions of the genome with features including PWMs (Figures 3C and 3F), chromatin states from the nine-state Drosophila genome model (Kharchenko et al., 2010), and the distance (in base pairs) to the nearest TSS. The x axis indicates peak category in the set of peaks used to test the model, and the y axis indicates the predictions from the classifier model.
(D) Boxplot of the number of occurrences of the pentamer “GAGAG” within 214 bp of random sites in the genome outside of peaks (gray) or peak summit of GAF (light red), CLAMP (light blue), or co-bound (purple) peaks.
(E) Boxplot of the number of occurrences of the pentamer “GAGAG” within 214 bp of random sites on the X chromosome (gray), CES (blue), or peak summit of GAF (light red) or CLAMP (light blue) peaks.
To define which additional features alter GAF and CLAMP occupancy, we used a machine learning approach based on the gradient boosting algorithm as implemented in the R package xgboost (Chen and Guestrin, 2016). Our preliminary analysis showed that both CLAMP and GAF are enriched at the transcription start site (TSS) of expressed genes (Figure S2A). Furthermore, CLAMP and GAF are enriched at specific chromatin states as defined using multiple profiles of histone marks and chromatin-associated proteins (Kharchenko et al., 2010) (Figure S2B). Therefore, in addition to the motif PWMs, the following features were used to develop a predictive model: (1) chromatin state identity, (2) distance to the nearest TSS, and (3) the frequency of the GAGAG pentamer within binding loci (Table S2). We also tested a number of other features describing CLAMP and GAF binding sites, such as the “GANNGAGA” pattern seen in CLAMP motif logos (Figure 3), but they were of low predictive value and therefore were not included in the final analysis (Table S2). Our model successfully classified 85.6% of GAF peaks and 79.5% of CLAMP peaks (Figure 4B), outperforming the direct classification of GAF and CLAMP peaks on the basis of the ChIP-seq PWMs alone (Figure 4A).
We next applied the same machine learning approach to a more diverse set of genomic loci, including GAF peaks, CLAMP peaks, shared GAF and CLAMP peaks, and randomly sampled regions of the genome without substantial GAF or CLAMP binding. Using this approach, GAF peaks were predicted with 63.7% success, CLAMP peaks with 65.5% success, and peaks with both proteins bound with 56.8% success (Figure 4C), significantly outperforming a 25% success rate expected by random chance.
Analysis of the model features in terms of their importance for successfully distinguishing CLAMP from GAF bindings sites revealed that the frequency of the simple GAGAG pentamer exhibited the highest predictive power. This feature is more predictive than both chromatin state and proximity to TSS. Moreover, the presence of a GAGAG pentamer is more predictive of GAF occupancy than either the ChIP-seq- or PBM-derived GAF motifs (Tables S2). Therefore, we directly investigated the frequency of GAGAG occurrence at GAF and CLAMP binding sites genome-wide and observed that GAF sites show a higher frequency of such pentamers than CLAMP binding sites (Figure 4D; p < 2.2 × 10−16). Notably, a similar analysis of the frequency of GAGAG occurrence on the X chromosome revealed that CESs and CLAMP sites both have a low GAGAG pentamer frequency (Figure 4E). Therefore, although both CLAMP and GAF are capable of binding to GAGAG pentamers, these pentamers are more frequently present at GAF binding sites than at CLAMP and MSL binding sites. Overall, we conclude that features beyond PWMs such as the frequency of GAGAG pentamers affect the relative occupancy of CLAMP versus GAF at specific sites.
CLAMP and GAF Alter Each Other’s Occupancy to Promote Specific Targeting of MSL Complex to CESs
Because the CLAMP binding motif is more similar to the MRE than the GAF binding motif, we hypothesized that CLAMP out-competes GAF at sites of high MSL complex occupancy, such as CESs. To test our hypothesis, we measured how GAF and CLAMP alter each other’s occupancy in vivo. We quantified the relationship among CLAMP, GAF, and MSL complex by performing RNAi targeting clamp and Trl (GAF), followed by ChIP-seq for each factor in male S2 cells. We confirmed efficient knockdown for clamp and Trl by western blotting (Figures S3A and S3B), consistent with previous studies using these same RNAi constructs (Fuda et al., 2015; Soruco et al., 2013).
We next examined enrichment for all CLAMP and GAF peaks under gfp (control, blue), Trl (pink), or clamp (green) RNAi conditions (Figure 5A). As expected, average CLAMP occupancy was reduced after clamp RNAi, and average GAF occupancy at most peaks was reduced after Trl RNAi. We investigated GAF localization after clamp RNAi and noted that, on average, GAF occupancy increased, consistent with in vitro competition between the two factors. The shaded region surrounding each line represents the 95% confidence interval across multiple biological replicates. CLAMP occupancy also increased after Trl RNAi on average, although more modestly. Next, we plotted average gene profiles centered at CES peaks for all three factors to determine the relationships between CLAMP and GAF at CES and their impact on MSL complex occupancy (Figure 5B). Unlike other binding sites in the genome that most often have occupancy for both CLAMP and GAF, GAF enrichment is very low at CES under control RNAi conditions (Figures 1E and S1F), consistent with in vivo polytene staining (Figure 1A).
Figure 5. CLAMP and GAF Alter Each Other’s Occupancy to Promote Specific Targeting of MSL Complex to CESs.
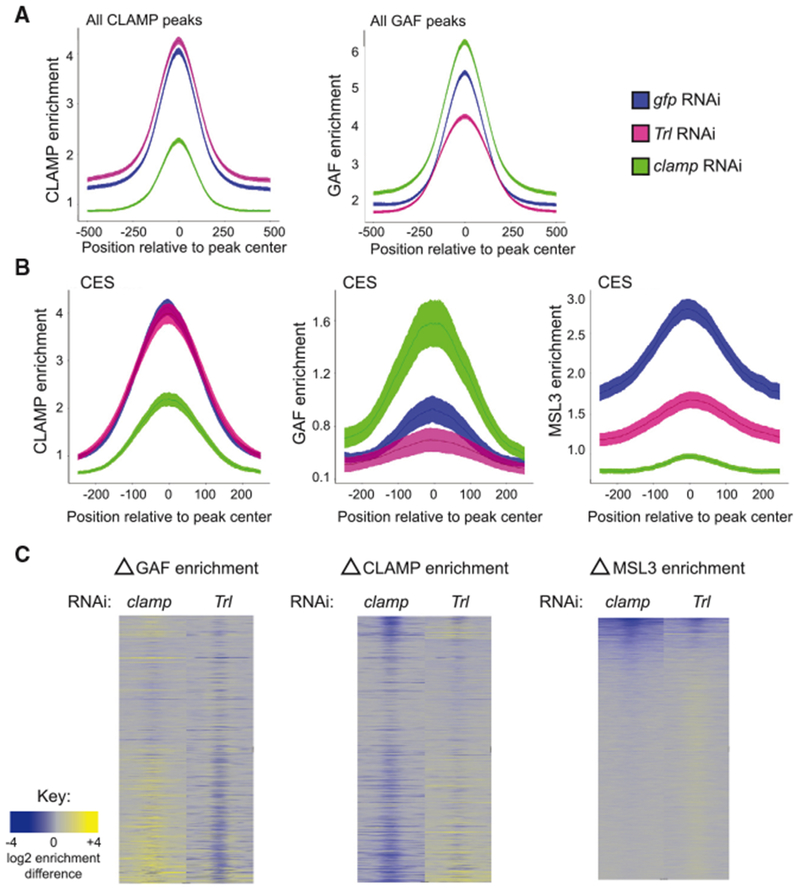
(A) Average enrichment of CLAMP (left) and GAF (right) at CLAMP or GAF peaks centered on peak summit under gfp (blue), Trl (pink), and clamp (green) RNAi conditions. Lines indicate mean enrichment, with shaded areas representing 95% confidence intervals.
(B) Average enrichment of CLAMP (left), GAF (middle), and MSL3 (right) at CES under gfp (blue), Trl (pink), and clamp (green) RNAi conditions. Lines indicate mean enrichment with shaded areas representing 95% confidence intervals.(
C) Heatmap of the change in GAF enrichment (left), CLAMP enrichment (middle), and MSL3 enrichment (right) of the 1,000 bp regions on the X chromosome ordered as depicted in Figure 1F under clamp RNAi (left column) and Trl RNAi (right column). Gray regions indicate background enrichment, while yellow and blue represent increases and decreases in enrichment, respectively.
In order to define the different types of occupancy changes that occur after RNAi treatments, we generated heatmaps to measure the difference in CLAMP, GAF, or MSL complex occupancy between control and clamp or Trl RNAi treatment at all sites on the X chromosome that were bound by any factor after any RNAi treatment (Figure 5C). We rank-ordered these by MSL occupancy under wild-type conditions (as in Figure 1F), and the full set of sites is inclusive of 1 kb regions centered over peaks. As expected from the average profile plots, Trl RNAi decreased GAF signal, and clamp RNAi decreased CLAMP signal (Figure 5C, columns 2 and 3). In contrast, Trl RNAi caused both increases and decreases in CLAMP occupancy throughout the genome (Figure 5C, column 4), accounting for the modest overall difference observed in the average gene profiles analysis (Figures 5A and 5B). Similarly, clamp RNAi causes diverse changes on GAF occupancy (Figure 5C, column 1) but increases GAF occupancy at more sites, leading to the overall increase observed in the average gene profiles. Overall, we observed both competitive and synergistic interactions between GAF and CLAMP occupancy at different genomic locations, but competition occurs more often than synergy.
In addition to the global analysis above, we also identified specific examples of the diverse binding relationships using genome browser views of RNAi ChIP-seq tracks (Figures S3C–S3F). Included are interdependent (eve gene) and independent (lolal gene) autosomal binding sites. On the X chromosome, we show a site where both factors compete for binding leading to an increase in occupancy after RNAi treatment against the other factor (Smr gene), and a site where GAF reduction partially decreases CLAMP binding despite no stable GAF occupancy (Sta gene).
Because of the larger DNA binding domain of CLAMP and its ability to outcompete GAF in vitro (Figure 2), we were not surprised to find sites where CLAMP outcompetes GAF. What was intriguing was the ability of GAF to compete with CLAMP in vivo, resulting in some shared competitive binding sites, such as the Smr example. To further investigate the competitive capacity of GAF, we focused on the GAGAG pentamers, on the basis of our machine learning analysis (Figure 4). We measured the frequency of GAGAG pentamers at GAF and CLAMP binding sites before and after RNAi treatments. Interestingly, the frequency of GAGAG pentamers at CLAMP binding sites increases significantly after Trl RNAi but not at the remaining GAF binding sites (p < 1.2 x 10−8 and p < 0.454, respectively), consistent with the ability of GAF to bind more strongly to these pentamers than CLAMP (Figure S3G).
Next, we determined how clamp and Trl RNAi treatments alter MSL complex recruitment. We observed that both clamp and Trl RNAi decrease MSL complex occupancy (Figure 5C). However, clamp RNAi has a stronger effect on MSL complex occupancy than Trl RNAi consistent with previous in vivo reports (Greenberg et al., 2004; Soruco et al., 2013). Polytene chromosome analysis of Trl hypomorphs had not detected a strong role for GAF in promoting MSL complex occupancy across the X chromosome, likely because of the lower of resolution of polytene staining compared with ChIP-seq. We specifically tested the CES 3′ of roX2 and identified binding relationships similar to those observed globally (Figure S3H), although the GAF IP signal exhibited variability between replicates. Consistent with genome-wide analysis, MSL3 IP after Trl RNAi was significantly reduced, suggesting reduced MSL complex occupancy. Therefore, even though GAF is not stably bound at most MSL complex binding sites, it contributes to MSL complex recruitment.
Synergy between CLAMP and GAF Promotes Chromatin Accessibility
Both CLAMP and GAF regulate chromatin accessibility by recruiting the NURF chromatin remodeler (Fuda et al., 2015; Tsukiyama and Wu, 1995; Urban et al., 2017a). CLAMP recruits NURF to CESs to open the chromatin environment on the male X chromosome and alters positioning of the nucleosome-depleted region at promoters of active genes (Urban et al., 2017a). Similarly, GAF can maintain nucleosome-free promoter regions at paused genes (Fuda et al., 2015). Therefore, we hypothesized that GAF and CLAMP functionally cooperate through the regulation of chromatin accessibility.
To understand the relationship between CLAMP and GAF on chromatin in vivo, we first examined genetic interactions between clamp and Trl mutants using a position-effect variegation (PEV) assay with the white-mottled 4 (wm4) eye color reporter system (Figure 6A) (Gerasimova et al., 1995; Judd, 1955). In this assay, the white gene, which encodes red eye pigment, is inverted such that it is adjacent to a heterochromatin boundary. Spreading of heterochromatin can be inferred from a change in the level of mosaicism of the eye color from red (open chromatin) to white (closed chromatin) as the white gene becomes inactivated (Eissenberg, 1989). We used flies heterozygous for mutant alleles because of lethality of homozygous mutants. The clamp2 allele is a protein null frameshift mutation that results in an early stop codon and causes homozygous lethality in both males and females (Urban et al., 2017b). The GAF mutant allele Trl13C is a hypomorphic allele caused by a P element insertion that has previously been reported to be an E(var) (enhancer of variegation) that increases the spread of heterochromatin in the mutant state (Katokhin et al., 2001).
Figure 6. Synergy between CLAMP and GAF Promotes Chromatin Accessibility.
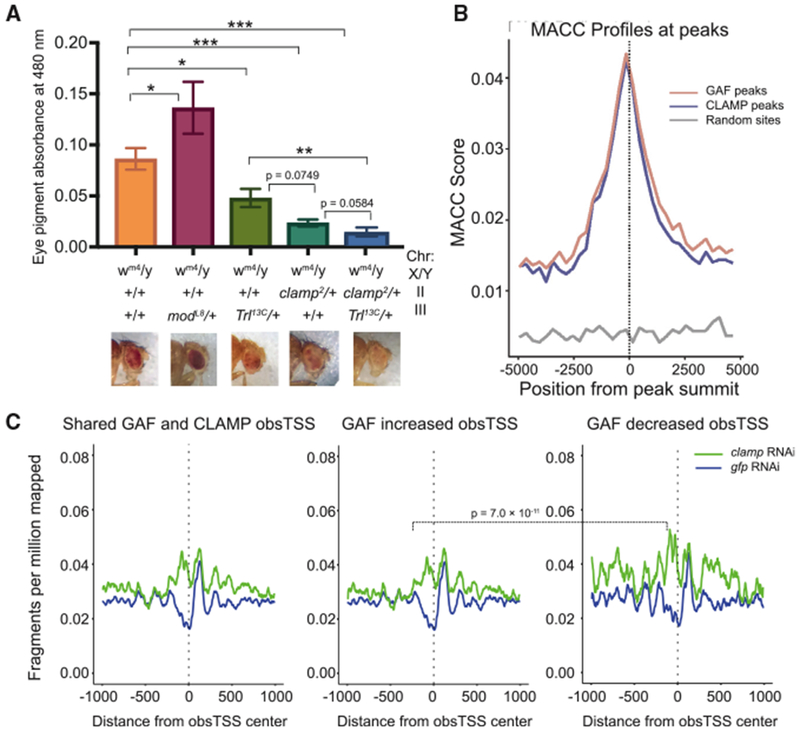
(A) Eye pigmentation assay to quantify PEV of control wm4 flies and crosses of wm4 with modL8, Trl13C, clamp2, and clamp2;Trl13C heterozygous mutants. Absorbance measured at 480 nm was plotted with SE for at least six replicates, five male fly heads per replicate. From left to right, n = 55, 45, 45, 30, and 30. Representative images of eye color are included below the appropriate genotype. Mann-Whitney U test p values are displayed for mutants versus control flies and comparisons between Trl13C, clamp2, and clamp2;Trl13C flies. *p < 0.5, **p < 0.01, and ***p < 0.001.
(B) MNase accessibility (MACC) scores around peak centers are plotted for GAF (light red) and CLAMP (light blue) as well as random control sites (gray).
(C) Nucleosome occupancy plots show chromatin accessibility in S2 cells under gfp control and clamp RNAi conditions for regions surrounding observed TSS (obsTSS) that are co-occupied by both GAF and CLAMP under control RNAi conditions. Three profiles are shown: (1) average profile for all co-occupied genes (left), (2) an average gene profile for genes where GAF ChIP-seq occupancy increases after clamp RNAi (middle), and (3) an average gene profile for genes where GAF ChIP-seq occupancy decreases after clamp RNAi (right).
We measured eye pigment using absorbance of homogenized heads from male mutant flies, with representative images shown for each genotype (Figure 6A). We determined that both clamp2 and Trl13C single mutants are enhancers of PEV, or E(var), on the basis of their whiter eye color relative to wm4 flies and to a control suppressor of variegation (Su[var]) mutant allele of modulo (ModL8) (Graba et al., 1994). Double heterozygous mutant flies (clamp2/+;Trl13C/+) have a stronger E(var) phenotype compared with single mutants, consistent with a synthetic genetic interaction between the clamp2 and Trl13C alleles (Figure 6A). Therefore, we hypothesized that CLAMP and GAF function synergistically as activators that open chromatin because their mutants cause a spreading of heterochromatin.
In order to test our hypothesis that CLAMP and GAF function synergistically to regulate chromatin at the same locations at the molecular level, we analyzed our previously generated micro-coccal nuclease sequencing (MNase-seq) data from S2 cells treated with control or clamp RNAi (Urban et al., 2017a). We were first interested in the accessibility of regions bound by GAF or CLAMP under control conditions. As expected, GAF and CLAMP peaks were more accessible compared with random sites when measured using the MNase accessibility (MACC) score calculated from the slope of MNase titrations (Figure 6B) (Mieczkowski et al., 2016).
We next chose to analyze nucleosome occupancy at the genomic locations surrounding TSS of GAF- and CLAMP-bound genes, because these loci are likely candidates for chromatin regulation by these proteins. First, we analyzed how clamp RNAi alters nucleosome occupancy surrounding observed TSS (obsTSS) (Henriques et al., 2013) of genes that are normally occupied by both GAF and CLAMP and observed an increase in nucleosome occupancy in the clamp RNAi sample (Figure 6C, left). From this subset of genes, we compared genes in which clamp RNAi increases GAF ChIP-seq occupancy with those in which clamp RNAi decreases GAF ChIP-seq occupancy (Figure 6C). When GAF occupancy increases after clamp RNAi treatment, changes in nucleosome occupancy are not as dramatic compared with when GAF ChIP-seq occupancy decreases after clamp RNAi treatment (p < 7.0 × 10−11). Thus, these results reveal that a reduction in both CLAMP and GAF recruitment causes a greater change in nucleosome occupancy than a reduction in CLAMP occupancy alone. Overall, our data are consistent with a model in which CLAMP and GAF synergistically alter nucleosome occupancy.
DISCUSSION
Variation of sequence composition within transcription factor bindings sites is widespread among individuals and across species. How does this variation alter the recruitment of the transcription complexes that regulate gene expression? Here, we provide key insight into how sequence variation within GA-rich transcription factor binding sites changes the relative occupancy of two transcription factors with similar binding sites, CLAMP and GAF, to specifically target the MSL complex to the X chromosome. By combining in vivo, in vitro, and computational approaches, we determined that CLAMP and GAF compete with each other in vitro (Figure 2) and alter each other’s occupancy in vivo (Figure 5). We show that variation of the sequence composition within the GA-rich motifs drives the relative occupancy of CLAMP versus GAF (Figure 3). By using machine learning and direct occupancy measurements, we show that a precise GAGAG pentamer is predictive for GAF but not CLAMP binding. This difference in binding preferences drives the differential occupancy of GAF and CLAMP at MSL complex binding sites, which contain a low frequency of GAGAG pentamers (Figure 4). Therefore, we provide insight into how small changes in DNA sequence cause differential occupancy of two transcription factors with similar binding sites to promote the specific recruitment of a large transcription complex.
We further demonstrate that GAF and CLAMP have diverse interactions throughout the genome including both competitive and interdependent binding relationships. These interactions affect the recruitment of co-factors such as the MSL complex. Surprisingly, despite the absence of stable GAF occupancy at MSL complex binding sites, GAF also has a significant role in targeting MSL complex to its binding sites because reduction of GAF protein leads to a decrease in MSL occupancy. How does GAF promote MSL complex targeting in the absence of its stable binding to MSL complex binding sites?
It is possible that GAF may still bind to MSL complex binding sites transiently as a redundant mechanism to ensure that the binding sites remain accessible, because transcription factors cycle on and off their binding sites rapidly. A facilitated diffusion model for competition between transcription factors was recently proposed (Cartailler and Reingruber, 2015) that may be relevant to the relationship between GAF and CLAMP. In this model, transcription factors in solution can facilitate the binding and release of transcription factors bound to DNA. Also, both GAF and CLAMP functionally interact with the NURF chromatin-remodeling complex (Tsukiyama and Wu, 1995; Tsukiyama et al., 1994; Urban et al., 2017a). Therefore, it is possible that by cycling on and off of their binding sites, CLAMP and GAF ensure that NURF is able to maintain the high level of chromatin accessibility that is present at the sites of strongest MSL complex occupancy. Consistent with this model, CESs with the highest level of chromatin accessibility are not reduced in accessibility after depleting CLAMP (Urban et al., 2017a), suggesting redundancy with a similar factor, such as GAF.
The different binding specificities of GAF and CLAMP also provide insight into how CLAMP is specifically enriched at CESs. CLAMP has more tolerance for variation in sequence composition in the middle of its GA-rich motif and does not require the GAGAG pentamer nucleotide sequence that is present at lower frequency at CESs. In contrast, GAF requires the GAGAG pentamer for its binding. It is possible that there is more variability in GA-rich motifs with CESs because they recently inserted onto the X chromosome through transposon hopping (Ellison and Bachtrog, 2013; Joshi and Meller, 2017). The rapidly evolving MSL complex (Kuzu et al., 2016) could have then acquired the ability to physically associate with CLAMP and not GAF because of the enrichment of CLAMP at clusters of MREs over gene bodies that are optimal MSL complex target sites.
On the basis of an available crystal structure of the synthetic six zinc-finger protein Aart (Segal et al., 2006), proteins with six tandem zinc fingers such as CLAMP may wrap around the entire double helix, unlike GAF, which has a single zinc finger. Therefore, it is possible that additional zinc fingers promote more stable binding by CLAMP, while GAF may interact with DNA with lower affinity, consistent with the ability of CLAMP to outcompete GAF for binding in vitro (Figure 2). Also, multiple fingers may also allow CLAMP to be flexible and tolerate more changes in its cis-element binding sites. Interestingly, some MREs have a C at the third or fourth position with the core GA-rich element, which is thought to promote a low-affinity MSL2 interaction (Fauth et al., 2010; Zheng et al., 2014). Therefore, it is possible that after CLAMP and GAF open chromatin, CLAMP associates with MSL complex (Wang et al., 2013b) to increase the local concentration of MSL complex such that it can physically interact with DNA. Supporting this order of action, CLAMP and GAF are maternally loaded into the early embryo before zygotic genome activation (Harrison and Eisen, 2015; Rieder et al., 2017). Therefore, CLAMP and GAF are potentially bound to chromatin before the formation and recruitment of MSL complex at nuclear cycle 14 (2-hr-old embryos) (Strukov et al., 2011).
Here, we have demonstrated that small changes in sequence composition within GA-rich motifs alter the relative occupancy of CLAMP compared with that of GAF to promote the recruitment of the MSL dosage compensation complex specifically to the male X chromosome. We have provided insight into how sequence variation within similar cis elements generated over evolutionary time drives competition between two transcription factors with similar binding sites to specifically target a large transcription complex. Overall, we provide insight into how local and evolutionarily recent variation in cis-element sequences drives the formation of specialized transcriptional programs.
EXPERIMENTAL PROCEDURES
Fly Stocks
All flies were maintained in a 25°C incubator. Flies for PEV assay include the following stocks: white mottled 4 flies (In[1]wm4;+;+ Bloomington stock center #807), modulo mutant flies (w+;+;ModL8/TM3, Sb Bloomington stock center #38432), clamp mutant flies (w1118;clamp2/cyoGFP;+) (Urban et al., 2017b), GAF (Trl) mutant flies (w1118;+;Trl13C/TM6B, Tb, Sb Bloomington stock center #58473), and the double-heterozygous CLAMP and GAF mutants were generated from the above stocks to a final genotype of w1118;clamp2/cyoGFP;Trl13C/TM6B, Tb. Flies for polytene spreads are a control gfp RNAi line (w1118; P{UAS-GFP.dsRNA.R}142 Bloomington stock center #9330).
Immunostaining of Polytenes
Polytene squashes were prepared from male third-instar larvae as previously reported (Cai et al., 2010). Immunostaining was performed with a rabbit anti-CLAMP antibody (SDIX; Soruco et al., 2013) at a 1:1,000 dilution, a rabbit anti-GAF antibody (gift of G. Cavalli) at 1:1,000, and a goat anti-MSL3 serum (gift of M. Kuroda) at a dilution of 1:500. See Supplemental Experimental Procedures for additional details.
PEV Assay
Female white mottled flies were crossed to male mutant flies, and larvae and adult flies were sorted to select for male heterozygous mutants, with retention of mutation indicated by loss of balancer allele. After 2 days, images of eye colors were taken of flies on a CO2 pad using an OptixCam Summit D3K2-5 camera mounted on an Olympus SZX12 scope with an Amscope LED 144 ring light adaptor. At least six replicates were collected as a group of 5 flies, for a minimum total of 30 flies per genotype. Eye pigmentation assay was performed using a previously established protocol (Gerasimova et al., 1995; Judd, 1955) (see Supplemental Experimental Procedures).
Cloning, Expression, and Purification of MBP-GAF Protein
The GAF DNA binding domain (DBD) and surrounding sequence (aa 244–407) were ordered as E. coli codon-optimized cDNA (Genewiz gene synthesis, P2313-1/C78696). Restriction site cloning for Ndel and Xhol was then used to insert the GAF DBD into the pET-THMT expression vector (Peti and Page, 2007) containing a His-MBP fusion tag sequence. The vector was transformed into Bioline BL21 (DE3) cells for E. coli expression. MBP-GAF DBD was expressed and purified using standard protocols for His-tag purification and size exclusion chromatography (see Supplemental Experimental Procedures).
EMSA
The LightShift Chemiluminescent EMSA Kit (Thermo Fisher Scientific) was used according to manufacturer protocols and as previously reported (Kuzu et al., 2016). Biotin-labeled DNA probes containing (GA)n repeats were the same as used previously (Kuzu et al., 2016) and were added to a final concentration of 0.6 nM in each sample reaction. The first protein sample was the MBP-CLAMP DBD from the same protein preparation, as in previously published gel-shift experiments (Kuzu et al., 2016). The other protein was the MBP-GAF DBD prepared above. Images were captured using an Azure c600 imaging system.
GST-GAF Expression and PBMs
The GAF DBD (aa 244–407) cDNA was cloned into a GST-tag destination vector using the gateway cloning system (see Supplemental Experimental Procedures). The PureExpress in vitro transcription and translation kit (New England Biolabs) was used to express GST-GAF-DBD. The manufacturer’s protocol was altered by adding zinc acetate (0.05 mM final) prior to 2 hr expression at 37° C.
gcPBM experiments were performed using a custom-designed oligonucleotide array in 4 × 180K format (Agilent Technologies, AMADID #037964), described in detail previously (Kuzu et al., 2016). The array was converted to a double-stranded DNA array and used in PBM experiments essentially as described previously using PBS binding buffer with 50 μM zinc acetate (Berger et al., 2006), exceptthat here GST-GAF-DBD was applied to one fresh and one stripped array at a final concentration of 150 nM and either 525 or 600 nM MBP, respectively.
ChIP
S2 cells used for chromatin preparation were treated with previously validated RNAi targets (gfp [Hamada et al., 2005], clamp [Larschan et al., 2012; Soruco et al., 2013], Trl [Fuda et al., 2015]) for 6 days, as described in previous work (Kaye et al., 2017). Knockdown was validated via western blotting with the Western Breeze kit (Invitrogen). Antibodies used for detection were rabbit anti-CLAMP (SDIX; Soruco et al., 2013) at 1:1,000 and rabbit anti-GAF (gift of G. Cavalli) at 1:5,000. As a loading control, actin was detected with mouse anti-actin (Sigma-Aldrich) at a 1:50,000 dilution. Chromatin was prepared as previously described (Kaye et al., 2017). Immunoprecipitation was performed as previously described (Kaye et al., 2017) with 2 mL IPs of CLAMP antibody (2 μL/mL SDIX), GAF antibody (10 μL/mL, gift of J. Lis), or MSL3 serum (0.4 μL/mL, gift of M. Kuroda). Sequencing libraries were prepared using the NEBNext Ultra II DNA library preparation kit according to user manual protocol. For sequencing, NEBNext index adaptors 1–12 were used, with each replicate set as its own lane.
Computational Analysis
Detailed computational methods are provided in the Supplemental Information.
Supplementary Material
Highlights.
The GAF and CLAMP proteins have shared and unique GA-rich binding sites
Variation within DNA recognition elements drives differential occupancy
CLAMP and GAF both promote MSL recruitment for X chromosome dosage compensation
ACKNOWLEDGMENTS
We thank Dr. John Lisand Dr. Giacomo Cavalli for their gifts of GAF antibodies and Dr. Mitzi Kuroda for the gift of the MSL3 antiserum. We also thank Dr. Kristi Wharton for use of her microscope-mounted camera to image flies. Funding comes from the following grants: a Pew Biomedical Scholars grant, American Cancer Society (ACS) Research Scholar and NIH grant R01 GM098461-1 to E.L., a National Institute on Aging (NIA) T32 grant to E.G.K., NIH grant R01 HG005287 to M.L.B., and NIH grant R01GM118530 to N.L.F.
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession number for the data reported in this study is GEO: GSE110654.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, four figures, and four tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.02.098.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alekseyenko AA, Peng S, Larschan E, Gorchakov AA, Lee OK, Kharchenko P, McGrath SD, Wang CI, Mardis ER, Park PJ, and Kuroda MI (2008). A sequence motif within chromatin entry sites directs MSL establishment on the Drosophila X chromosome. Cell 734, 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X, Larschan E, Kwon SY, Badenhorst P, and Kuroda MI (2007). Regional control of chromatin organization by noncoding roX RNAs and the NURF remodeling complex in Drosophila melanogaster. Genetics 176, 1491–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrera LA, Vedenko A, Kurland JV, Rogers JM, Gisselbrecht SS, Rossin EJ, Woodard J, Mariani L, Kock KH, Inukai S, et al. (2016). Survey of variation in human transcription factors reveals prevalent DNA binding changes. Science 351, 1450–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belote JM, and Lucchesi JC (1980). Male-specific lethal mutations of Drosophila melanogaster. Genetics 96, 165–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benyajati C, Mueller L, Xu N, Pappano M, Gao J, Mosammaparast M, Conklin D, Granok H, Craig C, and Elgin S (1997). Multiple isoforms of GAGA factor, a critical component of chromatin structure. Nucleic Acids Res. 25, 3345–3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Philippakis AA, Qureshi AM, He FS, Estep PW 3rd, and Bulyk ML (2006). Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol 24, 1429–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W, Jin Y, Girton J, Johansen J, and Johansen KM (2010). Preparation of Drosophila polytene chromosome squashes for antibody labeling. J. Vis. Exp (36), 1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartailler J, and Reingruber J (2015). Facilitated diffusion framework for transcription factor search with conformational changes. Phys. Biol 12, 046012. [DOI] [PubMed] [Google Scholar]
- Chen T, and Guestrin C (2016). XGBoost: a scalable tree boosting system. arXiv, arXiv:1603.02754. https://arxiv.org/abs/1603.02754.
- Deng X, Hiatt JB, Nguyen DK, Ercan S, Sturgill D, Hillier LW, Schlesinger F, Davis CA, Reinke VJ, Gingeras TR, et al. (2011). Evidence for compensatory upregulation of expressed X-linked genes in mammals, Caenorhabditis elegans and Drosophila melanogaster. Nat. Genet 43, 1179–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Berletch JB, Ma W, Nguyen DK, Hiatt JB, Noble WS, Shendure J, and Disteche CM (2013). Mammalian X upregulation is associated with enhanced transcription initiation, RNA half-life, and MOF-mediated H4K16 acetylation. Dev. Cell 25, 55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissenberg JC (1989). Position effect variegation in Drosophila: towards a genetics of chromatin assembly. BioEssays 11, 14–17. [DOI] [PubMed] [Google Scholar]
- Ellison CE, and Bachtrog D (2013). Dosage compensation via transposable element mediated rewiring of a regulatory network. Science 342, 846–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas G, Gausz J, Galloni M, and Reuter G (1994). The Trithorax-like gene encodes the Drosophila GAGA factor. Nature 371, 806–808. [DOI] [PubMed] [Google Scholar]
- Fauth T, Muller-Planitz F, Konig C, Straub T, and Becker PB (2010). The DNA binding CXC domain of MSL2 is required for faithful targeting the dosage compensation complex to the X chromosome. Nucleic Acids Res. 38, 3209–3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuda NJ, Guertin MJ, Sharma S, Danko CG, Martins AL, Siepel A, and Lis JT (2015). GAGA factor maintains nucleosome-free regions and has a role in RNA polymerase II recruitment to promoters. PLoS Genet. 11, e1005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimova TI, Gdula DA, Gerasimov DV, Simonova O, and Corces VG (1995). A Drosophila protein that imparts directionality on a chromatin insulator is an enhancer of position-effect variegation. Cell 82, 587–597. [DOI] [PubMed] [Google Scholar]
- Graba Y, Laurenti P, Perrin L, Aragnol D, and Pradel J (1994). The modifier of variegation modulo gene acts downstream of dorsoventral and HOM-C genes and is required for morphogenesis in Drosophila. Dev. Biol 166, 704–715. [DOI] [PubMed] [Google Scholar]
- Greenberg AJ, Yanowitz JL, and Schedl P (2004). The Drosophila GAGA factor is required for dosage compensation in males and for the formation of the male-specific-lethal complex chromatin entry site at 12DE. Genetics 166, 279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada FN, Park PJ, Gordadze PR, and Kuroda MI (2005). Global regulation of X chromosomal genes by the MSL complex in Drosophila melanogaster. Genes Dev. 19, 2289–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison MM, and Eisen MB (2015). Transcriptional activation of the zygotic genome in Drosophila. Curr. Top. Dev. Biol 113, 85–112. [DOI] [PubMed] [Google Scholar]
- Henriques T, Gilchrist DA, Nechaev S, Bern M, Muse GW, Burkholder A, Fargo DC, and Adelman K (2013). Stable pausing by RNA polymerase II provides an opportunity to target and integrate regulatory signals. Mol. Cell 52, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inukai S, Kock KH, and Bulyk ML (2017). Transcription factor-DNA binding: beyond binding site motifs. Curr. Opin. Genet. Dev 43, 110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi SS, and Meller VH (2017). Satellite repeats identify X chromatin for dosage compensation in Drosophila melanogaster males. Curr. Biol 27, 1393–1402.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judd BH (1955). Direct proof of a variegated-type position effect at the white locus in Drosophila melanogaster. Genetics 40, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katokhin AV, Pindyurin AV, Fedorova EV, and Baricheva EM (2001). Molecular genetic analysis of the Drosophila melanogaster Trithorax-like gene coding for the GAGA transcription factor. Genetika 37, 368–374. [PubMed] [Google Scholar]
- Kaye EG, Kurbidaeva A, Wolle D, Aoki T, Schedl P, and Larschan EN (2017). Drosophila dosage compensation loci associate with a boundary forming insulator complex. Mol. Cell. Biol 37, e00253–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Alekseyenko A, Schwartz YB, Minoda A, Riddle NC, Ernst J, Sabo PJ, Larschan E, Gorchakov A, Gu T, et al. (2010). Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature 471, 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzu G, Kaye EG, Chery J, Siggers T, Yang L, Dobson JR, Boor S, Bliss J, Liu W, Jogl G, et al. (2016). Expansion of GA dinucleotide repeats increases the density of CLAMP binding sites on the X-chromosome to promote Drosophila dosage compensation. PLoS Genet. 12, e1006120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larschan E, Bishop EP, Kharchenko PV, Core LJ, Lis JT, Park PJ, and Kuroda MI (2011). X chromosome dosage compensation via enhanced transcriptional elongation in Drosophila. Nature 471, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larschan E, Soruco MML, Lee OK, Peng S, Bishop E, Chery J, Goebel K, Feng J, Park PJ, and Kuroda MI (2012). Identification of chromatin-associated regulators of MSL complex targeting in Drosophila dosage compensation. PLoS Genet. 8, e1002830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomaev D, Mikhailova A, Erokhin M, Shaposhnikov AV, Moresco JJ, Blokhina T, Wolle D, Aoki T, Ryabykh V, Yates JR 3rd., et al. (2017). The GAGA factor regulatory network: Identification of GAGA factor associated proteins. PLoS ONE 12, e0173602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon MF (1961). Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 190, 372–373. [DOI] [PubMed] [Google Scholar]
- Machanick P, and Bailey TL (2011). MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelitsch MD, and Weissman JS (2000). A census of glutamine/asparagine-rich regions: implications for their conserved function and the prediction of novel prions. Proc. Natl. Acad. Sci. USA 97, 11910–11915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieczkowski J, Cook A, Bowman SK, Mueller B, Alver BH, Kundu S, Deaton AM, Urban JA, Larschan E, Park PJ, et al. (2016). MNase titration reveals differences between nucleosome occupancy and chromatin accessibility. Nat. Commun 7, 11485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omichinski JG, Pedone PV, Felsenfeld G, Gronenborn AM, and Clore GM(1997). The solution structure of a specific GAGA factor-DNA complex reveals a modular binding mode. Nat. Struct. Biol 4, 122–132. [DOI] [PubMed] [Google Scholar]
- Peti W, and Page R (2007). Strategies to maximize heterologous protein expression in Escherichia coli with minimal cost. Protein Expr. Purif 51, 1–10. [DOI] [PubMed] [Google Scholar]
- Rieder LE, Koreski KP, Boltz KA, Kuzu G, Urban JA, Bowman SK, Zeidman A, Jordan WT 3rd, Tolstorukov MY, Marzluff WF, et al. (2017). Histone locus regulation by the Drosophila dosage compensation adaptor protein CLAMP. Genes Dev. 31, 1494–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal DJ, Crotty JW, Bhakta MS, Barbas CF 3rd, and Horton NC (2006). Structure of Aart, a designed six-finger zinc finger peptide, bound to DNA. J. Mol. Biol 363, 405–421. [DOI] [PubMed] [Google Scholar]
- Soruco MML, Chery J, Bishop EP, Siggers T, Tolstorukov MY, Leydon AR, Sugden AU, Goebel K, Feng J, Xia P, et al. (2013). The CLAMP protein links the MSL complex to the X chromosome during Drosophila dosage compensation. Genes Dev. 27, 1551–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub T, Grimaud C, Gilfillan GD, Mitterweger A, and Becker PB (2008). The chromosomal high-affinity binding sites for the Drosophila dosage compensation complex. PLoS Genet. 4, e1000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strukov YG, Sural TH, Kuroda MI, and Sedat JW (2011). Evidence of activity-specific, radial organization of mitotic chromosomes in Drosophila. PLoS Biol. 9, e1000574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tariq M, Wegrzyn R, Anwar S, Bukau B, and Paro R (2013). Drosophila GAGA factor polyglutamine domains exhibit prion-like behavior. BMC Genomics 14, 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukiyama T, and Wu C (1995). Purification and properties of an ATP-dependent nucleosome remodeling factor. Cell 83, 1011–1020. [DOI] [PubMed] [Google Scholar]
- Tsukiyama T, Becker PB, and Wu C (1994). ATP-dependent nucleosome disruption at a heat-shock promoter mediated by binding of GAGA transcription factor. Nature 367, 525–532. [DOI] [PubMed] [Google Scholar]
- Urban J, Kuzu G, Bowman S, Scruggs B, Henriques T, Kingston R, Adelman K, Tolstorukov M, and Larschan E (2017a). Enhanced chromatin accessibility of the dosage compensated Drosophila male X-chromosome requires the CLAMP zinc finger protein. PLoS ONE 12, e0186855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban JA, Doherty CA, Jordan WT 3rd, Bliss JE, Feng J, Soruco MM, Rieder LE, Tsiarli MA, and Larschan EN (2017b). The essential Drosophila CLAMP protein differentially regulates non-coding roX RNAs in male and females. Chromosome Res. 25, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CI, Alekseyenko AA, LeRoy G, Elia AE, Gorchakov AA, Britton LM, Elledge SJ, Kharchenko PV, Garcia BA, and Kuroda MI (2013a). Chromatin proteins captured by ChIP-mass spectrometry are linked to dosage compensation in Drosophila. Nat. Struct. Mol. Biol 20, 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CI, Alekseyenko AA, LeRoy G, Elia AE, Gorchakov AA, Britton LM, Elledge SJ, Kharchenko PV, Garcia BA, and Kuroda MI (2013b). Chromatin proteins captured by ChIP-mass spectrometry are linked to dosage compensation in Drosophila. Nat. Struct. Mol. Biol 20, 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins RC, and Lis JT (1999). DNA distortion and multimerization: novel functions of the glutamine-rich domain of GAGA factor. J. Mol. Biol 285, 515–525. [DOI] [PubMed] [Google Scholar]
- Zheng S, Villa R, Wang J, Feng Y, Wang J, Becker PB, and Ye K (2014). Structural basis of X chromosome DNA recognition by the MSL2 CXC domain during Drosophila dosage compensation. Genes Dev. 28, 2652–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zollman S, Godt D, Privd GG, Couderc JL, and Laski FA (1994). The BTB domain, found primarily in zinc finger proteins, defines an evolutionarily conserved family that includes several developmentally regulated genes in Drosophila. Proc. Natl. Acad. Sci. U S A 91, 10717–10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.