

Abstract
Polychlorinated biphenyls (PCBs) are persistent organic pollutants that contribute to inflammatory diseases such as atherosclerosis, and macrophages play a key role in the overall inflammatory response. Depending on specific environmental stimuli, macrophages can be polarized either to pro-inflammatory (e.g., M1) or anti-inflammatory (e.g., M2) phenotypes. We hypothesize that dioxin-like PCBs can contribute to macrophage polarization associated with inflammation. To test this hypothesis, human monocytes (THP-1) were differentiated to macrophages and subsequently exposed to PCB 126. Exposure to PCB 126, but not to PCB 153 or 118, significantly induced the expression of inflammatory cytokines, including TNFα and IL-1β, suggesting polarization to the pro-inflammatory M1 phenotype. Additionally, monocyte chemoattractant protein-1 (MCP-1) was increased in PCB 126-activated macrophages, suggesting induction of chemokines which regulate immune cell recruitment and infiltration of monocytes/macrophages into vascular tissues. In addition, oxidative stress sensitive markers including nuclear factor (erythroid-derived 2)-like 2 (NFE2L2; Nrf2) and down-stream genes, such as heme oxygenase 1 (HMOX1) and NAD(P)H quinone oxidoreductase 1 (NQO1), were induced following PCB 126 exposure. Since dioxin-like PCBs may elicit inflammatory cascades through multiple mechanisms, we then pretreated macrophages with both aryl hydrocarbon receptor (AhR) and NF-κB antagonists prior to PCB treatment. The NF-κB antagonist BMS-345541 significantly decreased mRNA and protein levels of multiple cytokines by approximately 50% compared to PCB treatment alone, but the AhR antagonist CH-223191 was protective to a lesser degree. Our data demonstrate the involvement of PCB 126 in macrophage polarization and inflammation, indicating another important role of dioxin-like PCBs in the pathology of atherosclerosis.
Keywords: dioxins, cardiovascular disease, PCB 126, inflammation, atherosclerosis, monocyte macrophage polarization
1. Introduction
Atherosclerosis is the result of chronic inflammation of blood vessels, leading to a progressive narrowing of the arterial wall because of the formation of an atheromatous plaque, which contains a necrotic core, calcified sites, lipids and many cell types (Ross, 1999). Polychlorinated biphenyls (PCBs) are a group of persistent and widespread environmental pollutants, which can contribute to a range of adverse health effects, including atherosclerotic cardiovascular diseases (Choi et al., 2003; Petriello et al., 2016; Wahlang et al., 2017). In fact, we recently observed that exposure to the dioxin-like PCB 126 increased systemic inflammation and accelerated atherosclerosis in a lean LDL receptor-deficient mouse model (Petriello et al., 2018). Numerous studies have also demonstrated that coplanar (or dioxin-like) PCBs can cause vascular endothelial dysfunction as evidenced by an upregulation of inflammatory mediators including the cytokine interleukin-6 (IL-6), vascular adhesion molecule-1 (VCAM-1), and monocyte chemoattractant protein-1 (MCP-1) (Eske et al., 2014; Helyar et al., 2009; Majkova et al., 2009). Furthermore, exposure to coplanar PCBs, such as PCB 77 and PCB 126, caused inflammation in endothelial cells via activation of the NF-κB signaling pathway (Liu et al., 2015).
In addition to endothelial cell activation and dysfunction, accumulating evidence has shown that atherosclerotic lesion formation and growth are effected by both innate and adaptive immune responses (Weber & Noels, 2011; Weber et al., 2008). Although many different types of cells are involved in the initiation and progression of atherosclerosis, macrophages are fundamental contributors, as they migrate from the blood and infiltrate into the intima, release pro-inflammatory cytokines and take up lipids leading to subsequent formation of foam cells. These inflammatory signals in turn result in continuous monocyte recruitment, which is necessary for atherosclerotic plaque maintenance and progression (Weber & Noels, 2011; Weber et al., 2008). Previous studies have shown that all atherosclerosis-related processes (initiation and expansion of lesions, necrosis and rupture, clinical manifestations, resolution and regression of lesions) are influenced by the presence of macrophages (Colin et al., 2014; Tabas & Bornfeldt, 2016).
Serving to protect the organism from infection both in innate and adaptive immune system, macrophages display high plasticity and versatility, in particular, the ability to promote inflammation when needed and switch off inflammatory responses when it is no longer necessary. Such events depend on the fact that macrophages can be activated by the combination of different environmental stimuli. In spite of a spectrum of inflammatory features and functional properties, there are two main groups, classically activated and alternatively activated macrophages, also termed M1 and M2, respectively (Mantovani et al., 2005; Mosser & Edwards, 2008). The macrophage phenotypes and functions can be assessed by their surface markers and secretome. For example, classical macrophage activation is a response to microbial products (e.g. lipopolysaccharide, LPS) or cytokines (e.g. interferon-γ, IFN-γ; tumor necrosis factor, TNFα). The M1 macrophage is classically characterized by the high secretion of inflammatory cytokines, which is regulated in part via nuclear factor NF-kB signaling (C. P. Liu et al., 2017). Such cytokines include interleukin-1 (IL-1 ), IL-6, IL-12, IL-23 and TNFα, which play a role in host defense role, and are also considered pro-atherogenic (Martinez & Gordon, 2014). Alternative macrophage polarization (e.g., M2) can be induced by IL-4 and IL-13, or IL-10 cytokines, and are involved in parasite killing, tissue remodeling, and immunoregulation (Martinez & Gordon, 2014).
In atherosclerotic lesions, macrophages are mainly derived from peripheral blood mononuclear cells, which have long been recognized to be heterogeneous. Recent studies show that there is a highly heterogeneous phenotype and environment in atherosclerotic lesions, such as platelet chemokine CXCL4-induced macrophages, M4 (Domschke & Gleissner, 2017; Erbel et al., 2015), oxidized phospholipid-induced macrophages, Mox (Kadl et al., 2010), hemoglobin (Hb)-stimulated macrophages, M(Hb) (Finn et al., 2012) and heme-induced macrophages, Mhem (Boyle et al., 2012). From a functional point of view, M1 and M4 macrophages display a pro-inflammatory profile and are thus pro-atherogenic (Mantovani et al., 2005; Domschke & Gleissner, 2017). Mox macrophages in mice were found to exhibit reduced phagocytic capacity and to express anti-oxidant genes (Kadl et al., 2010). M2, M(Hb), and Mhem showed anti-inflammatory function by preventing foam cell formation (De Paoli et al., 2014; Vinchi et al., 2014). Lesion-associated macrophages interact with a large variety of environmental stimuli, such as cytokines and oxidized phospholipid, which polarize macrophages to a certain phenotype and contribute to atherosclerosis (Chistiakov et al., 2015; Shapouri-Moghaddam et al., 2018).
We previously have shown that PCB 126, a coplanar or dioxin-like PCB and thus an agonist of the aryl hydrocarbon receptor (AhR), can cause vascular endothelial cell inflammation and accelerated atherosclerosis in preclinical models, but a mechanistic investigation into the effects of this pollutant on monocyte/macrophage polarization has been lacking. We hypothesized that PCB 126 can contribute to macrophage inflammation and polarization to a M1-like subtype. To test this hypothesis, THP-1 cells were differentiated to macrophages, and subsequently exposed to different types of PCBs, e.g., coplanar PCB 126, mixed type congener PCB 118, or non-coplanar PCB 153. THP-1 is a human monocytic cell line derived from an acute monocytic leukemia male patient, which is one of the most well established and extensively used cell models to mimic the function and mechanisms of monocytes and macrophages (Chanput et al., 2014; Qin, 2012). Inflammatory cytokines and genes associated with oxidative stress were examined by qPCR and multiplexed protein analysis. As in our previous studies using endothelial cell models, here we observed significant induction of multiple cytokines, chemokines, and inflammatory mediators in human macrophages exposed to PCB 126 but not to PCB 118 or PCB 153.
2. Materials and Methods
2.1. Chemicals and reagents
3,3’,4,4’,5-pentachlorobiphenyl (PCB 126), 2,3’,4,4’,5-pentachlorobiphenyl (PCB 118), and 2,2’,4,4’,5,5’-hexachlorobiphenyl (PCB 153) were purchased from AccuStandard Inc. (New Haven, CT, USA). Phorbol 12-myristate 13-acetate (PMA), dimethyl sulfoxide (DMSO), lipopolysaccharide (LPS), BMS-345541 (NF-κB antagonist) were purchased from Sigma (St. Louis, MO, USA). Recombinant human interferonγ (IFN-γ) was purchased from PeproTech (Rocky Hill, NJ, USA). CH-223191(AhR antagonist) was purchased from BioVision (Milpitas, CA, USA)
2.2. Cell culture and differentiation
The human monocytic leukemia cell line THP-1 was used as a model to study monocyte/macrophage functions. THP-1 cells were purchased from the American Type Culture Collection (ATCC® TIB-202™, Manassas, VA, USA), and were cultured in a complete growth medium of RPMI Medium 1640 (ATCC modification) (Gibco™, Grand Island, NY, USA), supplemented with 10% (v/v) ultimate grade fetal bovine serum (VWR Seradigm), 0.05 mM 2-mercaptoethanol (BME; TCI Co., Ltd., Tokyo, Japan), and antibiotics (100 U/mL penicillin and 100 µg/mL streptomycin, Gibco™). The cells were maintained in a humidified atmosphere containing 5 % CO2 at 37°C. Before PCB treatment, THP-1 cells were washed with serum free media (Macrophage-SFM; Gibco™) and seeded at a density of 1×106 cells/well in a 6 – well plate with 20 nM PMA in Macrophage-SFM for 24 h to differentiate.
ToxinSensor™ Chromogenic LAL Endotoxin Assay Kit (GenScript, Piscataway, NJ, USA) was used to evaluate the endotoxin level in cell culture reagents.
2.3. PCB treatment
Stock solutions of PCBs were prepared in DMSO, and differentiated THP-1 cells were treated with PCB 126, PCB 118, or PCB 153 at concentrations up to 5000 nM for 4–24 h as described within the figure legends. A cotreatment of 10 ng/mL LPS and 5 ng/mL IFN-γ acted as a positive control. As PCB 118 and PCB 153 elicited no response in initial proinflammatory cytokine induction studies, only PCB 126 was used moving forward with subsequent experimental settings (Supplemental Figure 1). The concentration of DMSO in the media was kept below 0.05%.
2.4. AhR and NF-κB antagonism/inhibition studies
After mature macrophages adhered to the cell culture plate (i.e., PMA treatment for 24 h), cells were pretreated with the AhR antagonist CH-223191 (5 µM) or the NF-kB antagonist BMS-345541 (5 µM) 2 h before PCB 126 (500 nM, 24 h) treatment. Total RNA and cell culture medium were processed and kept in −80 °C for gene expression and cytokine analysis, respectively. mRNA expression of TNFα, IL-1β, CYP1A1 were assessed by real-time PCR, and protein levels for cytokines TNFα, IL-1β, CCL2, IL-8, IL-1RA, CCL3, CCL4 IL-1, IP-10, and IFN-ɣ were measured utilizing Milliplex Map Human Cytokine/Chemokine Magnetic Bead Panel from cell culture medium as instructed.
2.5. Quantitative real-time PCR
Total RNA was extracted from the cells using the TRIzol reagent (Thermo Fisher Scientific Inc., Waltham, MA, USA). RNA purity and quantity were assessed with the Nanodrop 2000/2000c (Thermo Fisher Scientific Inc.) using the Nanodrop 2000 (Version 1.6.198) software. cDNA was synthesized from total RNA using the Quantabio qScrip cDNA SuperMix (QuantaBio Inc., Gaithersburg, MD, USA) and T 100™ Thermal Cycler (Bio-Rad, Hercules, CA, USA). Polymerase chain reaction (PCR) was performed on the CFX96 Touch Real-Time PCR Detection System (Bio-Rad) using the TaqMan® Fast Advanced Master Mix (Thermo Fisher Scientific Inc.). Primer sequences mixture from TaqMan Gene Expression Assays (Thermo Fisher Scientific Inc.) were as follows: actin beta (ACTB), Hs01060665_g1; interleukin-1 beta (IL1B), Hs01555410_m1; interleukin-6 (IL6), Hs00174131_m1; IL-8 (CXCL8), Hs00174103_m1; tumor necrosis factor alpha (TNF), Hs00174128_m1; C-C motif chemokine ligand 2 (CCL2), Hs00234140_m1; CCL3, Hs00234142_m1; CCL4, Hs99999148_m1; cytochrome P450 family 1 subfamily A member 1 (CYP1A1), Hs01054796_g1; nuclear factor, erythroid 2 like 2 (NFE2L2), Hs00978100_m1; Hs00971716_m1; NAD(P)H quinone dehydrogenase 1 (NQO1), Hs01045993_g1; heme oxygenase 1 (HMOX1), Hs01110250_m1; glutathione S-transferase theta 1 (GSTT1), Hs02512069_s1; glutathione-disulfide reductase (GSR), Hs00167317_m1; mannose receptor, C type 1 (MRC1), Hs00267207_m1; macrophage scavenger receptor 1 (MSR1), Hs00234007_m1.
The levels of mRNA were normalized relative to the amount of ACTB mRNA, and expression levels in cells treated with DMSO were set at 1. Gene expression levels were calculated according to the 2−∆∆Ct method (Livak & Schmittgen, 2001).
2.6. Cytokine/Chemokine assessment
The Milliplex Map Human Cytokine/Chemokine Magnetic Bead Panel (Millipore Corp, Billerica, MA, USA) was utilized to measure cell culture medium cytokines and chemokines Interferon-gamma (IFN-γ), interleukin-1 alpha (IL-1α), interleukin-1 beta (IL-1β), interleukin-1 receptor antagonist (IL-1RA), interleukin-6 (IL-6), interleukin-8 (IL-8), interleukin-17A (IL-17A), Interferon gamma-induced protein 10 (IP-10), macrophage chemoattractant protein-1 (MCP-1, or CCL2), macrophage inflammatory protein-1 alpha (MIP-1α, or CCL3), macrophage inflammatory protein-1 beta (MIP-1β, or CCL4), and tumor necrosis factor alpha (TNFα). Plates were run and analyzed on the Luminex Xmap MAGPIX system (Luminex Corp, Austin, TX, USA), according to the manufacturer’s instructions.
2.7. Statistical Analysis
The results of the cytokines are shown as mean value ± standard error (SEM) as error bars, respectively. Experiments were performed in triplicate. The statistical analysis was performed using GraphPad Prism-6. One-way ANOVA multiple comparison test (as a post-test analysis) was performed with the Tukey test (multiple comparison test comparing every group with every other group). Values are expressed as mean ± SEM. A probability value of p ≤ 0.05 was considered statistically significant.
3. Results
3.1. PCB 126 elicited inflammation and macrophage polarization
To examine the hypothesis that PCBs can influence macrophage polarization, PCB 126, PCB 118, and PCB 153 were chosen to treat THP-1 derived macrophages. These PCBs represent the three major classes of congeners that humans are exposed to (i.e., coplanar, mixed, and non-coplanar congeners). Initially, both concentration and temporal dose responses were examined. A concentration range of 5, 50, 500 and 5000 nM of the three representative PCBs was used, in addition to vehicle control DMSO and positive control LPS/IFN-γ treatment groups. After 16 h post exposure to PCBs, the expression of two acute inflammatory cytokines TNFα and IL-1 were increased significantly by doses of 50, 500 and 5000 nM of PCB 126 examined, but PCB 118 and PCB 153 had no effect (Figure S2). Then, to examine the effect of exposure duration on cytokine induction, macrophages were treated with two concentrations of PCB 126 (50 and 500 nM) for either 4, 8, 16 or 24 h. Increased IL-6 levels were detected as early as 4 h in PCB 126 (500 nM) treated group, while increased IL-1β levels were only observed at the 24 h time point group. CCL2 mRNA was not a sensitive marker of PCB 126 exposure, but did increase very rapidly in the LPS/IFN-ɣ groups. All exposure groups exhibited a time-dependent linear increase in expression of TNFα regardless of concentration, while IL-6 showed a biphasic response to PCB exposure at both concentrations (Figure S3). PCB 126 exposure for 24 h induced expression of inflammatory cytokines in macrophages in both 50 and 500 nM treatment groups, such as TNFα (up to 5 fold), IL-1β (up to 5 fold), CCL3 (up to 3 fold), CCL4 (up to 3 fold), and IL-8 (up to 7 fold) (Figure 1A). Chemokine (C-C motif) ligand 2 (CCL2), referred to as monocyte chemoattractant protein-1 (MCP1), which regulates immune cell recruitment and migration and infiltration of monocytes/macrophages into vascular tissues, also increased in PCB 126 treated macrophages (up to 2 fold) (Figure 1A). All the mentioned inflammatory cytokines increased significantly in PCB 126 and LPS/ IFN-γ co-treatment group. We also investigated two well established markers (MRC1 and MSR1) of the M2 macrophage phenotype, and PCB 126 elicited no significant response (data not shown).
Figure 1. PCB 126 elicited inflammation in macrophages.
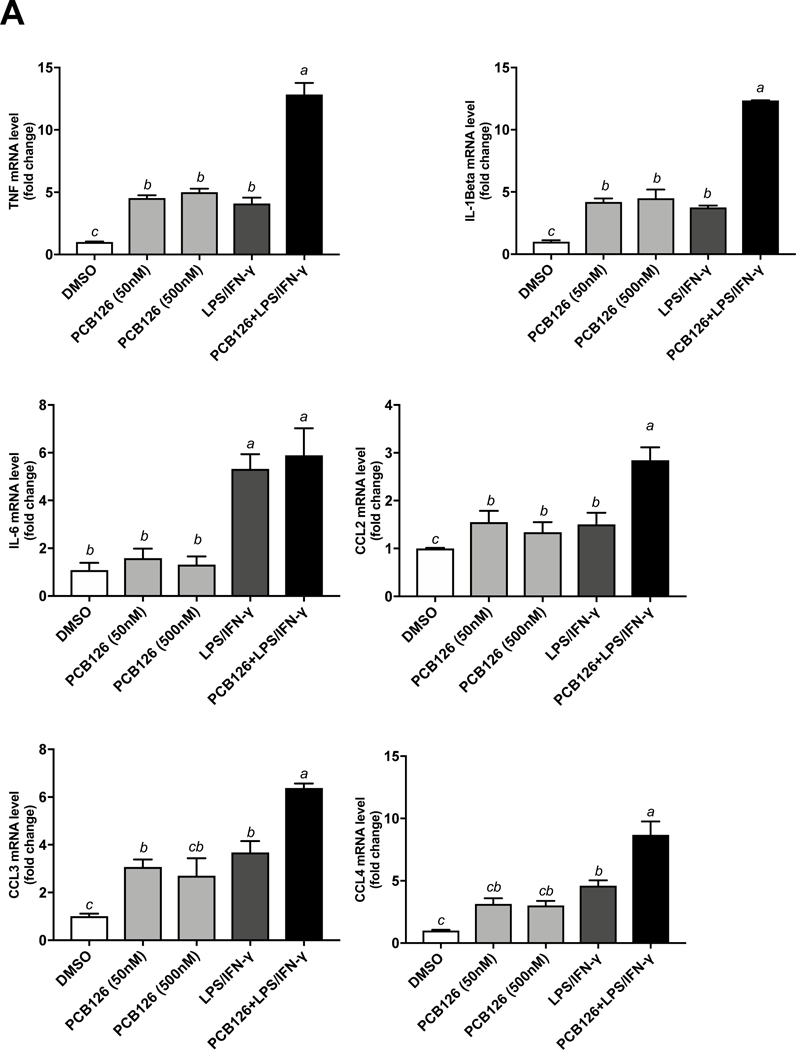


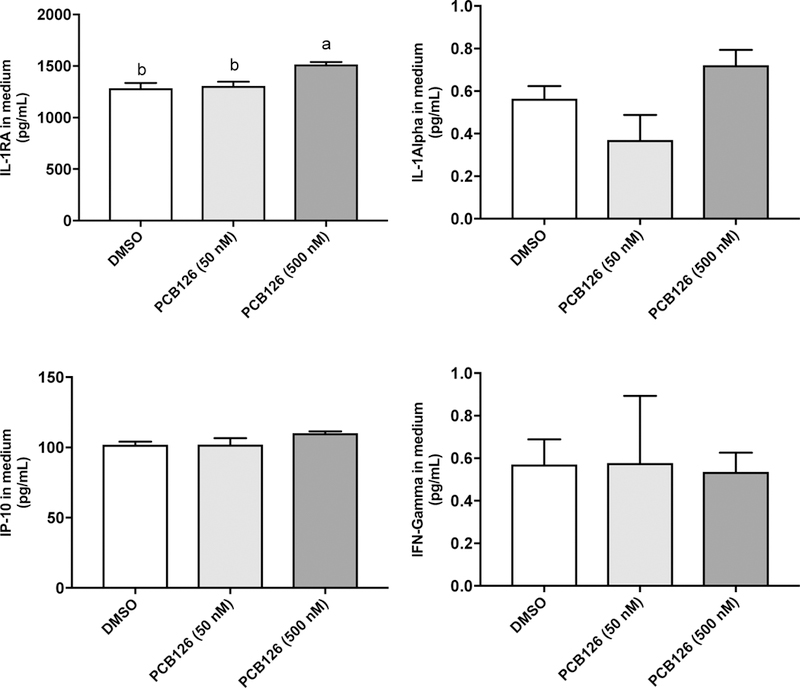
(A) THP-1 derived macrophage mRNA expression for TNF, IL-1β, IL-6, IL-8, CCL2 (MCP1), CCL3 (MIP-1α), CCL4 (MIP-1β) were assessed after 24 h treatment of PCB 126, PCB126 + LPS (10 ng/mL)/IFN-γ (5 ng/mL) and DMSO. (B) Protein levels for cytokines TNF, IL-1β, IL-8, CCL2, CCL3, CCL4, IL-1RA, IL-1, IP-10, and IFN-ɣ were measured utilizing Milliplex Map Human Cytokine/Chemokine Magnetic Bead Panel from cell culture medium after 24 h exposure of PCB 126. IL-6, IL-17A concentrations were below the lower limit of detection (<3.2 pg/mL) (data not shown). Values are mean ± SEM (n=3). Different letters indicate significant difference (P ≤ 0.05) between treatment groups. Tukey’s multiple comparisons test was used.
Furthermore, 12 cytokines and chemokines secreted into the cell culture medium, including IFN-γ, IL-1α, IL-1β, IL-1RA, IL-6, IL-8, IL-17A, IP-10, MCP-1(CCL2), MIP-1α (CCL3), MIP-1β (CCL4) and TNFα, were measured by the Milliplex Map Human Cytokine/Chemokine Magnetic Bead Panel. IL-1β, IL-1RA, IL-8, CCL2, CCL3, CCL4 and TNFα were significantly increased following PCB 126 (500 nM) exposure for 24 h (Figure 1B). IL-6, IL-17A concentrations were below the lower limit of detection (<3.2 pg/mL) (data not shown).
3.2. PCB 126 induced CYP1A1 and NFE2L2 in macrophages
Since it is well established that PCB 126 can activate the AhR and increase reactive oxygen species in other cell types, we next examined mRNA expression for cytochrome P450 1A1 (CYP1A1), nuclear factor (erythroid-derived 2)-like 2 (NFE2L2) and downstream genes, such as heme oxygenase 1 (HMOX1), NAD(P)H quinone oxidoreductase 1 (NQO1), glutathione S-transferase (GST) and glutathione-disulfide reductase (GSR) by real-time PCR after PCB 126 exposure for 24 h. CYP1A1 increased significantly in the 500 nM PCB 126 treated group and PCB 126+ LPS/ IFN-γ co-treatment group compared to all other groups (Figure 2). The expression of most antioxidant genes, such as Nrf2 (alias for NFE2L2), HMOX1, NQO1 expression were upregulatedd in PCB 126 treated groups and PCB 126+ LPS/ IFN-γ co-treatment group (Figure 2), indicating induction of xenobiotic-linked defensive mechanisms.
Figure 2. PCB 126 induced CYP1A1 and NFE2L2 in macrophages.

Cytochrome P450 1A1 (CYP1A1) increased significantly in PCB126 (500 nM), and PCB126 + LPS (10 ng/mL)/IFN-γ (5 ng/mL) treated groups compared to all other groups. Nuclear factor (erythroid-derived 2)-like 2 (NFE2L2) was induced with 500 nM PCB126, and PCB126 + LPS/IFN-γ compared to control, while down-stream genes, such as heme oxygenase 1 (HMOX1), NAD(P)H quinone oxidoreductase 1 (NQO1) were significantly increased following 500 nM PCB126 or PCB126 + LPS /IFN-γ exposure compared to all other groups, indicating induction of xenobiotic-linked defensive mechanisms. Values are mean ± SEM (n=3). Different letters indicate significant difference (P ≤ 0.05) between treatment groups. Tukey’s multiple comparisons test was used.
3.3. PCB 126 exacerbated inflammation in LPS exposed monocytes
Since undifferentiated monocytes may come into contact with both PCBs and other inflammatory stressors such as LPS in the blood simultaneously (before they migrate to the intima or tissues, and differentiate to macrophages), we next examined the 24 h treatment effect of PCB 126, LPS/IFN-ɣ, or PCB 126 + LPS/IFN-ɣ on THP-1 cell activation. Unlike with the macrophage studies described above, PCB 126 alone had minimal effect on TNFα or IL-1β mRNA expression in undifferentiated monocytes. Interestingly, and as expected, the positive control group of LPS/IFN-γ displayed increased expression of these pro-inflammatory cytokines, but this was synergistically increased when PCB 126 was concomitantly added (Figure 3A). CYP1A1 was induced by PCB 126 in monocytes, but there was no synergistic increase in the presence of LPS/IFN-ɣ (Figure 3B), confirming our previous findings that coplanar PCBs, like PCB 126, upregulate CYP1A1 expression (Hennig et al., 2002).
Figure 3. PCB 126 exacerbated inflammation in LPS exposed monocytes and induced CYP1A1.
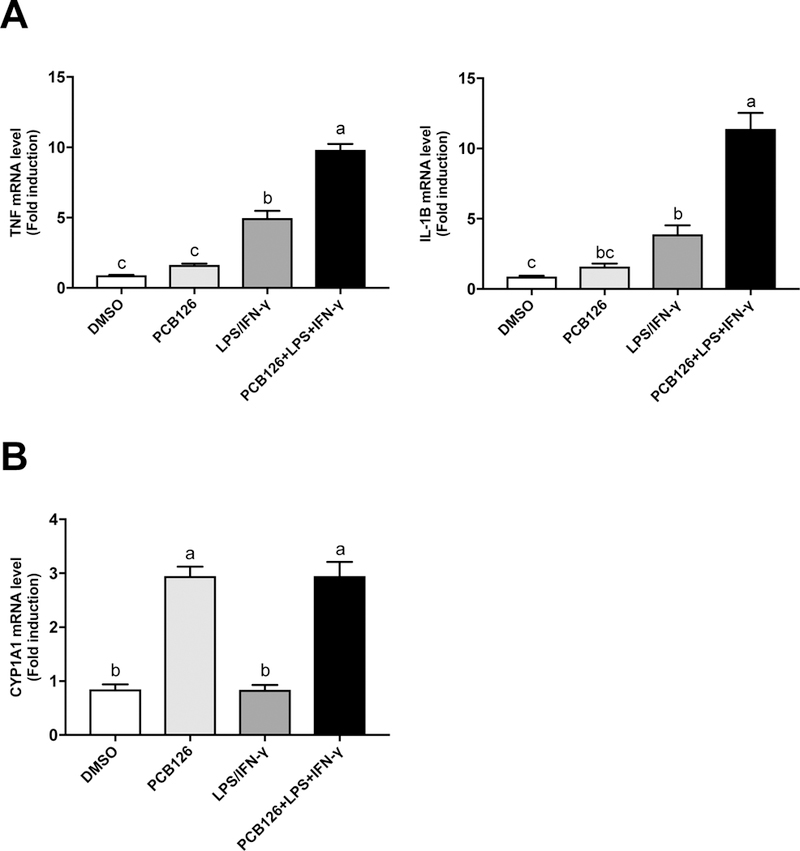
THP-1 cells were treated with PCB126, LPS/IFN-ɣ, and PCB126 + LPS/IFN-ɣ respectively for 24 h, and the results showed that the inflammatory cytokines, such as TNF and IL-1β, increased in LPS/IFN-ɣ and PCB126 + LPS/IFN-ɣ treated groups. However, CYP1A1 increased significantly in PCB126 and PCB126 + LPS/IFN-ɣ treated groups. Values are mean ± SEM (n=3). Different letters indicate significant difference (P ≤ 0.05) between treatment groups. Tukey’s multiple comparisons test was used.
3.4. PCB 126-induced oxidative stress and inflammation involved both AhR and NF-κB signaling pathways
To examine signaling mechanisms that may be responsible for the observed PCB 126-induced inflammation of macrophages, both the AhR antagonist (CH-223191, 5 M) and NF B antagonist (BMS-345541, 5 M) were utilized. mRNA expression for TNFα, IL-1β, CYP1A1 were assessed by real-time PCR (Figure 4A), and protein levels of cytokines TNFα, IL-1β, IL-8, CCL2, CCL3, CCL4, IL-1RA, IL-1, IP-10, and IFN-ɣ were measured utilizing Milliplex Map Human Cytokine/Chemokine Magnetic Bead Panel from cell culture medium (Figure 4B). The mRNA expression of TNFα and IL-1β were reduced by 2–3 fold in both AhR and NF-κB antagonist groups co-treated with PCB 126 (Figure 4A). At the protein level, concentrations of 6 inflammatory cytokines (TNFα, IL-1β, IL-8, IL-1RA, CCL3, CCL4) were decreased in the presence of the NF-κB antagonist while only 2 cytokines (TNFα, IL-8) were reduced in the presence of the AhR antagonist (Figure 4B).
Figure 4. PCB 126 induced inflammation and oxidative stress involved both AhR and NF B signaling pathway in macrophages.
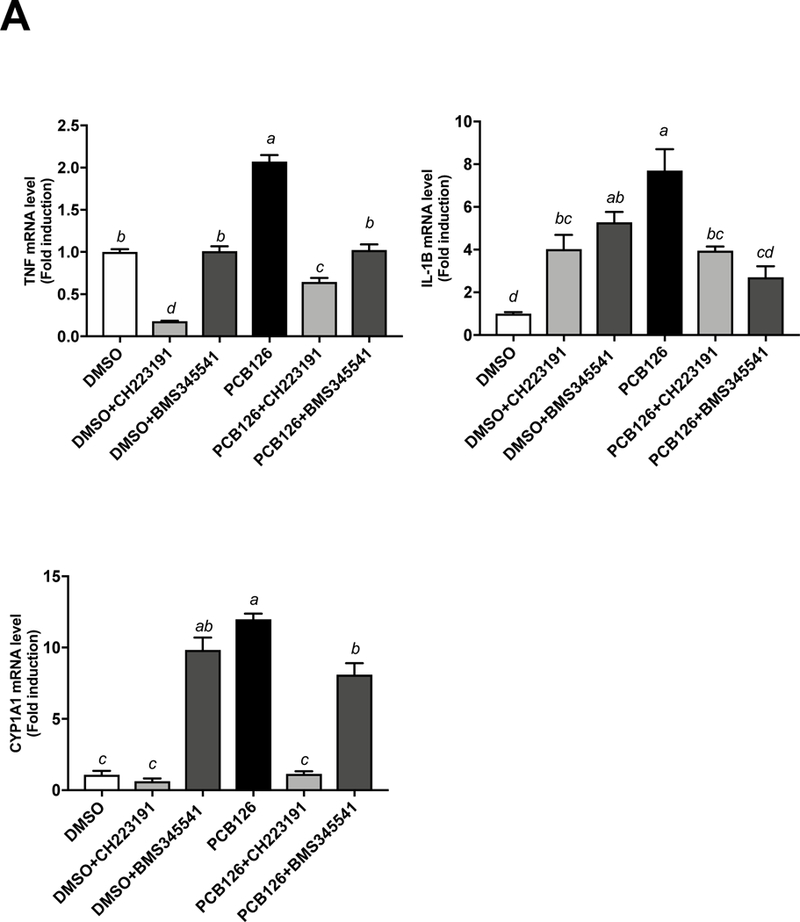
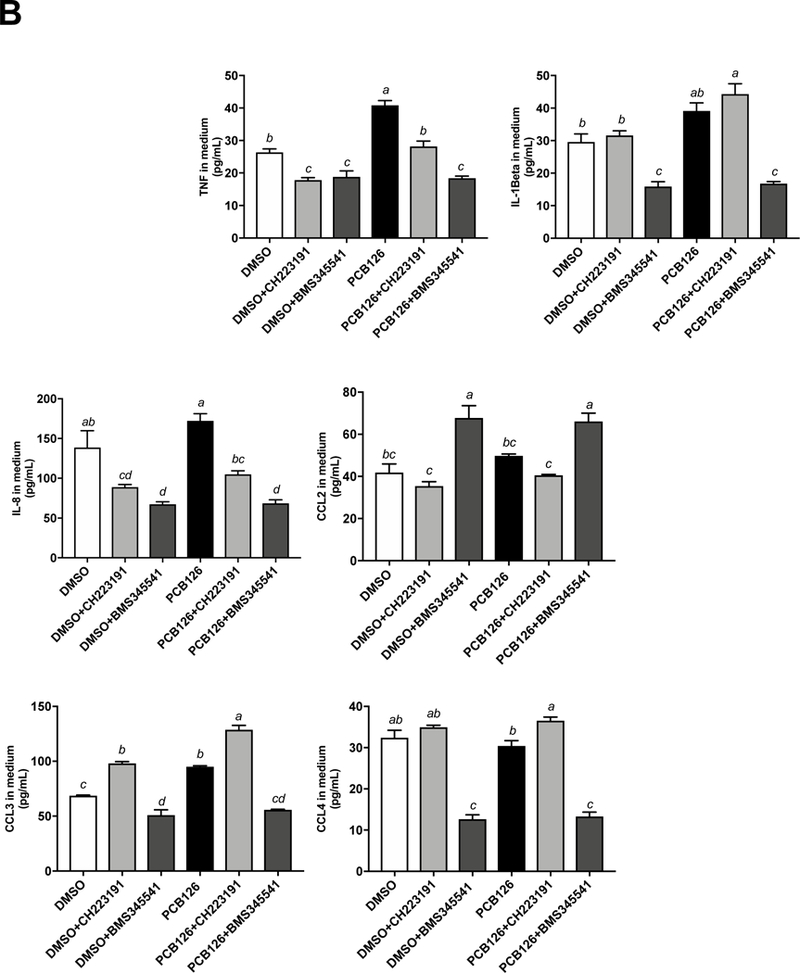

THP-1 derived macrophages were pretreated with the AhR antagonist CH-223191 (5 M) or the NF B antagonist BMS-345541 (5 M) for 2 h before PCB 126 (500 nM, 24 h) exposure. (A) mRNA expression for TNF, IL-1β, CYP1A1 were assessed. (B) Protein levels for cytokines TNF, IL-1β, IL-8, CCL2, CCL3, CCL4, IL-1RA, IL-1, IP-10, and IFN-ɣ were measured utilizing Milliplex Map Human Cytokine/Chemokine Magnetic Bead Panel from cell culture medium. IL-6, IL-17A concentrations were below the lower limit of detection (<3.2 pg/mL) (data not shown). Values are mean ± SEM (n=3). * P ≤ 0.05. Tukey’s multiple comparisons test was used.
4. Discussion
It is well documented that macrophage accumulation within the arterial wall is a hallmark of atherosclerosis (Geovanini & Libby, 2018; Z. Liu et al., 2017; Moore & Tabas, 2011; Zeller & Srivastava, 2014). A chronic inflammatory response in the vascular wall, linked to uptake of low-density lipoprotein by macrophages and their subsequent transformation into foam cells, are characteristic events in the pathology of atherosclerosis. Monocyte-derived macrophages are a homogeneous cell population and present a spectrum of phenotypes according to different micro-environmental stimuli. Accumulating evidence suggests that macrophages can show high plasticity in atherosclerotic plaques, and more macrophage subtypes have been profiled, such as M4 (Domschke & Gleissner, 2017; Erbel et al., 2015), Mox (Kadl et al., 2010), M(Hb) (Finn et al., 2012) and Mhem (Boyle et al., 2012). Numerous stimuli, such as inflammatory cytokines or oxidized phospholipids, can polarize macrophages to a certain phenotype and contribute to atherosclerosis (Chistiakov et al., 2015; Shapouri-Moghaddam et al., 2018). The current study focuses on the pro-inflammatory macrophage phenotype M1, a major mediator of inflammation during atherogenesis (Martinez & Gordon, 2014).
There is evidence that NF-kB signaling pathways are involved in monocyte activation and macrophage polarization (Brasier, 2010; C. P. Liu et al., 2017; Majdalawieh & Ro, 2010). In fact, NF-kB comprises a family of transcription factors that are oxidative stress sensitive and involved in the pathology of various inflammatory diseases, such as atherosclerosis (Pamukcu et al., 2011; Robbesyn et al., 2004; Yamamoto & Gaynor, 2001). Thus, inhibition of the NF-kB signaling pathways may reduce inflammation, attenuate atherosclerosis and prevent its complications (Pamukcu et al., 2011). There is also evidence that AhR plays a role in the pathogenesis of cardiovascular diseases (Yi et al., 2018). This may be due to its ability to modulate biological responses or other signaling pathways within different cells types, e.g., macrophages, in the cardiovascular system (Hewitson et al., 2004; Ogando et al., 2016; Wang et al., 2017). Interestingly, at least in mice, it appears that loss of AhR signaling can also lead to dysregulated inflammatory signaling. For example, AhR whole body knockouts display increased expression of TNF-α, IL-6 and IL-1β in an experimental model of autoimmune Uveitis, and peritoneal macrophages isolated from AhR null mice also displayed increased levels of these cytokines (Climaco-Arvizu et al., 2016; Huang et al., 2018). Importantly, these studies utilized mice that were deficient in AhR from birth and thus may have exhibited overall developmental differences of the immune system (Li et al., 2017). Also, as has been shown using cell-based assays, murine and human AhR can differ in ligand binding capabilities (Bock, 2018). Future work utilizing conditional or inducible knockouts will be critical to investigate AhR’s role in inflammation in adult mice. Interestingly, interactions between AhR and NF-kB pathways could be critical in regulating inflammatory responses and associated diseases (Vogel & Matsumura, 2009).
PCB 126 and other dioxin-like pollutants may also elicit toxic effects through additional signaling pathways. One possible explanation is the growing body of evidence of AhR crosstalk with multiple other inflammatory regulators, including NF-kB and Nrf2. For example, AhR appears to interact with two critical subunits of NF-kB, RelA and RelB, to modulate inflammation through the expression of many important cytokines (Vogel & Matsumura, 2009; Vondracek et al., 2011). In addition, there appears to be bidirectional interactions between AhR and the master regulator of antioxidant response, Nrf2. Interestingly, there appears to be AhR binding sequences within the Nrf2 promoter, and optimum upregulation of certain downstream genes, such as NQO1, requires both Nrf2 and AhR. (Kahan et al., 1988). Crosstalk between AhR and Nrf2 may explain in part the results presented in Figure 2, which indicate simultaneous upregulation of CYP1A1, NQO1 and HMOX1, following exposure to PCB 126.
There is accumulating evidence suggesting that exposure to certain environmental contaminants, such as polyhalogenated aromatic hydrocarbons, e.g., TCDD or PCBs (coplanar PCBs in particular), can be involved in the development of atherosclerosis. Previous studies have shown that exposure to PCBs can be pro-atherogenic by inducing pro-inflammatory reactions, causing endothelial cell dysfunction (Choi et al., 2003; Eske et al., 2014; Liu et al., 2015; Petriello et al., 2016). In the present study, we provide evidence that coplanar PCBs (PCB 126) can induce or exacerbate macrophage/monocyte inflammation, and thus can promote atherogenisis. Our findings add to this growing mechanistic understanding of how dioxin-like pollutants may contribute to the pathologies of cardiovascular disease, such as atherosclerosis (Mohsenzadeh et al., 2018; Petriello et al., 2018). Historically, the toxicity equivalents of PCB 126 and related dioxin-like pollutants have been determined using cell-based assays of rat or mouse origin, and it is critical for proper risk assessment paradigms to utilize human cell-based approaches since PCB 126 may be a less efficient AhR ligand as once thought (Larsson et al., 2015).
The finding that PCB 126 causes an inflammatory response in human macrophages through AhR and NF-κB transcriptional factors has not been previously reported. Our study confirmed the hypothesis that dioxin-like PCBs (e.g., PCB 126) can contribute to macrophage inflammation, and thus polarize monocytes to a M1-like phenotype, in contrast to PCB 118 and PCB 153. PCB 126-induced inflammation was both concentration and time dependent. Our data also suggest that PCB 126 is a ligand for the AhR within macrophages as evidenced by induction of CYP1A1 expression. PCB 126 also appears to modulate redox signaling, as the antioxidant regulator Nrf2 as well as its target genes such as HMOX1 and NQO1 were upregulated accordingly. Besides, earlier studies demonstrated that expression of Nrf2 can be directly modulated via ligand-activated AhR, and vice versa (Miao et al., 2005; Shin et al., 2007). AhR-Nrf2 crosstalk also plays a key role in drug metabolism (Haarmann-Stemmann et al., 2012). All these findings suggest that PCB 126 causes oxidant stress and inflammation in macrophages. In the NF-κB and AhR blocking experiments, most of the tested inflammatory cytokines were reduced due to NF-κB inhibition, and two notable acute phase proteins, TNFα and IL-8, were downregulated as a result of AhR blocking.
In concordance with our findings, a recent study showed that PCB 126 activates a vigorous pro-inflammatory state in fat cell precursors (preadipocytes) depending on AhR activation, although induction of the response was delayed compared to upregulation of CYP1A1, and an inhibitor (Bay11–7082, 4 M) of NF-κB partially protected preadipocytes from the effects of PCB 126 (Gourronc et al., 2018). Our previous studies demonstrated that coplanar PCB-induced endothelial cell inflammation is regulated in part through epigenetic modification of the NF-κB subunit p65 (Liu et al., 2015). Nutritional protection against PCB126 induced inflammation can also occur through AhR inhibition (Han et al., 2012) and epigenetic modifications of NF-κB target genes in human endothelial cells (Liu et al., 2016). These previous studies all suggest that both AhR and NF-κB signaling pathways are important in regulating PCB 126-mediated inflammation. Our present study provides evidence that macrophages can significantly contribute to a PCB-mediated inflammatory response through AhR and NF-κB signaling pathways.
In conclusion, we demonstrated that PCB 126 can contribute to macrophage polarization and inflammation, indicating another possible role of dioxin-like PCBs in the pathology of atherosclerosis. We also provide evidence that the observed PCB 126-induced inflammation is regulated in part through both AhR and NF-κB signaling pathways, i.e., critical pathways involved in the overall pathology of inflammatory diseases, such as atherosclerosis. Our findings have translational implications, suggesting that environmental insults can contribute to chronic diseases by modulating inflammatory cascades within immune cells creating an advantageous environment for cardiovascular disease progression.
Supplementary Material
Highlights.
Macrophages can be polarized either to pro-inflammatory or anti-inflammatory phenotypes.
Dioxin-like PCBs can contribute to macrophage polarization associated with inflammation.
PCB 126-induced inflammation is regulated in part through both AhR and NF-ĸB signaling pathways.
5. Acknowledgements
Research reported in this publication was supported in part by the National Institute of Environmental and Health Sciences at the National Institute of Health [P42ES007380], Michael C. Petriello was supported by National Institute of Health [K99ES028734].
Abbreviations:
- PCBs
polychlorinated biphenyls
- AhR
aryl hydrocarbon receptor
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- CYP1A1
cytochrome P450 family 1 subfamily A member 1
- NFE2L2
nuclear factor, erythroid 2 like 2
- NQO1
NAD(P)H quinone dehydrogenase 1
- HMOX1
heme oxygenase 1
- GST
glutathione S-transferase
- GSR
glutathione-disulfide reductase
- IFN-γ
interferon-γ
- LPS
lipopolysaccharide
- PMA
Phorbol 12-myristate 13-acetate
- BME
2-mercaptoethanol
- TNFα
tumor necrosis factor alpha
- IL-1α
interleukin-1 alpha
- IL-1β
interleukin-1 beta
- IL-1RA
interleukin-1 receptor antagonist
- IL-6
interleukin-6
- IL-8
interleukin-8
- IL-17A
interleukin-17A
- IP-10
Interferon gamma-induced protein 10
- MCP-1
(CCL2) macrophage chemoattractant protein-1
- MRC1
mannose receptor, C type 1
- MSR1
macrophage scavenger receptor 1
- MIP-1α
(CCL3) macrophage inflammatory protein-1 alpha
- MIP-1β
(CCL4) macrophage inflammatory protein-1 beta
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. References
- Bock KW (2018). From TCDD-mediated toxicity to searches of physiologic AHR functions. Biochem Pharmacol, 155, 419–424. doi: 10.1016/j.bcp.2018.07.032 [DOI] [PubMed] [Google Scholar]
- Boyle JJ, Johns M, Kampfer T, Nguyen AT, Game L, Schaer DJ, … Haskard DO (2012). Activating transcription factor 1 directs Mhem atheroprotective macrophages through coordinated iron handling and foam cell protection. Circ Res, 110(1), 20–33. doi: 10.1161/CIRCRESAHA.111.247577 [DOI] [PubMed] [Google Scholar]
- Brasier AR (2010). The nuclear factor-kappaB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc Res, 86(2), 211–218. doi: 10.1093/cvr/cvq076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanput W, Mes JJ, & Wichers HJ (2014). THP-1 cell line: an in vitro cell model for immune modulation approach. Int Immunopharmacol, 23(1), 37–45. doi: 10.1016/j.intimp.2014.08.002 [DOI] [PubMed] [Google Scholar]
- Chistiakov DA, Bobryshev YV, Nikiforov NG, Elizova NV, Sobenin IA, & Orekhov AN (2015). Macrophage phenotypic plasticity in atherosclerosis: The associated features and the peculiarities of the expression of inflammatory genes. Int J Cardiol, 184, 436–445. doi: 10.1016/j.ijcard.2015.03.055 [DOI] [PubMed] [Google Scholar]
- Choi W, Eum SY, Lee YW, Hennig B, Robertson LW, & Toborek M (2003). PCB 104-induced proinflammatory reactions in human vascular endothelial cells: relationship to cancer metastasis and atherogenesis. Toxicol Sci, 75(1), 47–56. doi: 10.1093/toxsci/kfg149 [DOI] [PubMed] [Google Scholar]
- Climaco-Arvizu S, Dominguez-Acosta O, Cabanas-Cortes MA, Rodriguez-Sosa M, Gonzalez FJ, Vega L, & Elizondo G (2016). Aryl hydrocarbon receptor influences nitric oxide and arginine production and alters M1/M2 macrophage polarization. Life Sci, 155, 76–84. doi: 10.1016/j.lfs.2016.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colin S, Chinetti-Gbaguidi G, & Staels B (2014). Macrophage phenotypes in atherosclerosis. Immunol Rev, 262(1), 153–166. doi: 10.1111/imr.12218 [DOI] [PubMed] [Google Scholar]
- De Paoli F, Staels B, & Chinetti-Gbaguidi G (2014). Macrophage phenotypes and their modulation in atherosclerosis. Circ J, 78(8), 1775–1781. [DOI] [PubMed] [Google Scholar]
- Domschke G, & Gleissner CA (2017). CXCL4-induced macrophages in human atherosclerosis. Cytokine doi: 10.1016/j.cyto.2017.08.021 [DOI] [PubMed]
- Erbel C, Tyka M, Helmes CM, Akhavanpoor M, Rupp G, Domschke G, … Gleissner CA (2015). CXCL4-induced plaque macrophages can be specifically identified by co-expression of MMP7+S100A8+ in vitro and in vivo. Innate Immun, 21(3), 255–265. doi: 10.1177/1753425914526461 [DOI] [PubMed] [Google Scholar]
- Eske K, Newsome B, Han SG, Murphy M, Bhattacharyya D, & Hennig B (2014). PCB 77 dechlorination products modulate pro-inflammatory events in vascular endothelial cells. Environ Sci Pollut Res Int, 21(10), 6354–6364. doi: 10.1007/s11356-013-1591-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn AV, Nakano M, Polavarapu R, Karmali V, Saeed O, Zhao X, … Virmani R (2012). Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J Am Coll Cardiol, 59(2), 166–177. doi: 10.1016/j.jacc.2011.10.852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geovanini GR, & Libby P (2018). Atherosclerosis and inflammation: overview and updates. Clin Sci (Lond), 132(12), 1243–1252. doi: 10.1042/CS20180306 [DOI] [PubMed] [Google Scholar]
- Gourronc FA, Robertson LW, & Klingelhutz AJ (2018). A delayed proinflammatory response of human preadipocytes to PCB126 is dependent on the aryl hydrocarbon receptor. Environ Sci Pollut Res Int, 25(17), 16481–16492. doi: 10.1007/s11356-017-9676-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haarmann-Stemmann T, Abel J, Fritsche E, & Krutmann J (2012). The AhR-Nrf2 pathway in keratinocytes: on the road to chemoprevention? J Invest Dermatol, 132(1), 7–9. doi: 10.1038/jid.2011.359 [DOI] [PubMed] [Google Scholar]
- Han SG, Han SS, Toborek M, & Hennig B (2012). EGCG protects endothelial cells against PCB 126-induced inflammation through inhibition of AhR and induction of Nrf2-regulated genes. Toxicol Appl Pharmacol, 261(2), 181–188. doi: 10.1016/j.taap.2012.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helyar SG, Patel B, Headington K, El Assal M, Chatterjee PK, Pacher P, & Mabley JG (2009). PCB-induced endothelial cell dysfunction: role of poly(ADP-ribose) polymerase. Biochem Pharmacol, 78(8), 959–965. doi: 10.1016/j.bcp.2009.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitson KS, McNeill LA, & Schofield CJ (2004). Modulating the hypoxia-inducible factor signaling pathway: applications from cardiovascular disease to cancer. Curr Pharm Des, 10(8), 821–833. [DOI] [PubMed] [Google Scholar]
- Huang Y, He J, Liang H, Hu K, Jiang S, Yang L, … Hou S (2018). Aryl Hydrocarbon Receptor Regulates Apoptosis and Inflammation in a Murine Model of Experimental Autoimmune Uveitis. Front Immunol, 9, 1713. doi: 10.3389/fimmu.2018.01713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR, … Leitinger N (2010). Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res, 107(6), 737–746. doi: 10.1161/CIRCRESAHA.109.215715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahan BD, Didlake R, Kim EE, Yoshimura N, Kondo E, & Stepkowski S (1988). Important role of cyclosporine for the induction of immunologic tolerance in adult hosts. Transplant Proc, 20(2 Suppl 2), 438–450. [PubMed] [Google Scholar]
- Larsson M, van den Berg M, Brenerova P, van Duursen MB, van Ede KI, Lohr C, … Andersson PL (2015). Consensus toxicity factors for polychlorinated dibenzo-p-dioxins, dibenzofurans, and biphenyls combining in silico models and extensive in vitro screening of AhR-mediated effects in human and rodent cells. Chem Res Toxicol, 28(4), 641–650. doi: 10.1021/tx500434j [DOI] [PubMed] [Google Scholar]
- Li J, Phadnis-Moghe AS, Crawford RB, & Kaminski NE (2017). Aryl hydrocarbon receptor activation by 2,3,7,8-tetrachlorodibenzo-p-dioxin impairs human B lymphopoiesis. Toxicology, 378, 17–24. doi: 10.1016/j.tox.2016.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CP, Zhang X, Tan QL, Xu WX, Zhou CY, Luo M, … Zeng X (2017). NF-kappaB pathways are involved in M1 polarization of RAW 264.7 macrophage by polyporus polysaccharide in the tumor microenvironment. PLoS One, 12(11), e0188317. doi: 10.1371/journal.pone.0188317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Perkins JT, & Hennig B (2016). EGCG prevents PCB-126-induced endothelial cell inflammation via epigenetic modifications of NF-kappaB target genes in human endothelial cells. J Nutr Biochem, 28, 164–170. doi: 10.1016/j.jnutbio.2015.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Perkins JT, Petriello MC, & Hennig B (2015). Exposure to coplanar PCBs induces endothelial cell inflammation through epigenetic regulation of NF-kappaB subunit p65. Toxicol Appl Pharmacol, 289(3), 457–465. doi: 10.1016/j.taap.2015.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Zhu H, Dai X, Wang C, Ding Y, Song P, & Zou MH (2017). Macrophage Liver Kinase B1 Inhibits Foam Cell Formation and Atherosclerosis. Circ Res, 121(9), 1047–1057. doi: 10.1161/CIRCRESAHA.117.311546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, & Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 25(4), 402–408. doi: 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Majdalawieh A, & Ro HS (2010). Regulation of IkappaBalpha function and NF-kappaB signaling: AEBP1 is a novel proinflammatory mediator in macrophages. Mediators Inflamm, 2010, 823821. doi: 10.1155/2010/823821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majkova Z, Smart E, Toborek M, & Hennig B (2009). Up-regulation of endothelial monocyte chemoattractant protein-1 by coplanar PCB77 is caveolin-1-dependent. Toxicol Appl Pharmacol, 237(1), 1–7. doi: 10.1016/j.taap.2009.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Sica A, & Locati M (2005). Macrophage polarization comes of age. Immunity, 23(4), 344–346. doi: 10.1016/j.immuni.2005.10.001 [DOI] [PubMed] [Google Scholar]
- Martinez FO, & Gordon S (2014). The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep, 6, 13. doi: 10.12703/P6-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao W, Hu L, Scrivens PJ, & Batist G (2005). Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: direct cross-talk between phase I and II drug-metabolizing enzymes. J Biol Chem, 280(21), 20340–20348. doi: 10.1074/jbc.M412081200 [DOI] [PubMed] [Google Scholar]
- Mohsenzadeh MS, Zanjani BR, & Karimi G (2018). Mechanisms of 2,3,7,8-tetrachlorodibenzo-p-dioxin- induced cardiovascular toxicity: An overview. Chem Biol Interact, 282, 1–6. doi: 10.1016/j.cbi.2018.01.002 [DOI] [PubMed] [Google Scholar]
- Moore KJ, & Tabas I (2011). Macrophages in the pathogenesis of atherosclerosis. Cell, 145(3), 341–355. doi: 10.1016/j.cell.2011.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser DM, & Edwards JP (2008). Exploring the full spectrum of macrophage activation. Nat Rev Immunol, 8(12), 958–969. doi: 10.1038/nri2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogando J, Tardaguila M, Diaz-Alderete A, Usategui A, Miranda-Ramos V, Martinez-Herrera DJ, … Manes S (2016). Notch-regulated miR-223 targets the aryl hydrocarbon receptor pathway and increases cytokine production in macrophages from rheumatoid arthritis patients. Sci Rep, 6, 20223. doi: 10.1038/srep20223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamukcu B, Lip GY, & Shantsila E (2011). The nuclear factor--kappa B pathway in atherosclerosis: a potential therapeutic target for atherothrombotic vascular disease. Thromb Res, 128(2), 117–123. doi: 10.1016/j.thromres.2011.03.025 [DOI] [PubMed] [Google Scholar]
- Petriello MC, Brandon JA, Hoffman J, Wang C, Tripathi H, Abdel-Latif A, … Morris AJ (2018). Dioxin-like PCB 126 Increases Systemic Inflammation and Accelerates Atherosclerosis in Lean LDL Receptor-Deficient Mice. Toxicol Sci, 162(2), 548–558. doi: 10.1093/toxsci/kfx275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petriello MC, Hoffman JB, Sunkara M, Wahlang B, Perkins JT, Morris AJ, & Hennig B (2016). Dioxin-like pollutants increase hepatic flavin containing monooxygenase (FMO3) expression to promote synthesis of the pro-atherogenic nutrient biomarker trimethylamine N-oxide from dietary precursors. J Nutr Biochem, 33, 145–153. doi: 10.1016/j.jnutbio.2016.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z (2012). The use of THP-1 cells as a model for mimicking the function and regulation of monocytes and macrophages in the vasculature. Atherosclerosis, 221(1), 2–11. doi: 10.1016/j.atherosclerosis.2011.09.003 [DOI] [PubMed] [Google Scholar]
- Robbesyn F, Salvayre R, & Negre-Salvayre A (2004). Dual role of oxidized LDL on the NF-kappaB signaling pathway. Free Radic Res, 38(6), 541–551. [DOI] [PubMed] [Google Scholar]
- Ross R (1999). Atherosclerosis--an inflammatory disease. N Engl J Med, 340(2), 115–126. doi: 10.1056/NEJM199901143400207 [DOI] [PubMed] [Google Scholar]
- Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, … Sahebkar A (2018). Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol, 233(9), 6425–6440. doi: 10.1002/jcp.26429 [DOI] [PubMed] [Google Scholar]
- Shin S, Wakabayashi N, Misra V, Biswal S, Lee GH, Agoston ES, … Kensler TW (2007). NRF2 modulates aryl hydrocarbon receptor signaling: influence on adipogenesis. Mol Cell Biol, 27(20), 7188–7197. doi: 10.1128/MCB.00915-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, & Bornfeldt KE (2016). Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ Res, 118(4), 653–667. doi: 10.1161/CIRCRESAHA.115.306256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinchi F, Muckenthaler MU, Da Silva MC, Balla G, Balla J, & Jeney V (2014). Atherogenesis and iron: from epidemiology to cellular level. Front Pharmacol, 5, 94. doi: 10.3389/fphar.2014.00094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel CF, & Matsumura F (2009). A new cross-talk between the aryl hydrocarbon receptor and RelB, a member of the NF-kappaB family. Biochem Pharmacol, 77(4), 734–745. doi: 10.1016/j.bcp.2008.09.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vondracek J, Umannova L, & Machala M (2011). Interactions of the aryl hydrocarbon receptor with inflammatory mediators: beyond CYP1A regulation. Curr Drug Metab, 12(2), 89–103. [DOI] [PubMed] [Google Scholar]
- Wahlang B, Barney J, Thompson B, Wang C, Hamad OM, Hoffman JB, … Hennig B (2017). PCB126 exposure increases risk for peripheral vascular diseases in a liver injury mouse model. Toxicol Sci doi: 10.1093/toxsci/kfx180 [DOI] [PMC free article] [PubMed]
- Wang L, Zhai DS, Ruan BJ, Xu CM, Ye ZC, Lu HY, … Lu ZF (2017). Quaking Deficiency Amplifies Inflammation in Experimental Endotoxemia via the Aryl Hydrocarbon Receptor/Signal Transducer and Activator of Transcription 1-NF-kappaB Pathway. Front Immunol, 8, 1754. doi: 10.3389/fimmu.2017.01754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber C, & Noels H (2011). Atherosclerosis: current pathogenesis and therapeutic options. Nat Med, 17(11), 1410–1422. doi: 10.1038/nm.2538 [DOI] [PubMed] [Google Scholar]
- Weber C, Zernecke A, & Libby P (2008). The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat Rev Immunol, 8(10), 802–815. doi: 10.1038/nri2415 [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, & Gaynor RB (2001). Role of the NF-kappaB pathway in the pathogenesis of human disease states. Curr Mol Med, 1(3), 287–296. [DOI] [PubMed] [Google Scholar]
- Yi T, Wang J, Zhu K, Tang Y, Huang S, Shui X, … Lei W (2018). Aryl Hydrocarbon Receptor: A New Player of Pathogenesis and Therapy in Cardiovascular Diseases. Biomed Res Int, 2018, 6058784. doi: 10.1155/2018/6058784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller I, & Srivastava S (2014). Macrophage functions in atherosclerosis. Circ Res, 115(12), e83–85. doi: 10.1161/CIRCRESAHA.114.305641 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.