

Summary
During transcription, the nascent RNA strand can base pair with its template DNA, displacing the non-template strand as ssDNA and forming a structure called an R-loop. R-loops are common across many domains of life and cause DNA damage in certain contexts. In this review, we summarize recent results implicating R-loops as important regulators of cellular processes such as transcription termination, gene regulation and DNA repair. We also highlight recent work suggesting that R-loops can be problematic to cells as blocks to efficient transcription and replication that trigger the DNA damage response. Finally, we discuss how R-loops may contribute to cancer, neurodegeneration and inflammatory diseases, and compare the available next-generation sequencing-based approaches to map R-loops genome-wide.
Introduction
R-loops are nucleic acid structures that form when an RNA strand invades double-stranded DNA. This produces a Watson-Crick RNA-DNA hybrid and displaces the non-hybridized strand as single-stranded DNA (ssDNA). The term R-loop is analogous to a D-loop containing an RNA moiety (Thomas et al., 1976). While short RNA-DNA hybrids form transiently during transcription and lagging-strand DNA synthesis, R-loops are distinct from these structures and span 100–2000 base pairs (Santos-Pereira and Aguilera, 2015). Originally thought to be rare byproducts of transcription, R-loops are now known to form across the genomes of bacteria, yeast, and higher eukaryotes throughout the cell cycle (Lang et al., 2017; Stork et al., 2016; Tresini et al., 2015; Wellinger et al., 2006).
R-loops participate in a number of physiological processes, but they also present a source of DNA damage (Costantino and Koshland, 2015; Santos-Pereira and Aguilera, 2015; Skourti-Stathaki and Proudfoot, 2014). Cells must therefore carefully regulate R-loop formation. An ever-growing number of factors have been proposed to resolve or prevent R-loops, including RNA binding and processing factors, helicases, DNA replication and repair-associated factors, nucleases, signaling pathways, and introns (Bhatia et al., 2017; Bonnet et al., 2017; Santos-Pereira and Aguilera, 2015) (Figure 1). However, whether many of these factors act in a direct or indirect manner is still not clear, and the putative biochemical activity of several factors has also not been validated. Studies describing the R-loop interactome have identified additional proteins regulating R-loop metabolism (Cristini et al., 2018; Nadel et al., 2015; Wang et al., 2018), and may help to distinguish which factors act at the R-loop from those that act indirectly.
Figure 1.
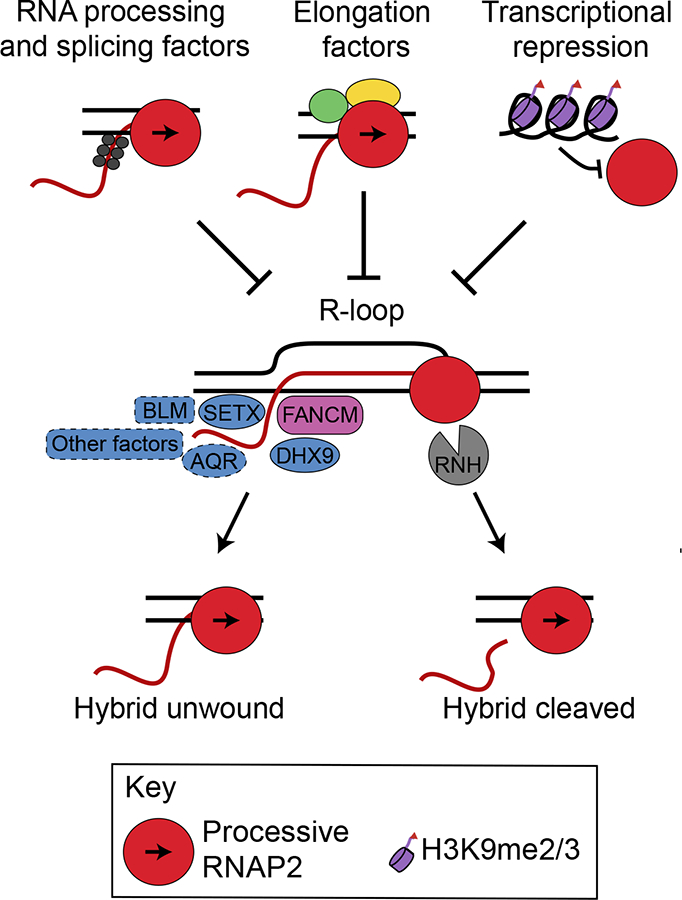
R-loop resolution and suppression mechanisms. R-loops form co-transcriptionally when nascent RNA (red) hybridizes with DNA (black), generating an RNA-DNA hybrid and displaced ssDNA. R-loops are suppressed by processing/splicing factors which coat nascent RNA, elongation factors that ensure processive transcription, or transcriptional repression at certain repeat sequences. Once formed, a variety of helicases (blue) and other factors such as the FANCM translocase, RNase H family nucleases (RNH), or the replisome (not shown) may remove the hybrid, restoring processive transcription. Not all factors implicated in this process have been illustrated for clarity, and many factors have not been examined in biochemical assays to support the proposed activity. Factors outlined in dotted lines show R-loop resolution in cellular assays, but have not been shown to biochemically resolve RNADNA hybrids in vitro.
In this review, we will focus on emerging themes in R-loop biology, an evolving field which has found diverse and highly context-sensitive roles for R-loops. We review the contexts in which R-loops have beneficial roles in transcriptional regulation and DNA repair, as well as potentially detrimental situations in which they block transcription and replication. We also describe how R-loops contribute to disease pathology through these roles and review emerging genomic techniques to map R-loops.
R-loops act at promoters to regulate gene expression
R-loops are transient, reversible structures that form in many regions of eukaryotic genomes, including regions transcribed by RNA polymerases I, II and III (El Hage et al., 2010; Tran et al., 2017). At RNA polymerase II (RNAPII) transcribed genes, genome-wide mapping studies indicate R-loops are abundant at promoters (Chen et al., 2017; Dumelie and Jaffrey, 2017; Ginno et al., 2012; Nadel et al., 2015; Sanz et al., 2016). While open chromatin at promoters may favor RNA-DNA hybrid formation, promoter R-loops actively regulate genes through several mechanisms.
At promoters, R-loops may facilitate transcription by protecting the underlying DNA from methylation (Ginno et al., 2012; Grunseich et al., 2018). DNA methyltransferases bind poorly to RNA-DNA hybrids, allowing R-loops to suppress methylation-associated silencing (Figure 2) (Grunseich et al., 2018). R-loops at promoters can also both promote and inhibit the binding of chromatin remodelers (Figure 2). In mouse embryonic stem cells, R-loops inhibit repressive chromatin modifying enzymes and recruit activating chromatin remodeling complexes to promote differentiation genes and facilitate a poised chromatin state (Chen et al., 2015). Additionally, R-loops can block transcription factor binding, though these results have not been generalized beyond a specific promoter locus (Boque-Sastre et al., 2015). R-loops arising from long non-coding RNAs that bind across promoters and coding regions in yeast can also displace repressors and promote transcription of nutrient utilization genes (Cloutier et al., 2016) (Figure 2). What distinguishes R-loops that favor, as opposed to inhibit protein binding is unknown, but in mouse embryonic stem cells CpG islands in the R-loop sequence favor protein recruitment, suggesting the process is sequence-dependent (Chen et al., 2015). Thus, R-loops can regulate gene expression through multiple context-dependent mechanisms. Importantly, although R-loops are thought to form transiently, they may act as an epigenetic mark, read by chromatin remodelers and other proteins to effect longer-lived changes in chromatin state. This characterization is consistent with their dynamic and prevalent nature across the genome (Chedin, 2016).
Figure 2.
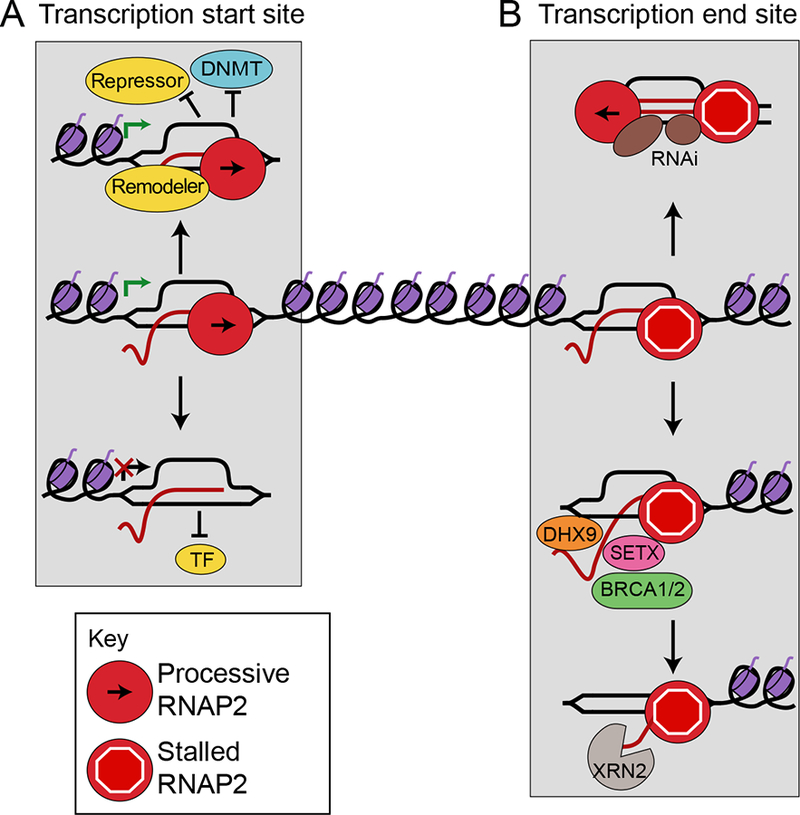
R-loops participate in gene regulation. A) At promoters, R-loops activate transcription by preventing binding of transcriptional repressors or DNA methylating enzymes (DNMT), or by acting as binding sites for transcription factors (top left). Alternatively, R-loops repress transcription by blocking transcription factor binding (bottom left). B) At terminators, R-loops facilitate efficient transcription termination by promoting RNAP II pausing and cleavage of the transcript from its template either by recruiting R-loop resolution helicases and RNases (bottom right), or by recruiting the RNAi silencing machinery (top right).
R-loops aid in transcriptional termination
Some studies suggest R-loops are also enriched at the 3’-end of some mammalian genes where they are proposed to mediate efficient transcription termination (Skourti-Stathaki et al., 2014; Skourti-Stathaki et al., 2011; Zhao et al., 2016). These R-loops stall RNAPII downstream of the poly-adenylation sequence. The RNA-DNA helicases senataxin (SETX) and DHX9 subsequently resolve these R-loops, releasing nascent RNA for degradation by exonucleases such as XRN2, leading to termination (Figure 2). Consistent with this idea, loss of SETX, DHX9 or XRN2 results in R-loop accumulation and defective termination (Cristini et al., 2018; Morales et al., 2016; Skourti-Stathaki et al., 2011). These studies do not specifically show that the enzymatic activities of these factors are necessary for R-loop resolution, which will be important in clarifying these mechanisms. Additionally, they focus on a few highly transcribed loci. Thus, genomic experiments are needed to clarify whether other classes of genes may require R-loops for termination. For example, convergent genes were found in one study to be associated with high levels of R-loops at terminators, suggesting that R-loops may be particularly important to prevent transcriptional read-through into adjacent genes (Sanz et al., 2016).
R-loops at some gene terminators may also trigger antisense transcription, generating dsRNA that recruits the RNA interference machinery and establishes repressive heterochromatin through the H3K9me2 mark to reinforce RNAPII pausing (Figure 2) (Skourti-Stathaki et al., 2014). However, a genome-wide study of R-loop mapping with histone modifications did not find correlations between other repressive chromatin marks and R-loop formation, instead finding a correlation with H3K4 methylation, a mark of active transcription (Sanz et al., 2016). The reasons for these apparently contradictory associations between R-loops and histone modifications are currently unclear. These results must also be carefully interpreted, since an R-loop disfavors nucleosome formation (Dunn and Griffith, 1980), meaning simultaneous occupancy of an R-loop and histone modification on a single allele is unlikely.
While R-loops play important roles in gene regulation and transcription termination, they are also problematic in certain contexts and lead to DNA damage when their turnover is deregulated (Sollier and Cimprich, 2015). This damage is strongly associated with blocks to replication fork progression and may also occur when R-loops interfere with productive transcription.
R-loops act as replication blocks
Transcription and replication share a common DNA template, but when replication forks encounter transcription machinery, the resulting transcription-replication collisions (TRC) can cause potentially lethal DNA damage. As the most prevalent source of R-loop-mediated damage seems to arise during S phase (Gan et al., 2011; Wellinger et al., 2006), R-loops are thought to exacerbate TRCs. Indeed, RNase H overexpression can reduce DNA damage (Kotsantis et al., 2016; Stork et al., 2016) and replication fork slowing (Kotsantis et al., 2016) under hormone or oncogene-induced replication stress, indicating that R-loops interfere with replication. Additionally, Fanconi Anemia pathway factors such as FANCA and FANCD2, which canonically act at replication forks to resolve intra-strand crosslinks, suppress R-loops to prevent damage arising from TRCs, and FANCM may directly resolve R-loops using its translocase activity (Figure 1) (Garcia-Rubio et al., 2015; Schwab et al., 2015).
Recently, the induction of TRCs in controlled systems has allowed for precise mechanistic studies of their causes and consequences. In studies on an engineered bacterial genome (Lang et al., 2017) and a mammalian episomal system (Hamperl et al., 2017), R-loops blocked replication and promoted severe genome instability when replication forks encountered them in the head-on (HO) orientation, but had more tolerable effects when replication and transcription were co-directional (CD). While genome instability from HO TRCs is likely a major challenge, bacteria may also exploit this instability to tune the rate of mutation at stress-responsive genes (Lang et al., 2017). Furthermore, in yeast R-loops are stabilized at short telomeres in S phase, such that ensuing TRCs promote recombination to maintain telomere length and prevent senescence (Graf et al., 2017).
R-loop levels are surprisingly affected by the orientation of TRCs. HO TRCs promote R-loop formation and CD TRCs reduce R-loops (Figure 3), suggesting important molecular differences in how replication forks encounter R-loops in the HO and CD orientations. In eukaryotic cells, the replicative helicase itself may unwind hybrids in the CD orientation, as it associates with the leading strand (Hamperl et al., 2017) and has biochemical RNA-DNA helicase activity (Shin and Kelman, 2006). However, in bacteria the replicative helicase travels on the lagging strand, raising questions about whether the molecular basis for this effect is evolutionarily conserved. R-loop resolution in both cases could instead be performed by a fork-associated factor that is not the replicative helicase.
Figure 3.
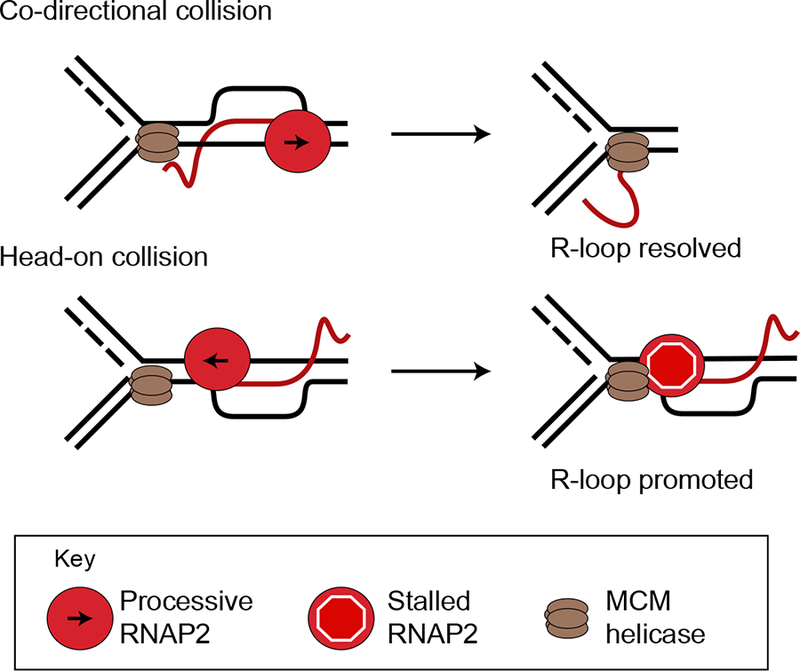
R-loops at transcription-replication collisions. Collisions of a replication fork with an R-loop can resolve the R-loop when replication forks are co-directional with transcription, or stabilize them when replication forks collide with the R-loop head-on. The MCM replicative helicase complex may directly unwind RNA-DNA hybrids in the co-directional orientation.
R-loops as effectors of transcriptional stress
R-loops may also promote toxicity by stalling transcription. Excessive pausing, arrest or backtracking of RNAP is a toxic condition known as transcription stress (Saponaro et al., 2014). R-loops block transcription in vitro (Belotserkovskii et al., 2017), and studies of transcription from R-loop forming loci show that the R-loop interferes with productive transcription (Bonnet et al., 2017; Hamperl et al., 2017; Lang et al., 2017). On the other hand, as already discussed, the transcription of some genes is facilitated by R-loops at their promoters. It is unclear what distinguishes R-loops promoting transcription from those blocking transcription. One possibility is that the regulatory R-loops are relatively rapidly turned over in cells, and do not persist long enough to block RNAP progression. As they are thought to recruit downstream factors to establish chromatin states, they may effect lasting changes while being short-lived themselves. It is also unclear whether problems with transcription derive from a RNAP stalled with the R-loop, or whether the R-loop acts as a transcriptional block to upstream polymerases (Figure 4A). In vitro experiments suggest that the second situation is at least possible (Belotserkovskii et al., 2017). Interestingly, the converse situation also appears to be possible in that persistent pausing of RNAP seems to promote R-loop formation from the nascent RNA (Shivji et al., 2018; Zhang et al., 2017).
Figure 4.
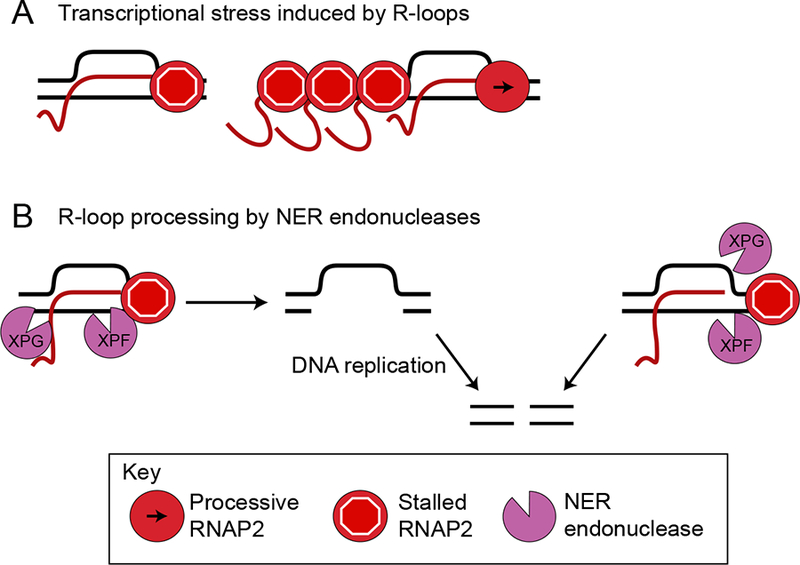
Transcription stress and R-loops. A) Transcription stress may arise from a stalled RNA polymerase associated with an R-loop (left) or upstream polymerases that are stalled from collisions with the R-loop (right). B) Two ways that NER processing may convert an R-loop to a DSB: either NER enzymes XPG and XPF cut the hybridized DNA, leading to a single-strand gap that is processed into a DSB by replication (top), or NER enzymes cut non-canonically to directly create a DSB at the 3’ or 5’ end of the R-loop (bottom).
R-loop resolution may involve factors that normally resolve other forms of transcription stress. Transcription-blocking, bulky DNA lesions are removed by transcription-coupled nucleotide excision repair (TC-NER). Although R-loops are much larger than typical transcription-blocking lesions, TC-NER factors may also act non-canonically to process R-loops, leading to DNA breaks (Shivji et al., 2018; Sollier et al., 2014; Yasuhara et al., 2018). What precisely occurs is not clear. The TC-NER nucleases XPG and XPF could excise R-loops that block transcription, leaving a ssDNA gap that could progress to a double-strand break (DSB) with additional strand breaks or DNA replication (Figure 4B) (Sollier et al., 2014). Possibly in a distinct manner, XPG and XPF were recently found to process R-loops forming over trinucleotide repeat sequences in yeast (Su and Freudenreich, 2017). While R-loop processing may cause DNA damage, it may prove an effective way to resolve R-loops throughout the cell cycle and in non-dividing cells, and relieve R-loop-mediated transcription stress.
Interestingly, the breast cancer susceptibility factors BRCA1 and BRCA2 help to resolve R-loops by mechanisms that are still poorly understood and may be cell-type specific (Bhatia et al., 2014; Hatchi et al., 2015; Shivji et al., 2018). It is clear that BRCA1 promotes the removal of R-loops by recruiting the RNA-DNA helicase SETX to termination sites, thereby preventing DNA damage and mutations (Hatchi et al., 2015; Shivji et al., 2018). Furthermore, while BRCA2 depletion increases R-loops at the 5’ end of genes, BRCA1 is reported to prevent R-loop accumulation at both the 3’ and 5’ ends of genes. Overall, as both BRCA1 and BRCA2 associate with RNAPII, they are key factors in suppressing R-loop-mediated transcriptional stress both by promoting elongation and by resolving R-loops that form after RNAP pausing (Figure 1) (Shivji et al., 2018; Zhang et al., 2017). Transcriptional stress from R-loops could also cause DNA damage by sequestering BRCA1, which contributes to both R-loop and DNA damage responses, with the stalled RNAPII (Gorthi et al., 2018). Moreover, related work demonstrates that overexpression of non-coding pericentromeric satellite RNAs sequesters BRCA1 in cells, making them vulnerable to R-loop-mediated damage (Zhu et al., 2018). R-loops associated with stalled transcription could thus represent a significant, if indirect, cause of DNA damage by preventing repair.
R-loops in canonical and non-canonical DNA damage responses
As central regulators of the DNA damage response (DDR), the ATR and ATM protein kinases orchestrate responses to replication fork slowdown or stalling (replication stress), and to DSBs respectively (Blackford and Jackson, 2017; Saldivar et al., 2017). R-loops activate the canonical DDR by stalling replication forks, triggering ATR activation, and they can also activate ATM by promoting DSB formation (Sollier and Cimprich, 2015).
However, not all R-loops activate both ATM and ATR. For example, R-loops induced by mutations in splicing factors cause replication stress and appear to solely activate ATR (Chen et al., 2018). Moreover, recent studies have shown that HO TRCs specifically activate ATR, while CD TRCs specifically activate ATM (Figure 5A,B) (Hamperl et al., 2017). Exactly how R-loops activate ATR vs ATM in different contexts is not known. ATM activation likely occurs when R-loops are converted to DSBs, either by replication through a gap or nick in the displaced ssDNA, or by nucleases acting on the R-loop or the adjacent fork. In bacteria, RNAP backtracking causes R-loop-mediated DSBs upon CD collisions (Dutta et al., 2011), and it is possible a similar mechanism occurs in eukaryotes (Figure 5A). In the case of ATR, a stalled fork at an R-loop may activate ATR in the classical manner by recruiting replication protein A (RPA) to ssDNA at the replication fork (Figure 5B). However, alternative ATR activation pathways may also be possible in this scenario.
Figure 5.
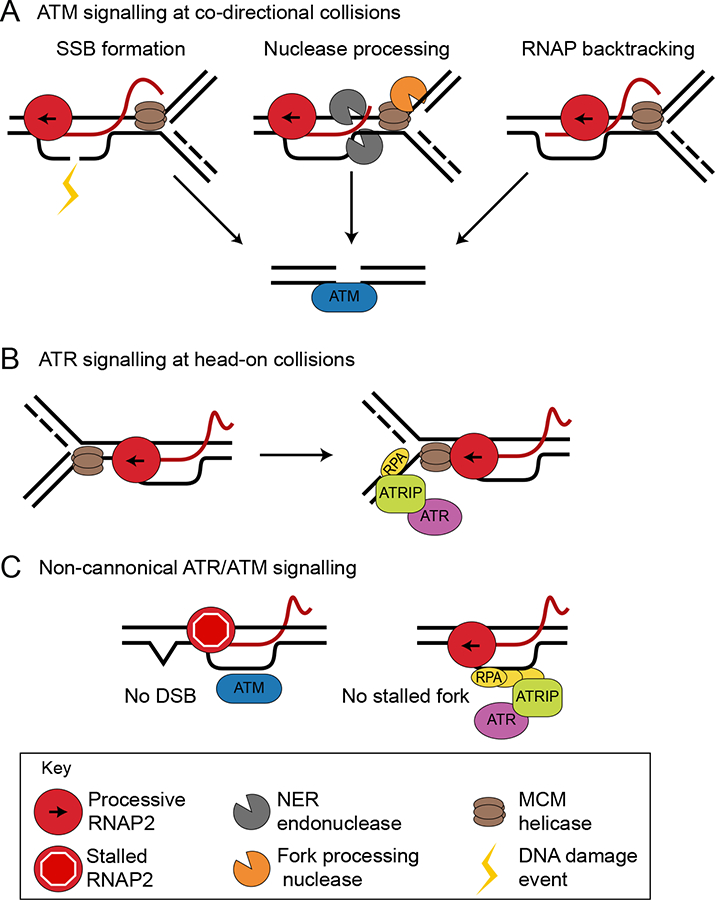
R-loops drive DNA damage responses. A) R-loops may cause DSBs and canonical ATM activation through three mechanisms: damage to the displaced ssDNA that is converted to a DSB by DNA replication, nucleolytic processing of the R-loop or fork stalled by R-loops, or collision of the replication fork with a backtracked RNA polymerase. B) R-loops may activate ATR in head-on collisions by stalling replication forks, which then accumulate RPA and signal to ATR through the canonical pathway. C) Non-canonical DNA damage response pathway activation could occur when polymerases stalled at R-loops activate ATM, or when RPA accumulates on the displaced ssDNA and activates ATR.
R-loops can also activate non-canonical ATM and ATR signaling in the absence of DSBs and outside of S phase. Transcription-blocking lesions in non-cycling cells induce R-loops, which activate non-canonical ATM signaling without DSB formation (Figure 5C) (Tresini et al., 2015). The ssDNA binding protein RPA, which recruits the ATR-ATRIP complex to ssDNA at replication forks, colocalizes with R-loops throughout the cell cycle (Nguyen et al., 2017). This could in principle directly promote ATR activity independent of DNA replication (Figure 5C). Indeed, ATR is activated by R-loops independent of DNA damage at centromeres during mitosis to promote chromosome segregation, and RPA is found at these sites as well (Kabeche et al., 2018). Whether R-loops activate ATR non-canonically in other contexts, and how ATR activation is restricted given that R-loops are present throughout the cell cycle, remain open questions.
Interestingly, the DDR also regulates R-loop resolution pathways. ATR and ATM activation promote the recruitment of the RNA-DNA helicase SETX to TRCs (Yuce and West, 2013). Moreover, ATR activation following DNA damage causes translocation of the RNA-DNA helicase DDX19 from the nuclear pore into the nucleus, where it unwinds RNA-DNA hybrids to relieve putative TRCs (Hodroj et al., 2017).
The fact that R-loops appear to be a potent source of DNA damage seems paradoxical in light of their prevalence and beneficial roles (Costantino and Koshland, 2015). One possibility is that most damage arises from a subset of relatively rare and “bad” R-loops that form in some contexts. In yeast, for example, HO TRCs are a strong, but not exclusive, determinant for which R-loops cause damage (Costantino and Koshland, 2018). Furthermore, R-loop-mediated genome instability in yeast occurs only if accompanied by histone H3 serine-10 phosphorylation, possibly owing to chromatin compaction near these R-loops (Garcia-Pichardo et al., 2017). In human cells, HO TRCs with an R-loop have been shown to be particularly detrimental to genome stability in controlled systems, and cellular perturbations that disrupt the replication program could cause damage by promoting HO collisions with R-loops (Hamperl et al., 2017). An alternative model posits that cells tolerate beneficial R-loops up to a critical threshold, at which point the pathways that resolve them and promote their turnover become saturated. This could cause “good” R-loops to abnormally persist and affect replication or transcription, causing damage more globally through multiple mechanisms. Multiple studies demonstrate RNase H-sensitive DNA damage under perturbations in which new R-loops are formed and globally increase across transcribed genes (Gorthi et al., 2018; Stork et al., 2016) and microscopy studies also suggest global R-loop increases in many situations (Bhatia et al., 2014; Sollier et al., 2014; Zhang et al., 2017). However, care must be taken in interpreting results from both approaches, as described below and in other reviews (Vanoosthuyse, 2018). In general, it is difficult to assess global changes in genome occupancy by sequencing without an internal standard and controls are crucial to account for potential off-target antibody staining.
Precisely which loci are responsible for the DNA damage resulting from R-loop perturbations is not clear, making it difficult at present to distinguish between so-called “good” and “bad” R-loops in sequencing studies. Furthermore, not all studies that have mapped R-loops under DNA-damage-inducing perturbations find a general increase in R-loops on the genome (Manzo et al., 2018). Emerging techniques to map DNA breaks, as well as DNA damage markers in systems where R-loops are induced should clarify how the replicative, transcriptional and chromatin landscape at R-loops can promote or repress damage and also what distinguishes potentially good and bad R-loops. In parallel, biochemical approaches will be critical to understand which specific R-loop-derived structures are able to differentially activate ATM and ATR.
R-loops and DNA double-strand break repair
While R-loops are a potential cause of DNA damage, recent studies show that RNA-DNA hybrids can also form after DNA damage, both inhibiting and promoting DNA repair. These hybrids could form by de novo transcription (Ohle et al., 2016; Yasuhara et al., 2018) or from hybridization of existing nascent RNA (Figure 6A) (Aguilera and Gomez-Gonzalez, 2017; Roy et al., 2010). Persistent hybrids may compromise DNA repair in multiple ways: impeding binding of DNA repair factors to DSBs (Aguilera and Gomez-Gonzalez, 2017), affecting the chromatin structure flanking DSBs (Cohen et al., 2018), and causing aberrant repair (Amon and Koshland, 2016; Cohen et al., 2018). Conversely, R-loops may also promote DSB repair (Ohle et al., 2016; Yasuhara et al., 2018) as excessive removal of R-loops reduces the efficiency of homologous recombination (HR) and non-homologous end joining (NHEJ) (Lu et al., 2018), the two main pathways of DSB repair. In summary, R-loop formation seems to be critical for efficient HR but not NHEJ in budding and fission yeast, while in human cells R-loops may facilitate both types of repair. The apparently contradictory positive and negative roles of RNA-DNA hybrids may depend on the type of repair, or reflect roles for RNA-DNA hybrids in multiple steps of the repair process.
Figure 6.
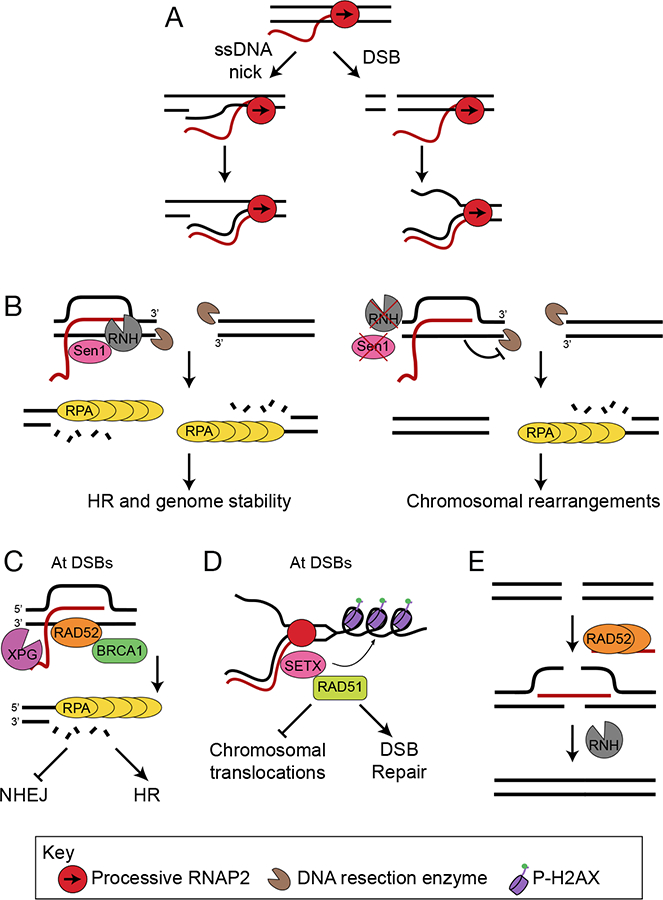
R-loops participate in DNA repair at breaks. A) DNA breaks, either ssDNA nicks (left) or DSBs (right), create free 3’-DNA ends, promoting the annealing of RNA to DNA to form hybrids. B) In yeast, R-loops form at DSBs and initiate repair by HR. Subsequent R-loop resolution by Sen1 or RNase H regulates the extent of DNA end resection and subsequent binding of RPA (left). In the absence of Sen1 or RNase H (right), RNA-DNA hybrids persist, blocking DNA resection and RPA binding and leading to chromosomal rearrangements. C) In human cells RNA-DNA hybrids formed at a subset of DSBs recruit Rad52, which further recruits XPG and BRCA1. XPG-mediated R-loop processing initiates DNA resection and repair by HR, and suppresses aberrant NHEJ. D) SETX is recruited to DSBs and resolves RNA-DNA hybrids at active genes. SETX regulates γH2AX spreading and Rad51 foci formation promoting DSB repair and suppressing chromosomal translocations. E) Rad52 promotes the formation of RNA- DNA hybrids that facilitate RNA-templated HR by bridging two DNA ends. The DNA ends are ligated, and the RNA strand is removed by RNase H.
One major influence R-loops have on DSB repair is to alter the efficiency of resection, a step that determines whether repair proceeds by HR or NHEJ (Figure 6B). While many studies link hybrids to resection efficiency, they have been reported to play conflicting roles. In fission yeast, R-loop formation prevents excessive resection at DSBs, but R-loop removal is also required for efficient RPA binding early after DSB formation (Ohle et al., 2016). In budding yeast, persistent R-loops also block DSB resection, with no detectable ssDNA formation on the break side adjacent to an R-loop (Costantino and Koshland, 2018). Additionally, budding yeast SAE2 and its ortholog in mammals CtIP, which mediate resection, have been shown to promote R-loop resolution, further complicating this relationship (Makharashvili et al., 2018). In a possibly alternative pathway in human cells, hybrids appear to enhance resection (Lu et al., 2018). R-loops also promote a specialized form of HR: transcription-associated homologous recombination repair (TA-HRR). During TA-HRR, RNA-DNA hybrids forming at a transcriptionally active subset of DSBs recruit Rad52. Subsequent recruitment of XPG and XPG-mediated R-loop processing initiates resection and HR (Figure 6C) (Yasuhara et al., 2018). In a possibly related mechanism deemed TC-HR, R-loops recruit the TC-NER factor CSB, which then recruits Rad52 to facilitate HR at sites of ROS-induced damage (Teng et al., 2018). R-loops can also affect other steps of DSB repair downstream of resection, such as Rad51 foci formation (Figure 6D) (Cohen et al., 2018). Therefore, R-loop processing, genomic location and transcriptional status are all potential determinants of how R-loops affect DSB repair.
RNA-DNA hybrids may also contribute to an alternative, RNA-templated form of HR during which a homologous RNA molecule is used instead of DNA as the template for DSB repair (Keskin et al., 2014; Mazina et al., 2017). In vitro, the recombination protein Rad52 promotes RNA-DNA hybrid formation by bridging DNA ends and facilitating their ligation (Figure 6E) (McDevitt et al., 2018). The prevalence of RNA-DNA hybrids acting via this mechanism in cells remains to be established.
R-loops and human disease
Molecular insights have revealed direct connections between R-loops and human disease (Table 1), opening possibilities for potential therapeutic modalities. We focus here on new developments and emerging themes in R-loops and disease and point the reader to other extensive reviews on this topic for a deeper understanding (Groh and Gromak, 2014; Richard and Manley, 2017).
Table 1.
R-loops and links to human disease.
| Disease | R-loop factor/locus | Proposed mechanism | References |
|---|---|---|---|
| Breast/Ovarian | Estrogen | Estrogen-induced R-loops cause DNA damage and genome instability. | Stork et al., 2016 |
| BRCA1 | BRCA1 interacts with SETX and suppresses R-loops and DNA breaks at gene terminators. | Hatchi et al., 2015 | |
| RNAP II pausing contributes to BRCA1-associated R-loop accumulation and breast cancer development. | Zhang et al., 2017 | ||
| BRCA1 is sequestered in cells expressing heterochromatin-associated noncoding RNAs, leading to genome instability. | Zhu et al., 2018 | ||
| BRCA2 | BRCA2 depletion elevates R-loop levels and causes genome instability. | Bhatia et al., 2014 | |
| Aldehydes deplete BRCA2 and cause R-loop-dependent genome instability. | Tan et al., 2017 | ||
| BRCA2 depletion causes transcription stress at gene promoters and R-loop-mediated DNA damage. | Shivji et al., 2018 | ||
| Ewing Sarcoma | EWS-FLI, BRCA1 | R-loops cause transcriptional stress, resulting in functional depletion of BRCA1 and subsequent DNA damage. | Gorthi et al., 2018 |
| Myelodysplastic syndromes (MDS) | SRSF2, U2AF1 | R-loops induced by splicing factor mutations cause replication stress and impair bone cell function. | Chen et al., 2018 |
| Multiple myeloma and Burkhitt’s lymphoma | TRD3-TOP3B | TRD3-TOP3B complex relieves negative supercoiling and reduces R-loop levels at c-MYC and Igh to suppress chromosomal translocations. | Yang et al., 2014 |
| Alternative lengthening of telomeres (ALT)-dependent cancers | Telomeric repeat-containing RNA (TERRA) | TERRA R-loops are upregulated in cancer cells and promote homologous recombination to preserve telomeres by the ALT pathway. | Arora et al., 2014 |
| Fanconi Anemia (FA) | FANCM, FANCD2 | FA factor deficiency leads to increased R-loop levels, exacerbating TRCs and causing genome instability. | Schwab et al., 2015, Garcia-Rubio et al., 2015 |
| AOA2 | SETX | SETX resolves R-loops in neuronal cells; R-loops are elevated in neural progenitor cells from AOA2 patients with SETX mutations. | Becherel et al., 2015 |
| ALS4 | Gain-of-function SETX mutation in ALS4 decreases R-loops levels, increases DNA methylation and upregulates inflammation genes. | Grunseich et al., 2018 | |
| Infertility | SETX | SETX is required for meiosis in mice; SETX−/− mice are infertile and accrue R-loops in germ cells. | Becherel et al., 2013 |
| Prader-Willi Syndrome | SNORD116 | Loss of R-loop formation at the SNORD116 locus in neurons leads to changes in expression of imprinted genes. | Powell et al., 2013 |
| Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) | C9ORF72 (C9) | R-loops form at repeat expansions, causing transcriptional interference and abortive transcripts, which sequester proteins and cause cellular stress. Alternatively, R-loop processing causes repeat instability. | Haeusler et al., 2014, Reddy et al., 2014 |
| Friedreich ataxia (FRDA) and Fragile X syndrome (FXS) | FXN, FMR1 | R-loops forming at repeat expansions impede RNAPII, causing gene silencing and promoting heterochromatin formation. | Groh et al., 2014 Colak et al., 2014, Loomis et al., 2014 |
| Aicardi Goutieres Syndrome (AGS) | RNaseH2, SAMHD1, TREX1 | Elevated R-loops are associated with decreased DNA methylation in AGS cells. | Lim et al., 2015 |
| Immunodeficiency, centromere instability and facial anomalies syndrome (ICF) | TERRA | Dysregulation of TERRA-R-loops causes telomere attrition. | Sagie et al., 2017 |
| AIDS-associated malignancies | TREX complex | Kaposi’s sarcoma-associated herpesvirus sequesters the transcription and export factor TREX, elevating R-loops and causing genome instability. | Jackson et al., 2014 |
Cancer
While cancers are a highly complex and diverse group of diseases, many rely on overactive growth factor signaling and exhibit high levels of DNA damage and mutagenesis (Hanahan and Weinberg, 2011). As R-loops form during transcription, and can cause DNA damage in some contexts, they provide a potential link between these two hallmarks of cancer.
In breast cancer cells subjected to high levels of estrogen signaling, R-loops were found to accumulate and drive DNA damage at genes induced by estrogen (Stork et al., 2016). Oncogenic mutations in HRAS similarly cause R-loop accumulation and subsequent DNA damage and replication stress (Kotsantis et al., 2016). These studies support that a cell’s transcriptional landscape may shape where DNA damage occurs through R-loop formation in active genes, and could even imply that R-loop formation at specific genes may drive a mutator phenotype (Loeb et al., 1974). Indeed, in human breast cancers translocations and structural variants are associated with genes induced by estrogen signaling (Stork et al., 2016). With increased sequencing of patient tumors and emerging technologies to map DNA damage in cells, it will be informative to compare patterns of DNA damage and mutation to transcription and R-loop formation. The established roles of R-loops in altering chromatin state will also be of interest as epigenomic instability becomes better characterized in human cancers.
Perturbations in pathways driven by the breast cancer susceptibility factors BRCA1 and BRCA2 can also cause R-loop driven DNA damage. Cell treated with carcinogenic aldehydes degrade BRCA2, and in turn accumulate DNA damaging R-loops (Tan et al., 2017). Mutations in BRCA1 also cause R-loops to accumulate at stalled transcription complexes. These R-loops are directly implicated in mammary tumorigenesis in mice (Zhang et al., 2017). While these cancer-causing perturbations cause BRCA1/2 insufficiency, which induces R-loops, a recent report indicates that the converse situation also occurs in that R-loops may prevent proper functioning of the BRCA1/2 pathway. In Ewing’s Sarcoma patient cells, R-loops induced by the oncogenic EWSFLI fusion protein sequester the available BRCA1, rendering these cancers functionally haploinsufficient for BRCA1 and preventing DNA repair (Gorthi et al., 2018). Given that BRCA2 also associates with RNAPII (Shivji et al., 2018), conditions which increase R-loops may sequester BRCA2 to confer vulnerabilities associated with both BRCA1 and 2-deficiency in other cancers. These vulnerabilities may provide therapeutic opportunities in cancers driven by R-loops. For example, functional haploinsufficiency in BRCA1 tumors could sensitize them to PARP inhibitors which are less toxic than conventional chemotherapy (Gorthi et al., 2018).
More directly, R-loops formed during oncogenesis could exert selective pressure on cancer cells by causing DNA damage. In a study of pre-leukemic myelodysplastic syndromes, a number of seemingly disparate splicing factor mutations were all found to induce R-loop formation, suggesting a common mechanism of action. These R-loops activate ATR and interfere with cellular proliferation. The authors also suggest that the cells which are able to proliferate in spite of this stress may ultimately become cancerous (Chen et al., 2018). In mature cancers, there may be an opportunity to re-activate the response to R-loop-induced replication stress and sensitize tumors to this endogenous damage. Indeed, in Synovial Sarcoma cells, ATR was found to suppress R-loops and DNA damage through an unknown mechanism. Inhibiting ATR caused these cells to accumulate R-loops and increased their sensitivity to chemotherapy (Jones et al., 2017). As R-loops seem to be a nearly universal byproduct of transcription, targeting R-loop tolerance in cancers could be a potent and specific way to treat certain otherwise intractable tumors.
Recent studies have also shown that genome instability and DNA damage trigger innate immune and pro-inflammatory responses, particularly via activation of the cGAS-STING axis (Ng et al., 2018). Given that R-loops forming upon oncogenic stimuli are a potent source of DNA damage, their formation may directly contribute to activation of innate immune responses in cancer cells. On one hand, activation of cGAS-STING is crucial for anti-tumor immunity, and immunomodulatory therapeutics are currently being explored to augment this pathway. However, cGAS-STING-mediated inflammation may also promote growth and metastasis of some tumors (Ng et al., 2018). How R-loop formation may affect cGAS-STING, or other innate immune signaling pathways, is therefore an open and exciting question.
Neurological Diseases
R-loops are also associated with several neurological diseases. R-loops forming at expanded trinucleotide DNA repeats are associated with heterochromatin formation and transcriptional repression of genes linked to neurological disorders, including Friedreich’s ataxia and fragile X syndrome (Table 1) (Colak et al., 2014; Groh et al., 2014; Loomis et al., 2014). In these cases, R-loop formation is limited to the expanded trinucleotides and primarily affects the repeat-containing gene (Groh et al., 2014).
Mutations in the R-loop resolving helicase SETX are linked to multiple neurological disorders (Groh et al., 2017), including ataxia with oculomotor apraxia 2 (AOA2) and amyotrophic lateral sclerosis type 4 (ALS4). Intriguingly, ALS4 patient cells have decreased R-loop levels due to a gain of helicase function in SETX. This results in increased promoter methylation and gene silencing, causing abnormal TGF-β signaling in ALS4 cells, and ultimately neuron dysfunction and death (Grunseich et al., 2018). Thus, while elevated R-loops are more commonly linked to disease, decreased R-loop levels can also be pathological. By contrast, AOA2 cells have increased R-loop levels and altered neuronal gene expression (Becherel et al., 2015), but how this relates to SETX has not been established.
Autoimmune Diseases
A role for R-loops in autoimmune diseases is also possible. Aicardi Goutieres Syndrome (AGS) is a rare inflammatory disorder that affects the brain, skin and immune system. It is attributed to mutations in nucleic acid-degrading enzymes, including TREX1, SAMHD1 and RNase H2, which cause endogenous nucleic acids to accumulate in the cytoplasm of cells and ultimately activate the cGAS-STING pathway and interferon responses (Crow and Manel, 2015). cGAS-STING activation in AGS cells has been directly linked to cytoplasmic DNA in micronuclei and DNA fragments released from stalled replication forks (Coquel et al., 2018; Mackenzie et al., 2017; Ng et al., 2018). However, a role for other cytoplasmic nucleic acids has not been ruled out. Indeed, RNA-DNA hybrids are elevated in the genome of AGS cells, and new sites of RNADNA hybrid formation overlap with sites of decreased DNA methylation. It has therefore been proposed that elevated R-loops may contribute to AGS pathology by altering gene expression or reactivating retroelements (Lim et al., 2015). While the presence of R-loops and DNA methylation-driven phenotypes in both AGS and ALS4 is suggestive of a common role for R-loops in both disease states, no specific link has been established. Beyond these phenotypes, hybrids may stall forks and release ssDNA through fork processing which directly activates cGAS-STING.
In summary, while R-loops have long been suspected to contribute to human disease, recent developments have directly implicated R-loops in disease pathology. In particular, these pathologies relate to the established roles of R-loops in inhibiting DNA methylation, and in their roles in promoting DNA damage. It will be interesting in the future to see if other R-loop related phenotypes, such as in establishing chromatin state, also contribute to disease pathology.
Emerging technologies to map genomic R-loops
Several techniques using next-generation sequencing now exist to map the positions of R-loops and answer questions about how their distribution changes between cell types or growth conditions. Broadly, these techniques detect R-loops by footprinting or direct pulldown, and each has advantages and disadvantages (Table 2). In footprinting, bisulfite treatment converts cytosine in the displaced ssDNA of the R-loop to uracil, which is read as thymine during sequencing. As with many R-loop driven phenotypes, exogenous RNase H treatment is used to ensure this signal depends on an RNA-DNA hybrid (Yu et al., 2003). Pulldown assays directly recognize the RNA-DNA hybrid, using either the hybrid-binding activity of RNase H or an antibody (S9.6) raised against RNA-DNA hybrids (Boguslawski et al., 1986). These tools do not recognize the ssDNA component of the hybrid, although it is possible that RNase H could be recognizing RPA-coated ssDNA in addition to the RNA-DNA hybrid (Nguyen et al., 2017). Here, we briefly review these genomic techniques but also refer the reader to another more detailed review on this topic for additional information (Vanoosthuyse, 2018).
Table 2:
Next-generation sequencing methods to map R-loops
| Method name | Fragmentation method | Detection method | Molecule sequenced | Advantages | Disadvantages | Primary reference |
|---|---|---|---|---|---|---|
| DRIP-seq | Restriction digest | S9.6 | dsDNA | Robust signal, widely adopted, easy to set up | Low resolution, no strand-specificity, not in situ | Ginno et al., 2012 |
| DRIVE-seq | Restriction digest | Catalytically inactive RNase H | dsDNA | Provides independent verification of some DRIP-seq results | Low enrichment, low resolution, no strand-specificity, reagent not commercially available, not in situ | Ginno et al., 2012 |
| S9.6-ChIP-seq | Sonication after cross-linking | S9.6 | dsDNA | May overcome bias and resolution issues in DRIP-seq | Not strand specific, cross-linking could effect results | El Hage et al., 2014 |
| S1-DRIP-seq | Sonication | S9.6 | dsDNA | Higher resolution than DRIP-seq | Not strand-specific, not in situ | Wahba et al., 2016 |
| DRIPc-seq | Restriction digest | S9.6 | RNA | Strand-specific, high resolution | Not in situ, requires lengthier sample preparation, S9.6 may recognize dsRNA | Sanz et al., 2016 |
| RDIP-seq | Sonication | S9.6 | RNA | Strand-specific, high resolution | Not in situ, lengthier preparation, S9.6 recognizes dsRNA | Nadel et al., 2015 |
| ssDRIP-seq | Sonication | S9.6 | ssDNA | Strand-specific, easy compared to other strand-specific techniques | Not in situ, low resolution | Xu et al., 2017 |
| Bis-DRIP-seq | Restriction digest | S9.6 | dsDNA with bisulfite conversions | Strand-specific, provides additional control to ensure S9.6 signal arises from an R-loop in situ | Requires many replicates | Dumelie and Jaffrey, 2017 |
| R-ChIP-seq | Sonication | Catalytically inactive RNase H | ssDNA | Strand specific, in situ capture | Cell line must be engineered to express catalytically inactive RNase H construct, inactive RNAseH may alter hybrid dynamics | Chen et al., 2017 |
DRIP-seq and its variants
The most widely adopted method for R-loop mapping is DRIP-seq (Ginno et al., 2012), which uses next generation sequencing to map R-loops isolated by S9.6 immunoprecipitation (El Hage et al., 2010). In DRIP-seq, nucleic acids are extracted from unfixed cells and gently fragmented using restriction enzymes. After immunoprecipitation, sequencing libraries are created using a standard dsDNA approach. Alternatively, the material can be used for targeted, higher precision quantification by qPCR.
DRIP-seq has proved to be a consistent, reproducible, and popular method for sequencing R-loops. However, there are some limitations. Fragmenting the genome with restriction enzymes introduces bias and limits resolution, particularly at the 5’-end of genes (Halasz et al., 2017). This can be addressed by sonicating DNA rather than using restriction enzymes (El Hage et al., 2014; Halasz et al., 2017). Sonication is also used in S1-DRIP-seq, where the displaced ssDNA is removed with S1 nuclease prior to sonication to stabilize the RNA-DNA hybrid (Wahba et al., 2016). DRIP-seq is also limited by its strand-insensitivity. This can be addressed by strand-specific DNA library preparation as in ssDRIP-seq (Xu et al., 2017). Alternatively, DRIP-RNA-seq (Chen et al., 2015), RDIP-seq (Nadel et al., 2015) and DRIPc-seq (Sanz et al., 2016) address both strand-specificity and resolution by sequencing the RNA component of the hybrid instead of the DNA. Finally, bis-DRIP-seq (Dumelie and Jaffrey, 2017) combines in situ ssDNA bisulfite footprinting with S9.6 hybrid pulldown, theoretically improving the specificity by targeting both the hybrids and ssDNA. While none of these methods are currently widely adopted, they provide workarounds for some of DRIP-seq’s limitations.
Moving beyond S9.6
Any variant of DRIP-seq presumes that S9.6 has an unbiased and specific affinity for hybrids. However, S9.6 has some affinity for double-stranded RNA (dsRNA) and shows biases in hybrid sequence recognition (Konig et al., 2017). As different DRIP-seq variants have found R-loops associated with homopolymeric dA:dT tracks (Wahba et al., 2016) as well as regions of high GC-skew (Ginno et al., 2012), it is unclear whether antibody binding biases are confounding results. Furthermore, as most R-loops are much longer than the 6 bp epitope of S9.6 (Phillips et al., 2013), biases in S9.6 may not strongly affect binding of most R-loops. Binding to dsRNA is mostly relevant to methods that sequence hybrid RNA like DRIPc-seq, and can be mitigated using RNase III treatment before pulldown (Hartono et al., 2018).
Catalytically-inactive RNase H also binds RNA-DNA hybrids, providing an alternative approach to S9.6-based methods. DRIVE-seq (Ginno et al., 2012) is conceptually similar to DRIP-seq, using tagged, catalytically-inactive RNase H to pull down hybrids. As it is less sensitive than DRIP-seq, its adoption has been limited.
An alternative approach uses RNase H to capture R-loops in their native context, limiting the opportunity for hybrids to dissociate or shift in position before immunoprecipitation. In R-ChIP-seq (Chen et al., 2017), catalytically-inactive RNase H is stably expressed in cells, and immunoprecipitated from cross-linked chromatin. Expressing the RNase H construct endogenously improves sensitivity and allows for in situ capture of hybrids, although its stable expression could in principle alter the turnover of R-loops.
Broad discrepancies remain between methods
Interestingly, there are some differences in where hybrids map depending on the method used. bis-DRIP-seq and R-ChIP-seq both involve an in situ step and show R-loops to be highly concentrated at gene promoters and almost entirely absent from the 3’ end of genes. Variants of DRIP-seq show high signal at promoters, but also appreciable signal in gene bodies and termination regions. As much of the DRIPc-seq and DRIP-seq signal is sensitive to RNase H (Chen et al., 2015; Ginno et al., 2012; Stork et al., 2016), the differences in signal outside of promoters are probably not due to off-target binding.
These differences more likely reflect differences between capturing hybrids in situ and after cell lysis. RNase H could recognize a promoter-proximal subset of hybrids in a chromatin context, reflecting an underlying biological role at these sites or increased accessibility within open chromatin at promoters. However, this does not necessarily explain why bis-DRIP-seq shows similar patterns (Vanoosthuyse, 2018). Alternatively, non-promoter associated signal could be artefactual. For example, the small RNA-DNA hybrids formed at transcription bubbles could hypothetically expand, forming a full length R-loop. This seems highly unlikely, as melting a kilobase length stretch of dsDNA to allow strand invasion from the nascent RNA would be a considerable kinetic barrier outside of highly denaturing conditions (Thomas et al., 1976). Nonetheless, bis-DRIP-seq has been used to test whether R-loops rehybridize in solution by delaying bisulfite treatment until after nucleic acid extraction. Under these conditions, some R-loops appear to be unstable, but there was no signal consistent with massive rehybridization throughout the gene body (Dumelie and Jaffrey, 2017). Thus, these differences probably do not derive from artefactual R-loops formed after extraction.
It must also be noted that in situ capture methods may have relatively low enrichment compared to solution capture: for example, bis-DRIP-seq requires high numbers of replicates to achieve consistent signal (Dumelie and Jaffrey, 2017). Therefore, some optimization of these protocols may refine the picture of where R-loops occur under in situ conditions. The development of new methods independent of S9.6 or RNase H could also clarify outstanding questions remaining about the observed differences between in situ and solution-based capture methods.
Conclusions and Future Questions
Recent work has successfully linked R-loops to cellular processes at the molecular level, but many open questions remain. While R-loops participate in a diverse array of processes, their roles can be strikingly different between systems and even different contexts within the same system. For example, R-loops appear to be able to both recruit and inhibit the binding of chromatin remodeling factors to alter transcription, inhibit or promote repair at double strand breaks, and in a more general sense both facilitate cellular processes and cause DNA damage. While the different contexts determining these roles are known in some cases, a major challenge for the field moving forward will be to resolve some of these apparent conflicts. As R-loops have complex interactions with replication, transcription and chromatin state, genomic studies to associate them with other factors may be limited by the number of potential confounding variables. Many of the fundamental conflicts in R-loop biology may be more cleanly resolved through the use of engineered systems to more precisely separate these variables.
Recent work demonstrating the formation of hybrids at DSBs and a role for them in repair requires further investigation as well. DSB-associated hybrids should now also be recognized as a factor complicating the analysis of hybrid formation in scenarios where DSBs and R-loops are both induced, as DNA breaks may lead to hybrid formation as well as the reverse. Additionally, it will be important to resolve which steps of repair are facilitated, as opposed to inhibited, by R-loops, as the current literature has some apparently contradictory effects of R-loops in modulating DNA repair.
The proliferation of R-loop mapping techniques also provides both opportunities and challenges to the field moving forward. As the distributions suggested by in situ capture methods are consistently different from those obtained by pulldown in solution, it will be important to understand what factors influence R-loop capture in different contexts. As data are acquired in more cell lines and conditions, comparisons between these techniques will become easier.
Finally, recent links between R-loops and disease raise the exciting possibility that manipulating R-loop levels could be exploited therapeutically. Studies have demonstrated that R-loops can be targeted using small molecules to suppress molecular phenotypes associated with disease (Colak et al., 2014; Powell et al., 2013). Given the complex roles of R-loops as both positive and negative regulators of various cellular functions, however, loci-specific and tunable strategies to manipulate R-loop levels will likely be needed.
R-loops are RNA-DNA hybrid structures prevalent in mammalian, yeast and bacterial genomes. Crossley et al review emerging themes in R-loop biology, including roles in regulating transcription and DNA damage repair, deleterious effects in blocking transcription and replication, and contributions to human diseases. Genomic R-loop mapping methods are also briefly compared.
Acknowledgments
We thank Hannah Long, Philippe Pasero and members of the Cimprich lab for critically reading this manuscript. This work was supported by a Fellow Award from the Leukemia and Lymphoma Society to MPC (5455–17), a Stanford Graduate Fellowship and NCI PHS grant (CA09302) to MB, an NIH grant to KAC (GM119334) and a V Foundation Grant (D2018–017) to KAC. KAC is an American Cancer Society Research Professor.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguilera A, and Gomez-Gonzalez B (2017). DNA-RNA hybrids: the risks of DNA breakage during transcription. Nat Struct Mol Biol 24, 439–443. [DOI] [PubMed] [Google Scholar]
- Amon JD, and Koshland D (2016). RNase H enables efficient repair of R-loop induced DNA damage. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora R, Lee Y, Wischnewski H, Brun CM, Schwarz T, and Azzalin CM (2014). RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat Commun 5, 5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becherel OJ, Sun J, Yeo AJ, Nayler S, Fogel BL, Gao F, Coppola G, Criscuolo C, De Michele G, Wolvetang E, et al. (2015). A new model to study neurodegeneration in ataxia oculomotor apraxia type 2. Hum Mol Genet 24, 5759–5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becherel OJ, Yeo AJ, Stellati A, Heng EY, Luff J, Suraweera AM, Woods R, Fleming J, Carrie D, McKinney K, et al. (2013). Senataxin plays an essential role with DNA damage response proteins in meiotic recombination and gene silencing. PLoS Genet 9, e1003435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belotserkovskii BP, Soo Shin JH, and Hanawalt PC (2017). Strong transcription blockage mediated by R-loop formation within a G-rich homopurine-homopyrimidine sequence localized in the vicinity of the promoter. Nucleic Acids Res 45, 6589–6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia V, Barroso SI, Garcia-Rubio ML, Tumini E, Herrera-Moyano E, and Aguilera A (2014). BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 511, 362–365. [DOI] [PubMed] [Google Scholar]
- Bhatia V, Herrera-Moyano E, Aguilera A, and Gomez-Gonzalez B (2017). The Role of Replication-Associated Repair Factors on R-Loops. Genes (Basel) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford AN, and Jackson SP (2017). ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol Cell 66, 801–817. [DOI] [PubMed] [Google Scholar]
- Boguslawski SJ, Smith DE, Michalak MA, Mickelson KE, Yehle CO, Patterson WL, and Carrico RJ (1986). Characterization of monoclonal antibody to DNA.RNA and its application to immunodetection of hybrids. J Immunol Methods 89, 123–130. [DOI] [PubMed] [Google Scholar]
- Bonnet A, Grosso AR, Elkaoutari A, Coleno E, Presle A, Sridhara SC, Janbon G, Geli V, de Almeida SF, and Palancade B (2017). Introns Protect Eukaryotic Genomes from Transcription-Associated Genetic Instability. Mol Cell 67, 608–621 e6. [DOI] [PubMed] [Google Scholar]
- Boque-Sastre R, Soler M, Oliveira-Mateos C, Portela A, Moutinho C, Sayols S, Villanueva A, Esteller M, and Guil S (2015). Head-to-head antisense transcription and R-loop formation promotes transcriptional activation. Proc Natl Acad Sci U S A 112, 5785–5790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chedin F (2016). Nascent Connections: R-Loops and Chromatin Patterning. Trends Genet 32, 828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chen JY, Huang YJ, Gu Y, Qiu J, Qian H, Shao C, Zhang X, Hu J, Li H, et al. (2018). The Augmented R-Loop Is a Unifying Mechanism for Myelodysplastic Syndromes Induced by High-Risk Splicing Factor Mutations. Mol Cell 69, 412–425 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chen JY, Zhang X, Gu Y, Xiao R, Shao C, Tang P, Qian H, Luo D, Li H, et al. (2017). R-ChIP Using Inactive RNase H Reveals Dynamic Coupling of R-loops with Transcriptional Pausing at Gene Promoters. Mol Cell 68, 745–757 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PB, Chen HV, Acharya D, Rando OJ, and Fazzio TG (2015). R loops regulate promoter-proximal chromatin architecture and cellular differentiation. Nat Struct Mol Biol 22, 999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloutier SC, Wang S, Ma WK, Al Husini N, Dhoondia Z, Ansari A, Pascuzzi PE, and Tran EJ (2016). Regulated Formation of lncRNA-DNA Hybrids Enables Faster Transcriptional Induction and Environmental Adaptation. Mol Cell 61, 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Puget N, Lin YL, Clouaire T, Aguirrebengoa M, Rocher V, Pasero P, Canitrot Y, and Legube G (2018). Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat Commun 9, 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak D, Zaninovic N, Cohen MS, Rosenwaks Z, Yang WY, Gerhardt J, Disney MD, and Jaffrey SR (2014). Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 343, 1002–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coquel F, Silva MJ, Techer H, Zadorozhny K, Sharma S, Nieminuszczy J, Mettling C, Dardillac E, Barthe A, Schmitz AL, et al. (2018). SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 557, 57–61. [DOI] [PubMed] [Google Scholar]
- Costantino L, and Koshland D (2015). The Yin and Yang of R-loop biology. Curr Opin Cell Biol 34, 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino L, and Koshland D (2018). Genome-wide Map of R-Loop-Induced Damage Reveals How a Subset of R-Loops Contributes to Genomic Instability. Mol Cell 71, 487–497 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristini A, Groh M, Kristiansen MS, and Gromak N (2018). RNA/DNA Hybrid Interactome Identifies DXH9 as a Molecular Player in Transcriptional Termination and R-Loop-Associated DNA Damage. Cell Rep 23, 1891–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, and Manel N (2015). Aicardi-Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol 15, 429–440. [DOI] [PubMed] [Google Scholar]
- Dumelie JG, and Jaffrey SR (2017). Defining the location of promoter-associated R-loops at near-nucleotide resolution using bisDRIP-seq. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn K, and Griffith JD (1980). The presence of RNA in a double helix inhibits its interaction with histone protein. Nucleic Acids Res 8, 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta D, Shatalin K, Epshtein V, Gottesman ME, and Nudler E (2011). Linking RNA polymerase backtracking to genome instability in E. coli. Cell 146, 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hage A, French SL, Beyer AL, and Tollervey D (2010). Loss of Topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev 24, 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hage A, Webb S, Kerr A, and Tollervey D (2014). Genome-wide distribution of RNADNA hybrids identifies RNase H targets in tRNA genes, retrotransposons and mitochondria. PLoS Genet 10, e1004716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan W, Guan Z, Liu J, Gui T, Shen K, Manley JL, and Li X (2011). R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev 25, 2041–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Pichardo D, Canas JC, Garcia-Rubio ML, Gomez-Gonzalez B, Rondon AG, and Aguilera A (2017). Histone Mutants Separate R Loop Formation from Genome Instability Induction. Mol Cell 66, 597–609 e5. [DOI] [PubMed] [Google Scholar]
- Garcia-Rubio ML, Perez-Calero C, Barroso SI, Tumini E, Herrera-Moyano E, Rosado IV, and Aguilera A (2015). The Fanconi Anemia Pathway Protects Genome Integrity from R-loops. PLoS Genet 11, e1005674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginno PA, Lott PL, Christensen HC, Korf I, and Chedin F (2012). R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol Cell 45, 814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorthi A, Romero JC, Loranc E, Cao L, Lawrence LA, Goodale E, Iniguez AB, Bernard X, Masamsetti VP, Roston S, et al. (2018). EWS-FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature 555, 387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf M, Bonetti D, Lockhart A, Serhal K, Kellner V, Maicher A, Jolivet P, Teixeira MT, and Luke B (2017). Telomere Length Determines TERRA and R-Loop Regulation through the Cell Cycle. Cell 170, 72–85 e14. [DOI] [PubMed] [Google Scholar]
- Groh M, Albulescu LO, Cristini A, and Gromak N (2017). Senataxin: Genome Guardian at the Interface of Transcription and Neurodegeneration. J Mol Biol 429, 3181–3195. [DOI] [PubMed] [Google Scholar]
- Groh M, and Gromak N (2014). Out of balance: R-loops in human disease. PLoS Genet 10, e1004630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh M, Lufino MM, Wade-Martins R, and Gromak N (2014). R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet 10, e1004318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunseich C, Wang IX, Watts JA, Burdick JT, Guber RD, Zhu Z, Bruzel A, Lanman T, Chen K, Schindler AB, et al. (2018). Senataxin Mutation Reveals How R-Loops Promote Transcription by Blocking DNA Methylation at Gene Promoters. Mol Cell 69, 426–437 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, et al. (2014). C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halasz L, Karanyi Z, Boros-Olah B, Kuik-Rozsa T, Sipos E, Nagy E, Mosolygo LA, Mazlo A, Rajnavolgyi E, Halmos G, et al. (2017). RNA-DNA hybrid (R-loop) immunoprecipitation mapping: an analytical workflow to evaluate inherent biases. Genome Res 27, 1063–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamperl S, Bocek MJ, Saldivar JC, Swigut T, and Cimprich KA (2017). Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 170, 774–786 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hartono SR, Malapert A, Legros P, Bernard P, Chedin F, and Vanoosthuyse V (2018). The Affinity of the S9.6 Antibody for Double-Stranded RNAs Impacts the Accurate Mapping of R-Loops in Fission Yeast. J Mol Biol 430, 272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatchi E, Skourti-Stathaki K, Ventz S, Pinello L, Yen A, Kamieniarz-Gdula K, Dimitrov S, Pathania S, McKinney KM, Eaton ML, et al. (2015). BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol Cell 57, 636–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodroj D, Recolin B, Serhal K, Martinez S, Tsanov N, Abou Merhi R, and Maiorano D (2017). An ATR-dependent function for the Ddx19 RNA helicase in nuclear R-loop metabolism. EMBO J 36, 1182–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson BR, Noerenberg M, and Whitehouse A (2014). A novel mechanism inducing genome instability in Kaposi’s sarcoma-associated herpesvirus infected cells. PLoS Pathog 10, e1004098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SE, Fleuren EDG, Frankum J, Konde A, Williamson CT, Krastev DB, Pemberton HN, Campbell J, Gulati A, Elliott R, et al. (2017). ATR Is a Therapeutic Target in Synovial Sarcoma. Cancer Res 77, 7014–7026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeche L, Nguyen HD, Buisson R, and Zou L (2018). A mitosis-specific and R loop-driven ATR pathway promotes faithful chromosome segregation. Science 359, 108–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keskin H, Shen Y, Huang F, Patel M, Yang T, Ashley K, Mazin AV, and Storici F (2014). Transcript-RNA-templated DNA recombination and repair. Nature 515, 436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig F, Schubert T, and Langst G (2017). The monoclonal S9.6 antibody exhibits highly variable binding affinities towards different R-loop sequences. PLoS One 12, e0178875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotsantis P, Silva LM, Irmscher S, Jones RM, Folkes L, Gromak N, and Petermann E (2016). Increased global transcription activity as a mechanism of replication stress in cancer. Nat Commun 7, 13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang KS, Hall AN, Merrikh CN, Ragheb M, Tabakh H, Pollock AJ, Woodward JJ, Dreifus JE, and Merrikh H (2017). Replication-Transcription Conflicts Generate R-Loops that Orchestrate Bacterial Stress Survival and Pathogenesis. Cell 170, 787–799 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YW, Sanz LA, Xu X, Hartono SR, and Chedin F (2015). Genome-wide DNA hypomethylation and RNA:DNA hybrid accumulation in Aicardi-Goutieres syndrome. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb LA, Springgate CF, and Battula N (1974). Errors in DNA replication as a basis of malignant changes. Cancer Res 34, 2311–2321. [PubMed] [Google Scholar]
- Loomis EW, Sanz LA, Chedin F, and Hagerman PJ (2014). Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet 10, e1004294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu WT, Hawley BR, Skalka GL, Baldock RA, Smith EM, Bader AS, Malewicz M, Watts FZ, Wilczynska A, and Bushell M (2018). Drosha drives the formation of DNA:RNA hybrids around DNA break sites to facilitate DNA repair. Nat Commun 9, 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. (2017). cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makharashvili N, Arora S, Yin Y, Fu Q, Wen X, Lee JH, Kao CH, Leung JW, Miller KM, and Paull TT (2018). Sae2/CtIP prevents R-loop accumulation in eukaryotic cells. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzo SG, Hartono SR, Sanz LA, Marinello J, De Biasi S, Cossarizza A, Capranico G, and Chedin F (2018). DNA Topoisomerase I differentially modulates R-loops across the human genome. Genome Biol 19, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazina OM, Keskin H, Hanamshet K, Storici F, and Mazin AV (2017). Rad52 Inverse Strand Exchange Drives RNA-Templated DNA Double-Strand Break Repair. Mol Cell 67, 19–29 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDevitt S, Rusanov T, Kent T, Chandramouly G, and Pomerantz RT (2018). How RNA transcripts coordinate DNA recombination and repair. Nat Commun 9, 1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales JC, Richard P, Patidar PL, Motea EA, Dang TT, Manley JL, and Boothman DA (2016). XRN2 Links Transcription Termination to DNA Damage and Replication Stress. PLoS Genet 12, e1006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadel J, Athanasiadou R, Lemetre C, Wijetunga NA, P OB, Sato H, Zhang Z, Jeddeloh J, Montagna C, Golden A, et al. (2015). RNA:DNA hybrids in the human genome have distinctive nucleotide characteristics, chromatin composition, and transcriptional relationships. Epigenetics Chromatin 8, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng KW, Marshall EA, Bell JC, and Lam WL (2018). cGAS-STING and Cancer: Dichotomous Roles in Tumor Immunity and Development. Trends Immunol 39, 44–54. [DOI] [PubMed] [Google Scholar]
- Nguyen HD, Yadav T, Giri S, Saez B, Graubert TA, and Zou L (2017). Functions of Replication Protein A as a Sensor of R Loops and a Regulator of RNaseH1. Mol Cell 65, 832–847 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohle C, Tesorero R, Schermann G, Dobrev N, Sinning I, and Fischer T (2016). Transient RNA-DNA Hybrids Are Required for Efficient Double-Strand Break Repair. Cell 167, 1001–1013 e7. [DOI] [PubMed] [Google Scholar]
- Phillips DD, Garboczi DN, Singh K, Hu Z, Leppla SH, and Leysath CE (2013). The sub-nanomolar binding of DNA-RNA hybrids by the single-chain Fv fragment of antibody S9.6. J Mol Recognit 26, 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell WT, Coulson RL, Gonzales ML, Crary FK, Wong SS, Adams S, Ach RA, Tsang P, Yamada NA, Yasui DH, et al. (2013). R-loop formation at Snord116 mediates topotecan inhibition of Ube3a-antisense and allele-specific chromatin decondensation. Proc Natl Acad Sci U S A 110, 13938–13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy K, Schmidt MH, Geist JM, Thakkar NP, Panigrahi GB, Wang YH, and Pearson CE (2014). Processing of double-R-loops in (CAG).(CTG) and C9orf72 (GGGGCC).(GGCCCC) repeats causes instability. Nucleic Acids Res 42, 10473–10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard P, and Manley JL (2017). R Loops and Links to Human Disease. J Mol Biol 429, 3168–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy D, Zhang Z, Lu Z, Hsieh CL, and Lieber MR (2010). Competition between the RNA transcript and the nontemplate DNA strand during R-loop formation in vitro: a nick can serve as a strong R-loop initiation site. Mol Cell Biol 30, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagie S, Toubiana S, Hartono SR, Katzir H, Tzur-Gilat A, Havazelet S, Francastel C, Velasco G, Chedin F, and Selig S (2017). Telomeres in ICF syndrome cells are vulnerable to DNA damage due to elevated DNA:RNA hybrids. Nat Commun 8, 14015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Cortez D, and Cimprich KA (2017). The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol 18, 622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Pereira JM, and Aguilera A (2015). R loops: new modulators of genome dynamics and function. Nat Rev Genet 16, 583–597. [DOI] [PubMed] [Google Scholar]
- Sanz LA, Hartono SR, Lim YW, Steyaert S, Rajpurkar A, Ginno PA, Xu X, and Chedin F (2016). Prevalent, Dynamic, and Conserved R-Loop Structures Associate with Specific Epigenomic Signatures in Mammals. Mol Cell 63, 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saponaro M, Kantidakis T, Mitter R, Kelly GP, Heron M, Williams H, Soding J, Stewart A, and Svejstrup JQ (2014). RECQL5 controls transcript elongation and suppresses genome instability associated with transcription stress. Cell 157, 1037–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab RA, Nieminuszczy J, Shah F, Langton J, Lopez Martinez D, Liang CC, Cohn MA, Gibbons RJ, Deans AJ, and Niedzwiedz W (2015). The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol Cell 60, 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, and Kelman Z (2006). DNA unwinding assay using streptavidin-bound oligonucleotides. BMC Mol Biol 7, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivji MKK, Renaudin X, Williams CH, and Venkitaraman AR (2018). BRCA2 Regulates Transcription Elongation by RNA Polymerase II to Prevent R-Loop Accumulation. Cell Rep 22, 1031–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skourti-Stathaki K, Kamieniarz-Gdula K, and Proudfoot NJ (2014). R-loops induce repressive chromatin marks over mammalian gene terminators. Nature 516, 436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skourti-Stathaki K, and Proudfoot NJ (2014). A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev 28, 1384–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skourti-Stathaki K, Proudfoot NJ, and Gromak N (2011). Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol Cell 42, 794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollier J, and Cimprich KA (2015). Breaking bad: R-loops and genome integrity. Trends Cell Biol 25, 514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollier J, Stork CT, Garcia-Rubio ML, Paulsen RD, Aguilera A, and Cimprich KA (2014). Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol Cell 56, 777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stork CT, Bocek M, Crossley MP, Sollier J, Sanz LA, Chedin F, Swigut T, and Cimprich KA (2016). Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su XA, and Freudenreich CH (2017). Cytosine deamination and base excision repair cause R-loop-induced CAG repeat fragility and instability in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 114, E8392–E8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SLW, Chadha S, Liu Y, Gabasova E, Perera D, Ahmed K, Constantinou S, Renaudin X, Lee M, Aebersold R, et al. (2017). A Class of Environmental and Endogenous Toxins Induces BRCA2 Haploinsufficiency and Genome Instability. Cell 169, 1105–1118 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y, Yadav T, Duan M, Tan J, Xiang Y, Gao B, Xu J, Liang Z, Liu Y, Nakajima S, et al. (2018). ROS-induced R loops trigger a transcription-coupled but BRCA1/2-independent homologous recombination pathway through CSB. Nat Commun 9, 4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M, White RL, and Davis RW (1976). Hybridization of RNA to double-stranded DNA: formation of R-loops. Proceedings of the National Academy of Sciences 73, 2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran PLT, Pohl TJ, Chen CF, Chan A, Pott S, and Zakian VA (2017). PIF1 family DNA helicases suppress R-loop mediated genome instability at tRNA genes. Nat Commun 8, 15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tresini M, Warmerdam DO, Kolovos P, Snijder L, Vrouwe MG, Demmers JA, van IWF, Grosveld FG, Medema RH, Hoeijmakers JH, et al. (2015). The core spliceosome as target and effector of non-canonical ATM signalling. Nature 523, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoosthuyse V (2018). Strengths and Weaknesses of the Current Strategies to Map and Characterize R-Loops. Noncoding RNA 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahba L, Costantino L, Tan FJ, Zimmer A, and Koshland D (2016). S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes Dev 30, 1327–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IX, Grunseich C, Fox J, Burdick J, Zhu Z, Ravazian N, Hafner M, and Cheung VG (2018). Human proteins that interact with RNA/DNA hybrids. Genome Res 28, 1405–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellinger RE, Prado F, and Aguilera A (2006). Replication fork progression is impaired by transcription in hyperrecombinant yeast cells lacking a functional THO complex. Mol Cell Biol 26, 3327–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Xu H, Li K, Fan Y, Liu Y, Yang X, and Sun Q (2017). The R-loop is a common chromatin feature of the Arabidopsis genome. Nat Plants 3, 704–714. [DOI] [PubMed] [Google Scholar]
- Yang Y, McBride KM, Hensley S, Lu Y, Chedin F, and Bedford MT (2014). Arginine methylation facilitates the recruitment of TOP3B to chromatin to prevent R loop accumulation. Mol Cell 53, 484–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuhara T, Kato R, Hagiwara Y, Shiotani B, Yamauchi M, Nakada S, Shibata A, and Miyagawa K (2018). Human Rad52 Promotes XPG-Mediated R-loop Processing to Initiate Transcription-Associated Homologous Recombination Repair. Cell. [DOI] [PubMed] [Google Scholar]
- Yu K, Chedin F, Hsieh CL, Wilson TE, and Lieber MR (2003). R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat Immunol 4, 442–451. [DOI] [PubMed] [Google Scholar]
- Yuce O, and West SC (2013). Senataxin, defective in the neurodegenerative disorder ataxia with oculomotor apraxia 2, lies at the interface of transcription and the DNA damage response. Mol Cell Biol 33, 406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chiang HC, Wang Y, Zhang C, Smith S, Zhao X, Nair SJ, Michalek J, Jatoi I, Lautner M, et al. (2017). Attenuation of RNA polymerase II pausing mitigates BRCA1-associated R-loop accumulation and tumorigenesis. Nat Commun 8, 15908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao DY, Gish G, Braunschweig U, Li Y, Ni Z, Schmitges FW, Zhong G, Liu K, Li W, Moffat J, et al. (2016). SMN and symmetric arginine dimethylation of RNA polymerase II C-terminal domain control termination. Nature 529, 48–53. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Hoong N, Aslanian A, Hara T, Benner C, Heinz S, Miga KH, Ke E, Verma S, Soroczynski J, et al. (2018). Heterochromatin-Encoded Satellite RNAs Induce Breast Cancer. Mol Cell 70, 842–853 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]