

Chromatin capture unveils a regulatory axis between metabolism, gene regulation, and proliferation in stem cells.
Abstract
Profiling the chromatin-bound proteome (chromatome) in a simple, direct, and reliable manner might be key to uncovering the role of yet uncharacterized chromatin factors in physiology and disease. Here, we have designed an experimental strategy to survey the chromatome of proliferating cells by using the DNA-mediated chromatin pull-down (Dm-ChP) technology. Our approach provides a global view of cellular chromatome under normal physiological conditions and enables the identification of chromatin-bound proteins de novo. Integrating Dm-ChP with genomic and functional data, we have discovered an unexpected chromatin function for adenosylhomocysteinase, a major one-carbon pathway metabolic enzyme, in gene activation. Our study reveals a new regulatory axis between the metabolic state of pluripotent cells, ribosomal protein production, and cell division during the early phase of embryo development, in which the metabolic flux of methylation reactions is favored in a local milieu.
INTRODUCTION
Most fundamental cellular functions rely on chromatin-based processes, such as transcription, replication, and inheritability of genetic and epigenetic information. In mammals, chromatin structures are spatially and functionally organized into compartments by the combination of strong and weak molecular interactions (1). To date, our knowledge on the distributions of chromatin-bound factors is limited to a reduced number of proteins and histone modifications (2, 3). Genomic, transcriptomic, and global proteomic profiling can only predict the proteomic composition of the cellular chromatin. Moreover, proteins lacking a chromatin/DNA recognition motif are difficult to foresee as chromatin-associated factors, thereby limiting their characterization. Hence, a simple, straightforward, and reliable “chromatomic” approach is in high demand to define the proteomic composition of the cellular chromatin and to discover unpredicted chromatin-associated factors de novo.
RESULTS
In the last decade, several methods have been designed to study the proteomic composition of the chromatin, the so-called chromatome (4) (table S1). However, most of these methodologies have notable limitations, resulting in the lack of a unique and solid surveyor assay that serves as a gold standard method for the systematic chromatin-bound proteome profiling. These limitations include (i) the relative methodological complexity, which makes them unsuitable as a routine laboratory technique; (ii) the presence of contaminants (such as antibodies) that hamper the detection of proteins by mass spectrometry (MS); and (iii) the incompatibility of the purified chromatin preparations as chromatin immunoprecipitation (ChIP)–able material for downstream functional analysis. In 2011, two independent laboratories developed a technology to capture nascent DNA labeled with the thymidine analog 5-ethynyl-2′-deoxyuridine (EdU) during DNA replication (5, 6). These two groups used the simple and highly efficient “Click chemistry” to covalently link biotin moieties to nascent EdU-labeled DNA that was further captured using streptavidin columns. Combined with high-resolution MS, the Click-assisted chromatin purification method allowed the definition of the first unbiased picture of the proteins associated to DNA during replication in cancer and pluripotent stem cells (7, 8). On the basis of the recent DNA-mediated chromatin pull-down (Dm-ChP) technology (6) and with the aim of surveying the chromatome of proliferating cells in a simple and robust manner, we designed an experimental strategy to capture the whole EdU-labeled genome under physiological conditions (Fig. 1A). Our strategy relies on (i) the efficient incorporation of the thymidine analog EdU during DNA replication in a noncytotoxic manner, (ii) the addition of a biotin moiety to the incorporated EdU under mild conditions, and (iii) streptavidin-biotin affinity binding to capture sheared EdU-labeled chromatin. As shown in Fig. 1B, the incubation of mouse embryonic stem cells (ESCs) with EdU for a short period of 10 min enables its incorporation into nascent DNA in living cells, displaying a punctuated distribution throughout the nucleus of ESCs in S phase, together with the replication-associated protein RPA32. In contrast, after incubation for 20 hours, EdU was uniformly distributed in the nuclei of virtually all ESCs in culture, indicating a global EdU incorporation into their genomes (Fig. 1B). As depicted in Fig. 1B, our strategy includes a formaldehyde cross-link step (1% FA, 10 min) to preserve protein-protein and protein-DNA interactions during the sample preparation, similarly to the ChIP procedure (step 1). After a mild cell permeabilization (0.1% Triton X-100), a biotin moiety is attached in situ to the incorporated EdU under mild conditions by Click chemistry (step 2). Chromatin is sheared by sonication to yield fragments of about 500 base pairs (bp) (step 3; Fig. 1A and fig. S1A), and streptavidin-biotin affinity columns are used to capture EdU-labeled chromatin (step 4; Fig. 1A). Captured chromatin is extensively washed, reverse cross-linked, and eluted by heating under reducing conditions [2% SDS and 0.1 M dithiothreitol (DTT); step 5]. Last, chromatin-bound proteins are further analyzed by conventional blotting methods or by MS techniques (step 6; Fig. 1A). Specificity of chromatin capture can be monitored by dot blot (Fig. 1C) or Western blot (fig. S1B). While histone H3 was efficiently captured from ESCs incubated with EdU (+EdU; Fig. 1C and fig. S1B), it remained undetectable on eluates from nonincubated ESCs (−EdU). In contrast, the cytoskeleton vinculin or β-actin proteins were absent in eluates from either +EdU- or −EdU-incubated cells (Fig. 1C and fig. S1B). We show that a similar experimental pipeline can be used for other cell types (fig. S1C) under distinct growth conditions (e.g., grown in suspension or adherent cultures) (fig. S1C).
Fig. 1. Dm-ChP for chromatin-bound proteome profiling.
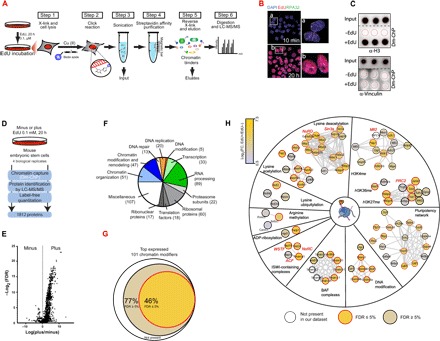
(A) Schematic representation of the Dm-ChP technique for chromatin-bound proteome profiling. Cells were incubated with a very low amount of EdU during a long pulse (0.1 μM EdU for 20 hours) to label the whole genome. The DNA and associated proteins were cross-linked with 1% formaldehyde (FA) for 10 min, and the cells were lysed (step 1). The EdU-labeled DNA was conjugated to a biotin group by a Click reaction (step 2) and then fragmented by sonication (step 3). Labeled DNA fragments were isolated using streptavidin magnetic beads (step 4) and eluted using Laemmli buffer (step 5). Eluted samples were analyzed by Western blot or high-resolution MS (step 6). μM, micromolar. LC-MS/MS, liquid chromatography–tandem mass spectrometry (B) ESCs were pulsed with EdU at the times indicated and then stained with an anti-RP32 antibody, and EdU was detected by a Click reaction. Insets show magnifications of the image. DAPI, 4′,6-diamidino-2-phenylindole. (C) Input material and eluates prepared by Dm-ChP from nonincubated (−EdU) and EdU-incubated mouse ESCs (+EdU) were analyzed in triplicate by dot blot using the indicated antibodies. Dashed red circles indicate the position of the dotted samples. (D) Experimental scheme for the proteomic survey of pluripotent ESCs using Dm-ChP with high-resolution MS (Dm-ChP–MS). (E) Volcano plot of the 1812 proteins identified in the proteomic analysis in ESCs. Proteins enriched in EdU-ESCs are shown on the right, and those enriched in (non-EdU) ESCs are shown on the left. FDR, false discovery rate. (F) Pie chart representing the functional groups, by manual curation, of the 488 chromatin-bound proteins found in ESCs by Dm-ChP–MS and Significance Analysis of INTeractome (SAINT). The number of proteins is indicated in parentheses. (G) Venn diagram indicating the overlap between the manually curated list of previously reported chromatin modifiers [from (63–65)] and our Dm-ChP–MS dataset. The list includes the top expressed chromatin modifiers in mouse ESCs (RPKM > 50). RPKM, Reads Per Kilobase Million. (H) Selected functional protein networks associated with chromatin organization and remodeling, DNA modification, and pluripotency. Related subnetworks are depicted separately according to their functional roles. The legend indicates the color code for the log2 of the fold changes in +EdU/−EdU conditions and the presence and significance of each protein in our dataset. Selected protein complexes are indicated in red. ACF (ID_925), adenosine 5′-triphosphate–dependent chromatin assembly factor complex; Mll2 (ID_6460), mixed-lineage leukemia 2 complex; NoRC (ID_5694), nucleolar remodeling complex; NuRD (ID_61), nucleosome remodeling deacetylase complex; PRC2 (ID_996), polycomb repressive complex 2; Sin3A (ID_283), paired amphipathic helix protein Sin3a complex; WSTF (ID_236), Williams syndrome transcription factor–containing complex (abbreviations for protein complexes are given with the identifier number of the archetypic complex at the CORUM database). ADP, adenosine 5′-diphosphate.
Incubation times of 48 hours or longer with 10 μM EdU have been shown to be cytotoxic in several human cancer cell lines (9–11). Although this concentration is a hundred times more concentrated than that we use in our experimental pipeline (0.1 μM EdU for 20 hours), we analyzed whether incorporation of EdU affects normal cell behavior. We did not observe any changes in the transcriptional program of EdU-incubated ESCs by RNA sequencing (RNA-seq) (fig. S2A). Further, phosphorylation levels of histone H2AX at serine 139 (γH2AX), an early marker of DNA damage, remained unchanged after EdU incorporation (fig. S2B). We additionally analyzed the subcellular distribution of γH2AX by immunofluorescence. In both nonincubated and EdU-incubated ESCs, γH2AX displays a typical cell-to-cell heterogeneous staining with the presence of discrete nuclear foci, which are characteristic of normally growing asynchronous ESCs (fig. S2C) (12). Last, cell cycle profile analysis showed equivalent proportions of cells in different phases of the cell cycle in EdU-ESCs and nonincubated cells, which is a signal of normal cell cycle progression (fig. S2D). In sum, all these results indicate that the incorporation of EdU in ESCs using our experimental scheme is compatible with normal physiological conditions, and that, in combination with Dm-ChP technology, it enables one to survey the intact chromatome of proliferating cells.
Pluripotent ESCs have a unique open chromatin with transcriptionally active and poised genomic regions established by the interplay between pluripotent transcription factors and chromatin remodeling complexes (13). To date, a partial proteomic view of the chromatin of ESCs has been gained by focusing on genomic regions occupied by the pluripotent factors (14), or decorated by specific modified histones (15), or by characterizing the strongly chromatin-bound insoluble nuclear material (16), or focusing on nascent chromatin (8). With the aim of uncovering both the strong and weakly chromatin-bound proteome of pluripotent ESCs in a global and unbiased manner, we combined Dm-ChP with high-resolution MS (Dm-ChP–MS; Fig. 1D). In total, we measured the relative abundance of 1812 proteins using label-free quantitative proteomics (Fig. 1D). The volcano plot distribution showed a large number of proteins enriched on chromatin captured from EdU-incubated ESCs, while the eluates from nonincubated control ESCs displayed very few enriched proteins (Fig. 1E). To provide a comprehensive dataset for the pluripotent chromatin-bound proteome, we used the Significance Analysis of INTeractome (SAINT) algorithm to identify the bona fide protein-chromatin interactions in the Dm-ChP–MS experiments (17). Using this approach, we identified a total of 488 confident chromatin-bound proteins of ESCs, of which nearly 60% primarily have a typical chromatin-based function, such as chromatin organization and remodeling, DNA repair, replication and modification, transcription, and RNA processes (i.e., splicing and capping) (Fig. 1F). Gene Ontology (GO) term enrichment analysis showed nucleic acid and chromatin binding as top category terms related with the molecular function of these 488 proteins (fig. S3A). Collectively, we found that about 75% of the top expressed ESC-specific chromatin modifiers described in the literature were present in our Dm-ChP samples (Fig. 1G). In particular, we purified the complete polycomb repressive complex 2 (PRC2) and the nearly complete mixed-lineage leukemia 2/COMplex of Proteins ASSociated with Set1 (MLL2/COMPASS)–related complex 2; these are two key protein complexes associated with transmission of the epigenetic cellular memory (Fig. 1H) (18, 19). In addition, we found factors that have been functionally linked with self-renewal and pluripotency capacity of ESCs, including the Brg-associated factors (esBAFs) (20) and a protein network centered around the pluripotency factors POU5F1 and SOX2.
Together, our results constitute the first broad characterization of the chromatin-bound proteome in pluripotent cells, and it encompasses major DNA factors and chromatin-associated complexes essential for sustaining ESC identity and the pluripotency transcriptional program.
In addition to relevant, well-known chromatin-associated proteins, our dataset included proteins with no obvious connections with chromatin (Fig. 1F). In addition to their primary function, increasing experimental evidence supports a chromatin function for some of these proteins, including proteasome subunits (21), ribosomal subunits (22, 23), and ribonuclear proteins (24). We aimed to validate the strength of our Dm-ChP–MS strategy to identify the potential role of chromatin-associated proteins de novo. Among the most biologically and pathologically relevant proteins from this group, we identified adenosylhomocysteinase (AHCY, also known as S-adenosyl-l-homocysteine hydrolase), a major one-carbon pathway metabolic enzyme and one of the most conserved enzymes across living organisms, including bacteria, nematodes, yeast, plants, insects, and vertebrates (25) (fig. S3, B and C). The homotetrameric structure of AHCY is a result of a dimer of dimers that catalyzes the reversible hydrolysis of S-adenosylhomocysteine (SAH) to adenosine and l-homocysteine (26) (Fig. 2A). The AHCY substrate SAH is the product of the S-adenosyl-l-methionine (SAM)–dependent methyltransferases (MTases), which transfer the methyl group from SAM to a variety of cellular substrates, including DNA, RNA, and proteins. The mammalian AHCY is the only enzyme capable of hydrolyzing SAH. Accordingly, a chromosomal deletion that includes Ahcy is embryonically lethal in mice at the peri-implantation stage (between 4.5 and 5.5 days post coitum) (27). Moreover, AHCY dysfunction has been associated with a wide variety of human disorders, including vascular disease, cancer, diabetic nephropathy, severe myopathy, and neurodevelopmental delay followed by childhood death (28–32). Despite the functional, pathological, and biological relevance of AHCY, and the presence of the methylation reactions in different subcellular compartments, no direct evidence exists for genomic recruitment or distribution of AHCY, and its role in stem cell biology remains unknown. We validated our MS data by Dm-ChP–Western blot using a specific antibody against AHCY (Fig. 2B). We confirmed the presence of a partial fraction of cellular pool of AHCY in the chromatin of ESCs growing in either serum-containing media plus leukemia inhibitory factor (LIF) or the so-called 2i-LIF media, which promotes the homogeneous expansion of preimplantation ground state–like ESCs. We confirmed the presence of AHCY in the nucleus of ESCs in culture, and the specificity of the nuclear signal was evaluated by reducing the expression of AHCY using short hairpin RNAs (shRNAs) against Ahcy (Fig. 2C). Using available single-cell RNA-seq data (33), we observed Ahcy expression during early mouse development (Fig. 2D). Ahcy expression levels increase with the acquisition of the pluripotency of blastomers, correlating with expression of the pluripotency marker Nanog (fig. S4) and the formation of the preimplantation blastocysts (Fig. 2D). Immunostaining and confocal microscopy analysis on mice embryos at the blastula stage revealed that AHCY is located in the cell nucleus (Fig. 2E), and it is most expressed in pluripotent NANOG-positive cells. These results indicate that AHCY has a nuclear function during early mammalian development.
Fig. 2. The AHCY enzyme is recruited to chromatin in pluripotent ESCs.
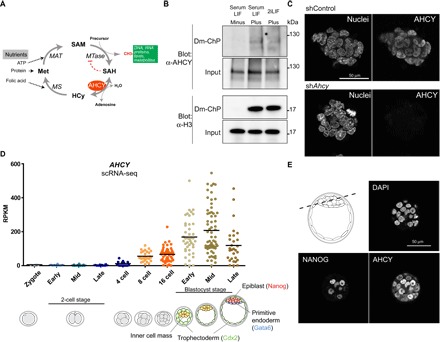
(A) Scheme of the methionine (Met) metabolic pathway. AHCY is depicted in red. MAT, methionine adenosyltransferase; MS, methionine synthase; MTase; methyltransferase; HCy, Homocysteine. (B) Input and chromatin isolated by Dm-ChP from ESCs, under serum-containing or 2iLIF culture conditions, with nonincubated (minus) or EdU-incubated cells (plus), were analyzed by Western blot using the indicated antibodies. H3 was used as a control of efficient chromatin purification by Dm-ChP. Note that specific AHCY bands correspond to the dimer in size that is resistant to reverse cross-linking by heating. (C) shControl and shAhcy-KD ESCs were stained with a specific anti-AHCY antibody and show the loss of a specific nuclear signal in all three shAhcy-KD cells. Nuclei were counterstained with DAPI. (D) Single-cell expression values of Ahcy during mouse development, from zygote to the late blastocyst stage. Each dot represents a cell from the original data of (33). scRNA-seq, single cell RNA-seq. (E) Representative confocal section covering the inner cell mass and the trophectoderm layers of mouse late preimplantation blastocysts (E4.5) immunostained for AHCY or NANOG.
We next studied the genomic occupancy of AHCY by ChIP followed by massive parallel sequencing (ChIP-seq). By combining the data from two independent ChIP-seq replicates, we identified 665 confident binding sites for AHCY in the mouse genome (Fig. 3A, fig. S5A, and table S3). We validated several AHCY peaks by ChIP and quantitative polymerase chain reaction (ChIP-qPCR; Fig. 3B) and confirmed that Ahcy knockdown led to reduction of the AHCY ChIP signal at specific promoters (Fig. 3B). Genome-wide analysis of ChIP-seq data revealed that AHCY is preferentially recruited to regions in close proximity to transcription start sites (TSS) (Fig. 3C), covering a narrow window around TSS of target genes (Fig. 3, D and E). We found that AHCY target genes are among the most highly expressed genes in ESCs (Fig. 3F) and are decorated with transcriptionally permissive histone posttranslational modifications (H3K36me3, H3K4me3, and H3K27ac) and only low levels of transcriptionally repressive marks (e.g., H3K27me3 and 5mC) (fig. S5). In addition, we found a high correlation between AHCY occupancy and key factors associated with the cancer-related ESC-specific Myc network, including Myc, TRIM28, and DMAP1 (Fig. 3G). The Myc network defined in ESCs is primary linked to proliferation and ribosomal activity and shares transcriptional activity in cancer cells (34), suggesting a potential role of AHCY in ESC proliferation. In agreement, we found that AHCY was enriched at TSS of a large number of ribosomal and mRNA processing genes (Fig. 3H), which are strongly expressed in ESCs, in line with their highly proliferative and transcriptional activity. In sum, these data indicate that AHCY associates at TSS of transcriptionally active genes in ESCs, with a potential direct impact in their transcriptional regulation at the chromatin level.
Fig. 3. AHCY occupies transcription start sites (TSS) of highly expressed genes in ESCs.
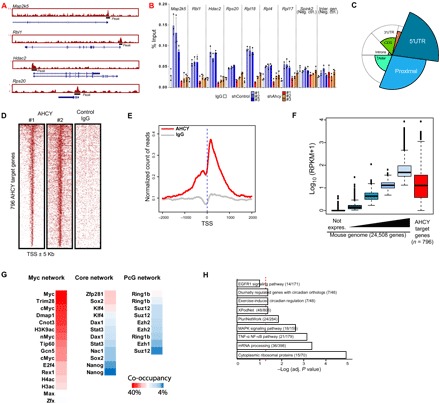
(A) Genomic visualization of AHCY ChIP-seq at several target sites. (B) AHCY ChIP-seq validation by ChIP-qPCR at TSS of AHCY target genes (Map2k5, Rbl1, Hdac2, Rps20, Rpl18, Rpl4, and Rpl17) and AHCY nontarget genomic regions (TSS of Spink2 and intergenic region at Chr15) as a negative control. ChIP-qPCRs were performed in shControl cells (n = 3) and shAhcy-KD ESCs with three independent shRNAs, showing the specific reduction in AHCY binding in shAhcy-KD ESCs (mean ± SD of three technical replicates). IgG, immunoglobulin G. (C) Genomic distribution of ChIP-seq peaks of AHCY. The spie chart represents the distribution of AHCY peaks corrected by the genome-wide distribution of each gene genomic feature (indicated in the background circle distribution). The spie charts indicate that AHCY preferentially occupies TSS neighborhood regions, including 5′ untranslated region (5′UTR) and proximal promoter regions. (D) Heat map showing the AHCY ChIP-seq occupancy profile around TSS (±5 kb) in two independent ChIP-seq replicates, with IgG as a negative control. (E) Meta-gene plot showing the AHCY ChIP-seq occupancy profile (red line) around TSS (±2 kb). IgG distribution is indicated by the gray line. (F) Box plots indicating the gene expression for the whole transcriptome in naïve ESCs, fractionated into five groups, from non-expressed genes (black box plot) to the highly expressed genes (light blue box plot). The red box plot indicates the expression of AHCY target genes. (G) Percentage of AHCY co-occupancy with chromatin factors from Myc, core, and PcG networks. (H) Pathway enrichment analysis of AHCY target genes. EGFR1, epidermal growth factor receptor 1; MAPK, mitogen-activated protein kinase; TNF-α, tumor necrosis factor–α; and NF-κB, nuclear factor κB.
The efficient knockdown of Ahcy had no effect on the levels of the DNA damage marker γH2AX, the formation of dome-shaped ESC colonies in culture (Fig. 4, A and B), or the expression levels of key pluripotency genes, such as Pou5f1, Nanog, and Sox2 (fig. S6), which are indications of normal stem cell identity. However, our unsuccessful attempts to generate full CRISPR knockout ESCs for Ahcy suggest that a low basal AHCY activity is essential for ESC fitness. Lowering the levels of AHCY resulted in a 40 to 50% reduction in the number of ESCs in culture (Fig. 4C). This reduction is in agreement with an increased number of cells in the G1 phase and a concomitant decrease of mitotically active S phase cells in shAhcy-KD ESC cultures (Fig. 4, D and E), thus indicating that AHCY functions to promote the proliferation of pluripotent cells.
Fig. 4. AHCY promotes the expression of ribosomal protein genes, protein synthesis, and proliferation in ESCs.
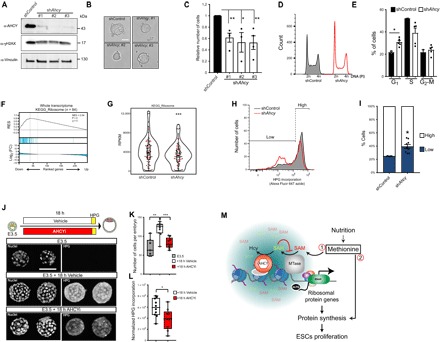
(A) Total cell extracts from shControl- or shAhcy (three independent shRNAs)–infected cells were analyzed by Western blot with the indicated antibodies. The protein vinculin was used as a loading control. (B) Images of ESC colonies growing under naïve culture conditions, displaying the typical dome shape in both shControl and shAhcy-KD ESCs. Scale bar, 100 μm. (C) Histogram indicating the relative number of shControl and shAhcy-KD ESCs after 48 hours of growing under naïve conditions (mean ± SE, n = 4 for shControl, shAhcy#1, and shAhcy#3 and n = 3 for shAhcy#2; *P ≤ 0.05, **P ≤ 0.01, two-tailed Student’s t test). (D) Cell cycle profile of shControl and shAhcy-KD ESCs analyzed by fluorescence-activated cell sorting (FACS). PI, propidium iodide. (E) Histogram indicating the percentage of shControl and shAhcy-KD ESCs in different phases of the cell cycle (mean ± SE; *P ≤ 0.05, two-tailed Student’s t test; n = 2 shControl and n = 5 shAhcy). (F) Gene set enrichment analysis (GSEA) plots for the gene expression data generated by RNA-seq from cells infected with shControl (n = 2) or shAhcy (n = 5). The complete mouse genome was ranked according to their log2 fold change expression between shControl- and shAhcy-infected cells. Genes down-regulated in shAhcy are distributed on the left of the plot, and those up-regulated are distributed on the right. The top significant KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway is shown together with the computed nominal P value, the q value, and the normalized enrichment score (NES). Distribution of ribosomal protein genes is indicated in blue lines, as well as the curve for the evolution of the gene density. (G) Violin plots indicating the expression of ribosomal protein genes calculated from RNA-seq. Each dot represents average expression of a ribosomal protein gene. Red dots indicate the ribosomal protein genes targeted by AHCY. ***P < 0.001, Wilcoxon matched pair signed-rank test. (H) Histogram indicates the relative incorporation of the methionine analog L-HPG in shControl and shAhcy-KD ESCs as analyzed by FACS. (I) Quantification of high and low percentage of L-HPG–incorporated cells (mean ± SE; *P ≤ 0.05, two-tailed Student’s t test; n = 3 shControl and n = 9 shAhcy). (J) Mouse preimplantation embryos (E3.5) were cultured ex vivo in the presence of 3-deazaadenosine (3-DZA) or DMSO (as vehicle) for 18 hours. Embryos were then pulsed once with L-HPG and labeled using the Click reaction. Developmental progression of embryos was evaluated by (K) counting the number of cells per embryo or (L) quantifying the total L-HPG incorporation per embryo and normalizing this by the number of cells. *P < 0.05, **P < 0.01, ***P < 0.001, Mann-Whitney test two-tailed; n = 3 for E3.5, and n = 10 for 18-hour vehicle and n = 9 for 18-hour AHCYi. (M) Model for the AHCY-dependent proliferation control of pluripotent cells. Nutrients are the primary source of cellular methionine, which, in turn, can be incorporated into the newly synthetized proteins (1) or it can be used to produce SAM as a methyl donor for MTases (2). Elevated levels of SAH produced by the transfer of the methyl group can inhibit the MTases. AHCY is recruited to highly transcribed genes, such as ribosomal protein genes, to favor the efficient methylation reactions at these loci and the production of mature transcripts in an efficient manner. Elevated ribosomal protein production sustains the rate of protein synthesis and proliferation in ESCs.
To gain insight into the mechanism of action of how AHCY promotes proliferation, we performed gene set enrichment analysis (GSEA) with transcriptome data from shControl and shAhcy-KD ESCs. We identified the ribosomal protein genes as the most prominent category of genes transcriptionally affected by AHCY depletion (Fig. 4, F and G). Proteomic analysis of shControl and shAhcy-KD ESCs indicated a consistent reduction in the ribosomal proteins encoded by AHCY target genes, further supporting their role in ribosome biogenesis (fig. S7, A and B). To test the impact of reducing ribosome production on protein synthesis upon AHCY depletion, we measured the efficiency of incorporating the methionine analog L-homopropargylglycine (L-HPG) in newly synthesized proteins at the single-cell level by fluorescence-activated cell sorting (FACS) (fig. S8, A and B). We discovered that the protein synthesis rate was remarkably reduced in shAhcy-KD ESCs as compared to shControl cells (Fig. 4, H and I). Similarly, the pharmacological blockage of AHCY activity with 3-deazaadenosine (3-DZA), a potent and high specific inhibitor, reduced the protein synthesis rate in ESCs in a dose-dependent manner (fig. S8, C and D). Mouse preimplantation embryos cultured in the presence of 3-DZA displayed severe developmental growth delay and a remarkable reduction in the protein synthesis rate (Fig. 4, J to L). Together, these findings support a functional role of AHCY in controlling the proliferation of pluripotent cells during early mammalian development by mechanistically regulating ribosomal protein gene expression and protein synthesis rate (Fig. 4M).
DISCUSSION
In summary, the Dm-ChP–MS pipeline presented in this study is a powerful method for chromatome profiling and for the identification of unpredicted chromatin-associated proteins. The experimental approach highlights its simplicity, its robustness, and its usability in a wide range of proliferating cells. In the last decade, several methodologies have been designed to study the proteomic composition of cells using different strategies. These methodologies fall into three major groups according to the material captured and the method used for capturing (table S1). Several of these strategies are relatively complex methodologies that include the genomic engineering of epitope-tagged proteins (e.g., HTB or TAP), the genomic insertion of DNA recognition sites at a specific locus (e.g., LexA), or the capture of specific genomic repetitive regions by oligo hybridization and pulldown (e.g., telomeres). Other chromatin-purifying methods result in a material with a remarkable abundance of contaminants, such as antibodies, that can mask the detection of low-abundant chromatin proteins. Last, traditional fractionation methodologies base their strategy on the inherent insolubility of the chromatin fraction, which is not an exclusive characteristic of the chromatin compartment. These strategies do not completely rule out the existence of contaminants, and even the more sophisticated one (as ChEP) required computational filtering based on complex machine-learning approaches to provide a probabilistic score for each protein identified as a putative chromatin factor. We believe that Dm-ChP methodology overcomes all these limitations by (i) being a simple experimental procedure easily implemented by researchers with limited experience in biochemical purification procedures, allowing a throughput approach; (ii) being a robust methodology with the absence of contaminants (e.g., antibodies), which could mask the detection of low-abundance chromatin proteins; and (iii) allowing the maintenance of the weak interactions of proteins due to in situ cross-link reaction. In addition, considering that the Dm-ChP relies on the incorporation of EdU during replication, we envision that Dm-ChP could be applied to purify chromatin from highly proliferative tumoral cells or from proliferating adult stem cells within a non-proliferative tissue, thereby highlighting important regulators in cancer and stem cell biology.
With the aim of testing the strength of Dm-ChP to identify relevant chromatin-associated proteins de novo, we have discovered the unexpected functional link of the metabolic enzyme AHCY with ribosomal protein gene transcription and stem cell proliferation. The biological and pathological relevance of AHCY is in line with its unique capacity to catabolize SAH metabolite. Elevated levels of SAH/SAM result in a competitive inhibition of SAM-dependent MTases (28). Several reports have highlighted a functional connection between AHCY and cancer cell fitness (35–38). However, the proposed mechanism of action are diverse, including the activation of the DNA damage response in human cancer cell lines (35–37) or attenuating the induction of mRNA cap methylation and protein synthesis upon ectopic expression of Myc in immortalized rat and human cells (38). By using Dm-ChP–MS, together with chromatin profiling by ChIP-seq, we have discovered that AHCY is a chromatin-associated protein that occupies TSS of active genes. We did not observe any sign of DNA damage response activation in shAhcy-KD ESCs, including comparable levels of γH2AX to control cells, the normal dome-shaped morphology of ESCs colonies, and the stable maintenance of pluripotent gene expression. This, together with the high correlation in genomic occupancy that we observed between AHCY and Myc-related factors, suggests that AHCY could be recruited to chromatin to facilitate an efficient mRNA cap methylation on specific genes in ESCs. Overall, our data point to a dual role of methionine for the control of protein synthesis: first, as a building block for protein production and, second, as a metabolic intermediate for the control of ribosome biogenesis (Fig. 4M).
By using Dm-ChP, we have provided additional data challenging the classical compartmentalization of cytosolic metabolic pathways and nuclear chromatin regulation (39). Recent studies propose that the chromatin recruitment of metabolic enzymes is considered an adaptive cell solution for the subcellular local demand of metabolites (40–42). Our findings suggest that AHCY recruitment to spatially confined genomic regions might favor a balanced SAM/SAH composition at SAH-sensitive chromatin subcompartments, thereby facilitating a transcription-permissive environment. Beyond the functional significance for the control of pluripotent cell proliferation, our discovery provides a mechanistic insight into the long-lasting factual connection between perinatal nutrition, which fuels the methionine cycle, and embryo development.
MATERIALS AND METHODS
Cell culture
E14TG2a from mouse embryo (male blastocyst, strain 129/Ola) (Sigma-Aldrich) were cultured on tissue culture plates coated with 0.1% gelatin (Millipore) under serum-free or serum conditions, as indicated. For serum-free (so-called 2iLIF) cultures, ESCs were grown in serum-free N2B27 medium [(Dulbecco’s modified Eagle’s medium (DMEM)/F12/Neurobasal 1:1, 0.5× N-2 supplement, 1× B-27 serum-free supplement, 50 μM 2-mercaptoethanol, bovine albumin fraction V 0.033%, 1× glutaMAX, 1× minimum essential medium (MEM) nonessential amino acids, and 1× penicillin-streptomycin; all from Gibco] supplemented with 1 μM PD0325901, 3 μM CHIR99021, and LIF. For serum-containing cultures, ESCs were cultured in knockout DMEM, 20% knockout serum replacement, 1× nonessential amino acids, 2 mM glutaMAX, 5 mM Hepes, and 0.5 mM 2-mercaptoethanol (all from Gibco) supplemented with LIF.
For stable depletion of gene expression, ESCs were incubated with media containing plasmid-based shRNA lentiviral particles for 24 hours. After 48 hours, infected cells were selected for 3 days with puromycin (2 μg/ml; Sigma).
For protein synthesis rate assays, cells were incubated with 50 μM L-HPG (Invitrogen) in serum-free and methionine-free DMEM (Gibco) supplemented with 0.5× N-2 supplement, 1× B-27 serum-free supplement, 50 μM 2-mercaptoethanol, bovine albumin fraction V 0.033%; 1× glutaMAX, 1× MEM nonessential amino acids, 1× penicillin-streptomycin, 1 μM PD0325901, 3 μM CHIR99021, and LIF. Additional leukemic and epithelial carcinoma human cell lines were cultured in suspension or under adherent conditions, as indicated in Table 1.
Table 1. List of human cell lines used in this study.
CIMA, Centro de Investigación Médica Aplicada, Spain; CRG, Center for Genomic Regulation, Spain; EC, epithelial carcinoma; FBS, fetal bovine serum (Hyclone); IMIM, Institut Hospital del Mar d’Investigacions Mèdiques, Spain; IRB, Research Institute Barcelona, Spain; LC, leukemic cells; NaPyr, sodium pyruvate (Gibco); P/S, penicillin-streptomycin (Gibco); Qx, glutaMAX (Gibco); RPMI 1640 (Gibco); GM-CSF, granulocyte-macrophage colony-stimulating factor; h.i., heat inactivated.
| Cell line name | Origin | Obtained from | Media |
Estimated doubling time |
| HEL | LC | Puri Fortes’ lab (CIMA) | 90% RPMI 1640 + 10 FBS + MEM + P/S + Qx + NaPyr | 35 hours |
| NB4 | LC | Luciano Di Croce’s lab (CRG) | 90% RPMI 1640 + 10 FBS + P/S + Qx | 35–45 hours |
| OCI-AML2 | LC | Puri Fortes’ lab (CIMA) | 90% DMEM +10 FBS + MEM + P/S + Qx + NaPyr | 30–50 hours |
| Skno-1 | LC | DSMZ (Deutsche Sammlung von Mikroorganismen und Zellkulturen) |
90% RPMI 1640 + 10% FBS + GM-CSF | 35–50 hours |
| K562 | LC | Luciano Di Croce’s lab (CRG) | 90% RPMI 1640 + 10 FBS + P/S + Qx | 30–40 hours |
| LoVo | EC | IMIM repository | 90% RPMI 1640 + 10 FBS + MEM + P/S + Qx + NaPyr | 48 hours |
| SW480 | EC | IMIM repository | DMEM + 10% FBS + 10 FBS + P/S + Qx | 25–30 hours |
| HT-29 | EC | IMIM repository | DMEM + 10% FBS + 10 FBS + P/S + Qx | 40–60 hours |
| MCF7 | EC | CRG repository | DMEM + 10% FBS + P/S + insulin | 30–72 hours |
| T47D | EC | Miguel Beato’s lab (CRG) | 90% RPMI 1640 + 10 h.i. FBS + MEM + P/S + Qx + NaPyr | 30–40 hours |
| Fadu | EC | Salvador Aznar-Benitah’s lab (IRB) | MEM + 10%FBS + P/S + PyrNa | 30 hours |
Lentivirus production and infection
Lentiviruses were produced as described previously (43). Human embryonic kidney (HEK) 293 T packaging cells were transfected using the calcium phosphate transfection method with 5 μg of pCMV-VSV-G, 6 μg of pCMVDR-8.91, and 7 μg of the pLKO-shRNA (Sigma-Aldrich) plasmid, together with either TRC2 MISSION pLKO.5 harboring nonmammalian shRNA control or shRNAs against three different regions of the Ahcy transcript (#1, GAGCAAATGTCACCAACTTTG; #2, CGGTGGAGAAAGTGAACATCA; #3, GTGGACCCACCCAGATAAATA). Cells were incubated with the transfection mix for 12 to 16 hours, after which the medium was replaced by ESC (LIF-free) fresh medium. After 48 hours, lentiviral particles were collected, filtered with a 0.45-μm filter, and stored at −80°C for further use. Lentiviral particles were prepared according to the Spanish National Biosafety Commission guidelines and the relevant European directives.
Western blotting
Samples were analyzed by Western blot, as described previously (44). The antibodies used are listed in Table 2.
Table 2. List of antibodies used in this study.
DB, dot blot; IF, immunofluorescence; WB, Western blot; HRP, horseradish peroxidase.
| Host | Source | Catalog no. | Use | Lot number | Dilution | |
| Primary antibody | ||||||
| NANOG | Rat | eBioscience | eBioMLC-51 | IF | E04352-1633 | 1:200 |
| γ-H2AX | Mouse | Abcam | ab22551 | WB/IF | – | 1:1000 |
| β-Actin | Mouse | Abcam | ab8226 | WB | 159477 | 1:10,000 |
| Vinculin | Mouse | Sigma | V9131 | WB/DB | 018M4779V | 1:10,000 |
| AHCY | Rabbit | MBL | RN126PW | WB/IF/ChIP | 001 | 1:1000–500 |
| RPA32 | Rat | Cell Signaling | 2208 | IF | — | 1:250 |
| H3 | Rabbit | Abcam | ab1791 | WB/DB | 825444 | 1:5000–10,000 |
| H3K9me3 | Rabbit | Abcam | ab8898 | WB | GR22415-1 | 1:2000 |
| IgG | Rabbit | Abcam | ab172730 | ChIP | GR262233-9 | 5 μg |
| Secondary antibody | ||||||
| HRP anti-mouse IgG | Goat | Dako | A-4416 | WB | 20030273 | 1:2000 |
| HRP anti-rabbit IgG | Goat | Dako | A-6667 | WB | 20042622 | 1:2000 |
| Anti-rat Alexa 488 | Donkey | Molecular Probes | A-21208 | IF | 1810471 | 1:500 |
| Anti-rabbit Alexa 555 | Donkey | Molecular Probes | A-21206 | IF | 1837922 | 1:500 |
Fluorescence-activated cell sorting
Cell cycle analysis
Cells were dissociated to single cells with trypsin (Gibco) and fixed with ethanol. DNA was stained with propidium iodide (Sigma-Aldrich). Cells were analyzed by FACS using FACScalibur (Becton Dickinson), and data were analyzed using FlowJo v10. The number of single cells collected for analysis was 10,000.
Protein synthesis
Cells were dissociated to single cells with trypsin, fixed with 4% paraformaldehyde (PFA), and analyzed by Click reaction [100 mM tris-HCl (pH 8), 2 mM CuSO4, 2 μM Alexa Fluor 647 azide (Invitrogen), and 100 mM ascorbic acid] for 30 min at room temperature (RT). Cells were analyzed by FACS using LSRII (Becton Dickinson), and data were analyzed using FlowJo v10. The number of single cells collected for analysis was between 2000 and 9500.
Chromatin immunoprecipitation–quantitative polymerase chain reaction
ESCs were cross-linked in 1% PFA for 10 min at RT in a shaker. The fixation reaction was stopped by adding a final concentration of 0.125 M glycine (pH 7.4) to the culture and incubating for 5 min. Cells were washed twice with 1× phosphate-buffered saline (PBS) at RT and harvested in ice-cold 1× PBS plus protease inhibitor cocktail (PIC; Roche). Pellets were resuspended in 1.3 ml of ice-cold IP buffer, 1× volume SDS buffer [100 mM NaCl, 50 mM tris-HCl (pH 8), 5 mM EDTA (pH 8), and 0.5% SDS], and 0.5 volume Triton dilution buffer [100 mM NaCl, 100 mM tris-HCl (pH 8.6), 5 mM EDTA (pH 8), and 5% Triton X-100] with PIC (Roche). Samples were sonicated for 3 × 10 min (30 s on/30 s off) in a Bioruptor (Diagenode) at maximum output. After sonication, samples were centrifuged at 4°C at maximum speed for 15 min. To check the yield of chromatin shearing, 1 to 5% of input material was reverse cross-linked for 3 hours at 65°C in a shaker (1000 rpm) in high-salt buffer (1× PBS, 500 mM NaCl) plus 0.2 μg of proteinase K (Invitrogen), followed by PCR purification using a kit (Qiagen). A total of 50 μg of sheared chromatin of 200 to 500 bp was immunoprecipitated with 5 μg of antibody (anti-AHCY or rabbit IgG, as control) to a final volume of 500 μl. ChIP reactions were incubated overnight at 4°C on rotation. The next day, 30 μl of blocked protein A agarose beads [0.05% bovine serum albumin (BSA) on IP buffer] was added to the ChIP reactions and incubated for 2 hours at 4°C. After incubation, beads were washed three times with 1 ml of salt buffer [140 mM NaCl, 50 mM Hepes (pH 7.5), and 1% Triton X-100] and once with 1 ml of high-salt buffer [500 mM NaCl, 50 mM Hepes, (pH 7.5), and 1% Triton X-100]. ChIP samples were eluted using 200 μl of freshly prepared elution buffer (1% SDS and 100 mM NaHCO3) and reverse cross-linked at 65°C overnight with shaking (1000 rpm). DNA was purified using a PCR purification kit (Qiagen). DNA was eluted with 100 μl of water.
Real-time PCR reactions were performed using SYBR Green I PCR Master Mix (Roche) and the Roche LightCycler 480. The oligonucleotides used in this study are given in Table 3.
Table 3. List of primers used in this study.
| Oligonucleotides (5′ to 3′) | |
| Map2k5_F | GCAGGATCGTCCGACTGAG |
| Map2k5_R | TTGGTTCCGGAGTAACAGCG |
| Rbl1_F | GGACCTCGGACTCTAACGGA |
| Rbl1_R | TCTCTCACGTCCGTCCCC |
| Hdac2_F | CGTAAGACCGAGGGGTGAAC |
| Hdac2_R | GGGTAGTCACACACAGTCCG |
| Rps20_F | GGGCGTCTTTCCGGTATCTTT |
| Rps20_R | CCACTTACGGGTCGCTGTTT |
| Rpl18_F | TCGGTCCTCCCACATACCAT |
| Rpl18_R | CTATCCGGAACTGGCGTCTC |
| Rpl4_F | TAGAGGAGGGCGAAGGTTCA |
| Rpl4_R | GGCCAGAAAAGGTGCTAGGT |
| Rpl17_F | GCTCGGCTTCTACGGTGAG |
| Rpl17_R | CAGGCTTAGGCACAGAGCAG |
| Spink2_F | CATATCCCGACGCATAGCCT |
| Spink2_R | TGGGGTGGAGAGGTAGCTTG |
| ChIP_neg_ctrl_F | CAGTCACCCAGGAAAGGCAA |
| ChIP_neg_ctrl_R | AAGGTAGGGCAAGAAGTGCC |
Chromatin immunoprecipitation sequencing
Chromatin material for ChIP-seq was prepared using the ChIP-IT High Sensitivity Kit from Active Motif, following the manufacturer’s instruction. ChIP-seq libraries were prepared with 2 to 10 ng of ChIP DNA material using the NEBNext Ultra DNA library Prep Kit for Illumina (New England Biolabs) following the manufacturer’s instruction. ChIP-seq libraries were amplified for 10 to 15 PCR cycles. Libraries were sequenced using a HiSeq 2500 sequencer (Illumina). Table 4 provides sequencing depth data.
Table 4. List of sequencing runs.
| Sample | Total reads |
Mapped reads |
Single/ Paired |
Read length |
|
GSM3262022 ChIPseq_Ahcy_Replicate1 |
117373523 | 77231344 | S | 50 |
|
GSM3262023 ChIPseq_Ahcy_Replicate2 |
65127303 | 49321533 | S | 50 |
|
GSM3262024 ChIPseq_IgG |
128209536 | 86504098 | S | 50 |
|
GSM3262025 RNAseq_shC_R1_shControl |
73466873 | 70314234 | S | 50 |
|
GSM3262026 RNAseq_shC_R2_shControl |
71083014 | 68659269 | S | 50 |
|
GSM3262027 RNAseq_sh4_R1_shAhcy |
71751488 | 68688907 | S | 50 |
|
GSM3262028 RNAseq_sh4_R2_shAhcy |
79931158 | 76726955 | S | 50 |
|
GSM3262029 RNAseq_sh5_R1_shAhcy |
71178447 | 68065093 | S | 50 |
|
GSM3262030 RNAseq_sh6_R1_shAhcy |
72046464 | 68723751 | S | 50 |
|
GSM3262031 RNAseq_sh6_R2_shAhcy |
72091988 | 69578603 | S | 50 |
|
GSM3262032 RNAseq_EdU_minus_R1 |
30894604 | 29693657 | S | 50 |
|
GSM3262033 RNAseq_EdU_minus_R2 |
47121290 | 45649660 | S | 50 |
|
GSM3262034 RNAseq_EdU_plus_R1 |
40075734 | 38383847 | S | 50 |
RNA extraction and library preparation
RNA extraction was performed using the RNeasy Mini Kit (Qiagen) following the manufacturer’s instruction. RNA samples were quantified, and quality was evaluated using a Bioanalyzer (RIN > 9.9). A total of 0.5 μg of RNA was used as starting material for library preparation with rRNA (ribosomal RNA) depletion using the TruSeq Stranded Total RNA Library Prep Kit (Ilumina) following the manufacturer’s instruction. Libraries were sequenced using a HiSeq2500 sequencer (50 bp, single end) with HiSeq v4 chemistry. Sequencing depth details are provided in Table 4.
Dot blot analysis
Samples were spotted in 1-μl dots onto a nitrocellulose membrane (Protan, 0.2 μM, Amersham) in triplicate, air-dried, and subjected to standard blotting procedures with the indicated antibodies.
Embryo collection and culture
All animal procedures were performed in accordance with Spanish and Catalan laws and overseen by the Institutional Animal Care and Use Ethics Committee of the Barcelona Biomedical Research Park. Embryos were collected from 5- to 8-week-old (C57BL/6J) superovulated females crossed with males (B6D2F1/J). Embryos were collected at 98 hours after human chorionic gonadotrophin injection (phCG) at the early blastocyst stage. Flushed embryos were cultured in M16 (SIGMA) drops under oil at 37°C and 5% CO2 in the presence of dimethyl sulfoxide (DMSO) or 3-DZA (200 nM; Cayman Chemical) for 18 hours. Embryos were then incubated for an additional hour with 50 μM L-HPG, at which point embryos were fixed in 4% paraformaldehyde for 10 min.
Immunofluorescence
Cells or flushed mouse embryos (CD1 strain, E4.5) were fixed in 4% PFA for 10 min, incubated for 30 min in blocking buffer [1% BSA, 10% fetal bovine serum (FBS), and 0.1% Triton X-100 in PBS], and then stained with primary antibodies for 2 hours at RT in blocking buffer with 5% FBS. Samples were washed three times for 5 min at RT with blocking buffer and incubated with conjugated secondary antibodies for 1 hour at RT. Where indicated, a Click reaction [100 mM tris-HCl (pH 8.0), 2 mM CuSO4, 2 μM Alexa Fluor 647 azide (Invitrogen), and 100 mM ascorbic acid] was performed for 30 min at RT. Samples were analyzed using a Leica TCS SPE inverted confocal microscope, and optical sections were captured. For quantification of signal intensity, the sum of the complete Z stack projection image was generated and quantified using ImageJ processing software (http://rsb.info.nih.gov/ij).
Dm-ChP–mass spectrometry
Sample processing
Eluted proteins were reduced, alkylated, and digested to peptide mixes according to the filter-aided sample preparation (45) method using LysC [1:10 (w/w), enzyme/substrate] at 37°C overnight followed by trypsin [1:10 (w/w), enzyme/substrate] at 37°C for 8 hours. Tryptic peptide mixtures were desalted using a C18 UltraMicroSpin column (46).
LC-MS analysis
Samples were analyzed in an LTQ-Orbitrap Velos Pro mass spectrometer (Thermo Fisher Scientific) coupled to nano-LC (Proxeon) equipped with a reversed-phase chromatography 25-cm column with an inner diameter of 75 μm, packed with 1.9-μm C18 particles (Nikkyo Technos Co.). Chromatographic gradients changed from 97% buffer A, 3% buffer B to 65% buffer A, 35% buffer B over 120 min at a flow rate of 250 nl/min, in which buffer A was 0.1% formic acid in water and buffer B was 0.1% formic acid in acetonitrile. The instrument was operated in DDA mode, and full MS scans with one microscan at a resolution of 60,000 were used over a mass range of mass/charge ratio (m/z) 350 to 2000 with detection in the Orbitrap. Following each survey scan, the top 20 most intense ions with multiple charged ions above a threshold ion count of 5000 were selected for fragmentation at a normalized collision energy of 35%. Fragment ion spectra produced via collision-induced dissociation were acquired in the linear ion trap. All data were acquired with Xcalibur software v2.2.
Data analysis
Acquired data were analyzed using the Proteome Discoverer software suite (v1.4, Thermo Fisher Scientific) and Mascot search engine (v2.5). Data were searched against SwissProt mouse for the mouse cell lines and against SwissProt human for cancer tissue samples; in both cases, the most common contaminants were added (47). A precursor ion mass tolerance of 7 parts per million (ppm) at the MS1 level was used, and up to three miscleavages for trypsin were allowed. The fragment ion mass tolerance was set to 0.5 Da. Oxidation of methionine and protein acetylation at the N terminus were defined as variable modification. Carbamidomethylation on cysteines was set as a fixed modification. The identified peptides were filtered using false discovery rate (FDR) < 5%.
Interactome analysis
Analysis of specific chromatin interactors was carried out with SAINT, as previously described (17) and using a threshold corresponding to FDR < 1%. Protein interaction data were retrieved from the STRING database (48). Only experimentally validated protein-protein interactions, curated databases, and text-mining information were considered. Functionally related protein networks were visualized using Cytoscape (v3.6.1) (49).
Shotgun mass spectrometry
Sample processing
Cells were lysed for 30 min in urea lysis buffer (6 M urea and 0.2 M ammonium bicarbonate) at RT. Then, samples were sonicated (Bioruptor, Diagenode) for 10 min at high intensity (30 s on/30 s off pulses) and clarified by centrifugation, and the protein concentration was quantified with a BCA Protein Assay Kit (Thermo Fisher Scientific). Equal amounts of protein sample were analyzed by MS, as follows. Samples were reduced with DTT (10 mM, 1 hour, 37°C), alkylated in the dark with iodoacetamide (20 mM, 30 min, 25 C), and digested. The resulting protein extract was first diluted 1:3 with 200 mM NH4HCO3 and digested using LysC [1:10 (w/w), enzyme/substrate] at 37°C overnight followed by trypsin [1:10 (w/w), enzyme/substrate] at 37°C for 8 hours. Tryptic peptide mixtures were desalted using a C18 UltraMicroSpin column.
LC-MS analysis
Peptide mixes were analyzed using an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific, San Jose, CA, USA) coupled to an EasyLC [Thermo Scientific (Proxeon), Odense, Denmark]. Peptides were loaded directly onto the analytical column and were separated by reversed-phase chromatography using a 50-cm column with an inner diameter of 75 μm, packed with 2-μm C18 particles spectrometer (Thermo Scientific, San Jose, CA, USA). Chromatographic gradients started at 95% buffer A and 5% buffer B with a flow rate of 300 nl/min and gradually increased to 22% buffer B in 79 min and then to 35% buffer B in 11 min. After each analysis, the column was washed for 10 min with 5% buffer A and 95% buffer B. Buffer A was 0.1% formic acid in water and buffer B was 0.1% formic acid in acetonitrile.
The mass spectrometer was operated in DDA mode, and full MS scans with one microscan at a resolution of 120,000 were used over a mass range of m/z 350 to 1500 with detection in the Orbitrap. Auto gain control was set to 2E5 and dynamic exclusion was set to 60 s. In each cycle of DDA analysis following each survey scan, top-speed ions charged 2 to 7 above a threshold ion count of 1e4 were selected for fragmentation at a normalized collision energy of 28%. Fragment ion spectra produced via high-energy collision dissociation were acquired in the ion trap. All data were acquired with Xcalibur software v3.0.63.
Data analysis
Proteome Discoverer software suite (v2.2, Thermo Fisher Scientific) and Mascot search engine [v2.5, Matrix Science (2)] were used for peptide identification and quantification. The data were searched against an in-house generated database containing all proteins corresponding to mouse in the SwissProt database plus a list of common contaminants and all the corresponding decoy entries (released, April 2018). A precursor ion mass tolerance of 7 ppm at the MS1 level was used, and up to three missed cleavages for trypsin were allowed. The fragment ion mass tolerance was set to 0.5 Da. Oxidation of methionine and protein acetylation at the protein N-terminus were defined as variable modification, whereas carbamidomethylation on cysteines was set as a fixed modification. Identified peptides were filtered using a 5% FDR. Protein abundance was estimated using the area under the chromatographic peak of the three most intense peptides per protein. Data were log-transformed, and fold changes, P values, and q values were calculated to assess protein relative quantification.
DNA-mediated chromatin pulldown
Cells were pulsed for 20 hours (for ESCs) or 48 hours (for human cancer cell lines) with 0.1 μM of the thymidine analog, EdU (Invitrogen). Subsequently, the cells were fixed in 1% PFA for 10 min at RT and quenched with 0.125 mM glycine (pH 7) for 5 min at RT. Cells were harvested, pelleted by centrifugation (720g, 10 min at 4°C), and lysed for 30 min at 4°C in lysis buffer A [10 mM Hepes (pH 7.9), 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% (v/v) glycerol, 1 mM DTT, 10 mM β-glycerol phosphate, 1 mM sodium orthovanadate, PIC (Roche), and 0.1% (v/v) Triton X-100]. Nuclei were pelleted by centrifugation (1300g, 4 min at 4°C), washed with PBS + PIC, and subjected to Click reaction for 30 min at RT with 0.2 mM biotin-azide (Invitrogen). In Click, an organic azide reacts with a terminal acetylene, and the nucleotide-exposed ethynyl residue of EdU is derivatized by a copper-catalyzed cycloaddition reaction, to form a covalent bond between EdU and biotin. Nuclei were repelleted by centrifugation (1300g, 4 min at 4°C), washed with PBS + PIC, and suspended in shearing buffer [20 mM tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 0.1% (w/v) SDS, 0.5% (w/v) sodium deoxycolate, 1% (v/v) Triton X-100, 10 mM β-glycerol phosphate, 1 mM sodium orthovanadate, and PIC]. Nuclei suspension was extensively sonicated (Bioruptor, Diagenode) for four to six cycles of 10 min at high intensity (30 s on/30 s off pulses). Lysates were centrifuged (20,800g, 20 min at 4°C), and supernatant was collected as input for further analysis. To analyze shearing efficiency, 5% of input material was reverse cross-linked by incubation overnight at 65°C with 250 mM NaCl and then digested with proteinase K (0.1 mg/ml) for 1 hour at 55°C. DNA was purified using the PCR purification kit from Qiagen, following the manufacturer’s instruction. For chromatin capture, input material was diluted 1:4 with blocking buffer [1% Triton X-100, 2 mM EDTA (pH 8), 150 mM NaCl, 20 mM tris-HCl (pH 8), 20 mM β-glycerol phosphate, 2 mM sodium orthovanadate, PIC, and salmon sperm DNA (10 mg/ml)] and then incubated with preblocked Dynabead M-280 streptavidin (Invitrogen) for 30 min at 4°C. Beads were washed twice with blocking buffer (without salmon sperm DNA), twice with high-salt blocking buffer (containing 500 mM NaCl), and once with tris-EDTA buffer (pH 8). Beads were suspended in modified Laemmli buffer [2% (v/v) SDS, 0.06 M tris-HCl (pH 6.5), and 0.1 M DTT] for 20 min at 95°C, and the supernatant was collected for analysis by dot blot, Western blot, or MS. For MS analysis of Dm-ChP samples, 30 × 106 to 40 × 106 ESCs were lysed in 5 ml of lysis buffer A, incubated in 1 ml of Click reaction buffer, sheared in 4 ml of shearing buffer, incubated with 0.5 ml of Dynabeads M-280 streptavidin, and resuspended in 150 ml of modified Laemmli buffer.
Bioinformatic analysis
Analysis of ChIP-seq data
ChIP-seq samples were mapped against the mm9 mouse genome assembly using BowTie with the option –m 1, to discard reads that could not be uniquely mapped to just one region (50). MACS (Model-based analysis of ChIP-Seq) was run with the default parameters, but the shift size was adjusted to 100 bp to perform the peak calling against the corresponding control sample (51). Peaks from two replicates were merged, and those with a fold-enrichment score above 4.50 were selected as confident AHCY binding sites. Genome distribution of each set of peaks was calculated by counting the number of peaks fitting on each class of region according to RefSeq annotations. Distal region is the region 2.5 kilo–base pairs (kbp) to 0.5 kbp upstream of the TSS; proximal region, from 0.5 kbp until the TSS; UTR, untranslated sequence; CDS, protein coding sequence; introns, intronic regions; and intergenic, the nonintronic regions of the genome. Peaks that overlapped with more than one genomic feature were proportionally counted the same number of times. To generate spie charts, the genome distribution of all features in the full genome was first calculated and then the R-caroline package was used to combine the spie chart of each set of peaks with the full genome distribution (52). Each set of target genes was retrieved by matching the ChIP-seq peaks in the region 2.5 kbp upstream of the TSS until the end of the transcripts as annotated in RefSeq. Heat maps displaying the density of ChIP-seq reads 5 kb around the TSS of each target gene set were generated by counting the number of reads in this region for each individual gene and normalizing this value with the total number of mapped reads of the sample. Genes on each ChIP heat map were ranked by the logarithm of the averaged number of reads on the same genomic region. The University of California Santa Cruz (UCSC) genome browser was used to generate the screenshots shown for each group of experiments (53).
GO term enrichment was performed using the DAVID (Database for Annotation, Visualization and Integrated Discovery) database with the total set of proteins against the entire mouse genome as the background (54). The Enrichr tool was used to analyze pathway enrichment (55). The Mouse Genome Informatics tool was used to retrieve the mammalian loss-of-function phenotype (www.informatics.jax.org) (56). The human disorders associated with selected genes were retrieved using the BioMart tool from Ensembl (57). Protein interaction data were retrieved from the STRING database (48) using the protein mode. Only interactions from protein-protein interaction databases, curated databases, and text-mining information were considered. The network was visualized using Cytoscape (v3.6.0) (49). Genomic co-occupancy was analyzed using the BindDB online tool (http://bind-db.huji.ac.il/). Published datasets used for co-occupancy analysis and histone mark level analysis are shown in Table 5.
Table 5. List of available datasets used in this study.
| Histone mark/DNA modification/protein | Sample accession number |
| H3K4me3 | GSM590112 |
| H3K27me3 | GSM590116 |
| H3K36me3 | GSM590120 |
| H3K27ac | GSM1355158 |
| 5mC | GSM1053460 |
| Myc | GSM286124 |
| Trim28 | GSM308532 |
| cMyc | GSM288356 |
| Dmap1 | GSM516405 |
| Cnot3 | GSM308533 |
| nMyc | GSM288357 |
| Tip60 | GSM516406 |
| Gcn5 | GSM516407 |
| cMyc | GSM286124 |
| E2f4 | GSM516408 |
| Max | GSM516404 |
| Rex1 | GSM286125 |
| Zfx | GSM288352 |
| Zfp281 | GSM286123 |
| Sox2 | GSM286121 |
| Klf4 | GSM286122 |
| Klf4 | GSM288354 |
| Dax1 | GSM286119 |
| Stat3 | GSM686673 |
| Dax1 | GSM286119 |
| Stat3 | GSM288353 |
| Nac1 | GSM286120 |
| Sox2 | GSM288347 |
| Nanog | GSM288345 |
| Nanog | GSM1090230 |
| Ring1b | GSM656523 |
| Ring1b | GSM1003596 |
| Suz12 | GSM1019771 |
| Suz12 | GSM288360 |
| Ezh2 | GSM480161 |
| Ezh2 | GSM475259 |
| Ring1b | GSM585229 |
| Ezh1 | GSM386202 |
| Suz12 | GSM480162 |
Analysis of RNA-seq data
RNA-seq samples were mapped against the mm9 mouse genome assembly using TopHat (58) with the option --g 1 to discard reads that could not be uniquely mapped to just one region. Cufflinks and Cuffdiff were run to quantify the expression in RPKMs of each annotated transcript in RefSeq and to identify the list of differentially expressed genes on each case (59). Qlucore Omics Explorer 3.4 software (Qlucore, Lund, Sweden) was used to perform GSEA with predefined gene sets, and GSEA software (www.broadinstitute.org/gsea/msigdb/) was used to perform GSEA with custom-defined gene sets (60, 61).
Accession numbers
Raw data and processed information of the ChIP-seq and RNA-seq experiments generated in this article were deposited in the National Center for Biotechnology Information Gene Expression Omnibus repository under the accession number GSE116799. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (62) partner repository with the dataset identifier PXD011670.
Supplementary Material
Acknowledgments
We thank P. Vizán and members of the Di Croce laboratory for critical reading of the manuscript and insightful discussions, V.A. Raker for scientific editing, S. Nakagawa for advice on mouse embryo manipulation, and the CRG Genomics Unit, CRG/UPF Flow Cytometry Unit, and the CRG Advanced Light Microscopy Unit for assistance with sequencing, FACS, and microscopy services, respectively. Funding: We acknowledge support from the Spanish Ministry of Economy, Industry and Competitiveness to the EMBL partnership, Centro de Excelencia Severo Ochoa, the CERCA Programme/Generalitat de Catalunya, the Secretary for Universities and Research of the Ministry of Economy and Knowledge of the Government of Catalonia (to S.A. and A.A.-C.), and the Lady Tata Memorial Trust (to S.A.). The CRG/UPF Proteomics Unit is a member of the ProteoRed PRB3 consortium that was supported by grant PT17/0019 of the PE I+D+i 2013–2016 from the Instituto de Salud Carlos III (ISCIII), ERDF, and “Secretaria d’Universitats i Recerca del Departament d’Economia i Coneixement de la Generalitat de Catalunya” (2017SGR595). The Di Croce Laboratory was supported by grants from the Spanish Ministerio de Educación y Ciencia (BFU2016-75008-P), AGAUR, and La Marato TV3. Author contributions: S.A. conceived and planned this project; collected, analyzed, and interpreted data; and wrote the manuscript with input from coauthors. A.A.-C. collected, analyzed, and interpreted data. E.Bl. performed the bioinformatics analysis of deep sequencing data. E.Bo. and E.S. performed and analyzed proteomic data. C.C. conducted and analyzed data. L.D.C. conceived and planned this project. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The accession number for the ChIP-seq and RNA-seq data reported here is GSE116799. MS proteomics data were deposited in the ProteomeXchange Consortium via the PRIDE (62) partner repository with the dataset identifier PXD011670. All other data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/3/eaav2448/DC1
Fig. S1. Chromatin capture from pluripotent ESCs by Dm-ChP.
Fig. S2. Culture conditions for Dm-ChP are compatible with normal physiology.
Fig. S3. Biological and pathologically relevant chromatin-associated proteins in ESCs.
Fig. S4. Expression of key developmental genes during early mammalian development.
Fig. S5. AHCY is recruited to the TSS of transcriptionally active genes.
Fig. S6. Expression of pluripotency genes upon Ahcy knockdown.
Fig. S7. Proteomic analysis of ribosomal protein encoded by AHCY targets.
Fig. S8. Inhibition of AHCY reduces rate of protein synthesis.
Fig. S9. Gating strategy for cell cycle and HPG incorporation analysis by FACS.
Table S1. Principal methods developed to identify protein components of the chromatin.
Table S2. Proteomic data.
Table S3. ChIP-seq data.
Table S4. RNA-seq data.
REFERENCES AND NOTES
- 1.Erdel F., Rippe K., Formation of chromatin subcompartments by phase separation. Biophys. J. 114, 2262–2270 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stunnenberg H. G.; International Human Epigenome Consortium, Hirst M., The International Human Epigenome Consortium: A blueprint for scientific collaboration and discovery. Cell 167, 1145–1149 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Skipper M., Eccleston A., Gray N., Heemels T., Le Bot N., Marte B., Weiss U., Presenting the epigenome roadmap. Nature 518, 313 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Wierer M., Mann M., Proteomics to study DNA-bound and chromatin-associated gene regulatory complexes. Hum. Mol. Genet. 25, R106–R114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sirbu B. M., Couch F. B., Feigerle J. T., Bhaskara S., Hiebert S. W., Cortez D., Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 25, 1320–1327 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kliszczak A. E., Rainey M. D., Harhen B., Boisvert F. M., Santocanale C., DNA mediated chromatin pull-down for the study of chromatin replication. Sci. Rep. 1, 95 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopez-Contreras A. J., Ruppen I., Nieto-Soler M., Murga M., Rodriguez-Acebes S., Remeseiro S., Rodrigo-Perez S., Rojas A. M., Mendez J., Muñoz J., Fernandez-Capetillo O., A proteomic characterization of factors enriched at nascent DNA molecules. Cell Rep. 3, 1105–1116 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aranda S., Rutishauser D., Ernfors P., Identification of a large protein network involved in epigenetic transmission in replicating DNA of embryonic stem cells. Nucleic Acids Res. 42, 6972–6986 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diermeier-Daucher S., Clarke S. T., Hill D., Vollmann-Zwerenz A., Bradford J. A., Brockhoff G., Cell type specific applicability of 5-ethynyl-2′-deoxyuridine (EdU) for dynamic proliferation assessment in flow cytometry. Cytometry A 75, 535–546 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Qu D., Wang G., Wang Z., Zhou L., Chi W., Cong S., Ren X., Liang P., Zhang B., 5-Ethynyl-2′-deoxycytidine as a new agent for DNA labeling: Detection of proliferating cells. Anal. Biochem. 417, 112–121 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Neef A. B., Luedtke N. W., Dynamic metabolic labeling of DNA in vivo with arabinosyl nucleosides. Proc. Natl. Acad. Sci. U.S.A. 108, 20404–20409 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahuja A. K., Jodkowska K., Teloni F., Bizard A. H., Zellweger R., Herrador R., Ortega S., Hickson I. D., Altmeyer M., Mendez J., Lopes M., A short G1 phase imposes constitutive replication stress and fork remodelling in mouse embryonic stem cells. Nat. Commun. 7, 10660 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orkin S. H., Hochedlinger K., Chromatin connections to pluripotency and cellular reprogramming. Cell 145, 835–850 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rafiee M.-R., Girardot C., Sigismondo G., Krijgsveld J., Expanding the circuitry of pluripotency by selective isolation of chromatin-associated proteins. Mol. Cell 64, 624–635 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ji X., Dadon D. B., Abraham B. J., Lee T. I., Jaenisch R., Bradner J. E., Young R. A., Chromatin proteomic profiling reveals novel proteins associated with histone-marked genomic regions. Proc. Natl. Acad. Sci. U.S.A. 112, 3841–3846 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Christoforou A., Mulvey C. M., Breckels L. M., Geladaki A., Hurrell T., Hayward P. C., Naake T., Gatto L., Viner R., Arias A. M., Lilley K. S., A draft map of the mouse pluripotent stem cell spatial proteome. Nat. Commun. 7, 8992 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi H., Larsen B., Lin Z.-Y., Breitkreutz A., Mellacheruvu D., Fermin D., Qin Z. S., Tyers M., Gingras A.-C., Nesvizhskii A. I., SAINT: Probabilistic scoring of affinity purification–mass spectrometry data. Nat. Methods 8, 70–73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aranda S., Mas G., Di Croce L., Regulation of gene transcription by Polycomb proteins. Sci. Adv. 1, e1500737 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schuettengruber B., Chourrout D., Vervoort M., Leblanc B., Cavalli G., Genome regulation by polycomb and trithorax proteins. Cell 128, 735–745 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Kadoch C., Crabtree G. R., Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci. Adv. 1, e1500447 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCann T. S., Tansey W. P., Functions of the proteasome on chromatin. Biomolecules 4, 1026–1044 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brogna S., Sato T.-A., Rosbash M., Ribosome components are associated with sites of transcription. Mol. Cell 10, 93–104 (2002). [PubMed] [Google Scholar]
- 23.Al-Jubran K., Wen J., Abdullahi A., Chaudhury S. R., Li M., Ramanathan P., Matina A., De S., Piechocki K., Rugjee K. N., Brogna S., Visualization of the joining of ribosomal subunits reveals the presence of 80S ribosomes in the nucleus. RNA 19, 1669–1683 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thompson P. J., Dulberg V., Moon K.-M., Foster L. J., Chen C., Karimi M. M., Lorincz M. C., hnRNP K coordinates transcriptional silencing by SETDB1 in embryonic stem cells. PLOS Genet. 11, e1004933 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kusakabe Y., Ishihara M., Umeda T., Kuroda D., Nakanishi M., Kitade Y., Gouda H., Nakamura K. T., Tanaka N., Structural insights into the reaction mechanism of S-adenosyl-L-homocysteine hydrolase. Sci. Rep. 5, 16641 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turner M. A., Yuan C.-S., Borchardt R. T., Hershfield M. S., Smith G. D., Howell P. L., Structure determination of selenomethionyl S-adenosylhomocysteine hydrolase using data at a single wavelength. Nat. Struct. Biol. 5, 369–376 (1998). [DOI] [PubMed] [Google Scholar]
- 27.Miller M. W., Duhl D. M., Winkes B. M., Arredondo-Vega F., Saxon P. J., Wolff G. L., Epstein C. J., Hershfield M. S., Barsh G. S., The mouse lethal nonagouti (a(x)) mutation deletes the S-adenosylhomocysteine hydrolase (Ahcy) gene. EMBO J. 13, 1806–1816 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tehlivets O., Malanovic N., Visram M., Pavkov-Keller T., Keller W., S-adenosyl-L-homocysteine hydrolase and methylation disorders: Yeast as a model system. Biochim. Biophys. Acta 1832, 204–215 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barić I., Inherited disorders in the conversion of methionine to homocysteine. J. Inherit. Metab. Dis. 32, 459–471 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Mita T., Kawazu I., Hirano H., Ohmori O., Janjua N., Shibata K., E1 mice epilepsy shows genetic polymorphism for S-Adenosyl-L-homocysteine hydrolase. Neurochem. Int. 38, 349–357 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Leal J. F., Ferrer I., Blanco-Aparicio C., Hernández-Losa J., Ramón y Cajal S., Carnero A., LLeonart M. E., S-adenosylhomocysteine hydrolase downregulation contributes to tumorigenesis. Carcinogenesis 29, 2089–2095 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Herrmann W., Schorr H., Obeid R., Makowski J., Fowler B., Kuhlmann M. K., Disturbed homocysteine and methionine cycle intermediates S-adenosylhomocysteine and S-adenosylmethionine are related to degree of renal insufficiency in type 2 diabetes. Clin. Chem. 51, 891–897 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Deng Q., Ramsköld D., Reinius B., Sandberg R., Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 343, 193–196 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Kim J., Woo A. J., Chu J., Snow J. W., Fujiwara Y., Kim C. G., Cantor A. B., Orkin S. H., A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell 143, 313–324 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Belužić L., Grbeša I., Belužić R., Park J. H., Kong H. K., Kopjar N., Espadas G., Sabidó E., Lepur A., Rokić F., Jerić I., Brkljačić L., Vugrek O., Knock-down of AHCY and depletion of adenosine induces DNA damage and cell cycle arrest. Sci. Rep. 8, 14012 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chayka O., D'Acunto C. W., Middleton O., Arab M., Sala A., Identification and pharmacological inactivation of the MYCN gene network as a therapeutic strategy for neuroblastic tumor cells. J. Biol. Chem. 290, 2198–2212 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park S. J., Kong H. K., Kim Y. S., Lee Y. S., Park J. H., Inhibition of S-adenosylhomocysteine hydrolase decreases cell mobility and cell proliferation through cell cycle arrest. Am. J. Cancer Res. 5, 2127–2138 (2015). [PMC free article] [PubMed] [Google Scholar]
- 38.Fernandez-Sanchez M. E., Gonatopoulos-Pournatzis T., Preston G., Lawlor M. A., Cowling V. H., S-adenosyl homocysteine hydrolase is required for Myc-induced mRNA cap methylation, protein synthesis, and cell proliferation. Mol. Cell. Biol. 29, 6182–6191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li X., Egervari G., Wang Y., Berger S. L., Lu Z., Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat. Rev. Mol. Cell Biol. 19, 563–578 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li X., Yu W., Qian X., Xia Y., Zheng Y., Lee J.-H., Li W., Lyu J., Rao G., Zhang X., Qian C.-N., Rozen S. G., Jiang T., Lu Z., Nucleus-translocated ACSS2 promotes gene transcription for lysosomal biogenesis and autophagy. Mol. Cell 66, 684–697.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mews P., Donahue G., Drake A. M., Luczak V., Abel T., Berger S. L., Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 546, 381–386 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boukouris A. E., Zervopoulos S. D., Michelakis E. D., Metabolic enzymes moonlighting in the nucleus: Metabolic regulation of gene transcription. Trends Biochem. Sci. 41, 712–730 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Beringer M., Pisano P., Di Carlo V., Blanco E., Chammas P., Vizán P., Gutiérrez A., Aranda S., Payer B., Wierer M., di Croce L., EPOP functionally links elongin and polycomb in pluripotent stem cells. Mol. Cell 64, 645–658 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Aranda S., Alvarez M., Turró S., Laguna A., de la Luna S., Sprouty2-mediated inhibition of fibroblast growth factor signaling is modulated by the protein kinase DYRK1A. Mol. Cell. Biol. 28, 5899–5911 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wiśniewski J. R., Zougman A., Nagaraj N., Mann M., Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Rappsilber J., Mann M., Ishihama Y., Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Bunkenborg J., Garciá G. E., Peña Paz M. I., Andersen J. S., Molina H., The minotaur proteome: Avoiding cross-species identifications deriving from bovine serum in cell culture models. Proteomics 10, 3040–3044 (2010). [DOI] [PubMed] [Google Scholar]
- 48.Szklarczyk D., Morris J. H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N. T., Roth A., Bork P., Jensen L. J., von Mering C., The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362–D368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T., Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Langmead B., Trapnell C., Pop M., Salzberg S. L., Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nussbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Feitelson D. G., Data on the distribution of cancer incidence and death across age and sex groups visualized using multilevel spie charts. J. Clin. Epidemiol. 72, 90–97 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Tyner C., Barber G. P., Casper J., Clawson H., Diekhans M., Eisenhart C., Fischer C. M., Gibson D., Gonzalez J. N., Guruvadoo L., Haeussler M., Heitner S., Hinrichs A. S., Karolchik D., Lee B. T., Lee C. M., Nejad P., Raney B. J., Rosenbloom K. R., Speir M. L., Villarreal C., Vivian J., Zweig A. S., Haussler D., Kuhn R. M., Kent W. J., The UCSC Genome Browser database: 2017 update. Nucleic Acids Res. 45, D626–D634 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang D. W., Sherman B. T., Lempicki R. A., Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 55.Kuleshov M. V., Jones M. R., Rouillard A. D., Fernandez N. F., Duan Q., Wang Z., Koplev S., Jenkins S. L., Jagodnik K. M., Lachmann A., McDermott M. G., Monteiro C. D., Gundersen G. W., Ma'ayan A., Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith C. L., Blake J. A., Kadin J. A., Richardson J. E., Bult C. J., the Mouse Genome Database Group , Mouse Genome Database (MGD)-2018: Knowledgebase for the laboratory mouse. Nucleic Acids Res. 46, D836–D842 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zerbino D. R., Achuthan P., Akanni W., Amode M. R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Girón C. G., Gil L., Gordon L., Haggerty L., Haskell E., Hourlier T., Izuogu O. G., Janacek S. H., Juettemann T., To J. K., Laird M. R., Lavidas I., Liu Z., Loveland J. E., Maurel T., McLaren W., Moore B., Mudge J., Murphy D. N., Newman V., Nuhn M., Ogeh D., Ong C. K., Parker A., Patricio M., Riat H. S., Schuilenburg H., Sheppard D., Sparrow H., Taylor K., Thormann A., Vullo A., Walts B., Zadissa A., Frankish A., Hunt S. E., Kostadima M., Langridge N., Martin F. J., Muffato M., Perry E., Ruffier M., Staines D. M., Trevanion S. J., Aken B. L., Cunningham F., Yates A., Flicek P., Ensembl 2018. Nucleic Acids Res. 46, D754–D761 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trapnell C., Pachter L., Salzberg S. L., TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roberts A., Trapnell C., Donaghey J., Rinn J. L., Pachter L., Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 12, R22 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P., Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mootha V. K., Lindgren C. M., Eriksson K.-F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstråle M., Laurila E., Houstis N., Daly M. J., Patterson N., Mesirov J. P., Golub T. R., Tamayo P., Spiegelman B., Lander E. S., Hirschhorn J. N., Altshuler D., Groop L. C., PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273 (2003). [DOI] [PubMed] [Google Scholar]
- 62.Vizcaino J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q.-W., Wang R., Hermjakob H., 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, 11033 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cheloufi S., Elling U., Hopfgartner B., Jung Y. L., Murn J., Ninova M., Hubmann M., Badeaux A. I., Euong Ang C., Tenen D., Wesche D. J., Abazova N., Hogue M., Tasdemir N., Brumbaugh J., Rathert P., Jude J., Ferrari F., Blanco A., Fellner M., Wenzel D., Zinner M., Vidal S. E., Bell O., Stadtfeld M., Chang H. Y., Almouzni G., Lowe S. W., Rinn J., Wernig M., Aravin A., Shi Y., Park P. J., Penninger J. M., Zuber J., Hochedlinger K., The histone chaperone CAF-1 safeguards somatic cell identity. Nature 528, 218–224 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gonzalez-Perez A., Jene-Sanz A., Lopez-Bigas N., The mutational landscape of chromatin regulatory factors across 4,623 tumor samples. Genome Biol. 14, r106 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shah M. A., Denton E. L., Arrowsmith C. H., Lupien M., Schapira M., A global assessment of cancer genomic alterations in epigenetic mechanisms. Epigenetics Chromatin 7, 29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/3/eaav2448/DC1
Fig. S1. Chromatin capture from pluripotent ESCs by Dm-ChP.
Fig. S2. Culture conditions for Dm-ChP are compatible with normal physiology.
Fig. S3. Biological and pathologically relevant chromatin-associated proteins in ESCs.
Fig. S4. Expression of key developmental genes during early mammalian development.
Fig. S5. AHCY is recruited to the TSS of transcriptionally active genes.
Fig. S6. Expression of pluripotency genes upon Ahcy knockdown.
Fig. S7. Proteomic analysis of ribosomal protein encoded by AHCY targets.
Fig. S8. Inhibition of AHCY reduces rate of protein synthesis.
Fig. S9. Gating strategy for cell cycle and HPG incorporation analysis by FACS.
Table S1. Principal methods developed to identify protein components of the chromatin.
Table S2. Proteomic data.
Table S3. ChIP-seq data.
Table S4. RNA-seq data.