

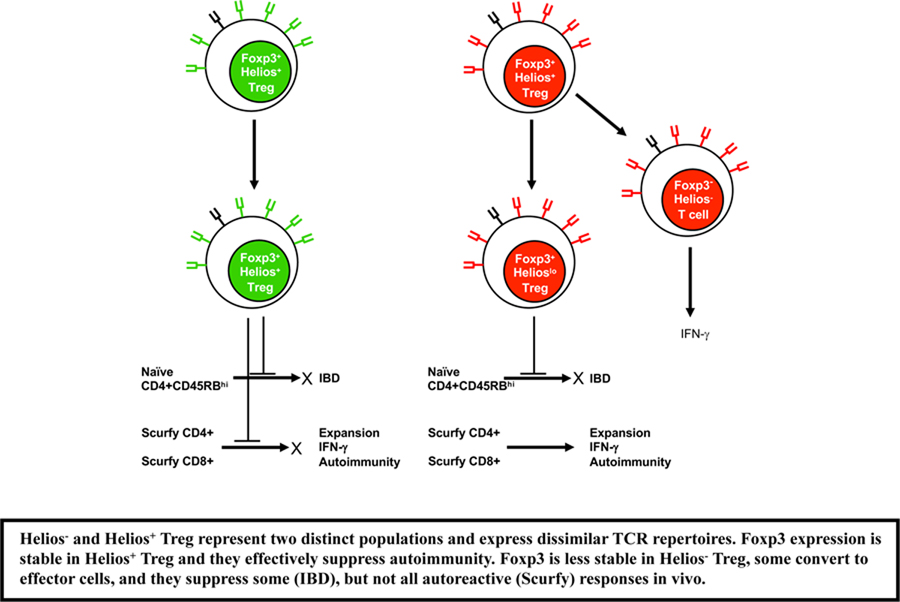
Abstract
The transcription factor Helios is expressed in a large subset of Foxp3+ Tregs. We previously proposed that Helios is a marker of thymic derived Treg (tTreg), while Helios− Treg were induced from Foxp3− T conventional (Tconv) cells in the periphery (pTreg). To compare the two Treg subpopulations, we generated Helios-GFP reporter mice and crossed them to Foxp3-RFP reporter mice. The Helios+ Treg population expressed a more activated phenotype, had a slightly higher suppressive capacity in vitro and expressed a more highly demethylated TSDR but were equivalent in their ability to suppress inflammatory bowel disease in vivo. However, Helios+ Treg more effectively inhibited the proliferation of activated, autoreactive splenocytes from scurfy mice. When Helios+ and Helios− Treg were transferred to lymphoreplete mice, both populations maintained comparable Foxp3 expression, but Foxp3 expression was less stable in Helios− Treg when transferred to lymphopenic mice. Gene expression profiling demonstrated a large number of differentially expressed genes and showed that Helios− Treg expressed certain genes normally expressed in CD4+Foxp3− T cells. TCR repertoire analysis indicated very little overlap between Helios+ and Helios− Treg. Thus, Helios+ and Helios− Treg subpopulations are phenotypically and functionally distinct and express dissimilar TCR repertoires.
Keywords: Animal models, Immune regulation, Regulatory T cells, Helios, TCR
Graphical Abstract
Introduction
CD4+Foxp3+ Regulatory T cells (Treg cells) suppress immune activation in a dominant manner and play a critical role in the maintenance of self-tolerance [1, 2]. Two major subsets of Treg cells have been defined in vivo; thymic-derived Treg cells (tTreg) develop in the thymus and peripherally induced Treg cells (pTreg) derived from conventional CD4+Foxp3− T cells that are converted in peripheral tissues to cells that express Foxp3 and acquire suppressive ability [3, 4]. Additionally, iTreg are conventional, naïve CD4+ T cells that can be induced in vitro to express Foxp3 and have suppressive functions both in vitro and in vivo. Although it has been demonstrated that Treg cells can be generated in vitro and in vivo in the periphery, the lack of a specific marker or markers has precluded an assessment of the relative numbers and percentages of pTreg vs tTreg and a direct comparison of their functional properties.
We previously demonstrated that Helios, a member of the Ikaros gene transcription factor family, is expressed in a subset of Foxp3+ Tregs in both mouse and man. Using an antibody to Helios, we characterized Helios expression in the thymus and in the periphery. We postulated that Helios is a marker of tTreg based on the findings that all Treg that arise from the thymus in the first weeks of life express Helios and, most importantly, that pTreg induced in vivo by oral tolerance lack Helios expression. [5]. A number of studies have supported this conclusion. Two groups have reported that the loss of the transcription factor Bach2 in Treg results in a fatal Th2 mediated inflammatory lung disease [6, 7]. Expression of Bach2 was required to suppress effector T cell programming and was also found to be essential for TGFβ induced Foxp3 expression and pTreg induction. Interestingly, Bach2 deficiency was associated with a specific loss of Helios− Treg. Second, Lathrop et al have shown that Treg from the colonic lamina propria of germ free mice, which lack pTreg, are almost exclusively Helios+ [8]. Third, Atarashi et. al demonstrated that the Clostridium induced Treg that accumulated in the colonic mucosa of germfree mice reconstituted with Clostridium, did not express Helios [9]. Furthermore, Luu et al. have shown that Treg deficient in the transcription factor c-rel lack thymic Treg, but accumulate pTreg in the periphery and are fully sufficient to generate iTreg in vitro. Interestingly, Helios+ Treg are specifically lacking in these mice and with age, only the Helios− Treg accumulate [10]. Lastly, mice with a T cell specific deficiency of Satb1 also lack thymic Treg, but develop Treg in the periphery and these pTreg do not express Helios [11]. On the other hand, several groups have concluded that Helios is not a marker of tTreg [12–14]. First, both Verhagen and Wraith and Gottschalk et al. have shown that iTreg generated in vitro, specifically with APC, express Helios. Gottschalk et al. further demonstrated that Helios was expressed in pTreg generated by low dose antigen administration in the absence of adjuvant. Lastly, Szurek et al. demonstrated that Aire deficient mice that lack some tissue specific self-peptides or mice that expressed an MHC class II molecule with a single covalently linked peptide (AbEp mice), that prevented binding of other self-peptides, developed Foxp3+Helioshi thymocytes normally. The significance of these observations is difficult to interpret as Helios expression is expressed very early during thymic development on all T cells at the DN2 and DN3 stages and there is no data to suggest that the strength of TCR signaling during development plays any role in the induction of Helios expression. In addition, mice that lacked the conserved non-coding sequence-1 (CNS-1) in the enhancer element of the Foxp3 locus, that contains the TGFβ-NFAT response element that regulates the generation of pTreg, [15] had a normal distribution of Helios+ and Helios− Treg in most sites, but a decreased percentage of Helios−Foxp3+ Treg in the colon [14]. However, it remains possible that some pTreg may be induced by pathways that do not involve signaling via CNS-1. None of these studies analyzed the functional or phenotypic properties of the Treg subsets induced in the genetically modified strains.
Further studies of the role of Helios in Treg function are hampered by the fact that Helios is a nuclear protein and it has not been possible to separate cells based on Helios expression using surrogate markers. In order to better study Helios, we have generated Helios-GFP reporter mice and crossed them to Foxp3-RFP reporter mice. Among the various lymphoid organs, Helios reporter mice show comparable expression patterns when compared to intracellular staining and accurately reflect Helios expression. Here, we demonstrate that the Helios− and Helios+ Treg populations are phenotypically very different and that Foxp3 expression, under lymphopenic conditions, is much more stable in Helios+ Treg. Although both populations have similar suppressive functions in vitro, in vivo they have differing capacities to control effector T cell activation and function. Detailed comparison of the transcriptomes of Helios+ and Helios− Treg revealed that the latter retain several features of non-Treg cells. Most importantly, Helios− Treg and Helios+ Treg populations show very little overlap in their TCR repertoires. These data indicate that two Treg populations are distinct and that Helios expression is a useful marker for stable Foxp3 expression.
Results
Helios+ Treg have an activated phenotype
In order to investigate the differences between Helios− and Helios+ Treg, we first analyzed splenocytes for the expression of a variety of Treg associated and activation markers. A higher percentage of Helios+ Treg expressed CD103, Nrp-1, OX40, TNFRII and CD69 when compared to Helios− Treg (Fig. 1A and 1C; all gating strategies shown in Supporting Information Fig. 1). Helios+ Treg also expressed Ki-67, indicating a higher proliferative rate. Smigiel et al have demonstrated that naïve Treg, characterized as CD44loCD62Lhi, express higher levels of CD25 while effector Treg, characterized as CD44hiCD62Llo, express lower levels of CD25 [16]. Helios+ Treg had a slightly lower percentage of cells that expressed CD25 and had a higher percentage of cells that were CD44hiCD62Llo, indicating that Helios+ Treg have an activated effector phenotype, but the expression of Helios does not distinguish naïve versus activated Treg based on the markers used by Smigiel et al. (Fig. 1A - C). Analysis of other lymphoid organs yielded similar results; Helios+ Treg are a heterogenous population of cells and none of the markers assayed distinguished Helios− Treg from Helios+ Treg.
Figure 1.

Helios+ Treg have an activated effector phenotype. Splenocytes were analyzed by flow cytometry and were gated on (A) CD4+Foxp3+ cells or (B) CD4+Foxp3+Helios− (left), CD4+Foxp3+Helios+ (right) cells. Data shown are representative of three independent experiments with n = 1 or 2 mice per experiment. (C) Summary data of (A) and (B) (paired Student’s t-test).
Functional properties of Helios− and Helios+ Treg
Further analysis of Helios− and Helios+ Treg was impeded by the fact that Helios is an intracellular transcription factor and we were unable to discover surrogate cell surface proteins for use in cell sorting. In order to investigate the functional differences between Helios− and Helios+ Treg, we generated a Helios-GFP reporter mouse (Supporting Information Fig. 2) and crossed it to a Foxp3-RFP reporter mouse [17]. The double reporter mouse exhibited a similar expression pattern of Helios and Foxp3 in multiple lymphoid organs (Fig. 2A) to that of cells analyzed by intracellular staining (Fig. 2B). Thus, we were able to use the double reporter mice to isolate Helios− and Helios+ Treg without using surrogate cell surface markers. However, it should be pointed out that both by intracellular staining and expression of the Helios reporter that there was a continuum of Helios expression with a population of Helioslo/int expressing cells evident using both techniques.
Figure 2.

In vitro properties of Helios− and Helios+ Treg. (A) The indicated lymphoid organs from double reporter mice were analyzed by flow cytometry and were gated on CD4+ cells or (B) were gated on CD4+ cells and Helios and Foxp3 expression were analyzed by intracellular staining. Data shown are representative of two independent experiments with n=2 mice per experiment. (C) Sorted Tconv cells (CD4+RFP−), Helios− Treg (CD4+RFP+GFP−) or Helios+ Treg (CD4+RFP+GFP+) from four pooled reporter mice were stimulated with T depleted splenocytes and anti-CD3 in the presence or absence of IL-2 for 3d. Cells were plated in triplicate. Data shown are representative of three independent experiments. (D) Sorted CD4+RFP− cells from reporter mice were stimulated with T depleted splenocytes and anti-CD3 in the presence or absence of sorted Helios− or Helios+ Treg for 3d. Cells were plated in triplicate. Data shown as the mean ± SD of three independent experiments expressed as percent proliferation compared to Tconv alone.
We first investigated the in vitro proliferative and suppressive capacities of Helios− and Helios+ Treg. The sorting strategy used for all experiments in shown in Supporting Information Fig. 2. Both Helios− and Helios+ Treg failed to proliferate in response to TCR stimulation alone but, were able to proliferate comparably to TCR stimulation in the presence of IL-2 (Fig. 2C). In addition, both subsets of Treg were suppressive in our in vitro suppression assay (Fig. 2D). However, Helios+ Treg suppressed slightly, and consistently, better. Similar results have been reported using a different double reporter mouse [18].
Helios− and Helios+ Treg stability
Previous studies have demonstrated that Helios expression in Treg is required to maintain effector Treg function [19]. Thus, we postulated that the Helios− Treg population in a normal mouse might be also be less stable. We first examined the stability of Helios− and Helios+ Treg under non-inflammatory conditions and transferred purified, congenically marked populations into WT recipients. When transferred into lymphoreplete WT recipients, most Helios+ Treg maintained Helios and Foxp3 expression (Fig. 3A and 3B). Moreover, both populations of transferred cells equally maintained Foxp3 expression. However, some Helios− Treg acquired Helios expression following transfer, but this level of Helios expression was always at lower levels than that observed in Helios+ Treg (Fig. 3B) and was similar to the Helioslo/int population present in total Treg population (Fig. 2A and B). Post sort analysis revealed that the purity of the transferred populations was not 100% and we cannot rule out a contribution of the expansion of the small percentage of contaminating cells (Supporting Information Fig. 3).
Figure 3.

Helios− Treg are unstable under lymphopenic conditions. Sorted Helios− or Helios+ Treg (1 × 106 cells) were injected r.o. into B6.SJL congenic recipients. At 4 weeks, spleen and LN from each mouse were pooled and enriched by autoMACS for CD4+ T cells. (A) Cells were gated on CD45.2 and CD4 and analyzed for GFP and RFP expression. Representative plots shown. (B) Summary data of (A) showing the percentage of the transferred cells that remain Foxp3+ (left). Data shown as mean ± SD, from three independent experiments with n=2–3 mice per experiment. Representative histogram for Helios expression gated on Foxp3+ Treg (right). (C) Sorted Foxp3−Helios− Tconv (CD4+RFP−GFP−) and Helios− or Helios+ Treg (7.5 × 105 cells) were injected r.o. into B6.SJL RAG−/− congenic recipients. The indicated lymphoid organs from each mouse were analyzed at 3 wk. Cells were gated on CD45.2 and CD4 and analyzed for GFP and RFP expression. Representative plots shown. Representative histogram for Helios expression gated on Foxp3+ Treg (right). (D) Summary data of (C) showing the percentage of the transferred cells that remain Foxp3+ (left). Data shown as mean ± SD, from three independent experiments with n=2–5 mice per experiment. (E) Tconv, Helios− Treg or Helios+ Treg were sorted from three pooled male reporter mice and were analyzed for CpG methylation at the Foxp3 TSDR region. Each CpG was analyzed in triplicate. Data shown as mean percent methylation of 11 combined CpG ± SD, representative of two independent experiments (left) or shown for each separate CpG (right).
We then transferred Helios− and Helios+ Treg to RAG2−/− recipients to examine stability under lymphopenic conditions. As the transferred cells proliferated extensively, we were able to examine the cells in different lymphoid organs. The stability of the transferred cells was similar in mLN and spleen, but was significantly higher in peripheral LN (pLN). Under lymphopenic conditions, a substantially larger percentage of the Helios− Treg lost Foxp3 expression in all organs examined (Fig. 3C and 3D). In contrast to the transfer to WT mice, most of the Helios− Treg that maintained Foxp3 expression acquired low levels of Helios expression. In addition, when CD4+ naïve cells (Foxp3−Helios− cells) were transferred as a control, a very small percentage of these cells converted to Foxp3+ pTreg in the mLN and pLN and also expressed intermediate levels of Helios. (Fig. 3C). Thus, Foxp3 expression is equally stable in both Helios− and Helios+ Treg upon transfer to WT mice, but under lymphopenic conditions, Helios− Treg more readily lose Foxp3 and acquire low levels of Helios expression.
The studies described above demonstrate that Helios− Treg have the propensity to lose Foxp3 expression and become ex-Tregs in a lymphopenic environment. One potential reason for decreased stability of the Helios− Treg population could be less demethylation of the conserved Foxp3 TSDR locus, that controls Treg stability [20]. Analysis of the Foxp3 TSDR in Helios− and Helios+ Treg revealed that although both Helios− and Helios+ Treg had highly demethylated TSDR regions, the TSDR of the Helios+ Treg was more demethylated than the TSDR of Helios− Treg (Fig. 3E). Differential demethylation was observed across all but one of the CpGs. Thus, the differences in demethylation might result in the increased loss of Foxp3 expression in Helios− Treg.
Suppressive properties of Helios− and Helios+ Treg in vivo
In order to study the suppressive capacity of Helios− and Helios+ Treg in vivo, we first examined Treg function in the IBD model of autoimmunity. Cell sorted naïve CD4+RFP−CD45RBhi cells were transferred into RAG−/− recipients in the presence or absence of congenically marked Treg cells. The transfer of CD4+RFP−CD45RBhi cells induced IBD in recipients as measured by weight loss (Fig. 4A), IFNγ production in CD4+ cells from the mLN (Fig. 4B), and infiltration and destruction of the colon (data not shown). Somewhat surprisingly, Helios− and Helios+ Treg were equally protective in all parameters assayed. We also examined the stability of the Treg under these inflammatory conditions. The absolute number of Treg (as measured by expression of the congenic marker) recovered after transfer was not statistically different (Fig. 4C). However, a significant percentage of Helios− Treg lost Foxp3 expression, while almost all Helios+ Treg maintained Foxp3 expression. Overall though, the total number of recovered Treg that maintained Foxp3 expression was not different between the two groups, which is consistent with the ability of the Helios− Treg to suppress disease as efficiently as the Helios+ Treg. Of those cells that maintained Foxp3 expression, Helios+ Treg maintained high Helios expression while most Helios− Treg acquired Helios, but at lower levels than that observed for Helios+ Treg (Fig. 4D).
Figure 4.

Helios− and Helios+ Treg protect mice from IBD. CD4+RFP−CD45RBhi T cells from 8–12 week old CD45.1 mice were injected r.o. into 8–10 week old B6.SJL RAG−/− recipients. Helios− or Helios+ Treg from congenically marked (CD45.2) reporter mice were co-injected where indicated. (A) Mice were monitored weekly for weight loss. Data shown as mean percent weight change from original weight, representative of four independent experiments with n=5 mice per experiment. (B) Effector cells (CD4+TCRβ+CD45.1+) from the mesenteric lymph node were analyzed at 10–11 wks by intracellular staining for cytokine production. Data shown as mean ± SD, representative of four independent experiments with n=5 mice per experiment. (C) Treg cells (CD4+ TCRβ+CD45.2+) were quantified at 10–11 wks from the mesenteric node (left), were analyzed for RFP (Foxp3) expression (middle) and total Foxp3+ Treg calculated (right). Data shown as mean ± SD, representative of four independent experiments with n=5 mice per experiment. (D) Foxp3+ Treg in (C) were analyzed for Helios expression. Data shown as the percent of CD4+ TCRβ+CD45.2+ Foxp3+ cells that express Helios. Data shown as mean ± SD, representative of four independent experiments with n=5 mice per experiment (left) or a representive histogram.
We then tested the ability of Helios− and Helios+ Treg to suppress activated effector cells in vivo. Scurfy mice contain a mutation in the Foxp3 gene and die 3–4 weeks after birth due to severe infiltration of lymphocytes into multiple organs [21]. The scurfy mutation is X-linked recessive, thus splenocytes from male scurfy mice contain large numbers of activated CD4+ and CD8+ autoreactive cells. In the scurfy transfer model, splenocytes from recently weaned, moribund male scurfy mice rapidly proliferate and cause organ-specific autoimmune infiltration upon transfer to RAG−/− recipients [22]. Splenocytes from scurfy mice were transferred to RAG−/− recipients in the presence or absence of congenically marked Helios− or Helios+ Treg cells and both the scurfy and Treg populations were analyzed six weeks later. Upon transfer, scurfy splenocytes expanded significantly and this expansion was efficiently inhibited by Helios+ Treg (Fig. 5A). Helios− Treg were less efficient at suppressing the expansion of both scurfy CD8+ and CD4+ cells. Additionally, transfer of scurfy cells alone resulted in a high percentage of CD4+ and CD8+ cells that produced IFNγ (Fig. 5B). While Helios+ Treg inhibited the percentage of scurfy CD4+ cells that produced IFNγ, Helios− Treg failed to do so. On a per cell basis, neither Helios− Treg nor Helios+ Treg inhibited the production of IFNγ by scurfy CD8+ cells. Finally, analysis of lymphocytic infiltrate into the liver and lungs of the mice revealed that Helios+ Treg were more protective than Helios− Treg (Fig. 5C).
Figure 5.

Helios− Treg mice fail to fully inhibit the expansion of effector scurfy cells. Splenocytes from 21d scurfy mice were injected r.o. into 8 wk old B6.SJL RAG−/− recipients. Helios− or Helios+ Treg cells were co-injected where indicated. After 6 weeks, splenocytes were (A) quantified (left panel), gated on scurfy CD45.1+TCRβ+ cells and CD8+ cells (middle panel) or CD4+ cells quantified (right panel) (B) Scurfy CD4+ cells, scurfy CD8+ cells or Treg (CD45.2+TCRβ+) were analyzed by intracellular staining for IFNγ expression. Data shown as the percent of the gated cells that express IFNγ. (C) Liver and lung tissue were taken at 6 wks, stained for H&E and the degree of infiltration scored. (D) Helios− or Helios+ Treg (CD45.2+TCRβ+) were quantified (left) analyzed for RFP (Foxp3) expression (middle) and the total Foxp3+ Treg calculated (right). All data shown as mean ± SD, from two of three independent experiments with n=3 to 5 mice per experiment.
We also examined the stability of the transferred Tregs under these inflammatory conditions. Similar to the results observed in the IBD model, the absolute number of Treg cells recovered, as measured by the congenic marker, was not significantly different between Helios− and Helios+ Treg (Fig. 5D). While expression of Foxp3 was lost in both Helios+ and Helios− Treg populations, expression of Foxp3 was much less stable in the Helios− population, but again, the absolute numbers of Foxp3+ T cells recovered from the Helios+ and Helios− Treg recipients were similar. Interestingly, we noted that some Helios− Treg produced IFNγ (Fig. 5B) and that the IFNγ producers had lost Foxp3 expression (Fig. 5E). Analysis of Helios expression again demonstrated that Helios+ Treg maintained Helios expression, while Helios− Treg acquired Helios expression but at a lower level. Thus, under lymphopenic conditions, Helios− Treg more readily lose Foxp3 expression, but those that remain Foxp3+ are able to acquire Helios expression.
Transcriptome analysis of Helios− and Helios+ Treg
To further characterize differences between Helios+ and Helios− Treg, we compared the transcriptomes of the two subpopulations by next-generation sequencing analysis. Sorted CD4+RFP+GFP− (Helios−) and CD4+RFP+GFP+ (Helios+) Treg cells from five individual mice were analyzed (Fig. 6A). A large number of genes (1029) were found to be differentially expressed by a greater than a two-fold difference (p< .05) and the majority of differentially expressed genes were upregulated in Helios+ Treg. 838 genes were more highly expressed in Helios+ Treg, while 191 genes were more highly expressed in Helios− Treg. GO analysis for genes involved in apoptosis, cytokines and receptors, and members of the TNF and TNF receptor superfamilies (TNFRSF) also revealed a similar ratio of genes more highly expressed in Helios+ Treg (Fig. 6B). The higher expression of several of the cell surface antigens (Itgae [CD103], Nrp-1, Tnfrsf4 [OX40] and Tnfrsf1b [TNFRII)] seen Fig. 1A was confirmed in the RNA-Seq analysis (Fig. 6C). Other genes more highly expressed in Helios+ Treg were primarily activation induced genes or genes already known to be associated with Treg cells (Itgb1 [CD29], Tnfrsf9 [CD137], CD101, Tnfrsf18 [GITR], Klrg1, IL1R2, IL1RL1, Pdcd1 [PD-1], Tnfrsf8 [CD30] and Tigit). Flow cytometry analysis confirmed the higher expression of the proteins encoded by these genes in Helios+ Treg, although the expression of these proteins was not uniform for all cells, but was limited to a subset of the Helios+ Treg (Supporting Information Fig. 4). Other genes of note that are selectively or more highly expressed in Helios+ Treg compared to conventional naïve (CD4+CD44lo cells [TN] or memory (CD4+CD44hi [TM]) non-Treg cells included Itgβ8, Ikzf4 (Eos) and Penk (Supporting Information Fig. 5). Notably, Ly6c and Ly6a are more highly expressed in Helios− Treg but are genes that are expressed in naïve cells and are down-regulated upon activation (Supporting Information Fig. 4). Very few genes were more highly expressed in Helios− Treg including Vipr1, Dapl1, Tgfβr3, Amigo2 and Gbp2b (Supporting Information Fig. 5). However, these genes, as well as other genes more highly expressed in Helios− Treg, were also found in other CD4+ cell subsets, with the exception of Gbp2b which appears to be exclusive to Helios− Treg.
Figure 6.

Differential gene expression between Helios− and Helios+ Treg. Sorted Helios− and Helios+ Treg from 5 individual double reporter mice were analyzed by RNA-seq. (A) Heat map of all differentially expressed genes equal or greater than two-fold different and p< 0.05. (B) Selected GO analysis of differentially expressed genes. (C) Scatter plot of all expressed genes. (D) Validation of selected differentially expressed genes by qPCR (TN: CD4+CD44−) and flow cytometry. Cells were gated on CD4+Foxp3+ cells. Data shown as the triplicate mean, representative of three independent experiments.
Differential expression of a number of genes (Pde3b, Nte5 [CD73], Rorc [Rorγt], and Satb1) offers potential insights into the differential function and origin of the Helios− Treg subpopulation. Expression of Pde3b was higher in Helios− Treg (Fig. 6D) compared to Helios+ Treg. Pde3b functions by hydrolyzing cAMP and its expression has been shown to be normally repressed in Tregs, as maintenance of high intracellular levels of cAMP is needed to facilitate the transfer of cAMP from Treg to target cells to mediate suppression [23, 24]. Helios− Treg also expressed lower levels of CD73 than Helios+ Tregs. CD73 expression plays a role in Treg stability and cleaves extracellular AMP to adenosine which can mediate suppression of target cells [23, 25, 26]. Thus, Helios− Tregs may be defective in two of the proposed pathways involved in Treg suppression. Importantly, Helios− Treg expressed higher levels of the genomic organizer and repressor, Satb1, than Helios+ Treg (Fig. 6D). Satb1 is expressed at high levels in conventional T cells and suppression of Satb1 is required for Treg function and stability. As Satb1 repression occurs during Treg development in thymus, the continued expression of Satb1 in Helios− Treg strongly suggests that they are not of thymic origin [11]. Lastly, both mRNA and protein for Rorc, the Th17 effector cell transcription factor were more highly expressed in Helios− Treg (Supporting Information Fig. 4 and 5), further supporting the findings that Helios− Treg are unstable and have the ability to become effector cells. Taken together, this transcriptome analysis offers a basis for both the defective function and instability of Helios− Treg.
TCR repertoire analysis of Helios− and Helios+ Treg
Although the suppressive function of the Helios− Treg and Helios+ Treg appear to be similar in vitro and in the IBD model, differences in their ability to suppress activated effector cells, in the stability of Foxp3 expression, and in the expression of Satb1 indicate that the two Treg populations are likely distinct. However, one explanation, given that Helios+ Treg have a more activated phenotype with enhanced suppressor function, is that Helios− Treg are merely precursors of Helios+ Treg. In order to determine if the two are distinct populations and if Helios− Treg are precursors of Helios+ Treg, we analyzed the TCR repertoire of the two cell types using high throughput TCRβ sequencing analysis. Due to the limitations of sequencing large numbers of TCR molecules from normal, unmanipulated mice, previous analyses of the Treg TCR repertoire have been performed using T cells from mice with limited TCR repertoires, such as those with a fixed Vβ gene. We wished to compare the TCR repertoires of Helios− and Helios− Treg from mice with an unbiased, normal repertoire. While deep sequencing provides the ability to completely sample the TCR repertoire of all Treg present in a single mouse, adequate sampling of the TCR repertoire of the Foxp3− TN population from a single mouse, as a control, was beyond our ability. Thus, we chose to purify Treg cells only from one location (mLN) in order to limit the number of control TN that needed to be sequenced, but allowing us to sequence all CD4+ T cells from that location. In our initial analysis, we focused only on the Helios− and Helios+ Treg populations. We sequenced 2 × 105 Helios− Treg cells and 4 × 105 Helios+ Treg cells from the mLN of two individual mice from the same litter (Supporting Information Table 1). Diversity index is a measure of population diversity where an index of 1.0 indicates the TCR sequences are all identical and an index of 0 indicates no sequence similarity. The diversity indices of both Treg subsets were exceedingly low, indicating that both Treg populations are very diverse (Fig. 7A). Comparison of the Helios+ and Helios− Treg repertoires from a single mouse indicated that there was very little overlap between the two populations (10%) and that Helios− Treg and Helios+ Treg are separate and distinct populations (Fig. 7B). Moreover, it appeared that each population was more similar to its counterpart in another mouse than Helios− and Helios+ Treg cells within the same mouse.
Figure 7.

Helios− and Helios+ Treg do not have similar TCR repertoires. Helios− and Helios+ Treg were sorted from the mLN from two individual mice and high-throughput sequencing for TCRB CDR3 regions was performed. (A) Diversity index (scale=0–1). (B) Pearson correlation and similarity. TN cells (CD4+RFP−CD44−), Helios− Treg or Helios+ Treg were sorted from the mLN of three individual reporter mice and high-throughput sequencing for TCRB CDR3 regions was performed. (C) Diversity index (scale=0–1). (D) Pearson correlation and similarity. Data shown as mean ± SD.
We then deep sequenced the entire TCR repertoires of Helios+ and Helios− Treg as well as TN (Foxp3−CD44+) from the mLN of three individual littermates (Supporting Information Fig. 6 and Supporting Information Table 1). The CDR3 length distribution was similar among the three populations (Supporting Information Fig. 7A). Examination of the Vβ usage revealed that there was a statistically significant difference in the usage of Vβ12–1 within the Helios+ Treg population, but all other Vβ genes were used in a similar manner (Supporting Information Fig. 7B and 7D). This was observed in the first experiment as well with mice from a different facility (data not shown). However, usage of Vβ12–1 only accounted for 4% of all TCRβ genes. VJ usage was no different between the populations (Supporting Information Fig. 7C and 7D).
Diversity analysis of the populations showed that all three populations were very diverse (Fig. 7C). We also compared the similarity of the populations. In agreement with our initial experiment, the correlation between Helios− Treg and Helios+ Treg was reproducible and again, was very low (Fig, 7D). Analysis of the common sequence overlap revealed a similar finding; Helios− Treg and Helios+ Treg were not similar. However, Helios+ Treg and TN had a higher percentage of common sequences than Helios− Treg and TN. This experiment was repeated with four individual mice at a lower depth of sequencing and reproduced the results shown here (data not shown). Thus, the TCR repertoire analysis demonstrates that the repertoires of Helios− and Helios+ Treg are quite different, but does not permit us to draw any conclusions as to the origins of the two populations.
Discussion
We have previously demonstrated, using a mAb to Helios, a member of the Ikaros transcription factor family, that both mouse and human Foxp3+ Treg can be divided into two major subpopulations composed of Foxp3+Helios+ (~70–90%) and Foxp3+Helios− (~10–30%) cells depending on the species and the location in vivo. We hypothesized that the Foxp3+Helios+ population represented tTreg, while the Foxp3+Helios− population represented pTreg. A number of correlative observations supported this hypothesis: 1) the exclusive presence of Foxp3+Helios+ Treg in the neonatal thymus and in the periphery until one week after weaning, 2) the observation that iTreg generated in vitro were uniformly Foxp3+Helios−, and 3) that antigen-specific Foxp3+ T cells (pTreg) generated in vivo in response to the oral administration of antigen were Helios−. Several subsequent studies have disputed this claim and have proposed that the expression of Helios is either not specific to tTreg and can be expressed in pTreg [12–14] or is expressed in CD4+ Tconv and is related to the activation state or to the strength of the antigenic signal delivered to the cells [14, 27–29]. While it is widely acknowledged that pTreg can be generated in vivo under different conditions, the overall size and functional importance of the pTreg pool remains unknown. As cellular biotherapy with in vitro expanded polyclonal Treg is now being used therapeutically to prevent GVHD [30] or treat autoimmunity [31] and studies are ongoing to enhance Treg numbers and function by administration of low-dose IL-2 [32], it is critical to understand which Treg subpopulation is targeted by different therapeutic approaches.
Three limited studies have compared the phenotype and function of Helios− and Helios+ Treg. Using CD103 and GITR as surrogate markers for mouse Helios+ Treg, Zabransky et al. demonstrated that Helios+ Treg have the highest suppressive capacity [33]. Using a single-cell cloning approach to define human Helios− and Helios+ Treg, Himmel et al. interestingly found that Helios− Treg produced more IFNγ and that Helios+ Treg were slightly more demethylated than Helios− Treg, [34]. However, the suppressive ability of each population was comparable. The study was limited in that the sort was restricted to CD45RA+ naïve Treg and Treg were cultured for 3 weeks after sorting before being defined by Helios expression and then assayed. Finally, using a single cell cloning approach and TIGIT and FCRL3 as surrogate markers for Helios+ human Treg, Bin Dhuban et al also demonstrate that Helios− Treg have less suppressive capacity and produce inflammatory cytokines [35]. Again, the two markers do not divide Treg into two distinct populations that encompass the total Treg population.
In the present report, we have performed a comprehensive series of studies whose goal was to compare Helios+ and Helios− Treg and to address whether the Helios+ and Helios− populations of Treg are distinct subpopulations or whether they represent different stages of differentiation of a single Treg population. As we were unable to identify surrogate cell surface markers that could clearly separate the Helios+ and Helios− Treg subpopulations, we generated a Helios/Foxp3 double reporter mouse which readily allowed isolation of the two subpopulations. However, Helios expression is a continuum and while Helios+ cells are high in expression, there are Treg cells that express Helios at an intermediate level and our gating strategy has mostly removed this population. Not surprisingly, when comparing Helios− and Helios+ Treg, the two populations only moderately differed in their expression of Treg-associated cell surface makers. While the Helios+ subpopulation displayed higher percentages of cells that expressed “Treg activation antigens” at a higher level, this phenotypic analysis does not really distinguish between the possibility of two distinct populations or merely a difference in differentiation status. It thus remains possible that Helios− Treg could represent a Treg precursor population that upon activation would express Helios and other activation markers and would develop into a fully functioning Treg.
Analysis of gene expression by RNA-seq was somewhat more informative in that the two subpopulations differed by ~1000 genes. The majority of differentially expressed genes were present in the Foxp3+Helios+ subpopulation and many encoded activation antigens consistent with the protein data. Although the differential expression of a large number of genes in the Foxp3+Helios− subpopulation could indicate that the two populations are distinct, it does not rule out the possibility that the large differences represent different stages of differentiation Foxp3+Helios− Treg expressed Satb1 which is normally expressed at high levels in T conventional cells, but is repressed in Foxp3+Helios+ Treg. Repression of Satb1, a chromatin remodeler and super-enhancer, has been shown to be critical for Treg function [11, 36, 37] and ectopic expression of SATB1 in human Treg reprogrammed the cells into T effector cells that produced a variety of effector cytokines even in the presence of Foxp3. Similarly, we could detect expression of Pde3b in Foxp3+Helios− Treg, but not in Foxp3+Helios+ Treg. Pde3b is normally expressed in naïve CD4+ T cells and its expression is maintained upon T cell activation. One potential reason it is normally strongly repressed in Treg [23] is that the physiologic function of Pde3b is to break down adenosine and cyclic adenosine 3’,5’-monophosphate (cAMP) both of which have been proposed to mediate Treg suppression [26]. Ectopic expression of PDE3B in human Treg cells impaired Treg homeostasis as measured by reduced cell number. Lastly, one of the Treg signature genes whose expression is reduced in Helios− Treg is Nt5e (CD73), a 5’ ectonucleotidase that metabolizes AMP to the immunosuppressive molecule adenosine. Taken together, the higher levels of Satb1 and Pde3b and the lower levels of NT5e present in the Helios− Treg are consistent with the possibility that the Helios− Treg subpopulation is composed of pTreg which contain residual expression of certain genes normally expressed in CD4+Foxp3− T cells and have not fully differentiated into the Treg lineage. This hypothesis is supported by a recent single-cell expression analysis of gene expression in Treg versus Tconv which identified a subpopulation (26%) of Treg that overlapped in expression with Tconv and was termed “furtive Tregs.” While this population expressed the same levels of Foxp3 transcripts as did other Treg subpopulations, the “furtive Treg” subpopulation expressed low levels of Helios [38]. Interestingly, “furtive” Treg also expressed higher levels of Itgb1, Tnfrsf1b, Tnfrsf4, Tnfrsf9, and Tnfrsf18 and expressed lower levels of Vipr1, Dapl1 and Igfbp4, similar to the differences that we have observed in in our comparison of Helios− vs. Helios+ Treg.
Helios− Treg were comparable in their capacity to suppress the proliferation of naive T cells in culture and to inhibit the ability of naïve T cells to induce IBD. However, Helios− Treg were deficient in their capacity to suppress activated T effector cells in a model in which T cells from moribund scurfy mice are transferred to immunodeficient recipients. While lower levels of cAMP or adenosine and could explain the reduced function that we observe in Helios− Treg, Helios− Treg also appear to also have a defect in stability. While iTreg generated in vitro in general have fully methylated TSDR regions of the Foxp3 enhancer, pTreg can display a fully demethylated TSDR [39]. Both the Helios+ and the Helios− populations had demethylated TSDRs, but the extent of demethylation was slightly greater in the Helios+ subset. Consistent with this, both subpopulations of Treg maintained stable expression of Foxp3 upon transfer to normal recipients but major differences in stability were observed when the subpopulations were transferred to immunodeficient recipients. While both subpopulations lost significant expression of Foxp3, the loss was 50% greater in the Helios− subpopulation. The reduced fitness of Helios− Treg correlates with our previous study and that of Kim et al where Treg deficient in Helios have a reduced fitness [19, 40]. Our studies concluded that Helios deficient Treg were unable to generate or maintain activated effector Treg due to a reduction in Bcl2, while the studies of Kim et al concluded that instability of Treg was due to decreased activation of the STAT5 pathway. However, our gene expression analysis did not indicate a difference in Bcl2 nor in molecules within the STAT5 pathway, thus, the instability we observe in Helios− Treg does not appear to be similar to the instability observed in Treg cells that are deficient in Helios.
The cell transfer studies also revealed that a subpopulation of Helios− Treg could acquire expression of Helios both when transferred to immunocompetent mice and to RAG−/− mice. The level of Helios expressed was always less than that expressed by the Foxp3+Helios+ subset. It remains possible that some of the previous reports that claimed that pTreg expressed Helios reflected this low level of expression. Importantly, Helios+ Treg never lost Helios expression to become Helios− Treg, as loss of Helios was concomitant with loss of Foxp3, suggesting that high Helios expression is a fixed phenotype in Helios+ Treg. While Helios expression appears to be a stable marker that distinguishes the two subsets under physiologic conditions, it is clear that under lymphopenic conditions Helios− Treg can acquire a low level of Helios expression. In addition, pTreg generated under lymphopenic conditions from naïve cells also acquire Helios. A similar case may occur in vitro during the induction of iTreg in the presence of TGF-β and IL-2 where Helios appears to be induced if dendritic cells are present during the induction phase [12, 13].
A direct approach to determining the relationship between Treg and Tconv and between tTreg and pTreg is a comparison of the their TCR repertoires. Previous studies have concluded that the TCR repertoire of Treg and that of CD4+Foxp3− Tconv cells are quite distinct and exhibit little overlap but with wide variations in overlap [41], [42], [43] [44]. However, all of these early studies were done in mice with restricted TCR repertoires and the results may not reflect the broader repertoires in normal mice. Wide variations in overlap (60% to 10%) were also noted in studies that attempted to address the issue of peripheral conversion and pTreg generation by comparing thymic Treg to peripheral Treg ([44], [45] [46]. Studies that directly compared the pTreg and tTreg repertoires by comparing the Foxp3+ T cells that have been generated after transfer of Foxp3− T cells to normal or lymphopenic recipients [41] showed very low levels (4–10%) of overlap. Similarly, a low level of overlap (<10%) was observed in a comparison of tTreg and pTreg generated in mice that received tTreg to protect from induction of IBD [47]. Two previous studies have specifically examined the TCR repertoires of Helios− and Helios+ Treg. Szurek et al. performed high throughput sequencing on cells from TCR mini-gene mice [14] but restricted the analysis to the top 50 most prevalent sequences from each group. They concluded that most TCRs were expressed by both populations at similar frequencies. Lord et al. have also deep-sequenced Helios− and Helios+ Treg, along with effector CD4+ T cells, from inflamed versus non-inflamed colon of patients with ulcerative colitis [48]. Little overlap (5–10%) of Helios− and Helios+ Treg sequences was observed and even less overlap between either Treg subpopulation and effector CD4+Foxp3− cells regardless of location [48] which is in agreement with our study.
With the ease and lower cost of high throughput sequencing, it is now possible to examine the repertoires of normal mice without having to restrict the repertoire. Thus, we did not use repertoire restricted mice and were able to eliminate concerns of adequate random sampling and were able to completely sample all CD4+ T cells from the mLN, generating 3 to 5 million sequences per mouse. This approach allowed us to conclude that the repertoires of Helios− and Helios+ Treg are not similar. However, we are unable to conclude that Helios− Treg can be generated from TN nor did this approach definitively rule out the possibility that Helios− Treg are precursors to Helios+ Treg. It remains possible that Helios− Treg in the mLN were generated from TN in the small intestine lamina propria that subsequently migrated to the mLN, and that the TN population we analyzed from the mLN did not contain the precursors of the Helios− Treg isolated from the mLN. Future studies to examine the similarity of Treg cells to other populations will require an extensive analysis of these populations from multiple locations, particularly from the thymus. However, it may not be possible to deduce the origin of pTreg using unmanipulated, conventional mice as the precursor frequency may be too low to detect significant overlap of the TCR repertoires.
Lastly, we strongly advocate the use of Helios as a marker of Treg stability in the mouse, but particularly in man [49]. One major difference between the species is that we have not been able to detect Helios expression in human Foxp3−CD4+ T cells even after activation. If the Foxp3+Helios− Treg population in man is also composed of pTreg, it would clearly be a potentially inappropriate population to use for cellular biotherapy or to expand in vivo with cytokines or other biologics as it would be poorly suppressive, lack stability, and be deficient in a TCR repertoire with self-antigen specificity. The combined use of the appropriate surrogate markers and Helios expression should yield an optimal population for use in biotherapy.
Materials and Methods
Animals
C57BL/6 mice were obtained from Charles River (Frederick, MD). Helios-GFP mice were generated by TaconicArtemis (Cologne, Germany). The targeting vector was generated using BAC clones from the C57BL/6J RPCI-23 BAC library. A cassette containing an Internal Ribosome Entry Site (IRES) and the open reading frame of eGFP was inserted immediately after the translation termination codon of Ikzf2 and a puromycin resistance cassette, flanked by F3 sites, was inserted into intron 7. The targeting vector was transfected into the C57BL/6N Tac ES cell line. Correctly targeted ES cell clones were injected into C57BL/6 blastocysts and chimeric mice were crossed to FLPe recombinase expressing deleter mice to remove the puromycin resistant cassette. The Helios-GFP reporter mice were then crossed to Foxp3-RFP mice [17] purchased from The Jackson Laboratory (Bar Harbor, ME) to generate double reporter mice. Double reporter mice were also crossed to CD45.1 congenic mice. B6.SJL congenic, B6.SJL RAG deficient and B6 RAG deficient mice were obtained by NIAID and maintained by Taconic under NIAID contract. Female heterozygous B6.Cg-Foxp3sf/J (Scurfy) mice were purchased from Jackson Laboratories and bred to C57BL/6 male mice to generate hemizygous male B6 [Cg-Foxp3sf/Y (Scurfy)] offspring. Scurfy mice were also bred to B6.SJL (CD45.1) congenic mice. All animal protocols used in this study were approved by the NIAID Animal Care and Use Committee.
Media, antibodies and reagents
Mouse cells were grown in RPMI 1640 (Lonza, Walkersville, MD) supplemented with 10% heat-inactivated FCS, penicillin (100 µg /ml), streptomycin (100 µg/ml), 2 mM L-glutamine, 10 mM HEPES, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate (all Lonza, Walkersville, MD) and 50 µM 2-ME (Sigma, St. Louis, MO). Purified anti-CD3 (2C11), PE-CD25 (PC61), APC-CD62L (MEL-14), PE-Ki-67 (B56), AF700-CD4 (RM4–5), PE-IL1R2 (4E2), PE-PD-1 (J43) and FITC-CD8 (53–6.7) were purchased from BD Biosciences (San Jose, CA). BV650-CD4 (RM4–5), PE-CD103 (2E7), PE-CD134/OX40 (OX-86), PE-CD120b (TR75–89), e780-CD45.2 (104), BV421-IFNγ (XMG1.2), APC-CD8 (53–6.7), BV570-TCRβ (H57–597), AF700-CD45.1 (A20), PerCP-Cy5.5-CD45.2 (104), APC-IL-17A (TC11–18H10.1), BV650-IL-17A (TC11–18H10.1), PE-CD29 (HMβ1–1), PE-CD137 (17B5), PE-KLRG1 (2F1/KLRG1), PE-TIGIT (Vstm3), PE-Ly6C (HK1.4), PE-Ly6A (D7) and APC-NRP-1 (3E12) were purchased from BioLegend (San Diego, CA). FITC-Helios (22F6), e450-Foxp3 (FJK-16s), PE-CD69 (H1.2F3), PE-CD44 (IM7), APC-CD44 (IM7), e450-CD4 (RM4–5), PE-Foxp3 (FJK-16s), APC-CD45RB (C363.16A), PerCP-Cy5.5-CD45.1 (A20), APC-CD45.1 (A20), AF700-CD45.2 (104), APC-Foxp3 (FJK-16s), PE-CD101 (Moushi101), PE-GITR (DTA-1), PE-CD30 (mCD30.1), PE-RORγt (B2D) and e450-CD73 (eBioTY/11.8) were purchased from eBioscience (San Diego, CA). PE-IL1RL1 (DJ8) was purchased from mdbioproducts (St. Paul, MN). CD4, CD90.2 and PE microbeads were purchased from Miltenyi Biotec, Inc. (Auburn, CA). Human rIL-2 was obtained from the Preclinical Repository of the Biological Resources Branch, National Cancer Institute (Frederick, MD).
Flow cytometry
Intracellular staining for transcription factors and cytokines was performed with the Foxp3 staining buffer kit according to the manufacturer’s protocol (eBioscience). Flow cytometry was performed on a BD LSR II (Special Order with 488nm, 532 nm, 633 nm and 405 nm lasers) and analyzed using FlowJo® software. Cell sorting was performed on a BD FACSAria sorter or, for TCR repertoire analysis, on a BD Influx. Gating strategies are shown in Supporting Information Fig. 1. For all flow cytometry and cell sorting, the EJI guidelines were followed.
Proliferation and suppression assay
CD4+RFP−GFP− (Tconv), CD4+RFP+GFP− and CD4+RFP+GFP+ FACS sorted cells were purified from pooled spleen and LN of four male and female double reporter mice and were cultured as previously described [50]. Briefly, to assess proliferative capacity, Tconv cells, Helios− Treg or Helios+ Treg (5 × 104) were stimulated with T depleted splenocytes (5 × 104) and anti-CD3 in the presence or absence of IL-2 for 3d in triplicate. The cells were then pulsed for 6h with 3H thymidine. To assess suppressive capacity, Tconv cells were stimulated with T depleted splenocytes and anti-CD3 in the presence or absence of sorted Helios− or Helios+ Treg for 3d in triplicate. The cells were then pulsed for 6h with 3H thymidine.
Adoptive transfer experiments
Spleen and LN from CD45.2 reporter mice were labeled with CD4 microbeads and enriched for CD4+ cells by autoMACS and were then FACS sorted to isolate CD4+RFP+GFP− and CD4+RFP+GFP+ cells. One million cells were transferred by r.o. injection into congenic B6.SJL or B6.SJL RAG−/− mice. When transferred into WT mice, pooled spleen and LN cells were harvested 3 to 4 weeks later and were enriched for CD4+ cells by autoMACS before flow cytometry analysis. When transferred into RAG−/− mice, cells from the indicated lymphoid organs were harvested at 3 weeks and analyzed by flow cytometry. Similar results were observed using either male or female mice.
Methylation analysis of Treg-specific demethylated region
Spleen and LN cells from male mice were sorted by flow cytometry to obtain CD4+RFP−GFP− (Tconv), CD4+RFP+GFP− (Helios− Treg) and CD4+RFP+GFP+ (Helios+ Treg) cells. The cells were then analyzed for methylation status of the TSDR of the Foxp3 gene by bisulfite sequencing as previously described [19].
Inflammatory bowel disease model
Spleen and LN from male reporter mice were labeled with CD4 microbeads and enriched by autoMACS (Miltenyi Biotec, Inc, Auburn, CA) for CD4+ cells and were then FACS sorted to isolate CD45.2 CD4+RFP−CD45RBhi and CD45.1 CD4+RFP+GFP− and CD4+RFP+GFP+ Treg cells. CD45RBhi cells (4 × 105) and Tregs (2 × 105) were transferred by retro-orbital (r.o.) injection into B6 RAG−/− mice. In some experiments, CD4+RFP−CD45RBhi cells were sorted from CD45.1 reporter mice and Treg cells from CD45.2 reporter mice. Mice were monitored for weight loss and analyzed at 10–11 wks.
Scurfy cell transfer model
Splenocytes (2 × 106) from 21–28d male CD45.2 scurfy mice were transferred r.o. to B6 RAG−/− mice alone or with 5 × 105 CD4+RFP+GFP− or CD4+RFP+GFP+ Treg cells from CD45.1 reporter mice that had been enriched by autoMACS for CD4+ cells and then FACS sorted. In some experiments, the congenic markers were switched. At 6 weeks, lung and liver tissues were fixed, processed and stained with H&E (Histoserv, Germantown, MD) and splenocytes were analyzed for CD4, CD8 and cytokine expression.
Gene expression profiling
CD4+RFP+GFP− or CD4+RFP+GFP+ Treg cells were FACS sorted from pooled spleen and LN of five individual 8wk old male double reporter littermates. Stringent sorting gates resulted in approximately 2.2 × 105 CD4+RFP+GFP− cells and 6.0 × 105 CD4+RFP+GFP+ cells per mouse. Cells were processed and RNA was sequenced by the Genomics Unit, RML Research Technologies Section, NIAID, NIH. Reads were mapped to the mouse genome assembly mm10 using Hisat2 [51]. Reads mapping to genes were counted using htseq-count [52]. Differential expression analysis was performed using the Bioconductor package DESeq2 [53]. Further analysis was performed using Partek Genomic Suite. Data were deposited in the GenBank under accession number GSE117989 (https://www.ncbi.nlm.gov/genbank/).
Quantitative PCR
Tnaive (TN)(CD4+RFP−CD44−), TM (CD4+RFP−CD44+), Helios− Treg (CD4+RFP+GFP−) and Helios+ Treg (CD4+RFP+GFP+) cells from pooled spleen and LN were obtained by cell sort. RNA was isolated from the cells using the RNeasy Mini Kit (Qiagen, Germantown, MD) followed by cDNA synthesis (Invitrogen SuperScript III First-Strand Synthesis SuperMix for qRT-PCR) (Waltham MA). RT-PCR was performed in triplicate using Applied Biosystems TaqMan Universal PCR Master Mix and run on the Applied Biosystems QuantStudio 7 Flex machine (Waltham, MA). dCt was calculated by subtracting the average value for the 18s rRNA (Applied Biosystems) from the average value for each of the other genes and changed from logarithmic to linear form using the formula 2^(-dCt). The following TaqMan Gene Expression Assays were obtained from Applied Biosystems: Tgfbr (Mm00803538_m1) Satb1 (Mm01268940_m1) Vipr1 (Mm00449214_m1) Dapl1 (Mm01271524_m1) Pde3b (Mm00691635_m1) Amigo2 (Mm00662105_s1) Gbp2b (Mm00657086) Rorc (Mm01261022_m1) Itgb8 (Mm00623991_m1) Ikzf4 (Mm01133256_m1) Penk (Mm01212875_m1) and 18s rRNA.
TCR repertoire analysis
CD4+RFP+GFP− or CD4+RFP+GFP+ cells were FACS sorted from mLN of two individual 8wk old double reporter female littermates (experiment 1). Approximately 2.0 × 105 CD4+RFP+GFP− cells and 5.3 × 105 CD4+RFP+GFP+ cells per mouse were recovered. Genomic DNA was purified from the sorted cells using Cells and Tissue DNA Isolation Kit or Cells and Tissue DNA Isolation Micro Kit (Norgen Biotek Corp., Thorold, Ontario). TCRB CDR3 regions were amplified and sequenced at the deep level (200,000 cells). Amplification and sequencing were carried out by Adaptive Biotechnologies, Inc. (Seattle, WA).
For a second experiment, CD4+RFP−CD44−GFP−(TN), CD4+RFP+GFP− (Helios− Treg) or CD4+RFP+GFP+ (Helios+ Treg) cells from mLN of four individual female littermates were FACS sorted on a BD Influx. Isolated cells were analyzed at the deep (200,000 cells) level (CD4+RFP+GFP−), the ultra deep (800,000 cells) level (CD4+RFP+GFP+) or the max depth (4 × 106) level (CD4+RFP−CD44−GFP−). The repertoire analyses were performed using ImmunoSEQ software from Adaptive Biotechnologies (http://adaptivebiotech.com/immunoseq). The sample overlap is the sum of the counts for all shared sequences observed in two samples divided by the total number of counts for all sequences (shared and unshared) in the two samples. Counts are defined by V gene, J gene and CDR3 residues.
Statistics
All data are presented as the mean values ± SD. Most comparisons between groups were analyzed using unpaired Student’s t tests (Prism GraphPad). Paired Student’s t tests were used where specifically indicted. Statistical significance was established at the levels of *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Supplementary Material
Acknowledgements
We thank K. Weng for flow sorting, V. Kapoor for use and operation of the BD Influx, Kishore Kanakabandi for RNA extractions and Stacy Ricklefs for the library preparation. This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Conflict of Interest Disclosure
The authors declare no commercial or financial conflict of interest.
References
- 1.Shevach EM, Biological functions of regulatory T cells. Adv Immunol 2011. 112: 137–176. [DOI] [PubMed] [Google Scholar]
- 2.Rudensky AY, Regulatory T cells and Foxp3. Immunol Rev 2011. 241: 260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Curotto de Lafaille MA and Lafaille JJ, Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity 2009. 30: 626–635. [DOI] [PubMed] [Google Scholar]
- 4.Shevach EM and Thornton AM, tTregs, pTregs, and iTregs: similarities and differences. Immunol Rev 2014. 259: 88–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y and Shevach EM, Expression of Helios, an Ikaros Transcription Factor Family Member, Differentiates Thymic-Derived from Peripherally Induced Foxp3(+) T Regulatory Cells. Journal of Immunology 2010. 184: 3433–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim EH, Gasper DJ, Lee SH, Plisch EH, Svaren J and Suresh M, Bach2 Regulates Homeostasis of Foxp3(+) Regulatory T Cells and Protects against Fatal Lung Disease in Mice. Journal of Immunology 2014. 192: 985–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roychoudhuri R, Hirahara K, Mousavi K, Clever D, Klebanoff CA, Bonelli M, Sciume G, Zare H, Vahedi G, Dema B, Yu ZY, Liu H, Takahashi H, Rao M, Muranski P, Crompton JG, Punkosdy G, Bedognetti D, Wang E, Hoffmann V, Rivera J, Marincola FM, Nakamura A, Sartorelli V, Kanno Y, Gattinoni L, Muto A, Igarashi K, O’Shea JJ and Restifo NP, BACH2 represses effector programs to stabilize T-reg-mediated immune homeostasis. Nature 2013. 498: 506-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, Peterson DA, Stappenbeck TS and Hsieh CS, Peripheral education of the immune system by colonic commensal microbiota. Nature 2011. 478: 250–U142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng GH, Yamasaki S, Saito T, Ohba Y, Taniguchi T, Takeda K, Hori S, Ivanov II, Umesaki Y, Itoh K and Honda K, Induction of Colonic Regulatory T Cells by Indigenous Clostridium Species. Science 2011. 331: 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luu M, Jenike E, Vachharajani N and Visekruna A, Transcription factor c-Rel is indispensable for generation of thymic but not of peripheral Foxp3(+) regulatory T cells. Oncotarget 2017. 8: 52678–52689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kitagawa Y, Ohkura N, Kidani Y, Vandenbon A, Hirota K, Kawakami R, Yasuda K, Motooka D, Nakamura S, Kondo M, Taniuchi I, Kohwi-Shigematsu T and Sakaguchi S, Guidance of regulatory T cell development by Satb1-dependent super-enhancer establishment. Nat Immunol 2017. 18: 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verhagen J and Wraith DC, Comment on “Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells”. J Immunol 2010. 185: 7129; author reply 7130. [DOI] [PubMed] [Google Scholar]
- 13.Gottschalk RA, Corse E and Allison JP, Expression of Helios in peripherally induced Foxp3+ regulatory T cells. J Immunol 2012. 188: 976–980. [DOI] [PubMed] [Google Scholar]
- 14.Szurek E, Cebula A, Wojciech L, Pietrzak M, Rempala G, Kisielow P and Ignatowicz L, Differences in Expression Level of Helios and Neuropilin-1 Do Not Distinguish Thymus-Derived from Extrathymically-Induced CD4+Foxp3+ Regulatory T Cells. PLoS One 2015. 10: e0141161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schlenner SM, Weigmann B, Ruan Q, Chen Y and von Boehmer H, Smad3 binding to the foxp3 enhancer is dispensable for the development of regulatory T cells with the exception of the gut. J Exp Med 2012. 209: 1529–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD and Campbell DJ, CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J Exp Med 2014. 211: 121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wan YY and Flavell RA, Identifying Foxp3-expressing suppressor T cells with a bicistronic reporter. Proc Natl Acad Sci U S A 2005. 102: 5126–5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sugita K, Hanakawa S, Honda T, Kondoh G, Miyachi Y, Kabashima K and Nomura T, Generation of Helios reporter mice and an evaluation of the suppressive capacity of Helios(+) regulatory T cells in vitro. Exp Dermatol 2015. 24: 554–556. [DOI] [PubMed] [Google Scholar]
- 19.Sebastian M, Lopez-Ocasio M, Metidji A, Rieder SA, Shevach EM and Thornton AM, Helios Controls a Limited Subset of Regulatory T Cell Functions. J Immunol 2016. 196: 144–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, Schlawe K, Chang HD, Bopp T, Schmitt E, Klein-Hessling S, Serfling E, Hamann A and Huehn J, Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol 2007. 5: e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF and Ramsdell F, Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet 2001. 27: 68–73. [DOI] [PubMed] [Google Scholar]
- 22.Sharma R, Jarjour WN, Zheng L, Gaskin F, Fu SM and Ju ST, Large functional repertoire of regulatory T-cell suppressible autoimmune T cells in scurfy mice. J Autoimmun 2007. 29: 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA and Rudensky AY, Foxp3-dependent programme of regulatory T-cell differentiation. Nature 2007. 445: 771–775. [DOI] [PubMed] [Google Scholar]
- 24.Klein M and Bopp T, Cyclic AMP Represents a Crucial Component of Treg Cell-Mediated Immune Regulation. Front Immunol 2016. 7: 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ and Mosmann TR, T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5’-adenosine monophosphate to adenosine. J Immunol 2006. 177: 6780–6786. [DOI] [PubMed] [Google Scholar]
- 26.Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, Chen JF, Enjyoji K, Linden J, Oukka M, Kuchroo VK, Strom TB and Robson SC, Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 2007. 204: 1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akimova T, Beier UH, Wang L, Levine MH and Hancock WW, Helios expression is a marker of T cell activation and proliferation. PLoS One 2011. 6: e24226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ross EM, Bourges D, Hogan TV, Gleeson PA and van Driel IR, Helios defines T cells being driven to tolerance in the periphery and thymus. Eur J Immunol 2014. 44: 2048–2058. [DOI] [PubMed] [Google Scholar]
- 29.Serre K, Benezech C, Desanti G, Bobat S, Toellner KM, Bird R, Chan S, Kastner P, Cunningham AF, Maclennan IC and Mohr E, Helios is associated with CD4 T cells differentiating to T helper 2 and follicular helper T cells in vivo independently of Foxp3 expression. PLoS One 2011. 6: e20731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blazar BR, MacDonald KPA and Hill GR, Immune regulatory cell infusion for graft-versus-host disease prevention and therapy. Blood 2018. 131: 2651–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, Herold KC, Lares A, Lee MR, Li K, Liu W, Long SA, Masiello LM, Nguyen V, Putnam AL, Rieck M, Sayre PH and Tang Q, Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med 2015. 7: 315ra189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klatzmann D and Abbas AK, The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat Rev Immunol 2015. 15: 283–294. [DOI] [PubMed] [Google Scholar]
- 33.Zabransky DJ, Nirschl CJ, Durham NM, Park BV, Ceccato CM, Bruno TC, Tam AJ, Getnet D and Drake CG, Phenotypic and functional properties of Helios+ regulatory T cells. PLoS One 2012. 7: e34547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Himmel ME, MacDonald KG, Garcia RV, Steiner TS and Levings MK, Helios+ and Helios- cells coexist within the natural FOXP3+ T regulatory cell subset in humans. J Immunol 2013. 190: 2001–2008. [DOI] [PubMed] [Google Scholar]
- 35.Bin Dhuban K, d’Hennezel E, Nashi E, Bar-Or A, Rieder S, Shevach EM, Nagata S and Piccirillo CA, Coexpression of TIGIT and FCRL3 identifies Helios+ human memory regulatory T cells. J Immunol 2015. 194: 3687–3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beyer M, Thabet Y, Muller RU, Sadlon T, Classen S, Lahl K, Basu S, Zhou X, Bailey-Bucktrout SL, Krebs W, Schonfeld EA, Bottcher J, Golovina T, Mayer CT, Hofmann A, Sommer D, Debey-Pascher S, Endl E, Limmer A, Hippen KL, Blazar BR, Balderas R, Quast T, Waha A, Mayer G, Famulok M, Knolle PA, Wickenhauser C, Kolanus W, Schermer B, Bluestone JA, Barry SC, Sparwasser T, Riley JL and Schultze JL, Repression of the genome organizer SATB1 in regulatory T cells is required for suppressive function and inhibition of effector differentiation. Nat Immunol 2011. 12: 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kondo M, Tanaka Y, Kuwabara T, Naito T, Kohwi-Shigematsu T and Watanabe A, SATB1 Plays a Critical Role in Establishment of Immune Tolerance. J Immunol 2016. 196: 563–572. [DOI] [PubMed] [Google Scholar]
- 38.Zemmour D, Zilionis R, Kiner E, Klein AM, Mathis D and Benoist C, Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat Immunol 2018. 19: 291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, Olek S, Hamann A, von Boehmer H and Huehn J, DNA methylation controls Foxp3 gene expression. Eur J Immunol 2008. 38: 1654–1663. [DOI] [PubMed] [Google Scholar]
- 40.Kim HJ, Barnitz RA, Kreslavsky T, Brown FD, Moffett H, Lemieux ME, Kaygusuz Y, Meissner T, Holderried TA, Chan S, Kastner P, Haining WN and Cantor H, Stable inhibitory activity of regulatory T cells requires the transcription factor Helios. Science 2015. 350: 334–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lathrop SK, Santacruz NA, Pham D, Luo J and Hsieh CS, Antigen-specific peripheral shaping of the natural regulatory T cell population. J Exp Med 2008. 205: 3105–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wong J, Obst R, Correia-Neves M, Losyev G, Mathis D and Benoist C, Adaptation of TCR repertoires to self-peptides in regulatory and nonregulatory CD4+ T cells. J Immunol 2007. 178: 7032–7041. [DOI] [PubMed] [Google Scholar]
- 43.Hsieh CS, Liang Y, Tyznik AJ, Self SG, Liggitt D and Rudensky AY, Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity 2004. 21: 267–277. [DOI] [PubMed] [Google Scholar]
- 44.Pacholczyk R, Ignatowicz H, Kraj P and Ignatowicz L, Origin and T cell receptor diversity of Foxp3+CD4+CD25+ T cells. Immunity 2006. 25: 249–259. [DOI] [PubMed] [Google Scholar]
- 45.Hsieh CS, Zheng Y, Liang YQ, Fontenot JD and Rudensky AY, An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nature Immunology 2006. 7: 401–410. [DOI] [PubMed] [Google Scholar]
- 46.Wong J, Mathis D and Benoist C, TCR-based lineage tracing: no evidence for conversion of conventional into regulatory T cells in response to a natural self-antigen in pancreatic islets. J Exp Med 2007. 204: 2039–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haribhai D, Williams JB, Jia S, Nickerson D, Schmitt EG, Edwards B, Ziegelbauer J, Yassai M, Li SH, Relland LM, Wise PM, Chen A, Zheng YQ, Simpson PM, Gorski J, Salzman NH, Hessner MJ, Chatila TA and Williams CB, A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity 2011. 35: 109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lord J, Chen J, Thirlby RC, Sherwood AM and Carlson CS, T-cell receptor sequencing reveals the clonal diversity and overlap of colonic effector and FOXP3+ T cells in ulcerative colitis. Inflamm Bowel Dis 2015. 21: 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim YC, Bhairavabhotla R, Yoon J, Golding A, Thornton AM, Tran DQ and Shevach EM, Oligodeoxynucleotides stabilize Helios-expressing Foxp3+ human T regulatory cells during in vitro expansion. Blood 2012. 119: 2810–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thornton AM, Piccirillo CA and Shevach EM, Activation requirements for the induction of CD4+CD25+ T cell suppressor function. Eur J Immunol 2004. 34: 366–376. [DOI] [PubMed] [Google Scholar]
- 51.Kim D, Langmead B and Salzberg SL, HISAT: a fast spliced aligner with low memory requirements. Nat Methods 2015. 12: 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anders S, Pyl PT and Huber W, HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015. 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Love MI, Huber W and Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014. 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.