

Abstract
Signaling complexes targeting the myofilament are essential in modulating cardiac performance. A central target of this signaling is cardiac troponin I (cTnI) phosphorylation. This review focuses on cTnI phosphorylation as a model for myofilament signaling, discussing key gaps and future directions towards understanding complex myofilament modulation of cardiac performance. Human heart cTnI is phosphorylated at 14 sites, giving rise to a complex modulatory network of varied functional responses. For example, while classical Ser23/24 phosphorylation mediates accelerated relaxation, protein kinase C phosphorylation of cTnI serves as a brake on contractile function. Additionally, the functional response of cTnI multi-site phosphorylation cannot necessarily be predicted from the response of individual sites alone. These complexities underscore the need for systematically evaluating single and multi-site phosphorylation on myofilament, cellular and in vivo contractile function. Ultimately, a complete understanding of these multi-site responses requires work to establish site occupancy and dominance, kinase / phosphatase signaling balance, and the function of adaptive secondary phosphorylation. As cTnI phosphorylation is essential for modulating cardiac performance, future insight into the complex role of cTnI phosphorylation is important to establish sarcomere signaling in the healthy heart as well as identification of novel myofilament targets in the treatment of disease.
Keywords: troponin I, phosphorylation, cardiac, contraction, relaxation, signaling
Cardiac performance is determined by rhythmic cycles of systolic contraction and diastolic relaxation, to pump blood to meet the energy demands of peripheral tissues1. Nearly 80% of protein within the contractile cardiac myocyte is organized into sarcomeres, assembled in parallel arrays of myofilaments. Each sarcomere is composed of myosin-containing thick filaments and regulatory thin filaments (TFs) ultimately responsible for contraction and relaxation in response to intracellular Ca2+ transients. The cardiac TF is composed of actin, tropomyosin and a heterotrimeric troponin (cTn), consisting of the calcium-binding subunit, troponin C (cTnC); the tropomyosin-binding subunit, troponin T; and the inhibitory subunit troponin I (cTnI). When intracellular Ca2+ levels are low, cTn and tropomyosin sterically inhibits actin interactions with myosin. With each heartbeat, action potential-induced increases in intracellular Ca2+ bind to cTn inducing TF conformational changes, allowing myosin to strongly bind actin and generate force. During relaxation, Ca2+ dissociates from cTn as intracellular Ca2+ levels decrease, and the interaction between actin and myosin is once again blocked by cTn2.
Signaling mechanisms are critical to modulate each component of enhanced cardiac pump performance in response to increased peripheral energy demand. Several earlier reviews have summarized modulatory mechanisms of myosin3 and Ca2+ 4. This review focuses on phosphorylation of the TF molecular switch protein cTnI, which was the first reported sarcomere-based cardiac post-translational modification (PTM)5, 6. As an increasing number of signaling- and stress- pathways targeting cTnI for phosphorylation at multiple residue sites are identified7, it has become appreciated that the phosphorylation of cTnI modulates contractile function to meet bodily demand much like a rheostat. Understanding cTnI-mediated modulation of contraction and relaxation has reached a crossroads that requires integrated insight into the functional role played by these multiple phosphorylation sites. The functional response of cTnI phosphorylation is accordingly expected to serve as a template for gaining insight into PTM of other TF proteins. We therefore review the established mechanisms and new directions of cTnI-mediated cardiac modulation mediated via phosphorylation. The influences of other TF PTMs are also important, and are covered in more detail in earlier reviews.7
Cardiac troponin I Ser23/24 phosphorylation: Historical perspective
Phosphorylation of cTnI was first described in the early 1970’s5, 6, and its physiological significance was established in studies showing cTnI phosphorylation closely correlated in time with the in vivo response to β-adrenergic receptor (β-AR) activation of protein kinase A (PKA) and the increase in cardiac pump performance8, 9. Later biochemical and biophysical work demonstrated PKA phosphorylates Ser23 and Ser24 residues in the cardiac-specific, N-terminal extension of human cTnI (Fig. 1A, B; all residues discussed are numbered according to the human cTnI sequence containing the initial methionine; Uniprot P19429), to increase Ca2+ dissociation from cTnC and reduce the Ca2+ sensitivity of myofilament force10–12. These changes in myofilament function translated into the important accelerated relaxation, or positive lusitropic response observed in response to PKA activation13. Extensive studies subsequently demonstrated cTnI Ser23/24 phosphorylation accelerates in vivo relaxation14–17. The replacement of endogenous cTnI with phospho-mimetic cTnI Ser23/24Asp in transgenic animal models provided direct evidence showing cTnI mediated Ser23/24 phosphorylation and the shift in myofilament Ca2+ sensitivity significantly contributes to the accelerated relaxation in response to β1-AR-mediated activation of cardiac PKA14, 15. Further support for this idea was demonstrated by the absence of a PKA-induced change in myofilament Ca2+ sensitivity of knock-in cTnISer23/24Ala mice18, 19. The transduction mechanism for cTnI Ser23/24 phosphorylation mediated accelerated relaxation resulted from the reduced ability of the cTnI H3 helix “switch peptide” to insert into the hydrophobic patch of cTnC in response to Ca2+ 16 which accelerates the release of cTnI from cTnC20 (Figure 1B)
FIGURE 1.
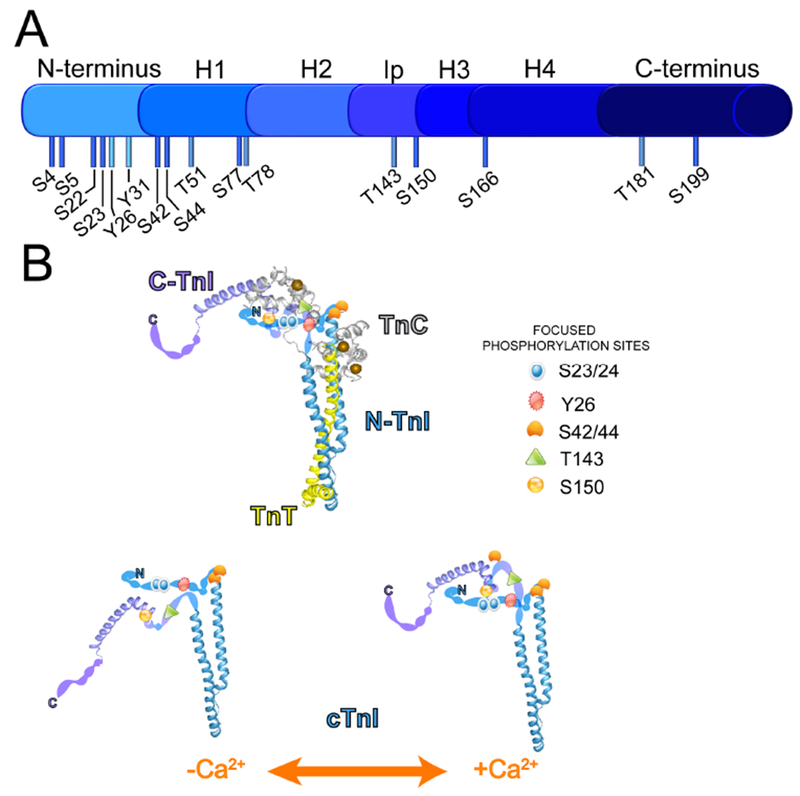
A. Location of known Ser (S), Thr (T), and Tyr (Y) phosphorylated residues on cardiac troponin I (cTnI). B. Location of phosphorylation sites within the troponin complex composed of cTnI, troponin T (TnT) and troponin C (TnC) and in the proposed TnI protein structure in the absence (−Ca2+) and presence (+Ca2+) bound to TnC. N-TnI refers to the amino-terminus and C-TnI to the carboxy-terminus of TnI. Structures in B are adapted from Takeda et al., 2003 (ref 72).
Under physiological conditions, cTnI Ser24 is more likely to be constitutively phosphorylated in the myocardium21,22 and is phosphorylated more quickly than Ser2312, 21. Until recently, accelerated relaxation was believed to be dependent on both Ser23 and Ser24 phosphorylation 12, 22. More recent work demonstrated this accelerated relaxation is possible when only one of the Ser sites is phosphorylated, however a complete understanding of the in vivo role of this single phosphorylation has yet to be determined23. In addition to physiological regulation, cTnI Ser23/24 phosphorylation is also important in pathophysiological conditions with Ser23/24 phosphorylation increasing during ischemia24, 25 but decreasing in human and animal models of decompensated heart failure26–28. In heart failure the down regulation of β-AR signaling reduces cTnISer23/24 phosphorylation which in turn slows myocardial relaxation to further deteriorate performance29, 30.
Dynamic modulation of the sarcomere: Multiple kinases and cTnI target residues
In addition to PKA, the cTnI Ser23/24 phosphorylation cluster also is a target for other AGC kinases PKG and PKC31, 32, as well as kinases from other kinome families, including PKD33, ROCKII34, RSK35, 36, and PAK137. There is also evidence AMPK and phosphorylase kinase phosphorylate the Ser23 residue38–40. Moreover, human cTnI residues are phosphorylated at 12 additional sites including Ser5, Ser6, Tyr26, Ser42, Ser44, Thr51, Ser77, Thr78, Thr143, Ser166, Thr181, Ser199, and residues equivalent to Tyr31 and Ser150 are phosphorylated in mammalian hearts39, 41, 42 (Figure 1A). Unlike Ser-23/24, the kinases responsible for phosphorylation at these sites are often not known or incomplete.
The functional output for most of these other cTnI phosphorylation sites remains to be thoroughly investigated, especially at the cellular and in vivo level. Classically, there is a dogma that cTnI phosphorylation functions as an “accelerator’ to enhance relaxation during periods of enhanced cardiac performance, which is largely based upon extensive work showing Ser23/24 phosphorylation accelerates diastolic relaxation13–15, 17. However, it is becoming increasingly clear that phosphorylation at other cTnI sites can act as a “brake” to ultimately slow and/or reduce pump function43–45. Thus, multiple kinases can target a single cTnI site, such as Ser23/24, and sites other than Ser23/24 within cTnI to produce different functional responses. As a result, there is growing recognition that cTnI modulation of the sarcomere is dynamic and requires an integrated understanding of the role that multiple phosphorylated sites play within cTnI as well as coordination at a single site by multiple kinases and phosphatases.
PKC phosphorylation of cTnI: An example of cTnI phosphorylation acting as a “brake”
In contrast to the accelerated relaxation produced by cTnI Ser23/24 phosphorylation, Ser42/44 or Thr143 phosphorylation responses are better defined as “brakes”. Protein kinase C (PKC) targets these 2 clusters in addition to Ser23/24 for phosphorylation32, 46(Figure 1B). Increased phosphorylation of these sites is reported under physiological and pathophysiological conditions41, 47–51, although technical issues have made it difficult to precisely establish Ser42 versus Ser44 phosphorylation levels. Experimental phosphorylation of cTnI Ser42/44 and phospho-mimetic substitutions each cause reduced myofilament Ca2+ sensitivity of tension, which is likely explained by conformational interactions between cTnI and cTnC that produce a higher dissociation constant (Kd) and faster Ca2+ off rate from cTn45, 52–54. In addition, phospho-mimetic cTnI Ser42/44 reduces peak tension and slows sliding speed in motility assays45. Studies on intact myocytes serve as an important bridge for integrating in vitro and in vivo studies, by showing that cTnI-Ser42Asp or –Ser44Asp each act as a brake to reduce the amplitude and rate of myocyte shortening and re-lengthening, although their influence is not additive43. Both cTnI- Ser42Asp and -Ser44Asp appear to dominantly reduce contractile function, even in the presence of equal amounts of phospho-mimetic Ser23/24Asp44.
Similar to Ser-42/44, biochemical studies demonstrate Thr143 phosphorylation decreases the Ca2+ sensitivity of in vitro sliding speed, but not isometric tension45, 55. In addition, phospho-mimetic Thr143 has little influence or decreases the thin filament Ca2+ off rate53, 55, indicating Thr143 works via a different mechanism than Ser42/44. Unlike Ser23/24, the conformational changes responsible for the responses to Ser42/44 or Thr143 are not well defined45, 55. Mechanistic insight often depends on the use of phospho-mimetic as well as phospho-null substitutions, but these approaches have proven challenging in the study of these sites as the typical use of phospho-null Ala substitutions at cTnI Ser42/44 is not functionally conservative and instead reduces myofilament Ca2+ sensitivity19, 54, 56. However, intact myocyte work shows that myofilament function is conserved when using a polar Asn substitution as a phospho-null substitution at Ser42/4456, which will be helpful for gaining mechanistic insight in the future. In addition, these results highlight the need to validate phospho-mimetic and phospho-null substitutions at each phosphorylated residue.
The myofilament responses to Ser42/44 and Thr143 phosphorylation predict that in vivo models with phospho-mimetic substitutions should develop a reduction in pump function. Instead, complex and divergent phenotypes develop in mice expressing combinations of phospho-mimetic or phospho-null substitutions at the PKC-targeted cTnI Ser23/24, Ser42/44, and Thr14357–59. Moreover, there is a wide range of exogenous cTnI replacement levels observed in these mice. For example, complete replacement with phospho-mimetic cTnISer23/24/42/44/Thr143 produces only minimal reductions in diastolic function while <10% replacement with the cTnI Ser42/44/Thr143 phospho-mimetic results in severe systolic and diastolic dysfunction58, 59. To date, there are no published models with only Ser42/44 or Thr143 phospho-mimetics, or functionally conservative phospho-null substitutions to resolve the ongoing debate about the roles these residues play in modulating cardiac performance.
Dynamic sarcomere modulation by cTnI phoshorylation: Current insights and gaps
To date, the majority of cTnI phosphorylation-mediated functional effects have been determined based on phosphorylation changes in isolation at an individual site, yet cTnI phosphorylation in the human heart does not solely occur at an individual site but rather occurs at multiple sites41. As the signaling cascades causing cTnI phosphorylation become better understood, we have begun to appreciate that kinase mediated signaling results in “phosphorylation clusters”, or phosphorylation of multiple sites by a given kinase. For example, PKC-associated cellular signaling can lead to the phosphorylation of cTnI Ser23/24, Ser42, Ser44 and Thr14332, 46. Understanding the effect of multiple cTnI site phosphorylation is further complicated by the presence of multiple signaling pathways that can become simultaneously activated and also result in phosphorylation at multiple clusters60, 61. For example, PKA mediated phosphorylation of cTnI Ser23/24 and AMPK-induced phosphorylation at Ser15039. Finally, each of these additional phosphorylation events occur on top of the basal cTnI phosphorylation. A significant portion of cTnI in the heart is basally phosphorylated and cTnI Ser23/24 comprises ~40% of this basal phosphorylation62, 63, any additional phosphorylation will occur at Ser23/24 as well as other sites. This integrated impact of basal TnI phosphorylation, together with transient signal- and stress-activated kinase activation and downstream targeting of cTnI phosphorylation clusters is important as it allows for optimal fine-tuning of cardiac performance. However, this flexibility also raises the need for studies investigating the complex myofilament, cellular, and in vivo responses and/or phenotypes produced by a phosphorylated cluster.
A difficulty towards understanding the integrated effects of multiple cTnI phosphorylation is that the functional response for a specific phosphorylation or cluster cannot necessarily be predicted based on the impact of each individual cTnI phosphorylation site. For example, phospho-mimetic cTnI Ser150Asp alone increases myofilament Ca2+ sensitivity, cooperativity and slows Ca2+ dissociation39, 40. Based on the similarly opposing impact of cTnI Ser23/24 and Ser150 phosphorylation on Ca2+ sensitivity and TF Ca2+ dissociation, cTnISer23/24/150Asp is not predicted to change Ca2+ sensitivity or Ca2+ dissociation from basal control values. However, while Ser150Asp attenuated the ability of cTnI Ser23/24Asp to decrease myofilament Ca2+ sensitivity, the Ca2+ dissociation remained accelerated similar to that of Ser23/24Asp alone 20, 39. A mechanism for Ser150 phosphorylation is not definitively established but likely involves a decrease of cTnI C-terminal binding to actin and increased binding to cTnC to slow Ca2+ dissociation that differs from the Ser23/24 mechanism and includes modified cooperativity20, 64 (Figure 1B). While the in vivo effect has yet to be determined, these results suggest that combination of cTnI Ser150 on top of basal Ser23/24 phosphorylation can enhance myocardial force production without altering Ca2+ transients and/or causing diastolic dysfunction in intact hearts, which could not be anticipated based on the individual effects of each phosphorylation site. Integrated cTnI Ser150 and Ser23/24 phosphorylation may therefore be significant during myocardial ischemia when phosphorylation at both sites is elevated25 and to improve cardiac performance during pathological systolic dysfunction.
Another example of integrated cTnI phosphorylation functional effects that would not be anticipated from the effects of the individual sites alone is observed from the function of combined cTnI Tyr26 and Ser23/24 phosphorylation. Isolated Tyr26 phosphorylation (Figure 1B), Tyr26Glu and Tyr26Asp each decreased myofilament Ca2+ sensitivity to a similar extent as Ser23/24Asp and Tyr26Glu accelerated TF Ca2+ dissociation similarly to Ser23/24Asp65. In contrast, the combination of the Ser23/24Asp/Tyr26Glu phospho-mimetic resulted in the expected non-additive reduction in Ca2+ sensitivity, but there was also a further doubling in the acceleration of TF Ca2+ dissociation. These findings suggest that the combination of cTnI Tyr26 on top of basal Ser23/24 phosphorylation further enhances diastolic relaxation beyond that of either phosphorylation alone, without further depressing systolic function. Increasing both cTnI Ser23/24 and Tyr26 phosphorylation therefore appears to be more effective at improving diastolic dysfunction in cardiac disease, such as heart failure with preserved ejection fraction, compared to either individual phosphorylation alone.
This integration of phosphorylation clusters is of further significance as it also extends beyond intra-cTnI interactions and includes evidence for communication between myofilament proteins44, 66. Together with the functional response to PKC –targeted cTnI presented above, these examples illustrate the important point that the independent functional effects of each phosphorylation site alone may not predict the response resulting from the integrated effects of multiple cTnI phosphorylations. The importance of this concept is further highlighted as the transduction mechanisms of phosphorylation integration within cTn is largely unknown for phosphorylation sites other than Ser23/24.
Our knowledge of integrated cTnI phosphorylation is further limited by our lack of understanding as to how phosphorylation occupancy level and functional dominance among phosphorylation sites modulate contractile function. While there is limited information about the functional dominance of one cTnI phosphorylation site compared to another, emerging evidence is describing the percent occupancy of individual cTnI phosphorylation sites under specific conditions41. An accurate quantification of the occupancy at any given site and the relative functional dominance of a site at a given occupancy are however challenging to determine and have yet to be systematically investigated. Moreover, it is unclear whether the same residues are phosphorylated on each cTnI or instead may be more heterogeneously dispersed either spatially or temporally across the population of cTnI within a sarcomere. Even though these important issues have yet to be resolved, it is clear that there is constitutive phosphorylation at multiple sites within cTnI that contributes to a basal state of contractile function41. Both kinase and phosphatase activity modulate contractile function by changing cTnI phosphorylation7, and the further participation of both kinases and phosphatases are essential for oscillatory or dynamic signaling67, 68. As an end target for multiple kinases and phosphatases, cTnI phosphorylation is therefore a central target for fine-tuning cardiac performance in response to physiological and environmental factors as well as during stress and pathophysiological conditions. Dynamic modulation via cTnI is essential for compensation during chronic dysfunction and a reduction in the ability of different cTnI phosphorylation sites to respond could accelerate or enhance contractile dysfunction. Thus, efforts to systemically address concepts such occupancy and dominance among the multiple cTnI phosphorylation sites is needed in the future.
A final issue when considering the integration of multiple phosphorylation sites is the growing evidence that sustained phosphorylation at one cTnI site triggers secondary signaling at other sites. This idea of a “secondary phosphorylation” response was first reported in mice expressing cTnISer42/44Ala69. Subsequently, the potential for secondary phosphorylation to play an adaptive functional role was demonstrated in isolated myocytes expressing cTnI Ser42Asp or Ser44Asp phospho-mimetics43, 44 In these studies, the initial reduction in shortening rate resulting from expression of these phospho-mimetics partially returned to baseline over time even though Ser42Asp or Ser44Asp replacement continued to increase. This partial return of contractile function concurrent with secondary phosphorylation at other cTnI sites and myofilament proteins43, 44 led to the conclusion that the onset of secondary phosphorylation serves an adaptive role to return function back toward a basal steady state or “set-point”, the absence of which produces a stress that results in disease70. This conclusion is consistent with the variable systolic and diastolic phenotypes reported in mice expressing cTnISer42/44 combined with Ser23/24 and/or Tl43 phosphomimetics57–59, 69. Adaptation of function in response to secondary cTnI phosphorylation adds another level of complexity, but also establishes cTnI as an important contributor to fine-tune sarcomere function. The consistent association between heart disease and elevated PKC activity, which targets Ser42/44 and Thr14332, 49, 54, 71 also suggests that this secondary phosphorylation may be particularly important during cycles of stress or under chronic pathological conditions. As a result, the absence or loss of the secondary phosphorylation response over time may push the sarcomere further from its set-point causing a faster and/or more severe deterioration in cardiac function. Alternately, maintaining or initiating additional secondary phosphorylation may restore the sarcomeric set-point and delay disease progression as demonstrated by expression of the desensitizing TnI Ser23/24Asp phospho-mimetic in a sensitized, genetic hypertrophic model70. Thus, future systematic studies are critical to understand spatial and temporal secondary phosphorylation and their impact on myocyte and in vivo adaptive function after primary cTnI phosphorylation events or pathological stress.
Future directions
The dynamic nature of cTnI phosphorylation discussed above shows that this molecular switch protein plays an essential role in modulating sarcomere function in both health and disease. Specifically, modulation via cTnI helps maintain basal contractile function under physiological conditions, and could also play a central role in adaptive phosphorylation during the compensatory phase of heart disease. Thus, it is critical to fully understand the impact of multiple integrated cTnI phosphorylation sites and clusters on cardiac performance. To achieve this goal requires future research utilizing multidisciplinary approaches to define the signaling cascades that target cTnI, their sites of phosphorylation and their temporal activation in response to environmental and/or pathophysiological conditions. The transduction mechanism(s) resulting from phosphorylation events and their functional affects also need to be defined at both the myofilament and cellular level. Insight into the integrated effects of multiple phosphorylations and the dynamic component of a cTnI signal is equally important and requires both cellular and in vivo approaches to evaluate the response to primary phosphorylation alone, the resulting changes in signaling and/or secondary phosphorylation events and pathological induced stress. These responses should also address whether a phosphorylation cluster observed under physiological conditions is changed under pathophysiological conditions or in response to therapeutic drugs and devices. The resulting work will fill significant gaps in our understanding of cTn and TF function, provide significant insight into the dynamic nature of the sarcomere and its regulation as well as ultimately make available a more complete understanding of the disease process and novel areas to intervene.
Acknowledgments
Funding
This work was supported by the National Institutes of Health NIH to B.J.B (HL114940).
Abbreviations
- TFs
thin filaments
- cTn
cardiac troponin
- cTnC
cardiac troponin C
- cTnI
cardiac troponin I
- PTM
post-translational modification
- β-AR
β-adrenergic receptor
- PKA
protein kinase A
- PKC
protein kinase C
Footnotes
Declarations of interest: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wilborn CD and Willoughby DS. The role of dietary protein intake and resistance training on Myosin heavy chain expression. J Int Soc Sports Nutr. 2004;1:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kobayashi T and Solaro RJ. Calcium, thin filaments, and the integrative biology of cardiac contractility. Annu Rev Physiol. 2005;67:39–67. [DOI] [PubMed] [Google Scholar]
- 3.Gordon aM , Homsher E and Regnier M Regulation of contraction in striated muscle. Physiol Rev. 2000;80:853–924. [DOI] [PubMed] [Google Scholar]
- 4.Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. 2 ed: Springer; Netherlands; 2001. [Google Scholar]
- 5.Perry SV and Cole HA. Phosphorylation of troponin and the effects of interactions between the components of the complex. Biochem J. 1974;141:733–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reddy YS, Ballard D, Giri NY and Schwartz A. Phosphorylation of cardiac native tropomyosin and troponin: inhibitory effect of actomyosin and possible presence of endogenous myofibrillar-located cyclic-AMP-dependent protein kinase. J Mol Cell Cardiol. 1973;5:461–71. [DOI] [PubMed] [Google Scholar]
- 7.Westfall MV. Post-translational modifications of troponin In: Jin JP, ed. Troponin:Regulator of muscle contraction New York: Nova; 2014: 163–202. [Google Scholar]
- 8.England PJ. Correlation between contraction and phosphorylation of the inhibitory subunit of troponin in perfused rat heart. FEBS Lett. 1975;50:57–60. [DOI] [PubMed] [Google Scholar]
- 9.Solaro RJ, Moir AJ and Perry SV. Phosphorylation of troponin I and the inotropic effect of adrenaline in the perfused rabbit heart. Nature. 1976;262:615–7. [DOI] [PubMed] [Google Scholar]
- 10.Robertson SP, Johnson JD, Holroyde MJ, Kranias EG, Potter JD and Solaro RJ. The effect of troponin I phosphorylation on the Ca2+-binding properties of the Ca2+-regulatory site of bovine cardiac troponin. J Biol Chem. 1982;257:260–3. [PubMed] [Google Scholar]
- 11.Swiderek K, Jaquet K, Meyer HE and Heilmeyer LM Jr. Cardiac troponin I, isolated from bovine heart, contains two adjacent phosphoserines. A first example of phosphoserine determination by derivatization to S-ethylcysteine. Eur J Biochem. 1988;176:335–42. [DOI] [PubMed] [Google Scholar]
- 12.Zhang R, Zhao J and Potter JD. Phosphorylation of both serine residues in cardiac troponin I is required to decrease the Ca2+ affinity of cardiac troponin C. J Biol Chem. 1995;270:30773–80. [DOI] [PubMed] [Google Scholar]
- 13.Kentish JC, McCloskey DT, Layland J, Palmer S, Leiden JM, Martin AF and Solaro RJ. Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circ Res. 2001;88:1059–65. [DOI] [PubMed] [Google Scholar]
- 14.Takimoto E, Soergel DG, Janssen PM, Stull LB, Kass DA and Murphy AM. Frequency- and afterload-dependent cardiac modulation in vivo by troponin I with constitutively active protein kinase A phosphorylation sites. Circ Res. 2004;94:496–504. [DOI] [PubMed] [Google Scholar]
- 15.Yasuda S, Coutu P, Sadayappan S, Robbins J and Metzger JM. Cardiac transgenic and gene transfer strategies converge to support an important role for troponin I in regulating relaxation in cardiac myocytes. Circ Res. 2007;101:377–86. [DOI] [PubMed] [Google Scholar]
- 16.Dong WJ, Jayasundar JJ, An J, Xing J and Cheung HC. Effects of PKA phosphorylation of cardiac troponin I and strong crossbridge on conformational transitions of the N-domain of cardiac troponin C in regulated thin filaments. Biochemistry. 2007;46:9752–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang R, Zhao J, Mandveno A and Potter JD. Cardiac troponin I phosphorylation increases the rate of cardiac muscle relaxation. Circ Res. 1995;76:1028–35. [DOI] [PubMed] [Google Scholar]
- 18.Pi Y, Kemnitz KR, Zhang D, Kranias EG and Walker JW. Phosphorylation of troponin I controls cardiac twitch dynamics: evidence from phosphorylation site mutants expressed on a troponin I-null background in mice. Circ Res. 2002;90:649–56. [DOI] [PubMed] [Google Scholar]
- 19.Pi Y, Zhang D, Kemnitz KR, Wang H and Walker JW. Protein kinase C and A sites on troponin I regulate myofilament Ca2+ sensitivity and ATPase activity in the mouse myocardium. J Physiol. 2003;552:845–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salhi HE, Hassel NC, Siddiqui JK, Brundage EA, Ziolo MT, Janssen PM, Davis JP and Biesiadecki BJ. Myofilament Calcium Sensitivity: Mechanistic Insight into TnI Ser-23/24 and Ser-150 Phosphorylation Integration. Front Physiol. 2016;7:567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keane NE, Quirk PG, Gao Y, Patchell VB, Perry SV and Levine BA. The ordered phosphorylation of cardiac troponin I by the cAMP-dependent protein kinase--structural consequences and functional implications. Eur J Biochem. 1997;248:329–37. [DOI] [PubMed] [Google Scholar]
- 22.Mittmann K, Jaquet K and Heilmeyer LM Jr., Ordered phosphorylation of a duplicated minimal recognition motif for cAMP-dependent protein kinase present in cardiac troponin I. FEBS Lett. 1992;302:133–7. [DOI] [PubMed] [Google Scholar]
- 23.Martin-Garrido A, Biesiadecki BJ, Salhi HE, Shaifta Y, Dos Remedios CG, Ayaz-Guner S, Cai W, Ge Y, Avkiran M and Kentish JC. Monophosphorylation of cardiac troponin-I at Ser-23/24 is sufficient to regulate cardiac myofibrillar Ca(2+) sensitivity and calpain-induced proteolysis. J Biol Chem. 2018;293:8588–8599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Avner Bs, Shioura KM, Scruggs SB, Grachoff M, Geenen DL, Helseth DL Jr., Farjah M, Goldspink PH and Solaro RJ. Myocardial infarction in mice alters sarcomeric function via post-translational protein modification. Mol Cell Biochem. 2012;363:203–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nixon BR, Walton SD, Zhang B, Brundage EA, Little SC, Ziolo MT, Davis JP and Biesiadecki BJ. Combined troponin I Ser-150 and Ser-23/24 phosphorylation sustains thin filament Ca(2+) sensitivity and accelerates deactivation in an acidic environment. J Mol Cell Cardiol. 2014;72:177–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bartel S, Stein B, Eschenhagen T, Mende U, Neumann J, Schmitz W, Krause EG, Karczewski P and Scholz H. Protein phosphorylation in isolated trabeculae from nonfailing and failing human hearts. Mol Cell Biochem. 1996;157:171–9. [DOI] [PubMed] [Google Scholar]
- 27.Bodor GS, Oakeley AE, Allen PD, Crimmins DL, Ladenson JH and Anderson PA. Troponin I phosphorylation in the normal and failing adult human heart. Circulation. 1997;96:1495–500. [DOI] [PubMed] [Google Scholar]
- 28.Zakhary DR, Moravec CS, Stewart RW and Bond M. Protein kinase A (PKA)-dependent troponin-I phosphorylation and PKA regulatory subunits are decreased in human dilated cardiomyopathy. Circulation. 1999;99:505–10. [DOI] [PubMed] [Google Scholar]
- 29.McConnell BK, Moravec CS and Bond M. Troponin I phosphorylation and myofilament calcium sensitivity during decompensated cardiac hypertrophy. Am J Physiol. 1998;274:H385–96. [DOI] [PubMed] [Google Scholar]
- 30.Wolff MR, Buck SH, Stoker SW, Greaser ML and Mentzer RM. Myofibrillar calcium sensitivity of isometric tension is increased in human dilated cardiomyopathies: role of altered beta-adrenergically mediated protein phosphorylation. J Clin Invest. 1996;98:167–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blumenthal DK, Stull JT and Gill GN. Phosphorylation of cardiac troponin by guanosine 3’:5’-monophosphate-dependent protein kinase. J Biol Chem. 1978;253:324–6. [PubMed] [Google Scholar]
- 32.Venema RC and Kuo JF. Protein kinase C-mediated phosphorylation of troponin I and C-protein in isolated myocardial cells is associated with inhibition of myofibrillar actomyosin MgATPase. J Biol Chem. 1993;268:2705–11. [PubMed] [Google Scholar]
- 33.Haworth RS, Cuello F, Herron TJ, Franzen G, Kentish JC, Gautel M and Avkiran M. Protein kinase D is a novel mediator of cardiac troponin I phosphorylation and regulates myofilament function. Circ Res. 2004;95:1091–9. [DOI] [PubMed] [Google Scholar]
- 34.Vahebi S, Kobayashi T, Warren CM, de Tombe PP and Solaro RJ. Functional effects of rho-kinase-dependent phosphorylation of specific sites on cardiac troponin. Circ Res. 2005;96:740–7. [DOI] [PubMed] [Google Scholar]
- 35.Cuello F, Bardswell SC, Haworth RS, Ehler E, Sadayappan S, Kentish JC and Avkiran M. Novel role for p90 ribosomal S6 kinase in the regulation of cardiac myofilament phosphorylation. J Biol Chem. 2011;286:5300–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Itoh S, Ding B, Bains CP, Wang N, Takeishi Y, Jalili T, King GL, Walsh RA, Yan C and Abe J. Role of p90 ribosomal S6 kinase (p90RSK) in reactive oxygen species and protein kinase C beta (PKC-beta)-mediated cardiac troponin I phosphorylation. J Biol Chem. 2005;280:24135–42. [DOI] [PubMed] [Google Scholar]
- 37.Sheehan KA, Ke Y, Wolska BM and Solaro RJ. Expression of active p21-activated kinase-1 induces Ca2+ flux modification with altered regulatory protein phosphorylation in cardiac myocytes. Am J Physiol Cell Physiol. 2009;296:C47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aj Moir and Sv Perry. Phosphorylation of rabbit cardiac-muscle troponin I by phosphorylase kinase. The effect of adrenaline. Biochem J. 1980;191:547–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nixon BR, Thawornkaiwong A, Jin J, Brundage EA, Little SC, Davis JP, Solaro RJ and Biesiadecki BJ. AMP-activated protein kinase phosphorylates cardiac troponin I at Ser-150 to increase myofilament calcium sensitivity and blunt PKA-dependent function. J Biol Chem. 2012;287:19136–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oliveira SM, Zhang YH, Solis RS, Isackson H, Bellahcene M, Yavari A, Pinter K, Davies JK, Ge Y, Ashrafian H, Walker JW, Carling D, Watkins H, Casadei B and Redwood C. AMP-activated protein kinase phosphorylates cardiac troponin I and alters contractility of murine ventricular myocytes. Circ Res. 2012;110:1192–201. [DOI] [PubMed] [Google Scholar]
- 41.Zhang P, Kirk JA, Ji W, dos Remedios CG, Kass DA, Van Eyk JE and Murphy AM. Multiple reaction monitoring to identify site-specific troponin I phosphorylated residues in the failing human heart. Circulation. 2012;126:1828–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.You B, Yan G, Zhang Z, Yan L, Li J, Ge Q, Jin JP and Sun J. Phosphorylation of cardiac troponin I by mammalian sterile 20-like kinase 1. Biochem J. 2009;418:93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lang SE, Schwank J, Stevenson TK, Jensen MA and Westfall MV. Independent modulation of contractile performance by cardiac troponin I Ser43 and Ser45 in the dynamic sarcomere. J Mol Cell Cardiol. 2015;79:264–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lang SE, Stevenson TK, Schatz TM, Biesiadecki BJ and Westfall MV. Functional communication between PKC-targeted cardiac troponin I phosphorylation sites. Arch Biochem Biophys. 2017;627:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burkart EM, Sumandea MP, Kobayashi T, Nili M, Martin AF, Homsher E and Solaro RJ. Phosphorylation or glutamic acid substitution at protein kinase C sites on cardiac troponin I differentially depress myofilament tension and shortening velocity. J Biol Chem. 2003;278:11265–72. [DOI] [PubMed] [Google Scholar]
- 46.Noland TA Jr and Kuo JF. Protein kinase C phosphorylation of cardiac troponin I or troponin T inhibits Ca2(+)-stimulated actomyosin MgATPase activity. J Biol Chem. 1991;266:4974–8. [PubMed] [Google Scholar]
- 47.Dong X, Sumandea CA, Chen YC, Garcia-Cazarin ML, Zhang J, Balke CW, Sumandea MP and Ge Y. Augmented phosphorylation of cardiac troponin I in hypertensive heart failure. J Biol Chem. 2012;287:848–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jideama NM, Noland TA Jr, Raynor RL, Blobe GC, Fabbro D, Kazanietz MG, Blumberg PM, Hannun YA and Kuo JF. Phosphorylation specificities of protein kinase C isozymes for bovine cardiac troponin I and troponin T and sites within these proteins and regulation of myofilament properties. J Biol Chem. 1996;271:23277–83. [DOI] [PubMed] [Google Scholar]
- 49.Noguchi T, Hunlich M, Camp PC, Begin KJ, El-Zaru M, Patten R, Leavitt BJ, Ittleman FP, Alpert NR, LeWinter MM and VanBuren P. Thin-filament-based modulation of contractile performance in human heart failure. Circulation. 2004;110:982–7. [DOI] [PubMed] [Google Scholar]
- 50.Walker LA, Fullerton DA and Buttrick PM. Contractile protein phosphorylation predicts human heart disease phenotypes. Am J Physiol Heart Circ Physiol. 2013;304:H1644–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walker LA, Walker JS, Ambler SK and Buttrick PM. Stage-specific changes in myofilament protein phosphorylation following myocardial infarction in mice. J Mol Cell Cardiol. 2010;48:1180–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hinken AC, Hanft LM, Scruggs SB, Sadayappan S, Robbins J, Solaro RJ and McDonald KS. Protein kinase C depresses cardiac myocyte power output and attenuates myofilament responses induced by protein kinase A. J Muscle Res Cell Motil. 2012;33:439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu B, Lopez JJ, Biesiadecki BJ and Davis JP. Protein kinase C phosphomimetics alter thin filament Ca2+ binding properties. PLoS One. 2014;9:e86279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Noland TA Jr, Raynor rL , Jideama NM, Guo X, Kazanietz MG, Blumberg PM, Solaro RJ and Kuo JF. Differential regulation of cardiac actomyosin S-1 MgATPase by protein kinase C isozyme-specific phosphorylation of specific sites in cardiac troponin I and its phosphorylation site mutants. Biochemistry. 1996;35:14923–31. [DOI] [PubMed] [Google Scholar]
- 55.Lu QW, Hinken AC, Patrick SE, Solaro RJ and Kobayashi T. Phosphorylation of cardiac troponin I at protein kinase C site threonine 144 depresses cooperative activation of thin filaments. J Biol Chem. 2010;285:11810–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lang SE, Stevenson TK, Xu D, O’Connell R and Westfall MV. Functionally conservative substitutions at cardiac troponin I S43/45. Arch Biochem Biophys. 2016;601:42–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bilchick KC, Duncan JG, Ravi R, Takimoto E, Champion HC, Gao WD, Stull LB, Kass DA and Murphy AM. Heart failure-associated alterations in troponin I phosphorylation impair ventricular relaxation-afterload and force-frequency responses and systolic function. Am J Physiol Heart Circ Physiol. 2007;292:H318–25. [DOI] [PubMed] [Google Scholar]
- 58.Kirk JA, MacGowan GA, Evans C, Smith SH, Warren CM, Mamidi R, Chandra M, Stewart AF, Solaro RJ and Shroff SG. Left ventricular and myocardial function in mice expressing constitutively pseudophosphorylated cardiac troponin I. Circ Res. 2009;105:1232–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sakthivel S, Finley NL, Rosevear PR, Lorenz JN, Gulick J, Kim S, VanBuren P, Martin LA and Robbins J. In vivo and in vitro analysis of cardiac troponin I phosphorylation. J Biol Chem. 2005;280:703–14. [DOI] [PubMed] [Google Scholar]
- 60.Solaro RJ. Multiplex kinase signaling modifies cardiac function at the level of sarcomeric proteins. J Biol Chem. 2008;283:26829–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Solaro RJ and Kobayashi T. Protein phosphorylation and signal transduction in cardiac thin filaments. J Biol Chem. 2011;286:9935–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ayaz-Guner S, Zhang J, Li L, Walker JW and Ge Y. In vivo phosphorylation site mapping in mouse cardiac troponin I by high resolution top-down electron capture dissociation mass spectrometry: Ser22/23 are the only sites basally phosphorylated. Biochemistry. 2009;48:8161–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang J, Guy MJ, Norman HS, Chen YC, Xu Q, Dong X, Guner H, Wang S, Kohmoto T, Young KH, Moss RL and Ge Y. Top-down quantitative proteomics identified phosphorylation of cardiac troponin I as a candidate biomarker for chronic heart failure. J Proteome Res. 2011; 10:4054–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ouyang Y, Mamidi R, Jayasundar JJ, Chandra M and Dong WJ. Structural and kinetic effects of PAK3 phosphorylation mimic of cTnI(S151E) on the cTnC-cTnI interaction in the cardiac thin filament. J Mol Biol. 2010;400:1036–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Salhi HE, Walton SD, Hassel NC, Brundage EA, de Tombe PP, Janssen PM, Davis JP and Biesiadecki BJ. Cardiac troponin I tyrosine 26 phosphorylation decreases myofilament Ca2+ sensitivity and accelerates deactivation. J Mol Cell Cardiol. 2014;76:257–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kooij V, Saes M, Jaquet K, Zaremba R, Foster DB, Murphy AM, Dos Remedios C, van der Velden J and Stienen GJ. Effect of troponin I Ser23/24 phosphorylation on Ca2+-sensitivity in human myocardium depends on the phosphorylation background. J Mol Cell Cardiol. 2010;48:954–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chaudhri VK, Kumar D, Misra M, Dua R and Rao KV. Integration of a phosphatase cascade with the mitogen-activated protein kinase pathway provides for a novel signal processing function. J Biol Chem. 2010;285:1296–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Samoilov M, Plyasunov S and Arkin AP. Stochastic amplification and signaling in enzymatic futile cycles through noise-induced bistability with oscillations. Proc Natl Acad Sci U S A. 2005;102:2310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Montgomery DE, Wolska BM, Pyle WG, Roman BB, Dowell JC, Buttrick PM, Koretsky AP, Del Nido P and Solaro RJ. alpha-Adrenergic response and myofilament activity in mouse hearts lacking PKC phosphorylation sites on cardiac TnI. Am J Physiol Heart Circ Physiol. 2002;282:H2397–405. [DOI] [PubMed] [Google Scholar]
- 70.Alves ML, Dias FA, Gaffin RD, Simon JN, Montminy EM, Biesiadecki BJ, Hinken AC, Warren CM, Utter MS, Davis RT 3rd, Sadayappan S, Robbins J, Wieczorek DF, Solaro RJ and Wolska BM. Desensitization of myofilaments to Ca2+ as a therapeutic target for hypertrophic cardiomyopathy with mutations in thin filament proteins. Circ Cardiovasc Genet. 2014;7:132–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bowling N, Walsh RA, Song G, Estridge T, Sandusky GE, Fouts RL, Mintze K, Pickard T, Roden R, Bristow MR, Sabbah HN, Mizrahi JL, Gromo G, King GL and Vlahos CJ. Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation. 1999;99:384–91. [DOI] [PubMed] [Google Scholar]
- 72.Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-satureated form. Nature. 2003;424:35–41. [DOI] [PubMed] [Google Scholar]