

Abstract
New antimalarial agents are identified and developed after extensive testing on Plasmodium falciparum parasites that can be grown in vitro. These susceptibility studies are important to inform lead optimisation and support further drug development. Until recently, little was known about the susceptibility of non-falciparum species as these had not been adapted to in vitro culture. The recent culture adaptation of P. knowlesi has therefore offered an opportunity to routinely define the drug susceptibility of this species, which is phylogenetically closer to all other human malarias than is P. falciparum. We compared the in vitro susceptibility of P. knowlesi and P. falciparum to a range of established and novel antimalarial agents under identical assay conditions. We demonstrated that P. knowlesi is significantly less susceptible than P. falciparum to six of the compounds tested; and notably these include three ATP4 inhibitors currently under development as novel antimalarial agents, and one investigational antimalarial, AN13762, which is 67 fold less effective against P. knowlesi. For the other compounds there was a less than two-fold difference in susceptibility between species. We then compared the susceptibility of a recent P. knowlesi isolate, UM01, to that of the well-established, older A1-H.1 clone. This recent isolate showed similar in vitro drug susceptibility to the A1-H.1 clone, supporting the ongoing use of the better characterised clone to further study drug susceptibility. Lastly, we used isobologram analysis to explore the interaction of a selection of drug combinations and showed similar drug interactions across species. The species differences in drug susceptibility reported by us here and previously, support adding in vitro drug screens against P. knowlesi to those using P. falciparum strains to inform new drug discovery and lead optimisation.
Keywords: Plasmodium falciparum, Plasmodium knowlesi, Drug susceptibility, Isobolograms
Graphical abstract
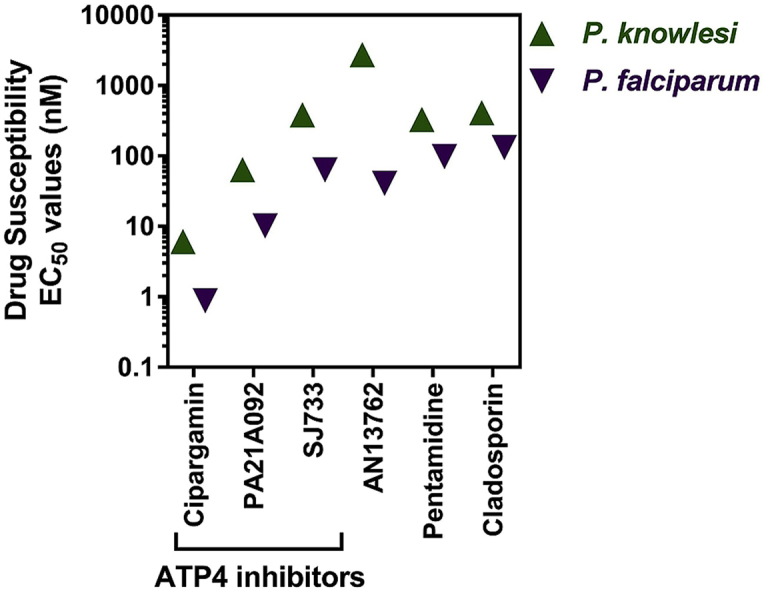
Highlights
-
•
P. knowlesi >6-fold less susceptible to several ATP4 inhibitors than P. falciparum.
-
•
P. knowlesi equally susceptible to artemisinins and synthetic endoperoxides.
-
•
Isobolograms show similar drug interactions for P. knowlesi and P. falciparum.
-
•
New P. knowlesi isolate (UM01) similarly susceptible to antimalarials as A1-H.1
1. Introduction
Since the turn of the century, advances in molecular approaches have revealed the existence of several zoonotic malaria species world-wide including; Plasmodium knowlesi (Singh et al., 2004), Plasmodium brasilianum (Lalremruata et al., 2015), Plasmodium cynomolgi (Ta et al., 2014) and Plasmodium simium (Brasil et al., 2017). Currently, P. knowlesi is the most important to human health as it has been shown to cause severe and fatal disease (Cox-Singh et al., 2008), has a 24 h life cycle, and is widespread in Asia, being the most prevalent cause of human malaria infection in Malaysia in 2014. (WHO, 2015a). An ideal antimalarial agent should kill all human-infecting malaria species. Current guidelines for non-falciparum malaria species recommend Artemisinin-based Combination Therapies (ACTs). Alternatively, chloroquine may also be used where a definitive species diagnosis can be made, and in areas where there is no evidence of chloroquine resistance (WHO, 2015b). Unfortunately, there is a dearth of information on drug susceptibility of non-falciparum malarias as we lack in vitro models to perform large-scale drug screens. However, P. knowlesi has recently been successfully adapted to long-term in vitro asexual blood stage culture (Gruring et al., 2014; Lim et al., 2013; Moon et al., 2013) making it possible to study drug susceptibility in detail, as is routinely done with P. falciparum.
Previous researchers have reported similar susceptibilities to antimalarial agents between P. falciparum and P. knowlesi in vitro (Arnold et al., 2016; Fatih et al., 2013; Paul et al., 2016). However, our recent study comparing P. falciparum and P. knowlesi in vitro under identical conditions identified significant differences in the susceptibility to dihydrofolate reductase (DHFR) inhibitors and dihydroorotate dehydrogenase (DHODH) inhibitors (van Schalkwyk et al., 2017). Several compounds are progressing through the Medicines for Malaria Venture (MMV) development pipeline and a number are now in human trials. Many of these were developed from hits identified through high-throughput screens on P. falciparum. The growth of P. knowlesi in vitro has opened the opportunity for more diverse screens of approved and experimental antimalarial agents. Importantly, P. knowlesi is more closely related phylogenetically to all the other human malaria species than is P. falciparum (Rutledge et al., 2017). P. knowlesi may thus be a suitable surrogate model for understanding the in vitro drug susceptibility of other non-falciparum malaria species.
In this study, we assess the susceptibility of the culture-adapted P. knowlesi A1-H.1 line to a range of current and emerging antimalarial agents, and compare these to the widely-available, drug susceptible P. falciparum 3D7 line. In addition, we report the drug susceptibility of a new culture-adapted P. knowlesi line, UM01, isolated from a human host in Malaysia in 2013 (Amir et al., 2016), and compare its susceptibility profile with our in vitro A1-H.1 line, which was isolated in the 1960s. Finally, we use isobologram analysis to demonstrate the in vitro interaction of selected antimalarial combinations against P. knowlesi and compare these to drug interactions in P. falciparum.
2. Material and methods
2.1. Parasite culture
P. falciparum (3D7) and P. knowlesi parasites (A1-H.1 and UM01) were maintained in culture in RPMI 1640 supplemented with 25 mM HEPES, 25 mM Na2HCO3, 10 mM D-glucose, 2 mM L-glutamine, 50 mg/L hypoxanthine, 25 mg/L gentamicin sulphate, 5 g/L Albumax II and 10% (v/v) equine serum (Thermo Fisher Scientific, 26-050-088). The P. falciparum (3D7) and P. knowlesi (A1-H.1) parasites were grown in human A+ red blood cells (National Health Blood and Transplant, UK). The UM01 was grown in erythrocytes from Macaca fascicularis provided by the National Institute for Biological Standards and Control (UK) in K2EDTA vacutainers (Becton Dickinson). Parasites were incubated in sealed flasks at 37 °C under a culture gas mixture of 96% N2, 3% CO2 and 1% O2.
2.2. Drug assays
Established antimalarial drugs and investigational compounds were supplied by the Medicines for Malaria Venture, Geneva, Switzerland. Chloroquine stocks were prepared in sterile distilled water and all other compounds were dissolved in DMSO.
All experiments were initiated using unsynchronised parasites with both the parasitaemia and haematocrit set to 1%. Drug susceptibility assays were set up in 96-well, flat-bottom microplates in a final volume of 200 μL, as described previously (van Schalkwyk et al., 2017). Controls were included for the background fluorescence (0% viability; parasites exposed to a supralethal 10 μM chloroquine concentration) and 100% growth (parasites in drug-free wells). The plates were incubated for one complete life cycle (27 h for P. knowlesi or 48 h for P. falciparum in vitro) at 37 °C in a modular incubation chamber (Billups-Rothenburg Inc.) under culture gas. Upon termination of the assay, the plates were stored at −20 °C overnight.
The SYBR green I fluorescent method was used to measure parasite survival (Bennett et al., 2004; Smilkstein et al., 2004). After the microplates were thawed, 100 μL from each well was transferred into a duplicate plate. To this was added 100 μL of SYBR green solution [SYBR green I (Thermo Fisher Scientific, S7563), diluted 1:5000 in a lysis buffer made of 20 mM Tris, 5 mM EDTA, 0.008% (w/v) saponin, 0.08% (v/v) Triton X-100, pH 7.5]. Plates were stored in the dark for 1 h, before fluorescence was read at 490 nm excitation and 520 nm emission wavelengths on a Spectramax M3 microplate reader (Molecular Devices) (van Schalkwyk et al., 2017). The background fluorescence (wells with parasites at 0% viability) was subtracted from all drug assay wells and the parasite proliferation determined as a percentage of the fluorescence in the drug-free control wells.
2.3. Drug combination studies
The in vitro interaction of a select number of drugs was examined using fixed-ratio isobolograms, prepared as described previously but using 3-fold dilutions instead of 2-fold dilutions (Fivelman et al., 2004; van Schalkwyk et al., 2008). These experiments were also performed with unsynchronised parasites at a final parasitaemia and haematocrit of 1%, and the SYBR green I fluorescent method used to measure parasite survival as described above. The Fractional Inhibitory Concentrations (FICs) were calculated as described previously (van Schalkwyk et al., 2008). Drug combinations with FICs less than 0.5 are considered synergistic, while FICs greater than 2 are antagonistic. FICs lying between 0.5 and 2 are considered additive (Bell, 2005; Odds, 2003).
2.4. Statistics
P values for comparisons between EC50 values were calculated using Student's two-tailed t-test for unpaired or paired samples as appropriate.
3. Results
3.1. Comparison of the susceptibility of P. knowlesi and P. falciparum to new and established antimalarial agents
Assays on drug-susceptible P. knowlesi A1-H.1 and P. falciparum 3D7 were performed in identical growth media, with drug exposure for a single life cycle duration (i.e., 27 h for P. knowlesi and 48 h for P. falciparum in vitro), and growth measured using the SYBR green I fluorescence method as reported previously (van Schalkwyk et al., 2017). The EC50 values for several experimental and established antimalarial agents are reported in Table 1. Small differences in susceptibility between species could be due to differences in growth rates or synchronicity of the cultures. Hence, we consider potentially important species differences only where there is a more than 3-fold difference in mean EC50 between the species.
Table 1.
Comparison of the in vitro susceptibility of Plasmodium knowlesi (A1-H.1) and Plasmodium falciparum (3D7) exposed to established and investigational antimalarial agents for one complete life cycle.
| Compound | EC50 values (nM) |
Fold difference (Pk/Pf) | p-valuea | |
|---|---|---|---|---|
|
P. knowlesi (A1-H.1) 27 h exposure |
P. falciparum (3D7) 48 h exposure |
|||
| P. knowlesi EC50 > 3 × P. falciparum EC50 | ||||
| Cipargamin (KAE609) | 6.1 ± 0.5 | 0.89 ± 0.08 | 6.83 | <0.0001 |
| SJ733 | 386 ± 34 | 64.3 ± 4.3 | 6.00 | <0.0001 |
| PA21A092 | 63.8 ± 7.6 | 10.2 ± 1.4 | 6.25 | 0.0002 |
| AN13762 | 2762 ± 296 | 41.3 ± 3.8 | 66.88 | <0.0001 |
| Pentamidine | 331 ± 20 | 99 ± 4 | 3.34 | 0.0003 |
| Cladosporin | 411 ± 134 | 133 ± 10 | 3.09 | 0.0831 |
| P. knowlesi EC50 < 1/3 × P. falciparum EC50 | ||||
| KAF156 | 1.7 ± 0.16 | 7.9 ± 0.12 | 0.22 | <0.0001 |
| Halofantrine | 0.92 ± 0.25 | 3.60 ± 0.40 | 0.31 | 0.0174 |
| NMT MMV884705 | 46.2 ± 16 | 206 ± 44 | 0.22 | 0.0266 |
| P. knowlesi EC50 ≈ P. falciparum EC50 | ||||
| New Isoquine | 15.9 ± 2.4 | 11.4 ± 2.38 | 1.39 | 0.2474 |
| Bisquinoline | 2.2 ± 1.40 | 2.3 ± 0.94 | 0.97 | 0.9715 |
| AQ-13 | 11.4 ± 2.82 | 5.1 ± 1.15 | 2.23 | 0.1091 |
| Quinidine | 38.4 ± 10.5 | 52.9 ± 10.7 | 0.73 | 0.3704 |
| Naphthoquine | 117 ± 83 | 111 ± 23 | 1.05 | 0.5544 |
| Methylene Blue | 5.38 ± 1.8 | 3.31 ± 0.7 | 1.63 | 0.3543 |
| MMV253 | 13.0 ± 1.9 | 7.4 ± 1.5 | 1.76 | 0.0810 |
| MMV048 | 17.2 ± 0.9 | 29.5 ± 2.5 | 0.58 | 0.0103 |
| Cyclohexamide | 117 ± 40 | 188 ± 35 | 0.62 | 0.2544 |
| Benzylquine | 31.53 ± 10.88 | 22.05 ± 2.02 | 1.43 | 0.3469 |
| WR194965 | 282 ± 111 | 539 ± 236 | 0.52 | 0.3633 |
| BIX-01294 | 22.6 ± 4.4 | 21.2 ± 4.1 | 1.07 | 0.8207 |
| Sitamaquine | 112 ± 25 | 72 ± 15 | 1.56 | 0.2350 |
| Methotrexate | 1991 ± 125 | 693 ± 48 | 2.87 | 0.0006 |
| MK-4815 | 47.6 ± 6 | 127 ± 16 | 0.37 | 0.0100 |
| MMV688558 | 17.5 ± 2.0 | 42.4 ± 1.6 | 0.41 | 0.0006 |
| Endoperoxides | ||||
| Dihydroartemisinin | 2.35 ± 0.23 | 5.16 ± 0.62 | 0.46 | 0.0017 |
| Artemisinin | 7.39 ± 1.87 | 11.00 ± 1.32 | 0.67 | 0.1667 |
| Artemisone | 0.47 ± 0.14 | 0.72 ± 0.15 | 0.65 | 0.2701 |
| Artesunate | 10.30 ± 1.9 | 8.28 ± 1.05 | 1.24 | 0.3552 |
| Artemether | 4.56 ± 0.55 | 6.93 ± 0.73 | 0.66 | 0.0413 |
| Arterolane (OZ277) | 2.27 ± 0.42 | 4.03 ± 0.59 | 0.56 | 0.0407 |
| Artefenomel (OZ439) | 4.76 ± 0.40 | 4.82 ± 0.62 | 0.99 | 0.9350 |
| Deoxyartemisinin | >10,000 | >10,000 | N.D. | N.D. |
| Delayed death compounds (multiple life cycle exposure)b | ||||
| Trans-mirincamycin | 2.49 ± 0.56 | 4.89 ± 1.17 | 0.51 | 0.1124 |
| Cis-mirincamycin | 2.51 ± 0.64 | 4.61 ± 1.01 | 0.54 | 0.1296 |
EC50 values report the mean ± SEM from at least 3 experiments, and some up to seven repeats, each performed in duplicate.
p values are calculated by comparing EC50 values for P. falciparum versus P. knowlesi using Student's two-tailed unpaired t-test.
Asynchronised parasites were exposed to these compounds for 68 h (P. knowlesi) or 120 h (P. falciparum).
Six compounds were shown to be more than three-fold less susceptible (i.e. higher EC50 values) when tested against P. knowlesi than P. falciparum (Table 1). Three of these (cipargamin, SJ733 and PA21A092) are inhibitors of the sodium channel ATP4 and all three were ∼6-fold less potent against P. knowlesi. Cladosporin and pentamidine are over 3-fold less potent against P. knowlesi than P. falciparum. However, the largest species difference was observed with AN13762, which was almost 67-fold less potent against P. knowlesi when compared to P. falciparum.
Conversely, we also identified three antimalarial compounds (KAF156, halofantrine and MMV884705) that were greater than 3-fold more potent (i.e., lower EC50 values) in P. knowlesi than P. falciparum (Table 1). In particular, KAF156 and MMV884705 were both around 4.5-fold more potent against P. knowlesi.
For most of the other compounds screened there was a less than 2-fold difference in susceptibility between species, and most were highly potent (EC50 < 100 nM: Table 1). Artemisinin-based Combination Therapies (ACTs), composed of a short-acting artemisinin derivative and a long-acting partner drug, are the current WHO recommended first-line treatments for uncomplicated P. falciparum malaria. Endoperoxides, including the aforementioned artemisinin derivatives, were all highly potent against both species (<11 nM; Table 1). Among the artemisinin derivatives, artemisone was the most potent with EC50 values below 1 nM. Furthermore, both synthetic endoperoxides, arterolane and artefenomel, exhibited EC50 values below 5 nM. As expected, deoxyartemisinin, an artemisinin lacking the endoperoxide bridge necessary for antimalarial potency, was inactive at 10 μM against both species.
Both isomers of the antibiotic mirincamycin were inactive after a single life cycle exposure, but were highly potent (<5 nM) when exposed for multiple life cycles of P. knowlesi and P. falciparum and were not significantly different between the two species (Table 1).
3.2. Susceptibility of UM01, a recent field-derived P. knowlesi line
Given that all comparisons have thus far focused on the use of a single culture-adapted P. knowlesi strain first isolated in the 1960s, we next sought to test the validity of these comparisons by additionally testing a more recent clinical P. knowlesi isolate. The susceptibility of the new P. knowlesi line UM01 (isolated in 2013 from a patient in Malaysia (Amir et al., 2016)) to a range of antimalarial agents is shown in Table 2, and compared to the human red blood cell adapted A1-H.1 line. The UM01 line had not been adapted to grow in human red blood cells at the time of this study but can be grown short-term in macaque red blood cells. Access to these cells was limited, resulting in only two experiments being performed in duplicate for each of the drugs, precluding statistical analysis in comparisons with the A1-H.1 line. For all but two antimalarial agents tested, and covering a diverse range of antimalarial targets, the EC50 values for the UM01 line fell within a two-fold difference when compared to the A1-H.1 line (Table 2). For those falling outside the two-fold difference in EC50 values, P218 (6.03-fold) and pyronaridine (2.18-fold), the UM01 line was more susceptible to both drugs. Importantly, the reduced susceptibility of P. knowlesi to the ATP4 inhibitors (compared with P. falciparum 3D7 in Table 1) is confirmed with the UM01 line being 3.5-fold and 4.3-fold less potent for PA21A092 and cipargamin, respectively. Furthermore, the UM01 line (in macaque red blood cells) is 88-fold less susceptible than 3D7 (in human red blood cells) to AN13762, compared with the 66-fold reduced susceptibility for the A1-H1 line relative to 3D7 (Table 1).
Table 2.
Comparison of the susceptibility of the culture-adapted P. knowlesi A1-H.1 line with a recent Malaysian P. knowlesi isolate, UM01.
| Compound | a UM01 (macaque RBC) EC50 (nM) (range/2) | b A1-H.1 (human RBC) EC50 (nM) ± SEM | Fold difference (A1-H.1/UM01) |
|---|---|---|---|
| Endoperoxides | |||
| Dihydroartemisinin | 2.1 (0.4) | 2.0 ± 0.3 | 0.95 |
| Artesunate | 13.8 (3.6) | 10.9 ± 1.7 | 0.79 |
| Artefenomel (OZ439) | 4.4 (1.6) | 6.6 ± 1.4 | 1.5 |
| Quinolines and amino-alcohols | |||
| Chloroquine | 21.5 (4.9) | 29.3 ± 4.7 | 1.36 |
| Mefloquine | 13.1 (2.5) | 10.9 ± 1.7 | 0.83 |
| Quinine | 40.3 (0.4) | 54.8 ± 3.0 | 1.36 |
| Ferroquine | 9.8 (2.32) | 12.2 ± 1.6 | 1.24 |
| Dihydrofolate reductase inhibitors | |||
| Pyrimethamine | 3.2 (0.5) | 5.1 ± 0.8 | 1.59 |
| Cycloguanil | 0.7 (0.1) | 1.3 ± 0.3 | 1.86 |
| Trimethoprim | 137 (54) | 265 ± 47 | 1.93 |
| P218 | 0.68 (0.04) | 4.1 ± 0.7 | 6.03 |
| Other | |||
| Atovaquone | 4.1 (0.04) | 2.6 ± 0.4 | 0.63 |
| Pyronaridine | 4.9 (0.84) | 10.7 ± 1.6 | 2.18 |
| DSM265 | 170 (66) | 303 ± 15 | 1.78 |
| DSM421 | 142 (71) | 194 ± 23 | 1.37 |
| Cipargamin (KAE609) | 3.8 (1.6) | 6.1 ± 0.5 | 1.61 |
| PA21A092 | 35.7 (2.8) | 63.8 ± 7.6 | 1.79 |
| AN13762 | 3618 (110) | 2762 ± 296 | 0.76 |
Parasites were exposed to the compounds for a single life cycle (27 h).
UM01 data are the mean of two independent experiments each performed in duplicate. The range/2, calculated for the two experiments, is shown in parentheses.
Data reported previously (van Schalkwyk et al., 2017) or from Table 1 above. Data are the mean ± SEM for at least three experiments each performed in duplicate.
3.3. Drug combination studies (isobolograms) comparing P. knowlesi and P. falciparum
In order to test whether there were differences in drug interactions between the two species, we tested five antimalarial combinations against the A1-H.1 strain, compared to Pf3D7, using fixed-ratio isobolograms (Fig. 1). Considering the 6-fold reduced susceptibility of P. knowlesi to ATP4 inhibitors – compared with P. falciparum, and reported in Table 1 – we included two ATP4 inhibitors (cipargamin and PA21A092) in our combination studies to assess whether their differing in vitro susceptibility might impact their interaction with other antimalarial agents. Previous reports, using different methodology to our fixed-ratio method, had suggested the chloroquine/quinine combination would be antagonistic in Pf3D7 (Skinner-Adams and Davis, 1999) and that an artemisinin/quinine combination would be additive (Gupta et al., 2002). For all of the combinations tested, the Fractional Inhibitory Concentrations (FICs) observed lay between 0.5 and 2.0 – cut-offs for synergistic and antagonistic interactions, respectively – suggesting all combinations were additive. However, three combinations (chloroquine/quinine (Fig. 1B and G), chloroquine/cipargamin (Fig. 1C and H), and dihydroartemisinin/cipargamin (Fig. 1E and J)) show a concave bend towards an antagonistic interaction. For the chloroquine/cipargamin interaction, higher FICs were observed for P. falciparum (range 1.331–1.800; Fig. 1C) than for P. knowlesi (range 1.165–1.428; Fig. 1H). The combination of dihydroartemisinin with quinine was very similar between species with all FICs lying between 0.887 and 1.047 for P. falciparum, and 1.001–1.072 for P. knowlesi (Fig. 1A and F, respectively). When examining the combination of the two selected ATP4 inhibitors, cipargamin and PA21A092, lower FICs were observed with P. falciparum (range 0.724–1.048; Fig. 1E) compared with P. knowlesi (range 0.939–1.024; Fig. 1J), suggesting an additive interaction against both species.
Fig. 1.

Comparison of the in vitro interaction of select antimalarial agents against Plasmodium falciparum (clone 3D7; panels A to E) and Plasmodium knowlesi (clone A1-H.1; panels F to J). Isobolograms demonstrate the interaction of quinine with dihydroartemisinin (Panels A and F), quinine with chloroquine (Panels B and G), the spiroindolone, cipargamin, with chloroquine (panels C to H), cipargamin with PA21A092 (panels D and I), and cipargamin with dihydroartemisinin (panels E and J). The FIC data are averaged from three or four independent experiments, each run in triplicate. Error bars represent standard error of the mean (SEM). FIC values lying between 0.5 and 2.0 are additive (i.e., no interaction). FIC values below 0.5 are considered synergistic interactions, while FIC values above 2.0 are considered antagonistic (cut-offs shown using dotted lines) (Odds, 2003).
4. Discussion
This study extends our previous observations defining the in vitro drug susceptibility of the zoonotic parasite P. knowlesi to a range of antimalarial agents and reveals key differences when compared to the P. falciparum line, 3D7 (van Schalkwyk et al., 2017). The high serum concentration (10%) in the RPMI 1640-based growth media needed to support our P. knowlesi growth can impact on the EC50 values of drugs that are highly protein bound. Hence, our testing of the widely available P. falciparum (3D7) control line under identical conditions is essential to compare species, and to relate our data to previously reported EC50 values obtained in other laboratories using different media composition, assay conditions, and readout.
We previously showed that P. knowlesi is less susceptible than P. falciparum to several inhibitors of the DHODH enzyme (DSM1, DSM265, DSM421) (van Schalkwyk et al., 2017). We now extend that observation to inhibitors of ATP4. Cipargamin (a spiroindolone), PA21A092 (pyrazoleamide) and SJ733 (dihydroisoquinolone) are chemically distinct entities shown to disrupt sodium homeostasis in P. falciparum (Jimenez-Diaz et al., 2014; Spillman et al., 2013; Vaidya et al., 2014). Mutations in ATP4 are associated with resistance to these compounds. Both our P. knowlesi lines (A1-H.1 and UM01) are 3.5–6.8-fold less susceptible to these compounds than our P. falciparum control line (Table 1, Table 2). Although the differences in the A1-H.1 line are significant and apply across the three chemical structures, the compounds can still be considered potent against P. knowlesi (EC50 < 100 nM for cipargamin and PA21A092), with the clinical significance of these differences so far unknown. Recent evidence has suggested that a further 28 compounds within the ‘Malaria Box’ may inhibit PfATP4 (Lehane et al., 2014). It will therefore be important to screen these other inhibitors of ATP4, from diverse chemical scaffolds, to establish if the reduced susceptibility in P. knowlesi is maintained relative to P. falciparum.
Furthermore, P. knowlesi is phylogenetically closer to all other human-infecting malaria species than is P. falciparum. If this reduced susceptibility to ATP4 inhibitors is present in other non-falciparum species then this may have important implications for drug development. Fortunately, studies so far have suggested P. vivax is similarly susceptible to cipargamin as P. falciparum ex vivo (Rottmann et al., 2010), and that PA21A092 was more potent against P. vivax than P. falciparum ex vivo (Vaidya et al., 2014). However, three rodent malaria species were less susceptible to SJ733 than P. falciparum (Jimenez-Diaz et al., 2014). Clearly, understanding the mechanism responsible for these species differences is vital, and is currently the subject of ongoing research in our laboratory.
Cladosporin targets the lysyl t-RNA synthetase and is > 100-fold more potent against P. falciparum than the human enzyme (Hoepfner et al., 2012). Attempts to optimise this target for antimalarial development should include screens against P. knowlesi which we show to be less susceptible in vitro than P. falciparum.
The large difference in the susceptibility to AN13762 of both P. knowlesi lines relative to 3D7 (67-fold and 88-fold for A1-H.1 and UM01 lines, respectively) suggest substantial differences in the potential target, or perhaps a species-specific resistance mechanism. The targets of AN13762 and pentamidine are unknown at present but identifying these will be crucial to understanding the mechanism and/or genetic target differences between the two species responsible for the reduced susceptibility of P. knowlesi, and whether this extends to other species.
Of less concern (from a therapeutic perspective) are the compounds showing increased susceptibility in P. knowlesi than P. falciparum. We have shown that DHFR inhibitors (pyrimethamine, cycloguanil and trimethoprim) are around 10-fold more potent against P. knowlesi than P. falciparum (van Schalkwyk et al., 2017). Here, we show also that the N-methyl transferase inhibitor, MMV884805, and KAF156 were both 4.5 fold more potent against P. knowlesi. Similar to our findings for mefloquine (van Schalkwyk et al., 2017), P. knowlesi was significantly more susceptible to halofantrine than P. falciparum. A previous study, using a short term ex vivo schizont maturation approach, reported that P. knowlesi (six isolates) was four times less susceptible than P. falciparum (one isolate) to mefloquine (Fatih et al., 2013). In contrast, our data using directly comparable in vitro assay conditions (parasitaemia, haematocrit, single life cycle exposure) and SYBR green I readout, suggest that P. knowlesi is more susceptible than P. falciparum to several amino-alcohols (mefloquine, halofantrine, and lumefantrine) (Table 1 and (van Schalkwyk et al., 2017)). Adapting more P. knowlesi isolates from the field (such as UM01) to in vitro culture will facilitate further tests to confirm this observation.
Most antimalarial compounds tested were equipotent against both species (Table 1). These compounds inhibit diverse targets such as haemozoin formation (AQ-13, MK4815, and quinidine), histone methyl transferase (BIX-01294), the V-type H+-ATPase (MMV253) and coenzyme A biosynthesis (MMV688558). Furthermore, all artemisinin derivatives and synthetic endoperoxides were potent against P. knowlesi.
We have shown previously that the lincosamide antibiotic, clindamycin, was highly potent against both P. knowlesi (15.9 nM) and P. falciparum (7.0 nM) when exposed across multiple life cycles, a phenomenon known as the delayed death effect (van Schalkwyk et al., 2017). Another lincosamide, mirincamycin, was reported to be more potent than clindamycin against P. falciparum lines and its activity shown to be independent of the isomers tested (Held et al., 2010). We confirm here that mirincamycin is more potent than clindamycin against P. falciparum 3D7, and demonstrate that it is also more potent against P. knowlesi A1-H.1 (all < 5 nM; Table 1). Again, this potency is independent of the isomers being screened.
There may be questions about the applicability of using the P. knowlesi A1-H.1 line, isolated from a human infection in 1963, to represent modern P. knowlesi parasites in drug susceptibility screens. In Table 2 we report the similarity in drug susceptibility between the “old” P. knowlesi parasites and our newer isolate, UM01. For most compounds tested, the EC50 values are lower for the UM01 line, perhaps because this line is not yet well-adapted to grow in vitro. UM01 only grows in macaque red blood cells and has 2-fold growth rate per life cycle compared with the 3–4 fold growth rate of the A1-H.1 line in human red blood cells. Nevertheless, the similar susceptibility profile of the UM01 line to the A1-H.1 line for endoperoxides and quinoline-based drugs is encouraging. Crucially, the reduced susceptibility – relative to P. falciparum – of the A1-H.1 line to the ATP4 inhibitors cipargamin and PA21A092, the DHODH inhibitors, DSM265 and DSM421 (reported in van Schalkwyk et al., 2017) and for AN13762 (Table 1) is confirmed here also for the UM01 line.
The similar susceptibility profile of our human red blood cell adapted A1-H.1 line to diverse antimalarial agents, when compared with a recent field isolate, UM01 (Table 2), supports its ongoing use in screens for antimalarial activity in spite of laboratory propagation since its origin in the 1960s. Once the UM01 line is culture-adapted to growth efficiently in in vitro human red cells, it will provide a viable alternative strain to more extensively compare drug susceptibility profiles. However, until that time, we are confident that the drug susceptibility reported for the A1-H.1 line faithfully represents the current drug susceptibility profiles of circulating P. knowlesi parasites, such as UM01.
To our knowledge, there have not been any published studies comparing the in vitro interaction of antimalarial agents in P. knowlesi with that of P. falciparum. Combinations of dihydroartemisinin and quinine were additive in both species similar to previous reports using other P. falciparum strains (Gupta et al., 2002). Three combinations; chloroquine/quinine, chloroquine/cipargamin, and dihydroartemisinin/cipargamin, all displayed a weak trend towards antagonism against both species. Perhaps surprisingly, the combination of two ATP4 inhibitors was additive, even tending toward synergism in P. falciparum. It might be expected that two drugs competing for the same target might elicit an antagonistic interaction, such as that between chloroquine and quinine (Skinner-Adams and Davis, 1999). However, it has been shown previously that mutations in ATP4 conferring resistance to PA21A092 do not elicit cross-resistance to another spiroindolone (NITD246). Taken together with the additive effect, this could suggest these drugs have different binding sites within this putative Na+-ATPase (Vaidya et al., 2014).
Tremendous strides have been made towards the eradication agenda with several countries gaining recent WHO certification for achieving malaria elimination (WHO, 2018). However, as progress is made in reducing the prevalence of P. falciparum and P. vivax malaria, the two species with the greatest burden of disease, other malarias are taking on a greater prominence. For example, P. knowlesi incidence was shown to increase in Malaysia after dramatic reductions in the number of cases due to P. falciparum and P. vivax (William et al., 2014). It is therefore imperative that the drug development community is able to test new compounds for activity against other human malarias to confirm equipotency and to ensure that new combinations are likely to be effective. The results presented here begin to address these issues, providing an in vitro pharmacological characterization of two strains of P. knowlesi and demonstrating the application of fixed ratio isobologram analysis for testing drug combinations in P. knowlesi as was done previously for P. falciparum (Fivelman et al., 2004). Additional testing of current and novel combinations will now be undertaken.
5. Conclusion
We have identified further species differences in drug susceptibility between P. knowlesi and P. falciparum to select antimalarial classes/targets (e.g., inhibitors of ATP4 and N-methyltransferase, and as yet unknown targets of KAF156 and AN13762), in addition to those we reported previously (van Schalkwyk et al., 2017). Furthermore, we have shown that the human adapted P. knowlesi A1-H.1 line, used by us to define susceptibility to new and established antimalarial agents, delivers EC50 values similar to a recently isolated P. knowlesi line, UM01, supporting the continued use of the A1-H.1 line as a model for testing drug susceptibility in P. knowlesi until new lines can be fully culture-adapted to human red blood cells. Finally, isobologram analysis showed that, for a limited selection of antimalarial combinations, drug interactions were similar between P. knowlesi and P. falciparum in vitro.
Competing interests
BB, RDN and DL are employees of the funder, MMV. All other authors: no competing interests to declare.
Author contributions
CJS, RM, DAvS conceived and designed the study. DAvS performed the experiments. DAvS, RM, BB, RDN, DL and CJS analysed the data. CJS and DAvS wrote the paper. All authors read and approved the final manuscript.
Funding
This project was supported by the Medicines for Malaria Venture [grant MMV RD/15/0017] awarded to DAvS. RWM is supported by the UK Medical Research Council (MRC) Career Development Award [MR/M021157/1] jointly funded by the UK MRC and the UK Department for International Development. CJS is supported by Public Health England.
Acknowledgements
We wish to thank Dr Neil Almond and Jo Hall (NIBSC, Potters Bar, UK) for the supply of blood samples from M. fascicularis used in this study.
References
- Amir A., Russell B., Liew J.W., Moon R.W., Fong M.Y., Vythilingam I., Subramaniam V., Snounou G., Lau Y.L. Invasion characteristics of a Plasmodium knowlesi line newly isolated from a human. Sci. Rep. 2016;6:24623. doi: 10.1038/srep24623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold M.S., Engel J.A., Chua M.J., Fisher G.M., Skinner-Adams T.S., Andrews K.T. Adaptation of the [3H]hypoxanthine uptake assay for in vitro-cultured Plasmodium knowlesi malaria parasites. Antimicrob. Agents Chemother. 2016;60:4361–4363. doi: 10.1128/AAC.02948-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell A. Antimalarial drug synergism and antagonism: mechanistic and clinical significance. FEMS Microbiol. Lett. 2005;253:171–184. doi: 10.1016/j.femsle.2005.09.035. [DOI] [PubMed] [Google Scholar]
- Bennett T.N., Paguio M., Gligorijevic B., Seudieu C., Kosar A.D., Davidson E., Roepe P.D. Novel, rapid, and inexpensive cell-based quantification of antimalarial drug efficacy. Antimicrob. Agents Chemother. 2004;48:1807–1810. doi: 10.1128/AAC.48.5.1807-1810.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasil P., Zalis M.G., de Pina-Costa A., Siqueira A.M., Junior C.B., Silva S., Areas A.L.L., Pelajo-Machado M., de Alvarenga D.A.M., da Silva Santelli A.C.F., Albuquerque H.G., Cravo P., Santos de Abreu F.V., Peterka C.L., Zanini G.M., Suarez Mutis M.C., Pissinatti A., Lourenco-de-Oliveira R., de Brito C.F.A., de Fatima Ferreira-da-Cruz M., Culleton R., Daniel-Ribeiro C.T. Outbreak of human malaria caused by Plasmodium simium in the Atlantic Forest in Rio de Janeiro: a molecular epidemiological investigation. Lancet Glob Health. 2017;5:e1038–e1046. doi: 10.1016/S2214-109X(17)30333-9. [DOI] [PubMed] [Google Scholar]
- Cox-Singh J., Davis T.M., Lee K.S., Shamsul S.S., Matusop A., Ratnam S., Rahman H.A., Conway D.J., Singh B. Plasmodium knowlesi malaria in humans is widely distributed and potentially life threatening. Clin. Infect. Dis. 2008;46:165–171. doi: 10.1086/524888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatih F.A., Staines H.M., Siner A., Ahmed M.A., Woon L.C., Pasini E.M., Kocken C.H., Singh B., Cox-Singh J., Krishna S. Susceptibility of human Plasmodium knowlesi infections to anti-malarials. Malar. J. 2013;12:425. doi: 10.1186/1475-2875-12-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fivelman Q.L., Adagu I.S., Warhurst D.C. Modified fixed-ratio isobologram method for studying in vitro interactions between atovaquone and proguanil or dihydroartemisinin against drug-resistant strains of Plasmodium falciparum. Antimicrob. Agents Chemother. 2004;48:4097–4102. doi: 10.1128/AAC.48.11.4097-4102.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruring C., Moon R.W., Lim C., Holder A.A., Blackman M.J., Duraisingh M.T. Human red blood cell-adapted Plasmodium knowlesi parasites: a new model system for malaria research. Cell Microbiol. 2014;16:612–620. doi: 10.1111/cmi.12275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S., Thapar M.M., Wernsdorfer W.H., Bjorkman A. In vitro interactions of artemisinin with atovaquone, quinine, and mefloquine against Plasmodium falciparum. Antimicrob. Agents Chemother. 2002;46:1510–1515. doi: 10.1128/AAC.46.5.1510-1515.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held J., Westerman R., Kremsner P.G., Mordmuller B. In vitro activity of mirincamycin(U24729A) against Plasmodium falciparum isolates from Gabon. Antimicrob. Agents Chemother. 2010;54:540–542. doi: 10.1128/AAC.01090-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoepfner D., McNamara C.W., Lim C.S., Studer C., Riedl R., Aust T., McCormack S.L., Plouffe D.M., Meister S., Schuierer S., Plikat U., Hartmann N., Staedtler F., Cotesta S., Schmitt E.K., Petersen F., Supek F., Glynne R.J., Tallarico J.A., Porter J.A., Fishman M.C., Bodenreider C., Diagana T.T., Movva N.R., Winzeler E.A. Selective and specific inhibition of the Plasmodium falciparum lysyl-tRNA synthetase by the fungal secondary metabolite cladosporin. Cell Host Microbe. 2012;11:654–663. doi: 10.1016/j.chom.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Diaz M.B., Ebert D., Salinas Y., Pradhan A., Lehane A.M., Myrand-Lapierre M.E., O'Loughlin K.G., Shackleford D.M., Justino de Almeida M., Carrillo A.K., Clark J.A., Dennis A.S., Diep J., Deng X., Duffy S., Endsley A.N., Fedewa G., Guiguemde W.A., Gomez M.G., Holbrook G., Horst J., Kim C.C., Liu J., Lee M.C., Matheny A., Martinez M.S., Miller G., Rodriguez-Alejandre A., Sanz L., Sigal M., Spillman N.J., Stein P.D., Wang Z., Zhu F., Waterson D., Knapp S., Shelat A., Avery V.M., Fidock D.A., Gamo F.J., Charman S.A., Mirsalis J.C., Ma H., Ferrer S., Kirk K., Angulo- Barturen I., Kyle D.E., DeRisi J.L., Floyd D.M., Guy R.K. (+)-SJ733, a clinical candidate for malaria that acts through ATP4 to induce rapid host-mediated clearance of Plasmodium. Proc. Natl. Acad. Sci. U. S. A. 2014;111:E5455–E5462. doi: 10.1073/pnas.1414221111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalremruata A., Magris M., Vivas-Martinez S., Koehler M., Esen M., Kempaiah P., Jeyaraj S., Perkins D.J., Mordmuller B., Metzger W.G. Natural infection of Plasmodium brasilianum in humans: man and monkey share quartan malaria parasites in the Venezuelan Amazon. EBioMedicine. 2015;2:1186–1192. doi: 10.1016/j.ebiom.2015.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehane A.M., Ridgway M.C., Baker E., Kirk K. Diverse chemotypes disrupt ion homeostasis in the Malaria parasite. Mol. Microbiol. 2014;94:327–339. doi: 10.1111/mmi.12765. [DOI] [PubMed] [Google Scholar]
- Lim C., Hansen E., DeSimone T.M., Moreno Y., Junker K., Bei A., Brugnara C., Buckee C.O., Duraisingh M.T. Expansion of host cellular niche can drive adaptation of a zoonotic malaria parasite to humans. Nat. Commun. 2013;4:1638. doi: 10.1038/ncomms2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon R.W., Hall J., Rangkuti F., Ho Y.S., Almond N., Mitchell G.H., Pain A., Holder A.A., Blackman M.J. Adaptation of the genetically tractable malaria pathogen Plasmodium knowlesi to continuous culture in human erythrocytes. Proc. Natl. Acad. Sci. U. S. A. 2013;110:531–536. doi: 10.1073/pnas.1216457110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odds F.C. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 2003;52:1. doi: 10.1093/jac/dkg301. [DOI] [PubMed] [Google Scholar]
- Paul A.S., Moreira C.K., Elsworth B., Allred D.R., Duraisingh M.T. Extensive shared chemosensitivity between malaria and babesiosis blood-stage parasites. Antimicrob. Agents Chemother. 2016;60:5059–5063. doi: 10.1128/AAC.00928-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottmann M., McNamara C., Yeung B.K., Lee M.C., Zou B., Russell B., Seitz P., Plouffe D.M., Dharia N.V., Tan J., Cohen S.B., Spencer K.R., Gonzalez-Paez G.E., Lakshminarayana S.B., Goh A., Suwanarusk R., Jegla T., Schmitt E.K., Beck H.P., Brun R., Nosten F., Renia L., Dartois V., Keller T.H., Fidock D.A., Winzeler E.A., Diagana T.T. Spiroindolones, a potent compound class for the treatment of malaria. Science. 2010;329:1175–1180. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge G.G., Bohme U., Sanders M., Reid A.J., Cotton J.A., Maiga-Ascofare O., Djimde A.A., Apinjoh T.O., Amenga-Etego L., Manske M., Barnwell J.W., Renaud F., Ollomo B., Prugnolle F., Anstey N.M., Auburn S., Price R.N., McCarthy J.S., Kwiatkowski D.P., Newbold C.I., Berriman M., Otto T.D. Plasmodium malariae and P. ovale genomes provide insights into malaria parasite evolution. Nature. 2017;542:101–104. doi: 10.1038/nature21038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B., Kim Sung L., Matusop A., Radhakrishnan A., Shamsul S.S., Cox-Singh J., Thomas A., Conway D.J. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet. 2004;363:1017–1024. doi: 10.1016/S0140-6736(04)15836-4. [DOI] [PubMed] [Google Scholar]
- Skinner-Adams T., Davis T.M. Synergistic in vitro antimalarial activity of omeprazole and quinine. Antimicrob. Agents Chemother. 1999;43:1304–1306. doi: 10.1128/aac.43.5.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smilkstein M., Sriwilaijaroen N., Kelly J.X., Wilairat P., Riscoe M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother. 2004;48:1803–1806. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillman N.J., Allen R.J., McNamara C.W., Yeung B.K., Winzeler E.A., Diagana T.T., Kirk K. Na+ regulation in the malaria parasite Plasmodium falciparum involves the cation ATPase PfATP4 and is a target of the spiroindolone antimalarials. Cell Host Microbe. 2013;13:227–237. doi: 10.1016/j.chom.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ta T.H., Hisam S., Lanza M., Jiram A.I., Ismail N., Rubio J.M. First case of a naturally acquired human infection with Plasmodium cynomolgi. Malar. J. 2014;13:68. doi: 10.1186/1475-2875-13-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidya A.B., Morrisey J.M., Zhang Z., Das S., Daly T.M., Otto T.D., Spillman N.J., Wyvratt M., Siegl P., Marfurt J., Wirjanata G., Sebayang B.F., Price R.N., Chatterjee A., Nagle A., Stasiak M., Charman S.A., Angulo-Barturen I., Ferrer S., Belen Jimenez-Diaz M., Martinez M.S., Gamo F.J., Avery V.M., Ruecker A., Delves M., Kirk K., Berriman M., Kortagere S., Burrows J., Fan E., Bergman L.W. Pyrazoleamide compounds are potent antimalarials that target Na+ homeostasis in intraerythrocytic Plasmodium falciparum. Nat. Commun. 2014;5:5521. doi: 10.1038/ncomms6521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Schalkwyk D.A., Moon R.W., Blasco B., Sutherland C.J. Comparison of the susceptibility of Plasmodium knowlesi and Plasmodium falciparum to antimalarial agents. J. Antimicrob. Chemother. 2017;72:3051–3058. doi: 10.1093/jac/dkx279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Schalkwyk D.A., Priebe W., Saliba K.J. The inhibitory effect of 2-halo derivatives of D-glucose on glycolysis and on the proliferation of the human malaria parasite Plasmodium falciparum. J. Pharmacol. Exp. Therapeut. 2008;327:511–517. doi: 10.1124/jpet.108.141929. [DOI] [PubMed] [Google Scholar]
- William T., Jelip J., Menon J., Anderios F., Mohammad R., Awang Mohammad T.A., Grigg M.J., Yeo T.W., Anstey N.M., Barber B.E. Changing epidemiology of malaria in Sabah, Malaysia: increasing incidence of Plasmodium knowlesi. Malar. J. 2014;13:390. doi: 10.1186/1475-2875-13-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (World Health Organization) 2015. World Malaria Report.http://www.who.int/malaria/publications/world-malaria-report/en/ [Google Scholar]
- WHO (World Health Organization) third ed. 2015. Guidelines for the Treatment of Malaria.http://www.who.int/malaria/publications/atoz/9789241549127/en/ [Google Scholar]
- WHO (World Health Organization) 2018. World Malaria Report.https://www.who.int/malaria/publications/world-malaria-report-2018/report/en/ [Google Scholar]