

Abstract
Adeno-associated virus (AAV) has emerged as a promising gene delivery vector because of its non-pathogenicity, simple structure and genome, and low immunogenicity compared to other viruses. However, its adoption as a safe and effective delivery vector for certain diseases relies on altering its tropism to deliver transgenes to desired cell populations. To this end, we have developed a protease-activatable AAV vector, named provector, that responds to elevated extracellular protease activity commonly found in diseased tissue microenvironments. The AAV9-based provector is initially inactive, but then it can be switched on by matrix metalloproteinases (MMP)-2 and -9. Cryo-electron microscopy and image reconstruction reveal that the provector capsid is structurally similar to that of AAV9, with a flexible peptide insertion at the top of the 3-fold protrusions. In an in vivo model of myocardial infarction (MI), the provector is able to deliver transgenes site specifically to high-MMP-activity regions of the damaged heart, with concomitant decreased delivery to many off-target organs, including the liver. The AAV provector may be useful in the future for enhanced delivery of transgenes to sites of cardiac damage.
Keywords: adeno-associated virus, AAV, gene therapy, gene delivery, viral vector, stimulus responsive, activatable, inflammation targeting, cardiac gene therapy, myocardial infarction, matrix metalloproteinase, provector
Graphical Abstract
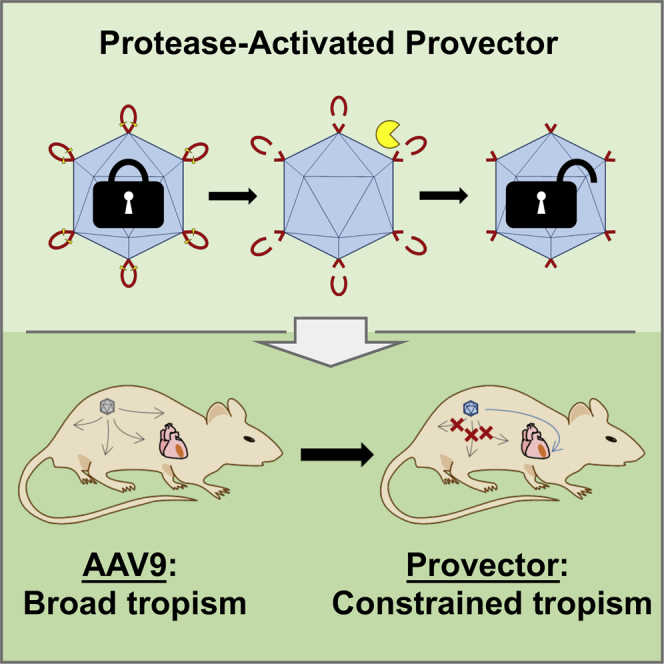
Guenther et al. have developed a protease-activatable AAV provector to target diseased tissue microenvironments exhibiting elevated extracellular protease activity. The provector delivers transgenes to high-MMP-activity regions of the damaged heart following a myocardial infarction, with concomitant decreased delivery to many off-target organs, including the liver.
Introduction
Cardiovascular disease (CVD) is one of the leading causes of death in the United States, with over 800,000 deaths attributable to CVD in 2013.1 Heart failure is particularly dangerous, and it is estimated that over 5 million Americans have this disease. While drugs and device therapies have had some success in treating the symptoms of heart failure, most have limitations, and research has turned to the field of gene therapy to provide potential treatments.2
Current cardiac gene therapy efforts have unfortunately been hampered by several limitations, including inefficient and/or nonspecific delivery of the vector to the target site, which leads to insufficient expression of the therapeutic gene at the site of disease and potentially negative side effects at sites of off-target gene delivery.3, 4 Many promising therapeutic genes would only be clinically viable if they could be delivered in a highly targeted manner to diseased cardiac tissues. This has led to the development of invasive delivery methods, such as direct myocardial injection or intracoronary perfusion.4, 5, 6 From a clinical standpoint, requiring the use of these administration procedures would not completely prevent the translation of cardiac gene therapies. However, the development of more tissue-targeted gene delivery vectors could greatly enhance gene therapy outcomes in general, with or with surgical administration methods.
Matrix metalloproteinases (MMPs) are a class of proteases normally involved in the remodeling of extracellular matrix, but they have been found to be upregulated in sites of cardiovascular disease, including myocardial infarction (MI),7 congestive heart failure,8, 9 and atherosclerosis.10 In particular, MMP-2 and MMP-9 have been found to be overexpressed post-MI in the infarcted tissue.11 Upregulation of MMPs can be used as a biomarker to target reporters and therapeutics to sites of disease. In the case of ischemic tissue damaged from MI and any resulting heart failure, MMPs could be leveraged to target delivery of cardiac gene therapies that may otherwise be unsafe or ineffective.
Previously, we developed a protease-activatable gene delivery vector based on AAV serotype 2 (AAV2).12, 13, 14 Short peptide locks were genetically inserted into the AAV2 capsid proximal to the binding domain of the primary cell surface receptor, heparan sulfate proteoglycan (HSPG). These locks consist of a tetra-aspartic acid motif flanked by two MMP-cleavable sequences, and they have been demonstrated to block heparin binding and prevent transduction until cleaved off by MMPs. The provector capsids also remain intact after exposure to MMPs, as visualized by electron microscopy.12 The initial provector prototype is limited by its parental AAV2 capsid’s native tropism and other characteristics, such as lower production titers compared to other AAVs. As more serotypes have been identified and characterized, other AAV variants have properties that may make them more suitable for in vivo gene delivery. AAV9 in particular demonstrates a number of characteristics that make it more amenable to clinical translation compared to AAV2, including higher production yields,15 lower prevalence of pre-existing neutralizing antibodies in patient populations,16, 17 and a more favorable biodistribution profile.18 For cardiac gene therapy applications in particular, AAV9 has been shown to provide the most robust gene delivery to heart tissue following systemic injection in mice and rats compared to other serotypes.19, 20
Because of AAV9’s superior characteristics for clinical translation compared to AAV2, we sought to expand the provector concept to AAV9. Here we describe the development of a provector based on AAV9 that responds to elevated protease activity commonly found in diseased tissue microenvironments. Using pseudo-rational design strategies, an AAV9 capsid site, located on the protrusions surrounding the icosahedral 3-fold axes of the capsid, was identified that allowed for successful insertion of an MMP-cleavable peptide lock. The generated provector variants effectively demonstrate switchable transduction in vitro when exposed to MMPs. We then tested the in vivo delivery performance of the provector variants in a murine model of MI. Following systemic injection, one of the provector variants displayed enhanced targeted transgene expression in high-MMP-activity regions of the damaged heart and decreased expression in several off-target organs as well as healthy regions of the heart.
Results
Generation and Structural Characterization of Protease-Activatable Variants of AAV9
AAV-based provectors were designed by genetically inserting small peptide locks containing MMP-cleavable domains into the capsid of AAV9 near its galactose-binding domain. We hypothesized the peptide locks would block the virus from binding and transducing cells. Exposure to MMPs would cleave the locks off the virus capsid, unmasking the receptor-binding domains on the capsid and allowing the virus to regain its receptor-binding and transduction capabilities. Four major design criteria were considered in creating the provector: (1) the capsid must be able to accommodate the peptide lock insertion and maintain capsid assembly and genome packaging, (2) the peptide lock must be accessible to MMPs for cleavage, (3) the inserted peptide lock must ablate transduction in the absence of MMPs, and (4) the provector must regain its transduction ability after MMP exposure. The same peptide lock (L001, amino acid sequence: AG-VPMSMRGG-G-D4-G-VPMSMRGG-GA) was inserted after a number of AAV9 capsid residues previously noted as being important for galactose binding21 to identify insertion sites that would satisfy the design criteria. The panel of viral variants was generated and screened for successful capsid assembly and genome packaging (Table S1). Most insertion sites tested either resulted in substantially lower viral titers, indicating capsid intolerance to peptide lock insertion, or viral variants whose transduction capabilities were permanently ablated and could not be switched back on after MMP exposure.
We then tested an insertion site slightly farther away from the galactose-binding domain but likely to tolerate peptide insertions. Specifically, we inserted the lock after capsid residue G453, which is surface exposed on the protrusions surrounding the 3-fold axes of the capsid and on the same wall of the protrusion as the galactose-binding residues. This capsid region has low sequence homology compared with other serotypes, suggesting that the site can accommodate changes. From here on, we refer to the AAV9 mutant with lock sequence L001 inserted after G453 as L001. Lock insertion appeared to be well tolerated at this site, as insertion of three different lock sequences resulted in titers similar to AAV9 (Table 1). L005 (amino acid sequence: AG-PLGLAR-G-D4-G-PLGLAR-GA) was chosen for insertion based on the peptide sequence used to construct the initial AAV2-based provector prototype.12 The provector variants exhibited a slight decrease in genome protection against nuclease digestion (Figure S1). These results suggest peptide lock insertion after G453 does not dramatically impact capsid assembly and genome packaging.
Table 1.
Primary Viral Variants Generated and Tested
| Virus Variant | Cleavage Sequencea | Titer (vg/mL) |
|---|---|---|
| AAV9 | – | 1.40E+12 |
| L001 | VPMSMRGG | 1.49E+12 |
| L005 | PLGLAR | 1.41E+12 |
| Scrambledb | SMVGMRPG | 8.56E+11 |
Locks containing indicated cleavage sequences were genetically inserted after residue G453.
Scrambled lock vector was generated by randomly scrambling the amino acids of the MMP cleavage sequence found in L001. This scrambled vector shares all characteristics of L001 except its cleavability by MMPs, thereby serving as a negative control.
To further characterize any changes in capsid structure due to lock insertion, we used cryoelectron microscopy and image reconstruction to determine the structure of the L001 vector. The study used vitrified L001 particles, purified from triple transfection of HEK293 cells, to 3.16-Å resolution (Figure S2). The viral protein (VP) monomer density was ordered from N-terminal residues 219–456 and residues 481–762 at a sigma (σ) threshold of 1 (Figure 1; Table S2). The VP1 unique sequence (residues 1–136, VP1u), the VP1/VP2 common region (residues 137–202), and the N terminus of VP3 (residues 203–218) are disordered, similar to reports for other AAVs, including AAV9.22 In addition, while the peptide insertion position was obvious in the L001 map, density was only observed for the main chain of 454-AGV-456 on the ascending side of the inserted peptide loop and 481-GQSQ-484 (equivalent to wild-type [WT] 455-GQSQ-458) of the descending side (Figure 1A). Side-chain density was only visible for residues (e.g., 452 and 485) at the base of the loop at ≥3.0σ (Figure 1A). Residues 457–480 were disordered. This is in contrast to the remainder of the map with density observed for the majority of the residue side chains within core secondary structure regions as well as surface loops (Figures 1D and 1E). The exceptions were acidic amino acids, as expected, because these residues are known to be sensitive to electron beam-induced radiation damage (data not shown).23
Figure 1.

Model Building for L001 and Comparison to AAV9
(A) The atomic model of L001 (gray) fitted into the L001 density shown in black mesh. The map is contoured at a sigma (α) threshold of 3.0. At this sigma, side-chain density is observed at the base of the inserted peptide. AAV9 is also shown in brown. (B and C) The same map and atomic model of L001 contoured at a sigma (α) threshold of 1.5 (B) and 1.0 (C), respectively, where side-chain density is not discernible but the map is extended to additional residues. (D and E) Density and atomic model of L001 VRIII (D) and the βC strand region (E), respectively, with the map contoured at a sigma (α) threshold of 2.5. The side chain for the amino acids in these regions was readily interpreted and identical between L001 and AAV9. (F) Superposition of the monomers of L001 and AAV9. The conserved eight-stranded anti-parallel β-barrel core with BIDG and CHEF sheet and variable regions (VRI–VRIX) are labeled. The icosahedral 2-fold, 3-fold, and 5-fold axes are denoted by an oval, a triangle, and a pentagon, respectively. The broken rectangle indicates the region of the insertion shown in (A)–(C). The images were generated using PyMol43 and Chimera.42
A superposition of the VP3 3D model generated for L001 onto the AAV9 structure shows the position of the peptide insertion (see broken rectangular box in Figure 1F). The ordered VP3 structure conserved the eight-stranded anti-parallel β-barrel core, with the BIDG sheet facing the virus interior and the αA helix forming the wall of the 2-fold axes, as reported for other parvovirus structures (Figure 1F). Large interconnecting loops between the secondary structure elements formed features observed at the capsid surface, including a channel at the 5-fold axes, protrusions at the 3-fold axes, and a depression of the 2-fold axes (Figure 2A). These capsid features were similar between L001 and AAV9 (Figures 2A and 2B), except at the 3-fold axes (Figures 2C and 2D). Here additional density was seen at the top of the protrusions, in a loop location known as variable region IV (VR-IV), as a result of the peptide insertion in L001 (see gray density). However, as stated above, this observed density represents the main chain for only a subset of residues, since the majority of the peptide was disordered. Overall, these results demonstrate the provector variants satisfy the first design criterion of accommodating lock insertion and maintaining capsid assembly and genome packaging.
Figure 2.

The L001 and AAV9 Capsid Structures
(A and B) Capsid surface density maps of AAV9 based on the X-ray structure coordinates (A) and capsid surface density from the cryo-reconstructed map of L001 (B), respectively. Both maps are radially colored from the center of the particle according to the color key shown. (C) Superposition of the AAV9 (brown) and L001 (gray) capsid surface density maps (shown in A and B). Gray density is observed on top of the 3-fold protrusions at the site of the peptide insertion. (D) Surface density (in gray) from a difference map subtracting AAV9 from L001 (in the Chimera program). A triangle, depicting the viral asymmetric unit, is shown, and the icosahedral 2-fold, 3-fold, and 5-fold axes are denoted by an oval, a triangle, and a pentagon, respectively, in (A)–(D). The maps were all contoured at a sigma (α) threshold of 1.0. The images were all generated in the Chimera program.42
Provectors Demonstrate Protease-Responsive Behavior after Exposure to MMPs
The next design criterion for a functional provector is that the peptide lock needs to be accessible to and cleavable by MMPs. We treated viruses with clinically relevant MMPs or a sham buffer, and the VPs and cleavage fragments were visualized on a silver stain (Figure 3A). AAV9 has no innate sensitivity to MMPs and the VPs remain intact regardless of MMP exposure. On the other hand, L001 has intact VPs when treated with sham buffer but nearly complete cleavage when treated with MMPs. The scrambled control shows almost no cleavage when treated with MMPs, indicating that the inserted scrambled lock is not significantly susceptible to MMP cleavage.
Figure 3.

In Vitro Functional Characterization of Provectors
(A) Proteolytic cleavage fragments of provector capsid subunits can be detected via silver stain. The vectors were treated with MMP-2, -7, -9, or sham buffer (S). Intact VPs are observed in the sham conditions. Conversely, N-terminal (VP1′, VP2′, and VP3′) and C-terminal (C-term) fragments are observed for L001 following treatment with MMPs. AAV9 and scrambled vectors do not yield VP cleavage fragments in response to MMPs. (B) Provectors demonstrate protease-activated transduction behavior. Vectors were treated with sham buffer or MMPs and then added to CHO-Lec2 cells (MOI: 5,000). Flow cytometry was then used to quantify GFP expression at 48 h post-transduction. The transduction index (TI) is calculated as the percent of GFP-positive cells multiplied by the geometric mean fluorescence intensity of the GFP-positive cells (TI = %GFP × gMFI). TI is a linear indicator of virus activity over a wider range of MOIs compared to the percent of GFP-positive cells alone.12 AAV9 demonstrates high transduction efficiency and is not affected by MMPs. L001 and L005 both demonstrate significantly higher transduction (∼5× and ∼2×, respectively) when treated with MMPs compared to sham treatment. Scrambled vector demonstrates low transduction efficiency regardless of MMP treatment. Error bars, SEM; n = 3; *p < 0.05, **p < 0.001. (C) Lectin competition assay demonstrates switchable galactose-binding activity by provector. Vectors were treated with either MMP-9 or sham buffer, then mixed with Erythrina cristagalli lectin and incubated with CHO-Lec2 cells (MOI: 5,000) for 1 h at 4°C. Media were removed, cells were washed with PBS, and fresh media were added. 48 h later, transduction was quantified using flow cytometry. AAV9 transduction is blocked by a high concentration of lectin, regardless of MMP treatment. In the locked (sham) state, L001 has a low transduction efficiency. After MMP exposure, L001 transduction is restored and is competitively inhibited by lectin. Results from two independent experiments are plotted for each lectin concentration, with the horizontal line indicating the average.
To determine if the provectors have protease-activatable transduction behavior, we transduced CHO-Lec2 cells with the AAV variants after exposure to MMPs, and we assayed for GFP transgene expression (Figure 3B). AAV9 showed a high transduction index (TI) regardless of MMP exposure. L001 showed a low TI when in the locked state following sham treatment (12% of AAV9 TI) and a 5.2-fold increase in TI after MMP exposure (to 66% of AAV9 TI). L005 showed less activatable transduction compared to L001; sham treatment led to higher background TI (31% of AAV9 TI) and only a maximum of 1.9-fold increase after MMP-9 exposure (to 60% of AAV9 TI). The scrambled control showed a low TI regardless of MMP exposure (17% of AAV9). Both provectors showed protease-responsive transduction, but L001 demonstrated an increased dynamic range compared to L005.
We hypothesized the peptide lock prevents transduction by blocking the virus’s ability to bind to galactose. To confirm, we performed a lectin competition assay by mixing sham- and MMP-9-treated viral variants with increasing concentrations of Erythrina cristagalli lectin (ECL) and applying the viruses to CHO-Lec2 cells (Figure 3C). Regardless of MMP exposure, AAV9 had a high initial transduction efficiency that decreased with a high concentration of ECL, indicating that its galactose-binding ability was not impacted by MMP exposure. Conversely, L001 exhibited low TI in the sham condition (i.e., locked state). When the provector was unlocked through exposure to MMP-9, its TI was initially high in the absence of ECL and then decreased with increasing concentrations of the competitor, similar to AAV9. As unlocked L001 demonstrated decreased transduction as ECL concentration increased, this suggests that unlocked L001 binds to the same galactose moieties as ECL and AAV9. Locked L001 also showed a slight decrease in transduction as ECL concentration increased. This suggests that the background transduction demonstrated by locked L001 was mediated by galactose as well, rather than an alternative transduction pathway. This result also suggests that the provector design could be optimized to further block galactose binding and decrease background transduction.
Intravenous Injection of Provector Leads to Targeted Transgene Expression in Damaged Cardiac Tissue and Decreased Delivery and Expression in Off-Target Organs
Since the provectors demonstrate protease-activatable behavior in vitro, we next investigated their protease-targeting ability in a murine model of MI created by ligation of the left anterior descending artery (LAD). Figure 4A shows representative images of heart cross-sectional slices of near-infrared fluorescence (NIRF) imaging agent targeting MMP activity24 and infrared fluorescent protein (iRFP) transgene expression delivered by AAV9, L001, L005, and scrambled provector in treated mice. AAV9, L005, and scrambled provector appeared to transduce indiscriminately throughout the myocardium, whereas there appeared to be distinct co-localization of MMP activity and iRFP expression for L001. The co-localization of iRFP with MMP activity was quantified via correlation analysis (Figures 4B and 4C). The iRFP and NIRF signal intensities for each pixel of heart tissue were extracted, and the associated Pearson’s correlation coefficients (PCCs) indicated that L001 demonstrated significantly greater co-localization of iRFP with MMP activity compared to AAV9.
Figure 4.

In Vivo Characterization of Provectors in Murine Model of Myocardial Infarction
(A) Heart cross-sectional images of MMP activity and vector transgene expression demonstrate co-localization of signal for provector-treated mice. MI was induced by ligating the left anterior descending coronary artery. At 2 days post-MI, mice were injected in the tail vein with 5 × 1010 vg AAV9, L001, L005, or scrambled vector packaging infrared fluorescent protein (iRFP) transgene. 10 days following injection, a peptide conjugated with IRDye 800CW (a near-infrared fluorescent marker dye) that binds to active MMPs was injected intravenously. Ex vivo slices of MI hearts were imaged to quantify co-localization of MMP activity (NIRF) and viral transduction (iRFP). Values reported in the scale bars represent pixel intensity values of the image. (B) Representative scatterplots of iRFP pixel intensity versus NIRF pixel intensity of AAV9, L001, L005, and scrambled vector show the correlation between these two signals for the pixels found within the heart slices. The spread of the scatterplot indicates the correlation between the signals, as quantified using Pearson’s correlation coefficient (PCC). (C) L001 provector displays the highest co-localization of transgene expression in cardiac tissue with high MMP activity, as quantified using PCC. L001 shows a significantly higher correlation of iRFP pixel intensity to NIRF pixel intensity than AAV9, indicating a stronger co-localization of viral transduction with MMP activity. Error bars, SEM; AAV9 and L001 n = 5, L005 N = 2, scrambled n = 4; *p < 0.05. (D) L001 provector has decreased viral genome delivery to the liver. 10 days following virus injection, off-target organs were harvested and genomic DNA was extracted. Viral genomes were quantified via qPCR using 100 ng DNA loaded in triplicate. Error bars, SEM; n = 3 except scrambled n = 4; **p < 0.001. (E) L001 provector yields significantly decreased transgene mRNA expression in the liver. 10 days following virus injection, off-target organs were harvested and RNA was extracted. iRFP mRNA was quantified via qPCR and normalized to 18 s mRNA in triplicate. Error bars, SEM; n = 3; *p < 0.05, **p < 0.001.
In addition to determining the targeting efficiency of the provectors to MMP-rich tissues in the heart, we assessed the viruses’ ability to detarget other organs. Off-target organs were harvested, and DNA and RNA were extracted to quantify the number of viral genomes delivered to the organs as well as the resulting transgene expression, respectively. AAV9 viral genomes were predominantly found in the liver and spleen (Figure 4D). L001 and L005 showed 153- and 400-fold decreases in viral genome delivery to the liver, respectively, compared to AAV9 (statistically significant); 6- and 4-fold decreases in the kidney, respectively; and a nearly 3-fold decrease in the spleen for both (not statistically significant). Interestingly, the scrambled vector showed a slightly higher viral genome per microgram (vg/μg) DNA compared to AAV9 in all organs except the liver, with a 58-fold increase in the spleen in particular (not statistically significant).
The observed decreases in viral genomes delivered to off-target organs by provector variants were reflected in decreases in transgene mRNA expression. AAV9 transgene mRNA indicated a high expression in the liver, brain, and muscle (Figure 4E). L001 and L005 showed 39- and 207-fold decreases in RNA expression in the liver compared to AAV9, respectively, as well as modest decreases in expression in the spleen, brain, and muscle (not statistically significant). The scrambled variant also showed decreases in RNA expression compared to AAV9 in nearly all organs, despite showing increases in viral genomes in those same organs.
We also investigated how the provectors’ blood circulation time may change in relation to AAV9 (Figure 5A). Variants were titered prior to injection to ensure equal dosing. L001 and the scrambled variant both demonstrated enhanced blood persistence compared to AAV9 through 24 h, but they then demonstrated faster clearance from the blood after that time point. As antibody neutralization is a significant concern for AAV-based vectors, we looked at the provectors’ ability to stimulate the production of antibodies that neutralize AAV9 transduction (Figure 5B). Mice were challenged with AAV9, L001, or the scrambled variant, and the resulting serum was tested for its ability to neutralize AAV9 transduction. The neutralizing antibody titer (i.e., the dilution at which 50% transduction is achieved) of serum raised against AAV9 was approximately 1:1,200, whereas the neutralizing antibody titers of serum raised against L001 and the scrambled variant were approximately 1:500 and 1:700, respectively, indicating that antibodies raised against L001 or the scrambled variant did not neutralize AAV9 as substantially.
Figure 5.

Blood Clearance and Neutralizing-Antibody Profile of Provector
(A) Provector has longer blood circulation time than AAV9. Mice were injected with 1 × 1011 vg AAV9, L001, or scrambled vector through the tail vein. At the indicated time points, blood was collected, and viral genomes were extracted and quantified via qPCR. L001 and scrambled vector show significantly increased blood persistence compared to AAV9 through 24 h and decreased persistence after that time point. (Inset) Early time points from 5 min to 1 h. Error bars, SEM; n = 3; +p < 0.05 AAV9 versus scrambled, +++p < 0.0001 AAV9 versus scrambled, ***p < 0.0001 AAV9 versus L001. (B) Antibodies raised against provector do not neutralize AAV9 transduction as significantly as antibodies raised against AAV9. Mice were injected with 1 × 1011 vg AAV9, L001, or scrambled vector through the tail vein, and 21 days later serum was collected. Serum dilutions were mixed with AAV9 and added to CHO-Lec2 cells (MOI: 1,000). Flow cytometry was then used to quantify GFP expression and data normalized to the TI achieved at the highest serum dilution condition. 50% transduction was achieved at a lower dilution of serum raised against L001 or scrambled vector compared to serum raised against WT. n = 3; ++p < 0.001 AAV9 versus scrambled, +++p < 0.0001 AAV9 versus scrambled, ***p < 0.0001 AAV9 versus L001.
Discussion
We successfully generated a panel of AAV9 variants that demonstrate switchable transduction in response to MMP exposure. Inserting the peptide lock after residue G453 resulted in vectors that meet all four necessary design criteria for a successful provector: (1) the vectors maintained near-WT levels of capsid assembly and genome protection with the lock insertions, (2) the locks were accessible and cleavable by MMPs, (3) the locks blocked galactose binding and transduction when not treated with MMPs, and (4) the vectors demonstrated a restoration of transduction following treatment with MMPs. Due to their clinical relevance in heart failure, MMP-2 and -9 were chosen for screening provectors’ in vitro susceptibility to proteolytic activation. However, it is likely the provectors may also be activated by a number of other MMPs in vivo because MMPs are highly promiscuous and share common substrate sequences.25
The panel of failed variants emphasizes the importance of the capsid insertion site on provector function. It is clear that cleavability by MMPs alone is not sufficient to generate the desired switchable transduction behavior. A number of variants built and tested do not show a restoration of transduction following proteolysis, despite successful lock cleavage (Figure S5). This indicates that insertion of the peptide lock after these unsuccessful residues may permanently ablate galactose binding through structural disruption of the galactose-binding pocket. It is also possible that the scar residues that remain after the inactivating motif of the lock has been cleaved off could still be sufficiently blocking galactose binding such that transduction cannot be restored.
Moreover, insertion of peptide locks into capsid hypervariable regions is not sufficient to yield successful provector designs. The capsid tertiary and quaternary structures need to be taken into careful consideration when picking lock insertion sites. An insertion site that is part of a hypervariable capsid region can still result in insertion intolerance, as shown with the N470, A472, V473, and S454 variants. Each galactose-binding pocket comprises residues from two different adjacent capsid subunits, and, interestingly, N470, A472, and V473 make up the floor of the binding pocket and are from a different subunit than the other residues in the binding pocket.21 It is possible that lock insertion after these residues disrupts subunit interactions necessary for capsid assembly, leading to the observed decreases in viral titers. The insertion intolerance of the S454 variant was especially surprising given its location in the middle of a known surface-exposed hypervariable region,22 and the insertion tolerance of G453 just one residue away. Insertion of other peptides unrelated to this project after S454 also results in low titers (data not shown). Additional exploration of the properties of this residue will be necessary to understand its insertion intolerance, particularly because the residue is tolerant to point mutations.26
The lack of ordering of the inserted peptide lock in the cryo-reconstruction is likely due to inherent flexibility at the N- and C-terminal ends as well as within the inserted sequence, 453-GAGVPMSMRGGGDDDDGVPMSMRGGGA-479, due to the glycine residues. This could result from the loop adopting different conformations, which would be incompatible with the icosahedral symmetry imposed during the structure determination. However, despite the lack of structure ordering, the inserted loop is functional, indicating that position 453 is a suitable location for the insertion of targeting peptides. This insertion does not cause any conformational changes in the overall capsid structure. This observation is similar to other chimeric AAVs generated by rational structure design, in which the loop of one serotype can be swapped with that of another serotype in order to improve transduction in a specific tissue or to escape the immune system.27
We demonstrated L001 provector’s switchable in vitro transduction behavior successfully translates to targeted gene delivery in vivo in a murine model of MI. It is interesting to note that L005, which displays inferior in vitro protease activatability compared to L001, demonstrates inferior in vivo targeting behavior as well, suggesting that in vitro activatability may be a good predictor of in vivo performance. Promisingly, the provector variants show decreased viral genome delivery and transgene expression in several off-target organs, as desired. In the future, depending on the transgene to be delivered, further optimization and a greater decrease in off-target expression may be needed. The locked L001 shows some basal level of transduction ability as well as a response to lectin concentration. These results indicate that galactose binding is not completely blocked and the lock can be further optimized to block receptor binding with greater efficiency. We can also combine the MMP-targeting capabilities of the provector with elements of transgene expression regulation, such as incorporating tissue-specific and/or inducible promoter systems, for potentially greater spatiotemporal control of transgene expression.
The different serological profile of L001 compared to AAV9 is perhaps not too surprising, considering that the 3-fold protrusions where the peptide lock is inserted is known to be a common site for neutralizing antibody binding and recognition, and G453-N457 has been identified as a potential neutralizing antibody epitope.26, 28, 29 Whether antibodies raised against AAV9 can still neutralize L001 remains to be determined, but it would be beneficial for clinical translation if pre-existing immunity against AAV9 did not also lead to immunity against L001.
Inclusion of the scrambled variant as a control demonstrates that insertion of a peptide after residue G453 alone does not confer targeted delivery of the provector to sites of upregulated MMP activity. The scrambled and L001 provectors share the exact same peptide lock amino acid composition, except the MMP cleavage sequence is in a random order in the scrambled vector. L001 and scrambled vectors show distinctly different biodistribution patterns, with the scrambled vector showing significantly less specificity and accuracy in targeting areas of high MMP activity. These results indicate cleavage of the peptide lock by MMPs is necessary for the observed targeted delivery. Furthermore, the scrambled vector has enhanced blood persistence similar to L001, thus delayed blood clearance alone cannot explain the targeted delivery shown by L001.
The observed discrepancy between the scrambled vector’s higher viral genome delivery but lower transgene expression suggests the scrambled variant may be sequestered in tissues without transducing them. A similar behavior was observed for an AAV9 mutant with its integrin-binding motif ablated (AAV9/NGA).30 This mutant demonstrates increased viral genome delivery but decreased transgene expression in the spleen, which the authors speculate could be due to sequestration of the AAV9/NGA mutant by the reticuloendothelial system. Blood clearance of the AAV9/NGA mutant shows more rapid clearance after 24 h, which the authors suggest supports the theory of nonspecific clearance and sequestration by the spleen and reticuloendothelial system. The scrambled vector tested here also shows a similar blood clearance profile, suggesting it may also be cleared and sequestered by the spleen and reticuloendothelial system. Since the average blood circulation time in a mouse is ∼15 s,31 the virus would have circulated ∼20 times by the 5-min time point. Thus, normalization to the first time point is not feasible.
The AAV9-based provectors demonstrate the design concept can be expanded to other serotypes besides AAV2, diversifying the applications and diseases that can be targeted for gene therapy. The properties of AAV9 may lead to superior therapeutic performance and clinical translation for the AAV9-based provectors compared to AAV2-based provectors, especially for applications in the cardiovascular system. Furthermore, AAV9-based provectors may be useful for the treatment of diseases in the CNS as well. Diseases such as Alzheimer’s,32, 33 Parkinson’s,34 and Huntington’s35, 36 demonstrate upregulation of extracellular proteases, and AAV9 demonstrates an enhanced ability to cross the blood-brain barrier compared to AAV2,37 so AAV9-based provectors could be better suited for CNS applications compared to AAV2-based provectors. Additional optimization of the provector platform may be required for clinical translation. Exploration of alternative protease cleavage motifs, peptide lock designs, and capsid insertion locations will enable us to generate a panel of provectors that demonstrate a range of sensitivities and specificities to different proteases that may be appropriate for various applications.
Materials and Methods
Cell Culture
HEK293T cells were used for AAV vector production, and CHO-Lec2 cells were used to characterize in vitro transduction behavior. HEK293T cells were maintained in DMEM (Lonza, Basel, Switzerland) supplemented with 10% fetal bovine serum (FBS, Atlanta-Biologicals, Flowery Branch, GA) and 1% penicillin-streptomycin (Life Technologies, Carlsbad, CA). CHO-Lec2 cells were maintained in MEM-alpha (Life Technologies, Carlsbad, CA) supplemented with 10% FBS and 1% penicillin-streptomycin. Cells were grown as adherent cultures in 5% CO2 at 37°C, subcultured after treatment with 0.25% trypsin-EDTA (Life Technologies, Carlsbad, CA) for 2−5 min at 37°C, and resuspended in complete medium.
Cloning and Production of AAV Vectors
All provector-packaging plasmids were generated by modifying the pAAV2/9 plasmid that encodes for the WT AAV9 capsid. Site-directed mutagenesis (SDM) was used first to remove one preexisting NgoMIV site after the ampR gene and two KasI restriction sites in the rep gene. SDM was then used to introduce NgoMIV and KasI restriction sites within the cap reading frame after the indicated amino acid residues (VP1 numbering). Peptide lock inserts were generated by annealing and phosphorylating synthesized oligonucleotides (see Table S3 for sequences; Integrated DNA Technologies, San Jose, CA) and ligating into the modified pAAV2/9 plasmids using the NgoMIV and KasI sites.
AAV vectors were produced as previously described.12 Briefly, triple transfection was performed in HEK293T cells with a packaging plasmid (pAAV2/9 or a provector derivative), a self-complementary GFP (scGFP) or self-complementary infrared fluorescent protein (sciRFP) transgene plasmid, and pXX6-80 helper plasmid. Virus was separated from cell lysate using an iodixanol step gradient. Virus was further purified using anion exchange chromatography (Q Sepharose, Pall, Port Washington, NY) and concentrated using Amicon 100-kDa molecular weight (MW) cutoff filters (Millipore, Burlington, MA) into GB-PF68 buffer (50 mM Tris [pH 7.6], 150 mM NaCl, 10 mM MgCl2, and 0.001% Pluronic F68). Viral titers were determined via qPCR using Power SYBR Green (Thermo Fisher Scientific, Carlsbad, CA) and primers against the cytomegalovirus (CMV) transgene promoter (see Table S4 for primer sequences) on a Bio-Rad CFX96.
For cryoelectron microscopy, recombinant AAV9_453_L001 vectors packaging the luciferase gene (rAAV9_453_L001_luc) were produced by triple transfection in HEK293 cells from three plasmids, pHelper, pTR-UF3-luc, and pAAV9_453_L001, in a ratio of 2:1:1. Plasmids (total of 40 μg) were used to transfect ∼75% confluent cells, in 15-cm cell culture dishes, grown in DMEM. The transfection reagent was 125 μL polyethyleneimine (1 mg/mL), incubated at room temperature (RT) for 15 min and added dropwise to the HEK293 cells. The following day, 5 mL fresh DMEM was added to each plate. The cells were harvested 72 h post-infection and collected by centrifugation at 1,500 rpm in a JA-20 rotor at 4°C for 20 min. The cell pellet was resuspended in 1× PBS, 1 mM MgCl2, and 2.5 mM KCl (TD buffer), and the supernatant was precipitated with 10% (w/v) polyethylene glycol 8000 (PEG8000). The cell pellet was subjected to three freeze-thaw cycles in a dry ice-ethanol bath and a 37°C water bath, respectively. After the third freeze-thaw cycle, the cell pellet was treated with Benzonase (250 U) and incubated at 37°C for 30 min. The PEG supernatant was spun at 10,000 rpm in a JA-10 rotor at 4°C for 90 min to recover the precipitated virus. The PEG pellet was resuspended in TD buffer, treated with Benzonase (250 U), and incubated at 37°C for 30 min. Both solutions were clarified by centrifugation at 10,000 rpm in a JA-20 rotor at 4°C for 20 min for further purification.
The clarified supernatant was diluted 1:1 with TD buffer and loaded onto a 1-mL gravity flow column packed with POROS CaptureSelect AAV9 Affinity resin (Thermo Scientific, Waltham, MA). The loaded column was washed with 20 mL TD buffer. The virus was eluted with 10 mL in 0.1 M NaAc and 0.75 M NaCl (pH 2.5) (elution buffer), and the peak fractions were collected and neutralized with 1 M Tris-HCl (pH 10) (neutralization buffer) at a 1:10 ratio. The elution fractions that showed VP1, VP2, and VP3 on an SDS-PAGE gel were pooled and concentrated, and the buffer was exchanged into 50 mM HEPES (pH 7.4), 100 mM NaCl, and 2 mM MgCl2. The sample purity and concentration were determined by SDS-PAGE and UV spectrometry for the L001 vector (E = 1.7 for concentration in mg/mL), respectively.
Genome Protection Assay
Virus samples were diluted 1:10 in a buffer of 1.5 mM MgCl2, 0.5 mg/mL BSA, and 50 mM Tris (pH 8.0). Samples were mixed with either benzonase (≥125 units; Sigma-Aldrich, St. Louis, MO) or vehicle (50% Glycerol, 50 mM Tris [pH 8.0], 20 mM NaCl, and 2 mM MgCl2) and incubated at 37°C for 30 min. The reactions were stopped by adding EDTA to a final concentration of 12 mM. The number of viral genomes was quantified using qPCR, and genome protection was calculated by normalizing benzonase-treated samples to vehicle-treated samples.
Cryoelectron Microscopy and Data Collection
3 μL purified rAAV9_453_L001_luc, at 0.5 mg/mL, was pipetted onto glow-discharged copper grids containing 2-nm carbon support over holes (Quantifoil R 2/4 200 mesh, Electron Microscopy Sciences, Hatfield, PA). The grids were immediately vitrified with a Mark IV vitrobot (FEI, Hillsboro, OR). The grids were screened for suitability for data collection on a Tecnai G2 F20-Twin transmission electron microscope operated at 200 kV and −20e−/Å2 dosage. The data were collected as movie frames on a Titan Krios electron microscope (FEI, Hillsboro, OR), operated at 300 kV with a DE20 direct electron detector, and the data collection parameters are listed in Table S2. Each frame was aligned with the DE_process_frames_software, with corresponding dark and bright reference images without radiation dose compensation.
Structure Determination, Model Building, and Refinement
Particle extraction from the aligned micrographs, data preprocessing, and structure determination were as previously described for human bocaviruses.38 The structure determination procedure followed the gold standard protocol, and the resolution was estimated at Fourier shell correlation (FSC) = 0.143. The high-resolution features of the map were sharpened with B-factors 1/50, 1/100, 1/150, and 1/200 Å2, and the maps were visually inspected in the program Coot.39 The 1/50 map showed the best features for the side and main chains, and it was selected for manual model building and refinement in the Coot and Phenix programs.39, 40
The AAV9_453_L001 map was interpreted with a model generated in the SWISS-MODEL online program41 from the structure of AAV9 (PDB: 3UX1) supplied as a template and the AAV9 VP3 sequence with the 26-residue sequence (AGVPMSMRGGGDDDDGVPMSMRGGGA) inserted at position 453. The model building and refinement steps were as previously reported for AAV2.5.27 The final refinement statistics are listed in Table S2. Figures were generated using the UCSF-Chimera Program42 and PyMol43 programs.
Proteolysis
MMP-2, MMP-7, and MMP-9 were purchased from Enzo Life Sciences (Farmingdale, NY). Protease activity was measured prior to each experiment to reduce variability due to enzyme storage, as previously described.12, 44 Briefly, the activity of 5 nM MMP on 5 mM of the fluorogenic substrate Mca-PLGL-Dpa-AR (Calbiochem, Burlington, MA) was measured using a Tecan M1000 plate reader in a buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 5 mM CaCl2, and 0.005% Brij-35. The amount of enzyme added to each proteolysis reaction was standardized to the initial reaction velocity, as previously described.12 The MMPs were diluted to 9× in MMP-9 storage buffer (50 mM Tris [pH 7.5], 1 mM CaCl2, 300 mM NaCl, 5 μM ZnCl2, 0.1% Brij-35, and 15% glycerol). Either MMP or equal-volume sham buffer (50 mM Tris [pH 7.5], 1 mM CaCl2, 300 mM NaCl, 5 μM ZnCl2, 0.1% Brij-35, and 15% glycerol) was added (final MMP concentration, 300 nM) to purified virus samples (1.5 × 1012 vg/mL) diluted in GB-PF68 buffer. The reactions were incubated at 37°C for 20 h and stopped with EDTA (25 mM final concentration).
Silver Stain
Virus samples (1.5 × 109 vg) were denatured in NuPAGE LDS Sample Buffer, run on a 4%–12% Bis-Tris NuPAGE gel for 2 h at 100V, and stained using the Silverquest staining kit (Life Technologies, Carlsbad, CA), according to the manufacturer’s instructions. Stained gels were imaged with an Epson scanner.
Transduction Assay
Viral transduction was tested using CHO-Lec2 cells. AAV vectors packaging an scGFP transgene were mixed with serum-free media, and they were added at an MOI of 5,000 to cells at 95% confluency. After an incubation period of 24 h, the media were replaced with serum-containing media. At 48 h post-transduction, the cells were harvested and GFP fluorescence was quantified using flow cytometry (FACSCanto II; BD Biosciences, Franklin Lakes, NJ).
Lectin Competition Assay
CHO-Lec2 cells at 95% confluency were incubated at 4°C for 30 min prior to transduction. ECL (reconstituted in PBS, 10 mg/mL; Vector Laboratories, Burlingame, CA) was diluted in cold serum-free media and mixed with AAV vectors packaging an scGFP transgene. The ECL-virus mixture was added at an MOI of 5,000 to cells and incubated at 4°C for 1 h. Virus-containing media were removed, the cells were washed with PBS, fresh media were added, and the cells were then incubated at 37°C for 48 h before being harvested for flow cytometry.
Animal Work
All animal work was carried out using 8- to 12-week-old female BALB/c mice purchased from Charles River Laboratories. Mice were maintained and treated in accordance with NIH guidelines and as approved by the University of Texas Health Science Center at Houston Animal Welfare Committee and Rice University Institutional Animal Care and Use Committee.
LAD Surgery
To create a myocardial infarct, the LAD was ligated according to published procedures.45 At 2 days after the induction of the myocardial infarct, AAV9, L001, L005, or scrambled provector (5 × 1010 vg/mouse) packaging an sciRFP transgene was injected through the tail vein in 0.1 mL GB-PF68 buffer. At 10 days after vector administration, 200 μCi 64-Cu-DOTA-IRDye800-MMP agent24 was administered in 0.1 mL through the tail vein for visualizing MMP activity in the infarct. 6 h later, animals were euthanized, and the heart tissues were collected for fluorescence imaging of iRFP transgene expression and NIRF-MMP activity.
iRFP and NIRF Imaging
Imaging was performed as previously described.46 Briefly, mice were sacrificed for iRFP and NIRF fluorescence imaging. Hearts were sliced cross-sectionally, and both iRFP and NIRF fluorescence imaging were performed on sliced hearts using a military-grade NIR-sensitive Gen III image intensifier and a frame-transfer 16-bit charge-coupled device (CCD) camera. For iRFP imaging, the tissues were excited with a 690-nm laser diode, and emission was collected with two 720-nm band-pass filters. For NIRF-MMP imaging, the tissues were excited with a 785-nm laser diode, and emission was collected with two 830-nm band-pass filters.
A custom MATLAB script was written to perform the image analysis as previously described.47 Image registration was performed to align corresponding NIRF and IRFP images. The heart slice regions of interest were then segmented to isolate the heart pixels by first normalizing any uneven background using top-hat algorithm followed by binarization using Otsu’s algorithm (Figure S4). The scatterplots were generated by plotting raw NIRF pixel intensities against the corresponding raw IRFP pixels for the heart slice pixels identified by the segmentation. The PCC was calculated from the scatterplot.
Tissue Processing for RNA and DNA Extraction and Quantification
Tissues were harvested and snap frozen in liquid nitrogen for storage. At the time of processing, tissues were thawed and homogenized using a mortar and pestle. 25 mg of each tissue was processed for genomic DNA extraction using DNeasy Blood and Tissue Kit (QIAGEN, Hilden, Germany), according to the manufacturer’s protocols. Viral genomes were quantified using 100 ng genomic DNA/well loaded in triplicate via qPCR using primers against the CMV transgene promoter. RNA was extracted using TRIzol (Thermo Fisher Scientific, Carlsbad, CA) according to the manufacturer’s protocol. 1 μg extracted RNA was reverse transcribed into cDNA according to the manufacturer’s protocol, using a 3:1 blend of random hexamers and anchored oligo-dT (Verso cDNA Synthesis Kit; Thermo Fisher Scientific, Carlsbad, CA). Vector transgene cDNA was then quantified by qPCR using 200 ng cDNA/well loaded in triplicate and primers against iRFP (PowerUp SYBR Green; Thermo Fisher Scientific, Carlsbad, CA) or the housekeeping gene 18 s (Power SYBR Green; Thermo Fisher Scientific, Carlsbad, CA) (see Table S4 for primer sequences). Transgene mRNA expression was normalized to that of 18 s.
Blood Circulation Assay
Healthy mice were injected with 1 × 1011 vg AAV9, L001, or scrambled vector packaging an scGFP transgene through the tail vein in 0.1 mL GB-PF68 buffer. At 5 min, 20 min, 40 min, 1 h, 4 h, 24 h, 48 h, and 72 h after injection, 10 μL blood was drawn by nicking the saphenous vein in the left leg, switching to the right leg after the 1-hr time point. Blood was mixed with 10 μL 3.2% sodium citrate solution (Greiner Bio-One, Kremsmunster, Austria). Viral DNA was extracted from blood samples using the DNeasy Blood and Tissue Kit (QIAGEN, Hilden, Germany) and quantified by qPCR using primers against the CMV transgene promoter.
In Vitro Neutralizing-Antibody Assay
Healthy mice were injected with 1 × 1011 vg AAV9, L001, or scrambled vector packaging an scGFP transgene through the tail vein in 0.1 mL GB-PF68 buffer. At 21 days post-injection, blood was collected via cardiac puncture and was allowed to clot for 30 min at room temperature. The clotted blood was centrifuged at 1,000 × g for 10 min, and the supernatant serum was collected. CHO-Lec2 cells were cultured and maintained as described above. 2-fold serial dilutions of sera were prepared in MEM-alpha. Diluted sera were mixed with an equal volume of AAV9 packaging an scGFP transgene and incubated for 30 min at 37°C. Diluted sera and virus mixtures were then added to cells at an MOI of 1,000, and the transduction assay was performed as described above.
Statistical Analysis
Data were analyzed using GraphPad Prism. One-way ANOVA was performed with Dunnett’s post hoc multiple comparison test to compare provector variants to AAV9. Unpaired t test was used to compare L001 PCCs to AAV9 PCCs for statistical significance.
Structure Accession Numbers
The L001 cryo-EM reconstructed density map and capsid coordinates are deposited in the Electron Microscopy Data Bank (EMDB): EMD-0535 and EMD-6NXE, respectively.
Author Contributions
Conceptualization, C.M.G. and J.S.; Methodology, C.M.G., M.J.B., M.L.H., W.C., B.Z., S.K., E.M.S.-M., M.A.-M., and J.S.; Investigation, C.M.G., M.J.B., M.L.H., W.C., M.Y., M.L., B.Z., A.D.B., S.K., A.C.E., and J.V.; Writing – Original Draft, C.M.G. and J.S.; Writing – Review & Editing, C.M.G., M.J.B., M.L.H., W.C., M.Y., M.L., B.Z., A.D.B., S.K., A.C.E., J.V., E.M.S.-M., M.A.-M., and J.S.; Supervision, E.M.S.-M., M.A.-M., and J.S.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This material is based on work supported by the NIH (grant numbers R21HL126053, R01HL138126, and R01CA207497), the American Heart Association (grant numbers 13GRNT14420044 and 15GRNT23070007), and a Dunn Foundation grant to J.S.; an American Heart Association Predoctoral Fellowship (16PRE30690006) to C.M.G.; the NIH (R01GM082946) to M.A.-M.; and National Science Foundation Fellowships to A.C.E. (2015197891) and W.C. (2018253392). We acknowledge the University of North Carolina at Chapel Hill Gene Therapy Center Vector Core for providing us with pXX6-80 and scAAV2-CMV-GFP and the University of Pennsylvania Vector Core for providing us with pAAV2/9. We thank the Electron microscopy core of the University of Florida (UF) Interdisciplinary Center for Biotechnology Research (ICBR) for access to electron microscopes utilized for negative stain electron microscopy and cryoelectron microscopy (cryo-EM) data collection. The Spirit and TF20 cryoelectron microscopes were provided by the UF College of Medicine (COM) and Division of Sponsored Programs (DSP). Data collection at Florida State University was made possible by NIH grants S10 OD018142-01 (purchase of a direct electron camera for the Titan-Krios at FSU, P.I. Taylor), S10 RR025080-01 (purchase of an FEI Titan Krios for 3-D EM, P.I. Taylor), and U24 GM116788 (The Southeastern Consortium for Microscopy of MacroMolecular Machines, P.I. Taylor).
Footnotes
Supplemental Information includes five figures and four tables and can be found with this article online at https://doi.org/10.1016/j.ymthe.2019.01.015.
Supplemental Information
References
- 1.Mozaffarian D., Benjamin E.J., Go A.S., Arnett D.K., Blaha M.J., Cushman M., Das S.R., de Ferranti S., Després J.P., Fullerton H.J., Writing Group Members. American Heart Association Statistics Committee. Stroke Statistics Subcommittee Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133:e38–e360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 2.Wolfram J.A., Donahue J.K. Gene therapy to treat cardiovascular disease. J. Am. Heart Assoc. 2013;2:e000119. doi: 10.1161/JAHA.113.000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pacak C.A., Byrne B.J. AAV vectors for cardiac gene transfer: experimental tools and clinical opportunities. Mol. Ther. 2011;19:1582–1590. doi: 10.1038/mt.2011.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Müller O.J., Katus H.A., Bekeredjian R. Targeting the heart with gene therapy-optimized gene delivery methods. Cardiovasc. Res. 2007;73:453–462. doi: 10.1016/j.cardiores.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 5.Pleger S.T., Brinks H., Ritterhoff J., Raake P., Koch W.J., Katus H.A., Most P. Heart failure gene therapy: the path to clinical practice. Circ. Res. 2013;113:792–809. doi: 10.1161/CIRCRESAHA.113.300269. [DOI] [PubMed] [Google Scholar]
- 6.Tilemann L., Ishikawa K., Weber T., Hajjar R.J. Gene therapy for heart failure. Circ. Res. 2012;110:777–793. doi: 10.1161/CIRCRESAHA.111.252981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peterson J.T., Li H., Dillon L., Bryant J.W. Evolution of matrix metalloprotease and tissue inhibitor expression during heart failure progression in the infarcted rat. Cardiovasc. Res. 2000;46:307–315. doi: 10.1016/s0008-6363(00)00029-8. [DOI] [PubMed] [Google Scholar]
- 8.Thomas C.V., Coker M.L., Zellner J.L., Handy J.R., Crumbley A.J., 3rd, Spinale F.G. Increased matrix metalloproteinase activity and selective upregulation in LV myocardium from patients with end-stage dilated cardiomyopathy. Circulation. 1998;97:1708–1715. doi: 10.1161/01.cir.97.17.1708. [DOI] [PubMed] [Google Scholar]
- 9.Spinale F.G. Matrix metalloproteinases: regulation and dysregulation in the failing heart. Circ. Res. 2002;90:520–530. doi: 10.1161/01.res.0000013290.12884.a3. [DOI] [PubMed] [Google Scholar]
- 10.Newby A.C. Metalloproteinases and vulnerable atherosclerotic plaques. Trends Cardiovasc. Med. 2007;17:253–258. doi: 10.1016/j.tcm.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vanhoutte D., Schellings M., Pinto Y., Heymans S. Relevance of matrix metalloproteinases and their inhibitors after myocardial infarction: a temporal and spatial window. Cardiovasc. Res. 2006;69:604–613. doi: 10.1016/j.cardiores.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 12.Judd J., Ho M.L., Tiwari A., Gomez E.J., Dempsey C., Van Vliet K., Igoshin O.A., Silberg J.J., Agbandje-McKenna M., Suh J. Tunable protease-activatable virus nanonodes. ACS Nano. 2014;8:4740–4746. doi: 10.1021/nn500550q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ho M.L., Adler B.A., Torre M.L., Silberg J.J., Suh J. SCHEMA computational design of virus capsid chimeras: calibrating how genome packaging, protection, and transduction correlate with calculated structural disruption. ACS Synth. Biol. 2013;2:724–733. doi: 10.1021/sb400076r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson T.M., Judd J., Ho M.L., Suh J. Role of Tetra Amino Acid Motif Properties on the Function of Protease-Activatable Viral Vectors. ACS Biomater. Sci. Eng. 2016;2:2026–2033. doi: 10.1021/acsbiomaterials.6b00439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vandenberghe L.H., Xiao R., Lock M., Lin J., Korn M., Wilson J.M. Efficient serotype-dependent release of functional vector into the culture medium during adeno-associated virus manufacturing. Hum. Gene Ther. 2010;21:1251–1257. doi: 10.1089/hum.2010.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calcedo R., Vandenberghe L.H., Gao G., Lin J., Wilson J.M. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J. Infect. Dis. 2009;199:381–390. doi: 10.1086/595830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boutin S., Monteilhet V., Veron P., Leborgne C., Benveniste O., Montus M.F., Masurier C. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum. Gene Ther. 2010;21:704–712. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- 18.Zincarelli C., Soltys S., Rengo G., Rabinowitz J.E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 19.Bish L.T., Morine K., Sleeper M.M., Sanmiguel J., Wu D., Gao G., Wilson J.M., Sweeney H.L. Adeno-associated virus (AAV) serotype 9 provides global cardiac gene transfer superior to AAV1, AAV6, AAV7, and AAV8 in the mouse and rat. Hum. Gene Ther. 2008;19:1359–1368. doi: 10.1089/hum.2008.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inagaki K., Fuess S., Storm T.A., Gibson G.A., Mctiernan C.F., Kay M.A., Nakai H. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol. Ther. 2006;14:45–53. doi: 10.1016/j.ymthe.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bell C.L., Gurda B.L., Van Vliet K., Agbandje-McKenna M., Wilson J.M. Identification of the galactose binding domain of the adeno-associated virus serotype 9 capsid. J. Virol. 2012;86:7326–7333. doi: 10.1128/JVI.00448-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DiMattia M.A., Nam H.-J., Van Vliet K., Mitchell M., Bennett A., Gurda B.L., McKenna R., Olson N.H., Sinkovits R.S., Potter M. Structural insight into the unique properties of adeno-associated virus serotype 9. J. Virol. 2012;86:6947–6958. doi: 10.1128/JVI.07232-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ilyas M., Mietzsch M., Kailasan S., Väisänen E., Luo M., Chipman P., Smith J.K., Kurian J., Sousa D., McKenna R. Atomic Resolution Structures of Human Bufaviruses Determined by Cryo-Electron Microscopy. Viruses. 2018;10:22. doi: 10.3390/v10010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodenberg E., Azhdarinia A., Lazard Z.W., Hall M., Kwon S.K., Wilganowski N., Salisbury E.A., Merched-Sauvage M., Olmsted-Davis E.A., Sevick-Muraca E.M., Davis A.R. Matrix metalloproteinase-9 is a diagnostic marker of heterotopic ossification in a murine model. Tissue Eng. Part A. 2011;17:2487–2496. doi: 10.1089/ten.TEA.2011.0007. [DOI] [PubMed] [Google Scholar]
- 25.Turk B.E., Huang L.L., Piro E.T., Cantley L.C. Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nat. Biotechnol. 2001;19:661–667. doi: 10.1038/90273. [DOI] [PubMed] [Google Scholar]
- 26.Adachi K., Enoki T., Kawano Y., Veraz M., Nakai H. Drawing a high-resolution functional map of adeno-associated virus capsid by massively parallel sequencing. Nat. Commun. 2014;5:3075. doi: 10.1038/ncomms4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burg M., Rosebrough C., Drouin L.M., Bennett A., Mietzsch M., Chipman P., McKenna R., Sousa D., Potter M., Byrne B. Atomic structure of a rationally engineered gene delivery vector, AAV2.5. J. Struct. Biol. 2018;203:236–241. doi: 10.1016/j.jsb.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gurda B.L., DiMattia M.A., Miller E.B., Bennett A., McKenna R., Weichert W.S., Nelson C.D., Chen W.J., Muzyczka N., Olson N.H. Capsid antibodies to different adeno-associated virus serotypes bind common regions. J. Virol. 2013;87:9111–9124. doi: 10.1128/JVI.00622-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agbandje-McKenna M., Kleinschmidt J. Humana Press; 2012. AAV Capsid Structure and Cell Interactions; pp. 47–92. [DOI] [PubMed] [Google Scholar]
- 30.Shen S., Berry G.E., Castellanos Rivera R.M., Cheung R.Y., Troupes A.N., Brown S.M., Kafri T., Asokan A. Functional analysis of the putative integrin recognition motif on adeno-associated virus 9. J. Biol. Chem. 2015;290:1496–1504. doi: 10.1074/jbc.M114.608281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Debbage P.L., Griebel J., Ried M., Gneiting T., DeVries A., Hutzler P. Lectin intravital perfusion studies in tumor-bearing mice: micrometer-resolution, wide-area mapping of microvascular labeling, distinguishing efficiently and inefficiently perfused microregions in the tumor. J. Histochem. Cytochem. 1998;46:627–639. doi: 10.1177/002215549804600508. [DOI] [PubMed] [Google Scholar]
- 32.Leake A., Morris C.M., Whateley J. Brain matrix metalloproteinase 1 levels are elevated in Alzheimer’s disease. Neurosci. Lett. 2000;291:201–203. doi: 10.1016/s0304-3940(00)01418-x. [DOI] [PubMed] [Google Scholar]
- 33.Duits F.H., Hernandez-Guillamon M., Montaner J., Goos J.D., Montañola A., Wattjes M.P., Barkhof F., Scheltens P., Teunissen C.E., van der Flier W.M. Matrix Metalloproteinases in Alzheimer’s Disease and Concurrent Cerebral Microbleeds. J. Alzheimers Dis. 2015;48:711–720. doi: 10.3233/JAD-143186. [DOI] [PubMed] [Google Scholar]
- 34.Chung Y.C., Kim Y.-S., Bok E., Yune T.Y., Maeng S., Jin B.K. MMP-3 contributes to nigrostriatal dopaminergic neuronal loss, BBB damage, and neuroinflammation in an MPTP mouse model of Parkinson’s disease. Mediators Inflamm. 2013;2013:370526. doi: 10.1155/2013/370526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller J.P., Holcomb J., Al-Ramahi I., de Haro M., Gafni J., Zhang N., Kim E., Sanhueza M., Torcassi C., Kwak S. Matrix metalloproteinases are modifiers of huntingtin proteolysis and toxicity in Huntington’s disease. Neuron. 2010;67:199–212. doi: 10.1016/j.neuron.2010.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duran-Vilaregut J., del Valle J., Manich G., Camins A., Pallàs M., Vilaplana J., Pelegrí C. Role of matrix metalloproteinase-9 (MMP-9) in striatal blood-brain barrier disruption in a 3-nitropropionic acid model of Huntington’s disease. Neuropathol. Appl. Neurobiol. 2011;37:525–537. doi: 10.1111/j.1365-2990.2010.01157.x. [DOI] [PubMed] [Google Scholar]
- 37.Zhang H., Yang B., Mu X., Ahmed S.S., Su Q., He R., Wang H., Mueller C., Sena-Esteves M., Brown R. Several rAAV vectors efficiently cross the blood-brain barrier and transduce neurons and astrocytes in the neonatal mouse central nervous system. Mol. Ther. 2011;19:1440–1448. doi: 10.1038/mt.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mietzsch M., Kailasan S., Garrison J., Ilyas M., Chipman P., Kantola K., Janssen M.E., Spear J., Sousa D., McKenna R. Structural Insights into Human Bocaparvoviruses. J. Virol. 2017;91:e00261-17. doi: 10.1128/JVI.00261-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Afonine P.V., Grosse-Kunstleve R.W., Adams P.D., Urzhumtsev A. Bulk-solvent and overall scaling revisited: faster calculations, improved results. Acta Crystallogr. D Biol. Crystallogr. 2013;69:625–634. doi: 10.1107/S0907444913000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Gallo Cassarino T., Bertoni M., Bordoli L., Schwede T. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Z., Lasker K., Schneidman-Duhovny D., Webb B., Huang C.C., Pettersen E.F., Goddard T.D., Meng E.C., Sali A., Ferrin T.E. UCSF Chimera, MODELLER, and IMP: an integrated modeling system. J. Struct. Biol. 2012;179:269–278. doi: 10.1016/j.jsb.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delano, L.W. (2002). The PyMOL Molecular Graphics System. http://www.pymol.org at https://ci.nii.ac.jp/naid/10020095229/.
- 44.Ho M.L., Judd J., Kuypers B.E., Yamagami M., Wong F.F., Suh J. Efficiency of Protease-Activatable Virus Nanonodes Tuned Through Incorporation of Wild-Type Capsid Subunits. Cell. Mol. Bioeng. 2014;7:334–343. [Google Scholar]
- 45.Kolk M.V.V., Meyberg D., Deuse T., Tang-Quan K.R., Robbins R.C., Reichenspurner H., Schrepfer S. LAD-ligation: a murine model of myocardial infarction. J. Vis. Exp. 2009;(32):1438. doi: 10.3791/1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu B., Wu G., Robinson H., Wilganowski N., Hall M.A., Ghosh S.C., Pinkston K.L., Azhdarinia A., Harvey B.R., Sevick-Muraca E.M. Tumor margin detection using quantitative NIRF molecular imaging targeting EpCAM validated by far red gene reporter iRFP. Mol. Imaging Biol. 2013;15:560–568. doi: 10.1007/s11307-013-0637-8. [DOI] [PubMed] [Google Scholar]
- 47.Dunn K.W., Kamocka M.M., McDonald J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 2011;300:C723–C742. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.