

Abstract
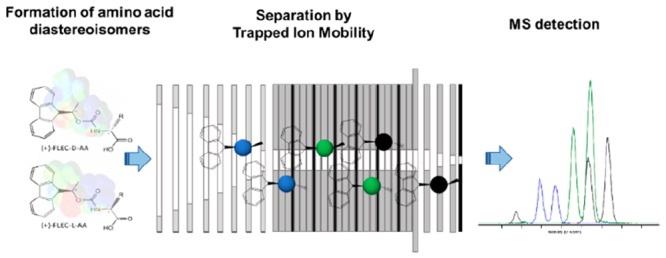
A novel analytical method based on hybrid trapped ion mobility spectrometry-time-of-flight mass spectrometry (TIMS-TOFMS) has been developed to achieve fast enantiomeric separation of amino acids (AAs). Resolution of chiral AAs was achieved by forming diastereomers through derivatization with the chiral agent (+)-1-(9-fluorenyl)ethyl chloroformate (FLEC), avoiding the use of reference compounds. Electrospray ionization (ESI) in positive mode yielded sodiated FLEC-AAs ions of which the diastereomers could be separated by TIMS. The effect of other alkali metal ions (such as Li and K) on the enantioselectivity was studied, but chiral discrimination was only observed for Na. TIMS conditions, including voltage ramp, ramp time, and accumulation time were optimized for each AA, and collision cross sections (CCSs) were determined for all diastereomers. The migration order of the DL enantiomers was found to be dependent on the structure of the AA. The resulting TIMS resolution (K0/ΔK0) for the FLEC-AA diastereomers on average was 115, requiring a mobility (K0) difference of about 0.009 cm2/(V s) to achieve 50%-valley separation. From the 21 AAs studied, enantiomer separation was achieved for 17 AAs with mobility differences ranging from 0.009 for lysine up to 0.061 cm2/(V s) for asparagine. Moreover, the presented methodology provided mutual separation of various AAs, allowing chiral analysis of multiple AAs simultaneously which may be challenging with previous enantioselective IMS approaches. It appeared possible to fully resolve all studied DL-AAs using three distinct TIMS methods, resulting in a total MS run time of about 3 min (1 min per method) and a total analysis time (including derivatization) of less than 15 min. The method demonstrated capable to determine enantiomeric ratios down to 2.5% with detection limits for the D enantiomers in the nanomolar range. This new TIMS-based methodology opens up possibilities for easy and fast analysis of AA enantiomers.
The intrinsic chiral environment of living systems makes enantiomers of a chiral compound often show mutually different chemical behavior or biological activity. In fact, when interacting with enzymes, proteins, or receptors, the enantiomeric configuration of the molecule is critical and changes may result in modulated biological function.1 Molecular chirality clearly plays an essential role in biological, medical, pharmaceutical, and food sciences, generating a strong requirement for enantioselective analytical methods.
Amino acids (AAs) are an important class of chiral molecules which constitute the building blocks of proteins and are key molecules in maintaining the physiology of the organism.2 Along with the 20 proteinogenic AAs, there are hundreds of AAs of nonprotein origin, which also have different key functions.3,4 Most AAs are chiral compounds, and although L-AAs are the predominant active form in mammalian biology, the presence of D-AAs in living organisms and in the environment has been reported extensively.1 Over the years, several analytical methods able to distinguish D and L AA enantiomers have been developed. Separation techniques, such as liquid chromatography (LC), gas chromatography (GC), and capillary electrophoresis (CE), are the most commonly employed techniques in chiral AA analysis.5−7 Their enantioseparation principle mostly relies on the selectivity provided by chiral stationary phases and/or selectors, and separation times typically range between 5 and 30 min.8−11
Mass spectrometry (MS), and in particular tandem MS, has emerged as an alternative technique for the fast chiral analysis of AAs. In this approach, enantiomers are first transformed into diastereomeric complexes using a chiral agent, and the chiral distinction is based on the different relative intensities of the fragment ions obtained for the respective AA enantiomers. However, interpretation of complex fragmentation patterns can be difficult, and as no actual separation of enantiomers takes place, enantiopure reference compounds are required.12,13
Ion mobility spectrometry (IMS) has demonstrated to be a powerful tool for the fast separation (milliseconds) of isobaric and isomeric compounds. In drift-tube IMS, ions are separated based on their mobility in an electric field through a neutral gas. As the mobility is a function of the ion collision cross section (CCS), which depends on the size and shape of the ion in the gas phase, compounds may be separated by IMS, even if their m/z ratio is the same.14 In 2011, Fernández-Lima et al. introduced trapped ion mobility spectrometry (TIMS)15,16 in which ions are carried into the drift cell using a nitrogen gas flow. An electric field gradient is applied in the opposite direction in order to trap and separate ions depending on their size, charge, and shape.17 This allows use of much shorter IM drift tubes,18 while potentially achieving a high resolving power (up to 300), providing the possibility to attain fast separations of isobaric and isomeric compounds.19 In TIMS, resolution (or resolving power) R is often defined as K/ΔK where K is the mobility and ΔK is the full width at half-maximum (fwhm) of a compound peak in the mobilogram.17
Compound enantiomers exhibit identical CCSs, making their direct separation by IMS not feasible. In order to achieve chiral separation of enantiomers by IMS, several strategies have been adopted, such as creating an asymmetric environment in the drift tube by doping the drift gas with a chiral agent20,12 or by inducing a conformational change with a complexing agent leading to different CCSs of the studied enantiomers.21,14,22 Chiral separation of AAs by IMS has been studied by few researchers. Prabha et al. achieved the enantioseparation of the AAs Ser, Met, Thr, Phe, and Trp by doping the drift gas with a volatile chiral reagent ((S)-(+)-2-butanol).12 Further works on the enantioseparation of AAs by IMS employ the formation of noncovalent diastereomeric complexes of the AAs with a metal-ion containing chiral reference agent prior to IMS analysis.22,14,21 Mie et al. first demonstrated the potential of this approach by the enantioseparation of six AAs using high-field asymmetric waveform ion mobility spectrometry (FAIMS). They formed metal-bound trimeric complexes of the form [M(II)(L-ref)2(D/L-AA)-H]+ where M was Ni, Zn, Mg, or Cu, AA was Trp, Pro, Phe, Val, Arg, and Lys, and L-ref is the AA acting as a chiral reference compound.22 Using traveling wave ion mobility spectrometry (TWIMS), Domalain et al. described the differentiation of seven AA enantiomers through their cationization with copper(II) and a reference AA. After screening 11 AAs, D-Phe was selected as the most suitable chiral reference compound.14 In a similar way, Xiangying Yu et al. studied the chiral separation of Trp, Gln, Tyr, Thr, His, Glu, Met, Phe, and Arg by forming binuclear copper bound tetrameric ions with L-Trp, L-Pro, L-Tyr, L-Phe, and L-His as reference.21 The main limitation of these approaches is the need for specific reference compounds for each AA, limiting general application. Moreover, the enantiomeric discrimination was only demonstrated for individual AAs, i.e., no mixtures of AAs were analyzed.
The present work proposes a new alternative and generic approach for achieving chiral separation of AAs by employing TIMS. To this end, prior to TIMS analysis the AA enantiomers were quickly converted to diastereomers with the chiral reagent (+)-1-(9-fluorenyl)ethyl chloroformate (FLEC). Subsequently, the diastereomers were efficiently separated based on their difference in ion mobility. In total, 22 proteogenic and nonproteogenic AAs were included in the study and parameters affecting IMS separation were evaluated. The novel enantioselective TIMS method is fast and applicable to any chiral AA, omits the need for AA-specific reference compounds, and allows one to resolve multiple AAs in one run.
Experimental Section
Chemicals
All reagents employed were of analytical grade. Sodium tetraborate, lithium carbonate, (+)-FLEC (18 mM in acetonitrile), 9-fluorenylmethoxycarbonyl chloride (FMOC-Cl), ammonium hydroxide (28%, w/v), acetic acid, and sodium hydroxide were purchased from Sigma-Aldrich (St. Louis, MO). Potassium carbonate was obtained from Acros Organics (New Jersey). Acetonitrile (ACN) was supplied by Fluka (Steinheim, Germany). Water was deionized and purified with a Milli-Q purification system (Millipore, Belford, NJ).
DL- and L-forms of tyrosine (Tyr), alanine (Ala), valine (Val), phenylalanine (Phe), tryptophan (Trp), histidine (His), arginine (Arg), lysine (Lys), isoleucine (Ile), methionine (Met), leucine (Leu), asparagine (Asn), serine (Ser), glutamic acid (Glu), proline (Pro), threonine (Thr), 2-Aminoadipic acid (Aad), and selenomethionine (SeMet), and D-glutamine (D-Gln), L-glutamine (L-Gln), D-pipecolic acid (D-Pipe), D-ornithine (D-Orn), and glycine (Gly) were purchased from Sigma-Aldrich (Madrid, Spain), whereas D-citrulline (D-Cit), L-citrulline (L-Cit), L-pipecolic acid (L-Pipe), and L-ornithine (L-Orn) were obtained from Fluka (Buchs, Switzerland).
Trapped Ion Mobility Spectrometry
TIMS-TOFMS analysis was performed on a TIMS-TOF instrument from Bruker Daltonik (Bremen, Germany) equipped with an electrospray ionization (ESI) source. A daily calibration was performed using the Agilent ESI low concentration Tune mix. Two types of calibrations were applied. In the first, a wide voltage scan allowed detection of all ions from the calibrant solution. Herewith a calibration from m/z 322.0212 to m/z 2121.9331 covering a 1/K0 range (K0 is the reduced mobility) from 0.7319 to 1.7286, allowing correction of small day-to-day variations in measured mobilities. For higher resolution IM analysis, calibration was performed over a smaller mobility ranges (1/K0, 0.7319–0.9848) using two or three m/z values (322.0481, 622.0290, 922.0098). In the latter case, the ramp voltage range was kept constant but shifted to lower or higher voltages to detect all the required calibrant ions. Samples were directly injected using a syringe pump from Cole-Parmer (Vernon Hill, IL) with a glass syringe of 1 mL from Hamilton (Bonaduz, Switzerland) at a flow rate of 180 μL/h. The TIMS was operated in positive-ion mode with an electrospray voltage of 4.5 kV. The mass range was set at m/z 100–3000. The ionization source conditions were end plate offset, 500 V; nebulizer pressure, 0.5 bar; drying gas, 4 L/min at 200 °C. The TOFMS settings were rf funnel 1, 150 Vpp; rf funnel 2, 300 Vpp; isCID energy, 0 eV; multipole rf, 150 Vpp; quadrupole ion energy, 5 eV at low mass of 50 m/z; collision energy, 8 eV; collision rf, 600 Vpp; transfer time, 55 μs; prepulse storage, 1 μs.
TIMS was operated using two different modes. First, samples were analyzed using a wide voltage scan (109–160 V) to provide a general overview of the elution voltages. This mode was operated using general conditions of ion mobility such as ramp start V, 109 V; ramp end V, 160 V; ramp time, 255 ms; accumulation time, 50 ms. Subsequently, TIMS parameters (accumulation, ramp voltages, and ramp time) were adjusted to obtain maximum resolving power for a particular set of DL-AAs. The duty cycle was 80%. Optimum conditions are reported in the Results and Discussion.
Sample Preparation
Stock solutions of each AA were prepared dissolving the appropriate amount of AA in a buffer solution. Sodium tetraborate buffer solution (26 mM in Milli-Q water, pH 9.2) was employed as buffer solution to dissolve the AAs before the derivatization step. Lithium carbonate and potassium carbonate solutions (26 mM each) were prepared in Milli-Q water and adjusted with 20% acetic acid to pH 9.2. These AA solutions were stored at 4 °C until the derivatization step with FLEC.
Derivatization of AAs was performed according to Prior et al.23 The 18 mM (+)-FLEC in ACN was freshly prepared each day. Briefly, 50 μL of a 90 μg/mL AA solution in 26 mM sodium tetraborate (pH 9.2) were mixed with 50 μL of 12 mM (+)-FLEC dissolved in ACN. The solution was kept at room temperature for 10 min to complete reaction, and the resulting solution was diluted 1:1 in Mili-Q water–ACN (50:50, v/v) before infusion into the ESI-TIMS-TOFMS system.
Data Analysis
Resolution (R) was determined from the measured mobilograms (1/K0 vs intensity) according R = K0/ΔK0 where K0 is the reduced mobility of the respective analyzed diastereomer and ΔK0 its peak width at half height (fwhm). The peak mobilities (1/K0), the fwhms and ion collision cross sections (CCS) were obtained using the Compass Data Analysis 5.0 software from Bruker Daltonik.
Results and Discussion
Chiral Resolution of FLEC-AAs by TIMS
Ions of enantiomers of an AA have identical charge, m/z, and CCS, making their direct separation by IMS not feasible. Up until now, separation of AA enantiomers by IMS has been achieved by using gas-phase ion complexation with volatile chiral agents or by forming metal-ion noncovalent complexes with reference compounds.12,22,24,25 Since TIMS has demonstrated to be a powerful tool for the fast separation of isobaric or isomeric compounds,19 we have explored the potential of TIMS to resolve chiral AAs by first forming diastereomers in solution. For that purpose, we selected the chiral agent (+)-FLEC which reacts covalently and fast with the amino group of an AA, resulting in two diastereomers per pair of enantiomers (Figure 1A). Initial experiments were performed using DL-SeMet and DL-Orn as model compounds. Orn contains two amine groups leading to two FLEC molecules per AA. No singly derivatized FLEC-Orn was observed when analyzed by MS. ESI of the FLEC-AAs resulted in a mixture of protonated and sodiated ions. The sodium ions largely originate from the buffer employed for derivatization. When analyzed by TIMS, for the protonated ions ([FLEC2-Orn + H]+, [FLEC-SeMet + H]+) and ions containing one Na ion per molecule ([FLEC2-Orn + Na]+, [FLEC-SeMet + Na]+), a single peak was observed for each AA (Figure S1). On the other hand, the extracted-ion traces of the disodiated ions [FLEC2-Orn + 2Na – H]+ and [FLEC-SeMet + 2Na – H]+ both revealed two baseline separated peaks, suggesting separation of the respective diastereomers (Figure 2A,C, respectively). As negative control, we also carried out the AA derivatization by FMOC-Cl. FLEC and FMOC have very similar structure, only differing a methyl group on the acyl moiety, rendering FMOC to be nonchiral (cf. parts A and B of Figure 1). Consequently, FMOC derivatization of AAs results in two pair of enantiomers and not diastereomers. When monitoring [FMOC2-Orn + 2Na – H]+ and [FMOC-SeMet + 2Na – H]+, no separation is observed (Figure 2), indicating that TIMS indeed provides the FLEC-AA diastereomer separation.
Figure 1.

Scheme of the derivatization reaction of D- and L-AAs with (A) FLEC and (B) FMOC.
Figure 2.

TIMS-TOFMS of FLEC-derivatized Orn and SeMet showing mobilograms corresponding to (A) [FLEC2-Orn + 2Na – H]+, (B) [FMOC2-Orn + 2Na – H]+, (C) [FLEC-SeMet + 2Na – H]+, and (D) [FMOC-SeMet + 2Na – H]+.
Theoretically, multiple analyte peaks in (T)IMS may result from different positions of the adduct charge (i.e., occurrence of protomers and/or sodium-adduct isomers).19,26 In order to exclude this possibility and to confirm that separation of diastereomers was achieved, L-Orn and L-SeMet, and D/L mixtures of these AAs (ratio D/L, 1:1 and 1:3) were analyzed. For both FLEC-AAs, only one peak was observed for the L enantiomer (Figure 3A,B), whereas two peaks were observed for the 1:1 and 1:3 D/L mixtures, with the former showing similar peak heights and the latter showing clearly higher peak intensities for the L-form. The analysis of the nonchiral AA Gly was also carried out after derivatization with FLEC. This yielded one single peak in the mobilogram (Figure S2), again supporting the conclusion that the separation observed in TIMS indeed is caused by structural difference of the AA diastereomers and not by difference in adduct-ion position in the molecule or another reason.
Figure 3.

TIMS-TOFMS of FLEC-derivatized Orn (A) and SeMet (B) showing mobiligrams obtained for L-AA, 1:1 D/L-AA, and 1:3 D/L-AA.
Effect of the Alkali Cation on the Enantioseparation
Our initial results indicated that sodium-adduct formation was essential for the separation of the AA enantiomers by TIMS. This observation is in line with previous research on separation of isomeric molecules by IMS, where the formation of alkali-cation adducts demonstrated to be a prerequisite for their efficient separation.14,19,21 We also studied the effect of potassium and lithium cations on the chiral separation of AAs. FLEC derivatives of SeMet and Orn were prepared in solutions containing potassium or lithium ions and then analyzed by TIMS. Figure 4 shows the mobilograms obtained for the FLEC-AAs in the presence of Li+, Na+, or K+. No separation of FLEC-AA diastereomers was obtained when Li+ was used, whereas with K+ partial separation was observed for FLEC-Orn. Table S1 (Supporting Information) lists the CCS values obtained for the different FLEC-AA adducts. For both AAs the CCS increase with the size of the metal ion. Looking at the CCS of the individual diastereomers it could be discerned that, when diastereomer separation is achieved, the first migrating diastereomer exhibits a deviation from this trend in comparison with the second diastereomer (e.g., CCS 240.3 for [FLEC2-Orn + 2Li – H]+, and 237.7 and 241.5 for the first and second diastereomer of [FLEC2-Orn + 2Na – H]+, respectively) (Figure S3). This most probably is the result of a conformational change of the first migrating diastereomer to a more compact form in the presence of Na+ in comparison to the second diastereomer.
Figure 4.

TIMS-TOFMS of FLEC-derivatized Orn (A) and SeMet (B) showing mobilograms obtained in the presence of lithium, potassium, and sodium ions.
As the formation of sodium adducts showed to be crucial for the separation, the amount of sodium ions in the derivatization buffer was increased (from 26 mM to 50 mM). No change in diastereomer separation and intensity was observed, and therefore the concentration of 26 mM was maintained for further experiments (Figure S4A). When the AA concentration was varied (0.1–0.7 mM), the relative intensity of sodium adduct ions formed remained constant. Furthermore, the ratio between ions containing one and two sodium ions was constant between experiments (Figure S4B).
Evaluation of TIMS Conditions and Applicability to Multiple AAs
In order to further evaluate the potential of TIMS-TOFMS for separating FLEC-AA diastereomers, 21 AAs (17 proteinogenic and 4 nonproteinogenic AAs) were analyzed. AA solutions prepared in an enantiomeric ratio (D/L) of 1:1 and 1:3 were derivatized with FLEC and analyzed by direct infusion TIMS-TOFMS. Ions exiting the glass capillary are transferred into the TIMS cell by a nitrogen gas flow, whereas an opposite electrical field gradient is applied to trap and separate the ions. This takes place first in a TIMS analyzer. After trapping, the ions are moved to a similar second TIMS analyzer section where after trapping the electric field is decreased gradually by lowering the exit potential over time so that the ions are sequentially liberated into the time-of-flight mass spectrometer. The exit voltage range and the speed at which this voltage is changed (ramp time) define the slope of the electric field gradient which affects the resolving power of TIMS.
Initially, TIMS analysis were carried out using a wide voltage scan (109–160 V) providing the ion mobilities for all analyzed AAs. AAs carrying one FLEC per molecule were eluted at lower voltages than the AAs which contained two FLEC per molecule, such as Orn and Lys. In order to achieve optimum separation for each pair of diastereomers, the voltage range was narrowed down to a specific range and the voltage ramp time was adjusted (Table 1). The TIMS sensitivity depends of the number of ions trapped in the first TIMS analyzer, which is determined by the time that ions are accumulated. Too long accumulation times can result in saturation of the trap and space-charge effects, and therefore the accumulation was studied between 10 and 100 ms. The FLEC-AA signal intensities increased with accumulation time until 50 ms, except for Ser, which showed the highest intensity at 25 ms. Further increase of the accumulation up to 100 ms did not result in higher signal intensities for most FLEC-AAs (except for His), suggesting saturation of the trap. Therefore, the accumulation time was set to 50 ms, but for His and Ser, 100 and 25 ms were selected, respectively. Table 1 summarizes the ramp voltage, ramp time, and accumulation time employed for each FLEC-AA as well as the mobility and mobility differences between the diastereomers obtained under these conditions. From the 21 chiral AAs studied, separation of the FLEC diastereomers of 17 AAs was achieved. For Glu, Pro, Thr, and Ala, no enantioresolution was obtained. Diastereomer separation was obtained in general for disodiated FLEC-AAs ([M + 2Na – H]+); however, for Ile, Leu, and Val, the diastereomer separation was observed for the singly sodiated species ([M + Na – H]+). Next to AAs containing two amino groups, Tyr also appeared to be derivatized with two FLEC molecules, presumably due to a reaction of FLEC with the Tyr hydroxyl group under the conditions employed in this study. Figure 5 shows the mobilograms obtained for the resolved AAs and Table S1 their calculated CCS. For a large part, baseline separation of the FLEC-AA diastereomers was achieved. In the case of Arg, Lys, His, Met, and Leu, the diastereomers were partially resolved. The resulting TIMS resolution (K0/ΔK0) for the FLEC-AA diastereomers on average was 115, requiring a mobility difference (ΔK0) of about 0.009 cm2/(V s) to achieve 50%-valley separation. Experimental ΔK0 determined for the analyzed diastereomers ranged from 0. 009 for Lys (only partially resolved) to 0.061 for Asn. The particular migration order of the D and L enantiomers seems to be dependent on the structure of the AA (Table 1). Interestingly, this observation is opposite to the behavior of diastereomeric FLEC-AAs in capillary electrophoresis, where D-AAs commonly migrated first.23
Table 1. Separation Conditions, Mobilities, Ion Mobility Resolution, and First Migrating Enantiomer for 21 FLEC-AAs Analyzed by TIMS-TOFMS.
| AA | observed m/z | observed ion | voltage range (V) | ramp time (ms) | accu. time (ms) | K0 diast1 (cm2/(V s)) | K0 diast2 (cm2/(V s)) | K01 – K02 (cm2/(V s)) | ΔK0 needed for 50% valley separation | R diast1 | R diast2 | first migrating enantiomer |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ile | 390.0901 | [FLEC-Ile + Na – H ]+ | 115–125 | 510 | 50 | 1.109 | 1.092 | 0.017 | 0.008 | 128.9 | 156.0 | L |
| Leu | 390.1680 | [FLEC-Leu + Na – H]+ | 109–160 | 255 | 50 | 1.099 | 1.081 | 0.018 | 0.011 | 91.0 | 108.1 | L |
| Val | 376.0901 | [FLEC-Val + Na – H ]+ | 109–160 | 255 | 50 | 1.139 | 1.120 | 0.019 | 0.011 | 97.6 | 112.0 | –a |
| Asn | 413.1086 | [FLEC-Asn + 2Na – H]+ | 109–130 | 510 | 50 | 1.126 | 1.065 | 0.061 | 0.010 | 111.0 | 118.3 | L |
| Glu | 428.1080 | [FLEC-Glu + 2Na – H]+ | 109–160 | 255 | 50 | 1.064 | 0.010 | 94.0 | –a | |||
| Orn | 649.2285 | [FLEC2-Orn + 2Na – H]+ | 146–160 | 510 | 50 | 0.861 | 0.849 | 0.012 | 0.008 | 116.1 | 94.3 | D |
| SeMet | 478.0505 | [FLEC-SeMet + 2Na – H]+ | 115–130 | 510 | 50 | 1.056 | 1.037 | 0.019 | 0.008 | 118.4 | 129.7 | D |
| Aad | 442.1237 | [FLEC-Aad + 2Na – H]+ | 115–135 | 510 | 50 | 1.057 | 1.037 | 0.020 | 0.007 | 135.1 | 148.2 | D |
| Pipe | 410.1338 | [FLEC-Pipe + 2Na – H]+ | 115–130 | 510 | 50 | 1.089 | 1.064 | 0.025 | 0.009 | 114.7 | 118.2 | D |
| Gln | 427.1244 | [FLEC-Gln + 2Na – H]+ | 105–130 | 510 | 50 | 1.068 | 1.042 | 0.027 | 0.010 | 104.0 | 115.7 | L |
| Pro | 396.1174 | [FLEC-Pro + 2Na – H]+ | 109–135 | 510 | 50 | 1.092 | 0.010 | 91.6 | –a | |||
| Thr | 400.1113 | [FLEC-Thr + 2Na – H]+ | 109–160 | 255 | 50 | 1.109 | 0.09 | 100.2 | –a | |||
| Arg | 455.1665 | [FLEC-Arg + 2Na – H]+ | 120–130 | 510 | 50 | 1.032 | 1.017 | 0.015 | 0.009 | 107.7 | 127.2 | D |
| Lys | 663.2454 | [FLEC2-Lys + 2Na – H]+ | 145–155 | 510 | 50 | 0.861 | 0.852 | 0.009 | 0.009 | 116.2 | 85.2 | D |
| Tyr | 698.2139 | [FLEC2-Tyr + 2Na – H]+ | 115–170 | 510 | 50 | 0.818 | 0.803 | 0.016 | 0.009 | 101.8 | 80.3 | L |
| Ala | 370.1008 | [FLEC-Ala + 2Na – H ]+ | 105–160 | 255 | 50 | 1.153 | 0.009 | 96.3 | –a | |||
| Met | 430.1047 | [FLEC-Met + 2Na – H ]+ | 110–125 | 510 | 50 | 1.089 | 1.075 | 0.014 | 0.008 | 114.8 | 153.6 | D |
| Phe | 446.1330 | [FLEC-Phe + 2Na – H ]+ | 115–130 | 510 | 50 | 1.083 | 1.049 | 0.034 | 0.009 | 115.4 | 131.2 | L |
| Ser | 386.0989 | [FLEC-Ser + 2Na – H]+ | 105–120 | 510 | 25 | 1.145 | 1.120 | 0.026 | 0.008 | 124.7 | 160.0 | L |
| Trp | 485.1446 | [FLEC-Trp + 2Na – H]+ | 109–135 | 510 | 50 | 1.024 | 0.995 | 0.029 | 0.010 | 97.7 | 99.5 | L |
| His | 672.2053 | [FLEC2-His + 2Na – H]+ | 140–155 | 510 | 100 | 0.833 | 0.823 | 0.010 | 0.008 | 120.0 | 82.3 | D |
Migration order was not addressed as pure enantiomers were not available. accu. time, accumulation time; diast, diastereomer.
Figure 5.

TIMS-TOFMS of 17 FLEC-AAs using conditions optimized for each AA.
Chiral Resolution of FLEC-AAs in Mixtures
The developed TIMS-TOF method allowed separation of FLEC-AA diastereomers in less than 1 min. As all AAs are derivatized under the same conditions, we addressed the possibility to resolve multiple AAs in one TIMS-TOFMS run while maintaining chiral selectivity. Considering that the FLEC-AAs require specific voltage ramps to be chirally resolved, no single method was able to resolve all AAs. Therefore, three different voltage ramps were applied in order to achieve separation of most of the FLEC-AAs. A mixture of Orn, Lys, His, and Tyr was analyzed using a voltage ramp from 135 to 170 V, a mixture of Asn, Ser, Pipe, and Gln using 100–130 V, and a mixture of Met, Phe, SeMet, and Arg using 109–135 V. For all mixtures, a ramp time of 510 ms was used. The mobilogram obtained for the analysis of the mixture of Asn, Ser, Pipe, and Gln (Figure 6) shows diastereomer separation for all four AAs. Some diastereomers comigrated, but still the individual AAs could be discerned by their specific m/z value. Similar results were obtained for the other mixtures of AAs (Figure S5A,B). This opens new possibilities for analysis of AAs in mixtures, which is not feasible with ion mobility approaches which require a pure enantiomer of a different amino acid as chiral reference compound.22,14,21
Figure 6.

Heat map and extracted-ion mobilograms obtained during TIMS-TOFMS of a mixture of DL-Asn (m/z, 413.1086), DL-Ser (m/z, 386.0989), DL-Pipe (m/z, 410.1338) and DL-Gln (m/z, 427.1244) using a voltage ramp of 100–130 V in 510 ms.
The TIMS-TOF method allowed detection of FLEC-AAs down to the low-nanomolar level, demonstrating similar sensitivity to previously described analysis of FLEC-AAs by ESI-MS.23,27 TIMS-TOF showed able to detect an DL enantiomeric ratio down to 2.5% D indicating the potential of the method for the determination of the enantiomeric excess in real samples (Figure S6). The method showed good reproducibility with RSDs between 0.09% and 0.8% for diastereomer mobilities, between 1.3% and 9.6% for diastereomer peak areas, and between 0.1% and 0.9% for the calculated CCS values.
Conclusions
This work presents a new and fast method for the separation of DL-AA enantiomers by TIMS after derivatization with the chiral reagent (+)-FLEC. By forming diastereomers before analysis, doping of the IMS drift gas with a chiral volatile complexing agent is not needed. When compared to earlier reported approaches, the proposed method simplifies and uniforms diastereomer formation, avoiding the use of specific reference compounds, and extending the applicability to a large number of AAs. Our study indicates that adduct formation with alkali ions is essential for achieving the separation of the diastereomeric FLEC-AAs. The new method has shown to be applicable to a variety of AAs, and chiral separation was achieved for 17 from the 21 AAs studied. The analyte ions are separated on the millisecond time scale, and TIMS analysis times of lower than 1 min are sufficient to attain adequate results. We also have demonstrated that the simultaneous separation of diastereomers in a mixture by TIMS is very well possible. Taking the derivatization time (only 10 min) into account, mixtures of AA enantiomers can be resolved in a total analysis time of less than 15 min. Limits of detection are in the nanomolar range, and determination of enantiomeric ratios down to 2.5% are feasible. Overall, the presented methodology provides a promising general approach for separating chiral compounds with potential applications in different fields, such as environmental or pharmaceutical analysis. Applications to more complex matrixes, such as food or biological fluids, will require further evaluation of the effect of varying sodium ion concentrations in the samples.
Acknowledgments
R.P.-M., M.C.-P., and M.L.M thank the Spanish Ministry of Economy and Competitiveness (Grant CTQ2016-76368-P) and the Comunidad of Madrid (Spain) and European Funding from the FEDER Program (Grants S2013/ABI-3028, AVANSECAL-CM) for financial support. R.P.-M. thanks the University of Alcalá for her predoctoral contract and the mobility grant to stay at Vrije Universiteit. M.C.-P. thanks the Spanish Ministry of Economy and Competitiveness for their “Ramón y Cajal” Contract (Grant RYC-2013-12688).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b03661.
Ions obtained during TIMS-TOFMS of FLEC-Orn and FLEC-SeMet (Figure S1); mobilogram of the nonchiral AA Gly after FLEC derivatization (Figure S2); CCSs obtained for Orn and SeMet in the presence of different cations (Table S1 and Figure S3); effect of the variation of the concentration of sodium tetraborate during derivatization (Figure S4); CCSs measured for all FLEC-AAs studied (Table S2); mobilograms obtained for two different mixtures of FLEC-AAs (Figure S5); and mobilogram showing a mixture of enantiomers containing 2.5% of D-AA (Figure S6) (PDF)
Author Present Address
§ E.D.-V.: Center for Proteomics and Metabolomics, Leiden University Medical Center, Postbus 9600, 2300 RC, Leiden, The Netherlands.
The authors declare no competing financial interest.
Supplementary Material
References
- Rocco A.; Aturki Z.; Fanali S. Chiral separations in food analysis. TrAC, Trends Anal. Chem. 2013, 52, 206–225. 10.1016/j.trac.2013.05.022. [DOI] [Google Scholar]
- Wu G. Amino acids: metabolism, functions, and nutrition. Amino Acids 2009, 37, 1–17. 10.1007/s00726-009-0269-0. [DOI] [PubMed] [Google Scholar]
- Pérez-Míguez R.; Castro-Puyana M.; Marina M. L. In Advances in Food Analysis Research; Haynes A., Ed.; Nova Science Publishers: New York, 2015; pp 89–120. [Google Scholar]
- Bell E. A. Non protein amino acids of plants: significance in medicine, nutrition, and agriculture. J. Agric. Food Chem. 2003, 51, 2854–2865. 10.1021/jf020880w. [DOI] [PubMed] [Google Scholar]
- Hare P. E.; Gil-Av E. Separation of D and L amino acids by liquid chromatography: use of chiral eluants. Science 1979, 204, 1226–1228. 10.1126/science.36662. [DOI] [PubMed] [Google Scholar]
- Frank H.; Nicholson G. J.; Bayer E. Rapid gas chromatographic separation of amino acid enantiomers with a novel chiral stationary phase. J. Chromatogr. Sci. 1977, 15, 174–176. 10.1093/chromsci/15.5.174. [DOI] [PubMed] [Google Scholar]
- Wan H.; Blomberg L. G. Chiral separation of amino acids and peptides by capillary electrophoresis. J. Chromatogr. A 2000, 875, 43–88. 10.1016/S0021-9673(99)01209-1. [DOI] [PubMed] [Google Scholar]
- Ilisz I.; Berkecz R.; Peter A. HPLC separation of amino acid enantiomers and small peptides on macrocyclic antibiotic-based chiral stationary phases: A review. J. Sep. Sci. 2006, 29, 1305–1321. 10.1002/jssc.200600046. [DOI] [PubMed] [Google Scholar]
- Schurig V. Gas chromatographic enantioseparation of derivatized amino acids on chiral stationary phases-Past and present. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2011, 879, 3122–3140. 10.1016/j.jchromb.2011.04.005. [DOI] [PubMed] [Google Scholar]
- Wan H.; Blomberg L. G. Chiral separation of amino acids and peptides by capillary electrophoresis. J. Chromatogr. A 2000, 875, 43–88. 10.1016/S0021-9673(99)01209-1. [DOI] [PubMed] [Google Scholar]
- Vera C. M.; Shock D.; Dennis G. R.; Farrell W.; Shalliker R. A. Comparing the selectivity and chiral separation of D- and L-fluorenylmethyloxycarbonyl chloride protected amino acids in analytical high performance liquid chromatography and supercritical fluid chromatography; evaluating throughput, economic and environmental impact. J. Chromatogr. A 2017, 1493, 10–18. 10.1016/j.chroma.2017.02.017. [DOI] [PubMed] [Google Scholar]
- Dwivedi P.; Wu C.; Matz L. M.; Clowers B. H.; Siems W. F.; Hill H. H. Gas-phase chiral separations by ion mobility spectrometry. Anal. Chem. 2006, 78, 8200–8206. 10.1021/ac0608772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X.; Yao Z. P. Chiral recognition and determination of enantiomeric excess by mass spectrometry: A review. Anal. Chim. Acta 2017, 968, 1–20. 10.1016/j.aca.2017.03.021. [DOI] [PubMed] [Google Scholar]
- Domalain V.; Hubert-Roux M.; Tognetti V.; Joubert L.; Lange C. M.; Rouden J.; Afonso C. Enantiomeric differentiation of aromatic amino acids using traveling wave ion mobility-mass spectrometry. Chem. Sci. 2014, 5, 3234–3239. 10.1039/C4SC00443D. [DOI] [Google Scholar]
- Fernandez-Lima F.; Kaplan D. A.; Park M. A. Integration of trapped ion mobility spectrometry with mass spectrometry. Rev. Sci. Instrum. 2011, 82, 126106. 10.1063/1.3665933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Lima F.; Kaplan D. A.; Suetering J.; Park M. Gas-phase separation using a trapped ion mobility spectrometer. Int. J. Ion Mobility Spectrom. 2011, 14, 93–98. 10.1007/s12127-011-0067-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez D. R.; DeBord J. D.; Ridgeway M. E.; Kaplan D. A.; Park M. A.; Fernandez-Lima F. Ion dynamics in a trapped ion mobility spectrometer. Analyst 2014, 139, 1913–1921. 10.1039/C3AN02174B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridgeway M. E.; Lubeck M.; Jordens J.; Mann M.; Park M. A. Trapped ion mobility spectrometry: A short review. Int. J. Mass Spectrom. 2018, 425, 22–35. 10.1016/j.ijms.2018.01.006. [DOI] [Google Scholar]
- Zietek B. M.; Mengerink Y.; Jordens J.; Somsen G. W.; Kool J.; Honing M. Adduct-ion formation in trapped ion mobility spectrometry as a potential tool for studying molecular structures and conformations. Int. J. Ion Mobility Spectrom. 2018, 21, 19–32. 10.1007/s12127-017-0227-6. [DOI] [Google Scholar]
- Tian H.; Zheng N.; Li S.; Zhang Y.; Zhao S.; Wen F.; Wang J. Characterization of chiral amino acids from different milk origins using ultra-performance liquid chromatography coupled to ion-mobility mass spectrometry. Sci. Rep. 2017, 7, 46289. 10.1038/srep46289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X.; Yao Z. P. Chiral differentiation of amino acids through binuclear copper bound tetramers by ion mobility mass spectrometry. Anal. Chim. Acta 2017, 981, 62–70. 10.1016/j.aca.2017.05.026. [DOI] [PubMed] [Google Scholar]
- Mie A.; Jörntén-Karlsson M.; Axelsson B. O.; Ray A.; Reimann C. T. Enantiomer separation of amino acids by complexation with chiral reference compounds and high-field asymmetric waveform ion mobility spectrometry: preliminary results and possible limitations. Anal. Chem. 2007, 79, 2850–2858. 10.1021/ac0618627. [DOI] [PubMed] [Google Scholar]
- Prior A.; Moldovan R. C.; Crommen J.; Servais A. C.; Fillet M.; De Jong G. J.; Somsen G. W. Enantioselective capillary electrophoresis-mass spectrometry of amino acids in cerebrospinal fluid using a chiral derivatizing agent and volatile surfactant. Anal. Chim. Acta 2016, 940, 150–158. 10.1016/j.aca.2016.08.040. [DOI] [PubMed] [Google Scholar]
- Troć A.; Zimnicka M.; Danikiewicz W. Separation of catechin epimers by complexation using ion mobility mass spectrometry. J. Mass Spectrom. 2015, 50, 542–548. 10.1002/jms.3560. [DOI] [PubMed] [Google Scholar]
- Pang X.; Jia C.; Chen Z.; Li L. Structural characterization of monomers and oligomers of D-amino acid-containing peptides using T-Wave Ion Mobility Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 110–118. 10.1007/s13361-016-1523-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnke S.; Seo J.; Boschmans J.; Sobott F.; Scrivens J. H.; Bleiholder C.; Bowers M. T.; Gewinner S.; Schöllkopf W.; Pagel K.; Von Helden G. Protomers of benzocaine: solvent and permittivity dependence. J. Am. Chem. Soc. 2015, 137, 4236–4242. 10.1021/jacs.5b01338. [DOI] [PubMed] [Google Scholar]
- Moldovan R. C.; Bodoki E.; Servais A. C.; Crommen J.; Oprean R.; Fillet M. A micellar electrokinetic chromatography-mass spectrometry approach using in-capillary diastereomeric derivatization for fully automatized chiral analysis of amino acids. J. Chromatogr. A 2016, 1467, 400–408. 10.1016/j.chroma.2016.08.035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.