

Abstract
Background
8‐Aminoguanosine and 8‐aminoguanine are K+‐sparing natriuretics that increase glucose excretion. Most effects of 8‐aminoguanosine are due to its metabolism to 8‐aminoguanine. However, the mechanism by which 8‐aminoguanine affects renal function is unknown and is the focus of this investigation.
Methods and Results
Because 8‐aminoguanine has structural similarities with inhibitors of the epithelial sodium channel (ENaC), Na+/H+ exchangers, and adenosine A1 receptors, we examined the effects of 8‐aminoguanine on ENaC activity in mouse collecting duct cells, on intracellular pH of human proximal tubular epithelial cells, on responses to a selective A1‐receptor agonist in vivo, and on renal excretory function in A1‐receptor knockout rats. These experiments showed that 8‐aminoguanine did not block ENaC, Na+/H+ exchangers, or A1 receptors. Because Rac1 enhances activity of mineralocorticoid receptors and some guanosine analogues inhibit Rac1, we examined the effects of 8‐aminoguanine on Rac1 activity in mouse collecting duct cells. Rac1 activity was significantly inhibited by 8‐aminoguanine. Because in vitro 8‐aminoguanine is a purine nucleoside phosphorylase (PNPase) inhibitor, we examined the effects of a natriuretic dose of 8‐aminoguanine on urinary excretion of PNPase substrates and products. 8‐Aminoguanine increased and decreased, respectively, urinary excretion of PNPase substrates and products. Next we compared in rats the renal effects of intravenous doses of 9‐deazaguanine (PNPase inhibitor) versus 8‐aminoguanine. 8‐Aminoguanine and 9‐deazaguanine induced similar increases in urinary Na+ and glucose excretion, yet only 8‐aminoguanine reduced K+ excretion. Nsc23766 (Rac1 inhibitor) mimicked the effects of 8‐aminoguanine on K+ excretion.
Conclusions
8‐Aminoguanine increases Na+ and glucose excretion by blocking PNPase and decreases K+ excretion by inhibiting Rac1.
Keywords: 8‐aminoguanine, 8‐aminoguanosine, diuretic, natriuretic, purine nucleoside phosphorylase, Rac1
Subject Categories: Nephrology and Kidney, Basic Science Research, Cell Signalling/Signal Transduction, Hypertension
Clinical Perspective
What Is New?
The present study identifies inhibition of purine nucleoside phosphorylase as the mechanism by which 8‐aminoguanine induces diuresis, natriuresis, and glucosuria.
The present study also identifies inhibition of Rac1 as the mechanism by which 8‐aminoguanine decreases K+ excretion.
8‐Aminoguanosine, a prodrug that is converted to 8‐aminoguanine in vivo, is an even more potent direct Rac1 inhibitor compared with 8‐aminoguanine.
What Are the Clinical Implications?
8‐Aminoguanine and its prodrug 8‐aminoguanosine possess a unique combination of pharmacological activities and therefore represent a novel class of potentially useful and naturally occurring cardiovascular drugs.
8‐Aminoguanine and its prodrug 8‐aminoguanosine could be benefical in cardiovascular and renal diseases because they are diuretics, natriuretics, and glucosuric agents that increase levels of protective purines (eg, inosine and guanosine), decrease levels of harmful purines (eg, hypoxanthine), while blocking the effects of Rac1 (mediates many adverse effects on the cardiovascular system and kidneys).
Guanine moieties are susceptible to modification in the 8 position by reactive nitrogen species such as peroxynitrite or reactive oxygen species such as superoxide anion. These reactions ultimately yield a set of endogenous 8‐substituted guanosine and guanine compounds1, 2, 3, 4 that include 8‐nitroguanosine,5 8‐hydroxyguanosine,6 8‐nitroguanine,1 8‐hydroxyguanine,7 8‐hydroxy‐2′‐deoxyguanosine,8 8‐aminoguanosine,9 and 8‐aminoguanine.10
Until recently, there was no information about the effects of these naturally occurring 8‐substituted guanosine and guanine compounds on the cardiovascular system and kidneys. To address this knowledge gap, we investigated the effects of these compounds in rat kidneys in vivo and discovered that 8‐aminoguanosine and 8‐aminoguanine induce diuresis, natriuresis, and glucosuria, yet reduce potassium excretion.11 In addition, we observed that chronic oral treatment of rats with 8‐aminoguanosine and 8‐aminoguanine attenuate the development of deoxycorticosterone/salt‐induced hypertension.11
Because both 8‐aminoguanosine and 8‐aminoguanine have similar effects on renal excretory function and 8‐aminoguanosine is metabolized to 8‐aminoguanine by purine nucleoside phosphorylase (PNPase),10 we hypothesized that the renal actions of 8‐aminoguanosine are not direct but instead are mediated by 8‐aminoguanine. That is to say, our concept was that 8‐aminoguanosine is an 8‐aminoguanine “pro‐drug.” Indeed, our most recent results confirm that the diuretic, natriuretic, and glucosuric effects of 8‐aminoguanosine are attributable to its conversion to 8‐aminoguanine.12 Surprisingly, however, 8‐aminoguanosine, like 8‐aminoguanine, has direct effects to reduce potassium excretion.12
Having established that 8‐aminoguanine has pharmacological activity on the kidneys to increase sodium and glucose excretion while reducing potassium excretion, our next objective was to explore the mechanism of action by which 8‐aminoguanine induces these effects. Inasmuch as inhibition of the epithelial sodium channel (ENaC),13 Na+/H+ exchangers,14, 15, 16 and adenosine A1 receptors17 can increase sodium excretion and 8‐aminoguanine has structural similarities to known inhibitors of these proteins, we assessed whether the renal effects of 8‐aminoguanine could be explained in part by inhibition of ENaC, Na+/H+ exchangers, or adenosine A1 receptors. Because Rac1 enhances activity of mineralocorticoid receptors18, 19 and some guanosine analogues inhibit Rac1,20 we also examined the effects of 8‐aminoguanine on Rac1 activity. Finally, because in vitro 8‐aminoguanine is a PNPase inhibitor,21 we investigated the possibility that inhibition of PNPase mediates some of the effects of 8‐aminoguanine on renal excretory function.
Methods
For raw data and for additional information on analytic methods or study materials, please contact the corresponding author.
Animals
The University of Pittsburgh Institutional Animal Care and Use Committee approved all procedures, and this investigation conforms to National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Assessment of Role of A1‐Receptor Antagonism as a Possible Explanation for the Renal Excretory Effects of 8‐Aminoguanine In Vivo
First, we examined whether 8‐aminoguanine blocks A1 receptors in vivo. Adult male Sprague‐Dawley rats (Charles River Laboratories, Wilmington, MA) were anesthetized with Inactin (90 mg/kg IP), and body temperature was monitored with a rectal temperature probe and maintained at 37°C with a temperature‐controlled and heated surgical table and heat lamp. A polyethylene‐240 cannula was placed in the trachea to facilitate respiration, and a polyethylene‐50 cannula was placed in the jugular vein for administration of test agents and for infusion of 0.9% saline (50 μL/min) to maintain hemodynamic stability. Next, a polyethylene‐50 cannula was inserted into the carotid artery for measurement of mean arterial blood pressure (MABP) and heart rate (HR) using a pressure transducer (Micro‐Med, Inc; Louisville, KY) connected to a blood pressure analyzer (Micro‐Med, Inc). Also, a 1‐mm transit‐time flow probe was placed on the left renal artery for measurement of renal blood flow (RBF) using a Transonic T402 transit‐time flowmeter (Transonic Systems Inc; Ithaca, NY), and the left ureter was cannulated with polyethylene‐50 tubing for collection of urine. Hemodynamic variables were recorded using the PowerLab data acquisition system and LabChart software (ADInstruments, Colorado Springs, CO).
Following the surgical procedures, rats were administered an intravenous bolus of either 8‐aminoguanine (33.5 μmoles/kg) or vehicle and allowed to stabilize for 30 minutes. The dose of 8‐aminoguanine was selected on the basis of our previous findings that 33.5 μmoles/kg of 8‐aminoguanine induces diuresis and natriuresis in rats.11 MABP, HR, and RBF were recorded during four 15‐minute experimental periods. During the first 15‐minute period, only the vehicle (saline) for 2‐chloro‐N6‐cyclopentyladenosine (CCPA; Tocris, Minneapolis, MN) was infused. During the second, third, and fourth experiment periods, CCPA was infused at 0.06, 0.2, and 0.6 μg/kg per minute, respectively. CCPA is a highly selective A1‐receptor agonist.22
As an additional test of whether 8‐aminoguanine alters renal excretory function by blocking A1 receptors, we examined the effects of 8‐aminoguanine on renal excretory function in a novel strain of knockout rats, that is, the A1‐receptor knockout rat on a Dahl salt‐sensitive background. We recently developed this colony of rats and have described these animals in detail.23 Adult wild‐type and A1‐knockout rats were anesthetized with Inactin (90 mg/kg IP) and instrumented as described above. After a 1‐hour stabilization period, urine was collected for 30 minutes (period 1: 0–30 minutes into the protocol). Next, rats received an intravenous bolus of 8‐aminoguanine (33.5 μmoles/kg). Ten minutes after the test agents were administered, urine was collected for 30 minutes (period 2: 40–70 minutes into the protocol) and 15 minutes later urine was collected again for 30 minutes (period 3: 85–115 minutes into the protocol). Sodium and potassium in urine was measured by flame photometry (Model IL‐943; Instrumentations Laboratory Inc, Lexington, MA) and glucose in urine was measured by a glucose colorimetric assay kit (Cayman Chemical).
Effects of 8‐Aminoguanine on ENaC Activity
These experiments were conducted in immortalized mouse cortical collecting duct cells, which were originally a gift from Dr Alain Vandewalle (INSERM, Paris, France). Cells were cultured on permeable membrane supports and mounted in modified Costar Ussing chambers, which were continuously short‐circuited by a voltage clamp amplifier (Physiologic Instruments, San Diego, CA) as previously described by Butterworth and coworkers.24 The bathing solution was Ringer's with the following composition: 120 mmol/L of NaCl, 25 mmol/L of NaHCO3, 3.3 mmol/L of KH2PO4, 0.8 mmol/L of K2HPO4, 1.2 mmol/L of MgCl2, 1.2 mmol/L of CaCl2, and 10 mmol/L of glucose. Ussing chambers were gassed with 95% O2/5% CO2 at 37°C and maintained at pH 7.4. Short‐circuit currents (Isc) were measured before and after adding increasing concentrations of 8‐aminoguanine (5 minutes at each concentration) to the apical side of the permeable membrane support. At the end of each experiment, amiloride (10 μmol/L) was added to the apical cell surface and Isc was measured. Amiloride and 8‐aminoguanine were added to the apical, rather than basolateral, cell surface because ENaC is an apical sodium channel.25 The effects of 8‐aminoguanine on Isc were normalized to the amiloride‐sensitive Isc for each culture well.
Effects of 8‐Aminoguanine on Na+/H+ Exchange Activity
To determine whether 8‐aminoguanine inhibits Na+/H+ exchangers, we examined the ability of 8‐aminoguanine to cause intracellular acidification. Ethylisopropyl amiloride (EIPA), a well‐characterized inhibitor of Na+/H+ exchangers,26 served as a positive control. Human renal proximal tubule epithelial cells (Lonza, Walkersville, MD) were cultured in REBM (Lonza) supplemented with REGM™ SingleQuots™(Lonza) and 10% fetal bovine serum at 37°C with 5% CO2. One day before the experiment, 1.0×105 cells were plated in 0.5 mL of growth media per well in 24‐well plates. On the day of the experiment, the cells were loaded with 10 μmol/L SNARF‐5F (Molecular Probes, Eugene, OR) in serum‐free medium for 30 minutes at 37°C. The cells were washed briefly after loading and maintained in working buffer (133 mmol/L of NaCl, 4.5 mmol/L of KCl, 1.5 mmol/L of CaCl2, 6 mmol/L of glucose, and 10 mmol/L of HEPES, pH 7.3). The dual 642/572 nm fluorescence emission after excitation at 514 nm was recorded on Victor 2 Microplate Reader (Perkin Elmer; Waltham, MA). The cells were then treated with 8‐aminoguanine (100 μmol/L) or EIPA (25 μmol/L) for 10 minutes at 37°C. The 642/572 nm fluorescence emission was recorded again. To quantitatively convert changes in SNARF‐5F 642/572 ratio to changes in intracellular pH, in situ calibrations were performed to calibrate the SNARF‐5F dye in the cells. The 642/572 nm fluorescence emission ratios were recorded from SNARF‐5F‐loaded cells exposed to the H+/K+ exchanger nigericin (10 μg/mL) in solutions of high K+ (150 mmol/L of KCl, 1 mmol/L of MgCl2, and 20 mmol/L of HEPES) with 8 different extracellular pH values (6.33, 6.66, 7.00, 7.33, 7.66, 8.00, 8.33, and 8.66). The pH values in the cells before and after treatment were calculated using the equation in the SNARF‐5F manual.
Effects of 8‐Aminoguanine on Rac1 Activity
The Rac1 activity assay was based on the concept of measuring the ratio of GTP‐bound Rac1 to total Rac1 following treatment with a Rac1 activator in the presence and absence of 8‐aminoguanine and, for comparison, its prodrug 8‐aminoguanosine. In this regard, immortalized mouse cortical collecting duct cells were cultured in modified DM medium (DMEM:Ham's F12, 1:1 vol/vol; 60 nmol/L sodium selenite; 5 μg/mL transferrin; 2 mmol/L glutamine; 50 nmol/L dexamethasone; 1 nmol/L triiodothyronine; 10 ng/mL epidermal growth factor; 5 μg/mL insulin; 20 mmol/L d‐glucose; 2% fetal bovine serum; and 20 mmol/L Hepes, pH 7.4) at 37°C in 5% CO2/95% air. Cells were seeded on 75‐cm2 flasks and grown until 60% confluent. Cells were then serum starved for 24 hours followed by pretreatment with 30 μmol/L of 8‐aminoguanine, 8‐aminoguanosine, or vehicle for 1 hour at 37°C in 5% CO2/95% air. The Rho/Rac/Cdc42 Activator I (catalogue # CN04; Cytoskeleton, Denver, CO) was subsequently added to the cells at 1 μg/mL and incubated for 3 hours at 37°C in 5% CO2/95% air. The Rho/Rac/Cdc42 Activator I enters cells and activates Rac1 by deamidating glutamine‐61 of Rac1 in the Switch II region.27, 28 This modification, which converts glutamine‐61 to glutamate, blocks GTPase activity of Rac1, thus resulting in constitutively active Rac1.29 The biological activity of Rac1 protein was analyzed using pull‐down assays (Thermo Scientific, Rockford, IL). Cells were washed with Tris‐buffered saline and lysed for 5 minutes in 1 mL of lysis buffer containing Tris·HCl (25 mmol/L, pH 7.2), NaCl (150 mmol/L), MgCl2 (5 mmol/L), 1% NP‐40, 5% glycerol, and protease inhibitor cocktail. Cell lysates were clarified by centrifugation at 16 000g for 15 minutes. Fifteen microliters of the supernatant was analyzed for total Rac1, and 700 μL of supernatant was incubated for 1 hour at 4°C with GST‐human Pak1‐PBD (20 μg) immobilized on glutathione resin. The beads were washed 3 times with lysis buffer, and eluted with 50 μL of sample buffer, and 25 μL of the eluant was analyzed for active Rac1. The levels of total Rac1 and GTP‐bound Rac1 were analyzed by SDS‐PAGE and western blotting with anti‐Rac1 antibody. Densitometry analysis was performed, and the level of GTP‐Rac1 was normalized against the total amount of Rac1 present in the cell lysate.
Effects of 8‐Aminoguanine on Urinary Purines
Adult male Sprague‐Dawley rats were anesthetized with Inactin (90 mg/kg IP) and instrumented similar to the method described above. After a 1‐hour stabilization period, urine was collected for 30 minutes (period 1: 0–30 minutes into the protocol). Next, rats received an intravenous bolus of either vehicle (0.9% saline containing 0.03 N HCl) or 8‐aminoguanine (33.5 μmoles/kg). Each group of rats (n=7) received only 1 treatment. Ten minutes after the test agents were administered urine was collected for 30 minutes (period 2: 40–70 minutes into the protocol), and 15 minutes later urine was collected again for 30 minutes (period 3: 85–115 minutes into the protocol). Urinary levels of guanosine, guanine, inosine, and hypoxanthine were measured by ultra‐performance liquid chromatography–tandem mass spectrometry as described below.
Ultra‐Performance Liquid Chromatography–Tandem Mass Spectrometry Assay for Urinary Purines
Purines in urine were measured by ultra‐performance liquid chromatography–tandem mass spectrometry using selected reaction monitoring as previously described30 but with modifications. Urine samples were diluted 1 to 30 with water, and heavy isotope internal standards were added to each sample. Purines were separated by reversed‐phase ultra‐performance liquid chromatography (Waters UPLC BEH C18 column, 1.7 μm beads; 2.1×150 mm; Milford, MA) and quantified by selected reaction monitoring using a triple quadrupole mass spectrometer (TSQ Quantum‐Ultra; ThermoFisher Scientific, San Jose, CA) with a heated electrospray ionization source. The mobile phase was a linear gradient flow rate (300 μL/min) of 1% acetic acid in water (pH, 3; mobile phase A) and 100% methanol (mobile phase B), and was delivered with a Waters Acquity ultra‐performance liquid chromatographic system. The gradient (A/B) settings were: from 0 to 2 minutes, 99.6%/0.4%; from 2 to 3 minutes, to 98.0%/2.0%; from 3 to 4 minutes, to 85.0%/15.0%; from 4 to 6.5 minutes, to 99.6%/0.4%. The instrument parameters were: sample tray temperature, 10°C; column temperature, 50°C; ion spray voltage, 4.0 kV; ion transfer tube temperature, 350°C; source vaporization temperature, 320°C; Q2 CID gas, argon at 1.5 mTorr; sheath gas, nitrogen at 60 psi; auxiliary gas, nitrogen at 35 psi; Q1/Q3 width, 0.7/0.7 units full‐width half‐maximum; scan width, 0.6 units; scan time, 0.01 seconds. The following 8 transitions (selected reaction monitoring) were obtained: guanosine (284→152 m/z, retention time [RT]=3.10 minutes); 13C10,15N5‐guanosine (299→162 m/z, RT=3.10 minutes); guanine (152→135 m/z, RT=1.56 minutes); 13C2,15N‐guanine (155→138 m/z, RT 1.56 minutes); inosine (269→137 m/z, RT=3.10 minutes); 15N4‐inosine (273→141 m/z, RT=3.10 minutes); hypoxanthine (137→119 m/z, RT=1.86 minutes); 13C5‐hypoxanthine (142→124 m/z, RT=1.86 minutes).
Comparison of the Renal Effects of 8‐Aminoguanine, 9‐Deazaguanine, and Nsc23766
Adult male Sprague‐Dawley rats were anesthetized with Inactin (90 mg/kg IP) and instrumented similar to the method described above, with the exception that mesenteric blood flow was also measured with a transit‐time flow probe. After a 1‐hour stabilization period, urine was collected for 30 minutes (period 1: 0–30 minutes into the protocol). Next, rats received an intravenous bolus of either vehicle (0.9% saline containing 0.03 N HCl), 8‐aminoguanine (33.5 μmoles/kg), 9‐deazaguanine (67 μmoles/kg), or Nsc23766 (9.4 μmoles/kg). Like 8‐aminoguanine, 9‐deazaguanine is a potent inhibitor of PNPase. Nsc23766, on the other hand, is a selective inhibitor of Rac1. Although there are no within‐study head‐to‐head comparisons of potency between 8‐aminoguanine and 9‐deazaguanine, 9‐deazaguanine has a reported half maximal inhibitory concentration against PNPase of 2.3 μmol/L31; whereas 8‐aminoguanine has a reported Ki against PNPase of 0.8 μmol/L.21 Therefore, in the current study, we selected a dose of 9‐deazaguanine that was twice as large as that for a natriuretic dose of 8‐aminoguanine. The dose of Nsc23766 was selected on the basis of the studies in rats by Hummler et al32 who showed that 5 mg/kg (9.4 μmoles/kg) exerted a number of effects in rats consistent with inhibition of Rac1. Also, in preliminary studies we observed that higher doses of Nsc23766 caused unacceptable hypotension in rats; that is, 9.4 μmoles/kg was the highest tolerated dose. Each group of rats (n=6) received only 1 treatment. Ten minutes after the test agents were administered, urine was collected for 30 minutes (period 2: 30–70 minutes into protocol). Sodium and potassium in urine were measured by flame photometry (Model IL‐943; Instrumentations Laboratory Inc, Lexington, MA) and glucose in urine was measured by a glucose colorimetric assay kit (Cayman Chemical).
Statistical Analysis
Experiments with A1 knockout and wild‐type animals were performed in both male and female rats and responses were not sex dependent, so other in vivo experiments were conducted in male rats. Animals were randomly assigned to experimental groups by block randomization. 8‐Aminoguanine and 9‐deazaguanine increase urine volume so blinding was not possible. Data were analyzed statistically using a 1‐factor or 2‐factor ANOVA with or without repeated measures, as appropriate, or by unpaired, 2‐tailed Student t test. Post hoc comparisons were performed using the Fisher least significant difference (LSD) test if, and only if, the overall effects in the ANOVA were significant. Statistical tests were conducted using the NCSS 2004 software (Number Cruncher Statistical Systems; Kaysville, UT).
Results
Assessment of A1‐Receptor Blocking Activity of 8‐Aminoguanine In Vivo
A1‐receptor antagonists are K+‐sparing natriuretics,17 and the chemical structure of 8‐aminoguanine has features reminiscent of selective A1‐receptor blockers; for example, both 8‐aminoguanine and some A1‐receptor blockers have a xanthine scaffold (Figure 1). Therefore, we entertained the hypothesis that some of the effects of 8‐aminoguanine are mediated by antagonism of A1 receptors. To test this, CCPA was infused intravenously in rats treated or not with a diuretic/natriuretic dose (33.5 μmoles/kg, intravenous injection) of 8‐aminoguanine.11, 12 In control rats, CCPA induced dose‐dependent reductions in MABP, HR, and RBF (Figure 2), effects that are known to be due to activation of A1 receptors; for example, such responses to CCPA are absent in A1‐receptor knockout rats.23 Notably, 8‐aminoguanine did not significantly alter the effects of CCPA on MABP, HR, or RBF. These data suggest that 8‐aminoguanine does not induce its renal excretory effects via blockade of A1 receptors.
Figure 1.
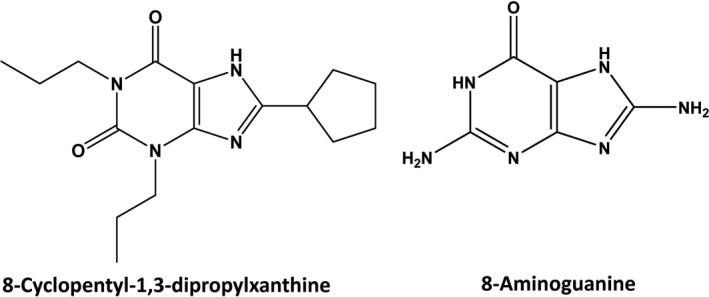
Comparison of the chemical structures of the selective A1 receptor antagonist 8‐cyclopentyl‐1,3‐dipropylxanthine and 8‐aminoguanine. Both 8‐cyclopentyl‐1,3‐dipropylxanthine and 8‐aminoguanine are K+‐sparing diuretics/natriuretics and share a xanthine scaffold, thus motivating the hypothesis that 8‐aminoguanine might be inducing its renal excretory effects by blocking A1 receptors.
Figure 2.
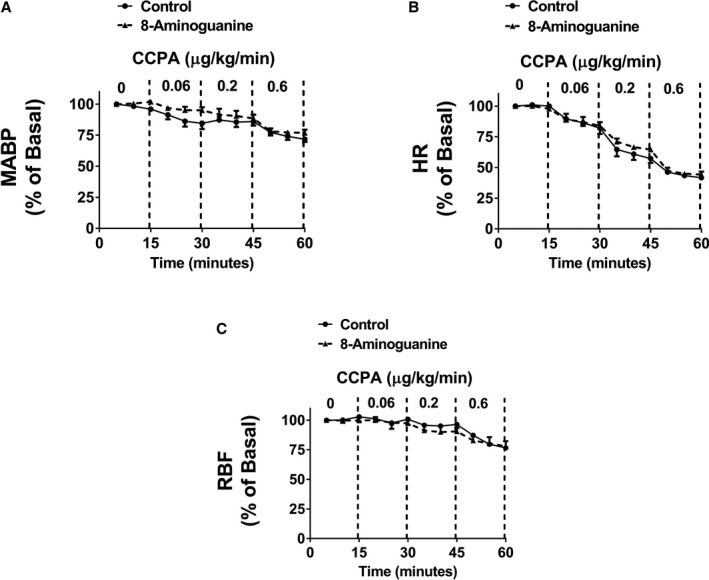
A diuretic/natriuretic dose of 8‐aminoguanine does not block A1 adenosine receptors in vivo. Figure illustrates the dose‐dependent effects of intravenous infusions of 2‐chloro‐N6‐cyclopentyladenosine (CCPA), a highly selective and potent A1‐receptor agonist, on mean arterial blood pressure (MABP; A), heart rate (HR; B), and renal blood flow (RBF; C) in anesthetized rats. Some rats were pretreated with a diuretic/natriuretic dose of 8‐aminoguanine (33.5 μmoles/kg, intravenous injection; n=3), whereas others receive only the vehicle for 8‐aminoguanine (control; n=3). Baseline MABP (mm Hg), HR (beats/min), and RBF (mL/min) were 133±3, 391±11 and 10.7±0.6, respectively, in the control group and 128±8, 362±22 and 11.4±1.6, respectively, in the 8‐aminoguanine‐treated group. Two‐factor ANOVA (treatment and time period were factors; interaction term included in model) indicated that CCPA induced a significant (P<0.0001) reduction in MABP, HR, and RBF. However, the interaction between the control group and 8‐aminoguanine‐treated group was not significant for any of the variables, thus indicating no involvement of A1 receptors in the renal excretory effects of 8‐aminoguanine. Values are means±SEM.
In the experiments with CCPA, we were unable to quantify the effects of CCPA on renal excretory function because of profound hypotension induced by this agonist. Therefore, to further test the hypothesis that 8‐aminoguanine affects renal function via antagonism of A1 receptors, we examined the effects of 8‐aminoguanine on urine volume and sodium, potassium, and glucose excretion in wild‐type versus A1‐receptor knockout rats. As shown in Figure 3, 8‐aminoguanine significantly and similarly increased urine volume, sodium excretion, and glucose excretion in wild‐type versus A1‐receptor knockout rats. Also, 8‐aminoguanine significantly and similarly reduced potassium excretion in wild‐type versus A1‐receptor knockout rats (Figure 3).
Figure 3.
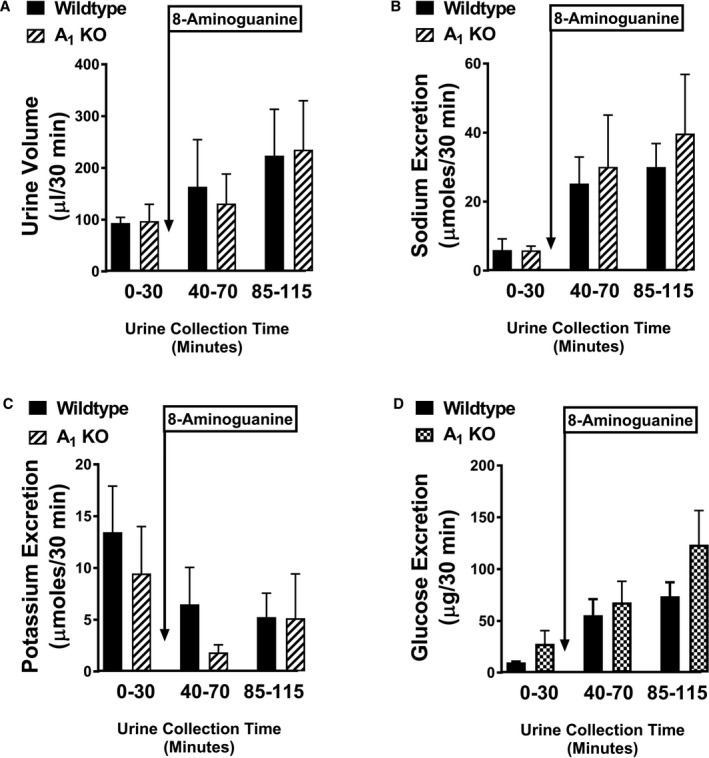
8‐Aminoguanine has similar effects in wild‐type and A1‐receptor knockout rats. Bar graphs show the effects of 8‐aminoguanine (33.5 μmoles/kg, intravenous injection) on urine volume (A), sodium excretion (B), potassium excretion (C), and glucose excretion (D) in wild‐type (n=6) vs A1‐receptor knockout rats (n=3). Two‐factor ANOVA (treatment and time period were factors; interaction term included in model) indicated that 8‐aminoguanine significantly increased urine volume (P=0.0236), sodium excretion (P=0.0032), and glucose excretion (P=0.0006) and significantly decreased potassium excretion (P=0.0010). However, the interaction between wild‐type and A1‐receptor knockout rats was not significant for any of the variables, thus indicating no involvement of A1 receptors in the renal excretory effects of 8‐aminoguanine. Values are means±SEM.
Effects of 8‐Aminoguanine on ENaC Activity
Molecular entities like 8‐aminoguanine exist in an equilibrium of multiple tautomeric forms that differ by the position of a proton. As shown in Figure 4, one tautomer of 8‐aminoguanine has structural features similar to amiloride, and another tautomer has structural features similar to triamterene. Also, like amiloride and triamterene,13 8‐aminoguanine is a K+‐sparing diuretic. These considerations suggest the hypothesis that 8‐aminoguanine exerts diuretic/natriuretic effects via inhibition of ENaC, which is the molecular target for amiloride and triamterene.13 However, over a large concentration range (0.1–100 μmol/L), 8‐aminoguanine had no detectable effect on amiloride‐sensitive short‐circuit current in mouse cortical collecting duct cells (Figure 5A). This was not due to inadequate concentrations of 8‐aminoguanine because a diuretic/natriuretic dose of 8‐aminoguanine provides renal cortical microdialysate levels of ≈10 μmol/L and urinary levels of 8‐aminoguanine of ≈100 μmol/L,12 thus indicating that the pharmacological levels of 8‐aminoguanine in vivo are in the vicinity of 10 to 100 μmol/L. Also, these negative results were not due to inadequate amiloride‐sensitive short circuit current in our preparation because 96±0.4% of the baseline short‐circuit current was inhibited by amiloride. Thus, the negative results with regard to inhibition of ENaC eliminate the possibility that 8‐aminoguanine induces its renal effects via inhibition of ENaC.
Figure 4.
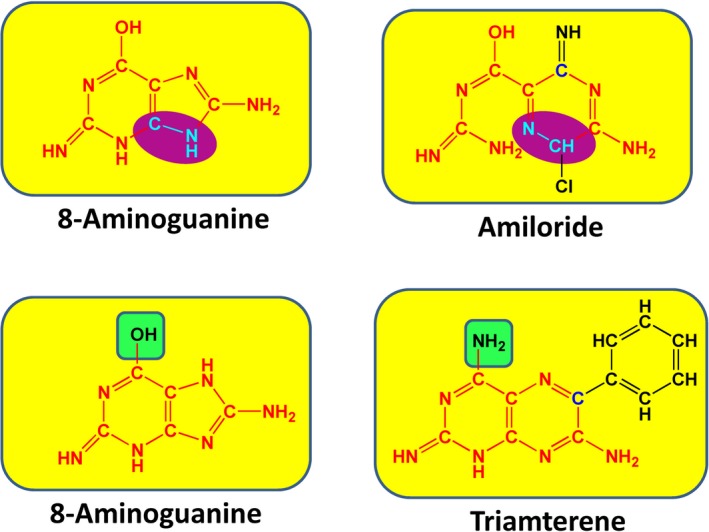
Comparison of the chemical structures of different tautomeric forms 8‐ aminoguanine vs amiloride and triamterene. The K+‐sparing diuretics/natriuretics amiloride and triamterene have structural features in common with 8‐aminoguanine. In this regard, the red bonds and atoms indicate structural identity, the functional groups highlighted in green are bioisosteres, and the structural features highlighted in purple are the same but in reverse orientation. These considerations motivated the hypothesis that 8‐aminoguanine might be inducing its renal excretory effects by inhibiting ENaC (which is the mechanism by which amiloride and triamterene induce diuresis/natriuresis) or inhibiting Na+/H+ exchangers (which is an effect of some amiloride derivatives).
Figure 5.
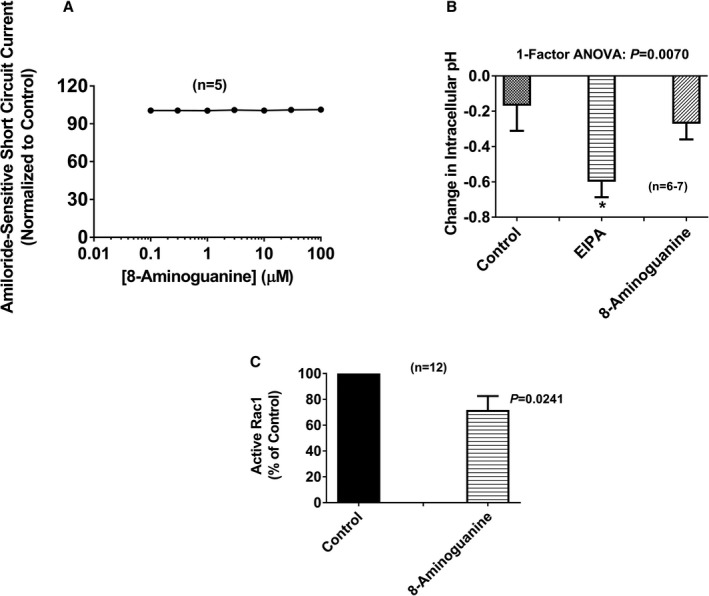
Concentrations of 8‐aminoguanine associated with diuresis/natriuresis do not block ENaC or Na+/H+ exchangers, but do inhibit Rac1. A illustrates the lack of effect of 8‐aminoguanine (0.1–100 μmol/L) on amiloride‐sensitive short‐circuit current in mouse collecting duct cells. The control amiloride‐sensitive short‐circuit current was 63±5 μA/cm2. In B, 8‐aminoguanine (100 μmol/L) does not alter intracellular pH in human renal proximal tubule epithelial cells, indicating that 8‐aminoguanine does not inhibit Na+/H+ exchangers. Ethylisopropyl amiloride (EIPA; 25 μmol/L), well‐known inhibitor of Na+/H+ exchangers, was employed as a positive control. In these experiments the baseline intracellular pH was 7.4±0.1. In the 1‐factor ANOVA, treatment group was the factor, and (*) indicates P<0.05 compared to Control. C demonstrates that 8‐aminoguanine (30 μmol/L) inhibits Rac1 activity in mouse collecting duct cells. Control active Rac1 (ratio of GTP‐bound Rac1 to total Rac1) was 2.5±0.7. P‐value in C is from unpaired, 2‐tailed Student t test. Values are means±SEM; however, in A the error bars reside within the symbols.
Effects of 8‐Aminoguanine on Na+/H+ Exchange Activity
Some amiloride derivatives, for example EIPA, inhibit Na+/H+ exchangers.26 Therefore, we also investigated whether 8‐aminoguanine has inhibitory activity against Na+/H+ exchangers. In this regard, we measured intracellular pH changes using 642/572‐nm fluorescence emission after excitation at 514 nm in SNARF‐5F‐loaded human renal proximal tubule epithelial cells following the addition of either EIPA (positive control; 25 μmol/L) or 8‐aminoguanine (100 μmol/L). As expected, EIPA caused a significant intracellular acidification, whereas 8‐aminoguanine did not (Figure 5B). These findings rule out the involvement of Na+/H+ exchangers in the diuretic/natriuretic effects of 8‐aminoguanine.
Effects of 8‐Aminoguanine on Rac1 Activity
Rac1 is known to activate mineralocorticoid receptors,18, 19 and some guanine derivatives, such as 8‐hydroxy‐2‐deoxyguanosine, inhibit Rac1.20 Therefore, it is conceivable that 8‐aminoguanine exerts effects on renal function via inhibition of Rac1. To test this hypothesis, we investigated whether 8‐aminoguanine (30 μmol/L) can inhibit Rac1 activation in mouse cortical collecting duct cells. These experiments were conducted in collecting duct cells, rather than in vivo, because our hypothesis was that inhibition of Rac1 activation in the collecting duct was mediating some of the effects of 8‐aminoguanine. Rac1 was activated using the reagent Rho/Rac/Cdc42 Activator I, and activation of Rac1 was assessed by the ratio of GTP‐bound Rac1 to total Rac1. A concentration of 8‐aminoguanine that is 3‐fold less than the urinary concentration of 8‐aminoguanine achieved after a diuretic/natriuretic dose of 8‐aminoguanine significantly (P=0.0241) inhibited Rac1 activation by ≈28% (Figure 5C). This finding leaves open the possibility that 8‐aminoguanine affects renal excretory function via inhibition of Rac1. This possibility was further evaluated in vivo (see below).
Effects of 8‐Aminoguanine on Urinary Purines
8‐Aminoguanine is known to inhibit PNPase,21 and this could be a part of the mechanism by which 8‐aminoguanine alters renal excretory function. However, whether a diuretic/natriuretic dose of 8‐aminoguanine is sufficient to block endogenous PNPase in vivo is unknown. Here, we examined the effects of a diuretic/natriuretic dose of 8‐aminoguanine (33.5 μmoles/kg, intravenous injection) on the urinary excretion rate of the 2 primary PNPase substrates, inosine and guanosine, and the 2 primary products of PNPase, hypoxanthine and guanine. Urine was collected during 3 periods: period 1, from 0 to 30 minutes; period 2, from 40 to 70 minutes; and period 3: from 85 to 115 minutes. The vehicle of either 8‐aminoguanine or 8‐aminoguanine was injected immediately after period 1. As shown in Figure 6A and 6B, intravenous injection of the vehicle for 8‐aminoguanine (control group) did not alter the urinary excretion of inosine or hypoxanthine during periods 2 and 3 compared to the baseline period 1. Similarly, vehicle injection did not alter the urinary excretion of guanosine (Figure 7A) or guanine (Figure 7B). In contrast, intravenous injection of a diuretic/natriuretic dose of 8‐aminoguanine caused a time‐dependent increase in the urinary excretion of inosine (Figure 6C). In this regard, the urinary excretion of inosine increased by ≈6‐fold during period 2 and by ≈11‐fold during period 3, compared with the baseline period 1. Correspondingly, 8‐aminoguanine decreased the urinary excretion of hypoxanthine by greater than 80% during periods 2 and 3 compared with period 1 (Figure 6D). Although not statistically significant, 8‐aminoguanine tended to increase the urinary excretion of guanosine (Figure 7C). Notably, 8‐aminoguanine strongly suppressed the urinary excretion of guanine by ≈84% and 89% during periods 2 and 3, respectively, compared with period 1 (Figure 7D). Together, these results indicate that a diuretic/natriuretic dose of 8‐aminoguanine blocks in vivo the PNPase‐mediated reactions: inosine→hypoxanthine and guanosine→guanine. This finding leaves open the possibility that 8‐aminoguanine affects renal excretory function via inhibition of PNPase. This possibility was further evaluated in vivo (see below).
Figure 6.
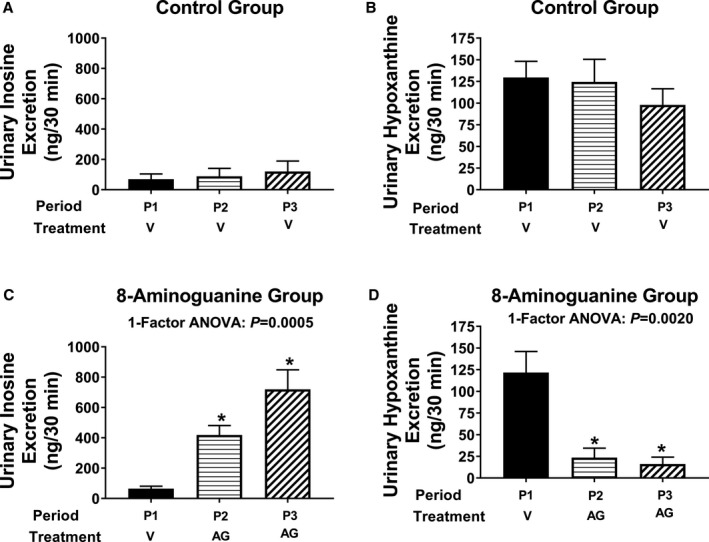
A diuretic/natriuretic dose of 8‐aminoguanine inhibits the metabolism of inosine to hypoxanthine in vivo. Urine was collected during 3 experimental periods: period 1 (P1, from 0 to 30 minutes); Period 2 (P2, from 40 to 70 minutes); and period 3 (P3, from 85 to 115 minutes). Either 8‐aminoguanine (AG; 33.5 μmoles/kg, a dose that induces diuresis/natriuresis; n=7) or its vehicle (V; n=7) was injected intravenously immediately after period 1. Vehicle did not affect the urinary excretion rate of inosine (A) or hypoxanthine (B). In contrast, 8‐aminoguanine increased the urinary excretion of inosine (C) while suppressing the urinary excretion of hypoxanthine (D). This indicates that a diuretic/natriuretic dose of 8‐aminoguanine inhibits the PNPase‐mediated reaction: inosine→hypoxanthine. For the 1‐factor ANOVAs, time period was the factor, and (*) indicates P<0.05 compared to period 1 (P1). Values are means±SEM.
Figure 7.
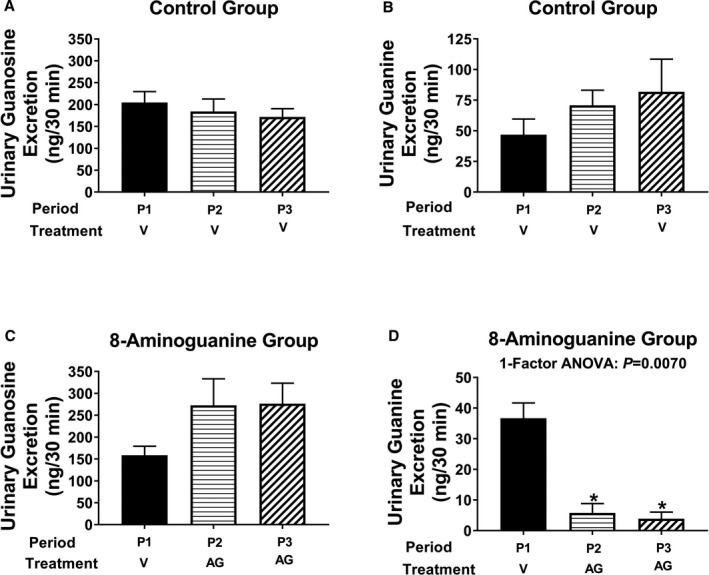
A diuretic/natriuretic dose of 8‐aminoguanine inhibits the metabolism of guanosine to guanine in vivo. Urine was collected during 3 experimental periods: period 1 (P1, from 0 to 30 minutes); period 2 (P2, from 40 to 70 minutes); and period 3 (P3, from 85 to 115 minutes). Either 8‐aminoguanine (AG; 33.5 μmoles/kg, a dose that induces diuresis/natriuresis; n=7) or its vehicle (V; n=7) was injected intravenously immediately after period 1. Vehicle did not affect the urinary excretion rate of guanosine (A) or guanine (B). In contrast, 8‐aminoguanine tended to increase the urinary excretion of guanosine (C) while significantly suppressing the urinary excretion of guanine (D). This indicates that a diuretic/natriuretic dose of 8‐aminoguanine inhibits the PNPase‐mediated reaction: guanosine→guanine. For the 1‐factor ANOVA, time period was the factor, and (*) indicates P<0.05 compared to period 1. Values are means±SEM.
Comparison of the Renal Effects of 8‐Aminoguanine, 9‐Deazaguanine, and Nsc23766
The results summarized above rule out the involvement of ENaC, Na+/H+ exchangers, and adenosine A1 receptors as mediators of the renal excretory effects of 8‐aminoguanine, yet suggest but do not prove that 8‐aminoguanine may alter renal excretory function via inhibiting PNPase and/or Rac1. To test this, we compared the renal excretory effects of 9‐deazaguanine (selective PNPase inhibitor), Nsc23766 (selective Rac1 inhibitor), and 8‐aminoguanine (endogenous K+‐sparing diuretic/natriuretic that increases urinary glucose excretion). The doses of 8‐aminoguanine and 9‐deazaguanine were carefully selected to approximate equal levels of PNPase inhibition21, 31 and the dose of Nsc23766 was selected based on previous reports that the selected dose inhibits Rac1 in vivo.32 Importantly, both 8‐aminoguanine and 9‐deazaguanine similarly increased urine volume (Figure 8A), Na+ excretion (Figure 8B), and glucose excretion (Figure 8C). However, 8‐aminoguanine, but not 9‐deazaguanine, suppressed K+ excretion (Figure 8D). Nsc23766 did not affect urine volume (Figure 8A), Na+ excretion (Figure 8B), or glucose excretion (Figure 8C). However, Nsc23766 mimicked the suppressive effects of 8‐aminoguanine on K+ excretion (Figure 8D). 8‐Aminoguanine, 9‐deazaguanine, and Nsc23766 had little to no effect on MABP, HR, RBF, or mesenteric blood flow (Figure 9A through 9D). These results corroborate the hypothesis that the effects of 8‐aminoguanine to increase urine volume, Na+ excretion, and glucose excretion are mediated by inhibition of PNPase, whereas the effects of 8‐aminoguanine on K+ excretion are due to inhibition of Rac1. We attempted experiments in which both Nsc23766 and 9‐deazaguanine were administered to rats; however, we observed that the combination of these 2 agents caused severe hypotension so we did not pursue this approach further.
Figure 8.
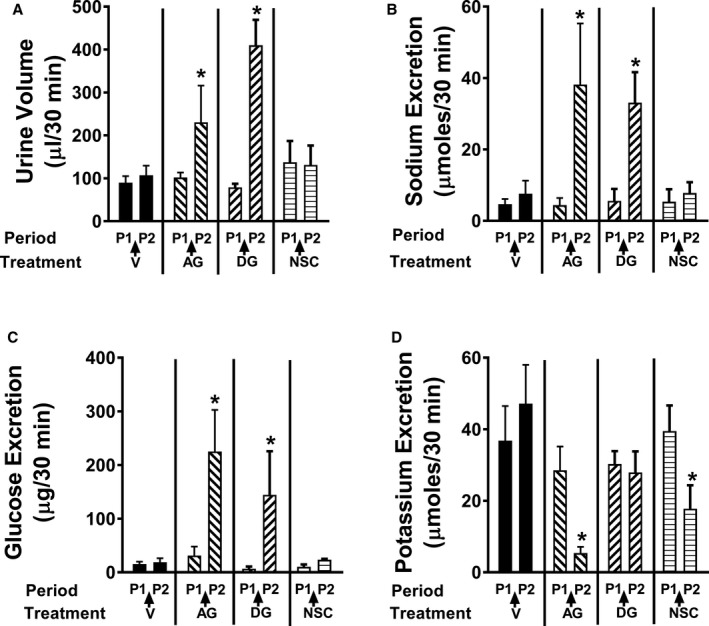
The diuretic (A), natriuretic (B), and glucosuric (C) effects of 8‐aminoguanine are mimicked by inhibition of PNPase, whereas the antikaluretic (D) effects of 8‐aminoguanine are mimicked by inhibition of Rac1. Urine was collected during 2 experimental periods: period 1 (P1, from 0 to 30 minutes) and period 2 (P2, from 40 to 70 minutes). Either 8‐aminoguanine (AG; 33.5 μmoles/kg; n=6) or its vehicle (V; n=6) or 9‐deazaguanine (DG, 67 μmoles/kg; PNPase inhibitor; n=6) or Nsc23766 (NSC, 9.4 μmoles/kg; Rac1 inhibitor; n=6) was injected intravenously immediately after period 1. Two‐factor ANOVA (treatment and time period were factors; interaction term included in model) indicated a significant interaction between treatment group and period (P=0.0005, 0.0455, 0.0194, and 0.0148 for urine volume, sodium excretion, glucose excretion, and potassium excretion, respectively), indicating that some treatments affected the corresponding variable and others did not. *P<0.05 compared to period 1. Values are means±SEM.
Figure 9.
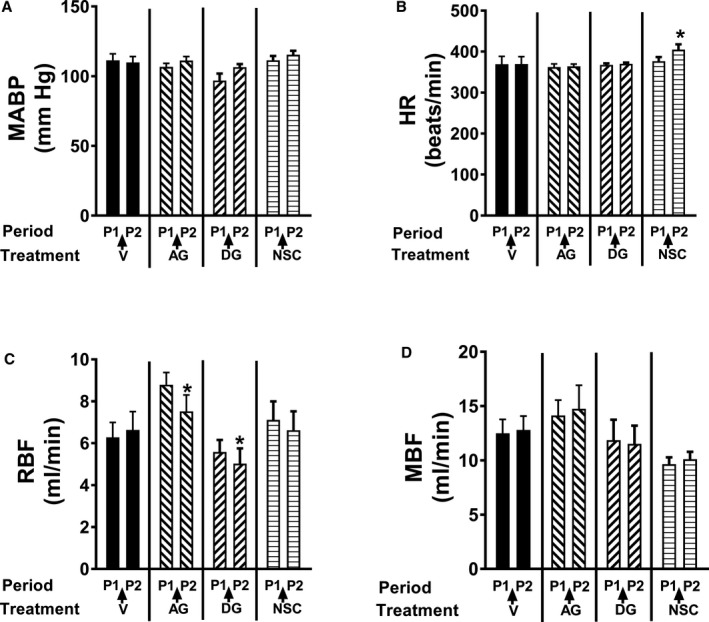
The test compounds 8‐aminoguanine, 9‐deazaguanine, and Nsc23766 had little to no effect on mean arterial blood pressure (MABP, A), heart rate (HR, B), renal blood flow (RBF, C), or mesenteric blood flow (MBF, D). Urine was collected during 2 experimental periods: period 1 (P1, from 0 to 30 minutes) and period 2 (P2, from 40 to 70 minutes). Either 8‐aminoguanine (AG; 33.5 μmoles/kg) or its vehicle (V) or 9‐deazaguanine (DG, 67 μmoles/kg; PNPase inhibitor) or Nsc23766 (NSC, 9.4 μmoles/kg; Rac1 inhibitor) was injected intravenously immediately after period 1. Although Nsc23766 significantly increased heart rate and 8‐aminoguanine and 9‐deazaguanine significantly reduced renal blood flow, these effects were minor and would not explain the renal excretory effects of these compounds. *P<0.05 compared to period 1 (P1). Values are means±SEM.
Effects of 8‐Aminoguanosine on Rac1 Activity
Recently, we reported that the effects of 8‐aminoguanosine on urine volume, Na+ excretion, and glucose excretion are not direct but rather are due to the metabolism of 8‐aminoguanosine to 8‐aminoguanine.12 Surprisingly, however, the antikaluretic effects of 8‐aminoguanosine did not require its conversion to 8‐aminoguanine.12 Because the effects of 8‐aminoguanine on K+ excretion are attributable to inhibition of Rac1 and 8‐aminoguanosine has direct effects on K+ excretion, we hypothesized that 8‐aminoguanosine must also be a direct Rac1 inhibitor. To test this, we examined the effects of 30 μmol/L of 8‐aminoguanosine on Rac1 activity in mouse cortical collecting duct cells using the same method as described for 8‐aminoguanine. As shown in Figure 10A and 10B, 8‐aminoguanosine directly and profoundly (>73%) inhibited Rac1 activity.
Figure 10.
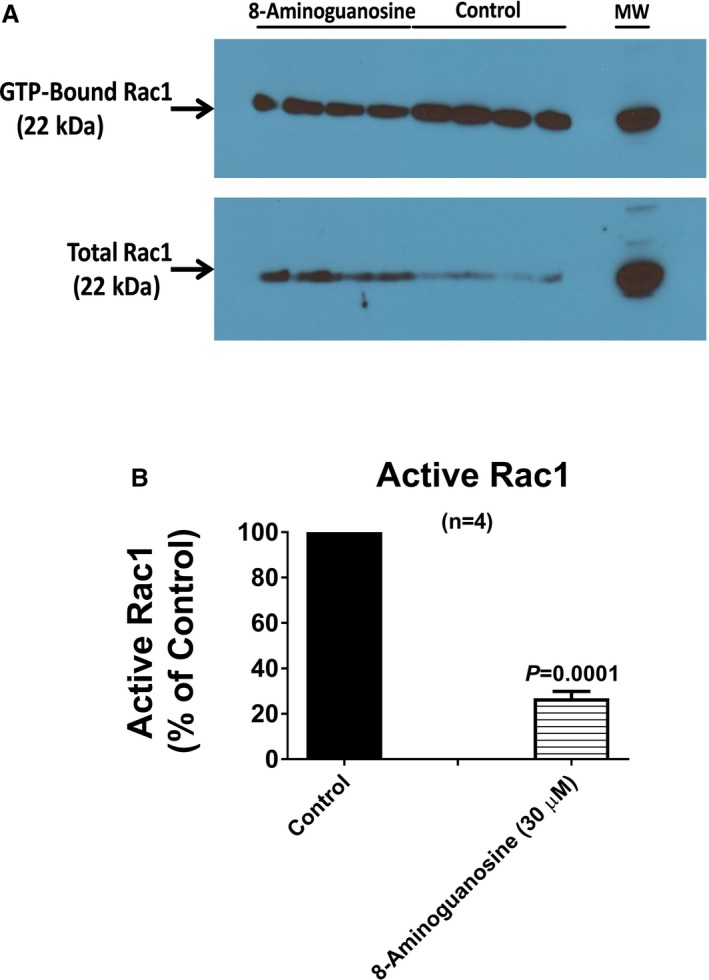
8‐Aminoguanosine is a potent Rac1 inhibitor. (A) An image of the western blot of proteins extracted from mouse collecting duct cells that were probed for GTP‐bound Rac1 and total Rac1. The ratio of GTP‐bound Rac1 to total Rac1 (by densitometry) was employed as a measure of active Rac1. Active Rac1 in the 8‐aminoguanosine‐treated cells was express as a percentage of the active Rac1 in control cells (B). Control active Rac1 (ratio of GTP‐bound Rac1 to total Rac1) was 5.1±0.6. These results demonstrate that 8‐aminoguanosine (30 μmol/L) inhibits Rac1 activity more so than does 8‐aminoguaine (compare with Figure 5C). P‐value in B is from unpaired, 2‐tailed Student t test. Values are means±SEM.
Discussion
Previously, we reported that 8‐aminoguanine is a K+‐sparing diuretic/natriuretic that, unlike most diuretics, increases urinary glucose excretion.11 The goal of the present study was to provide mechanistic understanding regarding how 8‐aminoguanine induces its renal excretory effects. In this regard, we considered 5 non–mutually exclusive possibilities, that is, inhibition of (1) A1 receptors, (2) ENaC, (3) Na+/H+ exchangers, (4) Rac1, or (5) PNPase. Our results clearly exclude inhibition of A1 receptors, ENaC, and Na+/H+ exchangers. However, our findings strongly support the conclusion that 8‐aminoguanine induces diuresis, natriuresis, and glucosuria by inhibiting PNPase, yet reduces K+ excretion by a separate mechanism, that is, inhibition of Rac1, a small G‐protein.
The evidence that 8‐aminoguanine induces diuresis, natriuresis, and glucosuria by inhibiting PNPase is based on 3 considerations. First, as previously published, in vitro 8‐aminoguanine is a potent PNPase inhibitor. Second, as shown in the present study, in vivo a dose of 8‐aminoguanine that influences renal excretory function also profoundly increases the urinary excretion of the PNPase substrate inosine (and to a lesser extent guanosine) and markedly decreases the urinary excretion of the PNPase products hypoxanthine and guanine. The 8‐aminoguanine–induced increase in the urinary ratio of inosine to hypoxanthine and guanosine to guanine is precisely what would be expected of a drug that blocks PNPase. If the dose of 8‐aminoguanine that causes renal effects had not influenced PNPase‐mediated reactions in vivo, this would have ruled out involvement of PNPase. Conversely, the fact that 8‐aminoguanine's renal excretory effects are concurrent with in vivo evidence of PNPase inhibition is consistent with the hypothesis that inhibition of PNPase mediates at least some of the renal excretory effects of 8‐aminoguanine.
It is possible, however, that the association between 8‐aminoguanine's effects on renal excretory function and PNPase inhibition is fortuitous; that is, the 2 events may be associated but mechanistically unrelated. To explore this possibility, we designed and conducted an experiment to compare the renal excretory effects of 8‐aminoguanine with the synthetic PNPase inhibitor 9‐deazaguanine. Our logic was that if 8‐aminoguanine induces renal effects via inhibition of PNPase, then an effective dose of another known PNPase inhibitor should mimic the renal actions of 8‐aminoguanine. Indeed, our results show that 8‐aminoguanine and 9‐deazaguanine produce nearly identical effects on urine volume, Na+ excretion, and glucose excretion. These findings strongly support the conclusion that inhibition of PNPase is the primary mechanism by which 8‐aminoguanine influences renal excretory function. This conclusion is further supported by our recent report that 8‐aminoguanosine increases urine volume, Na+ excretion, and glucose excretion via its conversion to 8‐aminoguanine.12 This would be expected if our hypothesis regarding PNPase is correct because 8‐aminoguanosine is a weak PNPase inhibitor, whereas 8‐aminoguanine is a potent PNPase inhibitor.21
Although the evidence supporting the conclusion that 8‐aminoguanine induces renal excretory effects via inhibition of PNPase is robust, exactly how PNPase inhibition induces diuresis/natriuresis/glucosuia is unknown and requires further experimentation to clarify. Our working hypothesis is that changes in kidney levels of PNPase substrates and products, particularly in the renal medulla, mediate the diuretic, natriuretic, and glucosuric effects of PNPase inhibition. Notably, 8‐aminoguanine profoundly reduces the urinary excretion of hypoxanthine. In the kidney, hypoxanthine, in collaboration with xanthine oxidase, produces reactive oxygen species such as superoxide anion,33 and superoxide anion decreases renal medullary blood flow and thereby suppresses Na+ excretion.34, 35 Indeed, the combination of hypoxanthine and xanthine oxidase decreases urine volume via a mechanism involving reactive oxygen species.33 Therefore, the profound reduction in hypoxanthine production that occurs when PNPase is inhibited by 8‐aminoguanine could contribute to the renal excretory effects of PNPase inhibitors such as 8‐aminoguanine.
In addition to decreasing the production of hypoxanthine, PNPase inhibition also increases renal levels of inosine and guanosine. Recent studies show that inosine can directly activate adenosine A2A receptors,36 and our previously published work demonstrates that guanosine increases extracellular levels of adenosine by inhibiting the disposition of extracellular adenosine.37, 38, 39, 40 Although adenosine per se is not considered a good substrate for mammalian PNPase,41 we have evidence (manuscript in preparation) that when PNPase is not in competition with adenosine deaminase for metabolism of adenosine, mammalian PNPase can metabolize adenosine to adenine. Therefore, it is conceivable that in the renal medulla, PNPase is an important determinant of extracellular levels of adenosine. Because adenosine increases renal medullary blood flow via A2 adenosine receptors and consequently increases Na+ excretion,42, 43 we postulate that A2 adenosine receptors mediate in part the diuretic, natriuretic, and glucosuric effects of PNPase inhibition.
It should be noted that there are several reports demonstrating renoprotective effects of exogenous inosine44, 45, 46, 47, 48, 49, 50, 51, 52 and guanosine.53 Because PNPase inhibition increases urinary levels and therefore, presumably, kidney levels of inosine and guanosine, PNPase inhibition may have utility as a renoprotective diuretic.
Inhibition of PNPase with 9‐deazaguanine increases urine volume, Na+ excretion, and glucose excretion; yet, unlike 8‐aminoguanine, 9‐deazaguanine has no effect on K+ excretion. This result implies that the antikaliuretic effects of 8‐aminoguanine are not mediated by inhibition of PNPase. Also, our previous studies show that 8‐aminoguanine does not reduce aldosterone levels,12 thus eliminating this mechanism as a possible explanation of the antikaliuretic effects of 8‐aminoguanine. Landmark studies by Shibata and colleagues demonstrate that Rac1 activates mineralocorticoid receptors,18, 19 and studies by Huh et al20 show that some guanine derivatives, such as 8‐hydroxy‐2‐deoxyguanosine, inhibit Rac1. The work of Shibata et al and Huh et al motivated our hypothesis that 8‐aminoguanine decreases K+ excretion by inhibiting Rac1. Three observations lend credence to this concept. First, inhibition of Rac1 with the selective Rac1 inhibitor Nsc23766 mimics the antikaliuretic effects of 8‐aminoguanine. Second, 8‐aminoguanine decreases active Rac1 (ie, GTP‐bound Rac1) in collecting duct cells. Third, the present findings show that 8‐aminoguanosine is a more potent Rac1 inhibitor compared with 8‐aminoguanine. Our previous work demonstrates that 8‐aminoguanosine's effects on urine volume, Na+ excretion, and glucose excretion are due to its conversion to 8‐aminoguanine12; that is, 8‐aminoguanosine is a prodrug. However, 8‐aminoguanosine has potent antikaliuretic effects independent of 8‐aminoguanine,12 a finding apparently at odds with the prodrug concept. The conclusion that 8‐aminoguanine's antikaliuretic effects are mediated by inhibition of Rac1 along with the observation that 8‐aminoguanosine is a more potent Rac1 inhibitor than 8‐aminoguanine provides a clear explanation for why 8‐aminoguanosine is a prodrug with regard to urine volume, Na+ excretion, and glucose excretion, yet is a direct antikaliuretic agent. With respect to inhibiting PNPase, 8‐aminoguanine is more potent than 8‐aminoguanosine; yet with respect to inhibiting Rac1, 8‐aminoguanosine is more potent than 8‐aminoguanine.
Notably, Nsc23766 decreased K+ excretion but did not increase Na+ excretion. The presumed mechanism by which Nsc23766 decreases K+ excretion is via inhibition of Rac1‐mediated activation of the mineralocorticoid receptor. However, inhibition of mineralocorticoid‐receptor signaling would be expected also to increase Na+ excretion. One explanation for the lack of effect of Nsc23766 on Na+ excretion is that the dose of Nsc23766 used in this study was simply not high enough to cause natriuresis. In this regard, we attempted to use higher doses of this compound; however, higher doses caused dramatic decreases in MABP. Because hypotension obscures natriuretic responses, we did not pursue this line of investigation further. Another possible explanation for the reduced K+ excretion with no change in Na+ excretion is that although Rac1 inhibition decreases K+ excretion by reducing mineralocorticoid‐receptor signaling, Rac1 inhibition engages additional mechanisms that offset any potential increases in Na+ excretion.
It is important to point out that the Ussing chamber experiments were designed to determine whether 8‐aminoguanine directly inhibits ENaC. Therefore, we did not add a Rac1 activator to this preparation because in the presence of a Rac1 activator, any changes in ENaC activity could have been due to either direct inhibition of ENaC by 8‐aminoguanine or via inhibition of Rac1 leading to reduced mineralocorticoid receptor‐induced expression of ENaC.
The fact that 8‐aminoguanosine and 8‐aminoguanine are diuretics, natriuretics, and glucosuric agents and inhibit Rac1 implies that these drugs may be particularly effective in preventing cardiovascular and renal diseases for 3 reasons. First, the diuretic and natriuretic effects of these compounds would provide antihypertensive activity, as previously reported by us in rats with deoxycorticosterone/salt‐induced hypertension.11 Second, the glucosuric effects of these compounds would reduce the risk of type 2 diabetes mellitus. Indeed, in unpublished studies, we have observed that chronic treatment (10 weeks) of ZDSD rats, a model of the metabolic syndrome, with 8‐aminoguanine lowered hemoglobin A1c levels from 11.3% to 8.4%. Third, there is a growing body of evidence that inhibiting Rac1 protects the cardiovascular system. For example, Rac1 inhibition (1) reduces vascular endothelium dysfunction54; (2) inhibits migration of stem cell antigen‐1 stem/progenitor cells, which may reduce neointima formation55; (3) decreases cardiac fibroblast migration, proliferation, and extracellular matrix production, which may protect against cardiac fibrosis56; (4) reduces store overload–induced calcium release and protects against ventricular arrhythmia57; and (5) inhibits the activation of some subtypes of NADPH oxidases.58 We have also observed protective effects of 8‐aminoguanosine/8‐aminoguanine in models of pulmonary hypertension and reduced stroke and increased life span of Dahl SS rats on a high‐salt diet (unpublished observations).
In the present study, we did not investigate whether the mechanism of action of 8‐aminoguanine involves inhibition of the sodium/glucose cotransporter 2. However, it is unlikely that the effects of 8‐aminoguanine are attributable to sodium/glucose cotransporter 2 inhibition because in the presence of sodium/glucose cotransporter 2 inhibitors, ≈50% of the filtered load of glucose is excreted in the urine, whereas in the presence of 8‐aminoguanine only 1% to 2% of the filtered load of glucose is excreted in the urine. Moreover, 8‐aminoguanine has no structural similarity whatsoever to any of the known sodium/glucose cotransporter 2 inhibitors.
Perspectives
8‐Aminoguanine, and its prodrug 8‐aminoguanosine, are naturally occurring purines that have diuretic, natriuretic, glucosuric, antikaliuretic, and antihypertensive activity. Thus, either 8‐aminoguanine or 8‐aminoguanosine might be effective in reducing the burden of hypertension and cardiovascular and renal diseases. However, before these agents can be further considered for development, it is critically important to understand the mechanisms underlying their effects. The present study provides evidence that 8‐aminoguanine exerts its diuretic, natriuretic, and glucosuric effects by inhibiting PNPase, yet decreases K+ excretion by inhibiting Rac1 (Figure 11). Thus, 8‐aminoguanine and its prodrug 8‐aminoguanosine possess a unique combination of pharmacological effects and therefore represent a novel class of potentially useful cardiovascular drugs.
Figure 11.

8‐Aminoguanine has a dual mechanism of action. The present findings support the view that 8‐aminoguanine engages 2 mechanism: inhibition of PNPase and inhibition of Rac1. Inhibition of PNPase mediates the effects of 8‐aminoguanine on urine volume and sodium and glucose excretion, whereas inhibition of Rac1 causes antikaliuresis. The mechanisms by which PNPase and Rac1 inhibition affect renal excretory function are currently unknown. However, since inhibition of PNPase increases renal levels of some purines (eg, inosine, guanosine, and perhaps adenosine) and decreases kidney levels of other purines (eg, hypoxanthine and guanine), it is likely that alterations in the renal concentrations of purines mediate in part the effects of 8‐aminoguanine on urine volume and sodium and glucose excretion. Also, since inhibition of Rac1 reduces mineralocorticoid signaling, it is possible that this effect mediates in part the ability of 8‐aminoguanine to reduce potassium excretion.
Sources of Funding
The work was supported by the National Institutes of Health (DK091190, HL069846, DK068575, HL109002 and DK079307).
Disclosures
The University of Pittsburgh has submitted a patent application (International Publication Number WO 2018/045045 A1) on uses for 8‐aminoguanine, 8‐aminoguanosine, and PNPase inhibitors. Edwin K. Jackson is listed as an inventor on this application. The remaining authors have no disclosures to report.
(J Am Heart Assoc. 2018;7:e010085 DOI: 10.1161/JAHA.118.010085.)
References
- 1. Ohshima H, Sawa T, Akaike T. 8‐Nitroguanine, a product of nitrative DNA damage caused by reactive nitrogen species: formation, occurrence, and implications in inflammation and carcinogenesis. Antioxid Redox Signal. 2006;8:1033–1045. [DOI] [PubMed] [Google Scholar]
- 2. Szabo C, Ohshima H. DNA damage induced by peroxynitrite: subsequent biological effects. Nitric Oxide. 1997;1:373–385. [DOI] [PubMed] [Google Scholar]
- 3. Yermilov V, Rubio J, Ohshima H. Formation of 8‐nitroguanine in DNA treated with peroxynitrite in vitro and its rapid removal from DNA by depurination. FEBS Lett. 1995;376:207–210. [DOI] [PubMed] [Google Scholar]
- 4. Misiaszek R, Crean C, Joffe A, Geacintov NE, Shafirovich V. Oxidative DNA damage associated with combination of guanine and superoxide radicals and repair mechanisms via radical trapping. J Biol Chem. 2004;279:32106–32115. [DOI] [PubMed] [Google Scholar]
- 5. Akaike T, Okamoto S, Sawa T, Yoshitake J, Tamura F, Ichimori K, Miyazaki K, Sasamoto K, Maeda H. 8‐Nitroguanosine formation in viral pneumonia and its implication for pathogenesis. Proc Natl Acad Sci U S A. 2003;100:685–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Park EM, Shigenaga MK, Degan P, Korn TS, Kitzler JW, Wehr CM, Kolachana P, Ames BN. Assay of excised oxidative DNA lesions: isolation of 8‐oxoguanine and its nucleoside derivatives from biological fluids with a monoclonal antibody column. Proc Natl Acad Sci U S A. 1992;89:3375–3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fraga CG, Shigenaga MK, Park JW, Degan P, Ames BN. Oxidative damage to DNA during aging: 8‐hydroxy‐2′‐deoxyguanosine in rat organ DNA and urine. Proc Natl Acad Sci U S A. 1990;87:4533–4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lam PM, Mistry V, Marczylo TH, Konje JC, Evans MD, Cooke MS. Rapid measurement of 8‐oxo‐7,8‐dihydro‐2′‐deoxyguanosine in human biological matrices using ultra‐high‐performance liquid chromatography‐tandem mass spectrometry. Free Radic Biol Med. 2012;52:2057–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sodum RS, Nie G, Fiala ES. 8‐Aminoguanine: a base modification produced in rat liver nucleic acids by the hepatocarcinogen 2‐nitropropane. Chem Res Toxicol. 1993;6:269–276. [DOI] [PubMed] [Google Scholar]
- 10. Osborne WR, Barton RW. A rat model of purine nucleoside phosphorylase deficiency. Immunology. 1986;59:63–67. [PMC free article] [PubMed] [Google Scholar]
- 11. Jackson EK, Gillespie DG, Mi Z. 8‐Aminoguanosine and 8‐aminoguanine exert diuretic, natriuretic, glucosuric, and antihypertensive activity. J Pharmacol Exp Ther. 2016;359:420–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jackson EK, Mi Z. 8‐Aminoguanosine exerts diuretic, natriuretic, and glucosuric activity via conversion to 8‐aminoguanine, yet has direct antikaliuretic effects. J Pharmacol Exp Ther. 2017;363:358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jackson EK. Drugs affecting renal excretory function In: Brunton LL, Hilal‐Dandam R, Knollmann BC, eds. Goodman & Gilman's the Pharmacological Basis of Therapeutics. New York: McGraw Hill; 2018:445–470. [Google Scholar]
- 14. Dos Santos EA, Dahly‐Vernon AJ, Hoagland KM, Roman RJ. Inhibition of the formation of EETs and 20‐HETE with 1‐aminobenzotriazole attenuates pressure natriuresis. Am J Physiol Regul Integr Comp Physiol. 2004;287:R58–R68. [DOI] [PubMed] [Google Scholar]
- 15. Noonan WT, Woo AL, Nieman ML, Prasad V, Schultheis PJ, Shull GE, Lorenz JN. Blood pressure maintenance in NHE3‐deficient mice with transgenic expression of NHE3 in small intestine. Am J Physiol Regul Integr Comp Physiol. 2005;288:R685–R691. [DOI] [PubMed] [Google Scholar]
- 16. Nishiki K, Tsuruoka S, Kawaguchi A, Sugimoto K, Schwartz GJ, Suzuki M, Imai M, Fujimura A. Inhibition of Rho‐kinase reduces renal Na‐H exchanger activity and causes natriuresis in rat. J Pharmacol Exp Ther. 2003;304:723–728. [DOI] [PubMed] [Google Scholar]
- 17. Kuan C‐J, Herzer WA, Jackson EK. Cardiovascular and renal effects of blocking A1 adenosine receptors. J Cardiovasc Pharmacol. 1993;21:822–828. [DOI] [PubMed] [Google Scholar]
- 18. Shibata S, Nagase M, Yoshida S, Kawarazaki W, Kurihara H, Tanaka H, Miyoshi J, Takai Y, Fujita T. Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat Med. 2008;14:1370. [DOI] [PubMed] [Google Scholar]
- 19. Shibata S, Mu S, Kawarazaki H, Muraoka K, Ishizawa K‐i, Yoshida S, Kawarazaki W, Takeuchi M, Ayuzawa N, Miyoshi J, Takai Y, Ishikawa A, Shimosawa T, Ando K, Nagase M, Fujita T. Rac1 GTPase in rodent kidneys is essential for salt‐sensitive hypertension via a mineralocorticoid receptor–dependent pathway. J Clin Invest. 2011;121:3233–3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huh JY, Son DJ, Lee Y, Lee J, Kim B, Lee HM, Jo H, Choi S, Ha H, Chung M‐H. 8‐Hydroxy‐2‐deoxyguanosine prevents plaque formation and inhibits vascular smooth muscle cell activation through Rac1 inactivation. Free Radic Biol Med. 2012;53:109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chern JW, Lee HY, Chen CS, Shewach DS, Daddona PE, Townsend LB. Nucleosides. 5. Synthesis of guanine and formycin B derivatives as potential inhibitors of purine nucleoside phosphorylase. J Med Chem. 1993;36:1024–1031. [DOI] [PubMed] [Google Scholar]
- 22. Kuan CJ, Herzer WA, Jackson EK. An experimental paradigm for investigating the role of endogenous adenosine/A1 receptor interactions in vivo. J Pharmacol Exp Ther. 1992;263:657–662. [PubMed] [Google Scholar]
- 23. Jackson EK, Gillespie DG, Mi Z, Cheng D. Adenosine receptors influence hypertension in Dahl Salt‐Sensitive Rats: dependence on receptor subtype, salt diet, and sex. Hypertension. 2018;72:511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Butterworth MB, Edinger RS, Johnson JP, Frizzell RA. Acute ENaC stimulation by cAMP in a kidney cell line is mediated by exocytic insertion from a recycling channel pool. J Gen Physiol. 2005;125:81–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rotin D, Kanelis V, Schild L. Trafficking and cell surface stability of ENaC. Am J Physiol Renal Physiol. 2001;281:F391–F399. [DOI] [PubMed] [Google Scholar]
- 26. Vigne P, Frelin C, Cragoe EJ Jr, Lazdunski M. Ethylisopropyl‐amiloride: a new and highly potent derivative of amiloride for the inhibition of the Na+/H+ exchange system in various cell types. Biochem Biophys Res Commun. 1983;116:86–90. [DOI] [PubMed] [Google Scholar]
- 27. Lerm M, Selzer J, Hoffmeyer A, Rapp UR, Aktories K, Schmidt G. Deamidation of Cdc42 and Rac by Escherichia coli cytotoxic necrotizing factor 1: activation of c‐Jun N‐terminal kinase in HeLa cells. Infect Immun. 1999;67:496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schmidt G, Sehr P, Wilm M, Selzer J, Mann M, Aktories K. Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor‐1. Nature. 1997;387:725–729. [DOI] [PubMed] [Google Scholar]
- 29. Flatau G, Lemichez E, Gauthier M, Chardin P, Paris S, Fiorentini C, Boquet P. Toxin‐induced activation of the G protein p21 Rho by deamidation of glutamine. Nature. 1997;387:729–733. [DOI] [PubMed] [Google Scholar]
- 30. Jackson EK, Ren J, Mi Z. Extracellular 2′,3′‐cAMP is a source of adenosine. J Biol Chem. 2009;284:33097–33106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Suver M, Zinic B, Portada T, Bzowska A, Ljubica Glavas‐Obrovac L. 9‐Deazaguanine and its methyl derivatives: synthesis, antitumor activity in vitro and effects on purine nucleoside phosphorylase gene expression. Croat Chem Acta. 2008;81:147–156. [Google Scholar]
- 32. Hummler JK, Dapaah‐Siakwan F, Vaidya R, Zambrano R, Luo S, Chen S, Kerr N, Vaccari JP, Keane RW, Dietrich WD, Bancalari E, Young KC, Wu S. Inhibition of Rac1 signaling downregulates inflammasome activation and attenuates lung injury in neonatal rats exposed to hyperoxia. Neonatology. 2017;111:280–288. [DOI] [PubMed] [Google Scholar]
- 33. Galat JA, Robinson AV, Rhodes RS. Oxygen free radical mediated renal dysfunction. J Surg Res. 1989;46:520–525. [DOI] [PubMed] [Google Scholar]
- 34. Majid DS, Nishiyama A. Nitric oxide blockade enhances renal responses to superoxide dismutase inhibition in dogs. Hypertension. 2002;39:293–297. [DOI] [PubMed] [Google Scholar]
- 35. Zou AP, Li N, Cowley AW Jr. Production and actions of superoxide in the renal medulla. Hypertension. 2001;37:547–553. [DOI] [PubMed] [Google Scholar]
- 36. Welihinda AA, Kaur M, Raveendran KS, Amento EP. Enhancement of inosine‐mediated A2AR signaling through positive allosteric modulation. Cell Signal. 2018;42:227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jackson EK, Cheng D, Jackson TC, Verrier JD, Gillespie DG. Extracellular guanosine regulates extracellular adenosine levels. Am J Physiol Cell Physiol. 2013;304:C406–C421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jackson EK, Mi Z. The guanosine‐adenosine interaction exists in vivo. J Pharmacol Exp Ther. 2014;350:719–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jackson EK, Gillespie DG. Regulation of cell proliferation by the guanosine‐adenosine mechanism: role of adenosine receptors. Physiol Rep. 2013;1:e00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jackson EK, Cheng D, Mi Z, Gillepsie DG. Guanosine regulates adenosine levels in the kidney. Physio Rep. 2014;2:e12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bzowska A, Kulikowska E, Shugar D. Purine nucleoside phosphorylases: properties, functions, and clinical aspects. Pharmacol Ther. 2000;88:349–425. [DOI] [PubMed] [Google Scholar]
- 42. Zou AP, Nithipatikom K, Li PL, Cowley AW. Role of renal medullary adenosine in the control of blood flow and sodium excretion. Am J Physiol Regul Integr Comp Physiol. 1999;276:R790. [DOI] [PubMed] [Google Scholar]
- 43. Zou AP, Wu F, Li PL, Cowley AW. Effect of chronic salt loading on adenosine metabolism and receptor expression in renal cortex and medulla in rats. Hypertension. 1999;33:511. [DOI] [PubMed] [Google Scholar]
- 44. Fernando AR, Gunter PA, Hendry WF, Smith AF, Watkinson LE, Wickham JE. An ultrastructural study on the effects of warm ischaemia on the inosine‐protected kidney. Br J Urol. 1979;51:167–172. [DOI] [PubMed] [Google Scholar]
- 45. Fitzpatrick JM, Wallace DM, Whitfield HN, Watkinson LE, Fernando AR, Wickham JE. Inosine in ischaemic renal surgery: long‐term follow‐up. Br J Urol. 1981;53:524–527. [DOI] [PubMed] [Google Scholar]
- 46. Wickham JE, Fernando AR, Hendry WF, Whitfield HN, Fitzpatrick JM. Intravenous inosine for ischaemic renal surgery. Br J Urol. 1979;51:437–439. [DOI] [PubMed] [Google Scholar]
- 47. Fitzpatrick JM, Marberger M, Koh SK, Wickham JE. A study of the effectiveness of inosine in renal transplantation. Eur Urol. 1981;7:16–18. [DOI] [PubMed] [Google Scholar]
- 48. Marberger M, Gunther R, Alken P, Rumpf W, Ranc M. Inosine: alternative or adjunct to regional hypothermia in the prevention of post‐ischemic renal failure? Eur Urol. 1980;6:95–102. [DOI] [PubMed] [Google Scholar]
- 49. Mathur VK, Ramsey EW. Comparison of methods for preservation of renal function during ischemic renal surgery. J Urol. 1983;129:163–165. [DOI] [PubMed] [Google Scholar]
- 50. Fernando AR, Armstrong DM, Griffiths JR, Hendry WF, O'Donoghue EP, Perrett D, Ward JP, Wickham JE. Enhanced preservation of the ischaemic kidney with inosine. Lancet. 1976;1:555–557. [DOI] [PubMed] [Google Scholar]
- 51. Rothwell D, Bartley J, James M. Preservation of the ischaemic canine kidney with inosine. Urol Res. 1981;9:75–78. [DOI] [PubMed] [Google Scholar]
- 52. Maggio AJ Jr, Das S, Smith RB, Kaufman JJ. Renal preservation with inosine. Urology. 1980;16:343–345. [DOI] [PubMed] [Google Scholar]
- 53. Kelly KJ, Plotkin Z, Dagher PC. Guanosine supplementation reduces apoptosis and protects renal function in the setting of ischemic injury. J Clin Invest. 2001;108:1291–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Carrizzo A, Vecchione C, Damato A, di Nonno F, Ambrosio M, Pompeo F, Cappello E, Capocci L, Peruzzi M, Valenti V, Biondi‐Zoccai G, Marullo AG, Palmerio S, Carnevale R, Spinelli CC, Puca AA, Rubattu S, Volpe M, Sadoshima J, Frati G, Sciarretta S. Rac1 pharmacological inhibition rescues human endothelial dysfunction. J Am Heart Assoc. 2017;6:e004746 DOI: 10.1161/JAHA.116.004746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yu B, Wong MM, Potter CM, Simpson RM, Karamariti E, Zhang Z, Zeng L, Warren D, Hu Y, Wang W, Xu Q. Vascular stem/progenitor cell migration induced by smooth muscle cell‐derived chemokine (C‐C Motif) ligand 2 and chemokine (C‐X‐C motif) ligand 1 contributes to neointima formation. Stem Cells. 2016;34:2368–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lavall D, Schuster P, Jacobs N, Kazakov A, Bohm M, Laufs U. Rac1 GTPase regulates 11beta hydroxysteroid dehydrogenase type 2 and fibrotic remodeling. J Biol Chem. 2017;292:7542–7553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang L, Lu X, Gui L, Wu Y, Sims SM, Wang G, Feng Q. Inhibition of Rac1 reduces store overload–induced calcium release and protects against ventricular arrhythmia. J Cell Mol Med. 2016;20:1513–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Elnakish MT, Hassanain HH, Janssen PM, Angelos MG, Khan M. Emerging role of oxidative stress in metabolic syndrome and cardiovascular diseases: important role of Rac/NADPH oxidase. J Pathol. 2013;231:290–300. [DOI] [PubMed] [Google Scholar]