

Abstract
Background
Left ventricular (LV) diastolic dysfunction often precedes heart failure with preserved ejection fraction, the dominant form of heart failure in postmenopausal women. The objective of this study was to determine the effect of oral estradiol treatment initiated early after ovariectomy on LV function and myocardial gene expression in female cynomolgus macaques.
Methods and Results
Monkeys were ovariectomized and randomized to receive placebo (control) or oral estradiol at a human‐equivalent dose of 1 mg/day for 8 months. Monkeys then underwent conventional and tissue Doppler imaging to assess cardiac function, followed by transcriptomic and histomorphometric analyses of LV myocardium. Age, body weight, blood pressure, and heart rate were similar between groups. Echocardiographic mitral early and late inflow velocities, mitral annular velocities, and mitral E deceleration slope were higher in estradiol monkeys (all P<0.05), despite similar estimated LV filling pressure. MCP1 (monocyte chemoattractant protein 1) and LV collagen staining were lower in estradiol animals (P<0.05). Microarray analysis revealed differential myocardial expression of 40 genes (>1.2‐fold change; false discovery rate, P<0.05) in estradiol animals relative to controls, which implicated pathways associated with better calcium ion homeostasis and muscle contraction and lower extracellular matrix deposition (P<0.05).
Conclusions
Estradiol treatment initiated soon after ovariectomy resulted in enhanced LV diastolic function, and altered myocardial gene expression towards decreased extracellular matrix deposition, improved myocardial contraction, and calcium homeostasis, suggesting that estradiol directly or indirectly modulates the myocardial transcriptome to preserve cardiovascular function.
Keywords: diastolic dysfunction, estrogen, fibrosis, menopause, transcriptome
Subject Categories: Animal Models of Human Disease, Gene Expression & Regulation, Heart Failure, Echocardiography
Clinical Perspective
What Is New?
Estradiol treatment initiated shortly after the onset of surgical menopause in monkeys improved postmenopausal diastolic function, inhibited myocardial extracellular matrix deposition, and led to myocardial expression profiles consistent with improved myocardial contraction and calcium homeostasis, all independent of measurable blood pressure effects.
What Are the Clinical Implications?
The results support the “timing hypothesis” of hormone therapy, indicating that estrogen treatment initiated early after menopause may have beneficial effects on the cardiovascular system, in this case by preventing left ventricular diastolic dysfunction, a precursor to heart failure with preserved ejection fraction.
Introduction
Cardiovascular disease is as prevalent in women as men, and the leading cause of mortality of both men and women.1 Women develop cardiovascular disease later in life than men, which in part has been attributed to cardioprotection by endogenous estrogens. The cardiovascular effects of estrogen treatment in menopause have been greatly debated since the controversial findings in the Women's Health Initiative, and the timing of introduction of therapy has a large impact on the potential risks and benefits.2 The ELITE (Early Versus Late Intervention Trial With Estradiol) study demonstrated that unopposed estrogen (17β‐estradiol) had beneficial effects on subclinical atherosclerosis progression3 and stress associated cortisol responses and working memory4 when administered early after the onset of menopause. However, there are few data regarding potential influence on cardiac function and risk of heart failure (HF) development. HF currently affects an estimated 6.5 million American adults, half of which are women.1 These numbers are expected to increase 46% by 2030 and cost an estimated $70 billion per year in direct and indirect costs.5 Approximately one half of patients with HF have preserved ejection fraction (HFpEF), which is twice as common in women as men, and for which there is no Food and Drug Administration–approved therapy.6, 7 In the Cardiovascular Health Study, over 90% of elderly women who developed HF over a 6‐year follow‐up developed HFpEF.8 The sexual dimorphism in HFpEF9, 10, 11 appears to be attributable to sex‐specific responses to stressors; women who develop HFpEF in response to aging, hypertension, and obesity/diabetes mellitus have normal‐sized left ventricles, whereas men tend to have mild left ventricular (LV) dilation and lower ejection fraction, generally related to history of myocardial infarction. Aortic banding in rat models to induce LV overload results in concentric LV hypertrophy, normal ejection fraction, and diastolic dysfunction in females, as opposed to dilated LV hypertrophy with thinner walls and reduced ejection fraction in males.12 Additionally, women develop HFpEF at an older age compared with men, particularly after the age of 55, corresponding with the menopausal period. The sharp rise in susceptibility to HF after menopause has led to suspicion that estradiol has protective effects on cardiac function. Recent studies in the Women's Health Initiative cohort have shown that among postmenopausal women, a shorter total reproductive duration, attributable largely to an earlier menopause, was associated with an increased risk of incident HF, and that nulliparity, often associated with menstrual dysfunction, was associated with increased risk of HFpEF (hazard ratio, 2.75; 95% confidence interval, 1.16–6.52).13 Earlier menopause was also associated with HF in women in the ARIC (Atherosclerosis Risk in Communities) study.14 Although the mechanisms underlying these associations are not known, these reproductive phenotypes are associated with reduced lifetime exposure to estrogens and progesterone. These observations are consistent with the hypothesis that the hormonal/physiologic changes associated with menopause trigger cardiovascular changes that contribute to diastolic dysfunction and eventually HFpEF.
Estrogen deficiency in postmenopausal women may be an important factor in the development of LV diastolic dysfunction (LVDD) and HFpEF, as estrogen influences a number of biological processes that have been implicated as having roles in the pathogenesis of LVDD. Estrogen maintains endothelial nitric oxide synthase activity, key to normal endothelial function, through genomic and nongenomic signaling. Classical estradiol signaling involves estradiol binding to intranuclear receptors estrogen receptor α (ERα) and/or estrogen receptor β (ERβ), which dimerize and bind to estrogen response elements and increase transcriptional activity related to endothelial nitric oxide synthase production. Nongenomic estradiol signaling may involve plasma membrane–bound ERα or plasma membrane– or endoplasmic reticulum–bound G protein–coupled estrogen receptor (GPER), resulting in activation of endothelial nitric oxide synthase through phosphorylation of serine 1177 via the PI3K/Phosphokinase B–mediated signaling pathway.15 Estradiol may also impede mechanisms associated with myocyte growth and the initiation of LV hypertrophy by blocking the activity of mitogen‐activated protein kinase pathways,16 as well as protrophic components of the cardiac renin‐angiotensin system.17, 18 Estradiol‐bound estrogen receptors have also been shown to modulate NFκB and other transcription factor activity through transcription factor cross‐talk.19 In addition, there is some evidence to suggest that estradiol may increase IκBα synthesis and inhibit NFκB transcriptional binding (and inflammation‐related gene expression) in vascular smooth muscle through ERβ.
The role of early estrogen intervention in the preservation of LV diastolic function in middle‐aged women after menopause has received little attention clinically, although much has been learned from rodent models. Groban and colleagues investigated the role of estrogen in the maintenance of cardiac structure and diastolic function in various rodent models, including the normal aging BNF344 rat,20 the estrogen‐sensitive hypertensive mRen2.Lewis rat,18, 21, 22, 23 and the spontaneously hypertensive rat and its normotensive Wistar Kyoto counterpart. In these studies, ovariectomy led to LVDD and remodeling, which were attenuated by estradiol or activation of the membrane estrogen receptor, G protein–coupled estrogen receptor, with G1.20, 22, 23
In the present study, we assessed the effects of exogenous oral estradiol replacement on cardiac structure and function in a translational model, the ovariectomized cynomolgus monkey (Macaca fascicularis). Cynomolgus monkeys have a similar reproductive system and menstrual cycle as women and are a well‐established nonhuman primate model of surgical menopause.24 The study was designed to determine the effects of oral estradiol replacement on cardiac structure and function and to explore myocardial gene expression profiles in order to provide insight into potential molecular pathways involved in estradiol effects on the heart and risk for HFpEF in female primates.
Methods
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedures. This information is available from the corresponding author on reasonable request.
Animals
Subjects were 23 older female cynomolgus monkeys (18–25 years of age estimated from dentition) obtained through the Institut Pertanian Bogor, Indonesia, and represent the “early” treatment cohort of a larger study to investigate the effects of estradiol on atherosclerosis and other phenotypes when administered early versus late in the post‐ovariectomy period.25 The design of this study was a 2‐group, parallel arm design with treatment lasting 8 months. All monkeys were fed a moderately atherogenic diet beginning 6 months before bilateral ovariectomy. Monkeys were assigned to control (n=12) or estradiol (n=11) treatment groups 1 month after ovariectomy using a stratified randomization scheme based on social group, body weight, and plasma cholesterol levels at the end of the baseline period. The diet contained 35% of calories from predominantly saturated fat, 46% of calories from carbohydrate, 19% of calories from protein (casein and lactalbumin), and 0.20 mg cholesterol per calorie. Estradiol animals received a human equivalent dose of 1 mg/day oral micronized estradiol (1 mg/1800 calories of diet) for 8 months before necropsy. All procedures were approved by the Wake Forest University Animal Care and Use Committee and were conducted in compliance with state and federal laws, standards of the US Department of Health and Human Services, and regulations and guidelines established by the Wake Forest University Animal Care and Use Committee. Wake Forest University is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care.
Serum Estradiol and Cytokine Measurements
Blood samples were obtained before ovariectomy at baseline, 1 month after ovariectomy before the beginning of estradiol administration, and periodically throughout the study. Serum estradiol was determined on ether extracted serum samples from fasted animals using a radioimmunoassay from Diagnostic Systems Laboratories as previously described.26 Serum cytokine levels of MCP1 (monocyte chemoattractant protein 1), interleukin 6, and vascular endothelial growth factor α were assessed in samples obtained immediately before necropsy using Simple Plex® assays on the Ella ELISA platform (Protein Simple, San Jose) according to manufacturer instructions.
Echocardiography
At the end of the 8‐month treatment period, transthoracic echocardiograms were performed on monkeys in the left lateral decubitus position under general endotracheal anesthesia (ketamine induction [15 mg/kg, intramuscular] followed by 2% isoflurane) using a Philips 5500 Sonos echocardiograph and 12‐MHz probe (Philips Medical Systems, Andover, MA) by a single echosonographer (LG) blinded to treatment group. Images were obtained and analyzed as described previously27 in accordance with American Society of Echocardiography recommendations.28 In some animals, some echocardiographic measurements were unable to be obtained due to difficulty in imaging. Oxygen saturation and systemic blood pressures (arm cuff) were recorded periodically throughout the procedure.
Necropsy
At necropsy, the animals were deeply sedated with pentobarbital, euthanized, flushed with lactated Ringer's solution, and all major organ systems were assessed and preserved. The heart was removed, photographed, and evaluated for gross lesions, and a segment of the apical left ventricle was dissected and immediately flash frozen in liquid nitrogen. The remaining heart and the coronary arteries were perfusion fixed for 1 hour at 100 mm Hg pressure using 4% paraformaldehyde.
Histopathologic Analysis of Cardiovascular Tissues
Coronary artery atherosclerosis was determined from 5 blocks (each 3 mm in length) cut perpendicular to the long axis of the arteries, 3 serial blocks each from the left circumflex and the left anterior descending coronary arteries. Atherosclerosis was evaluated as described previously.29 Briefly, vascular tissues were embedded in paraffin, and 5‐μm sections were made and stained with Verhoeff–van Gieson's stain. The extent of atherosclerosis was measured as the cross‐sectional area of intimal lesions in each of the sections of the artery segments. Atherosclerotic plaque area (mm2) was determined by computer‐assisted histomorphometry using Image Pro Plus software (Media Cybernetics, Inc, Silver Spring, MD).
A full‐thickness equatorial section of LV free wall was stained using Masson's trichrome to estimate fibrosis extent. Slides were digitally imaged using a BX61VS microscope and VS120 virtual slide imaging system (Olympus Corporation, Tokyo, Japan). Collagen fraction was quantified using automated algorithms developed on the Visiopharm Integrator System (Visiopharm, Broomfield, CO). Briefly, whole LV sections were outlined to determine the region of interest and total pixel area. The trichrome‐stained blue area representing collagen was quantified after defining the appropriate range of blue pixel values, this area was subsequently divided by total section area to calculate collagen area fraction and derive percent collagen area.
Myocardial RNA Isolation and Microarray Analysis of Gene Expression
RNA was isolated from frozen tissue sections (10–30 mg) using AllPrep ® DNA/RNA/miRNA kits (Qiagen, Valencia, CA) according to a modified manufacturer protocol using 1% β‐mercaptoethanol. After extraction, the total RNA concentration was determined via NanoDrop 2000 (Thermo Scientific, Waltham, MA). RNA quality was assessed using RNA integrity numbers obtained through using a RNA 6000 Nanokit for the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). A cutoff RNA integrity number of ≥7.0 was considered to be sufficient for evaluation of global gene expression using HT‐12 v4 Expression BeadChip microarrays (Illumina, San Diego, CA). Genes identified within enriched gene groups were analyzed for correlations with significant echocardiographic and histologic outcomes. Causal network analysis in Ingenuity Pathway Analysis (Qiagen, Valencia, CA)30 was used to identify potential upstream regulators for differentially expressed genes.
Quantitative Real‐Time Reverse Transcription Polymerase Chain Reaction
Complementary DNA (cDNA) was generated from total RNA isolated from the frozen tissue sections using a high‐capacity cDNA archive kit (Applied Biosystems, Foster City, CA) as previously described.25 Cynomolgus monkey–specific TaqMan FAM‐MGB probes for calmodulin 1 (CALM1); ryanodine receptor 2 (RYR2); and collagen, type I, α1 (COL1A1), the structural counterpart of collagen, type I, α2 (COL1A2) (Applied Biosystems, Foster City, CA) were used for quantitative real‐time reverse transcription polymerase chain reaction validation of differentially expressed genes identified via microarray analyses on an ABI Prism 7500 Fast system (Applied Biosystems, Foster City, CA). All quantitative real‐time reverse transcription polymerase chain reaction data were normalized to GAPDH.
Statistical Analysis
All data were analyzed for normality, found to be normally distributed, and are presented as mean±SEM. Body weight, body mass index, and hemodynamic and echocardiographic measures were analyzed using the Student t test. Estradiol levels between groups and time points were analyzed using repeated measures ANOVA and Bonferroni post hoc tests. Correlation analyses were performed using Pearson r correlation coefficient testing. The level of significance was set at P<0.05. All analyses, save gene expression analyses, were performed using Statistica version 13.0 (Dell Software, Round Rock, TX). Gene expression output was analyzed using GenomeStudio (Illumina) and Genesifter software (PerkinElmer, Waltham, MA). Gene expression differences of ≥1.2‐fold change with a Benjamini‐Hochberg31 false discovery rate <0.05 were considered significant. Gene ontology enrichment analysis using DAVID32 was used to obtain top enriched gene groups and are presented as an enrichment score based on an Expression Analysis Systematic Explorer score of P<0.05.
Results
Baseline and Posttreatment Measurements
At baseline, there were no differences between groups in traditional risk markers for cardiovascular disease, including total plasma cholesterol (TPC), high‐density lipoprotein cholesterol (HDLc), non–HDL‐C, triglycerides, and the TPC/HDL‐C ratio (all P>0.39).25 Ovariectomy resulted in significantly decreased serum estradiol concentrations in all animals measured 1 month after surgery (P<0.00002) (Table 1). Treatment of ovariectomized monkeys with a human equivalent dose of 1 mg estradiol/day resulted in mean estradiol concentrations comparable to that of postmenopausal women taking 1 mg micronized estradiol/day33 and significantly higher than that of the control monkeys in which estradiol levels were below the detection limit of the assay throughout the study. Estradiol treatment produced no significant effects on body weight, body mass index, systolic blood pressure, diastolic blood pressure, or heart rate (Table 2).
Table 1.
Effects of Ovariectomy (OVX) and Estradiol Treatment on Serum Estradiol Concentrations
| Group | Pre‐OVX (pg/mL) | Post‐OVX Pre‐Estradiol (pg/mL) | Post OVX 1‐Month Estradiol (pg/mL) | Post OVX 4‐Month Estradiol (pg/mL) | Post OVX 8‐Month Estradiol (pg/mL) |
|---|---|---|---|---|---|
| Control | 25.5±7.34 | 0.68±0.25 | 0.67±0.15 | 0.83±0.13 | 1.29±0.29 |
| Estradiol | 24.4±6.81 | 1.23±0.55 | 38.9±6.70 | 42.0±6.22 | 47.0±5.56 |
| P Value | 1.00 | 1.00 | <0.0001 | <0.0001 | <0.0001 |
There were no significant differences between ovariectomized (control) and ovariectomized animals receiving 1 mg/day oral estradiol (estradiol) before ovariectomy or at 1 month after ovariectomy before estradiol treatment initiation. Estradiol animals receiving 1 mg/day oral estradiol treatment significantly increased serum estradiol levels compared to control animals. Control (n=12), estradiol (n=11).
Table 2.
Effects of Estradiol Treatment on Body Weight, Hemodynamic, and Echocardiographic Parameters
| Measure | Control | n | Estradiol | n | P Value |
|---|---|---|---|---|---|
| Body weight, kg | 2.97 (0.17) | 12 | 3.08 (0.24) | 11 | 0.72 |
| Body mass index (body weight/trunk length)2 | 41.9 (1.96) | 12 | 42.5 (2.90) | 11 | 0.86 |
| Heart rate, beats/min | 150.0 (5.54) | 12 | 154.4 (6.47) | 11 | 0.61 |
| Systolic blood pressure, mm Hg | 90.8 (4.64) | 10 | 87.1 (3.91) | 10 | 0.55 |
| Diastolic blood pressure, mm Hg | 63.9 (10.6) | 10 | 53.5 (3.74) | 10 | 0.37 |
| Left atrial diameter, cm | 1.01 (0.03) | 11 | 0.98 (0.02) | 11 | 0.49 |
| Interventricular septal thickness, cm | 0.40 (0.02) | 8 | 0.39 (0.01) | 10 | 0.88 |
| Posterior wall thickness, cm | 0.41 (0.02) | 8 | 0.41 (0.02) | 10 | 0.80 |
| LV internal diameter end diastole, cm | 0.99 (0.05) | 6 | 1.12 (0.06) | 7 | 0.21 |
| LV internal diameter end systole, cm | 0.72 (0.06) | 6 | 0.80 (0.06) | 8 | 0.42 |
| Fractional shortening, % | 30.54 (4.15) | 11 | 31.16 (3.90) | 7 | 0.45 |
| Transmitral E velocity, cm/s | 38.56 (3.52) | 12 | 59.89 (3.71) | 11 | 0.0004 |
| E wave deceleration time, s | 0.087 (0.004) | 12 | 0.079 (0.004) | 11 | 0.20 |
| E wave deceleration slope, cm/s2 | 4.61 (0.52) | 12 | 7.99 (0.72) | 11 | 0.0009 |
| Transmitral A velocity, cm/s | 36.46 (5.13) | 12 | 54.44 (3.65) | 11 | 0.01 |
| E/A | 1.22 (0.15) | 12 | 1.15 (0.09) | 11 | 0.68 |
| Early mitral annular descent (e′) lateral annulus, cm/s | 3.41 (0.37) | 12 | 5.24 (0.27) | 11 | 0.0008 |
| Late mitral annular descent (a′) lateral annulus, cm/s | 3.22 (0.37) | 12 | 4.81 (0.41) | 11 | 0.008 |
| E/e′ lateral annulus | 11.78 (0.67) | 12 | 11.70 (0.69) | 11 | 0.94 |
| e′/a′ lateral annulus | 1.18 (0.12) | 12 | 1.18 (0.11) | 11 | 0.98 |
| Aortic diameter end diastole, cm | 0.86 (0.04) | 11 | 0.96 (0.03) | 10 | 0.08 |
| Aortic diameter end systole, cm | 0.84 (0.05) | 11 | 0.90 (0.02) | 10 | 0.27 |
Estradiol animals had higher transmitral early filling velocity (E) and deceleration slope, transmitral late filling velocity (A), early mitral annular descent velocity (e′), and late mitral annular descent velocity (a′) compared to control animals. Mean body weight, body mass index, heart rate, systolic blood pressure, and diastolic blood pressure were similar between groups. No differences were observed between groups in any echocardiographic structural or systolic functional parameter. LV indicates left ventricle. All data expressed as mean (SEM).
Doppler‐Echocardiographic Measures
Echocardiography was used to evaluate cardiac structural and functional differences between estradiol and control animals after 8 months of estradiol or vehicle treatment (Table 2). Doppler echocardiography showed that estradiol‐treated monkeys had higher transmitral early filling velocity (E) and deceleration slope, transmitral late filling velocity (A), early mitral annular descent velocity (e′), and late mitral annular descent velocity (a′) compared to control animals (all P<0.05) (Figure 1). E/e′, E/A, and e′/a′ ratios were not significantly different between groups (Table 2). No differences in left atrial diameter, interventricular septal thickness, posterior wall thickness, LV internal diameters, fractional shortening, or aortic diameters were observed between groups.
Figure 1.
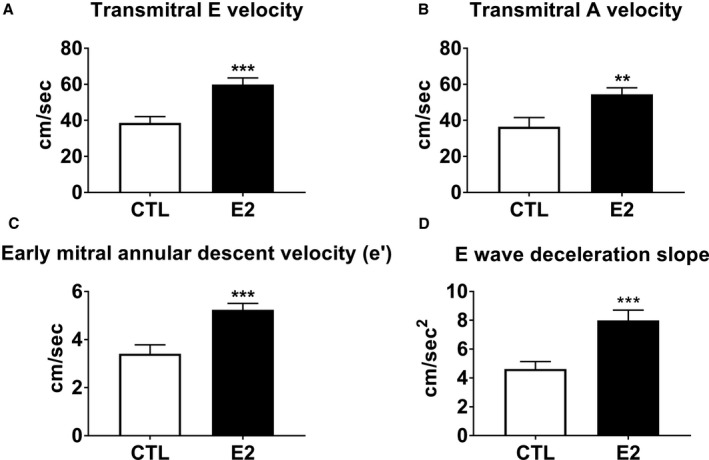
Effects of short‐term estradiol replacement on left ventricular diastolic function. Estradiol‐treated (E2) animals had significantly higher transmitral early (E) and late (A) transmitral velocities (A and B, respectively), early (e′) mitral annular velocities (C), and early (E) deceleration slope (D) than ovariectomized control (CTL) animals. Data represent mean±SEM. **P<0.01, ***P<0.0001 compared with CTL.
Histopathologic Analyses
Coronary artery atherosclerosis and myocardial collagen content were evaluated to assess the effects on the heart of estrogen treatment after ovariectomy. Coronary artery atherosclerosis (mean intimal area of the left anterior descending and left circumflex arteries) was not different between groups after the short, 8‐month course of estradiol exposure (P=0.63) (data not shown). Myocardial collagen content, assessed by histomorphometric analysis of Masson's trichrome‐stained LV free wall (Figure 2), was higher in control relative to estradiol‐treated monkeys (7.02±0.49% versus 5.52±0.45%, respectively; P=0.04) (Figure 3).
Figure 2.
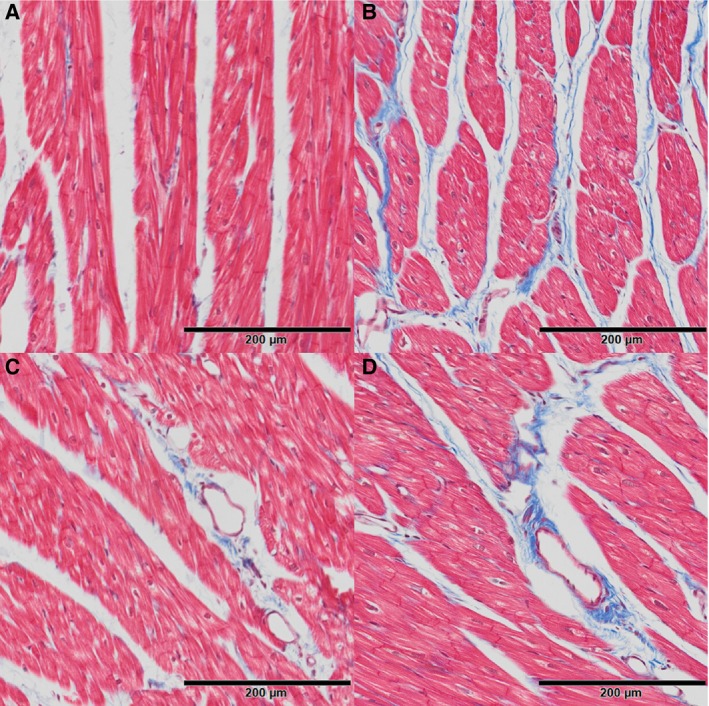
Effects of short term estradiol replacement on myocardial collagen deposition in estradiol and control animals. Estradiol treated animals had lower interstitial and perivascular collagen (blue) staining (A and C, respectively) compared to ovariectomized controls (B and D) on Masson's trichrome‐stained sections of left ventricular free wall.
Figure 3.
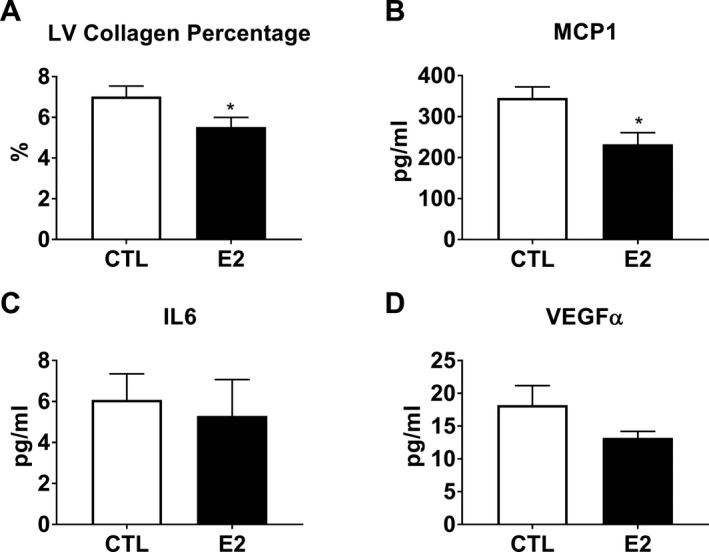
Effects of short term estradiol replacement on myocardial collagen content and circulating biomarkers. Estradiol treated (E2) animals had significantly lower left ventricular (LV) free wall collagen (A) and serum monocyte chemoattractant protein 1 (MCP1) levels (B) compared to ovariectomized controls (CTL). Interleukin‐6 (IL6) (C) and vascular endothelial growth factor alpha (VEGFα) (D) were not significantly different (P=0.73 and 0.22 respectively). Data represent mean±SEM. *P<0.05 compared with controls to controls.
Serum Cytokine Quantification
Selected serum cytokines were evaluated to assess the effects of estradiol on systemic inflammation. Estradiol‐treated monkeys had lower circulating levels of MCP1 compared to control animals (P=0.012), while there was no effect on serum interleukin‐6 and vascular endothelial growth factorα levels (P=0.73 and 0.22, respectively) (Figure 3).
Gene Expression
Global gene expression analysis of LV myocardium was performed to investigate potential pathways and mechanisms underlying echocardiographic and histomorphometric phenotypes. Analyses were conducted using a 1.2‐fold change threshold and a Benjamini‐Hochberg false discovery rate of P<0.05. Estradiol treatment resulted in differential expression of 40 unique genes (Table 3) that clearly delineated estradiol and control groups in an unsupervised Euclidian hierarchical cluster analysis (Figure 4). Gene ontology enrichment analysis of differentially expressed genes using DAVID version 6.7 revealed that the top 5 most significantly estradiol upregulated categories (P<0.05) were Regulation of Release of Sequestered Calcium Ion into Cytosol by Sarcoplasmic Reticulum based on calmodulin 1 (CALM1) and ryanodine receptor 2 (RYR2) gene expression; Muscle Contraction based on Aldolase A, fructose‐bisphosphate (ALDOA), Ryanodine receptor 2 (RYR2) and Smoothelin (SMTN) gene expression, Muscle System Process based on ALDOA, RYR2, and SMTN gene expression, and Cytoskeletal Protein Binding based on Enah/Vasp‐like (EVL), ALDOA, CALM1, and SMTN gene expression. The top 5 most significantly estradiol downregulated categories (highest enrichment scores, P<0.05) were Extracellular Matrix based on TIMP metallopeptidase inhibitor 1 (TIMP1), collagen, type I, α2 (COL1A2), matrix Gla protein (MGP), microfibrillar‐associated protein 5 (MFAP5), and thrombospondin 1 gene expression (THBS1); Structural Molecule Activity based on COL1A2, MGP, MFAP5, ribosomal protein S29 (RPS29), and THBS gene expression; Response to Calcium Ion based on MGP, thioredoxin interacting protein (TXNIP), and THBS1 gene expression; Extracellular Region Part based on TIMP1, chemokine (C‐X‐C motif) ligand 12 (CXCL12), COL1A2, MGP, MFAP5, and THBS1 gene expression; and Extracellular Matrix Structural Constituent based on COL1A2, MGP, and MFAP5 gene expression (Figure 4). Quantitative real‐time reverse transcription polymerase chain reaction analysis of CALM1, RYR2, and COL1A1 revealed similar magnitude and direction fold changes as those identified in the microarray analyses (Figure 5).
Table 3.
Estradiol Regulated Genes in LV Myocardium
| Gene | Gene ID | Fold Difference | Raw P | FDR |
|---|---|---|---|---|
| Brain expressed, X‐linked 1 | BEX1 | 1.45 | 0.003 | 0.038 |
| von Willebrand factor | VWF | 1.37 | 0.007 | 0.045 |
| CD74 molecule, major histocompatibility complex, class II invariant chain | CD74 | 1.34 | 0.000 | 0.022 |
| Ryanodine receptor 2 | RYR2 | 1.34 | 0.004 | 0.041 |
| Nucleobindin 1 | NUCB1 | 1.28 | 0.012 | 0.047 |
| Calcium and integrin binding family member 2 | CIB2 | 1.30 | 0.002 | 0.036 |
| Nuclear fragile X mental retardation protein interacting protein 2 | NUFIP2 | 1.26 | 0.011 | 0.047 |
| Aldolase A, fructose‐bisphosphate | ALDOA | 1.26 | 0.004 | 0.044 |
| Chromosome 14 open reading frame 180 | C14orf180 | 1.27 | 0.002 | 0.036 |
| Tetraspanin 9 | TSPAN9 | 1.23 | 0.011 | 0.047 |
| Acyl‐CoA synthetase short‐chain family member 2 | ACSS2 | 1.24 | 0.011 | 0.047 |
| AF4/FMR2 family, member 3 | AFF3 | 1.24 | 0.001 | 0.022 |
| Ankyrin repeat and SOCS box‐containing 10 | ASB10 | 1.25 | 0.005 | 0.044 |
| Sirtuin 4 | SIRT4 | 1.25 | 0.007 | 0.045 |
| Poly(rC) binding protein 2 | PCBP2 | 1.22 | 0.006 | 0.045 |
| Enah/Vasp‐like | EVL | 1.22 | 0.002 | 0.036 |
| Protein phosphatase 2 regulatory subunit B | PPP2R3B | 1.21 | 0.007 | 0.045 |
| Beta‐1,3‐N‐acetylgalactosaminyltransferase 2 | B3GALNT2 | 1.21 | 0.001 | 0.022 |
| Pyruvate kinase, muscle | PKM2 | 1.21 | 0.004 | 0.041 |
| Smoothelin | SMTN | 1.21 | 0.005 | 0.044 |
| Calmodulin 1 | CALM1 | 1.21 | 0.011 | 0.047 |
| Ribonuclease P/MRP 40 kDa subunit | RPP40 | 1.22 | 0.009 | 0.047 |
| TIMP metallopeptidase inhibitor 1 | TIMP1 | −1.20 | 0.007 | 0.045 |
| Ribosomal protein S26 | RPS26 | −1.22 | 0.011 | 0.047 |
| Actin, γ2, smooth muscle, enteric | ACTG2 | −1.22 | 0.006 | 0.045 |
| Thrombospondin 1 | THBS1 | −1.23 | 0.011 | 0.047 |
| Leptin receptor overlapping transcript‐like 1 | LEPROTL1 | −1.25 | 0.004 | 0.043 |
| Chromosome 11 open reading frame 54 | C11orf54 | −1.25 | 0.003 | 0.038 |
| Carnitine palmitoyltransferase 2 | CPT2 | −1.26 | 0.001 | 0.022 |
| Chemokine (C‐X‐C motif) ligand 12 | CXCL12 | −1.28 | 0.006 | 0.045 |
| S100 calcium binding protein A4 | S100A4 | −1.28 | 0.001 | 0.027 |
| Thioredoxin interacting protein | TXNIP | −1.32 | 0.007 | 0.045 |
| RAB12, member RAS oncogene family | RAB12 | −1.35 | 0.005 | 0.044 |
| Peptidase inhibitor 16 | PI16 | −1.39 | 0.011 | 0.047 |
| Microfibrillar associated protein 5 | MFAP5 | −1.40 | 0.009 | 0.047 |
| Collagen, type I, α2 | COL1A2 | −1.51 | 0.007 | 0.045 |
| Matrix Gla protein | MGP | −1.65 | 0.005 | 0.044 |
| Pyruvate dehydrogenase kinase, isozyme 4 | PDK4 | −1.88 | 0.011 | 0.047 |
| Ribosomal protein S29 | RPS29 | −2.78 | 0.000 | 0.008 |
Genes with a Benjamini‐Hochberg FDR <0.05 and an estradiol induced fold change greater than 1.2 were considered significant. FDR indicates false discovery rate.
Figure 4.
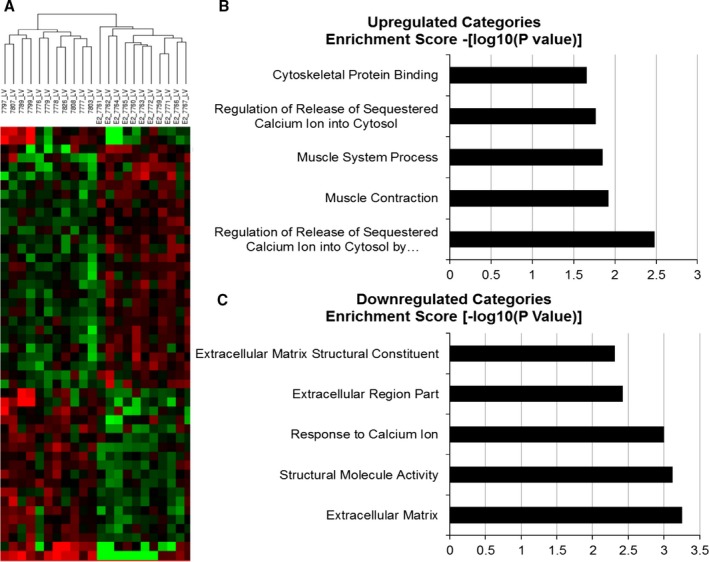
Effects of short term estradiol replacement on myocardial gene expression. Euclidian hierarchical clustering of animals using differentially expressed genes (1.2‐fold change with a Benjamini and Hochberg false discovery rate P<0.05) separated estradiol‐treated (estradiol) animals from ovariectomized control animals (A). Gene ontology enrichment analysis using DAVID indicated that the genes that were upregulated by estradiol treatment were categorized as being involved in calcium homeostasis and muscle contraction (B) and that genes downregulated by estradiol treatment were categorized as being involved in extracellular matrix deposition (C).
Figure 5.
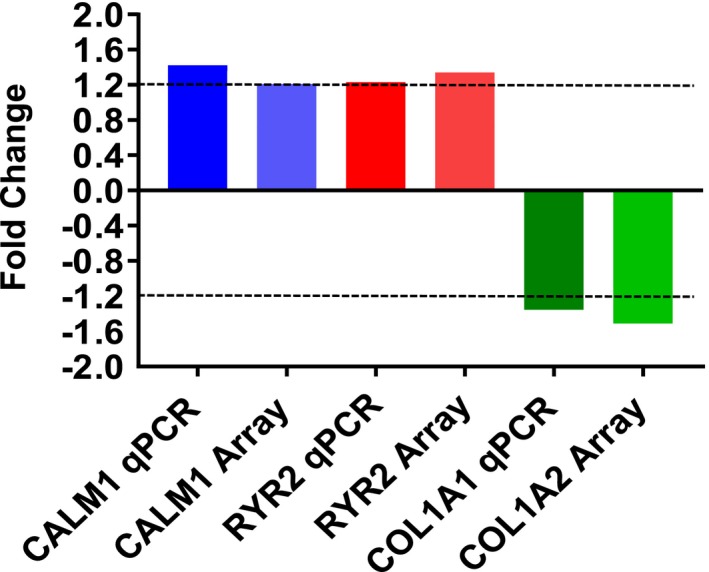
Comparison and validation of microarray (Array) differentially expressed genes using quantitative real‐time reverse transcription polymerase chain reaction (qPCR). Fold change differences of calmodulin 1 (CALM1), ryanodine receptor 2 (RYR2) and collagen, type I, α1 and 2 (COL1A1, COL1A2) between qPCR and microarray were of similar direction and magnitude.
Relationships Between Differentially Expressed Myocardial Genes and Key Phenotypes
Pearson correlation coefficient analysis across all animals using genes identified by gene ontology enrichment revealed significant correlations of ALDOA, CALM1, COL1A2, MGP, RPS29, SMTN, THBS1, TIMP1, and TXNIP with echocardiographic (E, E slope, A, e′, a′), histomorphometric (myocardial collagen %), and cytokine (serum MCP1) findings (Table 4 and Figure 6). Additionally, when each group was considered separately, CALM1 and SMTN were significantly correlated with Doppler echocardiographic (A, e′) and cytokine (MCP1) findings in control animals (Table 5) and COL1A2, RPS29, and RYR2 were significantly correlated with A, a′, and MCP1 in estradiol animals (Table 6). Scatterplots of variables with significant relationships are illustrated in Figures 7, 8, 9, 10, 11, 12 through 13.
Table 4.
Relationships Between Expression Levels of Differentially Expressed Genes and Key Phenotypes Across Control and Estradiol Subjects
| Variable | Gene | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ALDOA | CALM1 | COL1A2 | MGP | RPS29 | RYR2 | SMTN | THBS1 | TIMP1 | TXNIP | |
| Transmitral E velocity | 0.44a | 0.17 | −0.45a | −0.42a | −0.58b | 0.34 | 0.38c | −0.47a | −0.36c | −0.42c |
| Transmitral A velocity | 0.53a | 0.49a | −0.12 | −0.20 | −0.49a | 0.41c | 0.52a | −0.20 | −0.08 | −0.08 |
| E wave deceleration slope | 0.31 | 0.26 | −0.29 | −0.22 | −0.50a | 0.46a | 0.35c | −0.44a | −0.30 | −0.30 |
| Early mitral annular descent (e′) lateral annulus | 0.34 | 0.17 | −0.41c | −0.41c | −0.58b | 0.29 | 0.48a | −0.42 | −0.42c | −0.40c |
| Late mitral annular descent (a′) lateral annulus | 0.29 | 0.25 | −0.40c | −0.36c | −0.60b | 0.46a | 0.34 | −0.24 | −0.34 | −0.22 |
| LV collagen percentage, % | −0.21 | 0.02 | 0.31 | 0.58b | 0.22 | −0.13 | −0.17 | 0.33 | 0.25 | 0.47a |
| Serum MCP1 | −0.08 | 0.02 | 0.35c | 0.40c | 0.53a | −0.40c | −0.31 | 0.10 | 0.58b | 0.35c |
ALDOA indicates aldolase A, fructose‐bisphosphate; CALM1, calmodulin 1; CoA, coenzyme A; COL1A2, collagen, type I, α2; LV, left ventricular; MCP1, monocyte chemoattractant protein 1; MGP, matrix Gla protein; RPS29, ribosomal protein subunit 29; RYR2, ryanodine receptor 2; SMTN, smoothelin; THBS1, thrombospondin 1; TIMP1, TIMP metallopeptidase inhibitor; TXNIP, thioredoxin interacting protein.
P<0.05.
P<0.01.
P<0.10.
Figure 6.
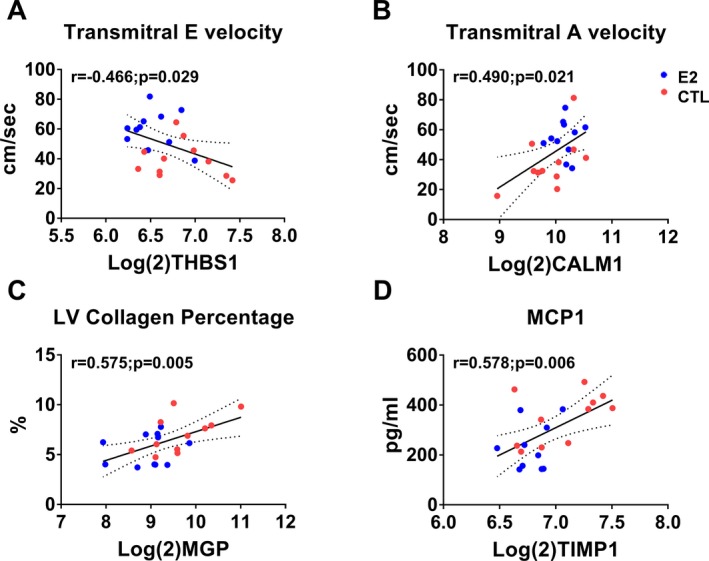
Scatterplots of expression levels of differentially expressed genes (DEGs) and echocardiographic and biomolecular phenotypes in estradiol and control (CTL) animals. Thrombospondin 1 (THBS1) negatively correlated with transmitral E velocity (A), Calmodulin 1 (CALM1) positively correlated with transmitral A velocity (B), matrix Gla protein (MGP) correlated positively with left ventricular collagen percentage (C), and TIMP metallopeptidase inhibitor 1 (TIMP1) correlated positively with monocyte chemoattractant protein 1 (MCP1) (D). Red symbols=control; blue symbols=estradiol treated.
Table 5.
Relationships Between Expression Levels of Differentially Expressed Genes and Key Phenotypes in Control Subjects
| Control Group Variable | Gene | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ALDOA | CALM1 | COL1A2 | MGP | RPS29 | RYR2 | SMTN | THBS1 | TIMP1 | TXNIP | |
| Transmitral E velocity | 0.39 | −0.06 | −0.30 | −0.18 | −0.03 | −0.23 | 0.51 | −0.27 | −0.04 | −0.20 |
| Transmitral A velocity | 0.53a | 0.53a | 0.25 | 0.10 | 0.07 | 0.11 | 0.66b | 0.01 | 0.35 | 0.18 |
| E wave deceleration slope | 0.30 | 0.09 | 0.08 | 0.30 | −0.09 | −0.22 | 0.40 | −0.22 | 0.20 | 0.17 |
| Early mitral annular descent | 0.31 | 0.07 | −0.15 | −0.21 | −0.07 | −0.27 | 0.63b | −0.10 | −0.06 | −0.09 |
| Late mitral annular descent | 0.06 | 0.29 | 0.01 | −0.05 | −0.13 | 0.14 | 0.50 | 0.08 | 0.07 | 0.11 |
| LV collagen percentage, % | −0.03 | 0.15 | 0.26 | 0.60 | −0.36 | 0.01 | −0.17 | 0.14 | 0.17 | 0.34 |
| Serum MCP1 | 0.37 | 0.62b | 0.19 | 0.32 | 0.48 | 0.51 | 0.10 | −0.13 | 0.52a | 0.54a |
ALDOA indicates aldolase A, fructose‐bisphosphate; CALM1, calmodulin 1; COL1A2, collagen, type I, α2; LV, left ventricular; MCP1, monocyte chemoattractant protein 1; MGP, matrix Gla protein; RPS29, ribosomal protein subunit 29; RYR2, ryanodine receptor 2; SMTN, smoothelin; THBS1, thrombospondin 1; TIMP1, TIMP metallopeptidase inhibitor; TXNIP, thioredoxin interacting protein.
P<0.05.
P<0.10.
Table 6.
Relationships Between Expression Levels of DEGs and Key Phenotypes in Estradiol Subjects
| estradiol Group Variable | Gene | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ALDOA | CALM1 | COL1A2 | MGP | RPS29 | RYR2 | SMTN | THBS1 | TIMP1 | TXNIP | |
| Transmitral E velocity | −0.02 | −0.13 | −0.17 | −0.23 | −0.32 | 0.15 | −0.32 | −0.22 | −0.07 | −0.06 |
| Transmitral A velocity | 0.13 | 0.05 | −0.28 | −0.17 | −0.60a | 0.33 | −0.10 | 0.11 | −0.16 | 0.30 |
| E wave deceleration slope | −0.22 | 0.04 | −0.28 | −0.21 | −0.04 | 0.40 | −0.16 | −0.25 | −0.25 | −0.14 |
| Early mitral annular descent | −0.38 | −0.44 | −0.32 | −0.06 | −0.41 | 0.12 | −0.31 | −0.40 | −0.47 | −0.20 |
| Late mitral annular descent | 0.08 | −0.21 | −0.76a | −0.49 | −0.60a | 0.34 | −0.22 | −0.15 | −0.54b | −0.03 |
| LV collagen percentage, % | 0.01 | 0.29 | −0.07 | 0.20 | 0.05 | 0.21 | 0.29 | 0.26 | −0.30 | 0.32 |
| Serum MCP1 | 0.04 | −0.51 | 0.08 | −0.07 | −0.12 | −0.69a | −0.40 | −0.29 | 0.28 | −0.48 |
ALDOA indicates aldolase A, fructose‐bisphosphate; CALM1, calmodulin 1; COL1A2, collagen, type I, α2; DEG, differentially expressed genes; LV, left ventricular; MCP1, monocyte chemoattractant protein 1; MGP, matrix Gla protein; RPS29, ribosomal protein subunit 29; RYR2, ryanodine receptor 2; SMTN, smoothelin; THBS1, thrombospondin 1; TIMP1, TIMP metallopeptidase inhibitor; TXNIP, thioredoxin interacting protein.
P<0.05.
P<0.10.
Figure 7.
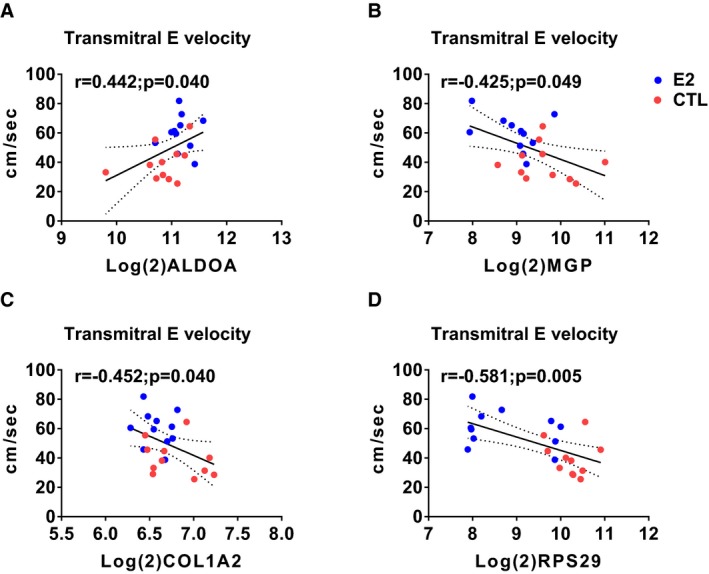
Scatterplots of expression levels of differently expressed genes (DEGs) and transmitral E velocity. Aldolase A, fructose‐bisphosphate (ALDOA) positively correlated with transmitral E velocity (A) whereas matrix Gla protein (MGP), collagen, type I, α2 (COL1A2), and ribosomal protein subunit 29 (RPS29) correlated negatively (B through D, respectively). Red symbols=control; blue symbols=estradiol treated.
Figure 8.
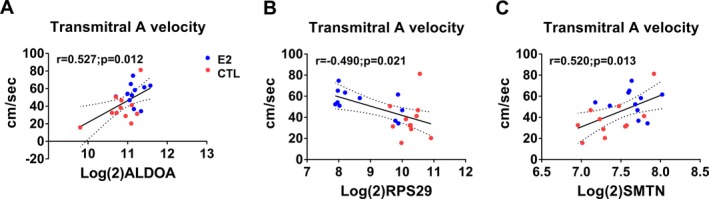
Scatterplots of expression levels of differently expressed genes (DEGs) and transmitral A velocity. Aldolase A, fructose‐bisphosphate(ALDOA) (A) and smoothelin (SMTN) (B) positively correlated with transmitral A velocity whereas ribosomal protein subunit 29 (RPS29) correlated negatively (C). Red symbols=control; blue symbols=estradiol treated.
Figure 9.
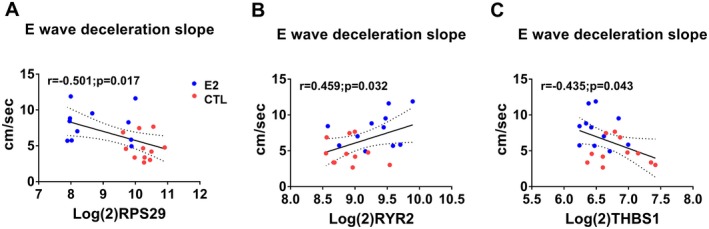
Scatterplots of expression levels of differently expressed genes (DEGs) and E wave deceleration slope. Ribosomal protein subunit 29 (RPS29) (A) and Thrombospondin 1 (THBS1) (C) negatively correlated with E wave deceleration slope whereas Ryanodine receptor 2 (RYR2) correlated positively (B). Red symbols=Control, Blue symbols=estradiol treated.
Figure 10.
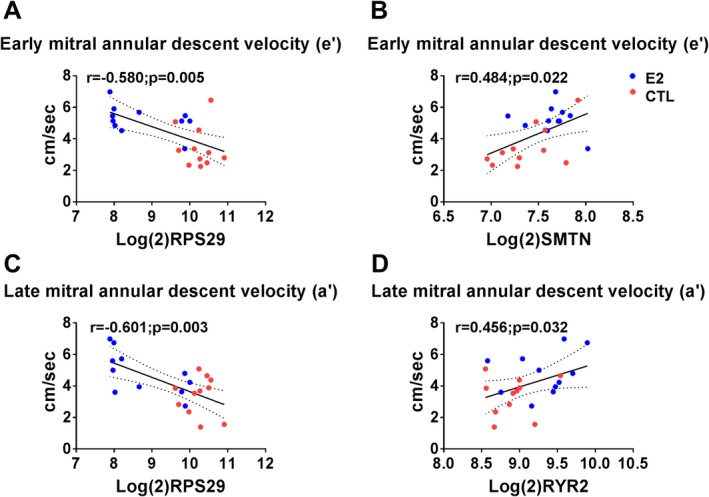
Scatterplots of expression levels of differently expressed genes (DEGs) and early and late mitral annular descent. Ribosomal protein subunit 29 (RPS29) correlated negatively with early (e′) and late (a′) mitral annular descent velocities (A and C, respectively). Smoothelin (SMTN) correlated positively with e′ (B) and Rryanodine receptor 2 (RYR2) correlated positively with a′ (D). Red symbols=control; blue symbols=estradiol treated.
Figure 11.
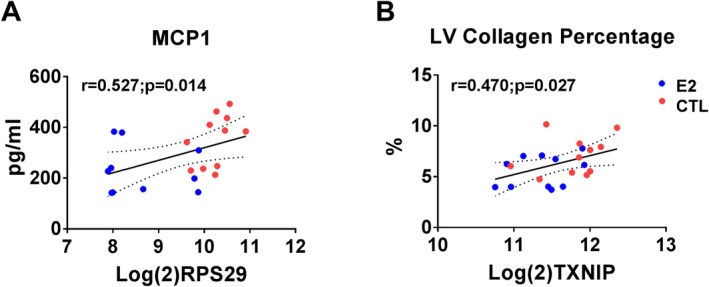
Scatterplots of expression levels of differently expressed genes (DEGs) and biomolecular phenotypes. Ribosomal protein subunit 29 (RPS29) positively correlated with monocyte chemoattractant protein 1 (MCP1) (A) and thioredoxin interacting protein (TXNIP) positively correlated with left ventricular (LV) collagen percentage (B). Red symbols=control; blue symbols=estradiol treated.
Figure 12.
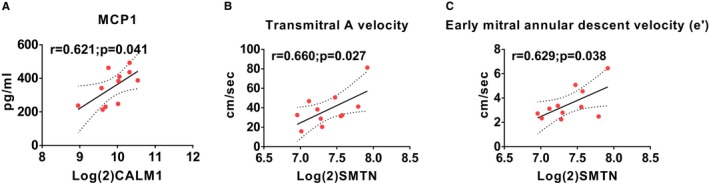
Scatterplots of expression levels of differently expressed genes (DEGs) and selected phenotypes in control animals. Calmodulin 1 (CALM1) correlated positively with monocyte chemoattractant protein 1 (MCP1) (A) and Smoothelin (SMTN) correlated positively with transmitral A velocity (B) and early mitral annular descent velocity (e′) (C).
Figure 13.
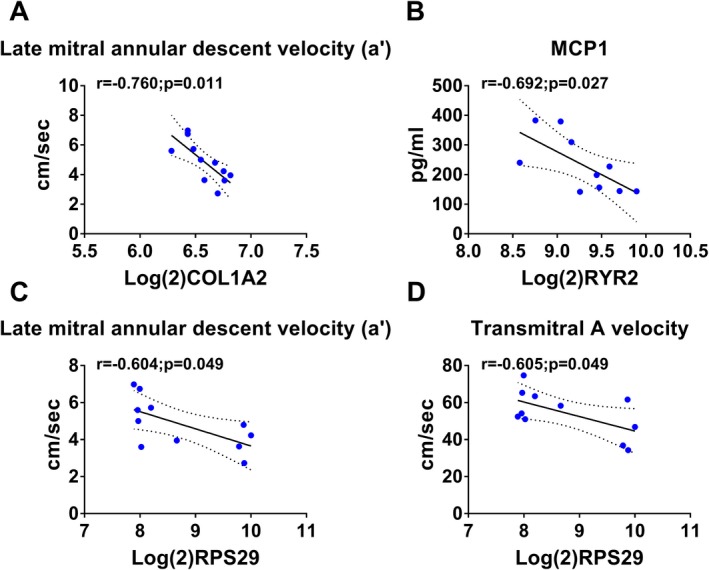
Scatterplots of expression levels of differently expressed genes (DEGs) and selected phenotypes in estradiol animals. Collagen, type I, α2 (COL1A2) (A) and ribosomal protein subunit 29 (RPS29) (C) correlated negatively with late mitral annular descent velocity (a′). Ryanodine receptor 2 (RYR2) correlated negatively with monocyte chemoattractant protein 1 (MCP1) (B) and RPS29 correlated negatively with transmitral A velocity (D).
Causal network analysis performed in Ingenuity Pathway Analysis using all differentially expressed genes identified NFκB inhibitor α as a significant mediator of estradiol effects on the LV myocardium based on differential gene expression of COL1A2, TIMP1, CXCL12, and MGP, between estradiol and control animals (Figure 14).
Figure 14.
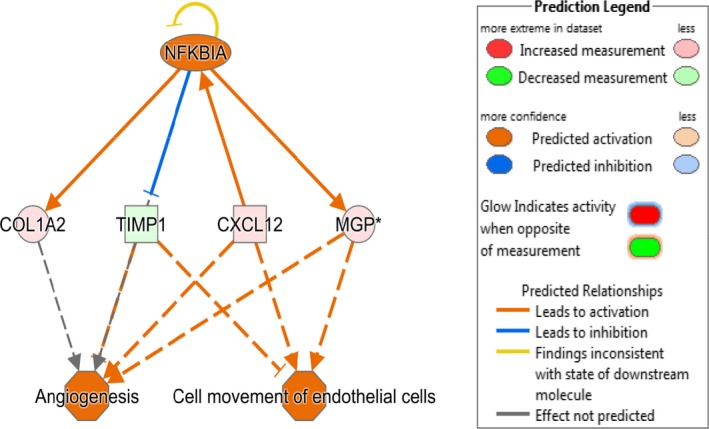
Causal network analysis of differentially expressed left ventricular (LV) myocardial genes. Ingenuity Pathway Analysis (IPA) was used to identify causal networks implicated in estradiol action. NFκB inhibitor αwas identified as an activated upstream regulator of differentially expressed genes collagen, type I, α2 (COL1A2), TIMP metallopeptidase inhibitor 1 (TIMP1), Chemokine (C‐X‐C motif) ligand 12, (CXCL12), and matrix Gla protein (MGP). The expected effects exerted by these genes include increased angiogenesis and movement of endothelial cells in estradiol treated (estradiol) animals compared to ovariectomized control (CTL) animals.
Discussion
Estrogen treatment initiated soon after induction of surgical menopause attenuated early LVDD in ovariectomized female monkeys. Estrogen improved Doppler indices of early and late filling and increased tissue Doppler velocities of early and late mitral annular descent. Interestingly, E/e′ was not significantly different, as the magnitude and direction of change was similar in both groups, suggesting no differences in filling pressures. Echocardiographic systolic and structural measures as well as systolic and diastolic blood pressure were not significantly different between groups, suggesting that mechanisms behind the lusitropic effects were independent of systolic function, blood pressure, and left ventricle size.
Estrogen and Cardiac Fibrosis
In order to investigate potential mechanisms underlying estrogen's effect on myocardial function, we assessed LV myocardial gene expression profiles. This experiment was originally designed with power to detect the effects of estradiol on arterial cell composition and inflammatory gene expression published elsewhere by our laboratory.25 However, this study's sample size is also considered sufficiently powered to detect changes using microarrays.34 Compared to control monkeys, estradiol‐treated monkeys had lower expression of genes encoding extracellular matrix (ECM) components (COL1A2, MFAP5, and MGP) and ECM deposition promoters (CXCL12, THBS1, TIMP1, S100A4, and RPS29). Expression of these genes was significantly correlated with numerous echocardiographic, histomorphometric, and cytokine phenotypes. Circulating biomarkers for both TIMP1 and COL1A2 expression have been documented as increased in serum/plasma from HFpEF patients; MGP expression is increased in cardiac myocytes and fibroblasts undergoing hypertrophy35; THBS1 is an activator of transforming growth factor β, and S100A4, also known as fibroblast specific protein 1, suppresses p53 in cardiac fibroblasts, resulting in cell proliferation and collagen production.36 These findings suggest that estradiol treatment inhibited cardiac fibrosis–promoting pathways, perhaps in part mediated by increased NFκB inhibitor α activity as identified by Ingenuity Pathway Analysis Causal Network Analysis. Image analysis of Masson's trichrome‐stained left ventricle sections revealed that control animals had significantly higher myocardial collagen staining than estradiol‐treated animals, consistent with the observed expression levels of key ECM components and modulators. Increased cardiac fibrosis is observed in patients with HFpEF and LVDD37 and has been demonstrated to contribute to diastolic dysfunction by increasing myocardial stiffness.37, 38, 39
Cardiac fibrosis develops when cardiac fibroblasts are activated to myofibroblasts. Putative mechanisms underlying this include reduced nitric oxide bioavailability40 and the presence of infiltrating monocytes and macrophages that secrete proinflammatory and profibrotic mediators including monocyte chemoattractant protein 1 (MCP1), tumor necrosis factor α, intercellular adhesion molecule‐1, and transforming growth factor β. Synthetic myofibroblasts increase production of fibrillar collagen, extracellular matrix, and tissue inhibitors of metalloproteinases and decrease production of degradative matrix metalloproteinases, causing unbalanced ECM turnover and ultimately leading to fibrosis. Estrogen has been shown to inhibit cardiac fibroblast proliferation and decrease cardiac fibroblast collagen production in vitro,41 perhaps through genomic (ERα, ERβ) and/or nongenomic (GPER) means via cAMP and protein kinase A signaling and downregulation of the cell cycle genes cyclin‐dependent kinase 1 and cyclin B1, respectively.42, 43
Estrogen is associated with protection from coronary heart disease and atherosclerosis in humans and animals, partially through beneficially influencing plasma lipoprotein levels.44, 45 We previously reported that there were no baseline differences in plasma triglycerides, HDL‐C, non–HDL‐C, TPC, or TPC/HDL‐C ratio, but that plasma TPC and non–HDL‐C concentrations were significantly decreased in estradiol treated animals compared to control animals after treatment (P<0.05).25 We found no significant differences in coronary artery atherosclerotic plaque area, consistent with the short study duration. Nevertheless, using this cohort of animals, we previously observed significant effects of estradiol on expression of key target genes in carotid arteries, including decreased expression of markers of macrophages, T cells, and other inflammatory cells in the arterial wall and lower levels of expression of proinflammatory genes such as MCP1, interferon‐γ, tumor necrosis factor α, collagen type I and matrix metalloproteinase 9.25 Atheroprotective effects of estrogen may be mediated in part through a reduction in vascular/endothelial MCP1 expression and decreased monocytic infiltration into arterial walls.46 Elevated serum MCP1 levels have been noted in patients with HFpEF and are thought to be a driver of macrophage infiltration and accumulation in the heart resulting in cardiac fibrosis.47 We demonstrated that estradiol‐treated animals had decreased serum MCP1 levels compared to controls, supporting the idea that estradiol treatment potentially reduced systemic and cardiac inflammation, and inhibited ECM deposition indicated by our LV Masson's trichrome analysis.
Estrogen and Cardiomyocyte Calcium Handling
The transcriptional profile in estradiol‐treated myocardium is consistent with maintenance of cardiomyocyte calcium homeostasis and contractility via increased expression of genes associated with sarcoplasmic reticulum calcium ion sensing, regulation (CALM1, PPP2R3B, CIB2 and NUCB1), handling (RYR2), and muscle contraction (ALDOA, EVL, SMTN). These findings are particularly interesting, as ventricular modeling studies by Adeniran48 suggested that in HFpEF, intracellular calcium levels are decreased in systole and diastole, resulting in reduced systolic contractility and impaired diastolic relaxation. RYR2 encodes a Ca2+ release channel, which opens to release Ca2+ during systole and closes during diastole to maintain normal contraction‐relaxation cycling. CALM1 encodes a regulatory protein of RYR2 and is responsible for inhibiting Ca2+ release from RYR2 in the sarcoplasmic reticulum to maintain a normal contraction‐relaxation cycle.49 Oxidative stress impairs binding of CALM1 to RYR2, causing aberrant Ca2+ release, resulting in cardiac hypertrophy and HF in murine and canine models.50, 51 Our results suggest that estradiol treatment led to tighter control of cardiac Ca2+ physiology and improved diastolic relaxation.
Molecular Mechanisms Underlying Estradiol Mediated Myocardial Gene Expression
Of the 22 genes upregulated by estradiol in myocardium in this study, we found evidence in the literature for the presence of estrogen response elements in 3 of them (EVL,52 RYR2,53 and VWF 53). A search for the list of 22 genes in the Signaling Pathways Project knowledgebase54 verified cistromic regulation of an additional 17 of these genes via estrogen receptors, the exceptions being PKM2 and PPP2R3B. Downregulated genes are likely modulated through transcription factor cross‐talk, such as occurs between estrogen receptors and NFκB, which was implicated in the causal network analysis findings noted in Figure 14.
Conclusion
To our knowledge, we show for the first time evidence for estradiol preservation of LV diastolic function and modulation of myocardial gene expression in ovariectomized female macaques, a well‐established translational nonhuman primate model of menopause. The pathway analysis findings relative to diastolic function are of particular importance in the aging female population given the strong female preponderance for development of HFpEF in humans and the lack of effective clinical therapies. We demonstrate that estradiol replacement, initiated shortly after the onset of menopause, improved postmenopausal diastolic function through its lusitropic benefits, which were associated with decreased ECM deposition, improved myocardial contraction, and calcium homeostasis, independent of measurable blood pressure effects. These results provide important insights into potential pathways underlying estrogen‐mediated cardioprotection and targets for prevention and/or treatment of diastolic dysfunction and perhaps HFpEF. The findings merit special consideration, especially with reemerging evidence supporting the “timing hypothesis” of hormone therapy, indicating that estrogen/hormone treatment initiated early after menopause may have beneficial effects on the cardiovascular system.55
Sources of Funding
The work described here was supported in part by NIH grants R01AG028641 (Register), R01HL122393 (Register), R01 AG033727 (Groban), R01 AG18915 (Kitzman) and T32‐OD010957 (Cline).
Disclosures
None.
Acknowledgments
The authors would like to thank J.D. Bottoms and Mary Ann Post for their technical assistance, and Dr Neil McKenna of the Baylor College of Medicine for providing assistance on utilization of the Signaling Pathways Project knowledgebase.
Coauthor Thomas B. Clarkson died on December 1, 2015.
(J Am Heart Assoc. 2018;7:e009769 DOI: 10.1161/JAHA.118.009769)
References
- 1. Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P. Heart disease and stroke statistics—2017 update: a report from the American Heart Association. Circulation. 2017;135:e146–e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Miller VM, Harman SM. An update on hormone therapy in postmenopausal women: mini‐review for the basic scientist. Am J Physiol Heart Circ Physiol. 2017;313:H1013–H1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hodis HN, Mack WJ, Henderson VW, Shoupe D, Budoff MJ, Hwang‐Levine J, Li Y, Feng M, Dustin L, Kono N, Stanczyk FZ, Selzer RH, Azen SP. Vascular effects of early versus late postmenopausal treatment with estradiol. N Engl J Med. 2016;374:1221–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Herrera AY, Hodis HN, Mack WJ, Mather M. Estradiol therapy after menopause mitigates effects of stress on cortisol and working memory. J Clin Endocrinol Metab. 2017;102:4457–4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Heidenreich PA, Albert NM, Allen LA, Bluemke DA, Butler J, Fonarow GC, Ikonomidis JS, Khavjou O, Konstam MA, Maddox TM, Nichol G, Pham M, Pina IL, Trogdon JG; American Heart Association Advocacy Coordinating Committee; Council on Arteriosclerosis Thrombosis and Vascular Biology; Council on Cardiovascular Randadiology and Intervention; Council on Clinical Cardiology; Council on Epidemiology and Prevention; Stroke Council . Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart Fail. 2013;6:606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Butler J, Fonarow GC, Zile MR, Lam CS, Roessig L, Schelbert EB, Shah SJ, Ahmed A, Bonow RO, Cleland JGF, Cody RJ, Chioncel O, Collins SP, Dunnmon P, Filippatos G, Lefkowitz MP, Marti CN, McMurray JJ, Misselwitz F, Nodari S, O'Connor C, Pfeffer MA, Pieske B, Pitt B, Rosano G, Sabbah HN, Senni M, Solomon SD, Stockbridge N, Teerlink JR, Georgiopoulou VV, Gheorghiade M. Developing therapies for heart failure with preserved ejection fraction current state and future directions. JACC Heart Fail. 2014;2:97–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee DS, Gona P, Vasan RS, Larson MG, Benjamin EJ, Wang TJ, Tu JV, Levy D. Relation of disease pathogenesis and risk factors to heart failure with preserved or reduced ejection fraction: insights from the Framingham Heart Study of the National Heart, Lung, and Blood Institute. Circulation. 2009;119:3070–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gottdiener JS, Arnold AM, Aurigemma GP, Polak JF, Tracy RP, Kitzman DW, Gardin JM, Rutledge JE, Boineau RC. Predictors of congestive heart failure in the elderly: the Cardiovascular Health Study. J Am Coll Cardiol. 2000;35:1628–1637. [DOI] [PubMed] [Google Scholar]
- 9. Bella JN, Palmieri V, Kitzman DW, Liu JE, Oberman A, Hunt SC, Hopkins PN, Rao DC, Arnett DK, Devereux RB. Gender difference in diastolic function in hypertension (the HyperGEN study). Am J Cardiol. 2002;89:1052–1056. [DOI] [PubMed] [Google Scholar]
- 10. Gerdts E, Okin PM, Omvik P, Wachtell K, Dahlof B, Hildebrandt P, Nieminen MS, Devereux RB. Impact of diabetes on treatment‐induced changes in left ventricular structure and function in hypertensive patients with left ventricular hypertrophy. The LIFE study. Nutr Metab Cardiovasc Dis. 2009;19:306–312. [DOI] [PubMed] [Google Scholar]
- 11. Vasan RS, Larson MG, Benjamin EJ, Evans JC, Reiss CK, Levy D. Congestive heart failure in subjects with normal versus reduced left ventricular ejection fraction: prevalence and mortality in a population‐based cohort. J Am Coll Cardiol. 1999;33:1948–1955. [DOI] [PubMed] [Google Scholar]
- 12. Douglas PS, Katz SE, Weinberg EO, Chen MH, Bishop SP, Lorell BH. Hypertrophic remodeling: gender differences in the early response to left ventricular pressure overload. J Am Coll Cardiol. 1998;32:1118–1125. [DOI] [PubMed] [Google Scholar]
- 13. Hall PS, Nah G, Howard BV, Lewis CE, Allison MA, Sarto GE, Waring ME, Jacobson LT, Manson JE, Klein L, Parikh NI. Reproductive factors and incidence of heart failure hospitalization in the Women's Health Initiative. J Am Coll Cardiol. 2017;69:2517–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Appiah D, Schreiner PJ, Demerath EW, Loehr LR, Chang PP, Folsom AR. Association of age at menopause with incident heart failure: a prospective cohort study and meta‐analysis. J Am Heart Assoc. 2016;5:e003769 DOI: 10.1161/JAHA.116.003769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Deschamps AM, Murphy E. Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am J Physiol Heart Circ Physiol. 2009;297:H1806–H1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. de Jager T, Pelzer T, Muller‐Botz S, Imam A, Muck J, Neyses L. Mechanisms of estrogen receptor action in the myocardium. Rapid gene activation via the ERK1/2 pathway and serum response elements. J Biol Chem. 2001;276:27873–27880. [DOI] [PubMed] [Google Scholar]
- 17. Brosnihan KB, Weddle D, Anthony MS, Heise C, Li P, Ferrario CM. Effects of chronic hormone replacement on the renin‐angiotensin system in cynomolgus monkeys. J Hypertens. 1997;15:719–726. [DOI] [PubMed] [Google Scholar]
- 18. Wang H, Jessup JA, Zhao Z, Da Silva J, Lin M, MacNamara LM, Ahmad S, Chappell MC, Ferrario CM, Groban L. Characterization of the cardiac renin angiotensin system in oophorectomized and estrogen‐replete mRen2.Lewis rats. PLoS One. 2013;8:e76992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, Scanlan TS. Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science. 1997;277:1508–1510. [DOI] [PubMed] [Google Scholar]
- 20. Alencar AK, da Silva JS, Lin M, Silva AM, Sun X, Ferrario CM, Cheng C, Sudo RT, Zapata‐Sudo G, Wang H, Groban L. Effect of age, estrogen status, and late‐life GPER activation on cardiac structure and function in the Fischer344xBrown Norway female rat. J Gerontol A Biol Sci Med Sci. 2017;72:152–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Groban L, Yamaleyeva LM, Westwood BM, Houle TT, Lin M, Kitzman DW, Chappell MC. Progressive diastolic dysfunction in the female mRen(2). Lewis rat: influence of salt and ovarian hormones. J Gerontol A Biol Sci Med Sci. 2008;63:3–11. [DOI] [PubMed] [Google Scholar]
- 22. Jessup JA, Wang H, MacNamara LM, Presley TD, Kim‐Shapiro DB, Zhang L, Chen AF, Groban L. Estrogen therapy, independent of timing, improves cardiac structure and function in oophorectomized mRen2.Lewis rats. Menopause. 2013;20:860–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang H, Jessup JA, Lin MS, Chagas C, Lindsey SH, Groban L. Activation of GPR30 attenuates diastolic dysfunction and left ventricle remodelling in oophorectomized mRen2.Lewis rats. Cardiovasc Res. 2012;94:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Register TC. Primate models in women's health: inflammation and atherogenesis in female cynomolgus macaques (Macaca fascicularis). Am J Primatol. 2009;71:766–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sophonsritsuk A, Appt SE, Clarkson TB, Shively CA, Espeland MA, Register TC. Differential effects of estradiol on carotid artery inflammation when administered early versus late after surgical menopause. Menopause. 2013;20:540–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Register TC, Wagner JD, Zhang L, Hall J, Clarkson TB. Effects of tibolone and conventional hormone replacement therapies on arterial and hepatic cholesterol accumulation and on circulating endothelin‐1, vascular cell adhesion molecule‐1, and E‐selectin in surgically menopausal monkeys. Menopause. 2002;9:411–421. [DOI] [PubMed] [Google Scholar]
- 27. Groban L, Kitzman DW, Register TC, Shively CA. Effect of depression and sertraline treatment on cardiac function in female nonhuman primates. Psychosom Med. 2014;76:137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise J, Solomon S, Spencer KT, St John Sutton M, Stewart W; American Society of Echocardiography's Nomenclature and Standards Committee; Task Force on Chamber Quantification; American College of Cardiology Echocardiography Committee; American Heart Association; European Association of Echocardiography; ESoC . Recommendations for chamber quantification. Eur J Echocardiogr. 2006;7:79–108. [DOI] [PubMed] [Google Scholar]
- 29. Clarkson TB, Anthony MS, Morgan TM. Inhibition of postmenopausal atherosclerosis progression: a comparison of the effects of conjugated equine estrogens and soy phytoestrogens. J Clin Endocrinol Metab. 2001;86:41–47. [DOI] [PubMed] [Google Scholar]
- 30. Kramer A, Green J, Pollard J Jr, Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30:523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B (Methodol). 1995;57:289–300. [Google Scholar]
- 32. da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
- 33. Lobo RA, Cassidenti DL. Pharmacokinetics of oral 17 beta‐estradiol. J Reprod Med. 1992;37:77–84. [PubMed] [Google Scholar]
- 34. Lin W‐J, Hsueh H‐M, Chen JJ. Power and sample size estimation in microarray studies. BMC Bioinformatics. 2010;11:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mustonen E, Pohjolainen V, Aro J, Pikkarainen S, Leskinen H, Ruskoaho H, Rysa J. Upregulation of cardiac matrix Gla protein expression in response to hypertrophic stimuli. Blood Press. 2009;18:286–293. [DOI] [PubMed] [Google Scholar]
- 36. Tamaki Y, Iwanaga Y, Niizuma S, Kawashima T, Kato T, Inuzuka Y, Horie T, Morooka H, Takase T, Akahashi Y, Kobuke K, Ono K, Shioi T, Sheikh SP, Ambartsumian N, Lukanidin E, Koshimizu TA, Miyazaki S, Kimura T. Metastasis‐associated protein, S100A4 mediates cardiac fibrosis potentially through the modulation of p53 in cardiac fibroblasts. J Mol Cell Cardiol. 2013;57:72–81. [DOI] [PubMed] [Google Scholar]
- 37. Zile MR, Baicu CF, Ikonomidis JS, Stroud RE, Nietert PJ, Bradshaw AD, Slater R, Palmer BM, Van Buren P, Meyer M, Redfield MM, Bull DA, Granzier HL, LeWinter MM. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation. 2015;131:1247–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gavras H, Kremer D, Brown JJ, Gray B, Lever AF, MacAdam RF, medina A, Morton JJ, Robertson JI. Angiotensin‐ and norepinephrine‐induced myocardial lesions: experimental and clinical studies in rabbits and man. Am Heart J. 1975;89:321–332. [DOI] [PubMed] [Google Scholar]
- 39. Burlew BS, Weber KT. Cardiac fibrosis as a cause of diastolic dysfunction. Herz. 2002;27:92–98. [DOI] [PubMed] [Google Scholar]
- 40. Vettel C, Lammle S, Ewens S, Cervirgen C, Emons J, Ongherth A, Dewenter M, Lindner D, Westermann D, Nikolaev VO, Lutz S, Zimmermann WH, El‐Armouche A. PDE2‐mediated cAMP hydrolysis accelerates cardiac fibroblast to myofibroblast conversion and is antagonized by exogenous activation of cGMP signaling pathways. Am J Physiol Heart Circ Physiol. 2014;306:H1246–H1252. [DOI] [PubMed] [Google Scholar]
- 41. Dubey RK, Gillespie DG, Jackson EK, Keller PJ. 17beta‐estradiol, its metabolites, and progesterone inhibit cardiac fibroblast growth. Hypertension. 1998;31:522–528. [DOI] [PubMed] [Google Scholar]
- 42. Watanabe T, Akishita M, He H, Miyahara Y, Nagano K, Nakaoka T, Yamashita N, Kozaki K, Ouchi Y. 17 beta‐estradiol inhibits cardiac fibroblast growth through both subtypes of estrogen receptor. Biochem Biophys Res Commun. 2003;311:454–459. [DOI] [PubMed] [Google Scholar]
- 43. Wang H, Zhao Z, Lin M, Groban L. Activation of GPR30 inhibits cardiac fibroblast proliferation. Mol Cell Biochem. 2015;405:135–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Adams MR, Kaplan JR, Manuck SB, Koritnik DR, Parks JS, Wolfe MS, Clarkson TB. Inhibition of coronary artery atherosclerosis by 17‐beta estradiol in ovariectomized monkeys. Lack of an effect of added progesterone. Arteriosclerosis. 1990;10:1051–1057. [DOI] [PubMed] [Google Scholar]
- 45. Grodstein F, Stampfer MJ, Manson JE, Colditz GA, Willett WC, Rosner B, Speizer FE, Hennekens CH. Postmenopausal estrogen and progestin use and the risk of cardiovascular disease. N Engl J Med. 1996;335:453–461. [DOI] [PubMed] [Google Scholar]
- 46. Pervin S, Singh R, Rosenfeld ME, Navab M, Chaudhuri G, Nathan L. Estradiol suppresses MCP‐1 expression in vivo: implications for atherosclerosis. Arterioscler Thromb Vasc Biol. 1998;18:1575–1582. [DOI] [PubMed] [Google Scholar]
- 47. Kuwahara F, Kai H, Tokuda K, Takeya M, Takeshita A, Egashira K, Imaizumi T. Hypertensive myocardial fibrosis and diastolic dysfunction: another model of inflammation? Hypertension. 2004;43:739–745. [DOI] [PubMed] [Google Scholar]
- 48. Adeniran I, MacIver DH, Hancox JC, Zhang H. Abnormal calcium homeostasis in heart failure with preserved ejection fraction is related to both reduced contractile function and incomplete relaxation: an electromechanically detailed biophysical modeling study. Front Physiol. 2015;6:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Belevych AE, Radwański PB, Carnes CA, Györke S. “Ryanopathy”: causes and manifestations of RyR2 dysfunction in heart failure. Cardiovasc Res. 2013;98:240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yamaguchi N, Takahashi N, Xu L, Smithies O, Meissner G. Early cardiac hypertrophy in mice with impaired calmodulin regulation of cardiac muscle Ca release channel. J Clin Invest. 2007;117:1344–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ono M, Yano M, Hino A, Suetomi T, Xu X, Susa T, Uchinoumi H, Tateishi H, Oda T, Okuda S, Doi M, Kobayashi S, Yamamoto T, Koseki N, Kyushiki H, Ikemoto N, Matsuzaki M. Dissociation of calmodulin from cardiac ryanodine receptor causes aberrant Ca(2+) release in heart failure. Cardiovasc Res. 2010;87:609–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kumar A, Dumasia K, Deshpande S, Balasinor NH. Direct regulation of genes involved in sperm release by estrogen and androgen through their receptors and coregulators. J Steroid Biochem Mol Biol. 2017;171:66–74. [DOI] [PubMed] [Google Scholar]
- 53. Bourdeau V, Deschenes J, Metivier R, Nagai Y, Nguyen D, Bretschneider N, Gannon F, White JH, Mader S. Genome‐wide identification of high‐affinity estrogen response elements in human and mouse. Mol Endocrinol. 2004;18:1411–1427. [DOI] [PubMed] [Google Scholar]
- 54. Ochsner S, Abraham D, Martin K, Ding W, McOwiti A, Wang Z, Andreano K, Hamilton R, Chen Y, Hamilton A, Gantner M, Dehart M, Qu S, Hilsenbeck S, Becnel L, Bridges D, Maayan A, Huss J, Stossi F, Foulds C, Kralli A, McDonnell D, McKenna N. The Signaling Pathways Project: an integrated ‘omics knowledgebase for mammalian cellular signaling pathways. bioRxiv. 2018. Available at: https://www.biorxiv.org/content/early/2018/08/27/401729.1. Accessed October 11, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lobo RA, Pickar JH, Stevenson JC, Mack WJ, Hodis HN. Back to the future: hormone replacement therapy as part of a prevention strategy for women at the onset of menopause. Atherosclerosis. 2016;254:282–290. [DOI] [PubMed] [Google Scholar]