

Taking its incidence and prognostic importance into account, acute ST‐segment–elevation myocardial infarction (STEMI) can be regarded as one of the most important challenges faced in the field of clinical cardiology. Coronary artery disease and particularly acute myocardial infarction (AMI) are the leading causes of death and disability worldwide.1 Despite remarkable progress in the fight, particularly in the past 3 decades, there is still room for improvement. Indeed, the standardized 1‐year death rate for STEMI has nearly halved over a 25‐year period.2 This decrease in mortality is attributable to outstanding achievement accomplished in the limitation of final myocardial infarct (MI) size by introduction of efficacious reperfusion methods such as fibrinolysis and primary percutaneous coronary intervention (pPCI) during that period. Currently, reopening of the occluded epicardial coronary artery by timely pPCI is widely accepted as the most effective treatment for patients presenting with an acute STEMI in limiting final MI size and preserving left ventricular (LV) function.3, 4 However, despite successful reperfusion by pPCI, mortality (15%)5 and particularly post‐MI morbidity still remain significant at 1 year.1, 6, 7 This disappointing course has been partly attributed to the potential detrimental effects of reperfusion itself. Indeed, reperfusion may lead to a further loss of cardiomyocytes that are succeeded to survive after initial ischemic insult in the subtended myocardial territory.
Hemodynamic manifestations of this postreperfusion process include “no‐reflow phenomenon”—severe myocardial malperfusion despite restoration of epicardial coronary patency,8, 9, 10 which has been reported to occur in up to 50% of patients with STEMI following pPCI despite restoration of thrombolysis in myocardial flow 3 in the epicardial coronary artery.11 In general, myocardial no‐reflow phenomenon refers to severe microvascular injury that is known to be associated with impaired LV function12, 13 and poor prognosis14, 15 in patients undergoing successful pPCI. In addition, the magnitude of the preserved microvasculature at the acute phase is one of the major determinants of the long‐term functional and structural myocardial recovery.16 Although it occurs in every patient undergoing pPCI at varying intensity, identification of coronary microvascular injury depends on the diagnostic capability of the method used in its detection. Considering this high incidence and its important clinical consequences,14, 15 a better understanding of the mechanisms underlying severe coronary microvascular injury resulting in myocardial malperfusion (myocardial tissue “no reflow”) after pPCI is mandatory to be able to develop efficacious therapeutic interventions for preventing this complication of STEMI. Nevertheless, our current knowledge on the pathophysiology of microvascular damage after pPCI is poor, and hence recommendation of risk prediction and therapeutic interventions could be premature and limited.
Notably, given the repeated failure of recent trials aiming to protect microcirculation during pPCI, it is obvious that identification of an appropriate therapeutic target linked to the underpinning mechanism has utmost clinical importance in the treatment of patients who develop extensive microvascular injury after pPCI. Although there is not yet a widely accepted and proven therapeutic intervention to limit postreperfusion microvascular injury, examination of microvascular integrity in the catheterization laboratory immediately upon completion of pPCI17 can provide a unique opportunity for timely identification of this patient subset with severe microvascular injury who may benefit from early adjunctive therapies within the therapeutic window.
The aim of this review is to provide an integrative perspective on the pathophysiological mechanisms underlying post‐pPCI coronary microvascular injury and does not consider discussing the potential cellular and molecular mechanisms as cardiomyocyte targets for cardioprotection. In this review, we propose an integrative categorization scheme, which can provide comprehensive pathophysiological insights into post‐pPCI microvascular injury and potentially pave the way for therapeutic targeting of central pathologies behind the myocardial no‐reflow phenomenon in the future.
Mechanisms of Microvascular Injury in Reperfused AMI
Obviously, reestablishing complete and sustained epicardial patency in a timely manner is the most critical step in salvaging ischemic myocardium from impending infarction. However, prompt restoration of coronary flow by reopening the occluded infarct‐related artery can itself paradoxically induce coronary microvascular injury and does not immediately terminate ongoing cardiomyocyte loss at the myocardial area at risk (AAR). During coronary occlusion and after reperfusion, dynamic pathological changes observed in both the microvascular and interstitial territories seem to contribute substantially to this progressive cardiomyocyte damage in the subtended myocardial area.18 Functional and structural consequences of the cortege of pathological changes temporally and spatially evolving at consecutive segments of myocardial circulation seem to determine the fate of the subtended myocardial territory (Table). In this regard, a new classification scheme using compartmental modeling (Figure 1) might help both to elucidate individual contributions of various factors to post‐pPCI microvascular injury and to reappraise any interplay between them.
Table 1.
Temporal and Spatial Changes Taking Place at the Consecutive Segments of Myocardial Circulation During Coronary Occlusion and After Mechanical Reperfusion
| Phase | Site | Total Coronary Resistance | Coronary Flow | |||
|---|---|---|---|---|---|---|
| Epicardial Coronaries | Arterioles | Capillaries | Cardiomyocytes and Interstitium | |||
| Early ischemia | Occluded | Reactive dilatation (adaptive response) | ··· | Functional abnormality, cellular edema | Immeasurably high (predominantly epicardial) | No‐flow |
| Prolonged ischemia | Occluded | Paralysis (both ischemic insult and adaptive response) | Increased permeability, loss of integrity, in situ thrombosis | Ischemic necrosis | Immeasurably high (predominantly epicardial) | No‐flow |
| Initial phase of reperfusion | Reopened | Paralysis | Obstructed by plugs and destructed | Interstitial edema | Low (predominantly microvasculara) | Overflow |
| Late phase of (established) reperfusion | Reopened | Partly recovered constrictor response | More plugged, more leaky | Deepened edema, even IMH | High (predominantly microvasculara) | Normal, slow or “no‐reflow” |
Total coronary resistance: epicardial resistance+microvascular resistance. IMH indicates intramyocardial hemorrhage.
Unless there is no additional epicardial lesion rather than the culprit one in the re‐opened infarct‐related artery.
Figure 1.

Compartmental schematization of the mechanisms behind post–primary percutaneous coronary intervention microvascular injury. Following reperfusion, factors contributing to microvascular injury at the subtended myocardial territory can be categorized under 2 major headings as intraluminal microvascular obstruction and extravascular compression of the microcirculation. Major pathologies contributing to luminal obstruction are distal thromboembolization, cellular plugging, in situ thrombosis, and vasospasm. External compression of microcirculation by cellular and/or interstitial edema, intramyocardial hemorrhage, and increased left ventricular filling pressures may also substantially contribute to microvascular impairment by generating an external compressive force on coronary microcirculation. LVEDP indicates left ventricular end‐diastolic pressure.
Indeed, duration of ischemia is considered as the most important determinant of the magnitude of the microvascular damage and its recovery after STEMI.19, 20 However, in a rat model of acute myocardial infarction, it has been recently demonstrated that ischemia alone induces only mild morphological changes in the coronary microcirculation with increased permeability. Nevertheless, ischemia followed by reperfusion has been shown to induce massive microvascular injury.21 Accordingly, on the temporal scale, beginning from the preocclusion period, interactions among the dynamic events evolved during both coronary occlusion and reperfusion seem to determine the eventual magnitude of microvascular damage and cardiomyocyte loss in the subtended microcirculatory territory over time (Figure 2). In addition, the functional status of the patient's coronary microcirculation before the STEMI (preexisting microvascular impairment),22 status of the infarct‐related artery beyond the culprit lesion, perfusion characteristics of the nonculprit vessels, presence of coexistent pathologies such as diabetes mellitus, and overall status and performance of the left ventricle and individual susceptibility23 contribute to the extent of microvascular damage at the subtended myocardial territory following reperfusion.
Figure 2.
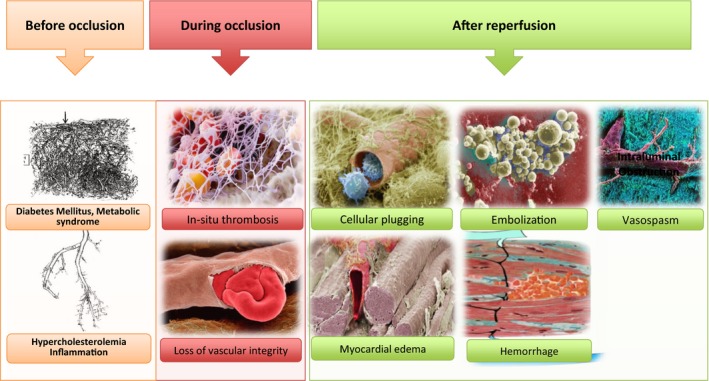
Intraluminal and extravascular factors of microvascular injury operating on the temporal scale (before, during, and after reperfusion). On the temporal scale, in addition to the preexisting microvascular impairment, dynamic events evolving both during coronary occlusion and after reperfusion seem to determine the final magnitude of microvascular damage in the subtended microcirculatory territory. In the preocclusive period, patients’ metabolic and inflammatory status and presence of diabetes mellitus, hypercholesterolemia, and hypertension may lead to a preexisting microvascular dysfunction. During epicardial occlusion, local procoagulant activity induced by local hypoxia in the downstream microcirculation may provide extremely suitable milieu for in situ microvascular thrombus formation. During this occlusive period, hypoxia‐induced endothelial disruption may also lead to loss of microvascular barrier function and microvascular leakage. After reperfusion, distal thromboembolization along with edema and intramyocardial hemorrhage developed in this destructed and leaky microcirculatory environment can markedly contribute to microvascular injury.
Following reperfusion, mechanisms involved in the development of microvascular impairment can categorically be classified under 2 major headings as (1) intraluminal microvascular obstruction and (2) extravascular compression of the microcirculation. Indeed, intraluminal obstructive and extravascular compressive pathologies both seem to operate interconnectedly in the development of post‐pPCI microvascular injury. Primary contributors to intraluminal microvascular obstruction are distal thromboembolization, cellular plugging, in situ thrombosis, and vasospasm. Besides these intraluminal factors, cellular and/or interstitial edema, intramyocardial hemorrhage, and increased LV filling pressures are the main pathologies that may exacerbate microvascular impairment by generating an external compressive force on coronary microcirculation (Figure 1).
Coronary Microvascular Obstruction
Coronary Intraluminal Plugging
In the setting of pPCI, coronary microvascular (intraluminal) obstruction can mainly be caused by distal thromboembolism, circulating blood cells plugging and in situ microvascular thrombosis. Intraluminal microvascular obstruction (MVO) is regarded to be the leading pathology behind post‐pPCI myocardial malperfusion. Pathologically, MVO first appears in the infarct core and evolves spatially and temporally, which corresponds with progressive myocardial damage occurring in the AAR after reopening the infarct‐related artery. Consistently, the MVO zone has been shown to increase for up to 48 hours after reperfusion.24 Indeed, after a period of ischemia, blood flow cannot be restored in more than half of the capillaries in the myocardial AAR after reestablishing epicardial patency.25 In ischemic‐reperfused myocardium, myocardial blood flow in certain microvascular areas is hyperemic during the first minutes of reperfusion. Subsequently, regional blood flow in the AAR rapidly and progressively declines26 and reaches a plateau within 2 to 8 hours after reperfusion, resulting in a nearly 3‐fold increase in the anatomic MVO (no‐reflow) zone.27 This delayed progressive fall in blood flow in myocardial areas that initially received adequate perfusion seems to occur due to concurrently activated intraluminal obstructive and extravascular compressive pathologies triggered by reperfusion itself. Experimental19 and clinical studies28 consistently demonstrated a close correlation between zones of anatomic MVO (no‐reflow) and myocardial necrosis. Furthermore, in a rat model of coronary occlusion and reperfusion, it was shown that the no‐reflow phenomenon may persist for 1 month after reperfusion and could predict infarct expansion.29
Factors that contribute to luminal MVO after pPCI will be reviewed below as classified in Figure 1.
Thromboembolism
In the setting of pPCI, atherothrombotic embolization from the culprit lesion, reported to occur in 11% to 14.5% of the procedures,30, 31, 32 is believed to be one of the leading contributors of MVO occurring during pPCI. Distal embolization of atherothrombotic particles during pPCI may cause both mechanical obstructions by their mass effect and activation of pathways that trigger in situ coagulation and inflammatory responses in the downstream microcirculation.
In both clinical and experimental trials, subjects with distal embolization early after reperfusion seem to have larger MI size and more extensive microvascular damage.32, 33 Consistently, in a study where embolized particles during pPCI were quantified using intracoronary Doppler wire as high‐intensity transient signals, it was demonstrated that distal embolization might transiently reduce coronary flow, but this may not have a marked influence on MI size and LV function.34 In addition, it has been reported that angiographically visible distal embolization occurring during pPCI may contribute to myocardial damage within hours after symptom onset, but it does not seem to have a major impact as ischemic time increases.32
In this context, it is expected for mechanical retrieval of epicardial thrombotic material using manual aspiration thrombectomy devices during pPCI to substantially limit distal embolization risk, resulting in significant reduction in the MVO zone and MI size. However, although significantly lower distal embolization rates seemed to be achieved with manual thromboaspiration compared with standard pPCI, this did not seem to translate into improved myocardial perfusion,35 contrary to what was initially observed in meta‐analyses.36 Even more importantly, most of the trials37, 38, 39, 40 evaluating the efficacy of manual thrombus aspiration during pPCI failed to show any benefit of the mechanical removal of epicardial thrombus on the limitation of MI size. Furthermore, either routinely41, 42 or selectively43 performed thromboaspiration did not show any clinical benefit in large‐scale clinical trials and in a recent meta‐analysis.44 All these negative results suggest that different approaches to tackling epicardial thrombus to initiate reperfusion, such as standard pPCI or thrombectomy followed by stenting, may not be associated with different pathological and clinical consequences. Moreover, reduction of distal embolization rates by manual thrombectomy does not necessarily translate into decreased infarct size and improved patient outcome. This makes the role of distal embolization from the proximal thrombotic occlusion in microvascular and myocardial damage during pPCI highly controversial and helps in understanding why the trials dealing with epicardial thrombus failed.
In light of the current data, it can be concluded that distal thromboembolism may not be sufficient to produce extensive microvascular injury and may contribute to MVO only to a limited extent. Thus, it does not appear to be a major therapeutic target for preserving capillary integrity during pPCI.
Cellular Plugging: Role of Circulating Blood Cells
Leukocyte and platelet plugging45 and red cell aggregation46 can intensify post‐pPCI MVO. After successful epicardial recanalization, neutrophils can worsen microvascular reperfusion by adhering to the endothelium with platelets47 and by releasing cytokines or other factors48 that may reduce microvascular blood flow. Capillaries in the no‐reflow zone have been shown to contain extensive leukocyte plugging (capillary trapping).25 This leukocyte plugging can also lead to erythrocyte packing and rouleaux formation upstream from mechanical obstruction. In addition, platelets and neutrophils act synergistically in provoking microvascular injury after reperfusion.45 Accordingly, significant relationships have been shown between the presence of MVO and monocyte counts49 and platelet activity.50 In patients who underwent successful pPCI, higher admission neutrophil count,51 platelet volumes51 and neutrophil‐to‐lymphocyte ratio52 were shown to be associated with increased coronary microvascular resistance, suggesting the obstructive role of circulating blood cells supplied by reperfusion in post‐PCI microvascular impairment.
In particular, maintenance of blood flow at the coronary microvascular segments, where capillary diameters are reduced under an erythrocyte diameter, largely depends on deformational capabilities of circulating blood cells and endothelial surface layer lubricity. However, following ischemia and reperfusion, the shedding of the glycocalyx layer, which covers the endothelial surface, makes the latter less permeable and more slippery and makes capillaries more prone to be obstructed by cellular plugging, leading to further increase in microvascular resistance.53, 54 Despite these potential negative effects of circulatory cell plugging on microvascular perfusion, several trials that aimed at complement inhibition,55 leukocyte integrin receptor antagonism,56 and increased local platelet inhibition57 all yielded negative results in acute MI patients. Although it seems that circulating blood cells could potentially be involved in intraluminal plugging following pPCI, these might only play a limited role in acute post‐pPCI coronary microvascular impairment. Leukocyte infiltration may be more important in infarct healing and remodeling rather than the determination of the extent of the MVO zone and infarct size. Therefore, the role of leukocytes in postreperfusion myocardial damage is contentious, and they do not seem to be a viable therapeutic target for limiting reperfusion‐related microcirculatory damage.
Humoral Factors (In Situ Thrombosis)
During epicardial coronary occlusion, the local procoagulant milieu in the downstream microcirculation is extremely suitable for de novo (autochthonous) microvascular thrombus formation. Local hypoxia can immediately precipitate local coagulation by triggering homeostatic mechanisms at the injured endothelium, which may consequently induce microvascular thrombosis and in situ fibrin generation at the site of local damage.58 During occlusion of an epicardial coronary artery, tissue factor expressed from mainly hypoxic and injured endothelial cells, together with stasis, can strongly stimulate the coagulation cascade and de novo fibrin formation at the microvascular level. Additionally, dysfunctional endogen fibrinolysis after reperfusion following an ischemic period, as evidenced by significantly impaired tissue plasminogen activator release from the endothelium,59 can also lead to inadequate removal of fibrin deposits from the coronary microcirculation. Moreover, after its formation, it would not be easy to sweep away this in situ formed fibrin from the lumen via mere restoration of blood flow to the infarcted region because of the former's active adherence to the vessel wall via intercellular cadherin receptors.60 Indeed, all of the formed blood cells are already prone to being attached to the fibrin mesh formed in the microvasculature both passively and actively via the specific fibrin receptors on their surfaces.61 Thus, even a small amount of intraluminal fibrin can constitute a sticky trap for the formed blood cells supplied by reperfusion62 and may impede reperfusion once the occluded epicardial artery is reopened (Figure 3). Although in situ fibrin formation in cardiac microvasculature after ischemia and reperfusion has received almost no attention, autochthonous fibrin depositions in the downstream microcirculation have already been clearly shown in cerebral,63, 64 intestinal,65 and renal66 ischemia‐reperfusion models.
Figure 3.
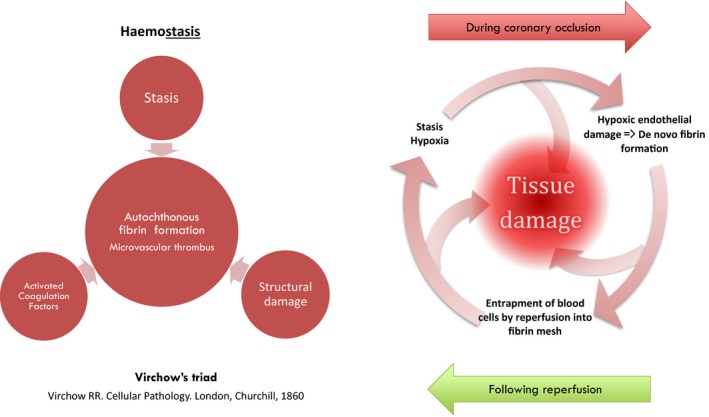
A vicious cycle of microvascular thrombosis: During epicardial occlusion, hypoxic endothelial damage, stasis, activated coagulation factors, and tissue factors trigger Virchow triad, which may lead to in situ microvascular fibrin generation. After reopening of the infarct‐related artery, formed blood cells supplied by reperfusion get entrapped in to the microvascular fibrin mesh, which may lead to further stasis and further fibrin generation.
In particular, in a condition of slow flow (low shear stress), fibrinogen also contributes to impeding microvascular flow via facilitating red blood cell aggregation,67 mediating leukocyte‐endothelium bridging,68 and facilitating postischemic leukocyte‐thrombocyte interaction.69
To this end, effective removal of fibrin(ogen) depositions from the microcirculation may result in better microvascular perfusion. In accordance with this perspective, adjunctive low‐dose intracoronary fibrinolytic drugs (streptokinase, urokinase) given immediately after successful pPCI were shown to be associated with significantly improved microvascular perfusion,70 decreased infarct size,71 preserved LV function71 and improved patient outcome.72 These encouraging results73 also served as a stimulus to further explore the effect of microvascular fibrin deposition removal using different fibrinolytic agent on patient outcome in a large‐scale ongoing clinical trial (T‐TIME [A Trial of Low‐Dose Adjunctive Alteplase During Primary PCI]).74
In this context, autochthonous fibrin generation at the downstream coronary microcirculation leading to in situ thrombotic MVO may play a pivotal role in the pathogenesis of microvascular impairment induced by ischemia and reperfusion. However, it is evident that large‐scale clinical trials targeting in situ microvascular thrombosis, such as the T‐TIME trial, are required to reach more conclusive results.
Vasospasm
Coronary microcirculation distal to the acute coronary occlusion is considered as maximally dilated due to exhausted autoregulatory function of arteriolar sphincters. However, even during severe myocardial ischemia, which is the most powerful stimulus known for vasodilation, a pharmacologically recruitable vasodilator reserve may persist.75 Furthermore, during myocardial ischemia/reperfusion (pPCI), the microcirculation remains highly responsive to α‐adrenergic coronary constrictor mediators. Impaired endothelial function by ischemia and reperfusion in conjunction with the release of soluble vasoconstrictor substances, such as serotonin and thromboxane A2 from ruptured plaque and platelet aggregates into the microcirculation may contribute to a vasospastic milieu during pPCI.76 Based on these rationales, the potential benefit of administration of adjunctive intracoronary vasodilators—in particular adenosine, which is a potent direct vasodilator of coronary microcirculation through stimulation of A2 receptors—in reducing microvascular injury in patients undergoing pPCI was examined in multiple clinical trials. Despite previous intriguing results,77, 78 in a recent clinical trial, local adjunctive intracoronary adenosine and sodium nitroprusside administration targeting vasodilation of the subtended microvascular bed were shown to be ineffective in reducing MVO in patients undergoing primary PCI.79 Furthermore, in this latest and most powerful study,79 high‐dose adenosine appeared to be associated with increased infarct size and reduced ejection fraction compared with the control group. These findings suggested that vasodilator agents should not be used in the setting of pPCI to prevent reperfusion injury. In particular, vasomotor function (vasodilator reserve) regulating distal pressure in the reperfused territory can be crucial in obviating an uncontrolled and abrupt increase in capillary pressure during reperfusion that might otherwise be a protective mechanism against myocardial edema and intramyocardial hemorrhage. Therefore, when the negative results of the recent studies using potent vasodilators administered as an adjunct to pPCI are taken into account, vasomotor function at arteriolar level seems to be protected and not be suppressed.
In light of the present data, at the current stage, the extent of contribution and potential role of vasospasm in coronary microvascular injury in the pPCI setting are highly controversial. Therefore, adjunctive pharmacological interventions targeting microvascular spasm to prevent microvascular injury seem not to be beneficial in the setting of pPCI.
Extravascular Compression of the Microcirculation
External compression of the capillary bed by interstitial and cellular myocardial edema and intramyocardial hemorrhage (IMH) developed in the surrounding myocardium after reperfusion substantially contributes to post‐pPCI myocardial malperfusion mainly by increasing total microvascular resistance. Both interstitial and cellular myocardial edema and IMH emerge as a consequence of prolonged ischemia and reperfusion and subsequently become the major contributors to microvascular injury by generating an external compressive force on coronary microvasculature (Figure 4).
Figure 4.
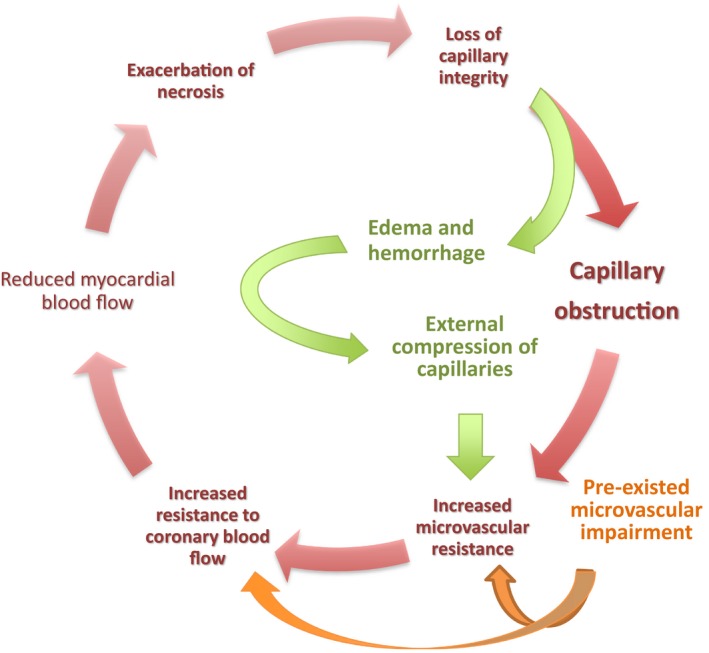
Myocardial edema and intramyocardial hemorrhage initially emerge as a consequence of prolonged ischemia and reperfusion, and they subsequently become one of the main contributors of the microvascular impairment by generating an external compressive force on coronary microvasculature.
The capillaries may not generate a significant resistance to myocardial blood flow in the healthy coronary circulation. However, the capillary network is also the most susceptible compartment to external compression generated by surrounding edema and IMH, as it is assumed to have the lowest radial force. The compressive force causes capillaries to shrink in diameter, with any such decrease resulting in an exponential increase in resistance, which may in turn lead to a substantial decrease in myocardial blood flow. Theoretically, extravascular pressure can simply be calculated as any extra volume added to the myocardial compartment divided by myocardial compliance. Situations like IMH and edema can lead to both an increase in the volume of interstitial space and a decrease in myocardial compliance80 and thus result in a marked increase in extravascular pressure that eventually causes substantial external microvascular compression.
During total coronary occlusion, depending on the duration and severity of the ischemia, hypoxia‐induced endothelial disruption leading to loss of microvascular barrier function results in microvascular leakage (MVL),81 which is the central anatomic substrate underlying myocardial edema and hemorrhage occurring after establishment of reperfusion. Consistently, clinical cardiac magnetic resonance (CMR) studies have implicated a loss of microvascular barrier function in AMI manifested as edema and IMH.82, 83 Therefore, edema and IMH, the main determinants of extravascular compressive force, can be regarded as a consequence of MVL. In a recent mouse I/R (Ischemia/Reperfusion) study, late gadolinium enhancement CMR, which is believed a measure of extravascular volume, and MVL quantified by histology revealed a high degree of spatial colocalization (r=0.959).84 This finding indicates that the microvascular barrier function is damaged after I/R, leading to MVL, and in the acute phase of I/R, late gadolinium enhancement CMR measures MVL. In the acute phase of I/R, within the risk zone, the area of MVL was even greater than that of infarct size or MVO zone and correlated with acute ventricular dilatation. In the chronic phase, the size of the MVL was shown to be correlated with a reduction in LV ejection fraction. Taken together, the findings of the study, for the first time, characterized the MVL as a major pathological consequence of reperfused AMI. Accordingly, when reperfusion starts after a period of occlusion, this leaky microvasculature constitutes the major structural background for the edema and hemorrhage compressing microvasculature externally.
Extravascular factors potentially associated with microvascular impairment after pPCI will be reviewed in an integrative perspective approach below.
Edema (Cellular and Interstitial)
At the early stages of ischemic injury, the energy‐dependent Na+/K+ pump breaks down and myocardial cellular edema develops. Subsequently, as ischemia deepens, endothelial cells, along with their glycocalyx cover and capillary membranes, get damaged, which renders microvasculature highly permeable (capillary leakage). In addition, during epicardial occlusion, due to severely increased myocardial demand (adaptive vasodilator response) and partly to ischemic insult (decreased contractile function), arteriolar sphincters are supposed to lose their regulatory vasoconstrictor function. Therefore, subtended microvascular segments are deprived of the protection of pressure‐regulating resistance arterioles at the time when reperfusion abruptly restarts. Upon reperfusion, before coronary autoregulation is restored, leaky and unguarded microcirculation is exposed to a pressure burst due to abruptly increased capillary hydrostatic pressure, which causes interstitial myocardial edema within early minutes during the reactive hyperemia phase.85 Indeed, myocardial edema is substantially exacerbated upon restoration of blood flow to the ischemic microvascular territory. Crucially, absence of reperfusion almost completely abolishes the initial wave of edema, indicating the direct link between reperfusion and myocardial edema formation. Contrary to the accepted view, recent experimental and clinical studies have stated that myocardial edema is not a stable phenomenon but follows a bimodal pattern.86, 87 The reperfusion (overperfusion)‐associated initial peak of edema seemed to occur rapidly at 120 minutes after reperfusion, and the deferred wave of edema seemed to occur due to the healing process on days 4 and 7 after reperfusion.86, 87 However, Carrick and colleagues88 reported that the severity of edema follows a bimodal time course only in patients with accompanying IMH. In this study, in patients without IMH, myocardial edema seemed to evolve progressively on a unimodal time course. In another report,89 which was a retrospective analysis of pooled data from 3 studies, it was concluded that no bimodal edema pattern was apparent. However, most patients in the evaluated studies included in this analysis underwent a single CMR scan at disparate times from each other, and there were no systematic serial examinations. Finally, in the most recent clinical and experimental studies,90, 91 which were specifically designed to provide a clear insight into the pattern of edematous response of myocardium to MI, it has been demonstrated that the post‐MI edematous reaction is not stable but follows a bimodal pattern regardless of the degree of IMH.
Myocardial edema has important consequences via triggering a cascade of events. First, edema itself creates an external compressive force on capillaries by increasing hydrostatic pressure within the interstitial space. Second, increased stiffness and reduced compliance of the edematous myocardium80 results in increased LV end‐diastolic pressure, which constitutes an additional external force compressing microcirculation externally. These mechanisms, by increasing the resistance to total myocardial blood flow, may drastically contribute to the extent of myocardial necrosis in the AAR.
Myocardial edema can, therefore, be an important therapeutic target in limiting postreperfusion microvascular damage. In this regard, interventions that may modulate the initial reperfusion‐driven wave of edema can be particularly appealing. To this end, during pPCI, controlled pressure or gradual reperfusion of epicardial occlusion, with the aim of preventing an abrupt increase in distal intracoronary and consequently capillary hydrostatic pressures during the reactive hyperemia phase may help to limit leakage from the capillaries and hence may substantially reduce interstitial edema formation.92
Intramyocardial Hemorrhage
IMH has been regarded as the hallmark of severe microvascular damage. IMH is observed in ≈30% to 45% of reperfused patients with STEMI.88, 93, 94 However, the temporal evolution of IMH has not yet been completely understood. Using T2* CMR imaging, which is thought to be relatively insensitive to the effects of edema and may provide a more objective assessment of hemorrhage, IMH was shown to increase progressively and to peak 2 days after reperfusion.95 Myocardial hemorrhage is associated with larger infarcts,96 adverse remodeling,97, 98, 99 persistent LV systolic dysfunction,88, 100 late arrhythmic risk,98, 101 and adverse clinical outcome.95, 102 In a multicenter observational cohort study, in which 54% of the patients undergoing pPCI had IMH, reported predictors of occurrence of IMH after reperfusion were anterior infarct location and periprocedural glycoprotein IIb‐IIIa inhibitor treatment.103
Hypoxia‐induced disruption in the endothelial barrier leads to loss of capillary integrity during prolonged coronary occlusion.58 During this occlusion period, adaptive vasodilatory response and ischemic insult of the arteriolar sphincters leave distal microcirculation defenseless to suddenly increased hydrostatic pressure during reperfusion. In this milieu, abrupt reperfusion by successful pPCI may generate a barotrauma‐like effect on the already‐destructed leaky capillaries that can result in the extravasation of erythrocytes from the leaking microvasculature into the interstitium, leading to IMH. However, occlusion without reperfusion leads only to intracellular edema but no hemorrhage.104 Consistently, IMH was observed significantly less frequently in patients in whom restoration of reperfusion could not be achieved105 and rarely observed in patients with untreated AMI.106 In the acute phase, interstitial erythrocyte accumulation creates an external compressive mass on capillaries. In the chronic phase, it triggers macrophage influx, generation of reactive oxygen species, inflammation, and fibrosis.99 In this context, IMH can be regarded as the more severe form of myocardial edema and the most severe manifestation of microvascular injury.
Imaging studies have repeatedly shown that almost all patients with IMH on T2‐weighted imaging have CMR‐defined MVO on contrast imaging.96, 107, 108 Concordantly, in a recent experimental study, overall regions of IMH and MVO zones in CMR were strikingly similar and corresponded to IMH confirmed by histopathology.96 This mutual relationship between IMH and severe MVO suggests that hemorrhage occurs only within regions of severe MVO/damage. Additionally, as an established invasive parameter of coronary microvascular status, the index of microvascular resistance measured immediately after pPCI has been demonstrated to be 2‐fold higher in reperfused patients with STEMI with IMH than in those without.109 Consistently, a high index of microvascular resistance value (>40 units) was recently shown to be an independent predictor of IMH presence in patients undergoing pPCI.110 All these findings concordantly imply that IMH may be a downstream consequence of severe microvascular damage. Once developed, IMH substantially potentiates progression of microvascular damage in the AAR by generating an external compressive mass on the surrounding capillaries. Additionally, an IMH‐driven decrease in myocardial compliance also leads to an increase in LV filling pressure that ultimately causes an increase in resistance to myocardial blood flow in the AAR, resulting in exacerbated necrosis.
Considering its paramount prognostic value,111 it is obvious that development of new therapeutic strategies aiming to limit IMH in reperfused AMI would definitely improve patient outcome. In this regard, first, it seems that post‐pPCI IMH may be amenable to therapeutic interventions aiming to preserve microvascular perfusion and integrity, as it is most likely preceded by MVO. Second, because capillary overpressurization is crucial for the development of myocardial edema and IMH, mechanical interventions to prevent uncontrolled pressure rise in the already‐injured capillary area during pPCI might be an appropriate strategy to prevent IMH. After reopening of the occluded epicardial coronary artery, following initial rise (hyperemic flow) and fall, myocardial blood flow in the area at risk is stabilized within ≈30 minutes in the AAR.26, 27 This finding implies that, in spite of the initial ischemic insult, coronary autoregulation in the reperfused myocardial territory recovers sometime after reperfusion. In this regard, regaining adaptive autoregulatory vasoconstrictor response of the pressure‐regulating resistance arterioles before full reperfusion is established by stenting may be critically important to prevent an uncontrolled rise in pressure in the injured microvascular territory. Therefore, to limit/prevent edema and IMH formation during pPCI, preventive measures should be taken before full, high‐pressure reperfusion established by stenting, which otherwise exposes unguarded capillaries to a pressure burst. These preventive measures may include gradual, controlled‐pressure reperfusion aiming to prevent an abrupt increase in distal intracoronary and consequently capillary hydrostatic pressure during reactive hyperemia phase by allowing enough time to arterioles until their adaptive vasoconstrictor response getting recovered.
Strikingly, intracerebral hemorrhage developed immediately after stenting of the high‐grade carotid stenosis,112, 113 namely, hyperperfusion syndrome, most likely shares the same mechanism with IMH. To maintain cerebral blood flow during a long‐lasting hypoperfusion period, cerebral autoregulation maximally dilates the arterioles distal to a subtotal occlusion. In conjunction with this adaptive vasodilatory response, the autoregulatory function of arteriolar sphincters is impaired in severely ischemic cerebral tissue. Therefore, cerebral arteriolar malfunction in the subtended microcirculatory territory results in incompetent vasoconstrictor response to a suddenly increased intravascular pressure abruptly reestablished by stenting of nearly occluded carotid arteries. Consequently, suddenly increased intracapillary hydrostatic pressure disrupts the tight junctions of the capillary endothelial cells and causes intracerebral hemorrhage. This common background strongly suggests the pivotal role of abrupt reopening of an occluded artery in the development of hemorrhage in the subtended microvascular territory.
Increased LV Filling Pressure
The proximity of the vascular and myocardial compartments makes intramyocardial vessels susceptible to mechanical and hemodynamic changes in the surrounding tissues and LV cavity. In patients with STEMI, increased diastolic filling pressures due to acute loss of contractile performance combined with increased muscle stiffness and reduced myocardial compliance due to edema and IMH can also contribute to a decrease in intramyocardial vascular capacitance that limits coronary flow in late diastole. Transmitted increased intracavitary pressure is partly responsible for external compression of coronary microcirculation, particularly in the subendocardium, which is the most susceptible region to elevation in LV filling pressure. Consistently, in patients with STEMI, LV end‐diastolic pressure has been shown to be correlated with zero flow pressure, an invasive parameter that mainly informs on the external pressure on the coronary microcirculation.114, 115 Thus, in the course of STEMI, diastolic LV dysfunction causing elevation in LV filling pressure may lead to a further restriction of the microvascular compartment. However, LV filling pressure is not the sole determinant of the myocardial interstitial pressure. In the setting of reperfused STEMI, intramyocardial edema and hemorrhage formed as a result of severe microvascular injury in the affected myocardial territory with limited compliance can cause a substantial increase in interstitial pressure. Furthermore, this new volume added to the low‐compliance chamber may increase interstitial pressure even above to the diastolic intraventricular pressure levels. In this situation, potential therapeutic interventions aiming to reduce ventricular filling pressure may not improve myocardial perfusion. Therefore, again, maximal benefit in limiting diastolic dysfunction developing in the reperfused myocardial segments can be expected from therapeutic applications, which may succeed in limiting edema/hemorrhage and no‐reflow.
Future Directions
As proposed in this review, multiple mechanisms underlying postreperfusion microvascular injury are not mutually exclusive and can act interconnectedly in concert. Furthermore, the individual contribution of these mechanisms to impaired myocardial reperfusion may vary temporally and spatially. Therefore, exclusive targeting of a particular mechanism in every patient may not be a logical approach to overcome the problem.
From an integrative perspective, after successful reperfusion with pPCI, microvascular plugging in conjunction with external compression of microcirculation with surrounding edema and IMH may significantly impede reperfusion flow via increasing total microvascular resistance, which may in turn result in reduction of myocardial blood flow and therefore propagation of necrosis in the subtended myocardial AAR. In the post‐pPCI microvascular injury spectrum, IMH, which is most likely preceded by MVO, is the most severe manifestation. Therefore, this most severe form of microvascular injury may be amenable to therapeutic interventions aiming to preserve microvascular perfusion and integrity first. Concurrently, mechanical interventions to modulate reperfusion during pPCI mainly by controlling pressure rise in the susceptible microvascular area might be an appropriate strategy to prevent interlinked edema and IMH.
Conclusions
Although timely reperfusion achieved by successful pPCI changed the overall course of AMI, it is not sufficient to terminate ongoing myocyte loss in case of severe microvascular injury. In this review, the potential roles of intra‐ and extravascular factors that may affect microvascular integrity and therefore cardiomyocyte survival in the ischemic area after successfully restored epicardial reperfusion were systematically overviewed in an integrative manner. It seems obvious that the interplay between intravascular obstructive factors and extravascular compressive forces underlies post‐pPCI microvascular injury (Figure 5). After occlusion of an epicardial artery and following reperfusion, multifactorial and multifaceted processes orchestrated by interlinked pathologies seem to cause progressive microvascular damage and cardiomyocyte loss at the subtended area (Figure 6). Overall, this progressive microvascular and myocardial damage following reperfusion might be attenuated by novel therapeutic interventions targeting mechanisms interconnected with both intraluminal obstruction (such as local pharmaceutical therapies for microvascular thrombolysis) and extravascular compression of the microcirculation (such as controlled‐pressure, gradual reperfusion).
Figure 5.

Microvascular impairment in reperfused acute myocardial infarction: integrated association of the factors related to intravascular obstruction and extravascular compression of the microcirculation. LV indicates left ventricular.
Figure 6.

Main stages of ischemia and reperfusion‐related events resulting in myocardial injury from a circulatory viewpoint.
Sources of Funding
Dr Sezer is supported by the Turkish Academy of Sciences (TUBA). Dr Hausenloy is supported by the British Heart Foundation (FS/10/039/28270); the National Institute for Health Research, University College London Hospitals Biomedical Research Centre; Duke‐National University Singapore Medical School; the Singapore Ministry of Health's National Medical Research Council under its Clinician Scientist‐Senior Investigator scheme (NMRC/CSA‐SI/0011/2017) and Collaborative Centre Grant scheme (NMRC/CGAug16C006); and the Singapore Ministry of Education Academic Research Fund Tier 2 (MOE2016‐T2‐2‐021).
Disclosures
None.
J Am Heart Assoc. 2018;7:e009949 DOI: 10.1161/JAHA.118.009949.
References
- 1. Yeh RW, Sidney S, Chandra M, Sorel M, Selby JV, Go AS. Population trends in the incidence and outcomes of acute myocardial infarction. N Engl J Med. 2010;362:2155–2165. [DOI] [PubMed] [Google Scholar]
- 2. Schmidt M, Jacobsen JB, Lash TM, Bøtker HE, Sørensen HT. 25 year trends in first time hospitalization for acute myocardial infarction, subsequent short and long term mortality, and the prognostic impact of sex and comorbidity: a Danish nationwide cohort study. BMJ. 2012;344:e356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. O'Gara PT, Kushner FG, Ascheim DD, Casey DE Jr, Chung MK, de Lemos JA, Ettinger SM, Fang JC, Fesmire FM, Franklin BA, Granger CB, Krumholz HM, Linderbaum JA, Morrow DA, Newby LK, Ornato JP, Ou N, Radford MJ, Tamis‐Holland JE, Tommaso CL, Tracy CM, Woo YJ, Zhao DX; American College of Cardiology Foundation, American Heart Association Task Force on Practice Guidelines, American College of Emergency Physicians, Society for Cardiovascular Angiography and Interventions . 2013 ACCF/AHA guideline for the management of ST‐elevation myocardial infarction: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the American College of Emergency Physicians and Society for Cardiovascular Angiography and Interventions. Catheter Cardiovasc Interv. 2013;82:E1–E27. [DOI] [PubMed] [Google Scholar]
- 4. Steg PG, James SK, Atar D, Badano LP, Blomstrom‐Lundqvist C, Borger MA, Di Mario C, Dickstein K, Ducrocq G, Fernandez‐Aviles F, Gershlick AH, Giannuzzi P, Halvorsen S, Huber K, Juni P, Kastrati A, Knuuti J, Lenzen MJ, Mahaffey KW, Valgimigli M, van ‘t Hof A, Widimsky P, Zahger D. ESC guidelines for the management of acute myocardial infarction in patients presenting with ST‐segment elevation. Eur Heart J. 2012;33:2569–2619. [DOI] [PubMed] [Google Scholar]
- 5. Hamm CW, Bassand JP, Agewall S, Bax J, Boersma E, Bueno H, Caso P, Dudek D, Gielen S, Huber K, Ohman M, Petrie MC, Sonntag F, Uva MS, Storey RF, Wijns W, Zahger D; ESC Committee for Practice Guidelines . ESC guidelines for the management of acute coronary syndromes in patients presenting without persistent ST‐segment elevation: the Task Force for the management of acute coronary syndromes (ACS) in patients presenting without persistent ST‐segment elevation of the European Society of Cardiology (ESC). Eur Heart J. 2011;32:2999–3054. [DOI] [PubMed] [Google Scholar]
- 6. Torabi A, Cleland JG, Khan NK, Loh PH, Clark AL, Alamgir F, Caplin JL, Rigby AS, Goode K. The timing of development and subsequent clinical course of heart failure after a myocardial infarction. Eur Heart J. 2008;29:859–870. [DOI] [PubMed] [Google Scholar]
- 7. Ezekowitz JA, Kaul P, Bakal JA, Armstrong PW, Welsh RC, McAlister FA. Declining in‐hospital mortality and increasing heart failure incidence in elderly patients with first myocardial infarction. J Am Coll Cardiol. 2009;53:13–20. [DOI] [PubMed] [Google Scholar]
- 8. Krug A, du Mesnil de Rochemont W, Korb G. Blood supply of the myocardium after temporary coronary occlusion. Circ Res. 1966;19:57–62. [DOI] [PubMed] [Google Scholar]
- 9. Kloner RA, Ganote CE, Jennings RB. The ‘‘no‐reflow’’ phenomenon after temporary coronary occlusion in the dog. J Clin Invest. 1974;54:1496–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ito H, Tomooka T, Sakai N, Yu H, Higashino Y, Fujii K, Masuyama T, Kitabatake A, Minamino T. Lack of myocardial perfusion immediately after successful thrombolysis. A predictor of poor recovery of left ventricular function in anterior myocardial infarction. Circulation. 1992;85:1699–1705. [DOI] [PubMed] [Google Scholar]
- 11. Durante A, Camici PG. Novel insights into an “old” phenomenon: the no‐reflow. Int J Cardiol. 2015;187:273–280. [DOI] [PubMed] [Google Scholar]
- 12. Lombardo A, Niccoli G, Natale L, Bernardini A, Cosentino N, Bonomo L, Crea F. Impact of microvascular obstruction and infarct size on left ventricular remodeling in reperfused myocardial infarction: a contrast‐enhanced cardiac magnetic resonance imaging study. Int J Cardiovasc Imaging. 2012;28:835–842. [DOI] [PubMed] [Google Scholar]
- 13. Weir RA, Murphy CA, Petrie CJ, Martin TN, Balmain S, Clements S, Steedman T, Wagner GS, Dargie HJ, McMurray JJ. Microvascular obstruction remains a portent of adverse remodeling in optimally treated patients with left ventricular systolic dysfunction after acute myocardial infarction. Circ Cardiovasc Imaging. 2010;3:360–367. [DOI] [PubMed] [Google Scholar]
- 14. de Waha S, Desch S, Eitel I, Fuernau G, Lurz P, Leuschner A, Grothoff M, Gutberlet M, Schuler G, Thiele H. Relationship and prognostic value of microvascular obstruction and infarct size in ST‐elevation myocardial infarction as visualized by magnetic resonance imaging. Clin Res Cardiol. 2012;101:487–495. [DOI] [PubMed] [Google Scholar]
- 15. van Kranenburg M, Magro M, Thiele H, de Waha S, Eitel I, Cochet A, Cottin Y, Atar D, Buser P, Wu E, Lee D, Bodi V, Klug G, Metzler B, Delewi R, Bernhardt P, Rottbauer W, Boersma E, Zijlstra F, van Geuns RJ. Prognostic value of microvascular obstruction and infarct size, as measured by CMR in STEMI patients. JACC Cardiovasc Imaging. 2014;7:930–939. [DOI] [PubMed] [Google Scholar]
- 16. Sezer M, Aslanger EK, Cimen AO, Yormaz E, Turkmen C, Umman B, Nisanci Y, Bugra Z, Adalet K, Umman S. Concurrent microvascular and infarct remodeling after successful reperfusion of ST‐elevation acute myocardial infarction. Circ Cardiovasc Interv. 2010;3:208–215. [DOI] [PubMed] [Google Scholar]
- 17. Bulluck H, Foin N, Tan JW, Low AF, Sezer M, Hausenloy DJ. Invasive assessment of the coronary microcirculation in reperfused ST‐segment‐elevation myocardial infarction patients: where do we stand? Circ Cardiovasc Interv. 2017;10:e004373. [DOI] [PubMed] [Google Scholar]
- 18. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. [DOI] [PubMed] [Google Scholar]
- 19. Reffelmann T, Hale SL, Li G, Kloner RA. Relationship between no‐reflow and infarct size as influenced by the duration of ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2002;282:H766–H772. [DOI] [PubMed] [Google Scholar]
- 20. Funaro S, Galiuto L, Boccalini F, Cimino S, Canali E, Evangelio F, DeLuca L, Paraggio L, Mattatelli A, Gnessi L, Agati L. Determinants of microvascular damage recovery after acute myocardial infarction: results from the acute myocardial infarction contrast imaging (AMICI) multicentre study. Eur J Echocardiogr. 2011;12:306–312. [DOI] [PubMed] [Google Scholar]
- 21. Hollander MR, de Waard GA, Konijnenberg LS, Meijer‐van Putten RM, van den Brom CE, Paauw N, de Vries HE, van de Ven PM, Aman J, Van Nieuw‐Amerongen GP, Hordijk PL, Niessen HW, Horrevoets AJ, Van Royen N. Dissecting the effects of ischemia and reperfusion on the coronary microcirculation in a rat model of acute myocardial infarction. PLoS One. 2016;11:e0157233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lerman A, Holmes DR, Herrmann J, Gersh BJ. Microcirculatory dysfunction in ST‐elevation myocardial infarction: cause, consequence, or both? Eur Heart J. 2007;28:788–797. [DOI] [PubMed] [Google Scholar]
- 23. Zalewski J, Undas A, Godlewski J, Stepien E, Zmudka K. No‐reflow phenomenon after acute myocardial infarction is associated with reduced clot permeability and susceptibility to lysis. Arterioscler Thromb Vasc Biol. 2007;27:2258–2265. [DOI] [PubMed] [Google Scholar]
- 24. Rochitte CE, Lima JAC, Bluemke DA, Reeder SB, McVeigh ER, Furuta T, Becker LC, Melin JA. Magnitude and time course of microvascular obstruction and tissue injury after acute myocardial infarction. Circulation. 1998;98:1006–1014. [DOI] [PubMed] [Google Scholar]
- 25. Engler RL, Schmid‐Schönbein GW, Pavelec RS. Leukocyte capillary plugging in myocardial ischemia and reperfusion in the dog. Am J Pathol. 1983;111:98–111. [PMC free article] [PubMed] [Google Scholar]
- 26. Ambrosio G, Weisman HF, Mannisi JA, Becker LC. Progressive impairment of regional myocardial perfusion after initial restoration of postischemic blood flow. Circulation. 1989;80:1846–1861. [DOI] [PubMed] [Google Scholar]
- 27. Reffelmann T, Kloner RA. Microvascular reperfusion injury: rapid expansion of anatomic no‐reflow during reperfusion in the rabbit. Am J Physiol Heart Circ Physiol. 2002;283:H1099–H1107. [DOI] [PubMed] [Google Scholar]
- 28. Ørn S, Manhenke C, Greve OJ, Larsen AI, Bonarjee VV, Edvardsen T, Dickstein K. Microvascular obstruction is a major determinant of infarct healing and subsequent left ventricular remodelling following primary percutaneous coronary intervention. Eur Heart J. 2009;30:1978–1985. [DOI] [PubMed] [Google Scholar]
- 29. Reffelman T, Hale SH, Dow JS, Kloner RA. No‐reflow phenomenon persists long‐term after ischemia/reperfusion in the rat and predicts infarct expansion. Circulation. 2003;108:2911–2917. [DOI] [PubMed] [Google Scholar]
- 30. Yunoki K, Naruko T, Inoue T, Sugioka K, Inaba M, Iwasa Y, Komatsu R, Itoh A, Haze K, Yoshiyama M, Becker AE, Ueda M. Relationship of thrombus characteristics to the incidence of angiographically visible distal embolization in patients with ST‐segment elevation myocardial infarction treated with thrombus aspiration. JACC Cardiovasc Interv. 2013;6:377–385. [DOI] [PubMed] [Google Scholar]
- 31. Lønborg J, Kelbæk H, Helqvist S, Holmvang L, Jørgensen E, Saunamäki K, Kløvgaard L, Kaltoft A, Bøtker HE, Lassen JF, Thuesen L, Terkelsen CJ, Kofoed KF, Clemmensen P, Køber L, Engstrøm T. The impact of distal embolization and distal protection on long‐term outcome in patients with ST elevation myocardial infarction randomized to primary percutaneous coronary intervention: results from a randomized study. Eur Heart J Acute Cardiovasc Care. 2015;4:180–188. [DOI] [PubMed] [Google Scholar]
- 32. Napodano M, Peluso D, Marra MP, Frigo AC, Tarantini G, Buja P, Gasparetto V, Fraccaro C, Isabella G, Razzolini R, Iliceto S. Time dependent detrimental effects of distal embolization on myocardium and microvasculature during primary percutaneous coronary intervention. JACC Cardiovasc Interv. 2012;5:1170–1177. [DOI] [PubMed] [Google Scholar]
- 33. Skyschally A, Walter B, Heusch G. Coronary microembolization during early reperfusion: infarct extension but protection by ischemic postconditioning. Eur Heart J. 2013;34:3314–3321. [DOI] [PubMed] [Google Scholar]
- 34. Okamura A, Ito H, Iwakura K, Kurotobi T, Koyama Y, Date M, Higuchi Y, Inoue K, Fujii K. Clinical implication of distal embolization during coronary interventional procedures in patients with acute myocardial infarction. JACC Cardiovasc Interv. 2008;1:268–276. [DOI] [PubMed] [Google Scholar]
- 35. Sharma V, Jolly SS, Hamid T, Sharma D, Chiha J, Chan W, Fuchs F, Bui S, Gao P, Kassam S, Leung RC, Horák D, Romppanen HO, El‐Omar M, Chowdhary S, Stanković G, Kedev S, Rokoss MJ, Sheth T, Džavík V, Overgaard CB. Myocardial blush and microvascular reperfusion following manual thrombectomy during percutaneous coronary intervention for ST elevation myocardial infarction: insights from the TOTAL trial. Eur Heart J. 2016;37:1891–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kumbhani DJ, Bavry AA, Desai MY, Bangalore S, Bhatt DP. Role of aspiration and mechanical thrombectomy in patients with acute myocardial infarction undergoing primary angioplasty. J Am Coll Cardiol. 2013;62:1409–1418. [DOI] [PubMed] [Google Scholar]
- 37. Lipiecki J, Monzy S, Durel N, Cachin F, Chabrot P, Muliez A, Morand D, Maublant J, Ponsonnaille J. Effect of thrombus aspiration on infarct size and left ventricular function in high‐risk patients with acute myocardial infarction treated by percutaneous coronary intervention. Results of a prospective controlled pilot study. Am Heart J. 2009;157:583. [DOI] [PubMed] [Google Scholar]
- 38. Stone GW, Maehara A, Witzenbichler B, Godlewski J, Parise H, Dambrink JH, Ochala A, Carlton TW, Cristea E, Wolff SD, Brener SJ, Chowdhary S, El‐Omar M, Neunteufl T, Metzger DC, Karwoski T, Dizon JM, Mehran R, Gibson CM; INFUSE‐AMI Investigators . Intracoronary abciximab and aspiration thrombectomy in patients with large anterior myocardial infarction: the INFUSE‐AMI randomized trial. JAMA. 2012;307:1817–1826. [DOI] [PubMed] [Google Scholar]
- 39. De Carlo M, Aquaro GD, Palmieri C, Guerra E, Giannini C, Lombardi M, Berti S, Petroino AS. A prospective randomized trial of thrombectomy versus no thrombectomy in patients with ST‐segment elevation myocardial infarction and thrombus‐rich lesions. JACC Cardiovasc Interv. 2012;5:1223–1230. [DOI] [PubMed] [Google Scholar]
- 40. Sim DS, Ahn Y, Kim YH, Lee D, Seon HJ, Park KH, Yoon HJ, Yoon NS, Kim KH, Hong YJ, Park HW, Kim JH, Jeong MH, Cho JG, Park JC. Effect of manual thrombus aspiration during primary percutaneous coronary intervention on infarct size: evaluation with cardiac computed tomography. Int J Cardiol. 2013;168:4328–4330. [DOI] [PubMed] [Google Scholar]
- 41. Fröbert O, Lagerqvist B, Olivecrona GK, Omerovic E, Gudnason T, Maeng M, Aasa M, Angerås O, Calais F, Danielewicz M, Erlinge D, Hellsten L, Jensen U, Johansson AC, Kåregren A, Nilsson J, Robertson L, Sandhall L, Sjögren I, Ostlund O, Harnek J, James SK; TASTE Trial . Thrombus aspiration during ST‐segment elevation myocardial infarction. N Engl J Med. 2013;369:1587–1597. [DOI] [PubMed] [Google Scholar]
- 42. Jolly SS, Cairns JA, Yusuf S, Meeks B, Pogue J, Rokoss MJ, Kedev S, Thabane L, Stankovic G, Moreno R, Gershlick A, Chowdhary S, Lavi S, Niemelä K, Steg PG, Bernat I, Xu Y, Cantor WJ, Overgaard CB, Naber CK, Cheema AN, Welsh RC, Bertrand OF, Avezum A, Bhindi R, Pancholy S, Rao SV, Natarajan MK, ten Berg JM, Shestakovska O, Gao P, Widimsky P, Džavík V; TOTAL Investigators . Randomized trial of primary PCI with and without routine manual thrombectomy. N Engl J Med. 2015;372:1389–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sirker A, Mamas M, Kwok CS, Kontopantelis E, Ludman P, Hildick‐Smith D. British Cardiovascular Intervention Society (BCIS) outcomes from selective use of thrombectomy in patients undergoing primary percutaneous coronary intervention for ST‐segment elevation myocardial infarction. JACC Cardiovasc Interv. 2016;9:126–134. [DOI] [PubMed] [Google Scholar]
- 44. Elgendy IY, Huo T, Bhatt DL, Bavry AA. Is aspiration thrombectomy beneficial in patients undergoing primary percutaneous coronary intervention? Meta‐analysis of randomized trials. Circ Cardiovasc Interv. 2015;8:e002258. [DOI] [PubMed] [Google Scholar]
- 45. Götz AK, Zahler S, Stumpf P, Welsch U, Becker BF. Intracoronary formation and retention of microaggregates of leukocytes and platelets contribute to post ischemic myocardial dysfunction. Basic Res Cardiol. 2005;100:413–421. [DOI] [PubMed] [Google Scholar]
- 46. Driesen RB, Zalewski J, Vanden Driessche N, Vermeulen K, Bogaert J, Sipido KR, Van de Werf F, Claus P. Histological correlate of a cardiac magnetic resonance imaged microvascular obstruction in a porcine model of ischemia‐reperfusion. Cardiovasc Pathol. 2012;21:129–131. [DOI] [PubMed] [Google Scholar]
- 47. Sheridan FM, Cole PG, Ramage D. Leucocyte adhesion to the coronary microvasculature during ischemia and reperfusion in an in vivo canine model. Circulation. 1996;93:1784–1787. [DOI] [PubMed] [Google Scholar]
- 48. Frangogianis NG, Youker KA, Rossen RD, Gwechenberger M, Lindsey MH, Mendoza LH, Michael LH, Ballantyne CM, Smith CW, Entman ML. Cytokines and microcirculation in ischemia and reperfusion. J Mol Cell Cardiol. 1998;30:2567–2576. [DOI] [PubMed] [Google Scholar]
- 49. van der Laan AM, Hirsch A, Robbers LF, Nijveldt R, Lommerse I, Delewi R, van der Vleuten PA, Biemond BJ, Zwaginga JJ, van der Giessen WJ, Zijlstra F, van Rossum AC, Voermans C, van der Schoot CE, Piek JJ. A proinflammatory monocyte response is associated with myocardial injury and impaired functional outcome in patients with ST‐segment elevation myocardial infarction: monocytes and myocardial infarction. Am Heart J. 2012;163:57–65. [DOI] [PubMed] [Google Scholar]
- 50. Aurigemma C, Scalone G, Tomai F, Altamura L, De Persio G, Stazi A, Lanza GA, Crea F. Persistent enhanced platelet activation in patients with acute myocardial infarction and coronary microvascular obstruction: clinical implications. Thromb Haemost. 2014;111:122–130. [DOI] [PubMed] [Google Scholar]
- 51. Sezer M, Okcular I, Goren T, Oflaz H, Nisanci Y, Umman B, Mercanoglu F, Bilge AK, Meric M, Umman S. Association of haematological indices with the degree of microvascular injury in patients with acute anterior wall myocardial infarction treated with primary percutaneous coronary intervention. Heart. 2007;93:313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee MJ, Park SD, Kwon SW, Woo SI, Lee MD, Shin SH, Kim DH, Kwan J, Park KS. Relation between neutrophil‐to‐lymphocyte ratio and index of microcirculatory resistance in patients with ST‐segment elevation myocardial infarction undergoing primary percutaneous coronary intervention. Am J Cardiol. 2016;118:1323–1328. [DOI] [PubMed] [Google Scholar]
- 53. Damiano ER. The effect of the endothelial‐cell glycocalyx on the motion of red blood cells through capillaries. Microvasc Res. 1998;55:77–91. [DOI] [PubMed] [Google Scholar]
- 54. Weinbaum S, Tarbell JM, Damiano ER. The structure and function of the endothelial glycocalyx layer. Annu Rev Biomed Eng. 2007;9:121–167. [DOI] [PubMed] [Google Scholar]
- 55. Armstrong PW, Granger CB, Adams PX, Hamm C, Holmes D Jr, O'Neill WW, Todaro TG, Vahanian A, Van de Werf F; APEX AMI Investigators . Pexelizumab for acute ST‐elevation myocardial infarction in patients undergoing primary percutaneous coronary intervention: a randomized controlled trial. JAMA. 2007;297:43–51. [DOI] [PubMed] [Google Scholar]
- 56. Faxon DP, Gibbons RJ, Chronos NAF, Gurbel PA, Sheehan F; HALT‐MI investigators . The effect of the CD11/CD18 integrin receptor on infarct size in patients with acute myocardial infarction treated with direct angioplasty: the result of the HALT‐MI study. J Am Coll Cardiol. 2002;40:1199–1204. [DOI] [PubMed] [Google Scholar]
- 57. Thiele H, Wöhrle J, Hambrecht R, Rittger H, Birkemeyer R, Lauer B, Neuhaus P, Brosteanu O, Sick P, Wiemer M, Kerber S, Kleinertz K, Eitel I, Desch S, Schuler G. Intracoronary versus intravenous bolus abciximab during primary percutaneous coronary intervention in patients with acute ST‐elevation myocardial infarction: a randomised trial. Lancet. 2012;379:923–931. [DOI] [PubMed] [Google Scholar]
- 58. French CJ, Zaman AK, Kelm RJ Jr, Spees JL, Sobel BE. Vascular rhexis: loss of integrity of coronary vasculature in mice subjected to myocardial infarction. Exp Biol Med (Maywood). 2010;235:966–973. [DOI] [PubMed] [Google Scholar]
- 59. Pedersen CM, Barnes G, Schmidt MR, Bøtker HE, Kharbanda RK, Newby DE, Cruden NL. Ischaemia‐reperfusion injury impairs tissue plasminogen activator release in man. Eur Heart J. 2012;33:1920–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bach TL, Barsigian C, Yaen CH, Martinez J. Endothelial cell VE‐cadherin functions as a receptor for the beta15‐42 sequence of fibrin. J Biol Chem. 1998;273:30719–30728. [DOI] [PubMed] [Google Scholar]
- 61. Béguin S, Kumar R, Keularts I, Seligsohn U, Coller BS, Hemker HC. Fibrin‐dependent platelet procoagulant activity requires GPIb receptors and von Willebrand factor. Blood. 1999;93:564–570. [PubMed] [Google Scholar]
- 62. Goel MS, Diamond SL. Adhesion of normal erythrocytes at depressed venous shear rates to activated neutrophils, activated platelets, and fibrin polymerized from plasma. Blood. 2002;100:3797–3803. [DOI] [PubMed] [Google Scholar]
- 63. Zhang ZG, Choopp M, Goussev A, Lu D, Morris D, Tsang W, Powers C, Ho KL. Cerebral microvascular obstruction by fibrin is associated with upregulation of PAI‐1 acutely after onset of focal embolic ischemia in rats. J Neurosci. 1999;19:10898–10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Adhami F, Liao G, Morozov YM, Schloemer A, Schmithorst VJ, Lorenz JN, Dunn RS, Vorhees CV, Wills‐Karp M, Degen JL, Davis RJ, Mizushima N, Rakic P, Dardzinski BJ, Holland SK, Sharp FR, Kuan CY. Cerebral ischemia‐hypoxia induces intravascular coagulation and autophagy. Am J Pathol. 2006;169:566–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schoots IG, Levi M, Roossink EH, Bijlsma PB, van Gulik TM. Local intravascular coagulation and fibrin deposition on intestinal ischemia reperfusion in rats. Surgery. 2003;133:411–419. [DOI] [PubMed] [Google Scholar]
- 66. Yamada K, Miwa T, Liu J, Nangaku M, Song W‐C. Critical protection from renal ischemia reperfusion injury by CD55 and CD591. J Immunol. 2004;172:3869–3875. [DOI] [PubMed] [Google Scholar]
- 67. Ami RB, Barshtein G, Zeltser D, Goldberg Y, Shapira I, Roth A, Keren G, Miller H, Prochorov V, Eldor A, Berliner S, Yedgar S. Parameters of red blood cell aggregation as correlates of the inflammatory state. Am J Physiol Heart Circ Physiol. 2001;280:H1982–H1988. [DOI] [PubMed] [Google Scholar]
- 68. Sriramarao P, Languino LR, Altieri DC. Fibrinogen mediates leukocyte‐endothelium bridging in vivo at low shear forces. Blood. 1996;88:3416–3423. [PubMed] [Google Scholar]
- 69. Kupatt C, Habazettl H, Hanusch P, Wichels R, Hahnel D, Becker BF, Boekstegers P. c7E3Fab reduces postischemic leukocyte‐thrombocyte interaction mediated by fibrinogen. Implications for myocardial reperfusion injury. Arterioscler Thromb Vasc Biol. 2000;20:2226–2232. [DOI] [PubMed] [Google Scholar]
- 70. Sezer M, Oflaz H, Gören T, Okçular I, Umman B, Nişanci Y, Bilge AK, Sanli Y, Meriç M, Umman S. Intracoronary streptokinase after primary coronary intervention. N Engl J Med. 2007;356:1823–1834. [DOI] [PubMed] [Google Scholar]
- 71. Sezer M, Cimen A, Aslanger E, Elitok A, Umman B, Buğra Z, Yormaz E, Türkmen C, Adalet IS, Nişanci Y, Umman S. Effect of intracoronary streptokinase administered immediately after primary percutaneous coronary intervention on long‐term left ventricular infarct size, volumes, and function. J Am Coll Cardiol. 2009;54:1065–1071. [DOI] [PubMed] [Google Scholar]
- 72. Greco C, Pelliccia F, Tanzilli G, Tinti MD, Salenzi P, Cicerchia C, Schiariti M, Franzoni F, Speziale G, Gallo P, Gaudio C. Usefulness of local delivery of thrombolytics before thrombectomy in patients with ST‐segment elevation myocardial infarction undergoing primary percutaneous coronary intervention (the delivery of thrombolytics before thrombectomy in patients with ST‐segment elevation myocardial infarction undergoing primary percutaneous coronary intervention [DISSOLUTION] randomized trial). Am J Cardiol. 2013;112:630–635. [DOI] [PubMed] [Google Scholar]
- 73. Armstrong PW. Pharmacotherapy: intracoronary streptokinase in acute myocardial infarction. Nat Rev Cardiol. 2010;7:67–69. [DOI] [PubMed] [Google Scholar]
- 74. Berry C. A trial of low‐dose adjunctive alTeplase During prIMary PCI (T‐TIME). NCT02257294.
- 75. Aversano T, Becker LC. Persistence of coronary vasodilator reserve despite functionally significant flow reduction. Am J Physiol. 1985;248:H403–H411. [DOI] [PubMed] [Google Scholar]
- 76. Heusch G. The coronary circulation as a target of cardioprotection. Circ Res. 2016;118:1643–1658. [DOI] [PubMed] [Google Scholar]
- 77. Niccoli G, Rigattieri S, De Vita MR, Valgimigli M, Corvo P, Fabbiocchi F, Romagnoli E, De Caterina AR, La Torre G, Lo Schiavo P, Tarantino F, Ferrari R, Tomai F, Olivares P, Cosentino N, D'Amario D, Leone AM, Porto I, Burzotta F, Trani C, Crea F. Open‐label, randomized, placebo‐controlled evaluation of intracoronary adenosine or nitroprusside after thrombus aspiration during primary percutaneous coronary intervention for the prevention of microvascular obstruction in acute myocardial infarction: The REOPEN‐AMI study (Intracoronary Nitroprusside Versus Adenosine in Acute Myocardial Infarction). JACC Cardiovasc Interv. 2013;6:580–589. [DOI] [PubMed] [Google Scholar]
- 78. Bulluck H, Sirker A, Loke YK, Garcia‐Dorado D, Hausenloy DJ. Clinical benefit of adenosine as an adjunct to reperfusion in ST‐elevation myocardial infarction patients: an updated meta‐analysis of randomized controlled trials. Int J Cardiol. 2016;202:228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nazir SA, McCann GP, Greenwood JP, Kunadian V, Khan JN, Mahmoud IZ, Blackman DJ, Been M, Abrams KR, Shipley L, Wilcox R, Adgey AA, Gershlick AH. Strategies to attenuate microvascular obstruction during primary PCI: the randomized reperfusion facilitated by local adjunctive therapy in ST‐elevation myocardial infarction trial. Eur Heart J. 2016;37:1910–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Desai KV, Laine GA, Stewart RH, Cox CS Jr, Quick CM, Allen SJ, Fischer UM. Mechanics of the left ventricular myocardial interstitium: effects of acute and chronic myocardial edema. Am J Physiol Heart Circ Physiol. 2008;294:H2428–H2434. [DOI] [PubMed] [Google Scholar]
- 81. Schwartz BG, Kloner RA. Coronary no‐reflow. J Moll Cell Cardiol. 2012;52:873–882. [DOI] [PubMed] [Google Scholar]
- 82. Payne AR, Berry C, Kellman P, Anderson R, Hsu LY, Chen MY, McPhaden AR, Watkins S, Schenke W, Wright V, Lederman RJ, Aletras AH, Arai AE. Bright‐blood T(2)‐weighted MRI has high diagnostic accuracy for myocardial hemorrhage in myocardial infarction: a preclinical validation study in swine. Circ Cardiovasc Imaging. 2011;4:738–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Dall'Armellina E, Piechnik SK, Ferreira VM, Si QL, Robson MD, Francis JM, Cuculi F, Kharbanda RK, Banning AP, Choudhury RP, Karamitsos TD, Neubauer S. Cardiovascular magnetic resonance by non contrast T1‐mapping allows assessment of severity of injury in acute myocardial infarction. J Cardiovasc Magn Reson. 2012;14:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gao XM, Wu QZ, Kiriazis H, Su Y, Han LP, Pearson JT, Taylor AJ, Du XJ. Microvascular leakage in acute myocardial infarction: characterization by histology, biochemistry, and magnetic resonance imaging. Am J Physiol Heart Circ Physiol. 2017;312:H1068–H1075. [DOI] [PubMed] [Google Scholar]
- 85. Kloner RA, Rude RE, Carlson N, Maroko PR, DeBoer LW, Braunwald E. Ultrastructural evidence of microvascular damage and myocardial cell injury after coronary artery occlusion: which comes first? Circulation. 1980;62:945–952. [DOI] [PubMed] [Google Scholar]
- 86. Fernández‐Jiménez R, Sánchez‐González J, Agüero J, García‐Prieto J, López‐Martín GJ, García‐Ruiz JM, Molina‐Iracheta A, Rosselló X, Fernández‐Friera L, Pizarro G, García‐Álvarez A, Dall'Armellina E, Macaya C, Choudhury RP, Fuster V, Ibáñez B. Myocardial edema after ischemia/reperfusion is not stable and follows a bimodal pattern: imaging and histological tissue characterization. J Am Coll Cardiol. 2015;65:315–323. [DOI] [PubMed] [Google Scholar]
- 87. Fernández‐Jiménez R, García‐Prieto J, Sánchez‐González J, Agüero J, López‐Martín GJ, Galán‐Arriola C, Molina‐Iracheta A, Doohan R, Fuster V, Ibáñez B. Pathophysiology underlying the bimodal edema phenomenon after myocardial ischemia/reperfusion. J Am Coll Cardiol. 2015;66:816–828. [DOI] [PubMed] [Google Scholar]
- 88. Carrick D, Haig C, Ahmed N, Rauhalammi S, Clerfond G, Carberry J, Mordi I, McEntegart M, Petrie MC, Eteiba H, Hood S, Watkins S, Lindsay MM, Mahrous A, Welsh P, Sattar N, Ford I, Oldroyd KG, Radjenovic A, Berry C. Temporal evolution of myocardial hemorrhage and edema in patients after acute ST‐segment elevation myocardial infarction: pathophysiological insights and clinical implications. J Am Heart Assoc. 2016;5:e002834 DOI: 10.1161/JAHA.115.002834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Nordlund D, Klug G, Heiberg E, Koul S, Larsen TH, Hoffmann P, Metzler B, Erlinge D, Atar D, Aletras AH, Carlsson M, Engblom H, Arheden H. Multi‐vendor, multicentre comparison of contrast‐enhanced SSFP and T2‐STIR CMR for determining myocardium at risk in ST‐elevation myocardial infarction. Eur Heart J Cardiovasc Imaging. 2016;17:744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Fernández‐Jiménez R, Barreiro‐Pérez M, Martin‐García A, Sánchez‐González J, Agüero J, Galán‐Arriola C, García‐Prieto J, Díaz‐Pelaez E, Vara P, Martinez I, Zamarro I, Garde B, Sanz J, Fuster V, Sánchez PL, Ibanez B. Dynamic edematous response of the human heart to myocardial infarction: implications for assessing myocardial area at risk and salvage. Circulation. 2017;136:1288–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Fernández‐Jiménez R, Galán‐Arriola C, Sánchez‐González J, Agüero J, López‐Martín GJ, Gomez‐Talavera S, Garcia‐Prieto J, Benn A, Molina‐Iracheta A, Barreiro‐Pérez M, Martin‐García A, García‐Lunar I, Pizarro G, Sanz J, Sánchez PL, Fuster V, Ibanez B. Effect of ischemia duration and protective interventions on the temporal dynamics of tissue composition after myocardial infarction. Circ Res. 2017;121:439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sezer M, Umman S. Gradual reperfusion and microvascular integrity and myocardial oedema patients undergoing primary PCI. (GUARD trial) NCT02732080.
- 93. Kidambi A, Biglands JD, Higgins DM, Ripley DP, Zaman A, Broadbent DA, McDiarmid AK, Swoboda PP, Al Musa T, Erhayiem B, Greenwood JP, Plein S. Susceptibility‐weighted cardiovascular magnetic resonance in comparison to T2 and T2 star imaging for detection of intramyocardial hemorrhage following acute myocardial infarction at 3 Tesla. J Cardiovasc Magn Reson. 2014;16:86 DOI: 10.1186/s12968-014-0086-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Betgem RP, de Waard GA, Nijveldt R, Beek AM, Escaned J, van Royen N. Intramyocardial haemorrhage after acute myocardial infarction. Nat Rev Cardiol. 2015;12:156–167. [DOI] [PubMed] [Google Scholar]
- 95. Carrick D, Haig C, Ahmed N, McEntegart M, Petrie MC, Eteiba H, Hood S, Watkins S, Lindsay MM, Davie A, Mahrous A, Mordi I, Rauhalammi S, Sattar N, Welsh P, Radjenovic A, Ford I, Oldroyd KG, Berry C. Myocardial hemorrhage after acute reperfused ST‐segment‐elevation myocardial infarction relation to microvascular obstruction and prognostic significance. Circ Cardiovasc Imaging. 2016;9:e004148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Robbers LF, Eerenberg ES, Teunissen PF, Jansen MF, Hollander MR, Horrevoets AJ, Knaapen P, Nijveldt R, Heymans MW, Levi MM, van Rossum AC, Niessen HW, Marcu CB, Beek AM, van Royen N. Magnetic resonance imaging‐defined areas of microvascular obstruction after acute myocardial infarction represent microvascular destruction and hemorrhage. Eur Heart J. 2013;34:2346–2353. [DOI] [PubMed] [Google Scholar]
- 97. Husser O, Monmeneu JV, Sanchis J, Nunez J, Lopez‐Lereu MP, Bonanad C, Chaustre F, Gomez C, Bosch MJ, Hinarejos R, Chorro FJ, Riegger GA, Llacer A, Bodi V. Cardiovascular magnetic resonance‐derived intramyocardial hemorrhage after STEMI: influence on long‐term prognosis, adverse left ventricular remodeling and relationship with microvascular obstruction. Int J Cardiol. 2013;167:2047–2054. [DOI] [PubMed] [Google Scholar]
- 98. Mather AN, Fairbairn TA, Ball SG, Greenwood JP, Plein S. Reperfusion hemorrhage as determined by cardiovascular MRI is a predictor of adverse left ventricular remodeling and markers of late arrhythmic risk. Heart. 2011;97:453–459. [DOI] [PubMed] [Google Scholar]
- 99. Bulluck H, Rosmini S, Abdel‐Gadir A, White SK, Bhuva AN, Treibel TA, Fontana M, Ramlall M, Hamarneh A, Sirker A, Herrey AS, Manisty C, Yellon DM, Kellman P, Moon JC, Hausenloy DJ. Residual myocardial iron following intramyocardial hemorrhage during the convalescent phase of reperfused ST‐segment‐elevation myocardial infarction and adverse left ventricular remodeling. Circ Cardiovasc Imaging. 2016;9:e004940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kidambi A, Mather AN, Motwani M, Swoboda P, Uddin A, Greenwood JP, Plein S. The effect of microvascular obstruction and intramyocardial hemorrhage on contractile recovery in reperfused myocardial infarction: insights from cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2013;15:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Cokic I, Kali A, Yang HJ, Yee R, Tang R, Tighiouart M, Wang X, Jackman WS, Chugh SS, White JA, Dharmakumar R. Iron‐sensitive cardiac magnetic resonance imaging for prediction of ventricular arrhythmia risk in patients with chronic myocardial infarction: early evidence. Circ Cardiovasc Imaging. 2015;8:e003642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Eitel I, Kubusch K, Strohm O, Desch S, Mikami Y, de Waha S, Gutberlet M, Schuler G, Friedrich MG, Thiele H. Prognostic value and determinants of a hypointense infarct core in T2‐weighted cardiac magnetic resonance in acute reperfused ST‐elevation‐myocardial infarction. Circ Cardiovasc Imaging. 2011;4:354–362. [DOI] [PubMed] [Google Scholar]
- 103. Amier RP, Tijssen RYG, Teunissen PFA, Fernández‐Jiménez R, Pizarro G, García‐Lunar I, Bastante T, van de Ven PM, Beek AM, Smulders MW, Bekkers SCAM, van Royen N, Ibanez B, Nijveldt R. Predictors of intramyocardial hemorrhage after reperfused ST‐segment elevation myocardial infarction. J Am Heart Assoc. 2017;6:e005651 DOI: 10.1161/JAHA.117.005651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Willerson JT, Scales F, Mukherjee A, Platt M, Templeton GH, Fink GS, Buja LM. Abnormal myocardial fluid retention as an early manifestation of ischemic injury. Am J Pathol. 1977;87:159–188. [PMC free article] [PubMed] [Google Scholar]
- 105. Waller BF, Rothbaum DA, Pinkerton CA, Cowley MJ, Linnemeier TJ, Orr C, Irons M, Helmuth RA, Wills ER, Aust C. Status of the myocardium and infarct‐related coronary artery in 19 necropsy patients with acute recanalization using pharmacologic (streptokinase, r‐tissue plasminogen activator), mechanical (percutaneous transluminal coronary angioplasty) or combined types of reperfusion therapy. J Am Coll Cardiol. 1987;9:785–801. [DOI] [PubMed] [Google Scholar]
- 106. Mathey DG, Schofer J, Kuck KH, Beil U, Klöppel G. Transmural, haemorrhagic myocardial infarction after intracoronary streptokinase. Clinical, angiographic, and necropsy findings. Br Heart J. 1982;48:546–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Bekkers SC, Smulders MW, Passos VL, Leiner T, Waltenberger J, Gorgels AP, Schalla S. Clinical implications of microvascular obstruction and intramyocardial haemorrhage in acute myocardial infarction using cardiovascular magnetic resonance imaging. Eur Radiol. 2010;20:2572–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Beek AM, Nijveldt R, van Rossum AC. Intramyocardial hemorrhage and microvascular obstruction after primary percutaneous coronary intervention. Int J Cardiovasc Imaging. 2010;26:49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Payne AR, Berry C, Doolin O, McEntegart M, Petrie MC, Lindsay MM, Hood S, Carrick D, Tzemos N, Weale P, McComb C, Foster J, Ford I, Oldroyd KG. Microvascular resistance predicts myocardial salvage and infarct characteristics in ST‐elevation myocardial infarction. J Am Heart Assoc. 2012;1:e002246 DOI: 10.1161/JAHA.112.002246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Carrick D, Haig C, Ahmed N, Carberry J, Yue May VT, McEntegart M, Petrie MC, Eteiba H, Lindsay M, Hood S, Watkins S, Davie A, Mahrous A, Mordi I, Ford I, Radjenovic A, Oldroyd KG, Berry C. Comparative prognostic utility of indexes of microvascular function alone or in combination in patients with an acute ST‐segment elevation myocardial infarction. Circulation. 2016;134:1833–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Hamirani YS, Wong A, Kramer CM, Salerno M. Effect of microvascular obstruction and intramyocardial hemorrhage by CMR on LV remodeling and outcomes after myocardial infarction: a systematic review and meta‐analysis. JACC Cardiovasc Imaging. 2014;7:940–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Xu Y, Feng WL, Miao Z, Ling F. Treatment and outcome of intracranial hemorrhage after carotid artery stenting. A ten year single center experience. Interv Neuroradiol. 2009;15:316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Okamura A, Nakaoka M, Ohbayashi N, Yahara K, Nabika K. Intraoperative idiopathic subarachnoid hemorrhage during carotid artery stenting: a case report and literature review. Interv Neuroradiol. 2015;21:592–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Van Herck PL, Carlier SG, Claeys MJ, Haine SE, Gorissen P, Miljoen H, Bosmans JM, Vrints CJ. Coronary microvascular dysfunction after myocardial infarction: increased coronary zero flow pressure both in the infarcted and in the remote myocardium is mainly related to left ventricular filling pressure. Heart. 2007;93:1231–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Ito H, Terai K, Iwakura K, Kawase I, Fujii K. Hemodynamics of microvascular dysfunction in patients with anterior wall acute myocardial infarction. Am J Cardiol. 2004;94:209–212. [DOI] [PubMed] [Google Scholar]