

Abstract
Background
Tetrahydrobiopterin is a cofactor of endothelial NO synthase (eNOS), which is critical to embryonic heart development. We aimed to study the effects of sapropterin (Kuvan), an orally active synthetic form of tetrahydrobiopterin on eNOS uncoupling and congenital heart defects (CHDs) induced by pregestational diabetes mellitus in mice.
Methods and Results
Adult female mice were induced to pregestational diabetes mellitus by streptozotocin and bred with normal male mice to produce offspring. Pregnant mice were treated with sapropterin or vehicle during gestation. CHDs were identified by histological analysis. Cell proliferation, eNOS dimerization, and reactive oxygen species production were assessed in the fetal heart. Pregestational diabetes mellitus results in a spectrum of CHDs in their offspring. Oral treatment with sapropterin in the diabetic dams significantly decreased the incidence of CHDs from 59% to 27%, and major abnormalities, such as atrioventricular septal defect and double‐outlet right ventricle, were absent in the sapropterin‐treated group. Lineage tracing reveals that pregestational diabetes mellitus results in decreased commitment of second heart field progenitors to the outflow tract, endocardial cushions, and ventricular myocardium of the fetal heart. Notably, decreased cell proliferation and cardiac transcription factor expression induced by maternal diabetes mellitus were normalized with sapropterin treatment. Furthermore, sapropterin administration in the diabetic dams increased eNOS dimerization and lowered reactive oxygen species levels in the fetal heart.
Conclusions
Sapropterin treatment in the diabetic mothers improves eNOS coupling, increases cell proliferation, and prevents the development of CHDs in the offspring. Thus, sapropterin may have therapeutic potential in preventing CHDs in pregestational diabetes mellitus.
Keywords: animal model cardiovascular disease, cardiac embryology, congenital cardiac defect, diabetes mellitus
Subject Categories: Animal Models of Human Disease, Basic Science Research, Congenital Heart Disease, Pharmacology
Clinical Perspective
What Is New?
Sapropterin treatment in the diabetic dams lowers the incidence of congenital heart defects in their offspring.
Pregestational diabetes mellitus reduces the commitment of second heart field progenitors to the outflow tract, endocardial cushions, and ventricular myocardium of the fetal heart.
Sapropterin treatment in the diabetic dams improves cell proliferation and cardiac transcription factor expression in the fetal heart.
Sapropterin treatment in the diabetic dams improves endothelial NO synthase function and lowers reactive oxygen species levels in the fetal heart.
What Are the Clinical Implications?
Sapropterin (Kuvan) is an orally active synthetic form of tetrahydrobiopterin and a US Food and Drug Administration–approved drug to treat phenylketonuria. Our study suggests that sapropterin may also have therapeutic potential in women with pregestational diabetes mellitus to prevent congenital heart defects in their children.
Congenital heart defects (CHDs) are the most common structural birth defect, occurring in 1% to 5% of live births, making them the leading cause of death in the first year of infant life.1, 2 The prevalence of CHDs has been rapidly increasing,3 and it is estimated that ≈2.4 million Americans, including 1 million children, are living with a congenital malformation of the heart.2 CHDs are formed when complex cellular and molecular processes underlying embryonic heart development are disturbed. The heart is developed from 3 pools of progenitor cells: the first heart field (FHF), the second heart field (SHF), and the cardiac neural crest (CNC).4 The FHF progenitors initially form the primary heart tube. SHF cells are then added to the heart tube to form the right ventricle (RV) and produce myocardial and endothelial cells of the outflow tract (OFT) and semilunar valves, as well as the vascular smooth muscle cells at the base of the aorta and pulmonary trunk. The CNC cells contribute to septation of the OFT and remodeling of semilunar valves, whereas the left ventricle (LV) is mainly formed from FHF progenitors. The SHF is particularly significant to CHDs because many common cardiac abnormalities, including atrial and ventricular septal defects (VSDs), cardiac valve malformation, double‐outlet RV, and truncus arteriosus, are caused by defects in SHF progenitors.4
Pregestational diabetes mellitus (type 1 or 2) in the mother increases the risk of a CHD in the child by >4‐fold.5, 6 Although good glycemic control in diabetic mothers lowers the risk, the incidence of CHDs in their children is still higher than in the general population.7, 8 The prevalence of pregestational diabetes mellitus has nearly doubled from 0.58% to 1.06% from 1996 to 2014 in Northern California,9 and has reached 4.3% in Saudi Arabia.10 As the prevalence of pregestational diabetes mellitus further increases in women during their reproductive age, more individuals will be born with CHDs, inevitably placing a large burden on the healthcare system.11, 12
Uncontrolled maternal diabetes mellitus is not conducive to proper gestation. Hyperglycemia leads to cellular oxidative stress through numerous pathways,13 which include increased electron transport chain flow, resulting in mitochondrial dysfunction, nonenzymatic protein glycosylation, and glucose auto‐oxidation, all contributing to reactive oxygen species (ROS) generation.14, 15 Increased oxidative stress can lead to the inactivation of many molecules and proteins necessary for proper heart development. Endothelial NO synthase (eNOS) is intimately regulated by oxidation‐reduction balance within the cell and is vital for cardiogenesis.16 eNOS expression in the embryonic heart regulates cell growth and protects early cardiac progenitors against apoptosis.17 The importance of eNOS in heart development has been demonstrated in eNOS−/− mice by a spectrum of cardiovascular anomalies, such as VSDs, valvular malformations, and hypoplastic coronary arteries.17, 18, 19
Tetrahydrobiopterin has antioxidant properties and is a critical cofactor for eNOS function.20 It is required for eNOS dimer stabilization and is an allosteric modulator of arginine binding to the active site.21 In states of oxidative stress, tetrahydrobiopterin levels decline, and eNOS is uncoupled, resulting in decreased NO synthesis and increased superoxide production, perpetuating the oxidative environment of the cell.22 The production of ROS is amplified by this feedback loop, further inducing eNOS uncoupling. Treatment with tetrahydrobiopterin has been shown to recouple eNOS and improve vascular endothelial function in diabetes mellitus.23, 24 However, the potential of tetrahydrobiopterin to reduce the severity and incidence of CHDs is not known. Sapropterin dihydrochloride (Kuvan) is an orally active, synthetic form of tetrahydrobiopterin and a US Food and Drug Administration–approved drug for the treatment of phenylketonuria.25 The present study was aimed to examine the effects of sapropterin in mice with pregestational diabetes mellitus. We hypothesized that sapropterin treatment during gestation recouples eNOS, improves cell proliferation in SHF‐derived cells, and reduces CHD incidence in the offspring of mice with pregestational diabetes mellitus.
Methods
The data, analytic methods, and study materials will be available from the corresponding author on reasonable request to other researchers for purposes of reproducing the results or replicating the procedure.
Animals
All procedures were performed in accordance with the Canadian Council on Animal Care guidelines and approved by the Animal Care Committee at Western University. C57BL/6 wild‐type and Rosa26 mTmG mice were purchased from Jackson Laboratory (Bar Harbor, ME). Mef2c cre/+ embryos were obtained from the Mutant Mouse Regional Resource Center (Chapel Hill, NC) and rederived. All animals were housed in a 12‐hour light/dark cycle and given ad libitum access to standard chow and water. A breeding program was established to generate embryonic, fetal, and postnatal mice.
Induction of Diabetes Mellitus and Sapropterin Treatment
A study flow chart in Figure 1 illustrates timelines of saline or streptozotocin injection, breeding, sapropterin or insulin treatment, and assessments of fetal hearts in 5 groups of mice. Female C57BL/6 mice, 8 to 10 weeks old, were made diabetic through 5 consecutive daily injections of streptozotocin (50 mg/kg body weight, IP; Sigma) freshly dissolved in sterile saline. Mice were randomly assigned to streptozotocin (n=37) or saline treatment (n=19) groups. One week after the last streptozotocin injection, nonfasting blood glucose levels were measured with a tail snip procedure using a glucose meter (One Touch Ultra2; LifeScan, Burnaby, BC, Canada). Mice were categorized as diabetic if blood glucose measurements exceeded 11 mmol/L and were subsequently bred to 10‐ to 12‐week‐old C57BL/6 male mice. In the morning, when a vaginal plug was observed indicating embryonic day (E) 0.5, the female diabetic mouse was placed in a separate cage with littermates. A cohort of diabetic and control female mice was treated with sapropterin dihydrochloride (Kuvan; BioMarin Pharmaceutical Inc) at a dose of 10 mg/kg body weight per day during gestation. Sapropterin was dissolved in water and mixed with a small amount of peanut butter in a weigh boat, ensuring it was fully consumed by the mouse. At the time of feeding, mice were separated and individually housed in cages for ≈15 minutes until the sapropterin or vehicle containing peanut butter mixture was fully consumed under an investigator's watch (A.E.). All mice were fed in the morning, once per day. Nonfasting blood glucose levels were monitored throughout pregnancy. To prevent hyperglycemia, a long‐acting form of insulin (Lantus; Sanofi Aventis) was administered SC to a cohort of diabetic dams (n=3) at a dose of 0.5 U/d.
Figure 1.

Experimental design to examine the effects of sapropterin (tetrahydrobiopterin) on congenital heart defects induced by pregestational diabetes mellitus. A study flow chart illustrates timelines of saline or streptozotocin injection, breeding, sapropterin or insulin treatment, and assessments of fetal hearts in 5 groups of mice. BG indicates blood glucose; E, embryonic day; eNOS, endothelial NO synthase; IP, intraperitoneal injection; OFT, outflow tract; PO, oral administration; ROS, reactive oxygen species.
Histological and Immunohistochemical Analysis
Fetal samples were harvested at E10.5, E12.5, and E18.5 for histological and immunohistochemical analysis. To diagnose CHDs in E18.5 hearts, fetuses were decapitated, and the isolated thorax was fixed overnight in 4% paraformaldehyde, dehydrated in ethanol, and paraffin embedded. Samples were divided into section (5‐μm slices), and they were stained with hematoxylin/eosin or toluidine blue to visualize morphological characteristics. Images were taken and analyzed using a light microscope (Observer D1; Zeiss, Germany). Embryonic samples at E10.5 and E12.5 were fixed in 4% paraformaldehyde for 1 and 2 hours, respectively, and processed as previously described. Immunostaining to analyze cell proliferation at E10.5 using anti–phosphohistone H3 antibody (1:1000; Abcam) and sex determination at E18.5 using anti–sex‐determining region Y protein antibody (1:200; Santa Cruz) were performed after antigen retrieval in citrate buffer (10 mmol/L, pH 6). This was followed by incubation with biotinylated goat anti‐mouse IgG (1:300; Vector Laboratories) secondary antibody. The signal was amplified by the ABC reagent (Vector Laboratories), allowing for visualization through 3‐3′ diaminobenzidine tetrahydrochloride (Sigma) with hematoxylin as a counterstain. Blinded phosphohistone H3–positive cell counts within the OFT were taken from at least 3 heart sections per heart and normalized to OFT length.
Lineage Tracing the SHF
Fate mapping of SHF progenitors was performed using the SHF‐specific Mef2c cre/+ transgenic mouse and the global double fluorescent Cre reporter line Rosa26 mTmG that has LoxP sites on either side of a tomato‐red fluorescence membrane protein cassette, which is proceeded by a green fluorescent protein (GFP) cassette. In the Mef2c Cre/+ transgenic line with C57BL/6 background, elements of the Mef2c promoter drive Cre recombinase expression in all SHF‐derived cells. When crossed with the Rosa26 mTmG mice, this results in a GFP signal that is detected in all SHF‐derived cells.26, 27 In all other tissues, the absence of the Cre results in membrane protein expression and red fluorescence. Diabetes mellitus was induced in homozygous Rosa26 mTmG females (8–10 weeks old) by streptozotocin, as previously described. Hyperglycemic Rosa26 mTmG female mice were crossed with Mef2c cre/+;Rosa26 mTmG males to generate E9.5 and E12.5 Mef2c cre/+;Rosa26 mTmG embryos, which were fixed, embedded, and divided into sections in the same manner as previously described. Immunostaining for membrane‐bound GFP was conducted using an anti‐GFP (1:500; Abcam) primary antibody, followed by biotinylated goat anti‐rabbit IgG (1:300; Vector Laboratories) secondary antibody using 3‐3′ diaminobenzidine tetrahydrochloride for visualization. The SHF‐derived cells, which are GFP+, were blindly quantified and compared between control and diabetic groups.
Analysis of Superoxide Levels
Hearts collected from E12.5 fetuses from all 4 groups were cryosectioned (CM1950; Leica, Germany) into 8‐μm‐thick slices and placed onto slides. A subset of cryosectioned embryonic hearts from untreated and sapropterin‐treated diabetic dams was incubated with either 300 μmol/L Nω‐nitro‐l‐arginine methyl ester (Sigma), a NOS inhibitor, or 100 U/mL superoxide dismutase (Sigma), a recombinant antioxidant enzyme for 30 minutes. Samples were then probed with 2 μmol/L dihydroethidium (Invitrogen Life Technologies, Burlington, ON, Canada) for 30 minutes in a dark humidity chamber at 37°C. After cover glass was mounted, dihydroethidium fluorescence signals were visualized using a fluorescence microscope (Observer D1; Zeiss, Germany). A total of 5 to 8 images from each sample were captured at fixed exposure times for all groups, and the fluorescence intensity per myocardial area was blindly quantified using AxioVision software.
Measurement of Cardiac Function
Pregnant dams were anesthetized, and M‐mode echocardiography images of E18.5 embryonic hearts were recorded using the Vevo 2100 ultrasound imaging system with an MS 700 transducer (VisualSonics, Toronto, ON, Canada), as previously described.18, 19 Briefly, an incision was made to the abdominal wall of the pregnant dam to expose the uterine sacs containing the E18.5 fetuses. The transducer was aligned to the uterine sac to obtain a short‐axis view of the fetal heart. The end‐diastolic LV internal diameter and end‐systolic LV internal diameter were measured from the short‐axis M‐mode images to calculate LV ejection fraction and fractional shortening.
Western Blotting for eNOS Dimers and Monomers
Ventricular myocardial tissues from E12.5 hearts were dissected in PBS and used for measurement of eNOS dimerization. To not disrupt the dimer, proteins were isolated from 3 pooled hearts from each group via sonication using a nonreducing lysis buffer, on ice.28 Protein lysates without boiling were run on 8% SDS‐PAGE at 4°C, followed by transferring to a nitrocellulose membrane, and immunoblotted with anti‐eNOS polyclonal antibody (1:3000; Santa Cruz). This technique resulted in 2 distinct bands at 260 and 130 kDa, representing the eNOS dimer and monomer, respectively.28 Proteins isolated from cultured coronary artery microvascular endothelial cells were boiled and separated with the sample, acting as an eNOS monomer size control.
Real‐Time Reverse Transcription–Polymerase Chain Reaction
Total RNA was isolated from E12.5 hearts using TRIzol reagent (Invitrogen). A total of 200 ng of RNA was synthesized into cDNA with Moloney murine leukemia virus reverse transcriptase and random primers. Real‐time PCR was conducted on cDNA using Evagreen qPCR MasterMix (Applied Biological Systems, Vancouver, BC, Canada). Primers were designed for Gata4, Gata5, Nkx2.5, Tbx5, Notch1, GTP cyclohydrolase 1 (GCH1), and dihydrofolate reductase (DHFR) using the Primer3 software v 4.1.0. The primer sequences are listed in Table 1. Eppendorf Realplex (Eppendorf, Hamburg, Germany) was used to amplify samples for 35 cycles. Values were normalized to 28S ribosomal RNA, and mRNA levels were extrapolated through a comparative threshold cycle method.19
Table 1.
Specific Primer Sequences Used in Real‐Time PCR Analysis
| Gene | Accession No. | Product Size | Primer Sequence |
|---|---|---|---|
| Gata4 | NM_008092.3 | 134 | Forward: GCCTGCGATGTCTGAGTGAC |
| Reverse: CACTATGGGCACAGCAGCTC | |||
| Gata5 | NM_008093.2 | 167 | Forward: ACCCCACAACCTACCCAGCA |
| Reverse: GCCCTCACCAGGGAACTCCT | |||
| Nkx2.5 | NM_008700.2 | 162 | Forward: GACAGCGGCAGGACCAGACT |
| Reverse: CGTTGTAGCCATAGGCATTG | |||
| Tbx5 | NM_011537.3 | 103 | Forward: AGGAGCACAGTGAGGCACAA |
| Reverse: GGGCCAGAGACACCATTCTC | |||
| Notch1 | NM_008714.3 | 142 | Forward: CAGCTTGCACAACCAGACAGA |
| Reverse: TAACGGAGTACGGCCCATGT | |||
| Gch1 | NM_008102.3 | 117 | Forward: TCAAGAGCGCCTCACCAAAC |
| Reverse: TTCTGCACGCCTCGCATTAC | |||
| Dhfr | NM_010049.3 | 153 | Forward: GAATCAACCAGGCCACCTCA |
| Reverse: TTGATGCCTTTTTCCTCCTG | |||
| 28S | NR_003279.1 | 178 | Forward: GGGCCACTTTTGGTAAGCAG |
| Reverse: TTGATTCGGCAGGTGAGTTG |
PCR indicates polymerase chain reaction.
Determination of Biopterin Levels
Tissue and plasma biopterin levels were determined using ultraperformance liquid chromatography coupled to mass spectrometry. Briefly, dams were anesthetized with an intraperitoneal injection of ketamine and xylazine cocktail at E12.5. Whole embryos were harvested, followed by blood collections from the dams for ultraperformance liquid chromatography analysis. About 1 mL of blood was collected via cardiac puncture using a 23‐gauge heparinized needle inserted into the beating LV of the dam. Plasma samples were then diluted 1:4 with 150 μL of acetonitrile containing internal standard (100 μmol/L aminopimelic acid and 2.5 μmol/L chlorpromazine) before injection into the ultraperformance liquid chromatography instrument. Total tetrahydrobiopterin and 7,8‐dihydrobiopterin levels were assessed using a Waters Acuity I Class ultraperformance liquid chromatography system coupled to a XEVO G2‐S quadrupole time‐of‐flight mass spectrometer (Waters Corporation, Milford, MA), as previously described.29
Statistical Analysis
Data are presented as the mean±SEM. Statistical analysis was performed using GraphPad Prism, Version 5 (GraphPad Software, La Jolla, CA). For comparisons between 2 groups, an unpaired Student t test was used. Multiple group comparisons between diabetic and control dams with and without sapropterin treatment, and their interactions, were conducted using 2‐way ANOVA, followed by the Bonferroni post hoc test. Two‐way repeated‐measures ANOVA, followed by the Bonferroni post hoc test, was used to analyze blood glucose differences over time between control and diabetic dams with or without sapropterin treatment. The incidence of CHDs was assessed with Fisher's exact test. Differences were deemed significant at P<0.05.
Results
Effects of Sapropterin on Blood Glucose, Fertility, Litter Size, and Biopterin Levels in Pregestational Diabetes Mellitus
This study was conducted in the same pregestational diabetes mellitus model we recently used.30, 31 One week after the final administration of streptozotocin, female mice with random blood glucose levels >11 mmol/L were bred with normal adult males. During gestation, blood glucose levels in the diabetic dams were progressively increased from E0.5 to E18.5 compared with both sapropterin‐treated and untreated control mice (Figure 2A). Treatment with insulin, but not sapropterin, in the diabetic mice restored blood glucose to normal levels. Diabetic dams had a significant lower fertility rate (38%) compared with controls (82%; P<0.05), which was improved to 64% by sapropterin treatment (Figure 2B).
Figure 2.
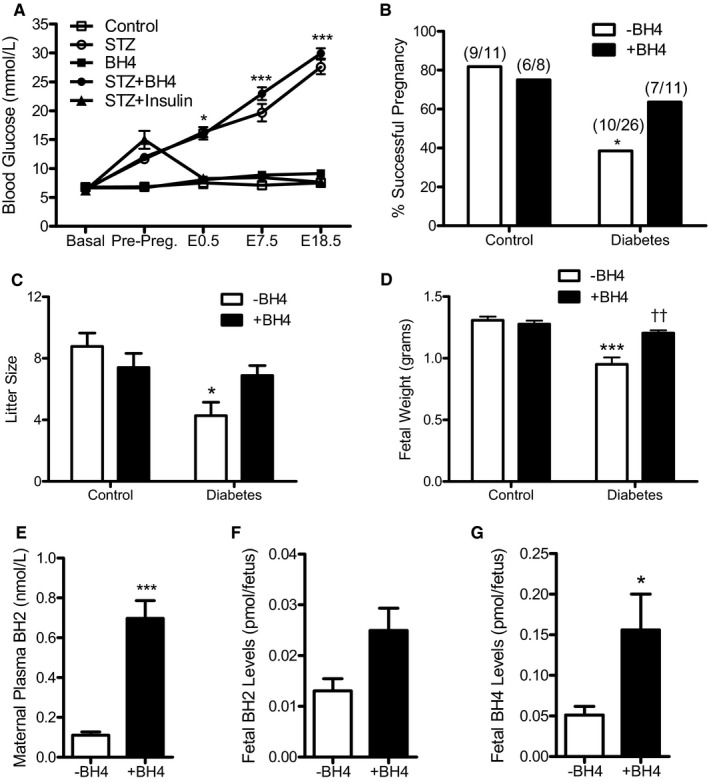
Blood glucose levels of pregnant mice, percentage of successful plugs, litter size, fetal body weight, and biopterin levels. A, Nonfasting blood glucose levels from before mating (basal) to embryonic day (E) 18.5 during pregnancy in streptozotocin‐treated and control female mice with and without sapropterin (tetrahydrobiopterin) administration (n=4–8 mice per group). B, The percentage of successful pregnancies. The numbers in the parentheses indicate the number of successful pregnancies at the day of fetus harvest/total plugs. C, The offspring litter size measured at the day of fetus harvest (n=5–11 litters per group). D, Fetal body weight at E18.5 (n=5–8 fetuses per group). E through G, Levels of 7,8‐dihydrobiopterin in maternal plasma, and fetal 7,8‐dihydrobiopterin and tetrahydrobiopterin levels at E12.5 with or without tetrahydrobiopterin treatment in the diabetic dams (n=3–9 mice per group). Two‐way repeated‐measures ANOVA (A) or two‐way ANOVA (C and D), followed by Bonferroni post hoc test, was used. B was analyzed by Fisher's exact test. E through G were analyzed by unpaired Student t test. Data are means±SEM (A and C through G). *P<0.05, ***P<0.001 vs corresponding controls; †† P<0.001 vs untreated diabetes mellitus.
Diabetic dams had a significantly smaller litter size (P<0.01), which was improved by sapropterin treatment (Figure 2C). Indeed, absorbed or dead fetuses were more commonly seen in utero in diabetic dams. Fetal body weight was significantly lower from diabetic dams than controls (P<0.001), and was restored to normal weight with sapropterin (P<0.001, Figure 2D). To assess biopterin levels after oral sapropterin administration, maternal blood and embryos were collected at E12.5. Our data show that sapropterin treatment significantly increased plasma 7,8‐dihydrobiopterin levels in the diabetic mothers (P<0.01, Figure 2E). 7,8‐Dihydrobiopterin levels in E12.5 embryos from sapropterin‐treated dams were elevated by 91.3% (P=0.0537, Figure 2F), and tetrahydrobiopterin levels in E12.5 embryos of diabetic dams were significantly increased (P=0.0433, Figure 2G).
Sapropterin Prevents CHDs Induced by Pregestational Diabetes Mellitus
CHDs were observed in 59.4% of offspring from diabetic dams (Table 2 and Figure 3). Normal control heart sections are shown in Figure 3A through 3C. Septal defects constituted a large proportion of the anomalies, with 47.4% atrial septal defects (Figure 3D) and 37.8% VSDs (Figure 3E). The diagnosis of atrial septal defects was based on a complete or partial absence of the septum secundum and/or primum in multiple serial heart sections. Hypoplastic left heart was seen in 21.1% of offspring from diabetic dams. One fetus had an atrioventricular septal defect. OFT defects were also common in offspring from diabetic mothers. A double‐outlet RV was present in one of the offspring (Figure 3F), and truncus arteriosus was seen in another fetus (Figure 3G). Maternal diabetes mellitus also resulted in cardiac valve defects, with 37.8% thickened aortic valves (Figure 3H) and 29.7% thickened pulmonary valves (Figure 3I). Finally, 24.3% of hearts from diabetic mothers displayed a narrowing aorta (Figure 3H versus 3B). Treatment with sapropterin to diabetic dams during pregnancy significantly decreased the overall incidence of CHDs to 26.5% (Table 2). Specifically, offspring from sapropterin‐treated diabetic mothers show a significantly lower incidence of atrial septal defects and thickened aortic valves, and did not display any VSD, atrioventricular septal defect, truncus arteriosus, double‐outlet RV, or hypoplastic left heart (Table 2, Figure 3J through 3O). Cardiac anomalies were seen in all litters from diabetic dams, and every dam in the sapropterin treatment group had at least one offspring with a CHD. No CHDs were seen in diabetic dams who received a daily dose of insulin to maintain normal blood glucose levels, indicating that CHDs seen in the diabetic offspring were induced through hyperglycemia (Table 2, Figure 3P through 3R). The incidence of CHDs was significantly higher in males than females (65% versus 47%; P<0.05). Furthermore, sapropterin treatment was more effective in reducing the incidence of CHDs to 16% in females compared with 35% in males (P<0.05, Figure 3S). Cardiac function of E18.5 fetuses was assessed through echocardiography. LV fractional shortening and ejection fraction were significantly decreased in offspring of diabetic dams, and were recovered by sapropterin treatment (P<0.001, Figure 3T and 3U). In addition, anterior wall thickness of the fetal heart during systole and diastole was reduced in offspring of diabetic dams, which was restored after sapropterin treatment (P<0.01, Figure 3V and 3W). Pregestational diabetes mellitus also induced neural tube defects (NTDs) in our model. A case of exencephaly in the offspring of a mother with pregestational diabetes mellitus is shown in Figure 3X.
Table 2.
Rate of CHDs in the Offspring of Diabetic and Control Mothers With and Without Sapropterin (Tetrahydrobiopterin) Treatment
| Control | Streptozotocin | Tetrahydrobiopterin | Streptozotocin+Tetrahydrobiopterin | Streptozotocin+Insulin | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| N | Dams | N | Dams | N | Dams | N | Dams | N | Dams | |
| 14 | 3 | 38 | 9 | 23 | 4 | 34 | 5 | 20 | 3 | |
| n | % | n | % | n | % | n | % | n | % | |
| Normal | 14 | 100 | 16 | 40.6** | 23 | 100 | 25 | 73.5† | 20 | 100 |
| Abnormal | 0 | 0 | 22 | 59.4** | 0 | 0 | 9 | 26.5† | 0 | 0 |
| ASD | 0 | 0 | 18 | 47.4** | 0 | 0 | 9 | 26.5 | 0 | 0 |
| VSD | 0 | 0 | 14 | 37.8* | 0 | 0 | 0 | 0.0† | 0 | 0 |
| Truncus arteriosus | 0 | 0 | 1 | 2.7 | 0 | 0 | 0 | 0.0 | 0 | 0 |
| AVSD | 0 | 0 | 2 | 5.4 | 0 | 0 | 0 | 0.0 | 0 | 0 |
| DORV | 0 | 0 | 1 | 2.7 | 0 | 0 | 0 | 0.0 | 0 | 0 |
| Pulmonary valve stenosis | 0 | 0 | 14 | 37.8* | 0 | 0 | 0 | 0.0† | 0 | 0 |
| Aortic valve stenosis | 0 | 0 | 11 | 29.7* | 0 | 0 | 1 | 2.9† | 0 | 0 |
| RV hypertrophy | 0 | 0 | 9 | 24.3 | 0 | 0 | 0 | 0.0† | 0 | 0 |
| Hypoplastic left heart | 0 | 0 | 8 | 21.1 | 0 | 0 | 0 | 0.0† | 0 | 0 |
| Narrowing of the aorta | 0 | 0 | 9 | 24.3 | 0 | 0 | 0 | 0.0† | 0 | 0 |
Data were analyzed using Fisher's exact test. ASD indicates atrial septal defect; AVSD, atrioventricular septal defect; CHD, congenital heart defect; Dams, litter numbers; DORV, double‐outlet right ventricle; N, total number of fetuses; RV, right ventricular; VSD, ventricular septal defect.
*P<0.05, **P<0.001 vs control, † P<0.001 vs streptozotocin.
Figure 3.
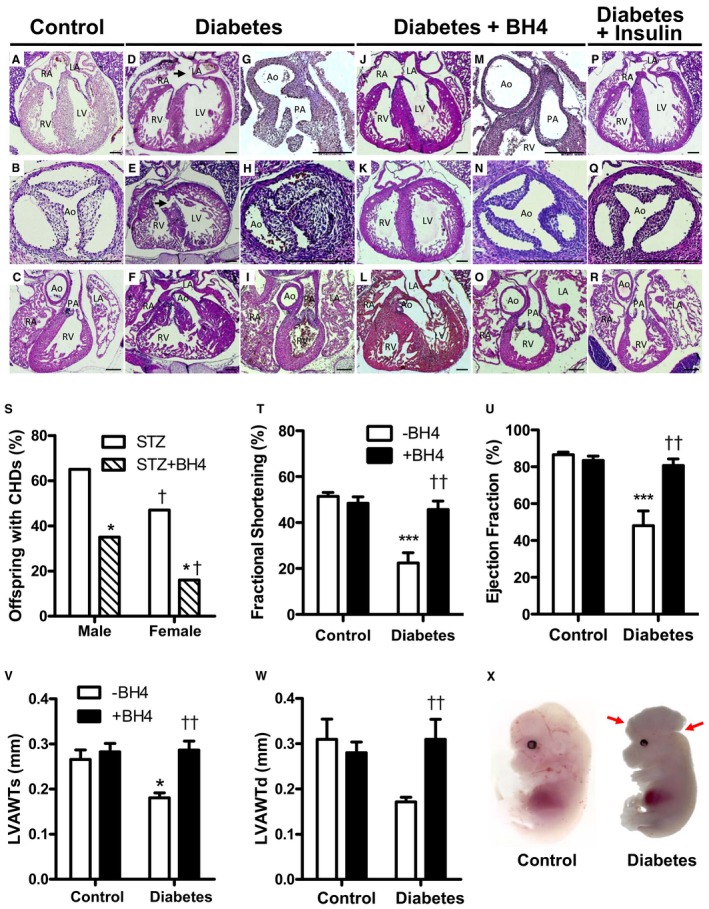
Effects of sapropterin (tetrahydrobiopterin) on congenital heart defects (CHDs) induced by pregestational diabetes mellitus. Representative histological sections of embryonic day (E) 18.5 hearts from offspring of diabetic mothers with and without tetrahydrobiopterin treatment. A through C, Heart sections from controls. Pregestational diabetes mellitus resulted in atrial septal defect (D), ventricular septal defect (E), double‐outlet right ventricle (F), truncus arteriosus (G), and thickened aortic (H) and pulmonary (I) valves. J through O, Tetrahydrobiopterin treatment in diabetic dams shows normal heart morphological characteristics. P through R, Diabetic dams receiving insulin therapy to control blood glucose levels show normal fetal heart morphological characteristics. Bar=200 μm. S, Incidence of CHDs as percentage of offspring separated by sex at E18.5 from diabetic dams with or without tetrahydrobiopterin treatment. *P<0.05 vs streptozotocin group of corresponding sex; † P<0.05 vs corresponding males. T through W, Left ventricular (LV) fractional shortening and ejection fraction and anterior wall thickness during systole (LVAWTs) and diastole (LVAWTd), assessed by echocardiography in E18.5 offspring (n=5–7 fetuses per group). *P<0.05, ***P<0.001 vs corresponding controls; †† P<0.001 vs untreated diabetes mellitus. X, An exencephaly was seen in the offspring of a mother with pregestational diabetes mellitus. A normal control fetus is shown on the right panel. Fetuses are harvested at E13.5. S through W were analyzed by 2‐way ANOVA, followed by Bonferroni post hoc test. Data are means±SEM (T‐W). LA indicates left atrium; PA, pulmonary artery; RA, right atrium; RV, right ventricle.
Sapropterin Prevents Myocardial and Valvular Abnormalities Induced by Pregestational Diabetes Mellitus
The free walls of the RV and LV at E18.5 were measured at the midventricular region (Figure 4A). The RV and LV myocardium was thinner in the offspring of diabetic mothers compared with those of control offspring, which was prevented by sapropterin treatment (P<0.001, Figure 4B and 4C). Additionally, because valve leaflets were thickened (Figure 3H and 3I), we assessed glycosaminoglycans, a component of extracellular matrix in the cardiac valves using toluidine blue staining. Our data show that glycosaminoglycans occupied a greater space in the leaflets of aortic and pulmonary valves (light purple color) from E18.5 fetal hearts of diabetic mothers compared with controls (Figure 5A and 5B). Treatment with sapropterin decreased these extracellular polysaccharides in both aortic and pulmonary valves (P<0.01, Figure 5D and 5E). There was no significant difference in glycosaminoglycan content in the mitral valve; however, the distal tip of the mitral valve was thicker in the offspring of diabetic dams (P<0.01, Figure 5C and 5F). Notably, the area of aortic orifice and the diameter of the pulmonary artery were significantly smaller in hearts from diabetic mothers compared with controls (P<0.01, Figure 5G and 5H).
Figure 4.

Effects of sapropterin (tetrahydrobiopterin) on fetal heart wall thickness of pregestational diabetes mellitus at embryonic day 18.5. A, Representative images of myocardial wall thickness in control and pregestational diabetes mellitus with and without tetrahydrobiopterin treatment. Arrows indicate compact myocardium boundary from which measurements were obtained. B and C, Quantification of left ventricular (LV) and right ventricular (RV) myocardial thickness, respectively. n=6 per group from 3 to 6 litters. Bar=200 μm. Data are means±SEM and analyzed using 2‐way ANOVA, followed by Bonferroni post hoc test (B and C). ***P<0.01 vs untreated control; † P<0.01 vs untreated diabetes mellitus.
Figure 5.
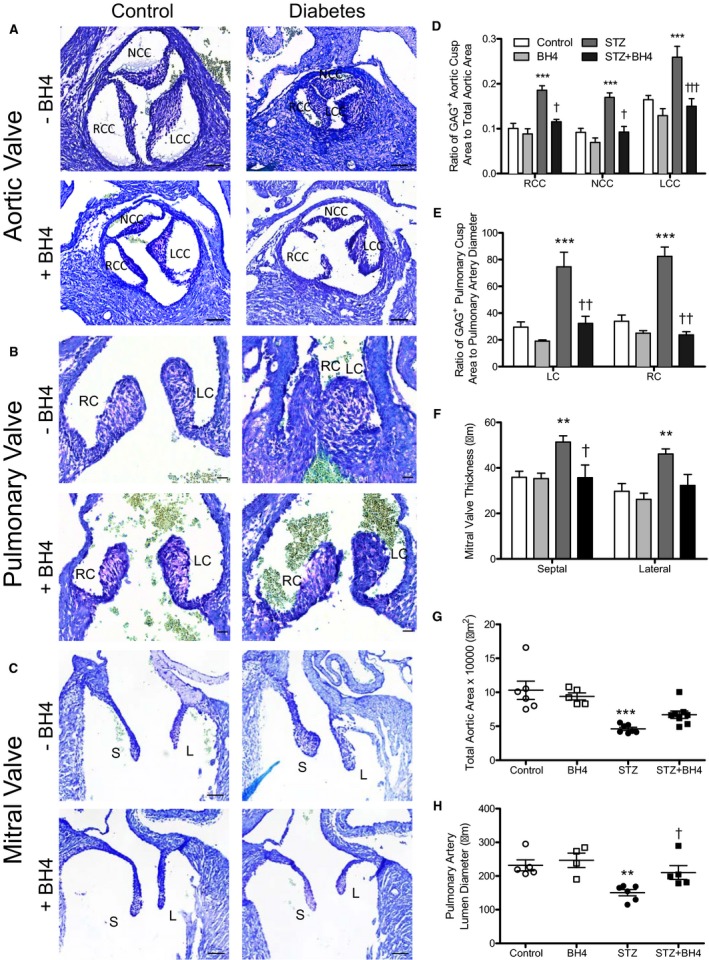
Effects of sapropterin (tetrahydrobiopterin) on aortic, pulmonary, and mitral valve defects induced by pregestational diabetes mellitus at embryonic day (E) 18.5. A through C, Representative images of toluidine blue staining of glycosaminoglycans in aortic, pulmonary, and mitral valves in E18.5 hearts. D, The ratio of glycosaminoglycan‐positive area/total valve leaflet area. E, Pulmonary valve leaflet thickness. F, Mitral valve leaflet thickness. G, Total aortic orifice area. H, Pulmonary artery luminal diameter at the base of the orifice. Bars=50, 20, and 20 μm (A, B, and C, respectively). n=4 to 7 hearts per group from 3 to 4 litters. Data are means±SEM and analyzed using 2‐way ANOVA, followed by Bonferroni post hoc test (B and C). LC indicates left cusp; LCC, left coronary cusp; NCC, noncoronary cusp; RC, right cusp; RCC, right coronary cusp. **P<0.01 vs controls; † P<0.01, †† P<0.01 vs untreated STZ group.
Effects of Sapropterin on OFT Length and Cell Proliferation in the Fetal Heart
Because offspring of diabetic mothers show OFT defects, we aimed to investigate changes in cell proliferation at a critical stage in OFT formation. At E10.5, sagittal sections of whole embryos reveal a shortened OFT in hearts from diabetic mothers compared with control (P<0.001, Figure 6A), which was restored with sapropterin treatment (P<0.05, Figure 6E). To further analyze this change, phosphorylated histone H3, a marker for the mitotic phase of cell division, was used to compare the levels of proliferating cells in the OFT at E10.5 using the same hearts (Figure 6B through 6D). Embryos from mice with pregestational diabetes mellitus had a significantly lower number of proliferating cells in the OFT (P<0.01, Figure 6F and 6G). Sapropterin treatment was able to prevent impaired cell proliferation in the OFT in hearts from diabetic dams (P<0.001, Figure 6F and 6G).
Figure 6.

Effects of sapropterin (tetrahydrobiopterin) on outflow tract (OFT) development and cell proliferation in embryonic day (E) 10.5 hearts of pregestational diabetes mellitus. A, Length of the OFT was measured via sagittal sections according to the line. B through D, Immunostaining for phosphohistone H3 (pHH3), marking proliferating cells in the OFT (B through C) and ventricular myocardium (D). C is the magnification of boxed area in B. E, Quantification of OFT length. F, Quantification of pHH3+ cells in the OFT. G, Quantification of pHH3+ cells in the ventricle. n=3 to 6 hearts per group from 2 to 4 litters. Bars=50 (in A and B) and 20 μm (in C and D). Data are means±SEM and analyzed using 2‐way ANOVA, followed by Bonferroni post hoc test. **P<0.01, ***P<0.001 vs untreated control; † P<0.05, †† P<0.01 vs untreated diabetes mellitus.
Fate Mapping of SHF‐Derived Cells in the Fetal Heart of Diabetic Mothers
To better understand the spectrum of malformations induced by maternal diabetes mellitus seen at birth, embryonic lineage tracing of the SHF was performed. Fate mapping using Mef2c‐Cre and the global double‐fluorescent Cre reporter line Rosa26 mTmG identified all SHF‐derived cells as GFP+. At E9.5, the number of GFP+ SHF cells and the total number of cells in the heart were significantly less than in control (P<0.01, Figure 7A, 7D, and 7E). Furthermore, significantly less GFP+ SHF cells were infiltrated into the endocardial cushion at E12.5 in diabetic embryos compared with control (P=0.0476, Figure 7B and 7F). Additionally, E12.5 hearts from diabetic mothers had thinner ventricular walls with significantly less myocardial cell layers than controls (P<0.01, Figure 7C and 7G).
Figure 7.
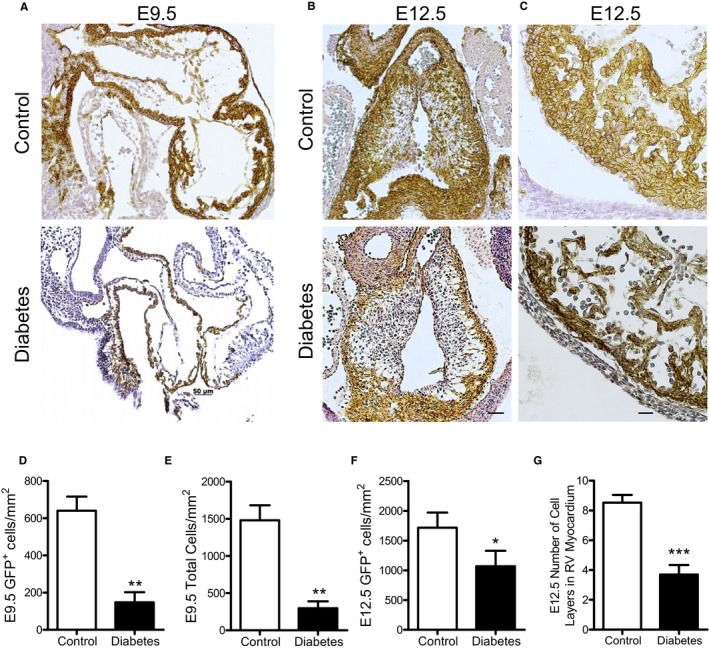
Effects of pregestational diabetes mellitus on second heart field (SHF) progenitor contribution to embryonic day (E) 9.5 and E12.5 hearts. Fate mapping using the mT/mG reporter showing green fluorescent protein–positive (GFP +) cells expressing Cre recombinase under the control of the anterior heart field–specific Mef2c transcription factor. Representative sections of outflow tract (OFT) of E9.5 (A) and E12.5 (B) hearts from control and diabetic mothers. C, Representative sections showing the cell layers of the right ventricular (RV) myocardium from E12.5 hearts. Quantification of SHF GFP + cells (D) and total number of cells (E) in the OFT cushions at E9.5. Quantification of SHF GFP + cells in the OFT cushions (F) and number of cell layers in the RV myocardium (G) at E12.5. N=3 per group from 2 litters (D and E). N=5 per group from 2 to 4 litters (F and G). Bar=50 μm. Data are means±SEM and analyzed using unpaired Student t test (D‐G). *P<0.05, **P<0.01, *** P<0.001 vs control.
Sapropterin Prevents Maternal Diabetes Mellitus–Induced Downregulation of Regulators of Heart Development
Pregestational diabetes mellitus induces changes in gene expression in the developing heart.32 To determine if key transcriptional regulators of heart development were altered under maternal diabetes mellitus and with sapropterin treatment, quantitative polymerase chain reaction analysis was performed from E12.5 hearts. Our data show that the mRNA levels of Gata4, Tbx5, Nkx2.5, Gata5, Bmp10, and Notch1 were significantly lower compared with controls (P<0.05, Figure 8A through 8F). Treatment with sapropterin significantly improved mRNA levels of these transcription factors (P<0.001, Figure 8A through 8F). Additionally, gene expression of tetrahydrobiopterin biosynthesis enzymes, including Gch1 and Dhfr, was significantly decreased in embryonic hearts from diabetic mothers (P<0.05, Figure 8G and 8H). Sapropterin treatment completely recovered the expression of Dhfr (P<0.001, Figure 8H).
Figure 8.
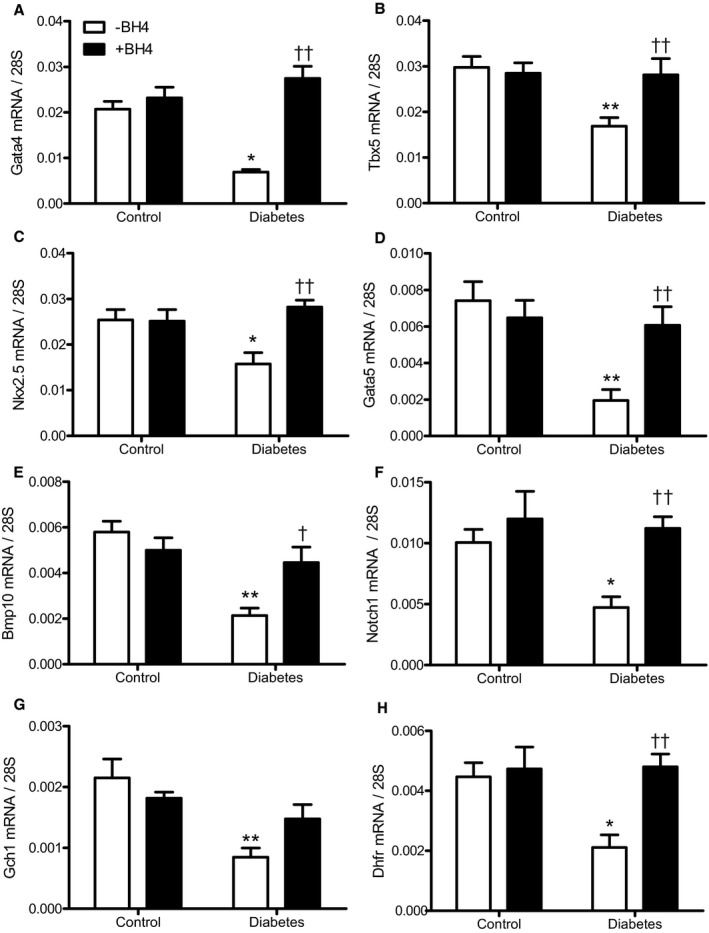
Effects of sapropterin (tetrahydrobiopterin) on molecular regulators of heart development at embryonic day (E) 12.5. Real‐time reverse transcription–polymerase chain reaction (RT‐PCR) of cardiac transcription factors and regulators in E12.5 hearts of offspring from control and diabetic mothers. A through F, The expression levels of Gata4, Tbx5, Nkx2.5, Gata5, Bmp10, and Notch1 were significantly decreased under maternal diabetes mellitus and restored with tetrahydrobiopterin treatment. G and H, The expression levels of Gch1 and Dhfr were significantly decreased with maternal diabetes mellitus. Tetrahydrobiopterin treatment rescues expression of DHFR. n=4 to 7 hearts per group. Data are means±SEM and analyzed using 2‐way ANOVA, followed by Bonferroni post hoc test. *P<0.05, **P<0.01 vs untreated control; † P<0.05, †† P<0.01 vs untreated diabetes mellitus.
Sapropterin Decreases Oxidative Stress and Improves eNOS Dimerization in Fetal Hearts of Pregestational Diabetes Mellitus
To examine the effects of sapropterin on ROS in the developing heart, a dihydroethidium probe was used to label the oxygen radical. Quantification of red fluorescence intensity indicates that superoxide generation was significantly elevated in embryonic hearts from mice with pregestational diabetes mellitus compared with control (P<0.05, Figure 9A and 9B). Sapropterin treatment significantly reduced myocardial ROS levels to basal conditions (P<0.05, Figure 9B). Pretreatment with Nω‐nitro‐l‐arginine methyl ester, an NOS inhibitor, and superoxide dismutase, an antioxidant enzyme, significantly decreased dihydroethidium fluorescence in embryonic hearts from diabetic dams, indicating eNOS uncoupling and superoxide generation, respectively (P<0.05 Figure 9B). Finally, eNOS uncoupling has been implicated as a mechanism for endothelial dysfunction in diabetes mellitus.23 To assess the effects of sapropterin on eNOS coupling, eNOS dimerization was analyzed using Western blotting in nonreducing conditions, which yields 2 bands at 260 and 130 kDa, representing the intact dimer and monomer of eNOS, respectively. Figure 9C shows a representative blot of the dimer and monomer of eNOS. In control conditions, there was a higher level of eNOS dimers than monomers, indicating a functional eNOS enzyme. However, embryonic hearts from diabetic dams show a significant decrease of dimer/monomer ratios compared with controls (P<0.05, Figure 9D), which was restored by sapropterin treatment (P<0.01, Figure 9D).
Figure 9.
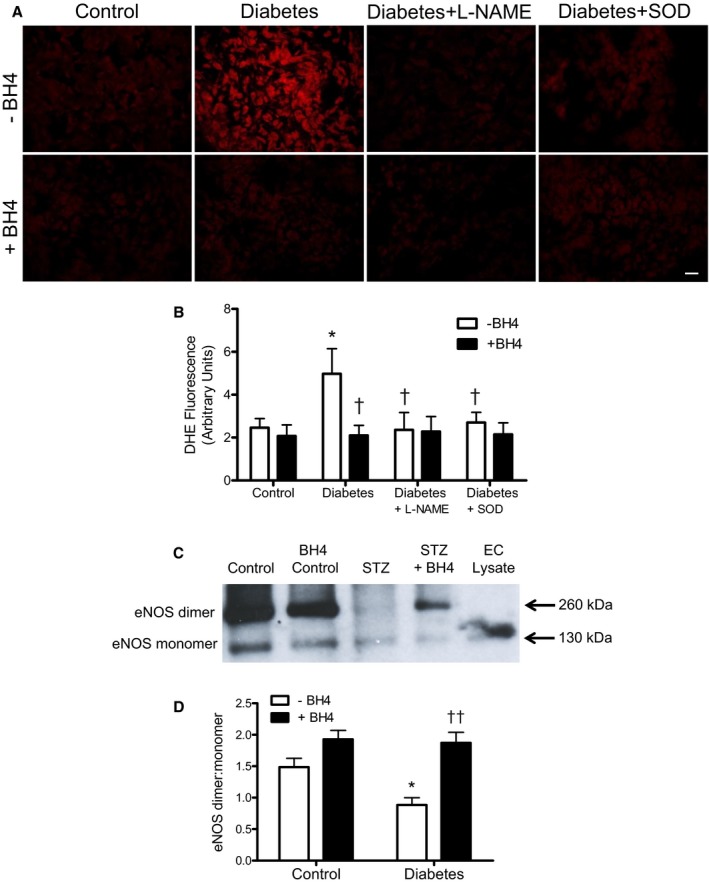
Effects of sapropterin (tetrahydrobiopterin) on superoxide production and endothelial NO synthase (eNOS) dimer/monomer protein levels in embryonic day (E) 12.5 hearts. A, Representative images of dihydroethidium staining in the ventricular myocardium of E12.5 hearts from control and diabetic dams with and without tetrahydrobiopterin administration. A subset of heart sections from diabetic dams were pretreated with Nω‐nitro‐l‐arginine methyl ester (L‐NAME; 300 μmol/L) or superoxide dismutase (SOD; 100 U/mL) for 30 minutes before dihydroethidium probing. B, Quantification of dihydroethidium fluorescence signals. C, Representative Western blotting showing eNOS dimer and monomer bands of E12.5 hearts from control and diabetic dams with and without tetrahydrobiopterin treatment. Denatured endothelial cell lysate validates the size of the eNOS monomer band. D, Densitometric analysis of eNOS dimer/monomer ratios. n=5 to 6 hearts per group from 2 to 4 litters. Data are means±SEM and analyzed using 2‐way ANOVA, followed by Bonferroni post hoc test. EC indicates endothelial cell. *P<0.05 vs untreated control; † P<0.05, †† P<0.01 vs untreated diabetes mellitus.
Discussion
The present study used a clinically relevant model of CHDs induced by pregestational diabetes mellitus that we recently established.30, 31 Consistent with our previous studies, a spectrum of CHDs was observed in the offspring of diabetic mothers. The CHDs range from atrial septal defects, VSD, and valve thickening, to major malformations, including atrioventricular septal defects, double‐outlet RV, truncus arteriosus, and hypoplastic left heart, as clinically categorized by Hoffman et al.33 Sapropterin treatment was able to prevent all major defects and significantly reduce the overall level of defects induced by pregestational diabetes mellitus. Other changes in the fetal heart, such as thinner ventricular myocardium, thickened valves, stenosis of the aorta and pulmonary artery, impaired cardiac function, and decreased cell proliferation and gene expression induced by maternal diabetes mellitus, were normalized with sapropterin treatment. Furthermore, sapropterin administration in the diabetic dams increased eNOS dimerization and decreased ROS levels in the fetal heart. Our study suggests that sapropterin improves eNOS coupling and prevents CHDs in pregestational diabetes mellitus in mice (Figure 10).
Figure 10.
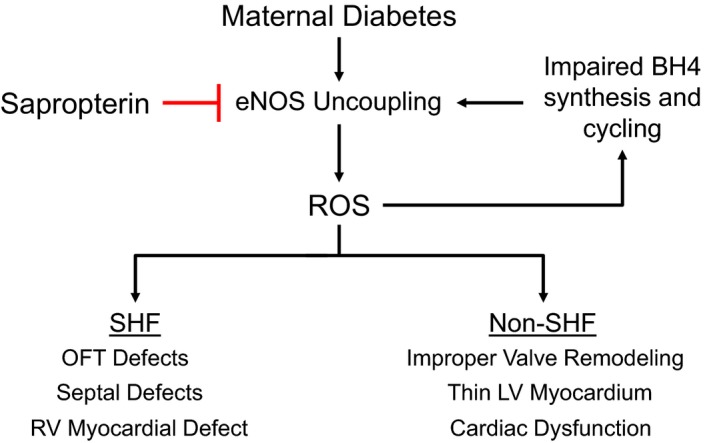
Schematic summary of endothelial NO synthase (eNOS) uncoupling and congenital heart defects induced by pregestational diabetes mellitus and the inhibitory effects by sapropterin treatment. Abnormalities of both second heart field (SHF) and non‐SHF derived structures are prevented by sapropterin treatment. LV indicates left ventricle; OFT, outflow tract; ROS, reactive oxygen species; and RV, right ventricle.
In this model, streptozotocin was used to induce hyperglycemia in female mice at least 7 days before they were bred with normal healthy males. Because streptozotocin has a short plasma half‐life of 10 minutes in rodents,34 it is unlikely that streptozotocin would cause any teratogenic effects in the fetus. To confirm that hyperglycemia is the cause of CHDs in this model, a group of mice were treated with insulin to lower glucose levels in diabetic dams. Our data show that insulin treatment abrogated CHDs in pregestational diabetes mellitus, which is consistent with clinical studies that show that good glycemic control in women with pregestational diabetes mellitus lowers the incidence of CHDs.7 The incidence of CHDs found in the diabetic groups with or without sapropterin treatment at E18.5 may be an underestimate because fetuses may have been absorbed in utero at ≈E12.5. To study the effects of sapropterin on maternal diabetes mellitus, blood glucose levels were assessed during pregnancy. Our data show that sapropterin treatment in the diabetic dams had no effect on the hyperglycemic state of the animal, suggesting that the beneficial effects of sapropterin on fertility (or percentage successful pregnancy), litter size, and heart development under pregestational diabetes mellitus are independent of blood glucose levels. In humans, sex‐related differences in the prevalence of CHDs have been reported. In a recent large cohort study with 9727 cases of CHDs, the male/female ratio was 55%:45%, indicating a significant predisposition of CHDs in the male sex.35 In agreement with clinical studies, our data show a male dominance of CHDs in the offspring of diabetic mothers. Notably, sapropterin treatment appears to be more effective in preventing CHDs in females than in males.
We have recently shown that defects in SHF signaling result in thin myocardium, septal defects, and OFT defects.26, 36 The defective SHF progenitor contribution may explain most heart malformations in the OFT, cardiac septum, and cardiac valves induced by pregestational diabetes mellitus. Because sapropterin treatment significantly prevented these CHDs in the offspring of diabetic dams, it is likely that impairment in SHF progenitors will be diminished by maternal sapropterin treatment, which still needs to be confirmed in future studies. Considering the presence of hypoplastic left heart and OFT septation defect (truncus arteriosus), it is possible that pregestational diabetes mellitus also impairs FHF and CNC cells in our model. Notably, these defects were all prevented in diabetic dams treated with sapropterin. These findings are consistent with previous studies showing that oxidative stress during diabetic pregnancy disrupts CNC migration and causes OFT defects in rodents,37, 38 which are prevented by treatment with vitamin E, an antioxidant.39 Our results suggest that sapropterin treatment improves the function of both SHF and non‐SHF (FHF and CNC) progenitors in pregestational diabetes mellitus (Figure 10).
Cell proliferation is critical to cardiac morphogenesis and the growth of the fetal heart.40 Decreased cell proliferation results in shortening of OFT length, thin myocardium, and CHDs.36, 41 In the present study, E10.5 hearts show less cell proliferation and a shorter OFT in embryos from diabetic dams. Furthermore, E18.5 hearts from diabetic dams are notably smaller and their ventricular walls are significantly thinner than control hearts. These abnormalities were all prevented by sapropterin treatment. Previous studies have shown that tetrahydrobiopterin increases DNA synthesis and cell proliferation in erythroid cells.42 Additionally, tetrahydrobiopterin mediates the proliferative effects of epidermal growth factor and nerve growth factor in PC12 cells.43 We have previously shown that eNOS promotes cardiomyocyte proliferation.17, 44 Because tetrahydrobiopterin increases eNOS coupling, as shown by higher dimer/monomer ratios in our study, it is possible that a normalized eNOS signaling may contribute to the improved cell proliferation by sapropterin treatment. Increases in cell apoptosis may also contribute to CHDs. However, we have previously shown that the incidence of cell apoptosis in fetal hearts of diabetic offspring is low (≈1%) and was not affected by antioxidant treatment, suggesting an insignificant role of apoptosis in our model.31 We, therefore, did not assess cell apoptosis in the present study.
Cardiac transcription factors, including Gata4, Gata5, Nkx2.5, and Tbx5, are critical to normal heart development, and genetic mutations of these transcription factors result in CHDs in humans.45 Interestingly, both eNOS and ROS can alter the expression of these transcription factors.16, 31 For example, deficiency in eNOS decreases the expression of cardiac transcription factors, including Gata4, during embryonic heart development.19 Additionally, the expression of cardiac transcription factors was downregulated in the fetal heart of offspring of diabetic mothers, which was restored by treatment with an antioxidant, N‐acetylcysteine.31 In agreement with our previous studies, we showed a downregulation of Gata4, Gata5, Nkx2.5, and Tbx5 in fetal hearts of offspring from diabetic mothers in the present study. Decreased expression was also seen in Bmp10, essential in cardiac growth and chamber maturation.46 Importantly, sapropterin administration to diabetic dams restored the expression profile of these factors to normal levels. We also assessed the expression of Gch1 and Dhfr, which are enzymes responsible for de novo tetrahydrobiopterin biosynthesis and recycling of 7,8‐dihydrobiopterin back to tetrahydrobiopterin, respectively. Both GCH1 and DHFR are sensitive to oxidative stress and NO signaling.21, 47 NO has been shown to stabilize DHFR protein from ubiquitination and degradation by S‐nitrosylation.48 During pregestational diabetes mellitus, GCH1 and DHFR transcript levels in the fetal heart were downregulated, which was normalized by sapropterin treatment. Our findings indicate a possible gene regulatory mechanism governing DHFR expression. Recent studies show a strong interaction between eNOS and Notch1, promoting semilunar valve and OFT development.49 Herein, we show that Notch1 mRNA is decreased in embryonic hearts from diabetic dams, which is rescued with sapropterin treatment. Our results indicate a perturbed NO‐Notch1 signaling pathway in the fetal hearts of diabetic dams. Together, these effects are consistent with the ability of sapropterin treatment to recouple eNOS and restore ROS balance in the embryonic heart of diabetic dams. Neuronal NOS and inducible NOS are dispensable for heart development because neuronal NOS−/− and inducible NOS−/− mice do not exhibit any developmental abnormalities of the heart.16
A major noncardiac malformation induced by pregestational diabetes mellitus is NTD, such as anencephaly, exencephaly, and spina bifida.50 It has been reported that 25% to 40% of offspring of diabetic dams have NTDs when the fetuses are examined at E10.5.51, 52 To analyze cardiac malformation, we examined the fetuses at E18.5 and may have missed most of the NTDs, which are likely absorbed beyond E10.5. This may be the reason that we only observed one exencephaly in the present study and a low incidence of NTDs (4.8%) in our previous study.31 Additionally, tetrahydrobiopterin biosynthesis is important to neural tube development. Inhibition of GCH1 activity, a rate‐limiting enzyme in the biosynthesis of tetrahydrobiopterin, interrupts neural tube closure, which can be prevented by tetrahydrobiopterin treatment in chick embryos.53 Furthermore, GCH1 haplotypes are significantly associated with a higher risk of NTD in infants.54 It is possible that sapropterin treatment may prevent NTD induced by pregestational diabetes mellitus, a hypothesis that needs to be tested in future studies. Surprisingly, the GCH1 knockout mice die at E13.5 because of bradycardia without any structural anomalies.55 Apart from being a cofactor of eNOS, tetrahydrobiopterin is also a substrate for aromatic amino acid hydroxylases.56 Tetrahydrobiopterin treatment at postnatal day 14 elevated dopamine levels in the brain and fully restored the loss of tyrosine hydroxylase protein caused by the tetrahydrobiopterin deficiency in infant mice,57 indicating an important role of tetrahydrobiopterin in the dopaminergic function of the brain. The effects of pregestational diabetes mellitus and sapropterin treatment on tyrosine hydroxylase expression and dopamine levels in the fetal brain are beyond the scope of the present investigation.
In summary, the present study demonstrates that treatment with sapropterin (Kuvan), an orally active synthetic form of tetrahydrobiopterin, during gestation improves eNOS coupling, reduces ROS, and increases cell proliferation in the embryonic heart of offspring of diabetic mothers. Notably, sapropterin treatment prevents the development of major CHDs induced by pregestational diabetes mellitus. Sapropterin is a US Food and Drug Administration–approved drug to treat phenylketonuria, a genetic disorder attributable to mutations of the phenylalanine hydroxylase (PAH) gene, leading to low levels of phenylalanine hydroxylase.25 Our study suggests that sapropterin may also have therapeutic potential in preventing CHDs in offspring of women with pregestational diabetes mellitus.
Author Contributions
Engineer, Lu, and Feng conceived the experiments. Engineer, Lu, Urquhart, Drysdale, Norozi, and Feng designed the experiments. Engineer, Saiyin, Lu, and Kucey performed the experiments and data analyses. Engineer and Feng wrote the manuscript. Engineer, Urquhart, Drysdale, Norozi, and Feng revised the manuscript. All authors contributed to the interpretation of results and proofreading of the manuscript.
Sources of Funding
This study was funded in part by grants from the Canadian Institutes of Health Research to Feng and Drysdale; and the Children's Health Foundation (London, ON, Canada) to Norozi, Drysdale, and Feng.
Disclosures
None.
Acknowledgments
We thank Dr Ben Rubin, Western University, for his advice on statistical analysis. Feng is a Richard and Jean Ivy Chair in Molecular Toxicology at University of Western Ontario.
(J Am Heart Assoc. 2018;7:e009624 DOI: 10.1161/JAHA.118.009624.)
References
- 1. Pierpont ME, Basson CT, Benson DW Jr, Gelb BD, Giglia TM, Goldmuntz E, McGee G, Sable CA, Srivastava D, Webb CL; American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young . Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115:3015–3038. [DOI] [PubMed] [Google Scholar]
- 2. Gilboa SM, Devine OJ, Kucik JE, Oster ME, Riehle‐Colarusso T, Nembhard WN, Xu P, Correa A, Jenkins K, Marelli AJ. Congenital heart defects in the United States: estimating the magnitude of the affected population in 2010. Circulation. 2016;134:101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marelli AJ, Ionescu‐Ittu R, Mackie AS, Guo L, Dendukuri N, Kaouache M. Lifetime prevalence of congenital heart disease in the general population from 2000 to 2010. Circulation. 2014;130:749–756. [DOI] [PubMed] [Google Scholar]
- 4. Meilhac SM, Lescroart F, Blanpain C, Buckingham ME. Cardiac cell lineages that form the heart. Cold Spring Harb Perspect Med. 2014;4:a013888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Oyen N, Diaz LJ, Leirgul E, Boyd HA, Priest J, Mathiesen ER, Quertermous T, Wohlfahrt J, Melbye M. Prepregnancy diabetes and offspring risk of congenital heart disease: a nationwide cohort study. Circulation. 2016;133:2243–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu S, Joseph KS, Lisonkova S, Rouleau J, Van den Hof M, Sauve R, Kramer MS; Canadian Perinatal Surveillance System (Public Health Agency of Canada) . Association between maternal chronic conditions and congenital heart defects: a population‐based cohort study. Circulation. 2013;128:583–589. [DOI] [PubMed] [Google Scholar]
- 7. Starikov R, Bohrer J, Goh W, Kuwahara M, Chien EK, Lopes V, Coustan D. Hemoglobin A1c in pregestational diabetic gravidas and the risk of congenital heart disease in the fetus. Pediatr Cardiol. 2013;34:1716–1722. [DOI] [PubMed] [Google Scholar]
- 8. Eriksen NB, Damm P, Mathiesen ER, Ringholm L. The prevalence of congenital malformations is still higher in pregnant women with pregestational diabetes despite near‐normal HbA1c: a literature review. J Matern Fetal Neonatal Med. 2017;28:1–5. [DOI] [PubMed] [Google Scholar]
- 9. Peng TY, Ehrlich SF, Crites Y, Kitzmiller JL, Kuzniewicz MW, Hedderson MM, Ferrara A. Trends and racial and ethnic disparities in the prevalence of pregestational type 1 and type 2 diabetes in Northern California: 1996–2014. Am J Obstet Gynecol. 2017;216:177.e1–177.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wahabi H, Fayed A, Esmaeil S, Mamdouh H, Kotb R. Prevalence and complications of pregestational and gestational diabetes in Saudi women: analysis from Riyadh Mother and Baby Cohort Study (RAHMA). Biomed Res Int. 2017;2017:6878263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Agarwal S, Sud K, Menon V. Nationwide hospitalization trends in adult congenital heart disease across 2003–2012. J Am Heart Assoc. 2016;5:e002330 DOI: 10.1161/JAHA.115.002330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Opotowsky AR, Siddiqi OK, Webb GD. Trends in hospitalizations for adults with congenital heart disease in the U.S. J Am Coll Cardiol. 2009;54:460–467. [DOI] [PubMed] [Google Scholar]
- 13. Ornoy A. Embryonic oxidative stress as a mechanism of teratogenesis with special emphasis on diabetic embryopathy. Reprod Toxicol. 2007;24:31–41. [DOI] [PubMed] [Google Scholar]
- 14. Eriksson UJ, Borg LA. Diabetes and embryonic malformations: role of substrate‐induced free‐oxygen radical production for dysmorphogenesis in cultured rat embryos. Diabetes. 1993;42:411–419. [DOI] [PubMed] [Google Scholar]
- 15. Zangen SW, Yaffe P, Shechtman S, Zangen DH, Ornoy A. The role of reactive oxygen species in diabetes‐induced anomalies in embryos of Cohen diabetic rats. Int J Exp Diabetes Res. 2002;3:247–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu Y, Feng Q. NOing the heart: role of nitric oxide synthase‐3 in heart development. Differentiation. 2012;84:54–61. [DOI] [PubMed] [Google Scholar]
- 17. Feng Q, Song W, Lu X, Hamilton JA, Lei M, Peng T, Yee SP. Development of heart failure and congenital septal defects in mice lacking endothelial nitric oxide synthase. Circulation. 2002;106:873–879. [DOI] [PubMed] [Google Scholar]
- 18. Liu Y, Lu X, Xiang FL, Lu M, Feng Q. Nitric oxide synthase‐3 promotes embryonic development of atrioventricular valves. PLoS One. 2013;8:e77611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu Y, Lu X, Xiang FL, Poelmann RE, Gittenberger‐de Groot AC, Robbins J, Feng Q. Nitric oxide synthase‐3 deficiency results in hypoplastic coronary arteries and postnatal myocardial infarction. Eur Heart J. 2014;35:920–931. [DOI] [PubMed] [Google Scholar]
- 20. Werner ER, Blau N, Thony B. Tetrahydrobiopterin: biochemistry and pathophysiology. Biochem J. 2011;438:397–414. [DOI] [PubMed] [Google Scholar]
- 21. Bendall JK, Douglas G, McNeill E, Channon KM, Crabtree MJ. Tetrahydrobiopterin in cardiovascular health and disease. Antioxid Redox Signal. 2014;20:3040–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ma S, Ma CC. Recent developments in the effects of nitric oxide‐donating statins on cardiovascular disease through regulation of tetrahydrobiopterin and nitric oxide. Vascul Pharmacol. 2014;63:63–70. [DOI] [PubMed] [Google Scholar]
- 23. Cai S, Khoo J, Mussa S, Alp NJ, Channon KM. Endothelial nitric oxide synthase dysfunction in diabetic mice: importance of tetrahydrobiopterin in eNOS dimerisation. Diabetologia. 2005;48:1933–1940. [DOI] [PubMed] [Google Scholar]
- 24. Heitzer T, Krohn K, Albers S, Meinertz T. Tetrahydrobiopterin improves endothelium‐dependent vasodilation by increasing nitric oxide activity in patients with type II diabetes mellitus. Diabetologia. 2000;43:1435–1438. [DOI] [PubMed] [Google Scholar]
- 25. Burnett JR. Sapropterin dihydrochloride (Kuvan/phenoptin), an orally active synthetic form of BH4 for the treatment of phenylketonuria. IDrugs. 2007;10:805–813. [PubMed] [Google Scholar]
- 26. Leung C, Lu X, Liu M, Feng Q. Rac1 signaling is critical to cardiomyocyte polarity and embryonic heart development. J Am Heart Assoc. 2014;3:e001271 DOI: 10.1161/JAHA.114.001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Verzi MP, McCulley DJ, De Val S, Dodou E, Black BL. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev Biol. 2005;287:134–145. [DOI] [PubMed] [Google Scholar]
- 28. Zou MH, Shi C, Cohen RA. Oxidation of the zinc‐thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Invest. 2002;109:817–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fukushima T, Nixon JC. Analysis of reduced forms of biopterin in biological tissues and fluids. Anal Biochem. 1980;102:176–188. [DOI] [PubMed] [Google Scholar]
- 30. Moazzen H, Lu X, Liu M, Feng Q. Pregestational diabetes induces fetal coronary artery malformation via reactive oxygen species signaling. Diabetes. 2015;64:1431–1443. [DOI] [PubMed] [Google Scholar]
- 31. Moazzen H, Lu X, Ma NL, Velenosi TJ, Urquhart BL, Wisse LJ, Gittenberger‐de Groot AC, Feng Q. N‐acetylcysteine prevents congenital heart defects induced by pregestational diabetes. Cardiovasc Diabetol. 2014;13:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vijaya M, Manikandan J, Parakalan R, Dheen ST, Kumar SD, Tay SS. Differential gene expression profiles during embryonic heart development in diabetic mice pregnancy. Gene. 2013;516:218–227. [DOI] [PubMed] [Google Scholar]
- 33. Hoffman JI, Kaplan S, Liberthson RR. Prevalence of congenital heart disease. Am Heart J. 2004;147:425–439. [DOI] [PubMed] [Google Scholar]
- 34. Schein PS, Loftus S. Streptozotocin: depression of mouse liver pyridine nucleotides. Cancer Res. 1968;28:1501–1506. [PubMed] [Google Scholar]
- 35. Hoang TT, Goldmuntz E, Roberts AE, Chung WK, Kline JK, Deanfield JE, Giardini A, Aleman A, Gelb BD, Mac Neal M, Porter GA Jr, Kim R, Brueckner M, Lifton RP, Edman S, Woyciechowski S, Mitchell LE, Agopian AJ. The congenital heart disease genetic network study: cohort description. PLoS One. 2018;13:e0191319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Leung C, Liu Y, Lu X, Kim M, Drysdale TA, Feng Q. Rac1 signaling is required for anterior second heart field cellular organization and cardiac outflow tract development. J Am Heart Assoc. 2016;5:e002508 DOI: 10.1161/JAHA.115.002508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Morgan SC, Relaix F, Sandell LL, Loeken MR. Oxidative stress during diabetic pregnancy disrupts cardiac neural crest migration and causes outflow tract defects. Birth Defects Res A Clin Mol Teratol. 2008;82:453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Molin DG, Roest PA, Nordstrand H, Wisse LJ, Poelmann RE, Eriksson UJ, Gittenberger‐De Groot AC. Disturbed morphogenesis of cardiac outflow tract and increased rate of aortic arch anomalies in the offspring of diabetic rats. Birth Defects Res A Clin Mol Teratol. 2004;70:927–938. [DOI] [PubMed] [Google Scholar]
- 39. Siman CM, Gittenberger‐De Groot AC, Wisse B, Eriksson UJ. Malformations in offspring of diabetic rats: morphometric analysis of neural crest‐derived organs and effects of maternal vitamin E treatment. Teratology. 2000;61:355–367. [DOI] [PubMed] [Google Scholar]
- 40. Sedmera D, Thompson RP. Myocyte proliferation in the developing heart. Dev Dyn. 2011;240:1322–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Roux M, Laforest B, Capecchi M, Bertrand N, Zaffran S. Hoxb1 regulates proliferation and differentiation of second heart field progenitors in pharyngeal mesoderm and genetically interacts with Hoxa1 during cardiac outflow tract development. Dev Biol. 2015;406:247–258. [DOI] [PubMed] [Google Scholar]
- 42. Tanaka K, Kaufman S, Milstien S. Tetrahydrobiopterin, the cofactor for aromatic amino acid hydroxylases, is synthesized by and regulates proliferation of erythroid cells. Proc Natl Acad Sci USA. 1989;86:5864–5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Anastasiadis PZ, Bezin L, Imerman BA, Kuhn DM, Louie MC, Levine RA. Tetrahydrobiopterin as a mediator of PC12 cell proliferation induced by EGF and NGF. Eur J Neurosci. 1997;9:1831–1837. [DOI] [PubMed] [Google Scholar]
- 44. Lepic E, Burger D, Lu X, Song W, Feng Q. Lack of endothelial nitric oxide synthase decreases cardiomyocyte proliferation and delays cardiac maturation. Am J Physiol Cell Physiol. 2006;291:C1240–C1246. [DOI] [PubMed] [Google Scholar]
- 45. McCulley DJ, Black BL. Transcription factor pathways and congenital heart disease. Curr Top Dev Biol. 2012;100:253–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen H, Shi S, Acosta L, Li W, Lu J, Bao S, Chen Z, Yang Z, Schneider MD, Chien KR, Conway SJ, Yoder MC, Haneline LS, Franco D, Shou W. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development. 2004;131:2219–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li H, Forstermann U. Pharmacological prevention of eNOS uncoupling. Curr Pharm Des. 2014;20:3595–3606. [DOI] [PubMed] [Google Scholar]
- 48. Cai Z, Lu Q, Ding Y, Wang Q, Xiao L, Song P, Zou MH. Endothelial nitric oxide synthase‐derived nitric oxide prevents dihydrofolate reductase degradation via promoting S‐nitrosylation. Arterioscler Thromb Vasc Biol. 2015;35:2366–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Koenig SN, Bosse K, Majumdar U, Bonachea EM, Radtke F, Garg V. Endothelial Notch1 is required for proper development of the semilunar valves and cardiac outflow tract. J Am Heart Assoc. 2016;5:e003075 DOI: 10.1161/JAHA.115.003075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Correa A, Gilboa SM, Besser LM, Botto LD, Moore CA, Hobbs CA, Cleves MA, Riehle‐Colarusso TJ, Waller DK, Reece EA. Diabetes mellitus and birth defects. Am J Obstet Gynecol. 2008;199:237.e1–239.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang P, Li X, Xu C, Eckert RL, Reece EA, Zielke HR, Wang F. Maternal hyperglycemia activates an ASK1‐FoxO3a‐caspase 8 pathway that leads to embryonic neural tube defects. Sci Signal. 2013;6:ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yang P, Zhao Z, Reece EA. Activation of oxidative stress signaling that is implicated in apoptosis with a mouse model of diabetic embryopathy. Am J Obstet Gynecol. 2008;198:130.e1–137.e1. [DOI] [PubMed] [Google Scholar]
- 53. Nachmany A, Gold V, Tsur A, Arad D, Weil M. Neural tube closure depends on nitric oxide synthase activity. J Neurochem. 2006;96:247–253. [DOI] [PubMed] [Google Scholar]
- 54. Lupo PJ, Chapa C, Nousome D, Duhon C, Canfield MA, Shaw GM, Finnell RH, Zhu H; National Birth Defects Prevention Study . A GCH1 haplotype and risk of neural tube defects in the National Birth Defects Prevention Study. Mol Genet Metab. 2012;107:592–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Douglas G, Hale AB, Crabtree MJ, Ryan BJ, Hansler A, Watschinger K, Gross SS, Lygate CA, Alp NJ, Channon KM. A requirement for Gch1 and tetrahydrobiopterin in embryonic development. Dev Biol. 2015;399:129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fitzpatrick PF. The aromatic amino acid hydroxylases. Adv Enzymol Relat Areas Mol Biol. 2000;74:235–294. [DOI] [PubMed] [Google Scholar]
- 57. Homma D, Katoh S, Tokuoka H, Ichinose H. The role of tetrahydrobiopterin and catecholamines in the developmental regulation of tyrosine hydroxylase level in the brain. J Neurochem. 2013;126:70–81. [DOI] [PubMed] [Google Scholar]