

Abstract
Background
Hereditary hemorrhagic telangiectasia (HHT) is a rare genetic vascular disorder caused by mutations in endoglin (ENG), activin receptor‐like kinase 1 (ACVRL1;ALK1), or SMAD4. Major clinical symptoms of HHT are arteriovenous malformations (AVMs) found in the brain, lungs, visceral organs, and mucosal surface. Animal models harboring mutations in Eng or Alk1 recapitulate all of these HHT clinical symptoms and have been useful resources for studying mechanisms and testing potential drugs. However, animal models representing SMAD4 mutations have been lacking. The goal of this study is to evaluate Smad4‐inducible knockout (iKO) mice as an animal model of HHT and compare the phenotypes with other established HHT animal models.
Methods and Results
Global Smad4 deletion was induced at neonatal and adult stages, and hemoglobin levels, gastrointestinal hemorrhage, and presence of aberrant arteriovenous connections were examined. Neonatal Smad4‐iKO mice exhibited signs of gastrointestinal bleeding and AVMs in the brain, intestine, nose, and retina. The radial expansion was decreased, and AVMs were detected on both distal and proximal retinal vasculature of Smad4‐iKOs. Aberrant smooth muscle actin staining was observed in the initial stage AVMs and their connecting veins, indicating abnormal arterial flow to veins. In adult mice, Smad4 deficiency caused gastrointestinal bleeding and AVMs along the gastrointestinal tract and wounded skin. HHT‐related phenotypes of Smad4‐iKOs appeared to be comparable with those found in Alk1‐iKO and Eng‐iKO mice.
Conclusions
These data further confirm that SMAD signaling is crucial for normal arteriovenous network formation, and Smad4‐iKO will be an alternative animal model of AVM research associated with HHT.
Keywords: animal model of human disease remodeling, arteriovenous fistula, arteriovenous malformation, hereditary hemorrhagic telangiectasia, transgenic model
Subject Categories: Vascular Disease, Peripheral Vascular Disease, Animal Models of Human Disease, Vascular Biology, Genetically Altered and Transgenic Models
Clinical Perspective
What Is New?
This study presents a novel mouse model for juvenile polyposis–hereditary hemorrhagic telangiectasia (HHT).
Mice with global Smad4 deletion in neonatal and adult stages reproduce phenotypes observed in patients with juvenile polyposis–HHT, including anemia and arteriovenous malformations in the brain, gastrointestinal tract, and skin.
The juvenile polyposis–HHT model recapitulates most of the HHT phenotypes, but the frequency and severity of the phenotypes in this model appear to be more variable in comparison with HHT1 and HHT2 models.
Results from this study strongly support the notion that SMAD4 is a central mediator of endoglin/activin receptor‐like kinase 1 signaling for the development of proper arteriovenous network.
What Are the Clinical Implications?
This study strengthens the premise that modulating SMAD4 activity is a therapeutic strategy for HHT1 and HHT2.
Smad4‐inducible knockout mouse model would be uniquely useful to test novel drugs that work with endoglin/activin receptor‐like kinase 1 at the plasma membrane to enhance collateral SMAD‐independent pathways to prevent or rescue HHT lesions in patients with juvenile polyposis–HHT.
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler‐Rendu‐Weber syndrome, is a familial vascular disease that affects ≈1 in 5000 individuals.1, 2, 3 Four clinical diagnosis criteria include family history, recurrent epistaxis, mucocutaneous telangiectases, and arteriovenous malformations (AVMs) in internal organs such as the lung, brain, and liver. Definite HHT is diagnosed if at least 3 of these 4 criteria are present.4 Heterozygous mutations of 2 genes, endoglin (ENG) and activin receptor‐like kinase 1 (ACVRL1; ALK1), encoding receptors for transforming growth factor‐β and bone morphogenetic proteins (BMPs) were found in >95% of people with definite HHT.5 A few cases of definite HHT were caused by SMAD4 mutations.6
SMAD4 is a crucial downstream signaling transducer of transforming growth factor‐β/BMP signaling. Mutations in SMAD4 or BMPR1A (BMP type I receptor; ALK3) were found in the juvenile polyposis (JP) syndrome, which is characterized by the presence of multiple hamartomatous gastrointestinal polyps with a family history of polyposis.7, 8 Gallione et al6 have identified the genetic linkages of SMAD4 mutations in patients with the combined syndrome of JP and HHT. Studies have shown that not all patients with SMAD4 mutations exhibit both JP and HHT phenotypes, but patients with HHT harboring SMAD4 mutations without apparent JP syndrome are at high risk of JP and early‐onset colorectal cancer.9 Smad4 haploinsufficiency was sufficient for intestinal tumor initiation and progression in mice,10 but HHT‐like vascular lesions have not been reported.
Animal models that reproduce HHT symptoms have been developed in the past 10 years. HHT phenotypes including anemia, visceral hemorrhages, and subdermal AVMs were recapitulated in adult mice by global or endothelial cell (EC)–specific conditional deletion of the Alk1 or Eng gene.11, 12 Development of AVMs were also shown in the brain and retina of neonatal mice in which Alk1 or Eng was deleted,11, 13, 14 or the ligands of these receptors (ie, BMP9 and BMP10) were blocked.15, 16 These mouse models have been pivotal for investigating pathogenetic mechanisms underlying AVM development and also for testing efficacy of potential drugs.17, 18 However, a HHT mouse model using Smad4 mutants has not been fully established to date.
In this study, we utilized Smad4‐floxed mice to examine HHT phenotypes at postnatal and adult stages and compare them with other established HHT mouse models.
Materials and Methods
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure. Mouse strains used in this study (Alk1‐, Eng‐, and Smad4‐floxed mice) can be distributed from Barrow Neurological Institute after acquiring proper approval and material transfer agreements from the institutes of original inventors.
Animals
All in vivo procedures were conducted in accordance with animal use guidelines established by the University of Florida and Barrow Neurological Institute (201401417)/St. Joseph Hospital Medical Center Institutional Animal Care and Use Committee (531). Establishment of the Alk1 2loxp, Eng 2loxP, and Smad4 2loxP allele in laboratory mice was previously described.19, 20, 21 Rosa26CreER (R26 CreER) mice were purchased from the Jackson Laboratory. Neonatal R26 CreER; Alk1 2loxP/2loxP (n=4), and control littermates (n=4) were given intragastric injection of 50 μg of tamoxifen at postnatal day 3, and physiological inspection and retina sampling were performed at postnatal day 5. Smad4 was deleted in neonatal pups by intragastric treatment of tamoxifen (100 μg) for 3 consecutive days from postnatal day 1, then tissues were harvested at postnatal day 7 (wild‐type, n=32; Smad4‐iKO, n=26). For adult mice, tamoxifen (0.1 mg/g of body weight) was intraperitoneally administered 3 times in adult wild‐type control (n=14), Smad4 2loxP/2loxP; R26 CreER (n=23), Alk1 2loxP/2loxP; R26 CreER (n=8) or Eng 2loxP/2loxP; R26 CreER (n=10) mice to delete the genes. All mutant mice used in this study were on a mixed (129Sv/C57BL6) background.
Hemoglobin Concentration and Gastrointestinal Hemorrhage Index
Hemoglobin in the blood collected from mouse tails was examined using a hemoglobin photometer (Hemopoint H2, Stanbio Laboratory).
Skin Wound Generation and Latex Perfusion
Full‐thickness skin in the mid‐dorsum were incised with a sterile disposable biopsy punch (4 mm diameter) in adult mice. Tamoxifen was also intraperitoneally treated at 0.1 mg/g of body weight from the day of wounding (day 0) to day 2. At day 6, physiological signs were measured and latex dye was infused. A detailed procedure for latex dye perfusion and image processing is the same as previously described.17, 18
Immunofluorescence and Immunohistochemistry
The following primary antibodies were used in the immunostaining: anti‐CD31 (Biocare Medical), anti‐SMA‐Cy3 (Sigma‐Aldrich Co.), and anti‐SMAD4 (Santa Cruz Biotechnology). For immunofluorescence of retinal vasculature, enucleated eyes were fixed in 4% paraformaldehyde, then retinas were isolated. Primary antibodies were applied in the retinas, then secondary antibodies conjugated with Alexa 405, 488, or 594 were used to detect primary antibodies. For immunohistochemistry, paraffin‐embedded tissues were sectioned and anti‐SMAD4 antibodies (Santa Cruz Biotechnology) were incubated on the rehydrated sections. Mouse‐on‐mouse AP polymer (Biocare Medical) was used as secondary antibodies according to the manufacturer's instruction. Images were captured using a BZ‐X700 microscope (Keyence).
Western Blot
Wounded skins or cultured cells were lysed in a RIPA buffer containing 50 mmol/L Tris‐HCl (pH 8.0), 150 mmol/L NaCl, 1% Nonidet P‐40, 0.5% Deoxycholate, 0.1% SDS, 2 mmol/L EDTA, 1X Halt Protease Inhibitor Cocktail (Pierce), and 1X Halt Phosphatase Inhibitor Cocktail (Pierce). Quantified proteins using DC Protein Assay kit (Bio‐Rad Laboratories) were subjected to SDS‐PAGE and membrane blotting. The blots were probed with the following primary antibodies: anti‐β‐Actin (1:5000; Sigma‐Aldrich Co.), anti‐phosphor‐SMAD 1/5/8 (1:1000; Cell Signaling Technology, Inc), anti‐SMAD1 (1:1000; Cell Signaling Technology, Inc), anti‐phosphor‐SMAD2/3 (1:1000; Cell Signaling Technology, Inc), and anti‐SMAD2 (1:1000; Cell Signaling Technology, Inc), anti‐SMAD4 (1: 250, Santa Cruz Biotechnology; used for Western blot and immunohistochemistry of mouse skin), anti‐SMAD4 (1: 5000, Abcam; used for Western blot of lung ECs), anti‐human endoglin (1:300, BD Biosciences), and anti‐mouse endoglin (1: 500, Santa Cruz Biotechnology), followed by species‐matched secondary antibody incubation. Band was detected by enhanced chemiluminescence reaction (Pierce), and the intensities were quantified with ImageJ (National Institutes of Health).
Cell Culture and SMAD4 Knockdown
Human umbilical vein ECs (HUVECs; LONZA) were cultured in EBM2‐Bulletkit (LONZA) on 2% gelatin‐coated culture plates. For knockdown experiments, HUVECs were transfected with 10 nmol/L of ON‐TARGETplus Non‐targeting Pool (Darmacon) or small interfering RNA against SMAD4 (Qiagen, mixture of 4 small interfering RNAs: SI03089527, SI03042508, SI00076041, SI00076020) using Lipofectamine RNAiMAX Reagent (Invitrogen) following manufacturer's instructions. Cells were starved for about 16 hours in serum‐free medium (EBM‐2 medium containing 0.1% fetal bovine serum) and then 10 ng/mL of human BMP9 (R&D systems) were treated for 2 hours.
Isolation of Pulmonary ECs From Neonatal Mice
Pulmonary ECs were sorted from PN7 wild‐type control or Smad4 2loxP/2loxP; R26 CreER mouse lungs using anti‐CD31 antibody‐conjugated magnetic beads (Invitrogen). Lungs were minced and digested with 1 mg/mL of Dispase/Collagenase (Roche) in DMEM at 37°C for 45 minutes. The crude cells were pelleted after filtering them through 70 μm nylon mesh. The cells were resuspended in PBS containing 0.1% BSA and then incubated with anti‐CD31 antibody‐coated Dynabeads (Invitrogen) for 12 minutes at room temperature. The bead‐bound cells were washed with PBS/0.1% BSA and the cells were placed on 2% gelatin‐coated plates and cultured in a formulated EC medium in which DMEM was complemented with 20% fetal bovine serum (HyClone), 0.5% heparin (200 mg/mL; Sigma‐Aldrich Co.), 1% endothelial mitogen (10 mg/mL; Biomedical Technologies, Inc), 1% nonessential amino acids (Mediatech, Inc), 1% sodium pyruvate (100 mmol/L; Invitrogen), and 0.4% penicillin‐streptomycin (Invitrogen). For Smad4 gene deletion, 4 hydroxyl‐tamoxifen (1 μmol/L; Sigma) were treated for 3 consecutive days. Cells were starved for about 16 hours in serum‐free medium (EBM‐2 medium containing 0.1% fetal bovine serum) and 10 ng/mL of mouse BMP9 (R&D systems) were treated for 2 hours.
Reverse Transcription Polymerase Chain Reaction Analysis
Total RNA were extracted from HUVECs using a Total RNA Purification Plus Kit (Norgen Biotek Corp.) and subjected to reverse transcription reaction using SuperScript IV (Invitrogen). Real‐time polymerase chain reaction was performed using a SYBR Premix (ABI), and the ΔΔCt method determined the relative levels of the transcripts. The relative expression represents the averages of the mRNA levels from 3 independent experiments. The primer sequences used for the reverse transcription polymerase chain reaction analyses are summarized in Table.
Table 1.
Real‐Time Polymerase Chain Reaction Primer Sequences
| Gene | Primer Name | Sequences (5′–3′) |
|---|---|---|
| ID1 | ID1‐F | CGGATCTGAGGGAGAACAAG |
| ID1‐R | CTGAGAAGCACCAAACGTGA | |
| ID3 | ID3‐F | GGAGCTTTTGCCACTGACTC |
| ID3‐R | TTCAGGCCACAAGTTCACAG | |
| JAG1 | JAGGED1‐F | AGGCCGTTGCTGACTTAGAA |
| JAGGED1‐R | GCAGAAGTGGGAGCTCAAAG | |
| HEY2 | HEY2‐F | TTCAAGGCAGCTCGGTAACT |
| HEY2‐R | CAGGCACTTACGAAACACGA | |
| ACTIN | ACTIN‐F | GAGCTGCCTGACGGCCAGG |
| ACTIN‐R | CATCTGCTGGAAGGTGGAC |
Statistical Analysis
Data are represented as mean±SD. One‐way ANOVA with post‐Tukey multiple comparison tests (GraphPad Prism 7) was used to analyze the data. Two‐tailed Student t test was used to determine a statistical significance between 2 groups. Paired t test was utilized to compare equal sample size groups, and independent sample t test (2‐sample equal variation test) was applied for unequal sample size groups. A value of P<0.05 was considered statistically significant.
Results and Discussions
To address whether SMAD4 is required for the formation of normal vascular network in early postnatal development, we generated tamoxifen‐inducible global Smad4 knockout mice (Smad4 2loxP/2loxP; R26 CreER: Smad4‐iKO hereafter), and tamoxifen was administered for 3 consecutive days from postnatal day 1 (Figure 1A). Most Smad4‐iKO mice were found moribund or dead by postnatal day 8, most likely attributable to gastrointestinal hemorrhages as described below. Body weights of tamoxifen‐treated Smad4‐iKO pups (n=26; 2.16±0.39) were lower than those of controls (n=34; 3.16±0.69) at postnatal day 7 (Figure 1B). The smaller body sizes and lower body weights of Smad4‐iKO mice are likely associated with problems in the gastrointestinal tract, as these mutants had hemorrhagic signs in overall intestines, especially in cecum and colon, and empty and shrunken stomachs, compared with controls (Figure 1C).
Figure 1.
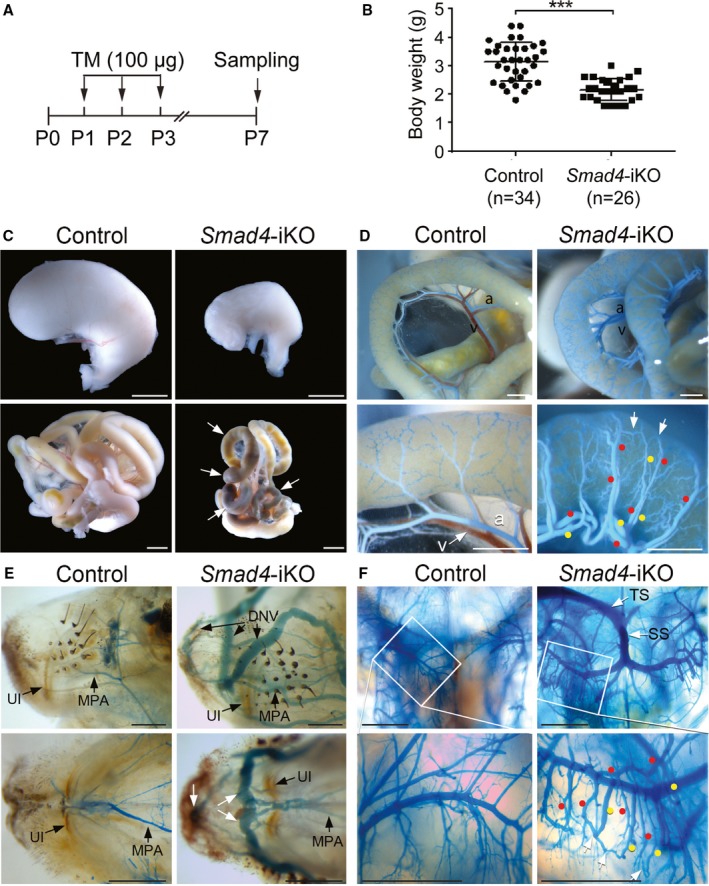
Smad4 deletion results in gastrointestinal hemorrhages and arteriovenous malformations in the intestines, head, and brain. A, Schematic overview of the experimental setup. 100 μg of tamoxifen (TM) was intragastrically injected for 3 consecutive days. B, Decreased body weight of Smad4‐inducible knockout (iKO) pups at postnatal day 7 (P7). All data represent mean±SD. ***P<0.001. C, Morphology of stomachs (upper panels) and intestines (lower panels) of control and Smad4‐iKO pups at P7. Arrows indicate the areas showing hemorrhagic signs. Scale bars: 2 mm. D, Mesenteric and intestinal vessels of controls and Smad4‐iKO visualized by latex dye perfusion via the left ventricle. Red and yellow dots indicate arteries and veins, respectively. White arrows indicate arteriovenous shunting vessels. Scale bars: 1 mm. E, Lateral (upper panels) and bottom‐up (lower panels) views of vasculature in snout areas of control and Smad4‐iKO mice. Apparent arteriovenous shunts were detected in Smad4 mutants. Scale bars: 1 mm. F, Vasculature in the area of the hippocampus of control and Smad4‐iKO visualized by latex dye. Numerous arteriovenous shunts and latex‐filled superior sagittal sinuses were detected in Smad4‐iKOs. Red and yellow dots indicate arteries and veins, respectively. White arrows mark arteriovenous shunts. Scale bars: 1 mm. a indicates artery; DNV, dorsal nasal vein; MPA, major palatine artery; P0, P1, P2, and P3, postnatal day 0, 1, 2, and 3, respectively; SS, straight sinus; TS, transverse sinus; UI, upper incisor; v, vein.
We then examined whether Smad4 deletion was sufficient for the formation of spontaneous AVMs by latex dye perfusion from the left ventricle. While the dye is restricted only in arteries of normal blood vessels without passing through capillary vessels, it crosses to veins in the presence of arteriovenous shunts. As shown in Figure 1D, the dye filled in both mesenteric arteries and veins of Smad4‐iKOs, which were dilated and tortuous, indicating the presence of multiple intestinal AVMs, whereas the dye was found only in the arteries of the controls. We also found arteriovenous shunts in nasal vessels in Smad4‐iKO mice (Figure 1E). Numerous AVMs found in the hippocampal areas of the brain of Smad4‐iKOs mice are reminiscent of the brain AVM phenotype in Alk1 mutants11 (Figure 1F).
We further investigated whether Smad4 deletion causes AVMs during the development of retinal vasculature. For comparison, we examined the retinal AVM formation in Alk1 knockout mice (Alk1 2loxP/2loxP; R26 CreER: Alk1‐iKO hereafter). A countless number of fully dilated AVMs were developed in postnatal day 5 Alk1‐iKO retinas (Figure 2B). This is well beyond the extent of retinal AVMs found in EC‐specific Alk1‐iKOs that were previously reported.14 We detected a varying degree of AVMs in developing retinal vasculature in Smad4‐iKO. While smooth muscle α actin (SMA) staining primarily marked arteries in the control retinas (Figure 2C through 2F), it appeared on the veins as well as the vessels connecting veins to arteries in Smad4‐iKO retinas (Figure 2G through 2N). Because of the complexity of retinal vascular networks and variable stages of AVMs, SMA staining was shown to be useful for identifying the number and locations of AVMs. Some of these SMA‐positive veins connected to arteries showed dilated, tortuous, and looping structures (Figure 2K through 2N), but some did not exhibit the distinctive AVM morphologies (Figures 2G and 2I, 3). We believe that the former cases represent mature AVMs and the later ones represent immature AVMs that are in the process of remodeling into AVMs. Not all SMAD4‐deficient veins were SMA‐positive, indicating that elevated SMA staining in the veins and arteriovenous shunting vessels of Smad4‐iKOs is likely a response to the increased flow. AVM counting per retina using CD31 and SMA‐double staining showed that 5.82±2.65 AVMs were present per Smad4‐iKO retina (Figure 2O), and percentages of retinas containing immature and mature AVMs were 33.3% and 66.7%, respectively (Figure 2P). In addition, radial expansion of retinal vasculature was decreased in Smad4‐iKO retinas compared with controls (Figure 2Q). Taken together, these data demonstrate that SMAD4 plays a critical role for the development of normal arteriovenous networks in the early postnatal stage, and SMAD4 deficiency results in AVMs in the brain, retina, and gastrointestinal tract.
Figure 2.
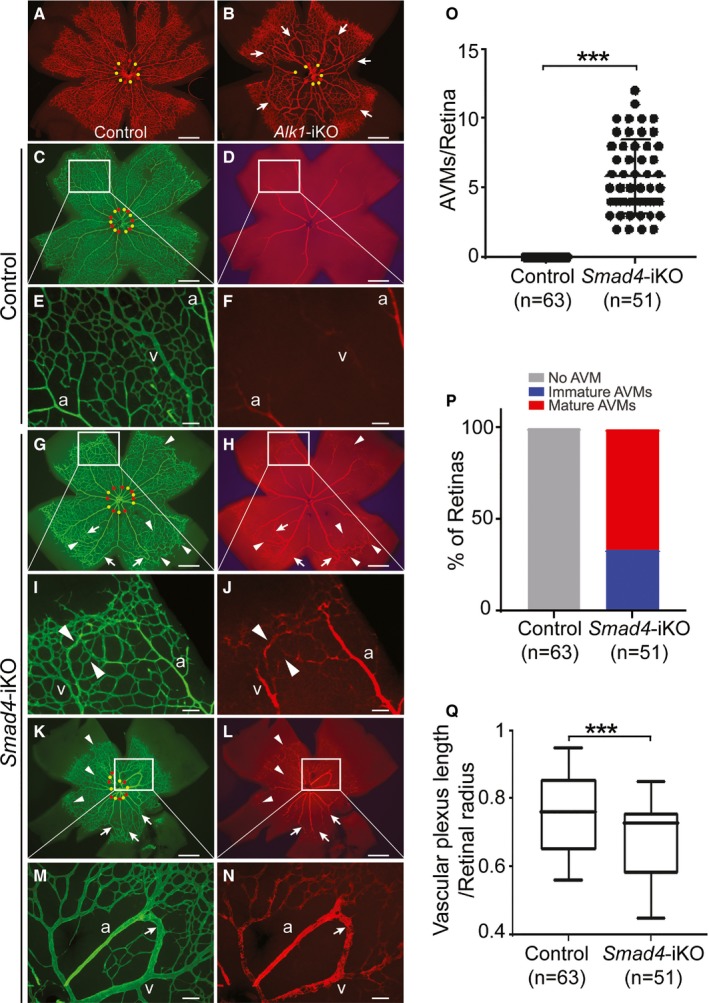
Smad4 deficiency induces spontaneous arteriovenous malformations (AVMs) in developing retina. A and B, Immunofluorescence staining of CD31 (red) in postnatal day 5 retinas of control (A) and Alk1‐inducible knockout (iKO) (B) mice, in which 50 μg of tamoxifen was intragastrically administrated to postnatal day 3 pups. C through N, CD31 (green) and smooth muscle α actin (red) staining of postnatal day 7 retinas from control (C through F) and Smad4‐iKO (G through N). 100 μg of tamoxifen was injected for 3 consecutive days from postnatal day 1. I and J, Magnified images of immature AVMs in (G and H). M and N, Magnified images of developed AVMs of Smad4‐iKO retina in the boxes of (K and L). In (C, G, and K), arteries and veins are indicated by red and yellow dots, respectively. Arrows and arrowheads indicate developed and immature AVMs, respectively. Scale bars in (A through D, G, H, K, and L) indicate 500 μm; (E, F, I, J, M, and N), 100 μm. O, Quantification of retinal AVMs in Smad4‐iKO pups. P, Percentage of retinas containing only immature or both immature and developed AVMs in Smad4‐iKO retinas. Q, Decreased progression of vascular plexus towards the edge of the retina. All data represent mean±SD. ***P<0.001. a indicates artery; v, vein.
Figure 3.
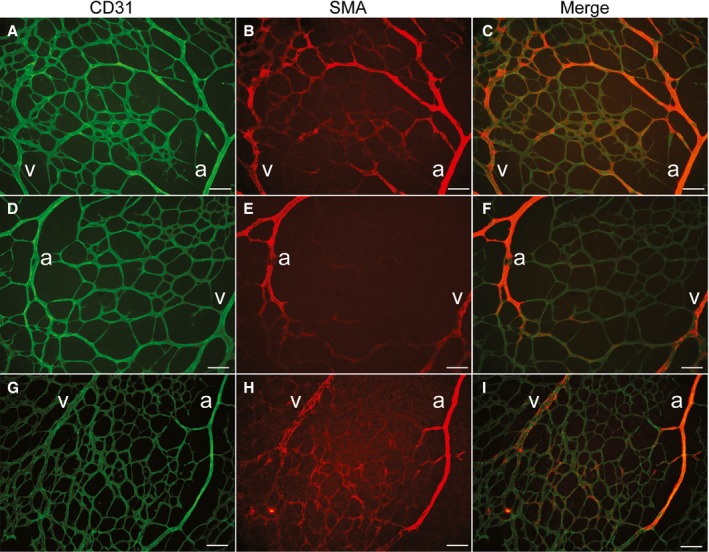
Immature arteriovenous malformation areas shown in a postnatal day 7 (P7) retina of Smad4‐inducible knockout (iKO). Immunofluorescence staining of CD31 (green; A, D, and G), smooth muscle α actin (SMA [red]; B, E, and H), and merged images (C, F, and I) of P7 retina of Smad4‐iKO. Aberrant SMA‐positive veins and connecting vessels to neighboring arteries indicate initial stages of arteriovenous shunting. Scale bars indicate 100 μm. a indicates artery; v, vein.
Next, we examined the consequence of global Smad4 deletion in adult mice for hemorrhage and development of AVMs in visceral organs and wounded skin and compared it with global Alk1 or Eng deletion. Tamoxifen was intraperitoneally administered for 3 days to Smad4‐iKO adult mice (older than 2 months). Approximately 32% of Smad4‐iKO mice (n=8/25) were found dead between 5 and 6 days after the first tamoxifen injection (day 0), and the mutants that survived at day 6 were mostly sick or moribund. To compare pathophysiological changes of Smad4‐iKO with other HHT models, similar experimental conditions were applied to Alk1‐iKO and Eng‐iKO (Eng 2loxP/2loxP; R26 CreER) mice. All 3 HHT models showed decreases in body weights (Figure 4A) and hemoglobin levels (Figure 4B) on day 6, compared with those on day 0. Reduced hemoglobin level was the greatest in Alk1‐iKO and the slightest in Smad4‐iKO among the 3 HHT models (Figure 4B). A varying degree of gastrointestinal hemorrhage was observed in all 3 HHT models, but the severity of bleeding correlated with reduced hemoglobin levels. The stomachs of Smad4‐iKO mice were enlarged with signs of bleeding, and the intestine and cecum appeared to be swollen and inflamed (Figure 4C). Vascular casting demonstrated dilated and tortuous AVMs in the stomach, intestines, and Peyer's patches in Smad4‐iKO mice as well as Alk1‐iKO and Eng‐iKO mice (Figure 4D). In spite of similar frequency of intestinal AVMs between Smad4‐iKO and Eng‐iKO mice, Smad4 mutants showed more severe AVMs and bleedings in the stomach (Figure 4E). It was shown that anemia observed from 4 weeks after deletion of Smad4 by interferon‐inducible Mx‐Cre resulted from gastric hemorrhages associated with multiple polyps in stomach but not from erythropoiesis.22 Considering a mild anemia phenotype, however, the early lethality of Sma4‐iKOs in this study is most likely caused by disruption of gastrointestinal homeostasis rather than hemorrhages.
Figure 4.
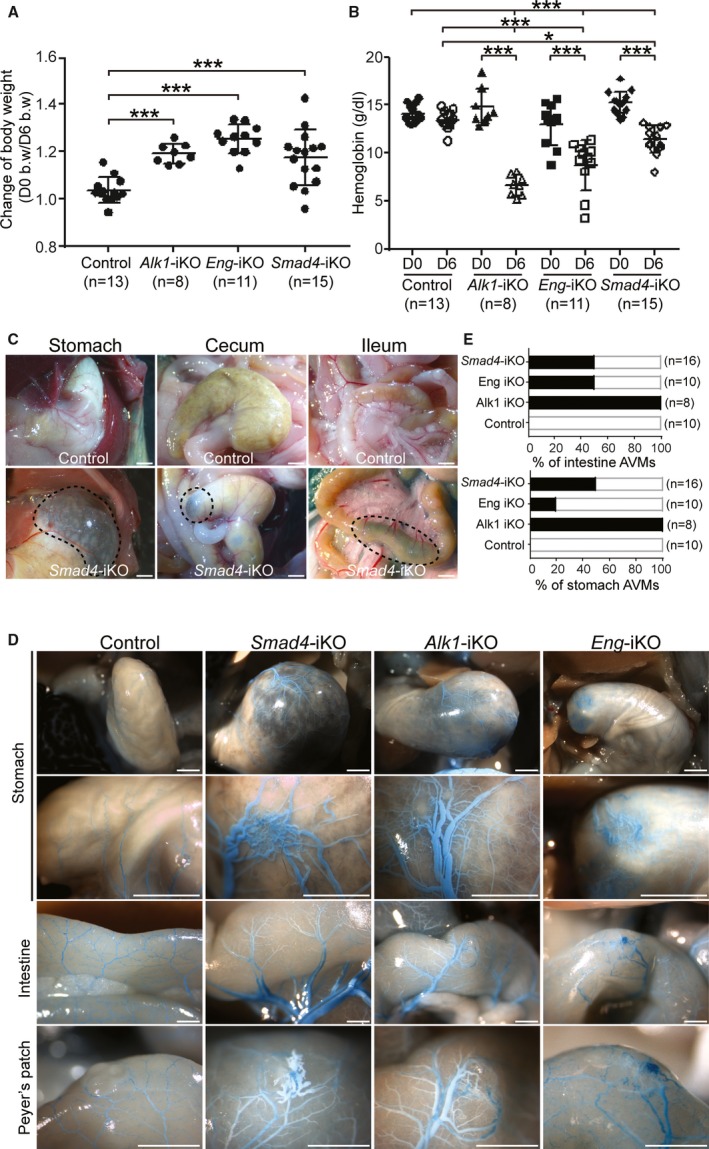
Global deletion of Smad4 in adult mice leads to gastrointestinal hemorrhages and arteriovenous malformations (AVMs). A and B, Reduction of body weight (b.w.; A) and hemoglobin levels (B) in Alk1‐, Eng‐, and Smad4‐inducible knockout (iKO) mice 6 days after tamoxifen treatment. *P<0.05, ***P<0.001. C, Hemorrhagic signs shown in gastrointestinal tracks of Smad4‐iKO mice. Scale bars indicate 1 mm. D, Visualized gastric and intestinal AVMs with latex dye perfusion. While the perfused dye was detectable only in arteries of control mice, it was observed in both dilated and tortuous arteries and veins in the stomachs, intestines, and Peyer's patches of Smad4‐, Alk1‐, and Eng‐iKO mice. Scale bars indicate 1 mm. E, Percentages of AVMs formed in intestine (upper panel) or stomach (lower panel) in Alk1‐, Eng‐, and Smad4‐iKO mice. D0 indicates day 0; D6, day 6.
Previously, we demonstrated that wounding or angiogenic stimulation is required for inducing AVMs in the skin of Alk1‐iKO and Eng‐iKO adult mice.11, 12, 17 To test whether wounding induces skin AVMs in Smad4‐iKO mice as well, a 4‐mm skip biopsy punch was placed on the dorsal skin on the first day of tamoxifen administration (day 0), and the blood vessels were visualized with latex perfusion on day 6. First, we confirmed the depletion of SMAD4 proteins in wounded skin and ear tissues with Western blot (Figure 5A) and immunohistochemistry (Figure 5B), respectively. Since Smad4‐iKO mice were lethal 6 days after tamoxifen treatment, Alk1‐iKO and Eng‐iKO mice were used as the controls to verify that 6 days were sufficient to form the subdermal AVMs, which can be visualized by latex dye perfusion. Vascular casting with latex clearly showed AVMs in the subdermal vessels surrounding of the wounds of Alk1‐iKO and Eng‐iKO mice (Figure 5D, 5E, and 5L), but not in controls (Figure 5C). Variable degrees of AVMs were observed in the wound areas of Smad4‐iKOs (Figure 5I): no AVMs (13%; Figure 5F), immature AVMs (39%; Figure 5G and 5G’), and fully developed AVMs (48%; Figure 5H and 5H’). Consistently, quantification on the vascular area containing the latex dye showed the presence of AVMs in all 3 HHT models, and Alk1‐iKO mice had the most robust and the least variable AVM development (Figure 5J and 5K). The reasons for the variability are unclear. Mixed strain background could be a reason. More likely is that the turnover rate of the SMAD4 protein might be slower than that of ALK1 or endoglin, or the gene deletion efficiency of Smad4 by Cre recombinase is lower than that of the Alk1 or Eng gene. Since the Smad4‐iKO mice died by day 7, the AVM phenotype by complete SMAD4 deficiency might not be fully exhibited in this model.
Figure 5.
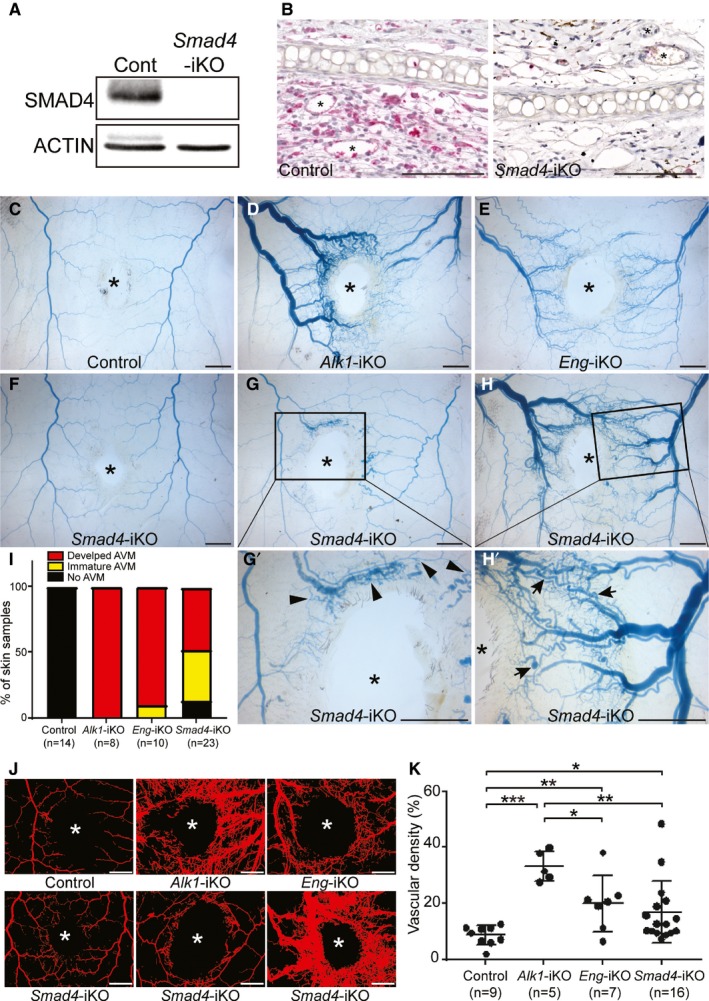
Smad4 deficiency causes skin arteriovenous malformations (AVMs) in response to wounding in adults. A and B, Western blot (A) of wounded skin and immunostaining of wounded ears with anti‐SMAD4 antibodies demonstrate that the SMAD4 protein is diminished in Smad4‐inducible knockout (iKO) mice. ACTIN was used as a loading control. Scale bars in B=100 μm. C through H’, Latex dye–perfused vasculature surrounding the wound in the dorsal skin of control (C), Alk1‐iKO (D), Eng‐iKO (E), and Smad4‐iKO (F through H’) mice 6 days after tamoxifen treatment. While most of the Alk1‐iKO and Eng‐iKO mice displayed well‐developed AVMs, Smad4‐iKO mice exhibited a range of severity of skin AVMs: no AVM (F), immature AVMs (G and G’), and developed AVMs (H and H’). Arrows indicate fully developed AVMs and arrowhead marks show immature AVMs. The wound sites are indicated by asterisks. The scale bars indicate 2 mm. I, Percentage of categorized wound‐induced AVMs in hereditary hemorrhagic telangiectasia model animals. J, Blood vessels containing the latex dye are processed for quantification. Representative converted images to quantify the vascular area containing latex. The wound sites are indicated by asterisks. The scale bars indicate 2 mm. K, Quantification of vascular density per a given area. All data represent mean±SD. *P<0.05, **P<0.01, and ***P<0.001. Cont indicates control.
To investigate the impact of SMAD4 deficiency in phosphorylation of receptor‐activated SMADs, we examined the phosphorylation status of SMAD1/5/8 and SMAD2/3 with or without BMP9 treatment in control and SMAD4‐deficient human and mouse ECs. Treatment of small interfering RNA targeting Smad4 nearly completely depleted SMAD4 in HUVECs (Figure 6A and 6B). Phosphorylation of SMAD1/5/8 in steady‐state conditions appeared to be elevated, while pSMAD2/3 was unchanged in SMAD4‐KD cells in normal culture media (Figure 6A and 6C). Endoglin protein level was reduced in SMAD4‐KD cells (Figure 6A and 6D). SMAD4‐deficiency did not affect BMP9‐responsive SMAD1,5,8 phosphorylation in serum‐starved HUVEC (Figure 6E through 6G). BMP9 treatment failed to normalize reduced endoglin protein levels in SMAD4‐KD cells (Figure 6E and 6H). The similar phenomenon was observed in primary pulmonary ECs isolated from Smad4‐iKO mice. 4 Hydroxyl‐tamoxifen treatment diminished SMAD4 by deleting the Smad4 gene in Smad4‐iKO cells (Figure 6I). 4 Hydroxyl‐tamoxifen treatment itself did not affect phosphorylation of R‐SMADs (Figure 6I). Consistent with the results in SMAD4‐KD HUVECs, pSMAD1/5/8 levels in a steady‐state condition were increased, but a similar BMP9 response was observed in 4 hydroxyl‐tamoxifen–treated Smad4‐iKO pulmonary ECs (Figure 6I through 6L). Endoglin protein level was markedly decreased in Smad4‐depeleted cells (Figure 6I, 6J, 6M, and 6N). Despite the elevated pSMAD1/5/8, expression of BMP9 responsive genes (ID1, ID3, JAG1, and HEY2) was blunted in SMAD4‐KD HUVECs (Figure 6O), indicating SMAD4 dependency in the regulation of these genes.
Figure 6.
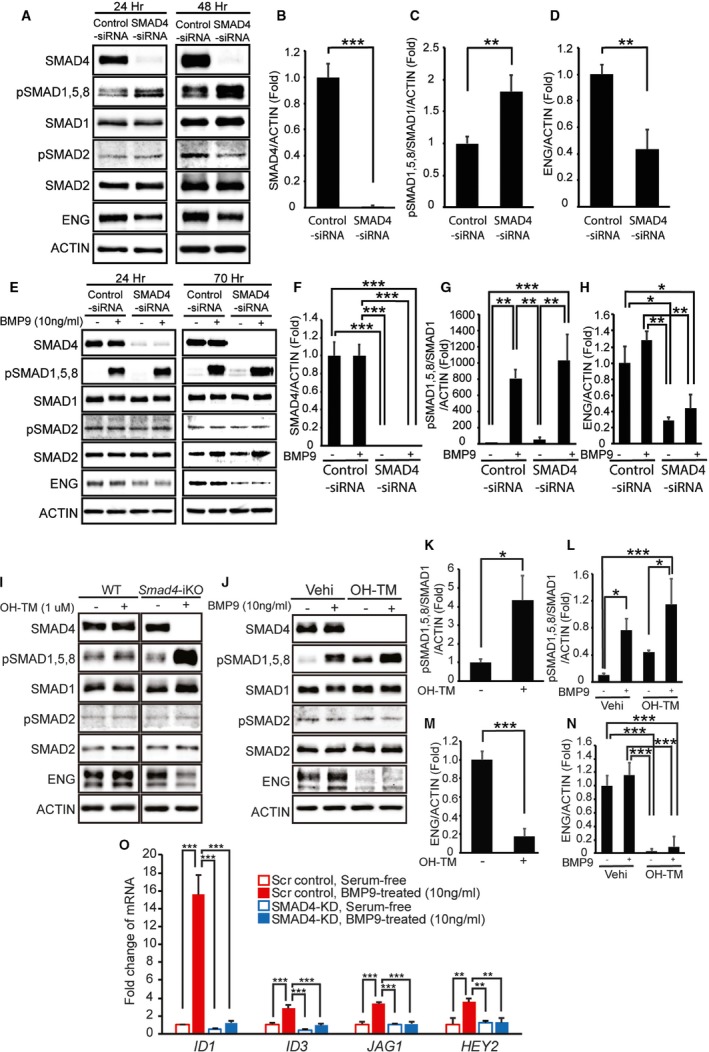
SMAD4 depletion causes loss of canonical ALK1/endoglin (ENG) signaling. A through D, Western blot analyses of SMAD4, phosphorylated and total R‐SMADs, and ENG in human umbilical vein endothelial cells (HUVECs) treated with scrambled control small interfering RNA (siRNA) or SMAD4‐siRNA for 24 and 48 hours in normal culture media (A). Quantification of SMAD4 proteins (B), pSMAD1, 5, and 8 levels (C), and ENG proteins (D) at 48 hours after siRNA transfection. E through H, Protein levels of SMAD4, phosphorylated and total R‐SMADs, and ENG in control and SMAD4‐KD HUVECs treated with or without bone morphogenetic protein 9 (BMP9) in serum‐starved condition at 24 and 70 hours after siRNA transfection (E). Quantification shows a >95% decrease of SMAD4 (F), responses to BMP9 (G), and reduction of ENG proteins (H) in SMAD4‐KD HUVECs 70 hours after transfection. 10 ng/mL of human BMP9 was treated for 2 hours after serum starvation for 16 hours. I through N, Protein levels of SMAD4, phosphorylated and total R‐SMADs, and ENG in primary pulmonary endothelial cells (pECs) isolated from wild‐type (WT) and Smad4‐inducible knockout (iKO) mice (I). Treatment with 4 hydroxyl‐tamoxifen (OH‐TM) did not affect SMAD4 and R‐SMAD expressions in WT cells but abolished SMAD4 expression and elevated pSMAD1, 5, and 8 levels in Smad4‐iKO cells (I and K). In serum‐free condition, SMAD1, 5, and 8 are activated by BMP9 treatment in both SMAD4‐depleted and normal pECs (J and L). ENG protein level is decreased in Smad4‐iKO pECs (I, J, M, and N). O, Quantitative reverse transcription polymerase chain reaction demonstrates that responses to BMP9 in upregulating BMP9‐ALK1 downstream genes ID1,ID3,JAG1, and HEY2 are markedly attenuated in SMAD4‐depleted HUVECs. ACTIN was used for normalization. All data represent mean±SD. *P<0.05, **P<0.01, ***P<0.001. Scr indicates scrambled‐siRNA; Vehi, vehicle.
We have shown here that global Smad4 deletion in neonates resulted in AVMs in the brain, retina, and visceral organs, and that Smad4 deletion in adults caused mild anemia and AVMs in the gastrointestinal tract and wounded skin. These HHT‐related phenotypes seen in Smad4‐iKO mice confirm the role of SMAD4 in the endoglin/ALK1 pathway for normal arteriovenous network formation. Since SMAD4 expression is ubiquitous, the cell type in which SMAD4 functions crucially for proper development of arteriovenous network is in question. Considering previous studies with Alk1 and Eng, 12, 13, 14 ECs are the most likely candidate. Consistent with this view, 2 recent reports have shown that endothelial‐specific Smad4 deletion caused defects in vascular development, including AVMs, in retina and visceral organs of postnatal stages similar to the global deletion shown in this study.23, 24
Both of these EC‐specific Smad4‐iKO studies showed increased EC proliferations, abnormal SMA expressions in venous vessels, decreased pericyte marker NG2 expressions, and disturbed expressions of artery‐ and vein‐specific genes in AVM‐associated vessels as common features.23, 24 Ola et al16 have previously shown that BMP9‐ALK1 signaling suppresses activation of phosphatidylinositol 3‐kinase (PI3K)‐protein kinase B (AKT) pathway, which is in part activated by vascular endothelial growth factor signaling. The same group recently reported that SMAD4 mediates BMP9‐ALK1 signaling for the suppression of flow‐induced AKT by repressing transcription of casein kinase 2, which inactivates phosphatase and tensin homolog (PTEN) deleted on chromosome 10.24 Since retinal AVMs in Alk1‐iEC KOs and Smad4‐iEC KOs were mostly found in the proximal area of vascular plexus where blood flow is high, it was suggested that this phenomenon may support the role of flow in AKT activation for AVM development.24 In the studies by us and Crist et al,23 however, the retinal AVMs formed in distal areas as well as proximal areas of Smad4‐iKOs. The difference might come from the timing of gene deletion, ie, postnatal day 0 versus postnatal day 1.23, 24 Crist et al have shown that reduction of vascular endothelial growth factor receptor 2 exacerbated the retinal AVM phenotype in Smad4‐iECKO mice.23 It indicates that the flow but not the vascular endothelial growth factor signaling is a primary inducer for the activation of PI3K‐AKT pathway in BMP9‐ALK1‐SMAD4‐deficient ECs.
Conclusions
Since global deletion of Smad4 results in an early lethality associated with gastrointestinal inflammations, and the variability of HHT‐related phenotypes appear to be high, it might not be the most suited preclinical model for HHT. However, the Smad4‐iKO model would be uniquely useful to test novel drugs that work with endoglin/ALK1 at the plasma membrane to enhance SMAD‐independent pathways to prevent or rescue HHT lesions in patients with JP and HHT.
Sources of Funding
This work was supported by Barrow Neurological Foundation, Department of Defense (PR161205), and the National Institutes of Health (HL128525) to Oh.
Disclosures
None.
Acknowledgments
We thank Dr Chu‐Xia Deng and Helen Arthur for providing the Smad4‐floxed and Eng‐floxed mice, respectively.
(J Am Heart Assoc. 2018;7:e009514 DOI: 10.1161/JAHA.118.009514)
References
- 1. Kjeldsen AD, Vase P, Green A. Hereditary haemorrhagic telangiectasia: a population‐based study of prevalence and mortality in Danish patients. J Intern Med. 1999;245:31–39. [DOI] [PubMed] [Google Scholar]
- 2. Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet. 2006;43:97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Govani FS, Shovlin CL. Hereditary haemorrhagic telangiectasia: a clinical and scientific review. Eur J Hum Genet. 2009;17:860–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, Kjeldsen AD, Plauchu H. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu‐Osler‐Weber syndrome). Am J Med Genet. 2000;91:66–67. [DOI] [PubMed] [Google Scholar]
- 5. McDonald J, Wooderchak‐Donahue W, VanSant Webb C, Whitehead K, Stevenson DA, Bayrak‐Toydemir P. Hereditary hemorrhagic telangiectasia: genetics and molecular diagnostics in a new era. Front Genet. 2015;6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S, Mitchell G, Drouin E, Westermann CJ, Marchuk DA. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet. 2004;363:852–859. [DOI] [PubMed] [Google Scholar]
- 7. Howe JR, Roth S, Ringold JC, Summers RW, Jarvinen HJ, Sistonen P, Tomlinson IP, Houlston RS, Bevan S, Mitros FA, Stone EM, Aaltonen LA. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science. 1998;280:1086–1088. [DOI] [PubMed] [Google Scholar]
- 8. Howe JR, Bair JL, Sayed MG, Anderson ME, Mitros FA, Petersen GM, Velculescu VE, Traverso G, Vogelstein B. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat Genet. 2001;28:184–187. [DOI] [PubMed] [Google Scholar]
- 9. Schwenter F, Faughnan ME, Gradinger AB, Berk T, Gryfe R, Pollett A, Cohen Z, Gallinger S, Durno C. Juvenile polyposis, hereditary hemorrhagic telangiectasia, and early onset colorectal cancer in patients with SMAD4 mutation. J Gastroenterol. 2012;47:795–804. [DOI] [PubMed] [Google Scholar]
- 10. Alberici P, Jagmohan‐Changur S, De Pater E, Van Der Valk M, Smits R, Hohenstein P, Fodde R. Smad4 haploinsufficiency in mouse models for intestinal cancer. Oncogene. 2006;25:1841–1851. [DOI] [PubMed] [Google Scholar]
- 11. Park SO, Wankhede M, Lee YJ, Choi EJ, Fliess N, Choe SW, Oh SH, Walter G, Raizada MK, Sorg BS, Oh SP. Real‐time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J Clin Invest. 2009;119:3487–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Garrido‐Martin EM, Nguyen HL, Cunningham TA, Choe SW, Jiang Z, Arthur HM, Lee YJ, Oh SP. Common and distinctive pathogenetic features of arteriovenous malformations in hereditary hemorrhagic telangiectasia 1 and hereditary hemorrhagic telangiectasia 2 animal models–brief report. Arterioscler Thromb Vasc Biol. 2014;34:2232–2236. [DOI] [PubMed] [Google Scholar]
- 13. Mahmoud M, Allinson KR, Zhai Z, Oakenfull R, Ghandi P, Adams RH, Fruttiger M, Arthur HM. Pathogenesis of arteriovenous malformations in the absence of endoglin. Circ Res. 2010;106:1425–1433. [DOI] [PubMed] [Google Scholar]
- 14. Tual‐Chalot S, Mahmoud M, Allinson KR, Redgrave RE, Zhai Z, Oh SP, Fruttiger M, Arthur HM. Endothelial depletion of Acvrl1 in mice leads to arteriovenous malformations associated with reduced endoglin expression. PLoS One. 2014;9:e98646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ruiz S, Zhao H, Chandakkar P, Chatterjee PK, Papoin J, Blanc L, Metz CN, Campagne F, Marambaud P. A mouse model of hereditary hemorrhagic telangiectasia generated by transmammary‐delivered immunoblocking of BMP9 and BMP10. Sci Rep. 2016;5:37366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ola R, Dubrac A, Han J, Zhang F, Fang JS, Larrivee B, Lee M, Urarte AA, Kraehling JR, Genet G, Hirschi KK, Sessa WC, Canals FV, Graupera M, Yan M, Young LH, Oh PS, Eichmann A. PI3 kinase inhibition improves vascular malformations in mouse models of hereditary haemorrhagic telangiectasia. Nat Commun. 2016;7:13650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Han C, Choe SW, Kim YH, Acharya AP, Keselowsky BG, Sorg BS, Lee YJ, Oh SP. VEGF neutralization can prevent and normalize arteriovenous malformations in an animal model for hereditary hemorrhagic telangiectasia 2. Angiogenesis. 2014;17:823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim YH, Kim MJ, Choe SW, Sprecher D, Lee YJ, Oh PS. Selective effects of oral antiangiogenic tyrosine kinase inhibitors on an animal model of hereditary hemorrhagic telangiectasia. J Thromb Haemost. 2017;15:1095–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang X, Li C, Herrera PL, Deng CX. Generation of Smad4/Dpc4 conditional knockout mice. Genesis. 2002;32:80–81. [DOI] [PubMed] [Google Scholar]
- 20. Park SO, Lee YJ, Seki T, Hong KH, Fliess N, Jiang Z, Park A, Wu X, Kaartinen V, Roman BL, Oh SP. ALK5‐ and TGFBR2‐independent role of ALK1 in the pathogenesis of hereditary hemorrhagic telangiectasia type 2. Blood. 2008;111:633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Allinson KR, Carvalho RL, van den Brink S, Mummery CL, Arthur HM. Generation of a floxed allele of the mouse Endoglin gene. Genesis. 2007;45:391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pan D, Schomber T, Kalberer CP, Terracciano LM, Hafen K, Krenger W, Hao‐Shen H, Deng C, Skoda RC. Normal erythropoiesis but severe polyposis and bleeding anemia in Smad4‐deficient mice. Blood. 2007;110:3049–3055. [DOI] [PubMed] [Google Scholar]
- 23. Crist AM, Lee AR, Patel NR, Westhoff DE, Meadows SM. Vascular deficiency of Smad4 causes arteriovenous malformations: a mouse model of Hereditary Hemorrhagic Telangiectasia. Angiogenesis. 2018;21:363–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ola R, Kunzel SH, Zhang F, Genet G, Chakraborty R, Pibouin‐Fragner L, Martin K, Sessa W, Dubrac A, Eichmann A. SMAD4 prevents flow induced arterial‐venous malformations by inhibiting casein kinase 2. Circulation. 2018. Available at: 10.1161/CIRCULATIONAHA.118.033842. Accessed October 30, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]