

Abstract
Background
Electrocardiographic (ECG) parameters are regarded as intermediate phenotypes of cardiac arrhythmias. Insight into the genetic underpinnings of these parameters is expected to contribute to the understanding of cardiac arrhythmia mechanisms. Here we used HXB/BXH recombinant inbred rat strains to uncover genetic loci and candidate genes modulating ECG parameters.
Methods and Results
RR interval, PR interval, QRS duration, and QTc interval were measured from ECGs obtained in 6 male rats from each of the 29 available HXB/BXH recombinant inbred strains. Genes at loci displaying significant quantitative trait loci (QTL) effects were prioritized by assessing the presence of protein‐altering variants, and by assessment of cis expression QTL (eQTL) effects and correlation of transcript abundance to the respective trait in the heart. Cardiac RNA‐seq data were additionally used to generate gene co‐expression networks. QTL analysis of ECG parameters identified 2 QTL for PR interval, respectively, on chromosomes 10 and 17. At the chromosome 10 QTL, cis‐eQTL effects were identified for Acbd4, Cd300lg, Fam171a2, and Arhgap27; the transcript abundance in the heart of these 4 genes was correlated with PR interval. At the chromosome 17 QTL, a cis‐eQTL was uncovered for Nhlrc1 candidate gene; the transcript abundance of this gene was also correlated with PR interval. Co‐expression analysis furthermore identified 50 gene networks, 6 of which were correlated with PR interval or QRS duration, both parameters of cardiac conduction.
Conclusions
These newly identified genetic loci and gene networks associated with the ECG parameters of cardiac conduction provide a starting point for future studies with the potential of identifying novel mechanisms underlying cardiac electrical function.
Keywords: bioinformatics, electrophysiology, rats
Subject Categories: Electrophysiology; Arrhythmias; Genetic, Association Studies; Gene Expression & Regulation; Computational Biology
Clinical Perspective
What Is New?
The study adds novel insights regarding the complex genetics underlying variability in ECG traits, and identifies 5 novel genes that potentially modulate cardiac conduction.
The study furthermore demonstrates the unique opportunities offered by combining genetic studies in rodents with gene expression and cardiac electrophysiological phenotypes to yield novel candidates in an unbiased, data‐driven way.
What Are the Clinical Implications?
Findings from this study provide insight into the molecular underpinnings of cardiac conduction, which may ultimately facilitate development of novel diagnostic and therapeutic strategies for patients with cardiac conduction disease and related arrhythmias.
The surface ECG records the electrical potential of the heart at the surface of the body as the electrical impulse travels throughout the heart with each heartbeat. The RR interval represents an entire cycle of cardiac electrical activity (1 heartbeat), with the number of cycles per minute being designated as the heart rate (beats per minute). The P wave denotes depolarization of the atria, which spreads from the sino‐atrial node toward the atrioventricular node, and from the right to the left atrium. The PR interval reflects the time the electrical impulse takes to travel from the sinus node through the atrioventricular node. The QRS complex represents the rapid depolarization of the ventricles and the T wave represents the repolarization of the ventricles.
The biological processes that underlie or impinge on the different parameters of the ECG are considered likely mediators of cardiac arrhythmia.1 As such, the different ECG parameters are considered important intermediate phenotypes (endophenotypes) of arrhythmia and dissecting the genetic underpinnings of these traits may therefore uncover pathways relevant for arrhythmia. This concept is supported by population‐based studies that demonstrated that ECG parameters are associated with risk of arrhythmia or sudden cardiac death,2 as well as by genome‐wide association studies (GWAS) that uncovered loci that are involved in both modulation of ECG traits and arrhythmia risk (eg, common variants at the SCN5A/SCN10A locus modulates both the PR interval as well as susceptibility to atrial fibrillation).3, 4
GWAS conducted in recent years in large samples of the general population have identified multiple loci harboring common genetic variants that modulate the different ECG parameters.5, 6, 7, 8 Yet, the understanding of the causal genes and underlying mechanisms of these loci has lagged behind. Furthermore, also when considered in aggregate, the identified loci explain only a small fraction of the population variance in these traits and therefore other loci remain to be identified. Genetic studies in rodents could be complementary to these human genetic studies and lead to the identification of new loci. In this respect, genetic studies in rodents overcome some limitations of human genetic studies in that they allow the integration of different levels of “omics” data with the phenotype, and thus provide new inroads for identification of genes underlying cardiac electrical function. This approach was previously used successfully by our group, resulting in the identification of Tnni3k as a novel gene for cardiac conduction (PR interval).9
The HXB/BXH panel of recombinant inbred (RI) rat strains derived by reciprocal crossing of the Brown Norway (BN) rat and the spontaneously hypertensive rat (SHR) has been a leading model in rodent genetic studies of complex traits, in particular cardiovascular.10, 11, 12 In the construction of an RI panel,13 2 inbred genetically distant progenitor strains are mated to produce F2 hybrids that carry a unique combination of maternal and paternal loci. Subsequent inbreeding of randomly chosen pairs of F2 animals and brother–sister mating for >20 generations yields individual homozygous RI strains. Thus, an RI panel offers a controllable, renewable resource of genetically identical (within strains) yet diverse (between strains) rats. The possibility to study genetically identical biological replicates optimizes estimation of trait heritability by reducing environmental variance, making RI panels statistically powerful for genetic studies even at relatively small sample sizes. Furthermore, the constant genetic background within each RI strain allows for the accumulation of genetic, omics, and phenotypic data over time. This is particularly the case for the HXB/BXH RI rat panel for which the parental strains have both been fully sequenced,14, 15 and genotyping of the RI strains has led to the identification of 1384 strain distribution patterns of single nucleotide polymorphisms (SNPs) for use in genetic mapping.16 Additionally, RNA‐seq‐based gene expression data are available for multiple tissues across the panel,17 as well as many cardiac physiological phenotypes such as blood pressure and cardiac mass.18 The panel therefore lends itself well to systems genetics studies by means of genome‐wide mapping of expression traits combined with physiological phenotypes and has proven to be pivotal in the identification of genes underlying mechanisms in complex diseases such as diabetes mellitus, metabolic syndrome, and cardiovascular disease.10, 19, 20
We here for the first time used the HXB/BXH RI rat panel to map genetic loci impacting on heart rate and ECG indices of cardiac conduction indices. By integrating ECG measurements with genotypic and cardiac transcriptomic data, we identify novel candidate genes and gene networks associated with cardiac conduction.
Methods
HXB/BXH RI Rat Strains
The HXB/BXH recombinant inbred rat strain panel, derived by intercrossing BN and SHR strains, has been described in detail previously.11, 21 In this study, 29 RI strains at >F60 are used, which constitute the same strains as in previous work.22 Rats were bred and housed at the Institute of Physiology, Academy of Sciences of the Czech Republic (Prague, Czech Republic). All of the animal studies were performed in agreement with the Animal Protection Law of the Czech Republic (311/1997) and approved by the Ethics Committee of the Czech Academy of Sciences Institute of Physiology.
Acquisition of ECG Data
ECGs were measured in 6 rats (all males, 6 weeks old) from each of the HXB/BXH RI strains. A 3‐lead surface ECG was recorded from subcutaneous 23‐gauge needle electrodes attached to each limb of rats in the prone position using the PowerLab acquisition system (ADInstruments). Signals from each channel (channels 1 and 2) were analyzed separately. ECG traces from each channel were signal averaged and analyzed for heart rate (RR interval) and for PR‐, QRS‐, and QT‐interval duration using the LabChart Pro software (ADInstruments). PR‐, QRS‐, and QT‐intervals were measured in 2 ways (Figure S1): (1) with the start of the QRS complex defined as the very first deflection from the baseline (typically a downward slope), and (2) with the start of the QRS complex defined as the start of its fast upstroke. For these parameters this yielded 4 measurements: measurements “a” and “b” for both channels. The QT interval was corrected for heart rate using the formula QTc=QT/(RR/100).
Heritability of ECG Traits
To assess the heritability of the ECG traits (PR, QRS, QTc, and RR), the narrow‐sense h2 was estimated in the set of RI lines using the formula h2=0.5VA/(0.5VA+VE).23 Within this formula, VA represents the additive genetic component (variance across strains, ie, the variance of the strain means) and VE represents the average environmental component (variance within strains, ie, the mean of the strain variances).24
Genotypic and Cardiac Transcriptome Data Sets
Previous genome‐wide genotyping (>20 000 SNPs) in the RI strains yielded 1384 strain distribution patterns, tagged by 1384 informative polymorphic markers, which were used for genetic mapping in this study.16 The acquisition of left ventricular transcriptomic data by RNA‐seq has been described in detail elsewhere.25 Briefly, mRNA was extracted from left ventricular tissue of 6‐week‐old male rats and sequenced on an Illumina HiSeq2000 using standard protocols. Reads were aligned to the BN reference genome RGSC 5.0 using TopHat v1.2.0.26 Gene expression levels were estimated using all read counts mapping to gene bodies. Gene expression values were normalized across samples using VST normalization27 and log2 transformed. Only genes expressed at sufficient levels (at least 1 FPKM28 in 5% of the samples) were considered for further analysis, amounting to a total of 11 221 genes. Principal component analysis of the normalized gene expression data revealed a confounding effect accounting for 40% of the variance observed between strains. The gene expression data were therefore corrected for the first principal component and the resulting data used in all subsequent analyses.
Quantitative Trait Loci Analysis
All 1384 informative genetic polymorphic markers were checked for association with each ECG parameter, taking together the 2 measurements (“a” and “b,” Figure S1) of a given channel in a multiresponse model using the Empirical Stochastic Search procedure.29 This Bayesian procedure30 evaluates all potential models, including multimarker models, and yields a posterior likelihood of association of each genetic marker conditional on all other markers: a marginal posterior probability of inclusion. Fixed effects on each strain were added as covariates in the analysis to account for potential outliers or genotyping errors. The significance threshold for marginal posterior probability of inclusion was set at a 5% false discovery rate assessed empirically using the procedure as described previously,31 corresponding to a marginal posterior probability of inclusion of 0.4 for PR interval in both channel 1 and 2.
Expression Quantitative Trait Locus Analysis and Quantitative Trait Transcript Analysis
For each genetic marker associated with an ECG trait, cis expression quantitative trait locus (eQTL) effects were tested for all genes located within 1 Mb of the genetic marker using linear modeling. For each gene, the expression values were first normal‐transformed and subsequently tested for association with the genetic marker of the locus using linear modeling.
Correlation between gene expression level and ECG traits was tested by calculating Spearman's correlation coefficient (ρ), referred to as quantitative trait transcript analysis. This was done separately for each channel (using the average of the 2 measurements “a” and “b” in the case of PR, QRS, and QT). P values were estimated using asymptotic T approximation. A nominal P value of 0.05 was considered significant.
Co‐expression Network Construction, Functional Annotation
Co‐expression networks were constructed for all genes using WGCNA32 with default settings as described previously.22 Networks were constructed with WGCNA β values of 7 and 8, with the latter being the preferred but more conservative value, leading to smaller but theoretically more robust networks.32 Of the candidate genes identified through integration of the ecgQTL, eQTL, and quantitative trait transcript analysis results, 4 (Acbd4, Fam171a2, Cd300lg, and Nhlrc1) were not part of any of the networks generated using the conservative settings. Hence the networks from the less conservative setting (β=7) were used for these genes.
The eigengene of each network was defined as the first principal component of the expression matrix of all genes in a network. Correlation of the eigengene with PR interval was calculated using Spearman's ρ. P values were estimated using asymptotic T approximation. A nominal P value of 0.01 was considered significant. Functional annotation was added to the networks based on enrichment for Gene Ontology terms (Biological Process, Cellular Component and Molecular Function) in the topGO package33 in R, using the parentChild method34 and a recommended minimum node‐size of 5.
Human Left Ventricular RNA‐Seq Gene Expression Data
Left ventricular human heart sample acquisition and processing has been described in detail elsewhere.35 Briefly, mRNA was isolated from left ventricular tissue of 108 nondiseased donors of European descent at the AMC in Amsterdam. All samples were subsequently sequenced on an Illumina HiSeq 2000 sequencer at the Max Delbrück Centre for Molecular Medicine in Berlin. Reads were aligned to human genome build HG19 using TopHat v1.2.0.26 Gene expression levels were estimated using all read counts mapping to gene bodies. Genes with a total read count of <10 reads were not considered. Data were normalized using VST36 and log2‐transformed. Orthologue mapping from rat to human was achieved using homologene data from Ensembl (release 91).
Testing for Enrichment for Electrophysiological Trait GWAS Genes in Co‐Expression Networks
Genes located within 1 Mb upstream and downstream of SNPs associated with electrophysiological traits by GWAS in humans were first identified. For this we searched for common genetic variants associated with ECG traits and atrial fibrillation susceptibility using the online catalog of GWAS hosted by the National Human Genome Research Institute (accessed October 201737). Briefly, the catalog, which is updated regularly, includes all published studies that have performed genome‐wide evaluations of human traits and disease phenotypes with a minimum of 100 000 SNPs and associations with P≤10−5. We focused on those associations that had reached genomewide statistical significance (P<5×10−8). In addition, we performed a literature search using the PubMed database that focused on peer‐reviewed publications in English. To ensure that we used independent SNPs, we pruned the SNP list thus generated by using a linkage disequilibrium cut‐off of r 2=0.1.
Next, for each GWAS trait (ECG parameters [heart rate, heart rate variability, PR interval, QRS duration, QTc interval, and ST‐T wave amplitudes] or arrhythmia [atrial fibrillation]), we identified all human genes within 1 Mb upstream or downstream of each associated SNP, by combining the SNP information from dbSNP version 144 with gene information from Biomart (Ensembl release 91). We refer to the thus identified genes as “GWAS genes” for brevity. For each GWAS gene, the corresponding homologue in rat was retrieved from Ensembl (release 91). This resulted in a list of GWAS gene rat homologues for each GWAS trait. Finally, for each co‐expression network generated from the RI gene expression data (see above), enrichment for GWAS gene homologues was tested for the set of genes comprising the network. Enrichment was calculated using the set of 11 221 expressed genes. This resulted in an enrichment P value for every co‐expression network and every GWAS trait.
Data Availability
The ECG data are available from the authors upon request; all other data are publicly available (please see cited references).
Results
Assessment of ECG Indices in the HXB/BXH RI Strains
ECGs were obtained in 6 male rats at 6 weeks of age from each of the 29 available HXB/BXH RI strains. At this age, gross cardiac structure and function is normal and cardiac hypertrophy is not yet evident,18, 38 allowing for the assessment of cardiac electrical function in the absence of structural remodeling. Signals acquired from 2 leads (from here on referred to as channel 1 and 2) were analyzed separately for RR interval (heart rate), PR interval (a measure of atrioventricular conduction), QRS duration (a measure of conduction time through the ventricles), and QTc interval (a measure of ventricular repolarization time). As expected, variability among rats within each RI strain was low, while considerable variability was observed across strains, suggesting a heritable genetic component underlying variation in these traits (Figure S2). Estimation of the narrow‐sense h2 23 revealed low heritability for QRS duration (h2=14%), moderate heritability for QTc interval (h2=23%) and high heritability for PR interval (h2=47%) and RR interval (h2=55%).
QTL Analysis Identifies 2 Loci Associated With PR Interval
Quantitative trait locus analysis was conducted for RR interval, PR interval, QRS duration, and QTc interval in the HXB/BXH RI panel using 1384 genetic markers. This was performed using the Empirical Stochastic Search procedure,29 which implements a Bayesian model selection approach where all potential single‐ and multimarker models underlying the variance observed in a quantitative phenotype are considered. This yields a marginal posterior probability of inclusion for each marker as a measure of association with an ECG parameter, conditional on all other markers, thereby effectively controlling for the genetic differences between the strains. Repeated data permutations were performed to assess the genome‐wide threshold for significant association, controlling the false discovery rate at 0.05. Two loci were found to be significantly associated with the PR interval (Figure 1). A locus on chromosome 10 was found to be significantly associated with PR interval in channel 2, with the same locus displaying suggestive association in channel 1. A locus on chromosome 17 was found to be associated with PR interval in channel 1, with a suggestive association in channel 2. Both identified QTL show similar trends overall in both channels (Table 1). No statistically significant QTL were identified for any of the other ECG indices.
Figure 1.
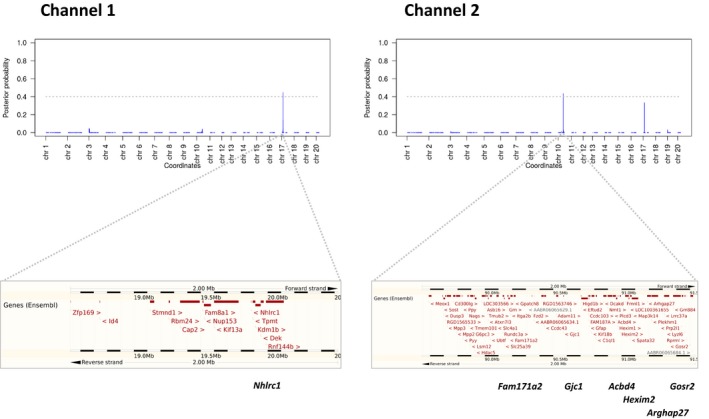
PR loci on chromosome 10 and chromosome 17. (top) Manhattan plots of association with PR interval as measured in channel 1 (left figure) and channel 2 (right figure). On the y‐axis is the Marginal Posterior Probability of Inclusion as determined using the sparse Bayesian modeling of the ESS software. The stronger the association, the higher the value. On the x‐axis are the genome coordinates of the interrogated markers. The gray dotted line signifies the 5% false discovery rate cut‐off value, determined by permutation analysis. Two loci were identified on chromosome 17 and chromosome 10 for PR interval in channel 1 and channel 2, respectively. Bottom: overview of the genes at the identified loci (source: Ensembl). Below each figure in bold are several genes discussed in detail within the article.
Table 1.
Genes With Significant eQTLs at Chromosome 10 and 17 Loci
| Chromosome | Position Rn3.4 | Position Rn5.0 | MPPI | FDR | ECG Trait | Ensembl Gene ID | Gene Symbol | eQTL P Value | QTT Trait | QTT p | QTT rho |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 10 | 91899984 | 90539744 | 0.43 | <5% | PR (Channel 2) | ENSRNOG00000003108 | Acbd4 | 5.24E‐08 | PR channel 1/2 | 0.0033/0.00052 | 0.53/0.61 |
| 10 | 91899984 | 90539744 | 0.43 | <5% | PR (Channel 2) | ENSRNOG00000020837 | Cd300lg | 7.49E‐06 | PR channel 1/2 | 0.0094/0.0084 | −0.48/−0.48 |
| 10 | 91899984 | 90539744 | 0.43 | <5% | PR (Channel 2) | ENSRNOG00000021041 | Fam171a2 | 4.49E‐06 | PR channel 1/2 | 0.026/0.023 | −0.41/−0.42 |
| 10 | 91899984 | 90539744 | 0.43 | <5% | PR (Channel 2) | ENSRNOG00000028569 | Arhgap27 | 6.13E‐04 | PR channel 1/2 | 0.0034/0.014 | −0.53/−0.46 |
| 17 | 24082890 | 19525138 | 0.45 | <5% | PR (Channel 1) | ENSRNOG00000026286 | Nhlrc1 | 3.38E‐04 | PR channel 1/2 | 0.064/0.033 | 0.35/0.4 |
eQTL indicates expression quantitative trait locus; FDR, false discovery rate; ID, identifier; MPPI, marginal posterior probability of inclusion; p, Spearman correlation P value; QTT, quantitative trait transcript; rho, Spearman correlation rho value.
Both QTL have corresponding regions in humans, with the chromosome 10 locus being syntenic to human 17q21.3 and the chromosome 17 locus syntenic to human 6p22.3. Each gene at each of the 2 rat loci has a corresponding homologue in humans.
Candidate Gene Identification at the Identified PR Interval–Associated Loci
Chromosome 10 QTL
The chromosome 10 QTL harbors 59 genes within 1 Mb upstream or downstream of the PR interval–associated genetic marker (Table S1). We first explored whether the observed effect on PR interval could be mediated via coding variants within the 59 genes present at the locus. A look‐up in the fully sequenced genomes of the 2 parental rat strains14, 15 identified a frameshift deletion in Hexim2 (c.472delG, p.Val158Metfs*22) in the SHR strain at the chromosome 10 locus, potentially leading to a loss of function. No other nonsynonymous coding variants were found in 58 genes located within the region.
Next, we explored whether the observed effect on PR interval could be mediated via transcriptional mechanisms. Using left ventricular gene expression data generated for the HXB/BXH RI panel by RNA‐seq,25 we tested for cis expression quantitative trait loci (cis eQTLs) for each gene in the locus using linear modeling (see Methods section for details; complete results are presented in Table S1). After correcting for multiple testing because of the number of candidate genes tested for cis‐acting regulation (P<0.001; 0.05/59 genes tested), significant cis eQTLs (P<0.001) were identified for 4 genes: Acbd4, Cd300lg, Fam171a2, and Arhgap27 (Figure 2 and Table 1). In addition, the cardiac expression level of all 4 eQTL genes was significantly correlated with PR interval among the strains in both ECG channels (Table 1).
Figure 2.
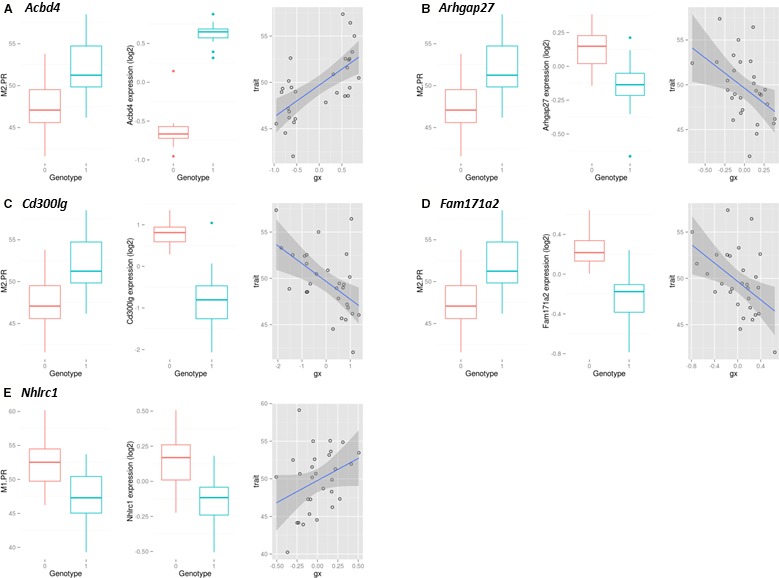
Genes with significant eQTL at chromosome 10 and 17 PR interval associated loci. For each gene (A: Acbd4, B: Arhgap27, C: Cd300lg, D: Fam171a2 and E: Nhlrc1), from left to right: (1) Boxplots of PR interval duration for the SHR allele (genotype “0”) and BN allele (genotype “1”); (2) Boxplots of gene expression data for the SHR allele (genotype “0”) and BN allele (genotype “1”); (3) Scatterplot of gene expression vs PR interval duration, with the blue line a linear least‐squares fit and the underlying gray shading the estimated error. Gene expression data were corrected for the first principal component; expression levels are relative. BN indicates Brown Norway rat; eQTL, expression quantitative trait locus; SHR, spontaneously hypertensive rat.
Among other genes at the locus, Gosr2 has been previously associated with QRS duration in humans,39 while Gjc1 encodes Connexin45, a gap junctional protein predominantly expressed in the cardiac conduction system; however, no significant eQTL effects or correlation of transcript abundance to PR interval was observed for Hexim2, Gosr2, or Gjc1.
Our approach identified 5 candidate genes (Hexim2, Acbd4, Cd300lg, Arhgap27, and Fam171a2) at the chromosome 10 locus. Hexim2 harbors a protein‐altering variant, whereas transcript abundance of Acbd4, Cd300lg, Arhgap27, and Fam171a2 (1) is under strong genetic control of the haplotype that modulates the PR interval between the 2 rat strains, and (2) is correlated with PR interval. An additional 2 genes at this locus may be of interest based on prior knowledge (Gosr2 and Gjc1).
Chromosome 17 QTL
The QTL identified on chromosome 17 contains 13 genes within 1 Mb upstream or downstream of the PR interval–associated genetic marker, none of which have been previously associated with cardiac traits. A look‐up in the fully sequenced genomes of the 2 parental rat strains14, 15 did not identify any protein‐altering variants within the region.
As with the chromosome 10 QTL, we next explored eQTL effects that could point to effects mediated by alterations in transcript abundance (complete results are presented in Table S1). After correcting for multiple testing, eQTL analysis revealed only a single significant cis‐eQTL for Nhlrc1 (P<0.004; 0.05/13 genes tested; Table 1). The transcript abundance of Nhlrc1 (Figure 2) correlates with PR interval in channel 2 (Spearman ρ=0.40, P=0.03) and to a somewhat lesser extent with PR interval in channel 1 (Spearman ρ=0.35, P=0.06).
Thus, our approach identified a single candidate gene, Nhlrc1, at the chromosome 17 locus, the transcript abundance of which (1) is under strong genetic control by the haplotype that modulates the PR interval among the 2 strains, and (2) is correlated with PR interval.
Expression of Candidate Genes in Rat and Human Cardiac Tissue
All identified candidate genes (Acbd4, Cd300lg, Arhgap27, Fam171a2, and Nhlrc1) have been previously reported to be expressed in rat and human left ventricular tissue.40 However, in a recent deep sequencing effort of human control left ventricular tissue, the expression of both FAM171A2 and NHLRC1 appeared negligible35 (Table S1). Additionally, a look‐up in the human GTEx database41 revealed robust expression of ACBD4, CD300LG, and ARHGAP27 in atrial appendage and left ventricular tissue, but low expression for FAM171A2 and NHLRC1. While atrial expression data are currently unavailable for the HXB/BXH RI strains, such data have been obtained from 4 normal adult male Sprague‐Dawley rats and is publicly available (GEO accession GSE5266 42). In this data set, 3 of the 5 candidate genes were included in the microarray design (Acbd4, Fam171a2, and Arhgap27), which all showed expression in rat atrial tissue.
Co‐Expression Network Analysis Reveals Networks Associated With ECG Traits
Co‐expressed genes (ie, genes that follow the same expression pattern) are likely to be part of the same complex or biological pathway.43 We first constructed co‐expression networks with the WGCNA package,32 and then investigated the association of multiple, potentially co‐regulated genes (ie, gene networks) with the ECG traits.
Transcriptome‐wide co‐expression network analysis of the cardiac RNA‐seq data identified 50 co‐expression networks (lists of all the genes per network are provided in Table S2). We then assessed the correlation between the eigengene of each network, a summary of the observed co‐variation of all genes in a network, with the different ECG traits (Table S3). Next, we performed Gene Ontology (GO) enrichment analysis33, 34 of the identified networks.
Eigengenes of 6 networks (M8, M12, M24, M26, M30, and M37) showed a significant correlation with one of the ECG traits (P<0.01 and |ρ|>0.4) (Table 2; complete results are shown in Table S3). Network M8 (n=40 genes), the eigengene of which was correlated with the PR interval (ρ=−0.515 [channel 1], −0.584 [channel 2]), is enriched for genes involved in the regulation of cell adhesion at the cell junction (Table S4). The eigengenes of networks M12 (n=21 genes) and M24 (n=22 genes) were correlated with the QRS duration in channel 1 (ρ=−0.488 [M12] and 0.484 [M24]). These networks are enriched for genes involved in RNA processing and splicing (Tables S5 and S6). Networks M26 (n=148 genes) and M30 (n=24 genes) were correlated with PR interval duration in channel 1 (ρ=−0.459 and −0.567, respectively) and channel 2 (ρ=−0.511 and −0.464, respectively). Network M26 is enriched for genes involved in cell adhesion and the extracellular component (Table S7). Network M37 (n=52 genes) was correlated with QRS duration in channel 2 (ρ=−0.519); networks M30 and M37 genes are enriched for genes involved in cell metabolism (Tables S8 and S9).
Table 2.
Co‐Expression Networks With Significant Correlation With ECG Traits
| Network | PR (1) | QRS (1) | QTc (1) | PR (2) | QRS (2) | QTc (2) | RR | Gene Ontology Enrichment | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rho | p | rho | p | rho | p | Rho | p | rho | p | rho | p | rho | p | ||
| M8 | −0.52 | 4.74E‐03a | −0.22 | 2.49E‐01 | −0.09 | 6.37E‐01 | −0.58 | 1.08E‐03a | −0.12 | 5.23E‐01 | 0.09 | 6.35E‐01 | 0.10 | 6.01E‐01 | Regulation of cell adhesion at the cell junction |
| M12 | −0.18 | 3.49E‐01 | −0.48 | 8.81E‐03a | −0.05 | 8.01E‐01 | −0.20 | 3.09E‐01 | −0.16 | 4.17E‐01 | 0.18 | 3.41E‐01 | 0.00 | 9.99E‐01 | RNA processing and splicing |
| M24 | 0.25 | 1.93E‐01 | 0.48 | 8.43E‐03a | 0.05 | 7.91E‐01 | 0.17 | 3.84E‐01 | 0.14 | 4.83E‐01 | −0.08 | 6.95E‐01 | 0.20 | 2.96E‐01 | RNA processing and splicing |
| M26 | −0.46 | 1.30E‐02a | 0.03 | 8.59E‐01 | 0.00 | 9.98E‐01 | −0.51 | 5.11E‐03a | −0.08 | 6.83E‐01 | 0.09 | 6.43E‐01 | 0.01 | 9.50E‐01 | Cell adhesion and the extracellular component |
| M30 | −0.57 | 1.61E‐03a | −0.50 | 6.16E‐03 | 0.03 | 8.81E‐01 | −0.46 | 1.21E‐02a | −0.12 | 5.21E‐01 | 0.36 | 5.33E‐02 | −0.08 | 6.85E‐01 | Cell metabolism |
| M37 | −0.19 | 3.14E‐01 | −0.24 | 2.05E‐01 | 0.24 | 2.03E‐01 | −0.17 | 3.82E‐01 | −0.52 | 4.43E‐03a | −0.10 | 6.21E‐01 | −0.34 | 7.35E‐02 | Cell metabolism |
P indicates Spearman correlation P value; rho, spearman correlation rho value.
P value < 0.05 (nominal significance).
Co‐Expression Network Analysis of Genes Under Transcriptional Control of PR Interval Loci
We next assessed whether genes at the PR interval QTLs and which had a significant cis‐eQTL were part of one of the co‐expression networks. Arhgap27 (chromosome 10 PR‐locus) is included in a co‐expression network consisting of 37 genes (network M47; Figure 3, Table S10), of which 8 show a significant correlation with PR interval (P<0.05). The eigengene of the co‐expression network correlated positively with PR interval (ρ=0.37 [channel 1], 0.36 [channel 2]; P<0.05). Gene Ontology enrichment analysis of the network revealed an overrepresentation of genes involved in cellular localization, multicellular organismal development, and RNA interference (Table 3 and Table S11).
Figure 3.
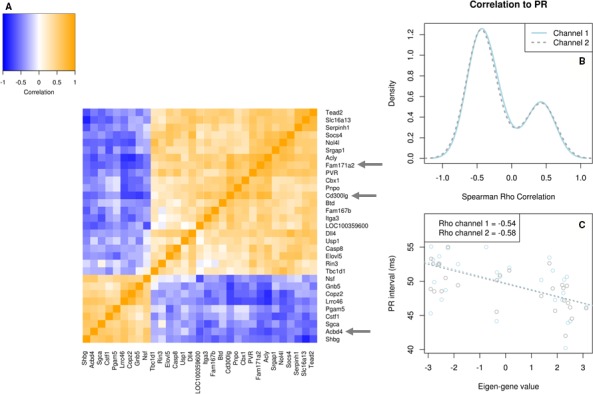
Co‐expression network containing the significant eQTL genes Acbd4, Fam171a2, and Cd300lg. Overview of the co‐expression network of 31 genes, containing 3 candidate genes identified through eQTL analysis. A, Heatmap showing the Spearman correlation between gene expression patterns of each gene pair. Blue signifies negative correlation, while orange signifies positive correlation. The 3 candidate genes, Fam171a2, Cd300lg, and Acbd4, are highlighted by gray arrows. B, Density distribution plot of the Spearman correlation values for each gene in the network with PR interval in channel 1 (solid line) and channel 2 (dashed line). This figure summarizes the correlation of the network with PR interval on gene level. C, Scatterplot of the network eigengene values vs PR interval. Dotted lines represent the linear least‐squares fits for channel 1 (light blue) and channel 2 (gray). Spearman Rho correlation values are given in the legend for each channel. This figure summarizes the correlation of the network with PR on network level. eQTL indicates expression quantitative trait locus.
Table 3.
GO Enrichment of Arhgap27 Co‐Expression Network
| Category | GO ID | Term | Count | % | P Value |
|---|---|---|---|---|---|
| Biological process | GO:0051641 | Cellular localization | 10 | 27.0 | 1.87E‐02 |
| GO:0007275 | Multicellular organismal development | 10 | 27.0 | 3.37E‐02 | |
| GO:0010608 | Posttranscriptional regulation of gene expression | 5 | 13.5 | 9.10E‐04 | |
| GO:0001101 | Response to acid chemical | 4 | 10.8 | 2.19E‐03 | |
| GO:1901699 | Cellular response to nitrogen compound | 4 | 10.8 | 2.37E‐03 | |
| GO:1901698 | Response to nitrogen compound | 4 | 10.8 | 4.88E‐02 | |
| GO:0071229 | Cellular response to acid chemical | 3 | 8.1 | 8.32E‐03 | |
| GO:0034660 | ncRNA metabolic process | 3 | 8.1 | 1.38E‐02 | |
| GO:0043068 | Positive regulation of programmed cell death | 3 | 8.1 | 3.86E‐02 | |
| GO:0043065 | Positive regulation of apoptotic process | 3 | 8.1 | 4.07E‐02 | |
| GO:0071417 | Cellular response to organonitrogen compound | 3 | 8.1 | 4.47E‐02 | |
| GO:0032008 | Positive regulation of TOR signaling | 2 | 5.4 | 2.36E‐03 | |
| Cellular compartment | GO:0044424 | Intracellular part | 24 | 64.9 | 2.72E‐02 |
| GO:0005622 | Intracellular | 24 | 64.9 | 4.84E‐02 | |
| GO:0044422 | Organelle part | 17 | 45.9 | 1.68E‐02 | |
| GO:0005789 | Endoplasmic reticulum membrane | 3 | 8.1 | 4.30E‐02 | |
| GO:0031902 | Late endosome membrane | 2 | 5.4 | 3.15E‐02 | |
| Molecular function | GO:0003723 | RNA binding | 6 | 16.2 | 3.80E‐02 |
| GO:0003725 | Double‐stranded RNA binding | 2 | 5.4 | 2.40E‐02 | |
| GO:0043566 | Structure‐specific DNA binding | 2 | 5.4 | 2.50E‐02 | |
| GO:0005085 | Guanyl‐nucleotide exchange factor activity | 2 | 5.4 | 3.60E‐02 |
GO indicates gene ontology; ncRNA, non‐coding RNA; TOR, target of rapamycin.
Using the default settings in WGCNA, none of the constructed co‐expression networks contained any of the remaining cis‐eQTL candidate genes. To avoid missing potentially interesting networks, we re‐ran WGCNA using less conservative parameterization (see Methods section for details) and focused our analysis on those networks containing the cis‐eQTL candidate genes. The 3 remaining genes with cis eQTLs at the chromosome 10 locus (Acbd4, Cd300lg, and Fam171a2) were found to be part of a single co‐expression network (Figure 4, Table S12, network M50). The network consists of 31 genes, of which 25 show a significant correlation with PR interval (P<0.05). The eigengene of the network correlates negatively with PR interval (ρ=−0.54 [channel 1], −0.58 [channel 2]; P<0.05). Gene Ontology enrichment analysis of the network highlights intracellular signal transduction, regulation of metabolic processes, and heart and cardiovascular system development as enriched biological processes (Table 4; complete results are presented in Table S13). A large portion of the proteins encoded by the genes in the network are localized in vesicles, extracellular organelles, and the membrane region.
Figure 4.
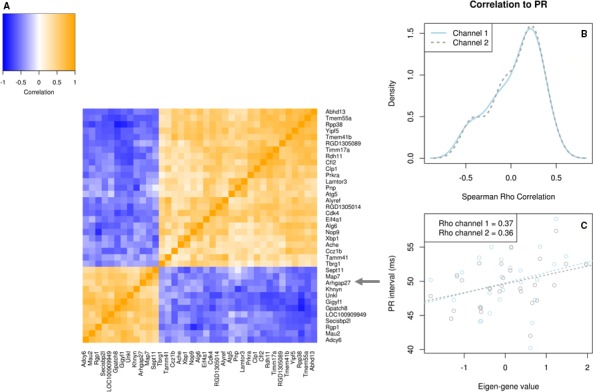
Co‐expression network containing significant eQTL gene Arhgap27. Overview of the co‐expression network of 37 genes, containing a candidate gene identified through eQTL analysis. A, Heatmap showing the Spearman correlation between gene expression patterns of each gene pair. Blue signifies negative correlation, while orange signifies positive correlation. The candidate gene, Arhgap27, is highlighted by gray arrows. B, Density distribution plot of the Spearman correlation values for each gene in the network with PR interval in channel 1 (solid line) and channel 2 (dashed line). This figure summarizes the correlation of the network with PR interval on gene level. C, Scatterplot of the network eigengene values vs PR interval. Dotted lines represent the linear least‐squares fits for channel 1 (light blue) and channel 2 (gray). Spearman Rho correlation values are given in the legend for each channel. This figure summarizes the correlation of the network with PR on network level. eQTL indicates expression quantitative trait locus.
Table 4.
GO Enrichment of Acbd4, Cd300lg, and Fam171a2 Co‐Expression Network
| Category | GO ID | Term | Count | % | P Value |
|---|---|---|---|---|---|
| Biological process | GO:0019222 | Regulation of metabolic process | 13 | 41.9 | 2.09E‐02 |
| GO:0009893 | Positive regulation of metabolic process | 9 | 29.0 | 3.83E‐02 | |
| GO:0048583 | Regulation of response to stimulus | 8 | 25.8 | 3.69E‐02 | |
| GO:0035556 | Intracellular signal transduction | 7 | 22.6 | 8.61E‐03 | |
| GO:0030163 | Protein catabolic process | 4 | 12.9 | 1.54E‐03 | |
| GO:0007507 | Heart development | 4 | 12.9 | 3.83E‐03 | |
| GO:0048598 | Embryonic morphogenesis | 4 | 12.9 | 8.83E‐03 | |
| GO:0048729 | Tissue morphogenesis | 4 | 12.9 | 9.54E‐03 | |
| GO:0030162 | Regulation of proteolysis | 4 | 12.9 | 1.25E‐02 | |
| GO:0035295 | Tube development | 4 | 12.9 | 1.40E‐02 | |
| GO:0009057 | Macromolecule catabolic process | 4 | 12.9 | 1.86E‐02 | |
| GO:1902580 | Single‐organism cellular localization | 4 | 12.9 | 3.47E‐02 | |
| GO:0006508 | Proteolysis | 4 | 12.9 | 3.47E‐02 | |
| GO:0009894 | Regulation of catabolic process | 4 | 12.9 | 3.60E‐02 | |
| GO:0051056 | Regulation of small GTPase‐mediated signal transduction | 4 | 12.9 | 4.11E‐02 | |
| GO:0072358 | Cardiovascular system development | 4 | 12.9 | 4.70E‐02 | |
| GO:0072359 | Circulatory system development | 4 | 12.9 | 4.70E‐02 | |
| Cellular component | GO:0043226 | Organelle | 21 | 67.7 | 4.92E‐02 |
| GO:0031982 | Vesicle | 11 | 35.5 | 2.78E‐02 | |
| GO:0043230 | Extracellular organelle | 9 | 29.0 | 3.15E‐02 | |
| GO:0098589 | Membrane region | 5 | 16.1 | 2.53E‐02 | |
| GO:0097458 | Neuron part | 5 | 16.1 | 4.32E‐02 | |
| GO:0016323 | Basolateral plasma membrane | 2 | 6.5 | 3.51E‐02 | |
| Molecular function | GO:0032403 | Protein complex binding | 6 | 19.4 | 5.50E‐03 |
| GO:0044877 | Macromolecular complex binding | 6 | 19.4 | 2.63E‐02 | |
| GO:0098772 | Molecular function regulator | 5 | 16.1 | 1.67E‐02 | |
| GO:0030234 | Enzyme regulator activity | 4 | 12.9 | 2.47E‐02 | |
| GO:0048037 | Cofactor binding | 3 | 9.7 | 1.07E‐02 | |
| GO:0051020 | GTPase binding | 3 | 9.7 | 4.58E‐02 | |
| GO:0016746 | Transferase activity, transferring acyl groups | 2 | 6.5 | 1.30E‐02 | |
| GO:0004197 | Cysteine‐type endopeptidase activity | 2 | 6.5 | 3.47E‐02 | |
| GO:0005518 | Collagen binding | 2 | 6.5 | 4.11E‐02 | |
| GO:0050839 | Cell adhesion molecule binding | 2 | 6.5 | 4.23E‐02 |
GO indicates gene ontology; GTPase, guanosine triphosphate hydrolase.
Nhlrc1, which showed a significant cis eQTL effect at the chromosome 17 QTL, is part of a co‐expression network with 80 genes (network M51, Table S14). Only 3 of these genes show a significant correlation with PR interval, which is reflected in the absence of correlation between the eigengene of the network and PR interval (Figure S3).
Enrichment for Cardiac Electrophysiological Trait GWAS Genes
We assessed whether genes comprising specific co‐expression networks were enriched in genes located at loci associated with ECG parameters (heart rate, heart rate variability, PR interval, QRS duration, QTc interval, and ST‐T wave amplitudes) or arrhythmia (atrial fibrillation) in GWAS in humans (hereafter referred to as “GWAS genes”). Results of enrichment analyses of these ECG/arrhythmia GWAS genes in the 49 rat heart gene networks are presented in Tables S15 and S16. A total of 13 gene networks were found enriched for ECG/arrhythmia GWAS genes at a nominal statistical significance (P<0.05). Of these, network M10, which was correlated with QTc interval in the rat (ρ=−0.416, P=2.56×10−2, Table S3), was enriched in GWAS genes from QTc interval GWAS (5/32 genes: Coro7; Flywch2; Trap1; Cluap1; Zfp213; P=9.88×10−3). Similarly, co‐expression network M22, which was correlated with QRS duration in rat (ρ=−0.423, P=2.32×10−2, Table S3), was enriched in GWAS genes from QRS GWAS (4/22 genes: Suox; Hadhb; Cs; Hadha; P=3.72×10−2). This suggests that the association between these gene co‐expression networks and cardiac electrophysiological traits identified in the RI strains is conserved in humans, emphasizing the potential value of these networks for studying the underlying mechanisms.
Discussion
We here used the HXB/BXH RI rat panel to map genetic loci modulating ECG parameters. We identified 2 QTLs on chromosomes 10 and 17 for PR interval, a measure of atrioventricular conduction. Genes at these 2 loci were prioritized by inspecting genes at the locus for protein‐altering genetic variation, by means of cis‐eQTL analysis and by correlating the abundance of their transcript with the PR interval. Furthermore, we constructed cardiac co‐expression networks and identified 6 gene networks that were correlated with PR interval or QRS duration, both parameters of cardiac conduction. Our study thus identifies novel candidate genes and gene networks for cardiac conduction, pointing to biological processes that may impact on this aspect of cardiac electrical function.
Slowing of conduction of the cardiac electrical impulse is a well‐established mediator of potentially lethal arrhythmia in the setting of various cardiac pathologies such as myocardial ischemia and heart failure.44, 45, 46 Cardiac conduction depends mainly on 3 factors, namely, the function of the cardiac sodium channel that mediates cardiomyocyte depolarization, cell–cell coupling by gap junctional channels, and tissue architecture including fibrosis. Each of these is, in turn, regulated by complex biological processes that remain largely unknown. The identification and understanding of these processes is essential to enable the development of therapeutic strategies to improve cardiac conduction and for the development of risk stratification strategies in patients with cardiac pathologies associated with sudden cardiac death. Rodent genetic studies such as ours can complement genetic studies in humans in dissecting the genetic underpinnings of cardiac conduction. In particular, they overcome some limitations of human genetic studies in that they allow the integration of different levels of “omics” data, such as cardiac transcriptomic data, with the phenotypic data, as done in this study.
Our genetic mapping approach identified 2 QTLs that modulate the PR interval, respectively, on rat chromosome 10 and chromosome 17. PR interval prolongation occurs with advancing age and in the setting of inherited or acquired cardiac disease, and is an established strong predictor of atrial fibrillation.42 The PR interval reflects the propagation of cardiac electrical impulse from the onset of atrial depolarization to the beginning of ventricular depolarization and as such encompasses conduction through the atria and atrioventricular node. Thus, genetic factors affecting the PR interval may act through biological processes that affect electrical function or tissue architecture of the atria, the atrioventricular node, or both. Furthermore, because the PR interval is modulated by autonomic tone, genetic factors may also affect PR interval duration through the nervous system.
Chromosome 10 PR Interval QTL
The chromosome 10 locus harbored the only gene containing a protein‐altering variant, namely, Hexim2, which contained a deletion. HEXIM2 negatively regulates the kinase activity of the cyclin‐dependent kinase P‐TEFb, which phosphorylates multiple target proteins to promote transcriptional elongation.47 While the deletion potentially leads to a loss of function, the latter is well tolerated in humans (probability of loss‐of‐function intolerance=0.37; source: ExAC database,48 accessed on February 2, 2018). HEXIM2 has no known association with human disorders according to the Genetic Association Database, the Atlas of Genetics and Cytogenetics in Oncology and Haematology, MalaCards, UniProtKB, and Genatlas (accessed on July 12, 2017). However, this does not rule out a role for Hexim2 in the modulating cardiac electrophysiology and as such it remains a potential candidate for further study.
At the chromosome 10 locus we detected an eQTL for 4 genes, namely, Acbd4, Cd300lg, Fam171a2, and Arhgap27. Furthermore, the transcript abundance of each of these genes was correlated with PR interval duration, making them relevant candidates. The Arhgap27 gene encodes a member of a large family of proteins that activate Rho‐type guanosine triphosphate metabolizing enzymes that may play a role in clathrin‐mediated endocytosis.49 Of note, atrioventricular conduction defects have been described secondary to alterations in Rho‐signaling.50, 51 More specifically, inhibition of Rho‐GTPase activity has been shown to slow atrioventricular conduction, mediated at least in part by a reduction in expression of Connexin40, a gap junction protein expressed in the cardiac conduction system.51 Overexpression of RhoA furthermore induced sinus node and atrioventricular dysfunction in mice, which could in part be secondary to cardiac structural abnormalities.52 A potential direct effect of RhoA on cardiac electrical activity was demonstrated by the observation that RhoA regulates the delayed rectifier potassium channel KV1.2.53 In humans, GWAS ARGHAP24, a paralog of Arghap27, is located at a locus that has been associated with PR interval duration.54
The other 3 genes at the chromosome 10 locus that display an eQTL effect (that is, Acbd4, Cd300lg, and Fam171a2) are part of the same gene expression network. While these 3 genes have been shown to be expressed in heart,39 they have not been implicated in cardiac electrophysiology or disease before. The Acbd4 gene encodes ACBD4 (acyl‐CoA binding domain protein 4), a peroxisomal membrane protein.55 A number of studies have linked peroxisome proliferator–activated receptor activity to ion channel remodeling, cardiac conduction disturbances, and arrhythmogenesis.56, 57, 58 Cd300lg encodes a type I cell surface glycoprotein that is localized in the endosome. Both Acbd4 and Cd300lg are target genes of the homeobox protein Nkx2.5, and Nkx2.5 mutations are associated with atrioventricular conduction disturbances.59 Furthermore, Acbd4 is reduced in the atrioventricular canal by Tbx3‐mediated repression,60 suggesting a role for both genes in heart development and function, particularly in relation to atrioventricular conduction. The function of the protein product of Fam171a2 is as yet unknown. The co‐expression network (M50, Table S12) containing Acbd4, Cd300lg, and Fam171a2 is significantly positively correlated with PR interval and shows enrichment for genes encoding proteins localized in vesicles, extracellular organelles, and the membrane region; one could speculate that 1 or more of these genes may impact on the PR interval by affecting vesicular ion channel transport to the membrane.
Although no cis‐eQTLs nor a coding region variant for this gene were found, a strong positional candidate for the effect of the chromosome 10 PR interval QTL is the Gjc1 gene. Gjc1 encodes Connexin45 (Cx45), a gap junctional protein predominantly expressed in the cardiac conduction system including the atrioventricular node and His‐bundle.61 Mice with inducible, cardiac‐specific knockdown of Cx45 display atrioventricular conduction delay.62 Another candidate at the chromosome 10 locus is Gosr2. In humans, GOSR2 overlaps a haplotype that has been associated with QRS duration (another parameter of cardiac conduction, namely, ventricular conduction) in GWAS40; GOSR2 mutations are furthermore associated with progressive myoclonus epilepsy.63 GOSR2 is a trafficking protein, involved in Golgi vesicle transport, which could theoretically be of relevance for ion channel trafficking in the cardiomyocyte and hence cardiac conduction. Although we found that Gjc1 and Gosr2 are expressed above background levels, as observed previously by others,62, 64 no eQTL for Gjc1 or Gosr2 was observed. Although this could be because of limited statistical power in our eQTL analysis, our results may also suggest that in the left ventricle of the studied HXB/BXH strains, the expression level of these genes is not under genetic control of the haplotype driving the effect at the locus. However, a gene dosage effect that is restricted to the cardiac conduction system (for example, in the atrioventricular node or the His‐bundle) cannot be ruled out. Testing this would necessitate eQTL analysis in these cardiac subcompartments, which is challenging because of their small size.
Chromosome 17 PR Interval QTL
The only candidate gene at the chromosome 17 locus based on eQTL analysis, Nhlrc1, encodes a single subunit E3 ubiquitin ligase, which polyubiquitinates the protein laforin and in complex with the latter promotes the degradation of misfolded proteins.65 Defects in NHLRC1 lead to an accumulation of laforin and onset of Lafora disease, also known as progressive myoclonic epilepsy type 2.66 Furthermore, NHLRC1 mutations have also been associated with polyglucosan storage myopathies, which present with skeletal and cardiac muscle weakness and wasting in addition to cardiomyopathy, conduction block, and arrhythmia.67 While the expression of Nhlrc1 significantly correlated with PR interval (ρ=0.4, P=0.03), the majority of the genes (78 out of 80) in the Nhlrc1 co‐expression network did not show a significant correlation with PR interval and hence any enrichment for biological processes would be uninformative. Based on these data, potential mechanisms for how Nhlrc1 could be affecting atrioventricular conduction are thus far unclear, but it may be speculated that accumulation of (toxic) misfolded proteins and/or glycogen may play a role.68
Expression of Candidate Genes in Rat and Human Cardiac Tissue
Since the PR interval mostly reflects conduction through the atria and the atrioventricular node, the functional relevance of the observed correlation with expression levels of the candidate genes in ventricular tissue may not be evident at first glance. It is, however, known that cis eQTLs are often shared across multiple tissues.41, 69 For human heart tissue, a previous study revealed an extensive correlation between cis eQTLs in left ventricle and atrial appendage, both absolute (Spearman ρ>0.70) as well as relative compared with other tissues.41 Indeed, according to the GTEx database, both FAM171A2 and ARHGAP27 have significant eQTLs that are shared between these cardiac tissues in humans. In addition, the eQTL for Acbd4 identified in this study is shared between left ventricular, kidney, and skeletal muscle tissues within the HXB/BXH RI strains, further demonstrating the presence of cis eQTLs across multiple tissues.69 Atrial expression data are currently unavailable for the HXB/BXH RI strains, but expression of Acbd4, Fam171a2, and Arhgap27 has been confirmed in atrial tissue of Sprague‐Dawley rats (GEO data set, accession GSE5266 42). In addition, RNA Seq data from the GTEx database demonstrate the presence of ACBD4, CD300LG, and ARHGAP27 in human atrial appendage. Taken together, these observations underscore the potential transferability of our current findings to atrial and/or atrioventricular tissue and consequent relevance for (atrio)ventricular conduction.
Conclusion
We here combined genetic and cardiac transcriptomic studies in RI rats to identify genetic loci and gene networks associated with the electrocardiographic PR interval. The integration of classical QTL mapping with eQTL and network analyses yielded candidate genes that are under strong transcriptional control, are correlated with the corresponding trait, and occur in co‐expression networks enriched for meaningful biological processes. The transcriptional analyses (ie, eQTL and co‐expression network analyses) proved key in identification of these candidate genes amidst the total list of 59 genes at the PR interval–associated loci on chromosomes 10 and 17. Conversely, 2 candidate genes with prior association with cardiac traits in human (Gosr2, Gjc1) showed no appreciable transcriptional differences between the strains. Without the transcriptional studies, these genes might have been selected as the sole prime candidates at the respective loci. Only combined, these data sets thus allowed a valid and unbiased starting point for future studies with the potential of identifying novel mechanisms underlying cardiac electrical function. This study therefore demonstrates the opportunities offered by combining genetic studies in rodents with gene expression and cardiac electrophysiological phenotypes to yield novel candidates in an unbiased way.
Sources of Funding
We acknowledge support from the Dutch Heart Foundation (CVON 2010‐12 PREDICT), the Netherlands Organization for Scientific Research (VICI project 016.150.610 and VIDI project 91714371), the InterUniversity Cardiology Institute of the Netherlands (061.02) and the Netherlands Heart Institute. Pravenec and Šilhavý were supported by grant 13‐10267S from the Czech Science Foundation. Adriaens’ work at the Maastricht Centre for Systems Biology is supported by the Dutch Province of Limburg. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosures
None.
Supporting information
Figure S1. Figure illustrating the principle of determining PR interval in an ECG in 2 ways: (A) with the start of the QRS complex defined as the very first deflection from the baseline (typically a downward slope), and (B) with the start of the QRS complex defined as the start of its fast.
Figure S2. Variability within and between strains for the different ECG parameters: RR‐interval, PR‐interval, QRS‐interval, and QT‐interval with and without correction for heart rate (corr.). Error bars indicate SEM.
Figure S3. Co‐expression network containing the significant eQTL gene Nhlrc1. Overview of the co‐expression network of 80 genes, containing a candidate gene identified through eQTL analysis. A, Heatmap showing the Spearman correlation between gene expression patterns of each gene pair. Blue signifies negative correlation, while orange signifies positive correlation. The candidate gene, Nhlrc1, is highlighted by a gray arrow. B, Density distribution plot of the Spearman correlation values for each gene in the network with PR interval in channel 1 (solid line) and channel 2 (dashed line). This figure summarizes the correlation of the network to PR interval on gene level. C, Scatterplot of the network eigengene values vs PR interval. Dotted lines represent the linear least‐squares fits for channel 1 (light blue) and channel 2 (gray). Spearman Rho correlation values are given in the legend for each channel. This figure summarizes the correlation of the network with PR on network level.
Table S1. Overview of Candidate Genes at the Identified Chromosome 10 and Chromosome 17 Loci
Table S2. Overview of All Genes That Are Part of a Co‐Expression Network
Table S3. Correlation of Co‐Expression Networks With ECG Traits
Table S4. Gene Ontology Enrichment Analysis Results for Co‐Expression Network M8
Table S5. Gene Ontology Enrichment Analysis Results for Co‐Expression Network M12
Table S6. Gene Ontology Enrichment Analysis Results for Co‐Expression Network M24
Table S7. Gene Ontology Enrichment Analysis Results for Co‐Expression Network M26
Table S8. Gene Ontology Enrichment Analysis Results for Co‐Expression Network M30
Table S9. Gene Ontology Enrichment Analysis Results for Co‐Expression Network M37
Table S10. Overview of All Genes That Are Part of the Arhgap27 Co‐Expression Network
Table S12. Overview of All Genes That Are Part of the Acbd4 Co‐Expression Network
Table S14. Overview of All Genes That Are Part of the Nhlrc1 Co‐Expression Network
Table S11. Gene Ontology Enrichment Analysis Results for the Arhgap27 Co‐Expression Network
Table S13. Gene Ontology Enrichment Analysis Results for the Acbd4 Co‐Expression Network
Table S15. Co‐Expression Network Enrichment for Cardiac Trait GWAS Genes
Table S16. Specification of Cardiac Trait GWAS Genes Present in Co‐Expression Networks M10 and M22
(J Am Heart Assoc. 2018;7:e009243 DOI: 10.1161/JAHA.118.009243.)
References
- 1. Kolder IC, Tanck MW, Bezzina CR. Common genetic variation modulating cardiac ECG parameters and susceptibility to sudden cardiac death. J Mol Cell Cardiol. 2012;52:620–629. [DOI] [PubMed] [Google Scholar]
- 2. Straus SM, Kors JA, De Bruin ML, van der Hooft CS, Hofman A, Heeringa J, Deckers JW, Kingma JH, Sturkenboom MC, Stricker BH, Witteman JC. Prolonged QTc interval and risk of sudden cardiac death in a population of older adults. J Am Coll Cardiol. 2006;47:362–367. [DOI] [PubMed] [Google Scholar]
- 3. Pfeufer A, van Noord C, Marciante KD, Arking DE, Larson MG, Smith AV, Tarasov KV, Muller M, Sotoodehnia N, Sinner MF, Verwoert GC, Li M, Kao WH, Kottgen A, Coresh J, Bis JC, Psaty BM, Rice K, Rotter JI, Rivadeneira F, Hofman A, Kors JA, Stricker BH, Uitterlinden AG, van Duijn CM, Beckmann BM, Sauter W, Gieger C, Lubitz SA, Newton‐Cheh C, Wang TJ, Magnani JW, Schnabel RB, Chung MK, Barnard J, Smith JD, Van Wagoner DR, Vasan RS, Aspelund T, Eiriksdottir G, Harris TB, Launer LJ, Najjar SS, Lakatta E, Schlessinger D, Uda M, Abecasis GR, Muller‐Myhsok B, Ehret GB, Boerwinkle E, Chakravarti A, Soliman EZ, Lunetta KL, Perz S, Wichmann HE, Meitinger T, Levy D, Gudnason V, Ellinor PT, Sanna S, Kaab S, Witteman JC, Alonso A, Benjamin EJ, Heckbert SR. Genome‐wide association study of PR interval. Nat Genet. 2010;42:153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Christophersen IE, Rienstra M, Roselli C, Yin X, Geelhoed B, Barnard J, Lin H, Arking DE, Smith AV, Albert CM, Chaffin M, Tucker NR, Li M, Klarin D, Bihlmeyer NA, Low SK, Weeke PE, Muller‐Nurasyid M, Smith JG, Brody JA, Niemeijer MN, Dorr M, Trompet S, Huffman J, Gustafsson S, Schurmann C, Kleber ME, Lyytikainen LP, Seppala I, Malik R, Horimoto A, Perez M, Sinisalo J, Aeschbacher S, Theriault S, Yao J, Radmanesh F, Weiss S, Teumer A, Choi SH, Weng LC, Clauss S, Deo R, Rader DJ, Shah SH, Sun A, Hopewell JC, Debette S, Chauhan G, Yang Q, Worrall BB, Pare G, Kamatani Y, Hagemeijer YP, Verweij N, Siland JE, Kubo M, Smith JD, Van Wagoner DR, Bis JC, Perz S, Psaty BM, Ridker PM, Magnani JW, Harris TB, Launer LJ, Shoemaker MB, Padmanabhan S, Haessler J, Bartz TM, Waldenberger M, Lichtner P, Arendt M, Krieger JE, Kahonen M, Risch L, Mansur AJ, Peters A, Smith BH, Lind L, Scott SA, Lu Y, Bottinger EB, Hernesniemi J, Lindgren CM, Wong JA, Huang J, Eskola M, Morris AP, Ford I, Reiner AP, Delgado G, Chen LY, Chen YI, Sandhu RK, Li M, Boerwinkle E, Eisele L, Lannfelt L, Rost N, Anderson CD, Taylor KD, Campbell A, Magnusson PK, Porteous D, Hocking LJ, Vlachopoulou E, Pedersen NL, Nikus K, Orho‐Melander M, Hamsten A, Heeringa J, Denny JC, Kriebel J, Darbar D, Newton‐Cheh C, Shaffer C, Macfarlane PW, Heilmann‐Heimbach S, Almgren P, Huang PL, Sotoodehnia N, Soliman EZ, Uitterlinden AG, Hofman A, Franco OH, Volker U, Jockel KH, Sinner MF, Lin HJ, Guo X; ISGC MCot, Neurology Working Group of the CC , Dichgans M, Ingelsson E, Kooperberg C, Melander O, Loos RJF, Laurikka J, Conen D, Rosand J, van der Harst P, Lokki ML, Kathiresan S, Pereira A, Jukema JW, Hayward C, Rotter JI, Marz W, Lehtimaki T, Stricker BH, Chung MK, Felix SB, Gudnason V, Alonso A, Roden DM, Kaab S, Chasman DI, Heckbert SR, Benjamin EJ, Tanaka T, Lunetta KL, Lubitz SA, Ellinor PT; Consortium AF . Large‐scale analyses of common and rare variants identify 12 new loci associated with atrial fibrillation. Nat Genet. 2017;49:946–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eppinga RN, Hagemeijer Y, Burgess S, Hinds DA, Stefansson K, Gudbjartsson DF, van Veldhuisen DJ, Munroe PB, Verweij N, van der Harst P. Identification of genomic loci associated with resting heart rate and shared genetic predictors with all‐cause mortality. Nat Genet. 2016;48:1557–1563. [DOI] [PubMed] [Google Scholar]
- 6. Arking DE, Pulit SL, Crotti L, van der Harst P, Munroe PB, Koopmann TT, Sotoodehnia N, Rossin EJ, Morley M, Wang X, Johnson AD, Lundby A, Gudbjartsson DF, Noseworthy PA, Eijgelsheim M, Bradford Y, Tarasov KV, Dorr M, Muller‐Nurasyid M, Lahtinen AM, Nolte IM, Smith AV, Bis JC, Isaacs A, Newhouse SJ, Evans DS, Post WS, Waggott D, Lyytikainen LP, Hicks AA, Eisele L, Ellinghaus D, Hayward C, Navarro P, Ulivi S, Tanaka T, Tester DJ, Chatel S, Gustafsson S, Kumari M, Morris RW, Naluai AT, Padmanabhan S, Kluttig A, Strohmer B, Panayiotou AG, Torres M, Knoflach M, Hubacek JA, Slowikowski K, Raychaudhuri S, Kumar RD, Harris TB, Launer LJ, Shuldiner AR, Alonso A, Bader JS, Ehret G, Huang H, Kao WH, Strait JB, Macfarlane PW, Brown M, Caulfield MJ, Samani NJ, Kronenberg F, Willeit J; Consortium CA, Consortium C , Smith JG, Greiser KH, Meyer Zu Schwabedissen H, Werdan K, Carella M, Zelante L, Heckbert SR, Psaty BM, Rotter JI, Kolcic I, Polasek O, Wright AF, Griffin M, Daly MJ; Dcct/Edic , Arnar DO, Holm H, Thorsteinsdottir U; e MC , Denny JC, Roden DM, Zuvich RL, Emilsson V, Plump AS, Larson MG, O'Donnell CJ, Yin X, Bobbo M, D'Adamo AP, Iorio A, Sinagra G, Carracedo A, Cummings SR, Nalls MA, Jula A, Kontula KK, Marjamaa A, Oikarinen L, Perola M, Porthan K, Erbel R, Hoffmann P, Jockel KH, Kalsch H, Nothen MM; Consortium H , den Hoed M, Loos RJ, Thelle DS, Gieger C, Meitinger T, Perz S, Peters A, Prucha H, Sinner MF, Waldenberger M, de Boer RA, Franke L, van der Vleuten PA, Beckmann BM, Martens E, Bardai A, Hofman N, Wilde AA, Behr ER, Dalageorgou C, Giudicessi JR, Medeiros‐Domingo A, Barc J, Kyndt F, Probst V, Ghidoni A, Insolia R, Hamilton RM, Scherer SW, Brandimarto J, Margulies K, Moravec CE, del Greco MF, Fuchsberger C, O'Connell JR, Lee WK, Watt GC, Campbell H, Wild SH, El Mokhtari NE, Frey N, Asselbergs FW, Mateo Leach I, Navis G, van den Berg MP, van Veldhuisen DJ, Kellis M, Krijthe BP, Franco OH, Hofman A, Kors JA, Uitterlinden AG, Witteman JC, Kedenko L, Lamina C, Oostra BA, Abecasis GR, Lakatta EG, Mulas A, Orru M, Schlessinger D, Uda M, Markus MR, Volker U, Snieder H, Spector TD, Arnlov J, Lind L, Sundstrom J, Syvanen AC, Kivimaki M, Kahonen M, Mononen N, Raitakari OT, Viikari JS, Adamkova V, Kiechl S, Brion M, Nicolaides AN, Paulweber B, Haerting J, Dominiczak AF, Nyberg F, Whincup PH, Hingorani AD, Schott JJ, Bezzina CR, Ingelsson E, Ferrucci L, Gasparini P, Wilson JF, Rudan I, Franke A, Muhleisen TW, Pramstaller PP, Lehtimaki TJ, Paterson AD, Parsa A, Liu Y, van Duijn CM, Siscovick DS, Gudnason V, Jamshidi Y, Salomaa V, Felix SB, Sanna S, Ritchie MD, Stricker BH, Stefansson K, Boyer LA, Cappola TP, Olsen JV, Lage K, Schwartz PJ, Kaab S, Chakravarti A, Ackerman MJ, Pfeufer A, de Bakker PI, Newton‐Cheh C. Genetic association study of QT interval highlights role for calcium signaling pathways in myocardial repolarization. Nat Genet. 2014;46:826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van der Harst P, van Setten J, Verweij N, Vogler G, Franke L, Maurano MT, Wang X, Mateo Leach I, Eijgelsheim M, Sotoodehnia N, Hayward C, Sorice R, Meirelles O, Lyytikainen LP, Polasek O, Tanaka T, Arking DE, Ulivi S, Trompet S, Muller‐Nurasyid M, Smith AV, Dorr M, Kerr KF, Magnani JW, Del Greco MF, Zhang W, Nolte IM, Silva CT, Padmanabhan S, Tragante V, Esko T, Abecasis GR, Adriaens ME, Andersen K, Barnett P, Bis JC, Bodmer R, Buckley BM, Campbell H, Cannon MV, Chakravarti A, Chen LY, Delitala A, Devereux RB, Doevendans PA, Dominiczak AF, Ferrucci L, Ford I, Gieger C, Harris TB, Haugen E, Heinig M, Hernandez DG, Hillege HL, Hirschhorn JN, Hofman A, Hubner N, Hwang SJ, Iorio A, Kahonen M, Kellis M, Kolcic I, Kooner IK, Kooner JS, Kors JA, Lakatta EG, Lage K, Launer LJ, Levy D, Lundby A, Macfarlane PW, May D, Meitinger T, Metspalu A, Nappo S, Naitza S, Neph S, Nord AS, Nutile T, Okin PM, Olsen JV, Oostra BA, Penninger JM, Pennacchio LA, Pers TH, Perz S, Peters A, Pinto YM, Pfeufer A, Pilia MG, Pramstaller PP, Prins BP, Raitakari OT, Raychaudhuri S, Rice KM, Rossin EJ, Rotter JI, Schafer S, Schlessinger D, Schmidt CO, Sehmi J, Sillje HHW, Sinagra G, Sinner MF, Slowikowski K, Soliman EZ, Spector TD, Spiering W, Stamatoyannopoulos JA, Stolk RP, Strauch K, Tan ST, Tarasov KV, Trinh B, Uitterlinden AG, van den Boogaard M, van Duijn CM, van Gilst WH, Viikari JS, Visscher PM, Vitart V, Volker U, Waldenberger M, Weichenberger CX, Westra HJ, Wijmenga C, Wolffenbuttel BH, Yang J, Bezzina CR, Munroe PB, Snieder H, Wright AF, Rudan I, Boyer LA, Asselbergs FW, van Veldhuisen DJ, Stricker BH, Psaty BM, Ciullo M, Sanna S, Lehtimaki T, Wilson JF, Bandinelli S, Alonso A, Gasparini P, Jukema JW, Kaab S, Gudnason V, Felix SB, Heckbert SR, de Boer RA, Newton‐Cheh C, Hicks AA, Chambers JC, Jamshidi Y, Visel A, Christoffels VM, Isaacs A, Samani NJ, de Bakker PIW. 52 genetic loci influencing myocardial mass. J Am Coll Cardiol. 2016;68:1435–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chambers JC, Zhao J, Terracciano CM, Bezzina CR, Zhang W, Kaba R, Navaratnarajah M, Lotlikar A, Sehmi JS, Kooner MK, Deng G, Siedlecka U, Parasramka S, El‐Hamamsy I, Wass MN, Dekker LR, de Jong JS, Sternberg MJ, McKenna W, Severs NJ, de Silva R, Wilde AA, Anand P, Yacoub M, Scott J, Elliott P, Wood JN, Kooner JS. Genetic variation in SCN10A influences cardiac conduction. Nat Genet. 2010;42:149–152. [DOI] [PubMed] [Google Scholar]
- 9. Lodder EM, Scicluna BP, Milano A, Sun AY, Tang H, Remme CA, Moerland PD, Tanck MW, Pitt GS, Marchuk DA, Bezzina CR. Dissection of a quantitative trait locus for PR interval duration identifies Tnni3k as a novel modulator of cardiac conduction. PLoS Genet. 2012;8:e1003113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heinig M, Petretto E, Wallace C, Bottolo L, Rotival M, Lu H, Li Y, Sarwar R, Langley SR, Bauerfeind A, Hummel O, Lee YA, Paskas S, Rintisch C, Saar K, Cooper J, Buchan R, Gray EE, Cyster JG, Cardiogenics C, Erdmann J, Hengstenberg C, Maouche S, Ouwehand WH, Rice CM, Samani NJ, Schunkert H, Goodall AH, Schulz H, Roider HG, Vingron M, Blankenberg S, Munzel T, Zeller T, Szymczak S, Ziegler A, Tiret L, Smyth DJ, Pravenec M, Aitman TJ, Cambien F, Clayton D, Todd JA, Hubner N, Cook SA. A trans‐acting locus regulates an anti‐viral expression network and type 1 diabetes risk. Nature. 2010;467:460–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hubner N, Wallace CA, Zimdahl H, Petretto E, Schulz H, Maciver F, Mueller M, Hummel O, Monti J, Zidek V, Musilova A, Kren V, Causton H, Game L, Born G, Schmidt S, Muller A, Cook SA, Kurtz TW, Whittaker J, Pravenec M, Aitman TJ. Integrated transcriptional profiling and linkage analysis for identification of genes underlying disease. Nat Genet. 2005;37:243–253. [DOI] [PubMed] [Google Scholar]
- 12. Printz MP, Jirout M, Jaworski R, Alemayehu A, Kren V. Genetic Models in Applied Physiology. HXB/BXH rat recombinant inbred strain platform: a newly enhanced tool for cardiovascular, behavioral, and developmental genetics and genomics. J Appl Physiol. 2003;94:2510–2522. [DOI] [PubMed] [Google Scholar]
- 13. Silver LM. Mouse Genetics: Concepts and Applications. Oxford, UK: Oxford University Press; 1995. [Google Scholar]
- 14. Atanur SS, Birol I, Guryev V, Hirst M, Hummel O, Morrissey C, Behmoaras J, Fernandez‐Suarez XM, Johnson MD, McLaren WM, Patone G, Petretto E, Plessy C, Rockland KS, Rockland C, Saar K, Zhao Y, Carninci P, Flicek P, Kurtz T, Cuppen E, Pravenec M, Hubner N, Jones SJ, Birney E, Aitman TJ. The genome sequence of the spontaneously hypertensive rat: analysis and functional significance. Genome Res. 2010;20:791–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Simonis M, Atanur SS, Linsen S, Guryev V, Ruzius FP, Game L, Lansu N, de Bruijn E, van Heesch S, Jones SJ, Pravenec M, Aitman TJ, Cuppen E. Genetic basis of transcriptome differences between the founder strains of the rat HXB/BXH recombinant inbred panel. Genome Biol. 2012;13:r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Consortium S , Saar K, Beck A, Bihoreau MT, Birney E, Brocklebank D, Chen Y, Cuppen E, Demonchy S, Dopazo J, Flicek P, Foglio M, Fujiyama A, Gut IG, Gauguier D, Guigo R, Guryev V, Heinig M, Hummel O, Jahn N, Klages S, Kren V, Kube M, Kuhl H, Kuramoto T, Kuroki Y, Lechner D, Lee YA, Lopez‐Bigas N, Lathrop GM, Mashimo T, Medina I, Mott R, Patone G, Perrier‐Cornet JA, Platzer M, Pravenec M, Reinhardt R, Sakaki Y, Schilhabel M, Schulz H, Serikawa T, Shikhagaie M, Tatsumoto S, Taudien S, Toyoda A, Voigt B, Zelenika D, Zimdahl H, Hubner N. SNP and haplotype mapping for genetic analysis in the rat. Nat Genet. 2008;40:560–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johnson MD, Mueller M, Adamowicz‐Brice M, Collins MJ, Gellert P, Maratou K, Srivastava PK, Rotival M, Butt S, Game L, Atanur SS, Silver N, Norsworthy PJ, Langley SR, Petretto E, Pravenec M, Aitman TJ. Genetic analysis of the cardiac methylome at single nucleotide resolution in a model of human cardiovascular disease. PLoS Genet. 2014;10:e1004813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pravenec M, Gauguier D, Schott JJ, Buard J, Kren V, Bila V, Szpirer C, Szpirer J, Wang JM, Huang H, Lezin ES, Spence MA, Flodman P, Printz M, Lathrop GM, Vergnaud G, Kurtz TW. Mapping of quantitative trait loci for blood pressure and cardiac mass in the rat by genome scanning of recombinant inbred strains. J Clin Invest. 1995;96:1973–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aitman TJ, Glazier AM, Wallace CA, Cooper LD, Norsworthy PJ, Wahid FN, Al‐Majali KM, Trembling PM, Mann CJ, Shoulders CC, Graf D, St Lezin E, Kurtz TW, Kren V, Pravenec M, Ibrahimi A, Abumrad NA, Stanton LW, Scott J. Identification of CD36 (Fat) as an insulin‐resistance gene causing defective fatty acid and glucose metabolism in hypertensive rats. Nat Genet. 1999;21:76–83. [DOI] [PubMed] [Google Scholar]
- 20. Morrissey C, Grieve IC, Heinig M, Atanur S, Petretto E, Pravenec M, Hubner N, Aitman TJ. Integrated genomic approaches to identification of candidate genes underlying metabolic and cardiovascular phenotypes in the spontaneously hypertensive rat. Physiol Genomics. 2011;43:1207–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pravenec M, Klir P, Kren V, Zicha J, Kunes J. An analysis of spontaneous hypertension in spontaneously hypertensive rats by means of new recombinant inbred strains. J Hypertens. 1989;7:217–221. [PubMed] [Google Scholar]
- 22. Moreno‐Moral A, Mancini M, D'Amati G, Camici P, Petretto E. Transcriptional network analysis for the regulation of left ventricular hypertrophy and microvascular remodeling. J Cardiovasc Transl Res. 2013;6:931–944. [DOI] [PubMed] [Google Scholar]
- 23. Hegmann JP, Possidente B. Estimating genetic correlations from inbred strains. Behav Genet. 1981;11:103–114. [DOI] [PubMed] [Google Scholar]
- 24. Belknap JK. Effect of within‐strain sample size on QTL detection and mapping using recombinant inbred mouse strains. Behav Genet. 1998;28:29–38. [DOI] [PubMed] [Google Scholar]
- 25. Rintisch C, Heinig M, Bauerfeind A, Schafer S, Mieth C, Patone G, Hummel O, Chen W, Cook S, Cuppen E, Colome‐Tatche M, Johannes F, Jansen RC, Neil H, Werner M, Pravenec M, Vingron M, Hubner N. Natural variation of histone modification and its impact on gene expression in the rat genome. Genome Res. 2014;24:942–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics. 2009;25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐Seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bottolo L, Chadeau‐Hyam M, Hastie DI, Langley SR, Petretto E, Tiret L, Tregouet D, Richardson S. ESS++: a C++ objected‐oriented algorithm for Bayesian stochastic search model exploration. Bioinformatics. 2011;27:587–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bottolo L, Petretto E, Blankenberg S, Cambien F, Cook SA, Tiret L, Richardson S. Bayesian detection of expression quantitative trait loci hot spots. Genetics. 2011;189:1449–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lewin A, Saadi H, Peters JE, Moreno‐Moral A, Lee JC, Smith KG, Petretto E, Bottolo L, Richardson S. MT‐HESS: an efficient bayesian approach for simultaneous association detection in OMICS datasets, with application to eQTL mapping in multiple tissues. Bioinformatics. 2016;32:523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alexa A, Rahnenfuhrer J. Topgo: enrichment analysis for gene ontology. R package. Version 2.20.0. 2010.
- 34. Grossmann S, Bauer S, Robinson PN, Vingron M. Improved detection of overrepresentation of gene‐ontology annotations with parent child analysis. Bioinformatics. 2007;23:3024–3031. [DOI] [PubMed] [Google Scholar]
- 35. Heinig M, Adriaens ME, Schafer S, van Deutekom HWM, Lodder EM, Ware JS, Schneider V, Felkin LE, Creemers EE, Meder B, Katus HA, Ruhle F, Stoll M, Cambien F, Villard E, Charron P, Varro A, Bishopric NH, George AL Jr, Dos Remedios C, Moreno‐Moral A, Pesce F, Bauerfeind A, Ruschendorf F, Rintisch C, Petretto E, Barton PJ, Cook SA, Pinto YM, Bezzina CR, Hubner N. Natural genetic variation of the cardiac transcriptome in non‐diseased donors and patients with dilated cardiomyopathy. Genome Biol. 2017;18:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, Junkins H, McMahon A, Milano A, Morales J, Pendlington ZM, Welter D, Burdett T, Hindorff L, Flicek P, Cunningham F, Parkinson H. The new NHGRI‐EBI Catalog of published genome‐wide association studies (GWAS Catalog). Nucleic Acids Res. 2017;45:D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Petretto E, Sarwar R, Grieve I, Lu H, Kumaran MK, Muckett PJ, Mangion J, Schroen B, Benson M, Punjabi PP, Prasad SK, Pennell DJ, Kiesewetter C, Tasheva ES, Corpuz LM, Webb MD, Conrad GW, Kurtz TW, Kren V, Fischer J, Hubner N, Pinto YM, Pravenec M, Aitman TJ, Cook SA. Integrated genomic approaches implicate osteoglycin (OGN) in the regulation of left ventricular mass. Nat Genet. 2008;40:546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sotoodehnia N, Isaacs A, de Bakker PI, Dorr M, Newton‐Cheh C, Nolte IM, van der Harst P, Muller M, Eijgelsheim M, Alonso A, Hicks AA, Padmanabhan S, Hayward C, Smith AV, Polasek O, Giovannone S, Fu J, Magnani JW, Marciante KD, Pfeufer A, Gharib SA, Teumer A, Li M, Bis JC, Rivadeneira F, Aspelund T, Kottgen A, Johnson T, Rice K, Sie MP, Wang YA, Klopp N, Fuchsberger C, Wild SH, Mateo Leach I, Estrada K, Volker U, Wright AF, Asselbergs FW, Qu J, Chakravarti A, Sinner MF, Kors JA, Petersmann A, Harris TB, Soliman EZ, Munroe PB, Psaty BM, Oostra BA, Cupples LA, Perz S, de Boer RA, Uitterlinden AG, Volzke H, Spector TD, Liu FY, Boerwinkle E, Dominiczak AF, Rotter JI, van Herpen G, Levy D, Wichmann HE, van Gilst WH, Witteman JC, Kroemer HK, Kao WH, Heckbert SR, Meitinger T, Hofman A, Campbell H, Folsom AR, van Veldhuisen DJ, Schwienbacher C, O'Donnell CJ, Volpato CB, Caulfield MJ, Connell JM, Launer L, Lu X, Franke L, Fehrmann RS, te Meerman G, Groen HJ, Weersma RK, van den Berg LH, Wijmenga C, Ophoff RA, Navis G, Rudan I, Snieder H, Wilson JF, Pramstaller PP, Siscovick DS, Wang TJ, Gudnason V, van Duijn CM, Felix SB, Fishman GI, Jamshidi Y, Stricker BH, Samani NJ, Kaab S, Arking DE. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat Genet. 2010;42:1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Safran M, Dalah I, Alexander J, Rosen N, Iny Stein T, Shmoish M, Nativ N, Bahir I, Doniger T, Krug H, Sirota‐Madi A, Olender T, Golan Y, Stelzer G, Harel A, Lancet D. GeneCards Version 3: the human gene integrator. Database (Oxford). 2010;2010:baq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. GTEx‐Consortium, Laboratory, Data Analysis Coordinating Center (LDACC)‐Analysis Working Group, Statistical Methods groups—Analysis Working Group, Enhancing GTEx (eGTEx) groups; NIH Common Fund; NIH/NCI; NIH/NHGRI; NIH/NIMH; NIH/NIDA; Biospecimen Collection Source Site—NDRI; Biospecimen Collection Source Site—RPCI; Biospecimen Core Resource—VARI; Brain Bank Repository—University of Miami Brain Endowment Bank; Leidos Biomedical—Project Management; ELSI Study; Genome Browser Data Integration & Visualization—EBI; Genome Browser Data Integration & Visualization—UCSC Genomics Institute, University of California Santa Cruz; Lead analysts; Laboratory, Data Analysis & Coordinating Center (LDACC); NIH program management; Biospecimen collection; Pathology; eQTL manuscript working group , Battle A, Brown CD, Engelhardt BE, Montgomery SB. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213.29022597 [Google Scholar]
- 42. McGrath MF, de Bold AJ. Transcriptional analysis of the mammalian heart with special reference to its endocrine function. BMC Genomics. 2009;10:254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome‐wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Janse MJ, Wit AL. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol Rev. 1989;69:1049–1169. [DOI] [PubMed] [Google Scholar]
- 45. Tomaselli GF, Zipes DP. What causes sudden death in heart failure? Circ Res. 2004;95:754–763. [DOI] [PubMed] [Google Scholar]
- 46. Cheng S, Keyes MJ, Larson MG, McCabe EL, Newton‐Cheh C, Levy D, Benjamin EJ, Vasan RS, Wang TJ. Long‐term outcomes in individuals with prolonged PR interval or first‐degree atrioventricular block. JAMA. 2009;301:2571–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Byers SA, Price JP, Cooper JJ, Li Q, Price DH. HEXIM2, a HEXIM1‐related protein, regulates positive transcription elongation factor b through association with 7SK. J Biol Chem. 2005;280:16360–16367. [DOI] [PubMed] [Google Scholar]
- 48. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell‐Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce‐Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano‐Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie‐Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG; Exome Aggregation Consortium . Analysis of protein‐coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sakakibara T, Nemoto Y, Nukiwa T, Takeshima H. Identification and characterization of a novel Rho GTPase activating protein implicated in receptor‐mediated endocytosis. FEBS Lett. 2004;566:294–300. [DOI] [PubMed] [Google Scholar]
- 50. Clerk A, Sugden PH. Small guanine nucleotide‐binding proteins and myocardial hypertrophy. Circ Res. 2000;86:1019–1023. [DOI] [PubMed] [Google Scholar]
- 51. Wei L, Taffet GE, Khoury DS, Bo J, Li Y, Yatani A, Delaughter MC, Klevitsky R, Hewett TE, Robbins J, Michael LH, Schneider MD, Entman ML, Schwartz RJ. Disruption of Rho signaling results in progressive atrioventricular conduction defects while ventricular function remains preserved. FASEB J. 2004;18:857–859. [DOI] [PubMed] [Google Scholar]
- 52. Sah VP, Minamisawa S, Tam SP, Wu TH, Dorn GW II, Ross J Jr, Chien KR, Brown JH. Cardiac‐specific overexpression of RhoA results in sinus and atrioventricular nodal dysfunction and contractile failure. J Clin Invest. 1999;103:1627–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cachero TG, Morielli AD, Peralta EG. The small GTP‐binding protein RhoA regulates a delayed rectifier potassium channel. Cell. 1998;93:1077–1085. [DOI] [PubMed] [Google Scholar]
- 54. Holm H, Gudbjartsson DF, Arnar DO, Thorleifsson G, Thorgeirsson G, Stefansdottir H, Gudjonsson SA, Jonasdottir A, Mathiesen EB, Njolstad I, Nyrnes A, Wilsgaard T, Hald EM, Hveem K, Stoltenberg C, Lochen ML, Kong A, Thorsteinsdottir U, Stefansson K. Several common variants modulate heart rate, PR interval and QRS duration. Nat Genet. 2010;42:117–122. [DOI] [PubMed] [Google Scholar]
- 55. Costello JL, Castro IG, Schrader TA, Islinger M, Schrader M. Peroxisomal ACBD4 interacts with VAPB and promotes ER‐peroxisome associations. Cell Cycle. 2017;16:1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Morrow JP, Katchman A, Son NH, Trent CM, Khan R, Shiomi T, Huang H, Amin V, Lader JM, Vasquez C, Morley GE, D'Armiento J, Homma S, Goldberg IJ, Marx SO. Mice with cardiac overexpression of peroxisome proliferator‐activated receptor gamma have impaired repolarization and spontaneous fatal ventricular arrhythmias. Circulation. 2011;124:2812–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xie Y, Gu ZJ, Wu MX, Huang TC, Ou JS, Ni HS, Lin MH, Yuan WL, Wang JF, Chen YX. Disruption of calcium homeostasis by cardiac‐specific over‐expression of PPAR‐γ in mice: a role in ventricular arrhythmia. Life Sci. 2016;167:12–21. [DOI] [PubMed] [Google Scholar]
- 58. Kistamas K, Szentandrassy N, Hegyi B, Ruzsnavszky F, Vaczi K, Barandi L, Horvath B, Szebeni A, Magyar J, Banyasz T, Kecskemeti V, Nanasi PP. Effects of pioglitazone on cardiac ion currents and action potential morphology in canine ventricular myocytes. Eur J Pharmacol. 2013;710:10–19. [DOI] [PubMed] [Google Scholar]
- 59. Chowdhury R, Ashraf H, Melanson M, Tanada Y, Nguyen M, Silberbach M, Wakimoto H, Benson DW, Anderson RH, Kasahara H. Mouse model of human congenital heart disease: progressive atrioventricular block induced by a heterozygous Nkx2‐5 homeodomain missense mutation. Circ Arrhythm Electrophysiol. 2015;8:1255–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. van den Boogaard M, Wong LY, Tessadori F, Bakker ML, Dreizehnter LK, Wakker V, Bezzina CR, ‘t Hoen PA, Bakkers J, Barnett P, Christoffels VM. Genetic variation in T‐box binding element functionally affects SCN5A/SCN10A enhancer. J Clin Invest. 2012;122:2519–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Coppen SR, Severs NJ, Gourdie RG. Connexin45 (alpha 6) expression delineates an extended conduction system in the embryonic and mature rodent heart. Dev Genet. 1999;24:82–90. [DOI] [PubMed] [Google Scholar]
- 62. Frank M, Wirth A, Andrie RP, Kreuzberg MM, Dobrowolski R, Seifert G, Offermanns S, Nickenig G, Willecke K, Schrickel JW. Connexin45 provides optimal atrioventricular nodal conduction in the adult mouse heart. Circ Res. 2012;111:1528–1538. [DOI] [PubMed] [Google Scholar]
- 63. Dibbens LM, Rubboli G. GOSR2: a progressive myoclonus epilepsy gene. Epileptic Disord. 2016;18:111–114. [DOI] [PubMed] [Google Scholar]
- 64. Vozzi C, Dupont E, Coppen SR, Yeh HI, Severs NJ. Chamber‐related differences in connexin expression in the human heart. J Mol Cell Cardiol. 1999;31:991–1003. [DOI] [PubMed] [Google Scholar]
- 65. Sanchez‐Martin P, Roma‐Mateo C, Viana R, Sanz P. Ubiquitin conjugating enzyme E2‐N and sequestosome‐1 (p62) are components of the ubiquitination process mediated by the malin‐laforin E3‐ubiquitin ligase complex. Int J Biochem Cell Biol. 2015;69:204–214. [DOI] [PubMed] [Google Scholar]
- 66. Kecmanovic M, Jovic N, Cukic M, Keckarevic‐Markovic M, Keckarevic D, Stevanovic G, Romac S. Lafora disease: severe phenotype associated with homozygous deletion of the NHLRC1 gene. J Neurol Sci. 2013;325:170–173. [DOI] [PubMed] [Google Scholar]
- 67. Hedberg‐Oldfors C, Oldfors A. Polyglucosan storage myopathies. Mol Aspects Med. 2015;46:85–100. [DOI] [PubMed] [Google Scholar]
- 68. Ortolano S, Vieitez I, Agis‐Balboa RC, Spuch C. Loss of gabaergic cortical neurons underlies the neuropathology of Lafora disease. Mol Brain. 2014;7:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Petretto E, Mangion J, Dickens NJ, Cook SA, Kumaran MK, Lu H, Fischer J, Maatz H, Kren V, Pravenec M, Hubner N, Aitman TJ. Heritability and tissue specificity of expression quantitative trait loci. PLoS Genet. 2006;2:e172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Figure illustrating the principle of determining PR interval in an ECG in 2 ways: (A) with the start of the QRS complex defined as the very first deflection from the baseline (typically a downward slope), and (B) with the start of the QRS complex defined as the start of its fast.
Figure S2. Variability within and between strains for the different ECG parameters: RR‐interval, PR‐interval, QRS‐interval, and QT‐interval with and without correction for heart rate (corr.). Error bars indicate SEM.
Figure S3. Co‐expression network containing the significant eQTL gene Nhlrc1. Overview of the co‐expression network of 80 genes, containing a candidate gene identified through eQTL analysis. A, Heatmap showing the Spearman correlation between gene expression patterns of each gene pair. Blue signifies negative correlation, while orange signifies positive correlation. The candidate gene, Nhlrc1, is highlighted by a gray arrow. B, Density distribution plot of the Spearman correlation values for each gene in the network with PR interval in channel 1 (solid line) and channel 2 (dashed line). This figure summarizes the correlation of the network to PR interval on gene level. C, Scatterplot of the network eigengene values vs PR interval. Dotted lines represent the linear least‐squares fits for channel 1 (light blue) and channel 2 (gray). Spearman Rho correlation values are given in the legend for each channel. This figure summarizes the correlation of the network with PR on network level.
Table S1. Overview of Candidate Genes at the Identified Chromosome 10 and Chromosome 17 Loci
Table S2. Overview of All Genes That Are Part of a Co‐Expression Network
Table S3. Correlation of Co‐Expression Networks With ECG Traits
Table S4. Gene Ontology Enrichment Analysis Results for Co‐Expression Network M8
Table S5. Gene Ontology Enrichment Analysis Results for Co‐Expression Network M12
Table S6. Gene Ontology Enrichment Analysis Results for Co‐Expression Network M24
Table S7. Gene Ontology Enrichment Analysis Results for Co‐Expression Network M26
Table S8. Gene Ontology Enrichment Analysis Results for Co‐Expression Network M30
Table S9. Gene Ontology Enrichment Analysis Results for Co‐Expression Network M37
Table S10. Overview of All Genes That Are Part of the Arhgap27 Co‐Expression Network
Table S12. Overview of All Genes That Are Part of the Acbd4 Co‐Expression Network
Table S14. Overview of All Genes That Are Part of the Nhlrc1 Co‐Expression Network
Table S11. Gene Ontology Enrichment Analysis Results for the Arhgap27 Co‐Expression Network
Table S13. Gene Ontology Enrichment Analysis Results for the Acbd4 Co‐Expression Network
Table S15. Co‐Expression Network Enrichment for Cardiac Trait GWAS Genes
Table S16. Specification of Cardiac Trait GWAS Genes Present in Co‐Expression Networks M10 and M22
Data Availability Statement
The ECG data are available from the authors upon request; all other data are publicly available (please see cited references).