

Abstract
We previously reported that pharmacological inhibition of a class of enzymes known as prolyl hydroxylase domain proteins (PHDs) has neuroprotective effects in various in vitro and in vivo models of Parkinson's disease (PD). We hypothesized that this was due to inhibition of the PHD2 isoform, preventing it from hydroxylating the transcription factor hypoxia inducible factor 1 α (HIF1α), targeting it for eventual proteasomal degradation. HIF1α itself induces the transcription of various cellular stress genes, including several involved in iron metabolism. Although all three isoforms of PHD are expressed within vulnerable dopaminergic (DAergic) substantia nigra pars compacta neurons, only select downregulation of the PHD2 isoform was found to protect against in vivo neurodegenerative effects associated with the mitochondrial neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. These findings were corroborated in induced pluripotent stem cell-derived neurons, providing validation in a pertinent human cell model. PHD2 inhibition was found to result in increased expression of ATP13A2, mutation of which is responsible for a rare juvenile form of PD known as Kufor-Rakeb syndrome. Knockdown of ATP13A2 expression within human DAergic cells was found to abrogate restoration of cellular iron homeostasis and neuronal cell viability elicited by inhibition of PHD2 under conditions of mitochondrial stress, likely via effects on lysosomal iron storage. These data suggest that regulation of ATP13A2 by the PHD2-HIF1α signaling pathway affects cellular iron homeostasis and DAergic neuronal survival. This constitutes a heretofore unrecognized process associated with loss of ATP13A2 function that could have wide-ranging implications for it as a therapeutic target for PD and other related conditions.
SIGNIFICANCE STATEMENT Reductions in PHD2 activity within dopaminergic neurons in vivo and in cultured human induced pluripotent stem cell-derived neurons protects against mitochondrial stress-induced neurotoxicity. Protective effects are dependent on downstream HIF-1α expression. Knockdown of ATP13A2, a gene linked to a rare juvenile form of Parkinson's disease and recently identified as a novel HIF1α target, was found to abrogate maintenance of cellular iron homeostasis and neuronal viability elicited by PHD2 inhibition in vivo and in cultured dopaminergic cells under conditions of mitochondrial stress. Mechanistically, this was due to ATP13A2's role in maintaining lysosomal iron stores. This constitutes a novel mechanism by which alterations in ATP13A2 activity may be driving PD-related neuropathology.
Keywords: ATP13A2, iron, MPTP, Parkinson's disease, prolyl hydroxylase domain enzymes, transgenic mice
Introduction
We previously demonstrated that broad pharmacological inhibition of prolyl hydroxylase domain proteins (PHDs) results in neuroprotective effects within vulnerable dopaminergic (DAergic) neurons in the substantia nigra pars compacta (SNpc) in several in vitro and in vivo models of Parkinson's disease (PD; Lee et al., 2009; Rajagopalan et al., 2014). This was found to coincide with upregulation of the master transcriptional regulator, hypoxia inducible factor 1 α (HIF1α; Lee et al., 2009; Rajagopalan et al., 2014). HIF1α is hydroxylated at conserved proline residues by these enzymes, resulting in its ubiquitination by the von Hippel Lindau E3 ubiquitin ligase and subsequent degradation by the proteasome (Bruick and McKnight, 2001; Harten et al., 2010; Miyata et al., 2011). Steady-state levels of HIF1α have been hypothesized to be preferentially limited by activity of the PHD2 isoform. PHD2 levels have been reported to be aberrantly elevated within affected DAergic neurons in postmortem SNpc tissues isolated from PD patients versus age-matched controls in conjunction with reductions in HIF1α levels (Elstner et al., 2011). HIF1α is itself part of a highly conserved complex that serves to coordinate expression of a number of neuroprotective genes involved in cellular stress responses, including several that aid in the control of iron homeostasis (Siddiq et al., 2005; Nakayama, 2009; Greer et al., 2012). Iron chelation has been shown in various PD models by several laboratories, including our own, to protect against increases in unsequestered iron within the nigrostriata, in conjunction with reduced neurodegeneration (Kaur et al., 2003; Mandel et al., 2004; Youdim et al., 2004; Lee et al., 2009). Based on the known role of the PHD2-HIF1α signaling pathway in the control of cellular iron levels, this suggests that the neuroprotective effects of PHD inhibition may be in part due to downstream effects on iron homeostasis.
Lysosomes are principle reservoirs of redox-reactive ferrous iron, defined as iron either in a “free” state or loosely bound to anion molecules, such as citrate (Kurz et al., 2006, 2008; Uchiyama et al., 2008; Terman and Kurz, 2013). Increases in lysosomal pH results in release of lysosomal iron into the cytosol, where it has been reported to contribute to subsequent cell death (Uchiyama et al., 2008). Lysosomal pH has recently been reported to be compromised in conjunction with mutations in a gene called ATP13A2 associated with PD and other closely related disorders, including a rare juvenile-onset form of PD known as Kufer-Rakeb syndrome (Park et al., 2011; Dehay et al., 2012; Podhajska et al., 2012). ATP13A2 is highly expressed within DAergic SNpc neurons and this expression is decreased in both postmortem PD tissues and in the brains of patients with Lewy body disorder (Murphy et al., 2013). The protein itself is a proton-pumping lysosomal ATPase and is suggested to play an important role in the autophagic lysosomal pathway (ALP), although its exact mechanistic role is still not completely understood. It has been suggested that losses in ALP dysfunction drives PD-associated neurodegeneration primarily via abnormal increases in levels of the protein α-synuclein. However, recent data in ATP13A2-deficent mice suggests that other independent neuropathological events may also be involved (Tsunemi et al., 2014; Kett et al., 2015).
Autosomal recessive mutations in ATP13A2 have been reported to coincide with increased levels of iron in the basal ganglia in conjunction with increased neuronal cell loss (Grünewald et al., 2012; Gusdon et al., 2012; Levi and Finazzi, 2014). The ATP13A2 promoter has recently been shown to contain a hypoxia response element (HRE), to which the HIF1α protein binds, resulting in increased ATP13A2 gene expression in cultured dopaminergic cells (Xu et al., 2012). PHD2 inhibition via subsequent increases in steady-state HIF1α protein levels would be predicted to increase ATP13A2 expression, which may in turn have positive effects on cellular iron homeostasis. Reducing ATP13A2 in this context could conversely cause disruptions in cellular iron homeostasis, resulting in increased susceptibility to neuronal cell loss. Here we report that not only is expression of ATP13A2 regulated by the PHD2-HIF1α signaling pathway, but that this is instrumental in the control of cellular iron homeostasis and DAergic neuronal survival, likely via maintenance of lysosomal iron storage. This has important implications for the potential mechanisms by which alterations in lysosomal ATP13A2 activity drives PD-related neuropathology and for future clinical applications toward enhancing ATP13A2 function.
Materials and Methods
Creation of DAergic PHD isoform-specific knockdown mouse models.
Mice in which PHD-specific isoforms are selectively knocked down within DAergic neurons were created by crossing floxed isoform-specific PHD lines provided to us by Professor Guo-Hua Fong (University of Connecticut; Takeda et al., 2008) with commercially available lines in which Cre recombinase is driven by the tyrosine hydroxylase (TH) promoter [B6.cg.-tg(Th-cre)1tmd/j 008601, Jackson Laboratory] to create pTH-CRE-floxed PHD lines. Floxed-only littermates were used as negative controls. All subsequent experiments were performed in 3–4-month-old mice of mixed gender unless otherwise noted. Studies were performed in adherence with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
PCR validation of select PHD isoform-specific deletions.
DNA was isolated from olfactory bulb (OB) and cerebellum (CB) tissues taken from heterozygous pTH-CRE-floxed PHD knockdowns versus analogous floxed-only controls using Extract-N-Amp Tissue PCR Kit (Sigma-Aldrich). The presence of the expected deletion for each PHD isoform was verified via PCR using specific primers. For PHD1, deletion of exon 3 was detected using the following primer: forward, GGAGCTGGAGTTCTAGGTCAGGTT; reverse, GGAGCTGGAGTTCTAGGTCAGGTT. This gave rise to a ∼1100 bp amplicon for the floxed allele and a ∼480 bp amplicon for the deleted allele. For PHD2, deletion of exon 2 was detected using the following primer: forward, CACTGACATGACATGGGAGGGGA; reverse, CCCGAAGAACGATACCGTCGAG. This gave rise to a ∼950 bp amplicon for the floxed allele and a ∼250 bp amplicon for the deleted allele. For PHD3, deletion of exon 2 was detected using the following primer: forward, CTCAGACCCCCTAAGTATGT; reverse, CCACGTTAACTCTAGAGCCACTGA. This gave rise to a ∼1228 bp amplicon for the floxed allele and a ∼1000 bp amplicon for the deleted allele.
Fluorescent immunocytochemistry.
For in vivo immunocytochemistry (ICC) analyses, mice were perfused with phosphate-buffered saline followed by 4% paraformaldehyde. Brains were sectioned at 40 μm with a cryostat and sections were incubated in primary anti-TH antibody (Millipore Bioscience Research Reagents) and in anti-PHD (Novus Biologicals) for 24 h, 4°C. Sections were then incubated with fluorescent goat anti-rabbit IgG-Alexa fluorophores (Life Technologies). Fluorescence intensity was analyzed using a Zeiss LSM 780. For in vitro ICC analyses, primary mesencephalic cultures were isolated as previously described (Kaur et al., 2003). After 4 d of in vitro culture, cells were exposed to the PHD2 inhibitor IOX2 (50 nm) in the absence or presence of the HIF1α inhibitor 2-methoxyestradiol (100 nm) for 4 h followed by 5 μm 1-methyl-4-phenylpyridium ion (MPP+) for 24 h. Cultures were immunostained with anti-TH antibody and Alexa Fluor 488 secondary antibody. Total numbers of TH-positive neurons were counted in 3–5 separate wells for each condition.
Western blot analysis.
Western blot analysis of SH-SY5Y and mouse striatal tissues were performed as previously described (Lee et al., 2009), using antibodies for PHD1 (Novus Biologicals), PHD2 (Cell Signaling Technology), PHD3 (Novus Biologicals), ATP13A2 (Sigma-Aldrich), HO-1 (Stressgen), and actin (Millipore).
Stereological TH+ cell counts following systemic in vivo 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration.
One-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) was given at a dosage of 20 × 2 mg/kg, 12 h apart, to elicit a ∼30% SN DAergic cell loss in control animals (Lee et al., 2009). Midbrain sections (40 μm) were incubated with polyclonal anti-TH antibody (1:1000; Millipore Bioscience Research Reagents) overnight at 4°C, followed by biotin-labeled secondary antibody and development using DAB (Vector Laboratories). Unbiased stereological cell counts were performed using the optical fractionator method as previously described (Lee et al., 2009).
Rearing behavior.
To evaluate spontaneous motor initiation, animals were placed individually in a cylinder to determine the number of rearing events over 45 s as described in previous reports (Willard et al., 2015). Each rearing session was video recorded. The average number of rears over two trials was taken. After testing, animals were returned back to group housing.
Measurements of cytosolic ferrous iron levels via calcein dequenching.
For quantitation of cytosolic ferrous iron levels in samples isolated from in vivo MPTP-treated versus saline-treated PHD2 knockdown and control mice, dopaminergic synaptosomes were prepared as previously described (Chinta et al., 2007; Choi et al., 2011). Isolated synaptosomes were resuspended in HEPES-buffered Krebs-like (HBK) buffer (Daniel et al., 2012) and plated onto clear bottom black plates (BD Biosciences) coated with 0.0033% (v/v) polyethyleneimine solution at a concentration of 5 μg protein in 100 μl HBK buffer. Synaptosomes were subsequently attached to plates by centrifugation at 1500 × g for 30 min, followed by incubation at 37°C in a 5% CO2 incubator for 45 min. Subsets of synatosomal aliquots were pretreated with bafilomycin (100 nm) for 15 min before performing measurements of cytosolic iron levels via the calcein dequenching method in the presence of the ferrous iron chelator salicylaldehyde isonicotinoyl hydrazone (SIH; gift of Katherine Franz), as previously described (Kaur et al., 2009). Dilution of calcein, SIH, and washes were all performed in HBK buffer. Values reported indicate the fraction of iron-bound calcein obtained by the ratio of the difference in total fluorescence after SIH was added to total fluorescence before SIH was added.
Induced pluripotent stem cell-derived DAergic neurons and treatments.
Human induced pluripotent stem cell (iPSC)-derived DAergic neurons were purchased from XCell Science. Cells were pretreated with IOX2 (2 μm) for 3 h before treatment in 1 mm MPP+ for an additional 24 h. Mitochondrial function was assessed by measurement of mitochondrial membrane potential via fluorescent tetramethylrhodamine methyl ester assay and cell viability via 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay.
Quantitative real-time RT-PCR.
A pure RNA tissue kit (Roche) was used to isolate RNA from mesencephalic cultures or from OB and ST tissues. Gene expression was determined via qPCR using SYBR Green master mix (Roche). The primer sets used were as follows: ATP13A2 forward: GGCCCTCTACAGCCTGACT; ATP13A2 reverse: CCAGGTTGGTGTTGATTGTG, HO-1 forward: GTCAAGCACAGGGTGACAGA; HO-1 reverse: ATCCCTGCAGCTCCTCAAA. Data were normalized to actin.
Generation and analysis of differentiated human SY5Y cells stably expressing human ATP13A2 shRNA.
Human SH-SY5Y neuroblastoma cells (ATCC) were grown to confluence in provided SHSY5Y media followed by subculture for 6 d in 10 μm retinoic acid followed by 3 d in 80 nm 12-O-tetradecanoyl-phorbol-13-acetate to allow their full differentiation into mature dopaminergic neurons (Presgraves et al., 2004). These cells were previously stably transfected with human ATP13A2 shRNA lentivirus from Sigma-Aldrich (TRCN0000050405) versus a scrambled shRNA control. RT-PCR was performed using human ATP13A2 primers (NM_001141973) and results normalized to the housekeeping gene actin. Reductions in ATP13A2 protein levels were verified by Western blotting. Following differentiation, cells were plated at a density of 15,000 cells per well in 96-well plates and treated with ±2.5 mm MPP+ and ±1 μm IOX2 for 24 h; a subset of cells was pretreated for 2 h with 100 nm bafilomycin before subsequent assays. Cytosolic ferrous iron levels were measured by calcein dequenching as above using a bottom-read plate reader from Biotek Instruments in the presence of the ferrous-specific iron chelator SIH. Cell viability was determined via a CyQuant fluorescent assay.
Statistics.
Results are presented as means ± SEM. Two-way ANOVA followed by Tukey's multiple-comparison test were performed to analyze statistical significance of in vivo data and one-way ANOVA followed by Tukey's multiple-comparison test for in vitro studies and iPSC-derived neurons. A value of p < 0.05 was considered significant.
Results
PHD2-HIF1α signaling affects DAergic cell viability
All three isoforms of PHD are expressed within DAergic as well as other cell types within the SN (Fig. 1A). To investigate whether effects of pharmacological PHD inhibition on DAergic neurons are isoform-specific and cell-autonomous, we generated mouse lines in which each PHD isoform was selectively knocked down specifically within DAergic neurons, including those of the SN. For these studies, individual floxed isoform-specific PHD lines were crossed with transgenics expressing Cre recombinase behind the TH promoter to create lines in which levels of each of the three PHD isoforms was individually knocked down versus floxed-only control littermates. Dopamine-select, PHD isoform-specific deletion was initially verified by PCR using DNA isolated from the OB, a tissue rich in DAergic neurons, versus the CB as negative control (Fig. 1B). PHD-isoform deletion was verified to occur only within the OB of Cre-floxed PHD knockdowns and not in the CB nor in either tissue in floxed-only controls. As expected due to the presence of both DAergic and non-DAergic cells within the OB, we observed the presence of both deleted and full-length gene products in our analysis. PHD isoform-specific protein knockdown was subsequently confirmed in nigrostriatal tissues from heterozygous pTH-CRE floxed PHD mice via Western blot analysis (Fig. 1C).
Figure 1.
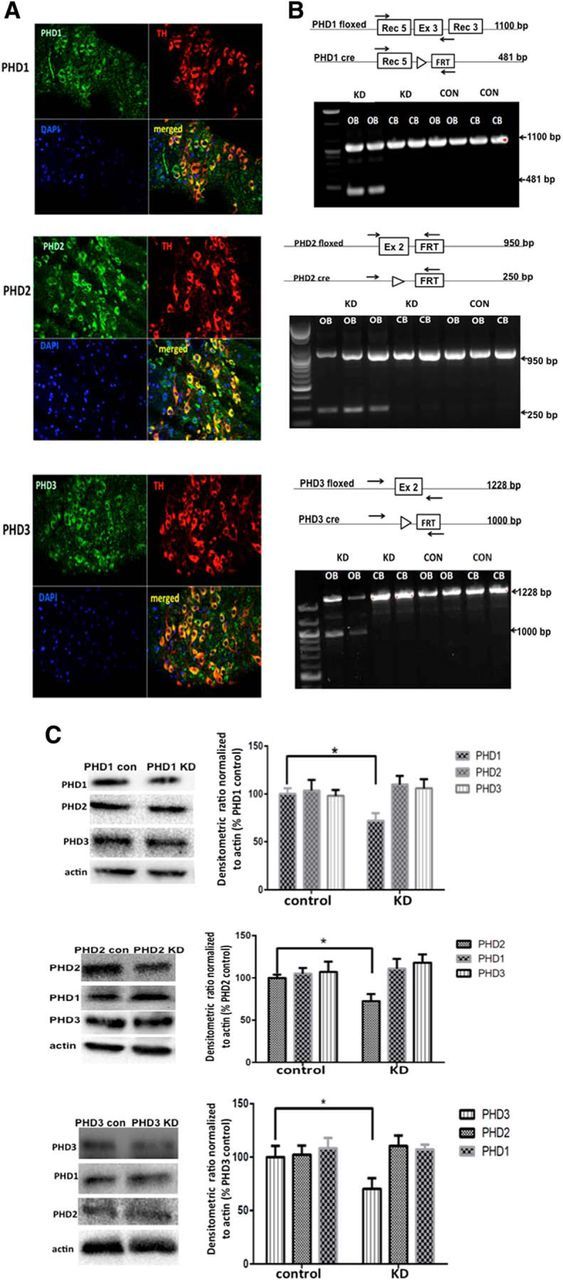
Dopamine-specific knockdown of individual PHD isoforms in vivo. A, Representative in vivo analyses of PHD1, PHD2, and PHD3 ICC demonstrating expression in both DAergic and non-DAergic cell types in SN. SN sections were separately stained for PHD isoforms (green, ×40) and TH (upper right; red, ×40), and nuclei counterstained with DAPI (lower left; blue, ×40); merged, lower right. B, Gene targeting strategy for generating pTH-PHD knockdown (KD) lines and confirmation of PHD isoform-specific deletions. Top, Schematic of genetic location of floxed PHD alleles (PHD floxed) and expected deletion products (PHD cre), forward and reverse primers used for the PCR analyses (arrows), and expected sizes of PCR products before and after Cre-mediated deletion. Bottom, PCR confirming deletions in DAergic-enriched OB versus CB negative controls isolated from pTH-CRE-floxed PHD mice (KD) versus floxed PHD controls (CON). C, Representative Western blots demonstrating select PHD isoform reduction in striatal tissues from pTH-CRE-floxed PHD lines (left); densitometric quantitation (right); *p < 0.05 for PHD con versus KD; Tukey's multiple-comparison test.
We next assessed the effects of PHD knockdown following systemic administration of the mitochondrial neurotoxin MPTP using the same low-dose administration regime previously used in our pharmacological PHD inhibitor studies for comparison (Lee et al., 2009). Genetic knockdown of PHD2 within DAergic neurons was found to significantly attenuate the loss of these neurons within the SN, to a degree similar to that observed using broad-acting PHD inhibitors or iron chelation (Fig. 2A; Kaur et al., 2003; Lee et al., 2009). In contrast, genetic reductions in DAergic levels of either the PHD1 or PHD3 isoform had no significant effect on restoration of cell viability (Fig. 2A). Knockdown of PHD2 was additionally shown to abrogate losses in motor function as assessed by rearing behavior, a measure that has been demonstrated to be more sensitive to dopamine loss than other motor tests, including the open field test and the pole test (Willard et al., 2015; Fig. 2B).
Figure 2.
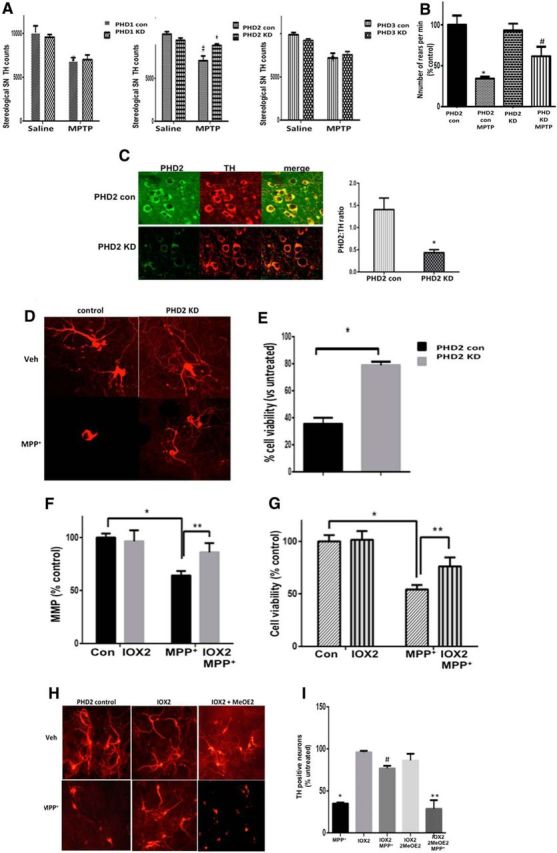
Reductions in PHD2 activity results in neuroprotection against the mitochondrial neurotoxin MPTP in vivo and its metabolite MPP+ in vitro in a HIF1α-dependent fashion. A, Stereological TH+ SN cell counts of pTH-CRE-floxed PHD mice (PHD KD) versus floxed-only controls (PHD con) intraperitoneally injected with MPTP demonstrates a statically significant neuroprotective effect when PHD2 levels were reduced, but not in the case of reductions in the other two isoforms. Data are expressed as average SN TH+ cells per animal, n = 5 per condition. B, Locomotor activity was monitored via the cylinder test in PHD2 knockdown (KD) mice versus littermate controls before the animals were killed. Values are reported as number of rears (% control); *p < 0.05, saline versus MPTP-treated control; #p < 0.05 MPTP-treated PHD2 control versus MPTP-treated PHD2 KD. C, Representative in vivo analysis of dual PHD2 and TH ICC in PHD2 controls (top) versus KD SN (bottom), demonstrating reductions in PHD2 protein expression within DAergic SN neurons in PHD2 KD mice. SN sections were separately stained for PHD2 (right; green, ×40) and TH (middle; red, ×40); merged, yellow (left). Quantitation is shown as PHD2/TH ratio, *p > 0.05 PHD2 KD versus PHD2 control. D, Representative TH immunostaining of mesencephalic cultures from genetic PHD2 KD mice ± MPP+. E, TH+ cell quantification (right) of DAergic PHD2 KD under conditions of mitochondrial stress. Data are expressed as percentage of cell loss versus controls. F, G, MPP+-mediated losses in mitochondrial function as monitored by mitochondrial membrane potential (MMP) levels (F) and cell viability (G) in human iPSC-derived neurons are abrogated by pharmacological PHD2 inhibition. Data are expressed as percentage untreated control. *p < 0.001 versus control; **p < 0.01 versus MPP+. H, Representative TH immunostaining of mesencephalic cultures from PHD2 control mice in the presence of vehicle or MPP+ ± IOX2, ± HIF1α inhibition via 2-methoxyestradiol (MeOE2). I, TH+ cell quantitation demonstrates that neuroprotection afforded by PHD2 inhibition in these cells is prevented by reductions in HIF1α activity. Data are expressed as percentage untreated controls. *p < 0.01 versus control; #p < 0.01 versus MPP+, **p < 0.01 versus IOX2 MPP+; Tukey's multiple-comparison test.
Primary mesencephalic cultures isolated from PHD2 knockdown animals, verified to display reductions in PHD2 protein levels selectively within dopaminergic SN neurons (Fig. 2C), also displayed similar neuroprotective effects in the presence of the MPTP metabolite MPP+ (Fig. 2D,E). Pharmacological PHD2 inhibition using the select inhibitor IOX2 was found to attenuate both reductions in mitochondrial function and neuronal cell loss in human iPSC-derived DAergic neurons treated with MPP+ (Fig. 2F,G), demonstrating that what we observe in the mice translates into analogous effects in a relevant nontransformed human cell model. Neuroprotective effects in mesencephalic cultures were found to be dependent on HIF1α activity (Fig. 2H,I).
The PHD2-HIF1α signaling pathway regulates expression of ATP13A2
Pharmacological PHD2 inhibition by IOX2 was found to result in increased expression of several HIF1α target genes in isolated mesencephalic cultures from floxed PHD2 control lines (data not shown), including that encoding the lysosomal ATPase ATP13A2 (Fig. 3A). Upregulation of ATP13A2 mRNA levels was also found to occur in both OB and ST tissues isolated from DAergic PHD2 knockdown mice versus floxed-only controls (Fig. 3B,C). This coincided with increased nigrostriatal levels of ATP13A2 protein in PHD2 knockdowns versus floxed-only controls (Fig. 3D). In contrast, no alterations in ATP13A2 levels were observed in either PHD1-specific or PHD3-specific knockdown lines (Fig. 3E). These data suggest that the PHD2-HIF1α pathway selectively acts to regulate levels of ATP13A2 within DAergic neurons.
Figure 3.

Reductions in PHD2 activity results in increases in expression of ATP13A2. A, RT-PCR analysis of ATP13A2 expression levels in mesencephalic cultures isolated from PHD2 control mice in the presence of pharmacological PHD2 inhibition via IOX2 demonstrates increased expression versus untreated controls. Data are reported as fold change. *p < 0.01 versus controls, unpaired t test. B, C, RT-PCR analyses of ATP13A2 levels in OB (B) and ST (C) tissues isolated from genetic PHD2 knock-out versus PHD2 control mice demonstrates upregulation gene expression in the context of in vivo reductions in DAergic PHD2 levels. The HIF1α target gene HO-1 was included as a positive control. Data are reported as fold change. *p < 0.01 versus controls, unpaired t test. D, Western blot analysis of ATP13A2 and HO-1 levels in striatal tissues isolated from PHD2 knockdowns (KDs) versus control mice demonstrates significant elevations in protein levels. Data are reported as percentage PHD2 control normalized to actin; *p < 0.01 versus PHD2, #p < 0.01 versus PHD2, unpaired t test. E, Western blot analysis of ATP13A2 levels in striatal tissues isolated from PHD1 and PHD3 KDs versus control mice demonstrates no significant change in protein levels. Data are reported as percentage PHD1 and PHD3 control normalized to actin, unpaired t test.
Regulation of ATP13A2 acts to maintain lysosomal iron stores under conditions of PD-related stress
Lysosomal pH has been found to be compromised in ATP13A2-deficient fibroblasts and DAergic cell lines, which can in turn result in disruptions in lysosomal iron storage (Dehay et al., 2012; Uchiyama et al., 2008). To explore the potential impact of reduced PHD2 on levels of lysosomal iron storage, we interrogated levels of cytosol ferrous iron in striatal dopaminergic synaptosomes isolated from PHD2 knockdown versus control mice, prepared via a magnetic bead column purification protocol previously described by our laboratory (Chinta et al., 2007; Choi et al., 2011). Levels of cytosolic iron were assessed using the calcein quenching assay in the absence and presence of the select ferrous iron chelator SIH (Tenopoulou et al., 2007; Kon et al., 2010). Ferrous cytosolic iron levels were found to be significantly reduced both basally and after MPTP treatment versus those from wild-type littermates, resulting in lower levels available within this cellular compartment to participate in subsequent neurotoxic redox reactions (Fig. 4A). After addition of the lysosomal ATPase inhibitor bafilomycin to induce release of lysosomal iron stores, cytosolic ferrous iron levels were not altered under basal conditions (data not shown). However, they were found to be significantly elevated in striatal dopaminergic synaptosomes from MPTP-treated PHD2 knockdowns versus MPTP-treated control mice (Fig. 4B). This suggests that in the context of PD-related stress, lysosomal iron storage is higher in the PHD2 knockdowns versus controls, possibly in part due to elevations in ATP13A2 levels in these animals.
Figure 4.

PHD2 inhibition in vivo abrogates basal and MPTP-mediated increases in cytosolic ferrous iron levels via increases in lysosomal iron storage. A, Cytosolic ferrous iron levels as measured by the calcein dequenching assay in striatal DAergic synaptosomes isolated from PHD2 knockdowns (KDs) versus PHD2 controls ± MPTP or saline, demonstrating that PHD2 KD results in reductions both basally and following MPTP treatment. Values are reported as percentage PHD2 control; *p < 0.05 versus PHD2 controls; #p < 0.05 versus PHD2 KD; &p < 0.05 versus MPTP-treated PHD2 controls; Tukey's multiple-comparison test. B, Cytosolic ferrous iron levels following treatment with bafilomycin reported as fold increases versus MPTP, demonstrating increases in lysosomal iron stores in PHD2 KDs; p̂ > 0.05 versus MPTP-treated PHD2 control; ^p̂ > 0.05 versus MPTP-treated PHD2 KD.
To validate a causative role for ATP13A2 in maintenance of lysosomal iron storage under conditions of PD-related stress, we interrogated the impact of its knockdown in differentiated human DAergic SY5Y cells. We found that stable lentiviral shRNA-mediated knockdown of ATP13A2 (Fig. 5A–C) resulted in elevations in cytosolic ferrous iron levels both basally and following MPTP treatment (Fig. 5D). Following administration of bafilomycin, while cytosolic ferrous iron levels within MPTP-treated control cells further increased, there were no additional increases within the MPTP-treated ATP13A2 knockdown cells. This suggests that, particularly under conditions of stress, maintenance of lysosomal iron storage is dependent in part on ATP13A2.
Figure 5.
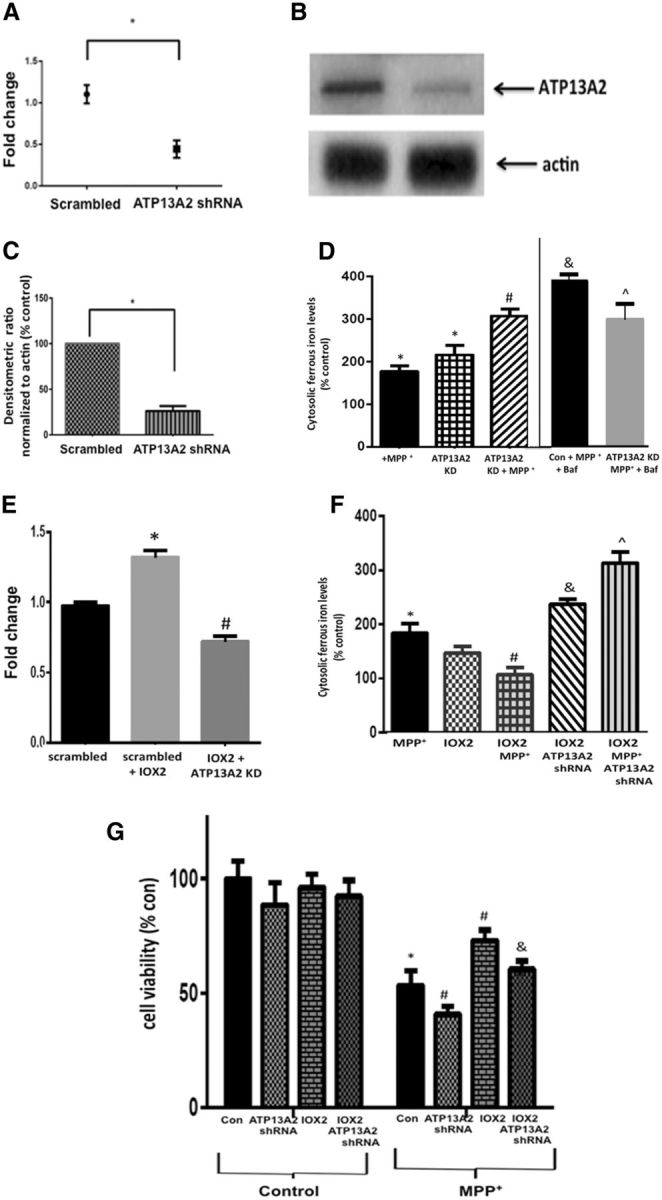
Knockdown (KD) of ATP13A2 expression in cultured human DAergic cells results in reduced ability to maintain lysosomal iron storage and abrogates PHD2 inhibition-mediated neuroprotection under conditions of stress. A, RT-PCR analysis of ATP13A2 levels in SH-SY5Y cells stably transfected with lentiviral ATP13A2 shRNA shows a significant reduction in ATP13A2 expression. Data are expressed as fold change; *p < 0.01 versus scrambled shRNA controls; unpaired t test. B, Representative Western blot of ATP13A2 levels in ATP13A2 shRNA transfected cells versus controls. C, Quantitation following normalization to actin (right) demonstrates corresponding reductions in ATP13A2 protein levels. Data are reported as percentage scrambled shRNA; *p < 0.01 versus scrambled; unpaired t test. D, Cytosolic ferrous iron measurements via calcein dequenching assay in stable differentiated ATP13A2 shRNA versus scrambled control SY5Y cells demonstrate that levels are increased following ATP13A2 KD both basally and following treatment with MPP+; bafilomycin results in significantly higher increases in cytosolic ferrous iron in MPP+ controls versus MPP+-treated ATP13A2 KD cells. Values are presented as percentage control; *p < 0.05 versus scrambled controls; #p < 0.05 versus MPP+, &p < 0.05 versus MPP+-treated controls; p̂ < 0.05 versus MPP+-treated KDs; Tukey's multiple-comparison test. E, RT-PCR analysis of ATP13A2 expression levels in the absence and presence of IOX2 demonstrates increased expression in scrambled controls, which is reduced by ATP13A2 shRNA. Data are reported as fold change; *p < 0.01 versus scrambled control; #p < 0.05 versus IOX2 alone; Tukey's multiple-comparison test. F, Cytosolic ferrous iron levels in MPP+-treated versus control cells in the absence and presence of IOX2 and ATP13A2 KD; values are presented as percentage controls. IOX2 prevents MPP+-mediated increases in cytosolic ferrous iron and this is reversed by knockdown of ATP13A2; *p < 0.05 versus controls; #p < 0.05 versus MPP+ alone; &p < 0.05 versus IOX2 alone; p̂ < 0.05 versus IOX2 KD; Tukey's multiple-comparison test. G, Cell viability as assessed by CyQuant fluorescence assay in differentiated control versus ATP13A2 KD cells in the absence or presence of IOX2 and MPP+; values are presented as percentage control. MPP+-mediated losses in cell viability are exacerbated by ATP13A2 KD both basally and in the presence of IOX2; *p < 0.05 versus controls, #p < 0.05 versus MPP+-control, &p < 0.05 versus MPP+, IOX2-treated control; Tukey's multiple-comparison test.
As previously demonstrated in the context of PHD2 knockdown in vivo (Fig. 3), select inhibition of PHD2 by IOX2 was found to result in upregulation of ATP13A2 expression, which was attenuated in the presence of ATP13A2 shRNA (Fig. 5E). At the concentrations used in this set of experiments, IOX2 on its own did not reduce cytosolic iron levels. However, it did abrogate MPP+-mediated increases and these effects were reversed in the presence of ATP13A2 shNA (Fig. 5F). While ATP13A2 knockdown alone did not have a significant impact on cell viability, it was found to abrogate IOX2-mediated neuroprotection against PD-related stress in these cells (Fig. 5G). Furthermore, ATP13A2 knockdown was found to partially abrogate neuroprotection elicited by IOX2 on MPP+-mediated neurotoxicity. This suggests that neuroprotection elicited by reductions in PHD2 levels can be abrogated by ATP13A2 knockdown and is therefore partially ATP13A2-dependent.
Discussion
HIF1α is part of a highly conserved complex that controls response to various cellular stressors via its ability to bind to HREs located within the promoters of HIF1α target genes, upregulating their expression. HIF1α can be inactivated via its hydroxylation by PHDs, resulting in its subsequent ubiquitination and degradation by the proteasome (Bruick and McKnight, 2001; Harten et al., 2010; Miyata et al., 2011). HIF1α knockdown in murine midbrain-derived neural precursor cells and in DAergic SN neurons in vivo has been reported to result in neuronal cell loss, suggesting that maintenance of HIF1α levels is important for survival of these cells (Milosevic et al., 2007). A better understanding the mechanistic role of the HIF1α signaling pathway in DAergic cell survival could provide us with important clues as to how its dysregulation promotes DAergic cell loss and may be prevented via its activation (Milosevic et al., 2007). Results from this current study support the notion that HIF1α signaling is an important pathway in the control of DAergic SNpc cell viability and therefore constitutes a credible therapeutic target for PD and other related disorders (Milosevic et al., 2007; Lee et al., 2009; Rajagopalan et al., 2014).
Our laboratory previously reported that broad-acting pharmacological PHD inhibition in vivo prevented increased nigral iron levels, mitochondrial dysfunction, and select nigrostriatal neurodegeneration associated with systemic MPTP administration in conjunction with enhanced HIF1α activation (Lee et al., 2009). In a separate study, we showed that pharmacological PHD inhibitors were also neuroprotective in an age-related genetic mouse model in which dopaminergic glutathione levels are downregulated, emulating biochemical changes reported to occur in the PD brain, including mitochondrial defects and selective age-related loss of DAergic SN neurons (Rajagopalan et al., 2014). However, the particular isoform(s) involved were not determined in these previous studies. Here we report that although all three PHD isoforms are expressed within DAergic neurons as well as other cell types in the SNpc, neuroprotective effects in the face of mitochondrial stress elicited by MPTP or its derivative MPP+ were found to be PHD2-specific and HIF1α-dependent. In addition to our own findings in the context of both differentiated human SY5Y cells and iPSC-derived neurons, relevance of these findings to the human disease state are further bolstered by previous postmortem data demonstrating the aberrant elevation of PHD2 levels and lowered HIF1α levels in affected DAergic neurons isolated from the patient SNpc tissues versus those from age-matched controls (Elstner et al., 2011). This suggests that the PHD2 isoform may be a select target for intervention in PD via its ability to regulate HIF1α protein levels in affected DAergic neurons.
HIF1α coordinates expression of a number of downstream neuroprotective genes, including several that aid in the preservation of cellular iron homeostasis under conditions of stress (Siddiq et al., 2005; Nakayama, 2009; Greer et al., 2012). In addition to PHD inhibition, iron chelation has also been demonstrated to protect against increases in unsequestered iron within the nigrostriata in association with various PD models including MPTP administration, in conjunction with reductions in accompanying neurodegeneration (Kaur et al., 2003; Mandel et al., 2004; Youdim et al., 2004; Lee et al., 2009). Several ongoing drug development studies, including in our own laboratory (data not shown), seek to identify novel iron-chelating agents for the treatment of PD. An important role of PHD2-HIF1α signaling may be its ability to regulate cellular iron metabolism and this pathway may therefore constitute an alternative target toward this same clinical aim.
The ATP13A2 gene has recently been reported to be a transcriptional target of HIF1α in cultured DAergic cell lines (Xu et al., 2012). The gene itself encodes a lysosomal ATPase that is highly expressed within DAergic SNpc neurons and reduced in postmortem SNpc tissues isolated from patients with PD and other related conditions (Murphy et al., 2013). The physiological function of ATP13A2, and hence the role of its dysfunction in PD, is still somewhat elusive. PD-linked mutations in ATP13A2 have been demonstrated to lead to alterations in ALP function, including decreased proteolytic processing of lysosomal enzymes, reduced degradation of lysosomal substrates, and diminished lysosomal-mediated clearance of autophagosomes in various cellular models in conjunction with increased DAergic cell death (Dehay et al., 2012). Conversely, restoration of ATP13A2 levels in ATP13A2 mutant or depleted cells restores lysosomal function and attenuates cell death. These data suggest that modulation of ATP13A2 levels may have therapeutic potential in the context of PD, although the precise mechanisms involved are unclear.
Lysosomal pH has been reported to be compromised in ATP13A2 mutant fibroblasts and in DAergic cell lines in which levels have been genetically reduced (Dehay et al., 2012). Based on studies in other cell types, this would result in disruption of lysosomal iron storage (Uchiyama et al., 2008). Levels of iron in the basal ganglia have been reported to be elevated in the context of mutations in ATP13A2 (Grünewald et al., 2012; Gusdon et al., 2012; Levi and Finazzi, 2014). While several previous publications have implicated a role for ATP13A2 in zinc homeostasis, its ability to affect cellular iron levels is unclear (Kong et al., 2014; Park et al., 2014; Tsunemi and Krainc, 2014; Rinaldi et al., 2015). Based on our results, one important consequence of inhibition of PHD2 may be the elevation of ATP13A2 expression, resulting in the preservation of cellular iron homeostasis in the face of mitochondrial stress. Mechanistically, this appears to involve the maintenance of lysosomal iron storage. Reductions in its levels reverse these effects, resulting in increased cytosolic iron levels and susceptibility to neuronal cell loss.
PHD2 knockdown may affect other potential HIF1α targets involved in the maintenance of cellular iron homeostasis, including transferrin receptor 1 (TfR1) and the divalent metal transporter (DMT1). Indeed, levels of TfR1 protein were found to be elevated while levels of DMT1 were reduced in striatal tissues from PHD2 knockdowns versus controls (Fig. 6). TfR1 elevations would be predicted to result in increased endocytosis of the TfR1-Tf-Fe3+ complex into the lysosome, which in the presence of low lysosomal pH would result in release of iron from the complex in the form of ferrous iron (Kurz et al., 2011). The HIF-responsive form of DMT1 present in dopaminergic cells is located on the lysosomal membrane, where it is believed to function in the export of ferrous iron from this organelle (Lis et al., 2005). In addition to presence of an HRE, lysosomal DMT1 also contains an iron response element (IRE) present in the isoform found to be upregulated following MPTP exposure in vivo and in affected human Parkinsonian SN tissues (Garrick et al., 2006; Salazar et al., 2008). This could explain why DMT1 protein levels are reduced in the face of PHD2 inhibition (i.e., an increase in HIF1α levels would initially result in increased DMT1 transcription), but in the face of high cellular iron levels associated with both PD and in PD models, presence of the IRE would drive increased degradation of the mRNA and subsequent reductions DMT1 protein levels. The net effect of lowered lysosomal DMT1 would be the same as that for elevations in TfR1 to retain ferrous iron within the lysosome. This may in part explain the elevated levels of lysosomal storage in the PHD2 knockdown animals. However, disruption of ATP13A2 results in reduced ability to maintain lysosomal iron stores, particularly under conditions of stress.
Figure 6.

Levels of TfR1 are elevated and DMT1 reduced in PHD2 knockdown (KD) mice. Western blot analysis of TfR1 and DMT1 levels in striatal tissues isolated from PHD2 KDs versus control mice. Data are reported as percentage PHD2 control normalized to actin; *p < 0.01 versus PHD2; Tukey's multiple-comparison test.
Taken in total, our data suggests that not only is expression of ATP13A2 regulated by the PHD2-HIF1α signaling pathway, but that this is instrumental in maintaining cellular iron homeostasis and cell viability in mitochondrially compromised DAergic neurons. This constitutes a novel function for ATP13A2 that could have important implications for mechanisms underlying the loss of cellular iron homeostasis associated with PD neuropathology. Based on these findings, further preclinical and clinical studies are strongly warranted to explore whether enhancement of ATP13A2 function constitutes a tractable therapeutic target for the clinical treatment of PD.
Footnotes
This work was supported by the National Institutes of Health (R01 NS047198, NS041264, and AG012141; to J.K.A. and S.R.).
The authors declare no competing financial interests.
References
- Bruick RK, McKnight SL. Conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- Chinta SJ, Kumar MJ, Hsu M, Rajagopalan S, Kaur D, Rane A, Nicholls DG, Choi J, Andersen JK. Inducible alterations of glutathione levels in adult dopaminergic midbrain neurons result in nigrostriatal degeneration. J Neurosci. 2007;27:13997–4006. doi: 10.1523/JNEUROSCI.3885-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SW, Gerencser AA, Lee DW, Rajagopalan S, Nicholls DG, Andersen JK, Brand MD. Intrinsic bioenergetic properties and stress sensitivity of dopaminergic synaptosomes. J Neurosci. 2011;31:4524–4534. doi: 10.1523/JNEUROSCI.5817-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel JA, Malladi CS, Kettle E, McCluskey A, Robinson PJ. Analysis of synaptic vesicle endocytosis in synaptosomes by high-content screening. Nat Protoc. 2012;7:1439–1455. doi: 10.1038/nprot.2012.070. [DOI] [PubMed] [Google Scholar]
- Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, Vital A, Vila M, Klein C, Bezard E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A. 2012;109:9611–9616. doi: 10.1073/pnas.1112368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elstner M, Morris CM, Heim K, Bender A, Mehta D, Jaros E, Klopstock T, Meitinger T, Turnbull DM, Prokisch H. Expression analysis of dopaminergic neurons in Parkinson's disease and aging links transcriptional dysregulation of energy metabolism to cell death. Acta Neuropathologica. 2011;122:75–86. doi: 10.1007/s00401-011-0828-9. [DOI] [PubMed] [Google Scholar]
- Garrick MD, Kuo HC, Vargas F, Singleton S, Zhao L, Smith JJ, Paradkar P, Roth JA, Garrick LM. Comparison of mammalian cell lines expressing distinct isoforms of divalent metal transporter 1 in a tetracycline-regulated fashion. Biochem J. 2006;398:539–546. doi: 10.1042/BJ20051987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer SN, Metcalf JL, Wang Y, Ohh M. The updated biology of hypoxia-inducible factor. EMBO J. 2012;31:2448–2460. doi: 10.1038/emboj.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünewald A, Arns B, Seibler P, Rakovic A, Münchau A, Ramirez A, Sue CM, Klein C. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol Aging. 2012;33:1843.e1–7. doi: 10.1016/j.neurobiolaging.2011.12.035. [DOI] [PubMed] [Google Scholar]
- Gusdon AM, Zhu J, Van Houten B, Chu CT. ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol Dis. 2012;45:962–972. doi: 10.1016/j.nbd.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harten SK, Ashcroft M, Maxwell PH. Prolyl hydroxylase domain inhibitors: a route to HIF activation and neuroprotection. Antioxid Redox Signal. 2010;12:459–480. doi: 10.1089/ars.2009.2870. [DOI] [PubMed] [Google Scholar]
- Kaur D, Yantiri F, Rajagopalan S, Kumar J, Mo JQ, Boonplueang R, Viswanath V, Jacobs R, Yang L, Beal MF, DiMonte D, Volitaskis I, Ellerby L, Cherny RA, Bush AI, Andersen JK. Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo. Neuron. 2003;37:899–909. doi: 10.1016/S0896-6273(03)00126-0. [DOI] [PubMed] [Google Scholar]
- Kaur D, Rajagopalan S, Andersen JK. Chronic expression of H-ferritin in dopaminergic midbrain neurons results in an age-related expansion of the labile iron pool and subsequent neurodegeneration: implications for Parkinson's disease. Brain Res. 2009;1297:17–22. doi: 10.1016/j.brainres.2009.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kett LR, Stiller B, Bernath MM, Tasset I, Blesa J, Jackson-Lewis V, Chan RB, Zhou B, Di Paolo G, Przedborski S, Cuervo AM, Dauer WT. α-Synuclein-independent histopathological and motor deficits in mice lacking the endolysosomal Parkinsonism protein Atp13a2. J Neurosci. 2015;35:5724–5742. doi: 10.1523/JNEUROSCI.0632-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kon K, Kim JS, Uchiyama A, Jaeschke H, Lemasters JJ. Lysosomal iron mobilization and induction of the mitochondrial permeability transition in acetaminophen-induced toxicity to mouse hepatocytes. Toxicol Sci. 2010;117:101–108. doi: 10.1093/toxsci/kfq175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong SM, Chan BK, Park JS, Hill KJ, Aitken JB, Cottle L, Farghaian H, Cole AR, Lay PA, Sue CM, Cooper AA. Parkinson's disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes α-synuclein externalization via exosomes. Hum Mol Genet. 2014;23:2816–2833. doi: 10.1093/hmg/ddu099. [DOI] [PubMed] [Google Scholar]
- Kurz T, Gustafsson B, Brunk UT. Intralysosomal iron chelation protects against oxidative stress-induced cellular damage. FEBS J. 2006;273:3106–3117. doi: 10.1111/j.1742-4658.2006.05321.x. [DOI] [PubMed] [Google Scholar]
- Kurz T, Terman A, Gustafsson B, Brunk UT. Lysosomes in iron metabolism, ageing and apoptosis. Histochem Cell Biol. 2008;129:389–406. doi: 10.1007/s00418-008-0394-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz T, Eaton JW, Brunk UT. The role of lysosomes in iron metabolism and recycling. Int J Biochem Cell Biol. 2011;43:1686–1697. doi: 10.1016/j.biocel.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Lee DW, Rajagopalan S, Siddiq A, Gwiazda R, Yang L, Beal MF, Ratan RR, Andersen JK. Inhibition of prolyl hydroxylase protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity: model for the potential involvement of the hypoxia-inducible factor pathway in Parkinson disease. J Biol Chem. 2009;284:29065–29076. doi: 10.1074/jbc.M109.000638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi S, Finazzi D. Neurodegeneration with brain iron accumulation: update on pathogenic mechanisms. Front Pharmacol. 2014;5:99. doi: 10.3389/fphar.2014.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis A, Paradkar PN, Singleton S, Kuo HC, Garrick MD, Roth JA. Hypoxia induces changes in expression of isoforms of the divalent metal transporter (DMT1) in rat pheochromocytoma (PC12) cells. Biochem Pharmacol. 2005;69:1647–1655. doi: 10.1016/j.bcp.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Mandel S, Weinreb O, Amit T, Youdim MB. Cell signaling pathways in the neuroprotective actions of the green tea polyphenol (-)-epigallocatechin-3-gallate: implications for neurodegenerative diseases. J Neurochem. 2004;88:1555–1569. doi: 10.1046/j.1471-4159.2003.02291.x. [DOI] [PubMed] [Google Scholar]
- Milosevic J, Maisel M, Wegner F, Leuchtenberger J, Wenger RH, Gerlach M, Storch A, Schwarz J. Lack of hypoxia-inducible factor-1 impairs midbrain neural precursor cells involving vascular endothelial growth factor signaling. J Neurosci. 2007;27:412–421. doi: 10.1523/JNEUROSCI.2482-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata T, Takizawa S, van Ypersele de Strihou C. Hypoxia. 1. Intracellular sensors for oxygen and oxidative stress: novel therapeutic targets. Am J Physiol Cell Physiol. 2011;300:C226–C231. doi: 10.1152/ajpcell.00430.2010. [DOI] [PubMed] [Google Scholar]
- Murphy KE, Cottle L, Gysbers AM, Cooper AA, Halliday GM. ATP13A2 (PARK9) protein levels are reduced in brain tissue of cases with Lewy bodies. Acta Neuropathol Commun. 2013;1:11. doi: 10.1186/2051-5960-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K. Cellular signal transduction of the hypoxia response. J Biochem. 2009;146:757–765. doi: 10.1093/jb/mvp167. [DOI] [PubMed] [Google Scholar]
- Park JS, Mehta P, Cooper AA, Veivers D, Heimbach A, Stiller B, Kubisch C, Fung VS, Krainc D, Mackay-Sim A, Sue CM. Pathogenic effects of novel mutations in the P-type ATPase ATP13A2 (PARK9) causing Kufor-Rakeb syndrome, a form of early-onset parkinsonism. Hum Mutat. 2011;32:956–964. doi: 10.1002/humu.21527. [DOI] [PubMed] [Google Scholar]
- Park JS, Koentjoro B, Veivers D, Mackay-Sim A, Sue CM. Parkinson's disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum Mol Genet. 2014;23:2802–2815. doi: 10.1093/hmg/ddt623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podhajska A, Musso A, Trancikova A, Stafa K, Moser R, Sonnay S, Glauser L, Moore DJ. Common pathogenic effects of missense mutations in the P-type ATPase ATP13A2 (PARK9) associated with early-onset parkinsonism. PLoS One. 2012;7:e39942. doi: 10.1371/journal.pone.0039942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presgraves SP, Ahmed T, Borwege S, Joyce JN. Terminally differentiated SH-SY5Y cells provide a model system for studying neuroprotective effects of dopamine agonists. Neurotox Res. 2004;5:579–598. doi: 10.1007/BF03033178. [DOI] [PubMed] [Google Scholar]
- Rajagopalan S, Chinta SJ, Andersen JK. Pharmacological prolyl hydroxylase domain inhibition as a therapeutic target for Parkinson's disease. CNS Neurol Disord Drug Targets. 2014;13:120–125. doi: 10.2174/18715273113126660131. [DOI] [PubMed] [Google Scholar]
- Rinaldi DE, Corradi GR, Cuesta LM, Adamo HP, de Tezanos Pinto F. The Parkinson-associated human P5B-ATPase ATP13A2 protects against the iron-induced cytotoxicity. Biochim Biophys Acta. 2015;1848:1646–1655. doi: 10.1016/j.bbamem.2015.04.008. [DOI] [PubMed] [Google Scholar]
- Salazar J, Mena N, Hunot S, Prigent A, Alvarez-Fischer D, Arredondo M, Duyckaerts C, Sazdovitch V, Zhao L, Garrick LM, Nuñez MT, Garrick MD, Raisman-Vozari R, Hirsch EC. Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson's disease. Proc Natl Acad Sci U S A. 2008;105:18578–18583. doi: 10.1073/pnas.0804373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiq A, Ayoub IA, Chavez JC, Aminova L, Shah S, LaManna JC, Patton SM, Connor JR, Cherny RA, Volitakis I, Bush AI, Langsetmo I, Seeley T, Gunzler V, Ratan RR. Hypoxia-inducible factor prolyl 4-hydroxylase inhibition: A target for neuroprotection in the central nervous system. J Biol Chem. 2005;280:41732–41743. doi: 10.1074/jbc.M504963200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Aguila HL, Parikh NS, Li X, Lamothe K, Duan LJ, Takeda H, Lee FS, Fong GH. Regulation of adult erythropoiesis by prolyl hydroxylase domain proteins. Blood. 2008;111:3229–3235. doi: 10.1182/blood-2007-09-114561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenopoulou M, Kurz T, Doulias PT, Galaris D, Brunk UT. Does the calcein-AM method assay the total cellular ‘labile iron pool’ or only a fraction of it? Biochem J. 2007;403:261–266. doi: 10.1042/BJ20061840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terman A, Kurz T. Lysosomal iron, iron chelation, and cell death. Antioxid Redox Signal. 2013;18:888–898. doi: 10.1089/ars.2012.4885. [DOI] [PubMed] [Google Scholar]
- Tsunemi T, Krainc D. Zn2 dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum Mol Genet. 2014;23:2791–2801. doi: 10.1093/hmg/ddt572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunemi T, Hamada K, Krainc D. ATP13A2/PARK9 regulates secretion of exosomes and α-synuclein. J Neurosci. 2014;34:15281–15287. doi: 10.1523/JNEUROSCI.1629-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama A, Kim JS, Kon K, Jaeschke H, Ikejima K, Watanabe S, Lemasters JJ. Translocation of iron from lysosomes into mitochondria is a key event during oxidative stress-induced hepatocellular injury. Hepatology. 2008;48:1644–1654. doi: 10.1002/hep.22498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willard AM, Bouchard RS, Gittis AH. Differential degradation of motor deficits during gradual dopamine depletion with 6-hydroxydopamine in mice. Neuroscience. 2015;301:254–267. doi: 10.1016/j.neuroscience.2015.05.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Guo H, Zhang X, Tang B, Cai F, Zhou W, Song W. Hypoxia regulation of ATP13A2 (PARK9) gene transcription. J Neurochem. 2012;122:251–259. doi: 10.1111/j.1471-4159.2012.07676.x. [DOI] [PubMed] [Google Scholar]
- Youdim MB, Stephenson G, Ben Shachar D. Ironing iron out in Parkinson's disease and other neurodegenerative diseases with iron chelators: a lesson from 6-hydroxydopamine and iron chelators, desferal and VK-28. Ann NY Acad Sci. 2004;1012:306–325. doi: 10.1196/annals.1306.025. [DOI] [PubMed] [Google Scholar]