

Abstract
Background
The tumor suppressor protein phosphatase and tensin homolog (PTEN) is a key regulator of the PI3K/AKT pathway which is frequently altered in a variety of tumors including a subset of acute B-lymphoblastic leukemias (B-ALL). While PTEN mutations and deletions are rare in B-ALL, promoter hypermethylation and posttranslational modifications are the main pathways of PTEN inactivation. Casein Kinase II (CK2) is often upregulated in B-ALL and phosphorylates both PTEN and DNA methyltransferase 3A, resulting in increased PI3K/AKT signaling and offering a potential mechanism for further regulation of tumor-related pathways.
Methods
Here, we evaluated the effects of CK2 inhibitor CX-4945 alone and in combination with hypomethylating agent decitabine on B-ALL proliferation and PI3K/AKT pathway activation. We further investigated if CX-4945 intensified decitabine-induced hypomethylation and identified aberrantly methylated biological processes after CK2 inhibition. In vivo tumor cell proliferation in cell line and patient derived xenografts was assessed by longitudinal full body bioluminescence imaging and peripheral blood flow cytometry of NSG mice.
Results
CX-4945 incubation resulted in CK2 inhibition and PI3K pathway downregulation thereby inducing apoptosis and anti-proliferative effects. CX-4945 further affected methylation patterns of tumor-related transcription factors and regulators of cellular metabolism. No overlap with decitabine-affected genes or processes was detected. Decitabine alone revealed only modest anti-proliferative effects on B-ALL cell lines, however, if combined with CX-4945 a synergistic inhibition was observed. In vivo assessment of CX-4945 in B-ALL cell line xenografts resulted in delayed proliferation of B-ALL cells. Combination with DEC further decelerated B-ALL expansion significantly and decreased infiltration in bone marrow and spleen. Effects in patient-derived xenografts all harboring a t(4;11) translocation were heterogeneous.
Conclusions
We herein demonstrate the anti-leukemic potential of CX-4945 in synergy with decitabine in vitro as well as in vivo identifying CK2 as a potentially targetable kinase in B-ALL.
Electronic supplementary material
The online version of this article (10.1186/s12885-019-5411-0) contains supplementary material, which is available to authorized users.
Keywords: Leukemia, CK2 inhibition, Hypomethylation, In vivo imaging, Methylome analysis
Background
In B cell acute lymphoblastic leukemia (B-ALL) distinct molecular aberrations contribute to leukemogenesis including mutations, chromosomal translocations or epigenetic dysregulation [1, 2]. The PI3K/AKT pathway induces proliferation, inhibits apoptosis and is involved in B-ALL pathogenesis suggesting potential therapeutic targets [3]. PTEN antagonizes AKT phosphorylation and subsequent pathway activation. As lately reviewed PTEN phosphorylation is considered the most common way of PTEN inactivation in B-ALL in contrast to mutations or deletions in other types of leukemia and solid tumors [4, 5]. Increased phosphorylation of PTEN reduces its phosphatase activity resulting in anti-apoptotic downstream signaling [6].
Apart from increased phosphorylation hypermethylation-induced decreased PTEN transcription has been reported in several tumors including B-ALL [7]. DNA methyltransferase 3A (DNMT3A) overexpression leads to PTEN promoter hypermethylation in chronic eosinophilic leukemia cells [8]. Hypomethylating agents (HMA) can restore the PTEN activity and inhibit PI3K/AKT downstream signaling [8]. In B-ALL inactivation of numerous tumor suppressor genes by aberrant methylation is associated with poor prognosis [9]. Recently we demonstrated that HMA induce apoptosis and cell cycle arrest and inhibit proliferation in human B-ALL [10].
Regulation of DNMT3A activity in B-ALL is not well explored as in contrast to other hematological neoplasms mutations do not seem to occur [11]. Deplus et al. reported that Casein Kinase II (CK2) is able to phosphorylate DNMT3A in osteosarcoma cells and thus mediate genomic methylation [12]. CK2 is a highly conserved serine/threonine kinase frequently overexpressed in solid and hematological neoplasms including B-ALL [13]. CK2 phosphorylates a variety of substrates across numerous pathways and cellular functions. Among those are apoptosis, cell cycle regulation, proliferation and transcription mediated via Wnt, hedgehog, JAK/STAT and NFkB pathways [14]. CK2 further phosphorylates and therefore inactivates tumor suppressor genes like IKZF1 and PTEN in human leukemia cells [15, 16]. In B-ALL cells, Gomes et al. demonstrated that CK2 inhibition by ATP-competitive inhibitor CX-4945 rescued PTEN phosphatase activity accompanied by apoptosis of B-ALL cells [6]. CK2 inhibition also restored IKZF1 tumor suppressor function [15]. Several studies further demonstrated anti-proliferative effects on solid and hematological tumor entities in vitro and in vivo [17–19]. The mechanisms behind CK2’s oncogenic effects and potential therapeutic interventions are currently widely discussed [20].
Here, we hypothesized that CK2 inhibitor CX-4945 alone and in combination with HMA decitabine (DEC) reduces PTEN and AKT phosphorylation and thus inhibits PI3K/AKT mediated proliferation in vitro and in vivo.
Methods
Cell lines and cell culture
Human B-ALL precursor cell lines SEM, RS4;11, REH and NALM-6 were purchased from German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany) and cultured as recommended by the manufacturer. Cell lines were selected in order to compare inhibitors in a variety of cell lines of distinct molecular and cytogenetic backgrounds to assess whether inhibitory effects were more prominent in certain subtypes. Media were supplemented with 10% heat-inactivated fetal calf serum (Biochrom, Berlin, Germany) and 100 μg/ml penicillin and streptomycin (Biochrom, Berlin, Germany) and cells were cultured at 37 °C and 5% CO2.
Inhibitory experiments
3.3 × 105 cells per ml were incubated with 5 μM CX-4945 (Hycultec, Beutelsbach, Germany) or 0.1 μM DEC (Selleckchem, Munich, Germany) dissolved in DMSO or respective concentrations of DMSO (controls) for up to 72 h. Apoptosis, proliferation and metabolic activity were assessed as previously described [21]. Experiments were carried out in biological triplicates. Results are described as mean ± standard deviation. Significance (p < 0.05) was estimated by 2-tailed student’s t test. Synergy of CX-4945 and DEC combination was calculated by the Bliss independence model [22]. In this test, the observed effect of a combinatory treatment is compared to a calculated expected effect. The expected effect (E) is calculated as follows: E = (A + B) − (A * B), where A and B represent the relative inhibition of single agents A and B. The difference between the observed and expected effect (Δ = O - E) of the combinatory treatment then determines the level of synergy. If the observed inhibitory effect is higher than the expected value (Δ > 0) the combination is referred to as synergistic while Δ < 0 is considered antagonistic. For the calculation, mean values of metabolic activity as well as proliferation after 48 h and 72 h incubation of three independent experiments were used.
Protein analysis
Cells were lysed using RIPA buffer (Cell Signaling, Danvers, MA, USA) and ultra sound exposure. Proteins were separated on Midi gels (Bio-Rad, Munich, Germany), blotted onto a PVDF membrane (Bio-Rad) using Trans-Blot® Turbo™ Transfer System (Bio-Rad, 2.5 A, 25 V, 10 min), blocked in LI-COR (Lincoln, NE, USA) blocking buffer and detected via LI-COR Odyssey Imaging System and Image Studio Lite software. Antibodies are listed in Additional file 1: Table S1. For quantification, band intensities were assessed using Image Studio Lite software. Values were normalized to respective GAPDH control bands run on each individual gel and ratios between phosphorylated and total protein are given below the images. Mean values and standard deviations of three individual experiments were calculated and displayed as bar charts.
Genome-wide methylation analysis
After CX-4945 and DEC combined incubation genomic DNA was extracted and bisulfite treated using the NucleoSpin® Tissue Kit (Macherey-Nagel, Dueren, Germany) and peqGOLD Bisulfite Conversion Kit (VWR Peqlab, Erlangen, Germany). The methylation status of the LINE-1 retrotransposon is a marker for global methylation [23]. LINE-1 Methylation specific qPCR (MsqPCR) was carried out in a final volume of 25 μl containing 20 to 50 ng bisulfite treated DNA, Quantitect SYBR Green PCR Master Mix (Qiagen, Hilden, Germany) and primers specific for either methylated or unmethylated DNA as follows: initial denaturation (15 min, 95 °C) followed by 45 cycles of denaturation (15 s, 94 °C), annealing (30 s, 55 °C) and elongation (30 s, 72 °C). Primer sequences are listed in Additional file 2: Table S2.
Whole methylome analyses were carried out at the Genomics & Proteomics Core Facility of the German Cancer Research Center (Heidelberg, Germany) using the Illumina Infinium MethylationEPIC BeadChip platform. Raw and processed data can be retrieved from Gene Expression Omnibus (GSE110454). Raw methylome data was normalized using quantile normalization [24] and summarized to calculate mean beta values [25]. Genes were considered hypomethylated based on a fold change (FC) < 0.25 compared to controls. Gene enrichment analysis was performed using the functional annotation tool DAVID [26] as previously described [27]. Gene ontology (GO) terms annotated to at least two genes and statistically over-represented within the hypomethylated gene set were evaluated by hyper-geometric testing using the entire genome as a universe set and a p-value threshold of 0.05. Heatmaps and Venn diagrams were generated using the built-in functions in R [28] and gene-GO term associations were visualized through Chord plots using the GOplot package [29].
Patients
Mononuclear cells from bone marrow (BM) aspirates of newly diagnosed ALL patients (Rostock University Medical Center) were isolated using Biocoll separating solution (Merck Millipore, Darmstadt, Germany) and previously characterized on a molecular level using next-generation sequencing (Cancer hotspot panel, Ion PGM System, Thermo Fisher Scientific, Schwerte, Germany) according to the manufacturer’s protocol [10]. All experiments were performed in accordance to the Declaration of Helsinki and the local ethical committee standards.
Animal studies
Stably GFP and enhanced firefly luciferase (ffLuc) transduced SEM and RS4;11 cell lines were kindly provided by Prof. Irmela Jeremias (Helmholtz Center Munich, Germany). SEM and RS4;11 were stably transduced with GFP and enhanced firefly luciferase (ffLuc) in the pCDH-EF1-MCS-T2A-copGFP vector (System Biosciences, Mountain View, CA, USA) using EcoRI and BamHI. Lentivirus production and cell transduction were carried out as described before [30].
NOD scid gamma mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, NSG, Charles River Laboratories, Sulzfeld, Germany) were bred and housed under specific pathogen-free conditions with access to water and standard chow ad libitum. All experiments were carried out in a laboratory setting and no intervention was performed within the animal housing and breeding rooms. Study group sizes were ten animals for cell line-derived in vivo studies based on biometric Power calculation and at least two animals per group for patient-derived (PDX) experiments [31]. Experiments were approved by the review board of the federal state Mecklenburg-Vorpommern, Germany (reference number: LALLF MV/7221.3–1.1-002/15). Study endpoints for all mice used are listed in Additional file 3 : Table S3 and Additional file 4 : Table S4.
Mice were i.v. injected with 2.5 × 106 cells (SEM-GFP-ffLuc, RS4;11-GFP-ffLuc or de novo B-ALL cells). Tumor cell engraftment was evaluated 7 days after injection via bioluminescence imaging (BLI) using the NightOWL LB 983 in vivo Imaging System (Berthold Technologies, Bad Wildbach, Germany) and Indigo software (Berthold Technologies, version 1.04). For detection animals were intraperitoneally injected with 4.5 mg D-Luciferin (GOLDbiotechnology, St. Louis, MO, USA), anesthetized with ketamine (75 mg/kg) and xylazine (5 mg/kg) and imaged in dorsal and ventral position (60 s exposure, 560 nm emission).
Therapeutic intervention and evaluation of induced effects
Mice were randomized based on sex, weight (19.1 g – 32.4 g), age (8–16 weeks) and tumor cell engraftment on d7. CX-4945 was dissolved in 0.9% saline containing 5% DMSO. DEC was dissolved in PBS. Animals were simultaneously treated with either vehicle (isotonic saline supplemented with 5% DMSO) or 50 mg/kg i.p. CX-4945 twice daily d7–12 based on previous dose finding studies. For combination studies 0.4 mg/kg DEC was applied i.p. once daily d7-d10. Results were compared to previously published DEC-treated animals [10]. Drug response was analyzed by BLI and peripheral blood (PB) flow cytometry (GFP+ or CD19-FITC+/CD45-PE+ (Becton Dickinson, Heidelberg, Germany)).
At d30 or when PB blast frequency reached 30% (PDX) mice were euthanized by cervical dislocation and leukemic blast frequency was analyzed in PB, BM and spleen. Means and standard deviation of BLI and blast frequency values of all mice of a study group were calculated. Student’s t-test was performed to compare study groups and values < 0.05 were considered significant. May-Grünwald Giemsa staining of PDX spleen cells was carried out as previously described and evaluated by EVOS xl core microscopy [21].
Results
CX-4945 affects CK2 activity, PI3K/AKT signaling and cell biology parameters
SEM and RS4;11 cells were incubated with 5 μM CX-4945 for up to 24 h. Quantification of western blot signals revealed most prominent reductions of phosphorylation of CK2 target sites (pS/pTDXE) [32] after 2 h (SEM) and 24 h (RS4;11) (Fig. 1a). Simultaneous analysis of PTEN, AKT and downstream signaling protein 4EBP1 also showed declined phosphorylation (Fig. 1b).
Fig. 1.

Effects of CX-4945 on CK2 activity and PI3K/AKT pathway phosphorylation in B-ALL cell lines. a CK2 activity in SEM and RS4;11 cells was assessed by western blot analysis of phosphorylated CK2 target sites following 2 h (SEM) or 24 h (RS4;11) incubation with CX-4945 (5 μM) (lower panel). For quantification Image Studio Lite software (Version 5.2) was used to determine signal intensities of phosphorylated CK2 target sites and total CK2 alpha 1 protein. Protein bands were normalized to the GAPDH band of the respective sample and the ratio of phosphorylated targets to total CK2A1 protein signal intensities was calculated. Means and SD of three independent experiments are depicted as bar charts in the upper panel. Individual ratios of one representative blot are stated below the respective bands. b Activity of PTEN, AKT and 4EBP1 proteins was analyzed by western blot of phosphorylated and total protein forms after 2 h (SEM) or 24 h (RS4;11) incubation with 5 μM CX-4945 (mean + SD; n = 3)
Compared to controls, proliferation and metabolic activity were significantly decreased in SEM, RS4;11, REH and NALM-6 after 72 h (p < 0.05) (Fig. 2a, b). Further, incubation with CX-4945 induced apoptosis with strongest effects in SEM and RS4;11 (Fig. 2c).
Fig. 2.

Biological effects of CK2 inhibition on B-ALL cell lines. a Proliferation was determined using trypan blue staining after 72 h incubation of increasing CX-4945 concentrations and compared to control cells set to 100% (mean + SD; n ≥ 2; * p < 0.05, ** p < 0.01, *** p < 0.005 for n ≥ 3 experiments). b The WST-1 assay was used to evaluate metabolic activity after 72 h incubation of increasing concentrations of CX-4945. Three technical replicates were performed for each sample (mean + SD; n ≥ 3; * p < 0.05, ** p < 0.01, *** p < 0.005). c The effect on apoptosis was measured by Annexin V-FITC/PI following 72 h CX-4945 incubation. Early apoptotic cells (Annexin V+/PI−) are located in the lower right quadrant while late apoptotic and necrotic cells (Annexin V+/PI+) are located in the upper right part. (n ≥ 2)
Combined inhibition of CK2 and DNMT3A increases effects synergistically
As CK2 is known to influence DNA methylation via DNMT3A phosphorylation CX-4945 was combined with DEC (0.1 μM) for up to 72 h. In DEC-sensitive cell lines SEM and NALM-6 simultaneous application of DEC and CX-4945 significantly reduced the metabolic activity to 33.6 and 33.5%, respectively compared to mono applications (Fig. 3a). The CX-4945 mono application-induced anti-metabolic effect was highest in DEC-resistant RS4;11 cells (37.8%) but addition of DEC did not further decrease metabolic activity. Potential synergistic effects of combined CK2 inhibition and HMA exposure were calculated with Bliss values above 0 indicating synergy. After 72 h, combined application of CX-4945 and DEC demonstrated synergistically reduced metabolic activity in SEM (0.194) and NALM-6 (0.240) but not in RS4;11 (− 0.027) cells. In contrast, no synergistic effect was detected analyzing proliferation. Similar results were obtained after 48 h of incubation (data not shown).
Fig. 3.

Effects of combined CK2 and DNMT3A inhibition in B-ALL cell lines. a The WST-1 metabolism assay was used to evaluate metabolic activity after 72 h incubation of CX-4945 (5 μM) and/or DEC (0.1 μM). Metabolic activity of control cells was set to 100%. Three technical replicates were performed for each sample (mean + SD; n ≥ 3; * p < 0.05, ** p < 0.01, *** p < 0.005). b Phosphorylation of PI3K/AKT pathway proteins was assessed by western blot analysis and subsequent quantification of bands as described before. For protein activity assessment the ratio between phosphorylated and total protein form was calculated and compared to control cells set to 100%. For CK2, phosphorylation of CK2 target sites was determined and compared to the total form of CK2 alpha (mean + SD after 24 h (SEM) or 2 h (RS4;11) incubation with 5 μM CX-4945 and/or 0.1 μM DEC; n = 3)
Synergistic effects are not evoked via PI3K/AKT signaling or intensified hypomethylation
Western blot analyses were performed to evaluate whether addition of DEC to CX-4945 would further influence PI3K/AKT signaling. While DEC alone had hardly any effect on protein phosphorylation combination of DEC and CX-4945 evoked results similar to CX-4945 alone (Fig. 3b). No additional PI3K or MAP kinase pathway regulation was observed (data not shown).
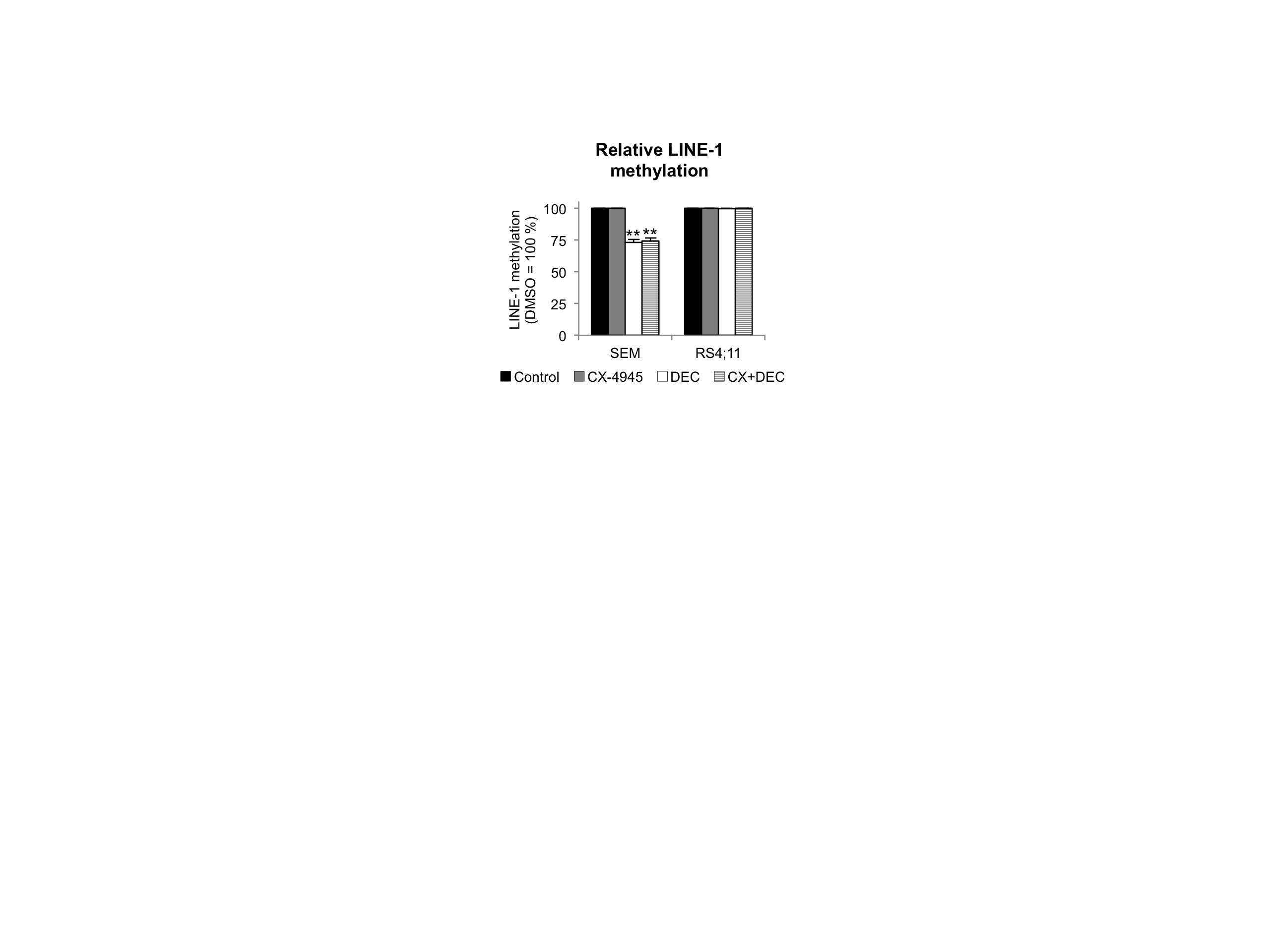
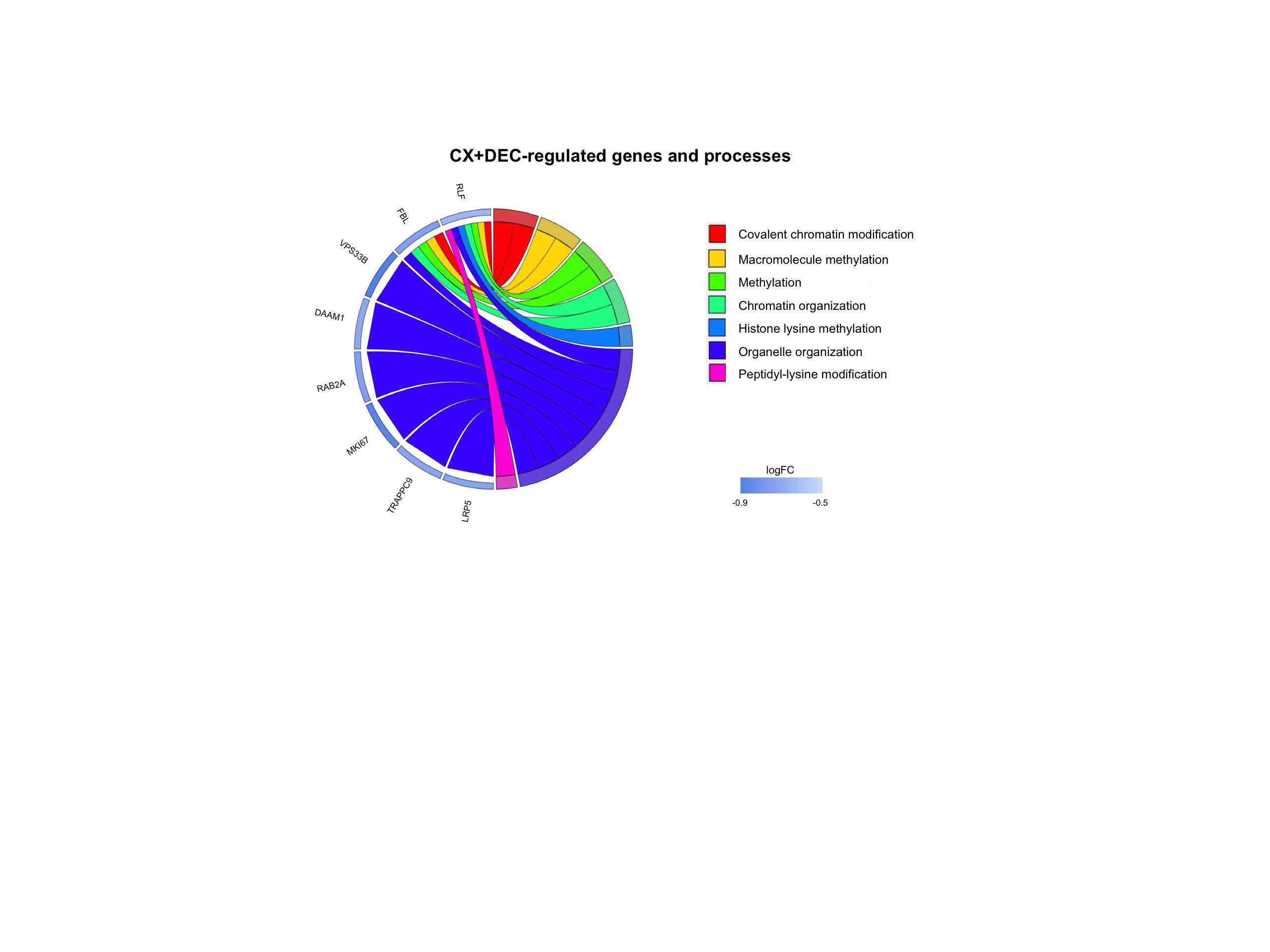
Changes in global methylation were assessed by LINE-1 retrotransposon MsqPCR and revealed no hypomethylating effect of CX-4945 (Additional file 5: Figure S1). To evaluate whether the observed synergistic anti-proliferative effect was evoked by potential gene-specific hypomethylation of tumor suppressor genes or signaling proteins genome-wide methylome analysis was performed. Compared to controls DEC reduced methylation of 1578 genes while the combination of both drugs induced hypomethylation in 927 genes (FC < 0.25). After CX-4945 incubation 54 genes were hypomethylated (Fig. 4a, Additional file 6: Table S5 and Additional file 7: Tables S6). Methylation of only 5 genes (BDNFOS, ZCCHC4, ABT1, BACH2, UBN1) was reduced by both mono applications as well as combined incubation. Among these were transcription factors ABT1 (activator of basal transcription 1) and BACH2 (basic leucine zipper transcription factor). CX-4945-hypomethylated genes mainly affected cellular process regulation, maturation and metabolism (Fig. 4b, c; Additional file 8: Table S7). The GO terms associated to genes regulated by combined CK2 inhibition and DEC exposure were chromatin modification and methylation (Additional file 9: Figure S2, Additional file 10: Table S8). Interestingly, there was marginal overlap between CX-4945, DEC or combined therapy affected processes (Fig. 4c) and none of the drugs changed the promoter methylation pattern of PTEN or CK2 (Additional file 11: Table S9).
Fig. 4.

Methylome analysis of SEM cells. a The Venn diagram shows the number of hypomethylated genes after 48 h incubation with 5 μM CX-4945, 0.1 μM DEC or both compared to DMSO. Methylation data was normalized using quantile normalization and only changes in methylation of at least 4-fold were considered relevant. b Hypomethylated genes were assigned to Gene Ontology (GO) Terms. The chord plot shows the association between the top 7 enriched GO terms and top 30 hypomethylated genes. Genes were further classified by their fold change compared to control cells (blue rectangle). CX-4945 incubation mainly influenced genes annotated by GO terms relating to metabolic processes. FBXO5 and RPS6KA2 have an association to all seven selected GO terms. c Average linking hierarchical clustering of the top-10 regulated biological processes of each treatment was performed and visualized in a heatmap based on statistical significance (−10log p-values)
Inhibition of CK2 alone and in combination with DEC delays tumor cell proliferation in vivo
The anti-leukemic activity of CX-4945 was investigated in vivo using orthotopic SEM-GFP-ffLuc- and RS4;11-GFP-ffLuc-derivded xenograft mouse models. These t(4;11)-harbouring cell lines were selected based on most promising results in vitro. Only healthy, age-, weight-, sex- and engraftment-matched animals were included in study groups. Therapeutic response was analyzed using longitudinal full body BLI and flow cytometric detection of GFP expression in PB, spleen and BM (Fig. 5). BLI signals directly correlate to the number of luciferase+ tumor blasts and are therefore used as a direct marker for tumor cell proliferation.
Fig. 5.

In vivo evaluation of CX-4945 and DEC. a Longitudinal observation of leukemic blast proliferation in four representative SEM-ffluc xenograft mice is displayed following vehicle (NaCl supplemented with 5% DMSO, BID d7–12), CX-4945 (50 mg/kg body weight (BW), BID d7–12), DEC (0.4 mg/kg BW, d7–10) or CX-4945 plus DEC treatment over a period of 30 days. Increasing luminescence (ph/s) is proportional to proliferation of luciferase-expressing SEM-ffluc cells. b Quantification of full body bioluminescence (ph/s) after treatment in SEM-ffluc and RS4;11-ffluc animals was performed by adding total luminescence signals of dorsal and ventral imaging. Significant differences are indicated for combined treatment of CX-4945 and DEC vs CX-4945 mono application. CX-4945 alone induced no significant change in full body bioluminescence compared to controls (mean + SD; * p < 0.05, ** p < 0.01, *** p < 0.005). Blast frequency in PB was assessed by flow cytometry measurement of GFP+ SEM-ffluc and GFP+ RS4;11-ffluc cells over a period of 30 days. Significant differences are indicated for combined therapy vs CX-4945 mono application (mean + SD; * p < 0.05, *** p < 0.005). Infiltration of leukemic blasts was analyzed in BM and spleen after euthanasia on day 30. The amount of GFP+ SEM-ffluc and RS4;11-ffluc blasts was determined by flow cytometry (mean + SD; * p < 0.05, *** p < 0.005)
In SEM-derived xenografts CX-4945 delayed B-ALL cell proliferation until day 24 (CX-4945 4.3 ± 2.2 × 109 ph/s; control 5.5 ± 0.8 × 109 ph/s) before reaching BLI signal levels similar to the controls on day 30. Animals also showed reduced PB blast counts throughout the entire examination period (d30, CX-4945 12.9 ± 7.2%; control 20.6 ± 9.5%). DEC alone induced stronger effects than CX-4945 mono therapy with significantly decreased BLI signals (d24, 2.1 ± 1.5 × 109 ph/s) as well as PB, spleen and BM blast frequencies (Fig. 5b). Combined CX-4945 and DEC therapy was more effective than DEC alone and significantly decreased bioluminescence signals and blast frequency compared to CX-4945 mono application (d24, 1.3 ± 1.3 × 109 ph/s). Further, leukemic infiltration of BM and spleen was significantly reduced compared to controls.
CX-4945 had no influence on RS4;11-derived xenografts (Fig. 5b). Addition of DEC resulted in significantly reduced B-ALL cell proliferation indicated by delayed BLI signal increase (d30, control 4.7 ± 0.4 × 109 ph/s; CX-4945 4.8 ± 0.6 × 109 ph/s; DEC 1.8 ± 0.2 × 109 ph/s; CX + DEC 1.4 ± 0.5 × 109 ph/s) as well as PB, BM and spleen blast counts.
CX-4945 in combination with DEC influences patient-derived B-ALL cell proliferation in vivo
To increase comparability of animal studies and human leukemia patients, xenografts of three B-ALL patients were established in NSG mice and treated with CX-4945, DEC or in combination until control animals reached a PB blast amount of 30%. To ensure comparability between cell line- and patient-derived xenografts, only MLL-rearranged samples were engrafted.
Treatment with CX-4945 did not decrease leukemic blasts in BM, spleen or PB in PDX mice (Fig. 6a). Combination with DEC as well as DEC incubation alone resulted in a strong reduction of tumor load in patient 0152-derived mice. Interestingly, basal AKT activity in spleen cells was reduced in patient 0152 xenografts compared to other PDX samples (Fig. 6b). Phosphorylation of PTEN and other PI3K pathway molecules was similar in all samples (data not shown). Cytospin preparations of spleen cells revealed no changes in blast morphology after CX-4945 or combined therapy (Fig. 6c, inset). However, PB blast frequency was decreased combinatory treatment (control 36.0%; CX + DEC 2.0%) (Fig. 6a).
Fig. 6.

Evaluation of CX-4945 and DEC in patient-derived xenograft mice. a Xenograft mice of three different B-ALL patients (0122, 0152, 0159; patient characteristics were reported previously) were treated with vehicle (NaCl supplemented with 5% DMSO; BID d7–12), CX-4945 (50 mg/kg BW, BID, d7–12) or CX-4945 plus DEC (0.4 mg/kg BW, d7–10) and euthanized when PB blast frequency in controls reached 25%. The amount of patient-derived B-ALL tumor cells in BM, spleen and PB was assessed by CD45-FITC/CD19-PE staining and subsequent flow cytometry (mean + SD; vehicle: n = 4–5 animals per patient, treatment groups: n = 1–2 animals per patient). b AKT and PTEN activity status was analyzed in PDX mice spleen cells by western blot of phosphorylated and total protein forms. c Cytospin preparations of cells isolated from spleens of patient 0152 derived mice. Slides were stained with Pappenheim and evaluated using the EVOS xl core microscope and 100x magnification)
In patient 0159-derived mice combined application of CX-4945 and DEC lowered spleen infiltration while the tumor burden in BM and PB remained constant. Only minor changes in leukemic blast frequency were observed in animals bearing patient 0122-derived tumors.
Discussion
In addition to mutations and structural changes of DNA several mechanisms and pathways are involved in B-ALL pathogenesis including posttranslational modifications and aberrant methylation. Thus combined targeting of tumor-initiating pathways is advantageous and currently evaluated in a variety of approaches in preclinical and clinical settings. Although CK2 inhibition and hypomethylation of tumor suppressor gene promoters show promising results in hematological neoplasms the effect of their combined application has not yet been addressed. Simultaneous CK2 inactivation and hypomethylation may shed light on potential crosstalk between both mechanisms and potential synergistic anti-proliferative effects on B-ALL cells.
In B-ALL increased promoter hypermethylation is a typical feature observed in relapse, often silencing tumor suppressor genes such as PTEN [5, 33]. PTEN is further inhibited by CK2-mediated phosphorylation. CK2 is frequently upregulated in hematological neoplasms and a key regulator of diverse cellular mechanisms including cell cycle, apoptosis and transcription [14, 34, 35].
In our study we found that CX-4945 inhibits CK2 resulting in decreased phosphorylation of PI3K pathway signaling proteins and subsequent reduction of B-ALL cell proliferation and metabolic activity as well as increased apoptosis rates. Other studies of CK2 inhibition on B-ALL cell lines also found PI3K pathway downregulation and reduced cell viability accompanied by apoptosis induction [6, 15, 36, 37]. Methylome analysis also revealed a strong influence of CX-4945 on the methylation of metabolism-related genes, matching the data obtained by WST-1 metabolic activity assay. Results within an in vivo human B-ALL xenograft model matched in vitro anti-proliferative results. Bioluminescence imaging also revealed delayed leukemic proliferation and reduced blast frequency in SEM-derived mice. Whether these effects were due to cytotoxicity or due to cell death induction has to be determined in further experiments. Matching our in vitro data Song et al. demonstrated reduced leukemic progression and an improved overall survival in a NALM-6-derived xenograft model after 10 days of treatment using higher concentrations than in our study [15]. Other groups also discovered anti-proliferative potential of CK2 inhibition in CLL [38, 39] and T-ALL in vivo studies [40].
We then added DEC to CX-4945 treatment in order to identify potential enhanced pathway and methylome modulation. We found that combined application of CX-4945 and DEC acted synergistically in vitro but while PI3K pathway phosphorylation mirrors CX-4945 mono treatment, methylome patterns of combined application show no overlap with either single application. This suggests that a previously unconsidered mechanism is responsible for the observed synergistic anti-proliferative effects. We hypothesized that hypomethylation and thus activation of PTEN might be responsible for decreased proliferation and metabolic activity but could not detect significantly lower methylation or phosphorylation after combined therapy. However, the transcription factor BACH2 is a potential linker between CX-4945 and DEC-induced effects. We found that BACH2 is hypomethylated by CX-4945, DEC and combined treatment while Ando et al. demonstrated that it can be phosphorylated by PI3K pathway proteins [41]. Regulating B cell development at the pre-B cell receptor checkpoint BACH2 functions as a tumor suppressor in B-ALL [42–44]. Further, Ge et al. recently demonstrated that both BACH2 and its counterpart BCL6 interact with CK2 target Ikaros (IKZF1) resulting in increased BACH2 expression and reduced B-ALL proliferation [45]. Epigenetic modulation of the BACH2/BCL6 axis may therefore be an additional mechanism to regulate tumor cell apoptosis and proliferation. However, the importance of CX-4945-mediated loss of CK2 kinase activity for the observed effects is not yet clear as DEC mono therapy was not significantly inferior to combined CX-4945 and DEC treatment during the observation period in vivo. Prolonged therapeutic intervention or higher CX-4945 doses might be beneficial for the treatment outcome as stated above. It further seems possible that, although no significant synergistic anti-proliferative effect was observed, CX-4945 alone or in combination with DEC influences biochemical, metabolic or other signaling pathways in tumor blasts.
As the PI3K/AKT signaling pathway can be aberrantly activated in B-ALL patients we further tested CX-4945 in three B-ALL patient-derived xenograft models all harboring a t(4;11) translocation [6. Independently of patients’ PTEN and AKT phosphorylation status or hotspot mutations no satisfactory anti-leukemic effect was achieved. This matches the findings of Prins et al. who incubated 56 primary pediatric and adult B-ALL samples with increasing concentrations of CX-4945 and found reduced tumor cell viability in only 3 (5.2%) samples compared to much higher response rates in T-ALL, CLL and lymphoma [38, 46]. In contrast Song et al. found a reduced number of blasts following CX-4945 therapy in high risk B-ALL xenografts. [15] This discrepancy might be explained by the fact that only patients with a t(4;11) were examined or by different application schemes. While Song et al. treated mice with 100 mg/kg daily for 22 days and analyzed organs directly after the last application we used a shorter application regime (50 mg/kg twice daily for 6 days) and evaluated the blast frequency after 30 days or more. It thus appears that CX-4945 delays leukemic blast proliferation during therapy while this effect diminishes when the drug is removed. This hypothesis also matches our findings from cell line-derived B-ALL xenograft observations. Evaluation of pharmacokinetic parameters demonstrated long half-life of CX-4945 in rats (14.7 h and 10.9 h for intravenous and oral administration, respectively) [47] while Siddiqui-Jain et al. found high clearance rates accompanied by rapidly decreasing plasma concentrations in mice [48]. Further pharmacokinetic evaluation is therefore necessary. Alternative application schemes might be beneficial and should be addressed in future studies.
Conclusion
Taken together we herein demonstrate that the novel strategy of combined CK2 inhibition and DEC application significantly decreases B-ALL proliferation. In this study we evaluated PI3K/AKT signaling, methylome patterns and therapeutic response in vivo after single and combined application. Although CK2 is known to interact with both, PI3K/AKT and DNMT3A-mediated methylation neither pathway can be made fully responsible for the observed synergistic effects [12]. Therefore, further experiments need to be conducted to identify the underlying mechanism of the demonstrated synergistic anti-tumor potential.
Additional files
Table S1. List of antibodies used for Western blot analyses. (DOCX 14 kb)
Table S2. List of primers used for LINE-1 methylation analysis. (DOCX 13 kb)
Table S3. List of cell line derived xenograft mice used (DOCX 16 kb)
Table S4. List of patient derived xenograft mice used (DOCX 16 kb)
{kind=link}
Figure S1. Evaluation of CX-4945- and DEC-induced effects on global methylation in SEM and RS4;11 cells. Global methylation was analyzed using methylation specific qPCR of the LINE-1 retrotransposon after 48 h incubation of CX-4945 (5 μM) and/or DEC (0.1 μM). LINE-1 methylation of control cells was set to 100 % (three technical replicates per sample; mean + SD; n=3; *p<0.05, **p<0.01, ***p<0.005). (JPG 16 kb)
Table S5. List of genes that were hypomethylated after CX-4945 incubation (FC<0.25) compared to controls. (XLSX 11 kb)
Table S6. List of genes that were hypomethylated after combined CX-4945 and DEC incubation (FC<0.25) compared to controls. (XLSX 48 kb)
Table S7. List of Biological processes regulated by CX-4945 incubation. (XLSX 15 kb)
{kind=link}
Figure S2. Genes and processes highly regulated by combined CX-4945 and DEC incubation. Hypomethylated genes were assigned to Gene Ontology (GO) Terms. The chord plot shows the association between the top 7 enriched GO terms and top 30 hypomethylated genes. Genes are further classified by their fold change compared to control cells (blue rectangle). (JPG 41 kb)
Table S8. List of Biological processes regulated by combined CX-4945 and DEC incubation. (XLSX 13 kb)
Table S9. Influence of CX and DEC on PTEN and CK2 promoter methylation beta values and respective fold changes (FC). (DOCX 14 kb)
Acknowledgements
We thank Irmela Jeremias and Binje Vick for lentiviral transduction of ALL cell lines and excellent collaboration.
We thank the microarray unit of the DKFZ Genomics and Proteomics Core Facility for providing the Illumina Human Methylation arrays and related services.
Funding
This work was supported by FORUN grant of Rostock University Medical Center (CR). AR received a scholarship of the German Federal State of Mecklenburg-Vorpommern, Germany.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Raw and processed whole-genome methylation data can be retrieved from Gene Expression Omnibus (GSE110454).
Abbreviations
- ALL
Acute lymphoblastic leukemia
- BLI
Bioluminescence imaging
- BM
Bone marrow
- CK2
Casein Kinase II
- DEC
Decitabine
- FC
Fold change
- ffluc
Firefly luciferase
- GFP
Green fluorescent protein
- GO
Gene ontology
- HMA
Hypomethylating agent
- MsqPCR
Methylation specific qPCR
- NSG
NOD scid gamma
- PB
Peripheral blood
- PDX
Patient-derived xenograft
Authors’ contributions
AR participated in study design, performed in vitro experiments, performed in vivo experiments, data analysis/interpretation, analyzed sequencing data, wrote the manuscript. CR designed study, established xenograft model, data analysis/interpretation, participated in writing the manuscript. SS performed in vivo experiments. CK performed in vitro experiments. GK provided technical assistance for in vivo experiments. AS provided technical assistance for in vitro experiments. MH analyzed methylome data. YG analyzed methylome data. GF analyzed methylome data, critically revised manuscript. BV participated in bioluminescence project design, participated in xenograft model establishment. HME designed sequencing experiments, critically revised manuscript. CJ designed study, participated in paper drafting and finalization. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The study was performed in accordance to the Declaration of Helsinki and the local ethical standards of the Rostock University Medical Center.
All animal experiments were approved by the review board of the federal state of Mecklenburg-Vorpommern, Germany (reference number: LALLF MV/7221.3–1.1-002/15).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Anna Richter, Email: Anna.Richter@med.uni-rostock.de.
Catrin Roolf, Email: Catrin.Roolf@med.uni-rostock.de.
Mohamed Hamed, Email: Mohamed.hamed@uni-rostock.de.
Yvonne Saara Gladbach, Email: Yvonne.Gladbach@uni-rostock.de.
Sina Sender, Email: Sina.Sender@med.uni-rostock.de.
Christoph Konkolefski, Email: Christoph.Konkolefski@uni-rostock.de.
Gudrun Knübel, Email: Gudrun.Knuebel@med.uni-rostock.de.
Anett Sekora, Email: Anett.Sekora@med.uni-rostock.de.
Georg Fuellen, Email: Fuellen@uni-rostock.de.
Brigitte Vollmar, Email: Brigitte.Vollmar@med.uni-rostock.de.
Hugo Murua Escobar, Email: Hugo.Murua.Escobar@med.uni-rostock.de.
Christian Junghanss, Phone: +49-381-494-7420, Email: Christian.Junghanss@med.uni-rostock.de.
References
- 1.Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–764. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 2.Bujko M, Musialik E, Olbromski R, Przestrzelska M, Libura M, Pastwińska A, et al. Repetitive genomic elements and overall DNA methylation changes in acute myeloid and childhood B-cell lymphoblastic leukemia patients. Int J Hematol. 2014;100:79–87. doi: 10.1007/s12185-014-1592-0. [DOI] [PubMed] [Google Scholar]
- 3.Neri LM, Cani A, Martelli a M, Simioni C, Junghanss C, Tabellini G, et al. targeting the PI3K/Akt/mTOR signaling pathway in B-precursor acute lymphoblastic leukemia and its therapeutic potential. Leukemia. Nat Publ Group; 2014;28:739–748. [DOI] [PubMed]
- 4.Milella M, Falcone I, Conciatori F, Cesta Incani U, Del Curatolo A, Inzerilli N, et al. PTEN: Multiple Functions in Human Malignant Tumors. Front Oncol. Frontiers Media SA; 2015;5:24. [DOI] [PMC free article] [PubMed]
- 5.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 6.Gomes AM, Soares MVD, Ribeiro P, Caldas J, Póvoa V, Martins LR, et al. Adult B-cell acute lymphoblastic leukemia cells display decreased PTEN activity and constitutive hyperactivation of PI3K/Akt pathway despite high PTEN protein levels. Haematologica Ferrata Storti Foundation. 2014;99:1062–1068. doi: 10.3324/haematol.2013.096438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Correia NC, Gírio A, Antunes I, Martins LR, Barata JT. The multiple layers of non-genetic regulation of PTEN tumour suppressor activity. Eur J Cancer. 2014;50:216–225. doi: 10.1016/j.ejca.2013.08.017. [DOI] [PubMed] [Google Scholar]
- 8.Nishioka C, Ikezoe T, Yang J, Udaka K, Yokoyama A. Imatinib causes epigenetic alterations of PTEN gene via upregulation of DNA methyltransferases and polycomb group proteins. Blood Cancer J Nat Publ Group; 2011;1:e48. [DOI] [PMC free article] [PubMed]
- 9.Stumpel DJPM, Schneider P, Van Roon EHJ, Boer JM, De Lorenzo P, Valsecchi MG, et al. Specific promoter methylation identifies different subgroups of MLL-rearranged infant acute lymphoblastic leukemia, influences clinical outcome, and provides therapeutic options. Blood. 2009;114:5490–5498. doi: 10.1182/blood-2009-06-227660. [DOI] [PubMed] [Google Scholar]
- 10.Roolf C, Richter A, Konkolefski C, Knuebel G, Sekora A, Krohn S, et al. Decitabine demonstrates antileukemic activity in B cell precursor acute lymphoblastic leukemia with MLL rearrangements. J Hematol Oncol. 2018;11:62. doi: 10.1186/s13045-018-0607-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang L, Rau R, Goodell MA. DNMT3A in haematological malignancies. Nat Rev Cancer. 2015;15:152–165. doi: 10.1038/nrc3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deplus R, Blanchon L, Rajavelu A, Boukaba A, Defrance M, Luciani J, et al. Regulation of DNA methylation patterns by CK2-mediated phosphorylation of Dnmt3a. Cell Rep. 2014;8:743–753. doi: 10.1016/j.celrep.2014.06.048. [DOI] [PubMed] [Google Scholar]
- 13.Gowda C, Soliman M, Kapadia M, Ding Y, Payne K, Dovat S. Casein Kinase II (CK2), Glycogen Synthase Kinase-3 (GSK-3) and Ikaros mediated regulation of leukemia. Adv Biol Regul. Elsevier Ltd; 2017;Aug:16–25. [DOI] [PMC free article] [PubMed]
- 14.Chua MMJ, Ortega CE, Sheikh A, Lee M, Abdul-rassoul H, Hartshorn KL, et al. CK2 in Cancer : cellular and biochemical mechanisms and potential therapeutic target. Pharmaceuticals. 2017;10. [DOI] [PMC free article] [PubMed]
- 15.Song C, Gowda C, Pan X, Ding Y, Tong Y, Tan BH, et al. Targeting casein kinase II restores Ikaros tumor suppressor activity and demonstrates therapeutic efficacy in high-risk leukemia. Blood. 2015;126:1813–1822. doi: 10.1182/blood-2015-06-651505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barata JT. The impact of PTEN regulation by CK2 on PI3K-dependent signaling and leukemia cell survival. Adv Enzym Regul. 2011;51:37–49. doi: 10.1016/j.advenzreg.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 17.Chon HJ, Bae KJ, Lee Y, Kim J. The casein kinase 2 inhibitor, CX-4945, as an anti-cancer drug in treatment of human hematological malignancies. Front Pharmacol Frontiers. 2015;6. [DOI] [PMC free article] [PubMed]
- 18.Agarwal M, Nitta RT, Li G. Casein Kinase 2: a novel player in glioblastoma therapy and cancer stem cells. J Mol Genet Med NIH Public Access; 2013;8. [DOI] [PMC free article] [PubMed]
- 19.Gray GK, McFarland BC, Rowse AL, Gibson SA, Benveniste EN, Gray GK, et al. Therapeutic CK2 inhibition attenuates diverse prosurvival signaling cascades and decreases cell viability in human breast cancer cells. Oncotarget Impact Journals. 2014;5:6484–6496. doi: 10.18632/oncotarget.2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buontempo F, McCubrey JA, Orsini E, Ruzzene M, Cappellini A, Lonetti A, et al. Therapeutic targeting of CK2 in acute and chronic leukemias. Leukemia Nature Publishing Group. 2018;32:1–10. doi: 10.1038/leu.2017.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kretzschmar C, Roolf C, Langhammer T-S, Sekora A, Pews-Davtyan A, Beller M, et al. The novel arylindolylmaleimide PDA-66 displays pronounced antiproliferative effects in acute lymphoblastic leukemia cells. BMC Cancer. BioMed Central; 2014;14:71. [DOI] [PMC free article] [PubMed]
- 22.Foucquier J, Guedj M. Analysis of drug combinations: current methodological landscape. Pharmacol Res Perspect. 2015;3:1–11. doi: 10.1002/prp2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miousse IR, Koturbash I. The Fine LINE: Methylation drawing the Cancer landscape. Biomed Res Int 2015;2015:131547. [DOI] [PMC free article] [PubMed]
- 24.Bolstad B. Probe Level Quantile Normalization of High Density Oligonucleotide Array Data. 2001:1–8.
- 25.Fortin JP, Triche TJ, Hansen KD. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics. 2017;33:558–560. doi: 10.1093/bioinformatics/btw691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 27.Hamed M, Trumm J, Spaniol C, Sethi R, Irhimeh MR, Fuellen G, et al. Linking hematopoietic differentiation to co-expressed sets of pluripotency-associated and imprinted genes and to regulatory microRNA-transcription factor motifs. PLoS One. 2017;12:1–20. doi: 10.1371/journal.pone.0166852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ihaka R, Gentleman R. R: a language for data analysis and graphics. J Comput Graph Stat. 1996;5:299–314. [Google Scholar]
- 29.Walter W, Sánchez-Cabo F, Ricote M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics. 2015;31:2912–2914. doi: 10.1093/bioinformatics/btv300. [DOI] [PubMed] [Google Scholar]
- 30.Terziyska N, Castro Alves C, Groiss V, Schneider K, Farkasova K, Ogris M, et al. In vivo imaging enables high resolution preclinical trials on patients’ leukemia cells growing in mice. PLoS One. 2012;7:e52798. doi: 10.1371/journal.pone.0052798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Townsend EC, Murakami MA, Christodoulou A, Christie AL, Köster J, DeSouza TA, et al. The public repository of xenografts enables discovery and randomized phase II-like trials in mice. Cancer Cell. 2016;29:574–586. doi: 10.1016/j.ccell.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meggio F. Pinna L a. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003;17:349–368. doi: 10.1096/fj.02-0473rev. [DOI] [PubMed] [Google Scholar]
- 33.Hogan LE, Meyer JA, Yang J, Wang J, Wong N, Yang W, et al. Integrated genomic analysis of relapsed childhood acute lymphoblastic leukemia reveals therapeutic strategies. Blood. 2011;118:5218–5226. doi: 10.1182/blood-2011-04-345595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rabalski AJ, Gyenis L, Litchfield DW. Molecular pathways: emergence of protein Kinase CK2 (CSNK2) as a potential target to inhibit survival and DNA damage response and repair pathways in Cancer cells. Clin Cancer Res. 2016;22:2840–2847. doi: 10.1158/1078-0432.CCR-15-1314. [DOI] [PubMed] [Google Scholar]
- 35.Pizzi M, Piazza F, Agostinelli C, Fuligni F, Benvenuti P, Mandato E, et al. Protein kinase CK2 is widely expressed in follicular, Burkitt and diffuse large B-cell lymphomas and propels malignant B-cell growth. Oncotarget. 2015;6:6544–6552. doi: 10.18632/oncotarget.3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buontempo F, Orsini E, Lonetti A, Cappellini A, Chiarini F, Evangelisti C, et al. Synergistic cytotoxic effects of bortezomib and CK2 inhibitor CX-4945 in acute lymphoblastic leukemia: turning off the prosurvival ER chaperone BIP/Grp78 and turning on the pro-apoptotic NF-κB. Oncotarget. 2015. [DOI] [PMC free article] [PubMed]
- 37.Mishra S, Pertz V, Zhang B, Kaur P, Shimada H, Groffen J, et al. Treatment of P190 Bcr/Abl lymphoblastic leukemia cells with inhibitors of the serine/threonine kinase CK2. Leukemia. 2007;21:178–180. doi: 10.1038/sj.leu.2404460. [DOI] [PubMed] [Google Scholar]
- 38.Martins LR, Mel a, Antunes I, Cardoso BA, Stansfield R, S Bertilaccio MT, et al. activity of the clinical-stage CK2-specific inhibitor CX-4945 against chronic lymphocytic leukemia. Leukemia. 2013;28:179–182. [DOI] [PubMed]
- 39.Martins LR, Lúcio P, Melao A, Cardoso BA, Stansfield R, Drygin D, et al. In vitro and in vivo targeting of chronic lymphocytic leukemia using CX-4945, a clinical-stage CK2-specific inhibitor. Blood American Society of Hematology; 2012;120:2897–2897.
- 40.Buontempo F, Orsini E, Martins LR, Antunes I, Lonetti A, Chiarini F, et al. Cytotoxic activity of the casein kinase 2 inhibitor CX-4945 against T-cell acute lymphoblastic leukemia: targeting the unfolded protein response signaling. Leukemia Macmillan Publishers Limited. 2014;28:543–553. doi: 10.1038/leu.2013.349. [DOI] [PubMed] [Google Scholar]
- 41.Ando R, Shima H, Tamahara T, Sato Y, Watanabe-Matsui M, Kato H, et al. The transcription factor bach2 is phosphorylated at multiple sites in murine B cells but a single site prevents its nuclear localization. J Biol Chem. 2016;291:1826–1840. doi: 10.1074/jbc.M115.661702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swaminathan S, Duy C, Müschen M. BACH2-BCL6 balance regulates selection at the pre-B cell receptor checkpoint. Trends Immunol. 2014;35:131–137. doi: 10.1016/j.it.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ye BH, Mai Y. A Bach2 link between pre-B cell receptor checkpoint and pre-B cell ALL. Cancer Cell Elsevier. 2013;24:282–284. doi: 10.1016/j.ccr.2013.08.017. [DOI] [PubMed] [Google Scholar]
- 44.Swaminathan S, Huang C, Geng H, Chen Z, Harvey R, Kang H, et al. BACH2 mediates negative selection and p53-dependent tumor suppression at the pre-B cell receptor checkpoint. Nat Med. 2013;19:1014–1022. doi: 10.1038/nm.3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ge Z, Zhou X, Gu Y, Han Q, Li J, Chen B, et al. Ikaros regulation of the BCL6 / BACH2 axis and its clinical relevance in acute lymphoblastic leukemia. Oncotarget. 2017;8:8022–8034. doi: 10.18632/oncotarget.14038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prins RC, Burke RT, Tyner JW, Druker BJ, Loriaux MM, Spurgeon SE. CX-4945, a selective inhibitor of casein kinase-2 (CK2), exhibits anti-tumor activity in hematologic malignancies including enhanced activity in chronic lymphocytic leukemia when combined with fludarabine and inhibitors of the B-cell receptor pathway. Leukemia. 2013;27:2094–2096. doi: 10.1038/leu.2013.228. [DOI] [PubMed] [Google Scholar]
- 47.Son YH, Song JS, Kim SH, Kim J. Pharmacokinetic characterization of CK2 inhibitor CX-4945. Arch Pharm Res. 2013;36:840–845. doi: 10.1007/s12272-013-0103-9. [DOI] [PubMed] [Google Scholar]
- 48.Siddiqui-Jain A, Drygin D, Streiner N, Chua P, Pierre F, O’Brien SE, et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res American Association for Cancer Research. 2010;70:10288–10298. doi: 10.1158/0008-5472.CAN-10-1893. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of antibodies used for Western blot analyses. (DOCX 14 kb)
Table S2. List of primers used for LINE-1 methylation analysis. (DOCX 13 kb)
Table S3. List of cell line derived xenograft mice used (DOCX 16 kb)
Table S4. List of patient derived xenograft mice used (DOCX 16 kb)
Figure S1. Evaluation of CX-4945- and DEC-induced effects on global methylation in SEM and RS4;11 cells. Global methylation was analyzed using methylation specific qPCR of the LINE-1 retrotransposon after 48 h incubation of CX-4945 (5 μM) and/or DEC (0.1 μM). LINE-1 methylation of control cells was set to 100 % (three technical replicates per sample; mean + SD; n=3; *p<0.05, **p<0.01, ***p<0.005). (JPG 16 kb)
Table S5. List of genes that were hypomethylated after CX-4945 incubation (FC<0.25) compared to controls. (XLSX 11 kb)
Table S6. List of genes that were hypomethylated after combined CX-4945 and DEC incubation (FC<0.25) compared to controls. (XLSX 48 kb)
Table S7. List of Biological processes regulated by CX-4945 incubation. (XLSX 15 kb)
Figure S2. Genes and processes highly regulated by combined CX-4945 and DEC incubation. Hypomethylated genes were assigned to Gene Ontology (GO) Terms. The chord plot shows the association between the top 7 enriched GO terms and top 30 hypomethylated genes. Genes are further classified by their fold change compared to control cells (blue rectangle). (JPG 41 kb)
Table S8. List of Biological processes regulated by combined CX-4945 and DEC incubation. (XLSX 13 kb)
Table S9. Influence of CX and DEC on PTEN and CK2 promoter methylation beta values and respective fold changes (FC). (DOCX 14 kb)
Data Availability Statement
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Raw and processed whole-genome methylation data can be retrieved from Gene Expression Omnibus (GSE110454).