

Keywords: nerve regeneration, Alzheimer's disease, synaptic plasticity, hippocampus, learning and memory, long-term potentiation, β amyloid, glutamate receptor, synaptic strength, neural regeneration
Abstract
Mounting evidence suggests that synaptic plasticity provides the cellular biological basis of learning and memory, and plasticity deficits play a key role in dementia caused by Alzheimer’s disease. However, the mechanisms by which synaptic dysfunction contributes to the pathogenesis of Alzheimer’s disease remain unclear. In the present study, Alzheimer’s disease transgenic mice were used to determine the relationship between decreased hippocampal synaptic plasticity and pathological changes and cognitive-behavioral deterioration, as well as possible mechanisms underlying decreased synaptic plasticity in the early stages of Alzheimer’s disease-like diseases. APP/PS1 double transgenic (5XFAD; Jackson Laboratory) mice and their littermates (wild-type, controls) were used in this study. Additional 6-week-old and 10-week-old 5XFAD mice and wild-type mice were used for electrophysiological recording of hippocampal dentate gyrus. For 10-week-old 5XFAD mice and wild-type mice, the left hippocampus was used for electrophysiological recording, and the right hippocampus was used for biochemical experiments or immunohistochemical staining to observe synaptophysin levels and amyloid beta deposition levels. The results revealed that, compared with wild-type mice, 6-week-old 5XFAD mice exhibited unaltered long-term potentiation in the hippocampal dentate gyrus. Another set of 5XFAD mice began to show attenuation at the age of 10 weeks, and a large quantity of amyloid beta protein was accumulated in hippocampal cells. The location of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor and N-methyl-D-aspartic acid receptor subunits in synaptosomes was decreased. These findings indicate that the delocalization of postsynaptic glutamate receptors and an associated decline in synaptic plasticity may be key mechanisms in the early onset of Alzheimer’s disease. The use and care of animals were in strict accordance with the ethical standards of the Animal Ethics Committee of Capital Medical University, China on December 17, 2015 (approval No. AEEI-2015-182).
Chinese Library Classification No. R453; R364
Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disease among older people. AD is defined pathologically by extracellular senile plaques of amyloid beta (Aβ) deposition (Braak and Braak, 1991; Thal et al., 2002; Huang and Mucke, 2012; Koehler and Williams, 2018; Zhang et al., 2018), and intracellular neurofibrillary tangles caused by hyperphosphorylation of Tau. Although the precise mechanisms underlying AD remain unclear, the “Aβ cascade hypothesis” (Hardy et al., 1991; Selkoe, 1991; Hardy and Selkoe, 2002) has been proposed to explain the pathogenesis of AD in recent years. This hypothesis suggests that aggregated Aβ may cause synaptic dysfunction, inflammation, and dementia (Selkoe and Hardy, 2016). However, in recent years, new drugs targeting Aβ for the treatment of AD have failed in clinical trials (Hitman et al., 1989; Small and Duff, 2008; Herrup, 2015), facilitating an effort to identify earlier treatable key events in the context of AD pathology. However, it remains unclear which mechanisms cause the onset of AD and boost its progression.
Progressive irreversible memory impairment is a feature of AD, primarily resulting from deficits in synaptic plasticity (Terry et al., 1991; Sheng., 2012; He et al., 2018), and is typically evaluated by long-term potentiation (LTP) and long-term depression (Briggs et al., 2017) in memory formation. In AD, LTP suppression and long-term depression facilitation were found to accompany memory loss, and are recognized as pathophysiological bases of dementia (Fitzjohn et al., 2001; Palop et al., 2007). Other studies reported that human-derived soluble Aβ oligomers inhibited LTP expression and attenuated learning and memory ability in rodents (Walsh et al., 2002; Shankar et al., 2008). While the relationship between Aβ and synaptic plasticity and cognitive behavior has been extensively studied (Walsh et al., 2002; Tsai et al., 2004; Cleary et al., 2005; Shankar et al., 2008; Li et al., 2011; Marsh and Alifragis, 2018), it would be useful to elucidate the possible molecular mechanisms involved in these events in the context of AD.
The development of various animal models of AD has provided a powerful tool for studying its pathogenesis and therapeutic basis. The 5XFAD transgenic mouse carrying human mutant APP695 with Swedish (K670N, M671L), Florida (I716V), and London (V717I) familial Alzheimer’s disease (FAD) mutations along with human PS1 harboring two FAD mutations, M146L and L286V (Oakley et al., 2006) is widely used in researching AD pathogenesis and therapeutic strategies. In the past several years, transgenic mouse model studies have produced mounting evidence for plaque- or soluble Aβ-related behavioral impairment and synaptic dysfunction (Wu et al., 2014; Iaccarino et al., 2016; Choi et al., 2018; Griñán et al., 2018). However, it may be valuable to explore the intracellular Aβ effects on synaptic function, since intracellular Aβ accumulation occurs before senile plaque formation (Oakley et al., 2006). In a previous study, we found that 5XFAD mice did not show spatial memory or learning impairment in the routine Morris water maze test until the age of 4.5 months, at which time Aβ-positive plaques had been developing extracellularly in the local field (Wang et al., 2014). These findings suggest that earlier AD-like phenomena occur in the AD mouse brain, which could potentially be discovered using sensitive measures. In the present study, we sought to detect an early time-point of decline in synaptic function in an AD-like process. We found that the decline in synaptic plasticity occurred ahead of extracellular Aβ accumulation in the hippocampus of 5XFAD transgenic mice at 10 weeks of age, while behavioral impairment was undetectable until 14 weeks of age, using a reversal learning protocol. Here, we propose that synaptic dysfunction might be a leading event in an AD-like process, even precede Aβ accumulation and obvious cognitive impairment.
Materials and Methods
Animals
APP/PS1 (5XFAD) double transgenic mice (stock No. 006554, Jackson Laboratory) and their non-transgenic wild-type littermates were randomly selected after identifying the genotype for FAD group and wild-type (WT) group (control) mice. We selected female animals to exclude the effects of sex on phenotype. There were seven WT mice and five FAD mice at 6 weeks old. There were eight WT mice and 11 FAD mice at 10 weeks old. There were seven WT mice and eight FAD mice at 12 weeks old.
Mice were specific-pathogen-free level and were housed in cages of up to five mice with a controlled environment of 22–25°C, 50% humidity and a 12-hour light/ dark cycle. The use and care of animals were in strict accordance with the ethical standards of the Animal Ethics Committee of Capital Medical University, China on December 17, 2015 (approval No. AEEI-2015-182).
Morris water maze
Mice at 12 weeks of age were subjected to the Morris water maze test. The experiment ran for 2 weeks, and the mice on the last day were 14 weeks old. The maze consisted of a circular stainless-steel tank (120 cm diameter) filled with water (20–22°C), and the tank was surrounded by a light blue curtain fixed with four visual cues inside. The circular swimming pool was divided into four quadrants: northeastern (NE), southeastern (SE), southwestern (SW) and northwestern (NW). The training task included two periods: acquisition training and reversal training. Twenty-four hours before acquisition training, mice were allowed to habituate in the pool containing a visible platform (5 cm diameter) for 60 seconds, the swim speed and the time to get to the platform (escape latency, seconds) were recorded. Mice that were unable to reach the visible platform within 60 seconds were excluded. In the acquisition training, mice were required to swim to find a hidden platform (in quadrant NE) submerged 0.5 cm below the water surface. Mice were trained in the acquisition task for four trials (four start points from six points: N, E, S, W, SE, NW) per day for five consecutive days. During each trial, the mouse was allowed to swim until the platform was found, and the escape latency was recorded. Mice that failed to find the hidden platform within 60 seconds were placed on the platform for 30 seconds by an experimenter and their escape latency was recorded as 60 seconds. After the acquisition training, the hidden platform was moved to the opposite quadrant (from NE to SW). We refer to this testing process as “reversal training”. This training was conducted in the same way as the acquisition training mentioned above, but with four new start points (four start points from six points: S, W, N, E, NW, SE). A probe trial (60 seconds) with withdrawal of the platform 24 hours after the last reversal training day was applied and the swimming time spent in the target quadrant and the other quadrants were recorded. Performance was automatically recorded with a video tracking system and analyzed using Smart software 2.0 (Panlab Harvard Apparatus, Barcelona, Spain).
Acute slice preparation
Mice were intraperitoneally anesthetized with 6% chloral hydrate (400 mg/kg) and perfused transcardially with ice-cold, high-sucrose modified artificial cerebrospinal fluid containing (in mM): 10 glucose, 213 sucrose, 3 KCl, 1 NaH2PO4·2H2O, 26 NaHCO3, 0.5 CaCl2·2H2O, 5 MgCl2·6H2O (pH7.4). Brains were rapidly removed into ice-cold high-sucrose artificial cerebrospinal fluid. The 400 μm-thick slices were cut on a vibratome (Leica, Bensheim, Germany) in high-sucrose artificial cerebrospinal fluid, and immediately transferred to an incubation chamber containing artificial cerebrospinal fluid (in mM): 10 glucose, 125 NaCl, 5 KCl, 1.2 NaH2PO4·2H2O, 26 NaHCO3, 2.6 CaCl2·2H2O, 1.3 MgCl2·6H2O (pH 7.4) (all reagents from Sigma, St. Louis, MO, USA). The slices were incubated to recover at 35°C for 40 minutes before being allowed to equilibrate at room temperature for a further 20 minutes. During slice preparation and recording, artificial cerebrospinal fluid was continuously mixed with the gas, including 95% O2 and 5% CO2.
Electrophysiological recording
A multi-electrode dish (MED 64 planar microelectrodes; Panasonic, Osaka, Japan) was prepared as described previously (Zhen et al., 2017). The perforant pathway in the dentate gyrus of the hippocampus was selected as the stimulation site; the recording site was determined by occurrence of field excitatory postsynaptic potential (fEPSP) response in dentate gyrus. The evoked fEPSPs were amplified by a 64-channel amplifier then digitized at a 20 kHz sampling rate. Synaptic responses were stabilized for 15 minutes; traces were obtained and analyzed using an MED64 System software program (Alpha Med Science Inc., Osaka, Japan). The input-output curve was determined by the measurement of fEPSP amplitude or slope in response to a series of stimulation intensities starting at 10 μA. The 30–50% maximum of input-output curve stimulation intensity was determined for subsequent LTP recording. Baseline responses were then recorded for an additional 15 minutes at 0.033 Hz of stimulation. LTP was then induced by theta-burst stimulation, which consisted of five bursts with interval of 10 seconds, each burst containing four pulses at 100 Hz with an inter-burst interval of 200 ms (5XTBS). After 5XTBS, the test stimulus was repeatedly delivered once every 30 seconds for 60 minutes to observe any changes in LTP magnitude and duration.
Immunohistochemistry
Half of each brain was subjected to electrophysiological recording as mentioned above. The other half of each brain was fixed in 4% paraformaldehyde in phosphate-buffered saline overnight at 4°C. After sucrose gradient dehydration, the brains were embedded with optimum cutting temperature compound and sectioned along the coronal plane on a freezing microtome (Model: 2165; Leica) at 30 μm. A standard avidin-biotin complex staining method was utilized to assess the distribution of Aβ signal. We used mouse monoclonal anti-Aβ antibody (6E10, 1:1000; 4°C, overnight, SIG-39300; Covance, Princeton, NJ, USA) and a secondary antibody, biotinylated goat anti-mouse IgG (1:200, room temperature, 2 hours; Vector Laboratories, Burlingame, CA, USA). After a sequential process of 3,3′-diaminobenzidine staining, air-drying, dehydration, and hyalinization, the sections were mounted on glass slides and cover-slipped. Specific Aβ-positive staining was observed with a light microscope (Olympus, Tokyo, Japan).
Subcellular fractionation of proteins
Subcellular fractionation of proteins was prepared as described previously (Gu et al., 2009). After slices were prepared as described above, the hippocampus was separated and homogenized in ice-cold lysis buffer (10 mL/g, pH 7.6) (in mM): 15 Tris, 250 sucrose, 1 phenylmethylsulfonyl fluoride, 2 ethylenediamine tetraacetic acid, 1 glycol-bis-(2-aminoethylether)-N,N,N′,N′-tetraacetic acid, 10 Na3VO4, 25 NaF, 10 sodium pyrophosphate, protease inhibitor tablet, and phosphatase inhibitor tablet. After centrifugation at 4°C and 800 × g for 10 minutes, the supernatant was taken, and 50 μL was left as the total proteins. The remaining supernatant was centrifuged at 4°C and 10,000 × g for 10 minutes. The supernatant and pellet were divided, and the supernatant was centrifuged at 4°C and 165,000 × g for 30 minutes to obtain the cytosolic fraction. The pellet was dissolved in lysis buffer containing 1% Triton X-100 and 300 mM NaCl, homogenized and centrifuged at 4°C and 16,000 × g for 30 minutes to obtain Triton soluble fraction, which includes cytosolic proteins in synapses and Triton insoluble fraction, which mainly includes membrane associated proteins in synapses and can be dissolved in 1% sodium dodecyl sulfate.
Western blot assay
Each fraction of proteins was separated on 10% sodium dodecyl sulfate-polyacrylamide gel, and transferred to nitrocellulose membranes (Millipore). After blocking with 5% nonfat dry milk for 1 hour at room temperature, membranes were incubated with the primary antibodies (NR1 monoclonal antibody, CST, Cat No. D65B7, 1:1000; GluA1 polyclonal antibody, Millipore, Cat No. AB1504 1:2000; tubulin monoclonal antibody, Sigma, Cat No. T8203, 1:10,000) at 4°C overnight. The primary antibody-labeled membranes were incubated with secondary antibodies (goat anti-mouse IgG, horseradish peroxidase conjugated, 1:10,000, Cat No. CW0102A or goat anti-rabbit IgG, horseradish peroxidase conjugated, 1:10,000, Cat No. CW0103A, CWBIO) at room temperature for 2 hours. The blots were then exposed to the chemiluminescence substrate (Thermo). The bands were then visualized using the gel imaging analysis system (Bio-Rad). The gray value was obtained from bands using ImageJ software (National Institutes of Health, Maryland, USA). Total protein fractions were normalized to their own tubulin of total protein. Triton soluble fraction or Triton insoluble fraction was normalized to its own total protein fractions.
Statistical analysis
Statistical analyses were performed using SPSS 19.0 software (IBM, Armonk, IL, USA) and GraphPad Prism 5.0 software (GraphPad Software Inc., La Jolla, CA, USA). All data are presented as the mean ± SEM. Data of escape latency curve, LTP curve and input-output curve were analyzed with repeated-measures analysis of variance. For two comparisons, the data were analyzed with two-tailed Student’s t-tests. A value of P < 0.05 was considered statistically significant.
Results
Behavioral impairment in 5XFAD mouse models at 12–14 weeks of age
The hippocampus is considered to be susceptible to early AD-related pathological factors (Anand and Dhikav, 2012; Song et al., 2016). The Morris water maze test is a commonly used approach for evaluating hippocampus-dependent spatial learning and memory ability (Moser and Moser, 1999; Dong et al., 2013). Here, reversal training was performed following routine acquisition training in the Morris water maze when mice were 12 weeks old. In acquisition training, from day 1 to day 5, escape latency was not significantly different between 5XFAD and WT mice (Figure 1A and B). In reversal training, 5XFAD mice had a significantly longer escape latency than WT mice at day 8 (P = 0.028), although they exhibited comparable performance to WT mice at any of the other training days (Figure 1B). In the reversal learning probe trial, 5XFAD mice spent less time in the target quadrant than WT mice (P = 0.042; Figure 1C) when the mice were 14 weeks of age. This finding suggested that a reversal learning impairment in 5XFAD mice was initiated at 12–14 weeks of age.
Figure 1.

Behavioral impairment in 5XFAD mouse models began at 12–14 weeks of age.
(A) Schematic diagram of two stages of the Morris water maze; (B) 12-week-old 5XFAD and WT mice were subjected to acquisition training and reversal training sequentially, and the time spent finding the hidden platform (escape latency, seconds) was recorded and used in statistical analysis (repeated- measures analysis of variance); (C) probe trials were conducted on day 12, and the percentage of swimming time spent in the target quadrant of total time (%) was recorded. *P < 0.05, vs. WT (mean ± SEM, Student’s t-test). WT: Wild-type; FAD: family Alzheimer’s disease; NW: northwest; NE: northeast; SE: southeast; SW: southwest.
Hippocampal LTP declines in 5XFAD mice aged 10 weeks
Since hippocampal synaptic function is necessary for Morris water maze learning and memory coding, investigating the timing of earlier synaptic functional decline may be valuable. The perforant pathway-dentate gyrus circuit was selected to determine hippocampal LTP expression. First, we examined LTP of 5XFAD mice and their littermates at 6 weeks of age. After applying 5XTBS to the perforant pathway, both the amplitude and slope of fEPSP recorded in the dentate gyrus in these two groups substantially increased over time (Figure 2A and E). Hippocampal synaptic plasticity occurs in multiple phases involving short-term and long-term changes in the synapse (Chakroborty et al., 2015). Changes of 0–2 minutes after theta-burst stimulation are thought to reflect post-tetanic potentiation associated with alterations in presynaptic neurotransmitter release properties (Fioravante and Regehr, 2011). The change of 15–20 minutes after theta-burst stimulation reflects early-LTP (E-LTP) attributed to the modification of pre-existing postsynaptic membrane proteins, particularly α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptors (Gu et al., 2009; Davies et al., 1989). Changes in the last 10 minutes of LTP are considered late phase-LTP (L-LTP), and are dependent on gene transcription and new protein synthesis (Frey et al., 1993). Analysis of these three phases revealed no statistical difference in either amplitude (Figure 2B–D) or slope (Figure 2F–H) between the FAD and WT groups in three phases at 6 weeks old. These results indicated normal hippocampal synaptic function in 5XFAD mice at 6 weeks of age.
Figure 2.

Hippocampal LTP is not damaged in 5XFAD mice at 6 weeks old.
(A) Summary plots of normalized fEPSP amplitude (%) in recording time. The fEPSPs were induced by 5XTBS (arrow). Inset traces: Representative traces of fEPSP before (black curve) and after (red curve) the induction of potentiation. (B) Normalized fEPSP amplitude (%) for PTP (1–2 minutes after theta-burst stimulation). (C) Normalized fEPSP amplitude (%) for E-LTP (15–20 minutes after theta-burst stimulation). (D) Normalized fEPSP amplitude (%) for the last 10 minutes. (E–H) Results of fEPSP slope (%) in the dentate gyrus of 6-week-old mice. All data are presented as the mean ± SEM (Student’s t-test). (B–D, F–H) or repeated-measures analysis of variance (A, E). WT: Wild-type; FAD: family Alzheimer’s disease; PTP: post-tetanic potentiation; LTP: long-term potentiation; E-LTP: early-LTP; fEPSP: field excitatory postsynaptic potential; TBS: theta-burst stimulation; 5XTBS: 5 theta-burst stimulation.
We next examined the LTP expression in 10-week-old mice. After 5XTBS, the fEPSP amplitude and slope of both groups increased over time. The increments of both amplitude and slope of fEPSP were slower in 5XAFD mice than WT controls (P = 0.035 for amplitude, P = 0.032 for slope) (Figure 3A and E). Normalized to baseline, the percentage of amplitude and slope values was lower in 5XFAD mice than WT controls in post-tetanic potentiation (P = 0.040 for amplitude, P = 0.046 for slope; Figure 3B and F), and the E-LTP phase (P = 0.029 for amplitude, P = 0.0253 for slope; Figure 3C and G). The current findings indicated that presynaptic neurotransmitter release properties and the modification of pre-existing proteins of postsynaptic membrane in 5XFAD mice at 10 weeks of age might be affected by AD-related pathological factors. Interestingly, there was no substantial difference in L-LTP phase between 5XFAD mice and WT mice (Figure 3D and H), although there was a tendency for a decreased slope in 5XFAD mice compared with WT mice (P = 0.077; Figure 3H). In general, hippocampal LTP declined in 5XFAD mice at 10 weeks old, at which time synaptic function might be recoverable, since there was no damage to the new protein synthesis associated with LTP expression.
Figure 3.

LTP declines in the hippocampus of 5XFAD mice at 10 weeks old.
(A) Summary plots of normalized fEPSP amplitude (%) in recording time. fEPSPs were induced by 5XTBS (arrow). Inset traces: Representative traces of fEPSP before (black curve) and after (red curve) the induction of potentiation. (B) Normalized fEPSP amplitude (%) for post-tetanic potentiation (1–2 minutes after theta-burst stimulation). (C) Normalized fEPSP amplitude (%) for E-LTP (15–20 minutes after theta-burst stimulation). (D) Normalized fEPSP amplitude (%) for the last 10 minutes. (E–H) Statistical analysis of fEPSP slope (%) of LTP in the dentate gyrus of 5XFAD and WT mice. After 5XTBS, the fEPSP amplitude and slope of both groups increased over time. The increments of both amplitude and slope of fEPSP were slower in 5XAFD mice than WT controls (*P < 0.05). All data are presented as the mean ± SEM (t-test for B–D, F–H or repeated-measures analysis of variance for A, E). *P < 0.05, vs. WT (mean ± SEM; Student’s t-test). WT: Wild-type; FAD: family Alzheimer’s disease; LTP: long-term potentiation; E-LTP: early-LTP; fEPSP: field excitatory postsynaptic potential; TBS: theta-burst stimulation; 5XTBS: 5 theta-burst stimulation.
Basal synaptic transmission is not altered in 5XFAD mice
To investigate whether the attenuation of hippocampal LTP of 5XFAD was induced by alteration of basal synaptic transmission, we recorded and analyzed the input-output curve of fEPSP, which represents basal synaptic transmission. We found no significant difference in fEPSP amplitude or slope in response to a series of stimulation intensities between the two groups at either 6 or 10 weeks old (Figure 4), suggesting that the function of hippocampal basal synaptic transmission was not impaired in 10-week-old 5XFAD mice. This result indicated that the attenuation of hippocampal LTP in 5XFAD mice could not be attributed to alteration of basal synaptic transmission. Moreover, these findings suggested that long-term synaptic strength was more susceptible to AD pathological context than basal synaptic function.
Figure 4.
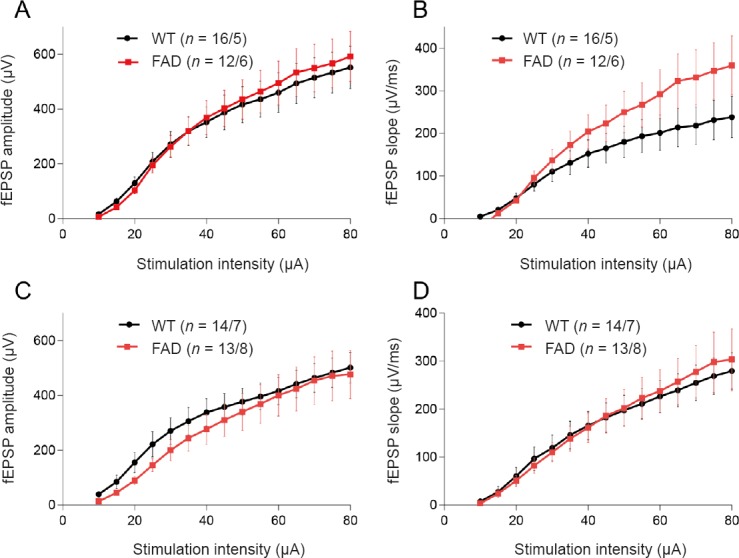
Basal synaptic transmission is intact in the hippocampus of 5XFAD mouse at 6 and 10 weeks of age.
(A, B) Input-output curves of fEPSP amplitude and slope in mice at 6 weeks of age. (C, D) Input-output curves of fEPSP amplitude and slope in mice at 10 weeks old. All data are presented as the mean ± SEM (repeated-measures analysis of variance). WT: Wild-type; FAD: family Alzheimer’s disease; fEPSP: field excitatory postsynaptic potential.
Distribution of postsynaptic glutamate receptor subunits GluA1 and NR1 at synapses is reduced in the hippocampus of 5XFAD mice
Considering the critical role of AMPA and N-methyl-D-aspartic acid (NMDA) receptors in LTP induction and expression (Nicoll, 2017), we explored the synaptic distribution of representative subunits (GluA1 for the AMPA receptor and NR1 for the NMDA receptor), in the hippocampus of 5XFAD mice at 10 weeks old. We did not find changes in the total protein level of GluA1 and NR1, while the synaptic location of both GluA2 and NR1 were altered in the hippocampus of 5XFAD mice compared with WT controls (Figure 5). For GluA1 location in the hippocampal synapses of 5XFAD mice at 10 weeks of age, we detected reduced Triton insoluble fraction and comparable Triton soluble fraction levels compared with WT controls (Figure 5A and B). However, for NR1 distribution, both Triton soluble fraction and Triton insoluble fraction levels were significantly decreased compared with WT controls (P = 0.017 for Triton soluble fraction, P = 0.045 for Triton insoluble fraction) (Figure 5A and C). These findings suggested that LTP impairment may be associated with postsynaptic delocalization of AMPA and NMDA receptors in the hippocampus of 5XFAD mice.
Figure 5.

Decreased subcellular distribution of glutamate receptors in the hippocampus of 5XFAD mice at 10 weeks old.
(A) Western blots showing the expression of glutamate receptors in different subcellular fractions in the hippocampus from wild-type and 5XFAD mice at 10 weeks of age. The α-tubulin was considered the internal control to normalize total protein level. (B) Normalization of AMPA-selective glutamate receptor 1 (GluA1) in different subcellular fractions in the hippocampus from 10-week-old WT and 5XFAD mice. (C) Normalization of NMDA receptor 1 (NR1) in different subcellular fractions in the hippocampus from 10-week-old WT and 5XFAD mice. *P < 0.05, vs. WT (mean ± SEM; n = 3 mice/genotype for GluA1, n = 4 mice/genotype for NR1; Student’s t-test). T: Total protein in tissue; S: cytosolic fraction; P1: triton-soluble fraction in the crude synaptosome fraction, which mainly includes cytosolic proteins in synapses; P2: triton-insoluble fraction in the crude synaptosome fraction, which mainly includes membrane-associated proteins in synapses. WT: Wild-type; FAD: family Alzheimer’s disease; AMPA: α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid; NMDA: N-methyl-D-aspartic acid.
Aβ accumulates intracellularly in 5XFAD mice at 10 weeks of age
The massive production and aggregation of Aβ are specific pathological features of AD, and are a key phenotype of 5XFAD mouse model (Oakley et al., 2006; Oh et al., 2018). Our previous study revealed that extracellular Aβ-positive plaques appeared in the cortex and hippocampus of transgenic mice at 5 months of age (Wang et al., 2014). Here, Aβ immunostaining was detected when hippocampal synaptic plasticity began to decline at 10 weeks of age. As shown in Figure 6, Aβ-positive signals were undetectable in brain sections of WT mice and little extracellular Aβ accumulation occurred in the 5XFAD mouse brain. We observed substantial intracellular Aβ aggregation in hippocampal neurons, indicating that decline in hippocampal LTP and postsynaptic translocation of glutamate receptor subunits were independent of extracellular Aβ deposition, and might be induced by aberrant intracellular overproduction of neuronal Aβ in the AD brain.
Figure 6.
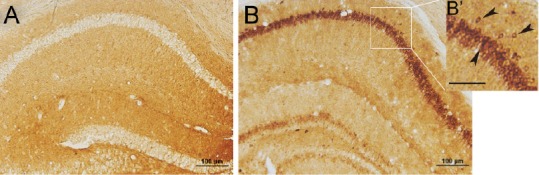
Amyloid beta intracellularly accumulates in the hippocampal CA1 region of 5XFAD mice at 10 weeks of age.
(A, B) Brain slices of wild-type and FAD mice were subjected to immunostaining with anti-amyloid beta specific antibody, 6E10 under the optical microscope (original magnification, 20×). Scale bar: 100 μm for A and B, 50 μm for B’. Arrows in B’ indicate neurons with intracellular amyloid beta-positive signals. FAD: Family Alzheimer’s disease.
Discussion
The mechanisms underlying AD are complex, and its precise pathogenesis remains unclear. The 5XFAD mouse model is a valuable and commonly used tool for tracking pathological changes, particularly the early events related to AD. Synaptic dysfunction plays a direct role in the occurrence of cognitive disorders, and is strongly associated with the onset of AD in the early pathophysiological processes of the disease (Olichney et al., 2011; Cukier et al., 2017). A previous study reported a significantly earlier synaptic impairment when only soluble Aβ or tau was present in 3xTg-AD mice (Baglietto-Vargas et al., 2018). Thus, evidence regarding the timing of the earliest stages of decline in synaptic function and the potential relationship between behavioral impairment, Aβ deposition and synaptic dysfunction could potentially be revealed with other commonly used AD transgenic models since earlier intervention strategies could ultimately benefit human AD (Lin et al., 2018). To the best of our knowledge, the current study is the first report of an early time point at which hippocampal LTP decline preceded hippocampus-dependent reversal learning and memory disability, accompanied by reduced synaptic distribution of glutamate receptor subunits, NR1 and GluA1 in the hippocampus. This finding may be associated with intracellular Aβ accumulation rather than extracellular Aβ deposition in the 5XFAD mouse model.
A previous study in our laboratory revealed that hippocampus-dependent spatial memory impairment in 5XFAD mice was initiated at 4.5 months of age, using the routine Morris water maze (Wang et al., 2014). This result is consistent with the finding that 5XFAD transgenic mice exhibited cognitive decline in the Y maze at 4 months old (Oakley et al., 2006). However, when 5-month-old model mice were treated with compound possessing β amyloid cleaving enzyme (BACE1), we did not observe an optimal effect, as observed in a parallel study with 3-month-old 5XFAD mice (Wang et al., 2014). These results suggest that an early window is available for studying AD pathogenesis and therapeutics. In the current study, we found cognitive impairment of 5XFAD mice at 12–14 weeks old, suggesting that the reversal learning test is more sensitive for detecting early hippocampus-dependent cognitive impairment than that routine Morris water maze protocol. Importantly, there is currently no evidence regarding the phenotype for this behavioral performance exhibited by the mouse model, prompting us to consider the time at which hippocampal synaptic transmission was functionally damaged.
As there is a long latent period before the onset of clinical symptoms of AD (Jack et al., 2018), synaptic dysfunction may occur at the asymptomatic stage of AD. As expected, hippocampal LTP in 10-week-old 5XFAD mice was suppressed compared with WT mice. Importantly, during the whole LTP stage, post-tetanic potentiation and E-LTP were damaged, while the last 10 minutes of LTP was intact in terms of both amplitude and slope of LTP compared with WT mice, implying that a compensatory mechanism is involved in memory coding of AD transgenic mice. This hypothesis may provide a reasonable explanation of why deficits in cognitive performance were only detectable using a much more difficult task in 5XFAD mice at this early stage of AD. This compensatory hypothesis was supported by findings of unchanged basal synaptic transmission, and our previous finding that theta-burst stimulation failed to induce LTP in the dentate gyrus of 6-month-old 5XFAD mice (Zhen et al., 2017).
Our current findings suggest that LTP decline in the early stages of AD is likely to be caused by attenuated presynaptic plasticity or reduced existing post synaptic membrane receptors, including the AMPA receptors and NMDA receptors (Gu et al., 2015) involved in LTP induction and expression (Davies et al., 1989; Frey et al., 1993; Fioravante and Regehr, 2011; Chakroborty et al., 2015). As expected, synaptic distribution of the AMPA receptor subunit, GluA1, and NMDA receptor subunit, NR1 in the 5XFAD hippocampus declined, while the total protein levels of these subunits remained similar in WT controls. Nevertheless, the possible molecular mechanisms underlying delocalization of postsynaptic glutamate receptors in the context of AD require in-depth exploration.
Although extracellular Aβ deposition has attracted much research attention in the past several decades, there are currently few available therapies targeting Aβ aggregation (Selkoe and Hardy, 2016). Thus, identifying events occurring before inexorable Aβ cascade and loss of neural network function could have far-reaching therapeutic implications. Indeed, intracellular Aβ accumulation has been reported in the brains of AD patients and animal models in the early stage (Gouras et al., 2000; Oakley et al., 2006; LaFerla et al., 2007). Our observation of intracellularly accumulated Aβ in the hippocampus of 5XFAD mice was consistent with these findings, indicating that intracellular overproduction of Aβ could trigger synaptic dysfunction, subsequently inducing cognitive impairment in the early stages of AD. Although our findings and observations using other types of AD model, such as 3xTg mice (Billings et al., 2005; Yamamoto et al., 2013), suggest a general chain of evidence in the early stages of the pathological process of AD, additional evidence requires validation in other AD animal models, and in AD patients.
Taken together, the current findings may provide an important clue toward the understanding of an early sequential pathological event in AD. In addition, our results provide a referable time point for studying mechanisms and therapies of AD using 5XFAD mouse models.
Additional file: Open peer review report 1 (93.9KB, pdf) .
Acknowledgments
We thank Hua Li and Hua Wei from Capital Medical University, China, for their technical support.
Footnotes
Conflicts of interest: The authors declare that there are no conflicts of interest associated with this manuscript.
Financial support: This study was supported by the National Natural Science Foundation of China, No. 81571038, 81771145 (both to YZ). The funding sources had no role in study conception and design, data analysis or interpretation, paper writing or deciding to submit this paper for publication.
Institutional review board statement: The use and care of animals were in strict accordance with ethical standards of the Animal Ethics Committee of Capital Medical University of China on December 17, 2015 (approval No. AEEI-2015-182). All experimental procedures described here were in accordance with the National Institutes of Health (NIH) guidelines for the Care and Use of Laboratory Animals.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Vasily Vorobyov, Russian Academy of Sciences, Institute of Cell Biophysics, Russia.
Funding: This study was supported by the National Natural Science Foundation of China, No. 81571038 , 81771145 (both to YZ).
P-Reviewer: Vorobyov V; C-Editor: Zhao M; S-Editors: Wang J, Li CH; L-Editors: Knight B, Robens J, Qiu Y, Song LP; T-Editor: Liu XL
References
- 1.Anand KS, Dhikav V. Hippocampus in health and disease: an overview. Ann Indian Acad Neurol. 2012;15:239–246. doi: 10.4103/0972-2327.104323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baglietto-Vargas D, Prieto GA, Limon A, Forner S, Rodriguez-Ortiz CJ, Ikemura K, Ager RR, Medeiros R, Trujillo-Estrada L, Martini AC, Kitazawa M, Davila JC, Cotman CW, Gutierrez A, LaFerla FM. Impaired AMPA signaling and cytoskeletal alterations induce early synaptic dysfunction in a mouse model of Alzheimer’s disease. Aging Cell. 2018 doi: 10.1111/acel.12791. doi: 10.1111/acel12791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 4.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 5.Briggs CA, Chakroborty S, Stutzmann GE. Emerging pathways driving early synaptic pathology in Alzheimer’s disease. Biochem Biophys Res Commun. 2017;483:988–997. doi: 10.1016/j.bbrc.2016.09.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chakroborty S, Kim J, Schneider C, West AR, Stutzmann GE. Nitric oxide signaling is recruited as a compensatory mechanism for sustaining synaptic plasticity in Alzheimer’s disease mice. J Neurosci. 2015;35:6893–6902. doi: 10.1523/JNEUROSCI.4002-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi SH, Bylykbashi E, Chatila ZK, Lee SW, Pulli B, Clemenson GD, Kim E, Rompala A, Oram MK, Asselin C, Aronson J, Zhang C, Miller SJ, Lesinski A, Chen JW, Kim DY, van Praag H, Spiegelman BM, Gage FH, Tanzi RE. Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science. 2018;361:eaan8821. doi: 10.1126/science.aan8821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 9.Cukier HN, Kunkle BK, Hamilton KL, Rolati S, Kohli MA, Whitehead PL, Jaworski J, Vance JM, Cuccaro ML, Carney RM, Gilbert JR, Farrer LA, Martin ER, Beecham GW, Haines JL, Pericak-Vance MA. Exome sequencing of extended families with Alzheimer’s disease identifies novel genes implicated in cell immunity and neuronal function. J Alzheimers Dis Parkinsonism. 2017;7:355. doi: 10.4172/2161-0460.1000355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davies SN, Lester RA, Reymann KG, Collingridge GL. Temporally distinct pre- and post-synaptic mechanisms maintain long-term potentiation. Nature. 1989;338:500–503. doi: 10.1038/338500a0. [DOI] [PubMed] [Google Scholar]
- 11.Dong Z, Bai Y, Wu X, Li H, Gong B, Howland JG, Huang Y, He W, Li T, Wang YT. Hippocampal long-term depression mediates spatial reversal learning in the Morris water maze. Neuropharmacology. 2013;64:65–73. doi: 10.1016/j.neuropharm.2012.06.027. [DOI] [PubMed] [Google Scholar]
- 12.Fioravante D, Regehr WG. Short-term forms of presynaptic plasticity. Curr Opin Neurobiol. 2011;21:269–274. doi: 10.1016/j.conb.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fitzjohn SM, Morton RA, Kuenzi F, Rosahl TW, Shearman M, Lewis H, Smith D, Reynolds DS, Davies CH, Collingridge GL, Seabrook GR. Age-related impairment of synaptic transmission but normal long-term potentiation in transgenic mice that overexpress the human APP695SWE mutant form of amyloid precursor protein. J Neurosci. 2001;21:4691–4698. doi: 10.1523/JNEUROSCI.21-13-04691.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frey U, Huang YY, Kandel ER. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- 15.Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griñán-Ferré C, Izquierdo V, Otero E, Puigoriol-Illamola D, Corpas R, Sanfeliu C, Ortuño-Sahagún D, Pallàs M. Environmental enrichment improves cognitive deficits, AD hallmarks and epigenetic alterations presented in 5xFAD mouse model. Front Cell Neurosci. 2018;12:224. doi: 10.3389/fncel.2018.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu W, Fukuda T, Isaji T, Hang Q, Lee HH, Sakai S, Morise J, Mitoma J, Higashi H, Taniguchi N, Yawo H, Oka S, Gu J. Loss of alpha1, 6-fucosyltransferase decreases hippocampal long term potentiation: implications for core fucosylation in the regulation of ampa receptor heteromerization and cellular signaling. J Biol Chem. 2015;290:17566–17575. doi: 10.1074/jbc.M114.579938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gu Z, Liu W, Yan Z. {beta}-Amyloid impairs AMPA receptor trafficking and function by reducing Ca2+/calmodulin-dependent protein kinase II synaptic distribution. J Biol Chem. 2009;284:10639–10649. doi: 10.1074/jbc.M806508200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 20.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 21.He J, Yan B, Song XZ, Wu XY. Effects of placental mesenchymal stem cell transplantation on behaviors and neurotransmitters in Alzheimer disease rats. Zhongguo Zuzhi Gongcheng Yanjiu. 2018;22:4650–4656. [Google Scholar]
- 22.Herrup K. The case for rejecting the amyloid cascade hypothesis. Nat Neurosci. 2015;18:794–799. doi: 10.1038/nn.4017. [DOI] [PubMed] [Google Scholar]
- 23.Hitman GA, Toms GC, Boucher BJ, Garde L, Baker P, Awad J, Festenstein H. 2’-5’ oligoadenylate synthetase and its relationship to HLA and genetic markers of insulin-dependent diabetes mellitus. Immunogenetics. 1989;30:427–431. doi: 10.1007/BF02421174. [DOI] [PubMed] [Google Scholar]
- 24.Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iaccarino HF, Singer AC, Martorell AJ, Rudenko A, Gao F, Gillingham TZ, Mathys H, Seo J1, 2, Kritskiy O, Abdurrob F, Adaikkan C, Canter RG, Rueda R, Brown EN, Boyden ES, Tsai LH. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature. 2016;540:230–235. doi: 10.1038/nature20587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jack CR., Jr NIA-AA Research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14:535–562. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koehler D, Williams FE. Utilizing zebrafish and okadaic acid to study Alzheimer’s disease. Neural Regen Res. 2018;13:1538–1541. doi: 10.4103/1673-5374.237111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 29.Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. 2011;31:6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin W, Yang LK, Zhu J, Wang YH, Dong JR, Chen T, Wang D, Xu XM, Sun SB, Zhang L. Deep brain stimulation for the treatment of moderate-to-severe Alzheimer’s disease: Study protocol for a prospective self-controlled trial. Clin Trials Degener Dis 2018. 2018;3:66–70. [Google Scholar]
- 31.Marsh J, Alifragis P. Synaptic dysfunction in Alzheimer’s disease: the effects of amyloid beta on synaptic vesicle dynamics as a novel target for therapeutic intervention. Neural Regen Res. 2018;13:616–623. doi: 10.4103/1673-5374.230276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moser EI, Moser MB. Is learning blocked by saturation of synaptic weights in the hippocampus? Neurosci Biobehav Rev. 1999;23:661–672. doi: 10.1016/s0149-7634(98)00060-8. [DOI] [PubMed] [Google Scholar]
- 33.Nicoll RA. A brief history of long-term potentiation. Neuron. 2017;93:281–290. doi: 10.1016/j.neuron.2016.12.015. [DOI] [PubMed] [Google Scholar]
- 34.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oh SJ, Lee HJ, Kang KJ, Han SJ, Lee YJ, Lee KC, Lim SM, Chi DY, Kim KM, Park JA, Choi JY. Early detection of abeta deposition in the 5xFAD mouse by amyloid PET. Contrast Media Mol Imaging. 2018;2018:5272014. doi: 10.1155/2018/5272014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olichney JM, Yang JC, Taylor J, Kutas M. Cognitive event-related potentials: biomarkers of synaptic dysfunction across the stages of Alzheimer’s disease. J Alzheimers Dis 26 Suppl. 2011;3:215–228. doi: 10.3233/JAD-2011-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 39.Selkoe DJ. Alzheimer’s disease. Cold Spring Harb Perspect Biol. 2011;3:a004457. doi: 10.1101/cshperspect.a004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sheng M, Sabatini BL, Südhof TC. Synapses and Alzheimer’s disease. Cold Spring Harb Perspect Biol. 2012;4:1–18. doi: 10.1101/cshperspect.a005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Small SA, Duff K. Linking Abeta and tau in late-onset Alzheimer’s disease: a dual pathway hypothesis. Neuron. 2008;60:534–542. doi: 10.1016/j.neuron.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Song Z, McDonough IM, Liu P, Lu H, Park DC. Cortical amyloid burden and age moderate hippocampal activity in cognitively-normal adults. Neuroimage Clin. 2016;12:78–84. doi: 10.1016/j.nicl.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;27:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 46.Thal DR, RHYPE, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 47.Tsai J, Grutzendler J, Duff K, Gan WB. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci. 2004;7:1181–1183. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]
- 48.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 49.Wang Q, Xiao B, Cui S, Song H, Qian Y, Dong L, An H, Cui Y, Zhang W, He Y, Zhang J, Yang J, Zhang F, Hu G, Gong X, Yan Z, Zheng Y, Wang X. Triptolide treatment reduces Alzheimer’s disease (AD)-like pathology through inhibition of BACE1 in a transgenic mouse model of AD. Dis Model Mech. 2014;7:1385–1395. doi: 10.1242/dmm.018218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu Z, Guo Z, Gearing M, Chen G. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s [corrected] disease model. Nat Commun. 2014;5:4159. doi: 10.1038/ncomms5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamamoto S, Ooshima Y, Nakata M, Yano T, Matsuoka K, Watanabe S, Maeda R, Takahashi H, Takeyama M, Matsumoto Y, Hashimoto T. Generation of gene-targeted mice using embryonic stem cells derived from a transgenic mouse model of Alzheimer’s disease. Transgenic Res. 2013;22:537–547. doi: 10.1007/s11248-012-9651-x. [DOI] [PubMed] [Google Scholar]
- 52.Zhang L, Yang J, Liu ZH. STEP61 negatively regulates amyloid beta-mediated ERK signaling pathway in Alzheimer’s disease cell model. Zhongguo Zuzhi Gongcheng Yanjiu. 2018;22:4507–4512. [Google Scholar]
- 53.Zhen J, Qian Y, Weng X, Su W, Zhang J, Cai L, Dong L, An H, Su R, Wang J, Zheng Y, Wang X. Gamma rhythm low field magnetic stimulation alleviates neuropathologic changes and rescues memory and cognitive impairments in a mouse model of Alzheimer’s disease. Alzheimers Dement (N Y) 2017;3:487–497. doi: 10.1016/j.trci.2017.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.