

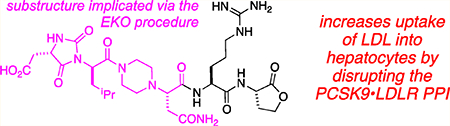
Abstract
The protein–protein interaction between proprotein convertase subtilisin/kexin type 9 (PCSK9) and low-density lipoprotein receptor (LDLR) is a relatively new, and extremely important, validated therapeutic target for treatment and prevention of heart disease. Experts in the area agree that the first small molecules to disrupt PCSK9·LDLR would represent a milestone in this field, yet few credible leads have been reported. This paper describes how side-chain orientations in preferred conformations of carefully designed chemotypes were compared with LDLR side chains at the PCSK9·LDLR interface to find molecules that would mimic interface regions of LDLR. This approach is an example of the procedure called EKO (Exploring Key Orientations). The guiding hypothesis on which EKO is based is that good matches indicate the chemotypes bearing the same side chains as the protein at the sites of overlay have the potential to disrupt the parent protein–protein interaction. In the event, the EKO procedure and one round of combinatorial fragment-based virtual docking led to the discovery of seven compounds that bound PCSK9 (SPR and ELISA) and had a favorable outcome in a cellular assay (hepatocyte uptake of fluorescently labeled low-density lipoprotein particles) and increased the expression LDLR on hepatocytes in culture. Three promising hit compounds in this series had dissociation constants for PCSK9 binding in the 20–40 μM range, and one of these was modified with a photoaffinity label and shown to form a covalent conjugate with PCSK9 on photolysis.
Graphical Abstract
INTRODUCTION
Heart disease, a leading cause of death, is frequently associated with plaque in the arteries causing atherosclerosis, which is attributed by elevated levels of low-density lipoprotein (LDL) in the blood. LDLRs (LDL receptors), on the surface of hepatocytes, are responsible for capturing LDL particles and importing them into the liver for destruction. Thus, LDLR·LDL complexes on the surface of hepatocytes undergo clathrin-mediated endocytosis into endosomes, wherein the acidic environment triggers rearrangement of the LDLR extracellular domain, releasing bound lipoproteins, leaving the LDLR to be recycled to the plasma membrane (Figure 1).1
Figure 1.

In the absence of PCSK9, LDL·LDLR is endocytosed into hepatocytes, and then the LDLR is recycled and LDL is digested. In the presence of PCSK9, LDLR is not recycled and the whole complex is decomposed. Thus, PCSK9·LDLR interaction suppresses recycling of LDLR and diminishes uptake of LDL particles by the liver.
Removal of LDL particles by hepatocytes is negatively modulated by a chaperone called proprotein convertase subtilisin/kexin type 9 (PCSK9)2,3 that recognizes plasma membrane LDLR.4 PCSK9 inhibits the rearrangement of LDLR in LDLR·LDL complexes, so that the receptor is not recycled to the plasma membrane but is instead routed to lysosomes, where LDL, LDLR, and PCSK9 are degraded (Figure 1).5–8 Plasma LDL cholesterol levels thus can be decreased by inhibiting the PCSK9·LDLR interaction, because that increases the display of LDLR on the hepatocytes.1,9,10 This effect was discovered after genetic studies had correlated LDL levels in some patients with mutations associated with gain or loss of function of PCSK9;11,12 two loss-of-function mutations correlate with significantly reduced risk of coronary heart disease.11 Indeed, an individual with no detectable PCSK9, and extremely low LDL levels, was healthy, suggesting that suppression of PCSK9 for lipid lowering is safe.13 Conversely, individuals with gain-of-function mutations in PCSK9 have a higher risk of coronary heart disease.11,12
Multiple clinical studies have shown that injectable antibody therapeutics that impede the PCSK9·LDLR protein–protein interaction (PPI) significantly decrease circulating LDL levels.14 Subsequently, two antibody drugs that disrupt PCSK9·LDLR were FDA approved (Repatha from Amgen and Praluent from Sanofi/Regeneron); they appear to be tolerated well, with no serious side effects, and are efficacious.15–20 PCSK9·LDLR, therefore, is a validated target for medicinal chemistry.
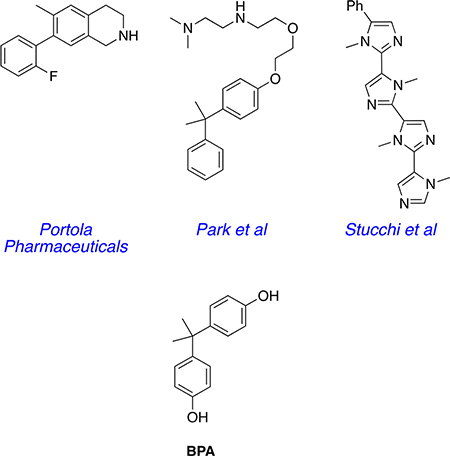
The preferred modality for disruption of PCSK9·LDLR, however, is small-molecule drugs, not monoclonal antibodies (mAbs), on the basis of mode of administration, cost, shelf life, and immunogenic response issues. Thus, in 2014 GenenTech stated, “Undoubtedly, an orally available small molecule inhibitor of PCSK9, due to lower cost and ease of administration, would be a highly desirable alternative therapeutic agent.”21 Peptides have been used to mimic either component in the PCSK9·LDLR interaction,22–28 but they have poor efficacy in plasma. There are limited reports of nonpeptidic small molecules that effectively inhibit the PCSK9·LDLR interaction, despite of intense efforts to find such compounds. Portola Pharmaceuticals have reported tetrahydroisoquinolines that increased both LDL uptake into liver cells and LDLR cell surface populations.29 Similarly, Park and co-workers reported compounds that had the same types of activities and reduced LDL in wild-type mice but not the corresponding in a PCSK9 knock out murine model.30,31 The Park compounds have fragments that structurally resemble the plasticizer bisphenol A (BPA). Both the Portola and Park studies do not report any direct evidence that these small molecules bind PCSK9; the compounds could act via another mechanism. However, Stucchi et al.32 reported an oligo N-methylimidazole that induced concentration-dependent disruption of PCSK9· LDLR based on an ELISA assay and increased LDL uptake in HepG2 cells. Curiously, the IC50 reported for the binding of this compound to PCSK9 (11.2 ± 0.2 μM) was greater than the EC50 for increased LDL uptake into HepG2 cells (6.04 μM).
Our laboratories have developed a technique for accelerated discovery of small molecules that inhibit PPIs: Exploring Key Orientations (EKO).33,34 EKO features relatively rigid, usually nonpeptidic, chemotypes with three amino acid side chains. Preferred conformations of these chemotypes are simulated and then systematically compared, via a data-mining algorithm, with side-chain orientations of protein–protein interface amino acids in solid-state structures. A chemotype that overlays well on one protein at the interface is a candidate to displace that same protein from the PPI.
Research featured here illustrates how the EKO approach was used to discover small molecules that bind PCSK9 with low micromolar affinities, which prevent the PCSK9·LDLR interaction in LDL uptake by hepatocytes.
RESULTS AND DISCUSSION
Small Molecule Design and Syntheses.
Several candidate chemotypes were conceived and screened using the EKO approach. In EKO, preferred conformations of the featured chemotypes with three methyl side chains are simulated, but they may overlay on any three residues of a protein in a PPI. Chemotypes with side chains corresponding to the protein at the overlay sites must then be prepared and assayed to test the validity of the EKO prediction.
In the event, molecules in series A gave several overlays on LDLR of the PCSK9·LDLR complex (crystal structure PDB ID 3gcx).35 Specifically, EKO indicated that preferred conformations of structures A overlaid Cα−Cβ atoms well in the following sets of interface side chains of LDLR in the PCSK9·LDLR crystal structure: Asp299, Leu298, Asp301; Cys297, Asn301, Asp299; Leu298, Asp299, Asn301; Val307, Cys308, Leu318; and Asn309, Cys308, Leu318. Thus, the residues implicated in total were Cys297, Leu298, Asp299, Asp301, Val307, Cys308, Asn309, and Leu318. On the basis of Ala-scan studies, Horton and co-workers conclude the LDLR side chains implicated in PCSK9·LDLR hot-spots are Asn295, Glu296, Asp310, and Tyr315,8 while crystallographic evidence led others to suggest that Asp299, Leu318, and Asn309 were implicated.36 Throughout we have underlined residues that have been postulated to be hot spots that were also overlaid via preferred conformations of A. In our view, the presence or lack of overlap with hot spots is insufficient grounds for “go” or “no-go” decisions in EKO analyses. There are two reasons for this: (i) the uncertainties of predicting hot spots,37 even with experimental data, and (ii) the notion of isolated hot-spots is being superseded by hot segments in which disruption of one residue in a tightly packed arrangement alters the contributions of the others.38–40
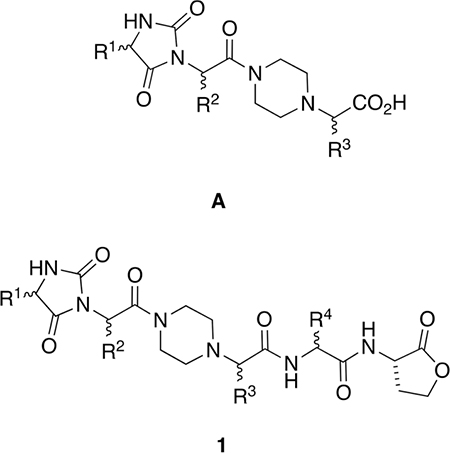
Several compounds in series A were prepared and tested using a commercial TR-FRET assay (BPS Bioscience Inc., San Diego, CA); however, the results were inconclusive [Figure S1, Supporting Information (SI)]. We were unsure if this negative result was because of poor affinity of the compounds A, insufficient sensitivity in the TR-FRET assay, or both. In any event, it seemed clear that modifications to improve the affinity of the core A toward PCSK9 would be desirable. Consequently, an iterative docking and energy minimization procedure was used to virtually modify chemotypes A to include additional pharmacophores that would increase binding efficiencies. Specifically, the preferred conformations of A were overlaid on LDLR in the 3gcx structure, LDLR was removed, then the chemotypes were docked in place using Glide within the Schrodinger package.41–43 That procedure gave “baseline energies” for interactions of A with PCSK9 to which data from docking virtually modified analogs could be compared;44 this procedure was performed in the Schrodinger software package using the CombiGlide routine.
Figure 2a shows a docked and minimized structure of A PCSK9 generated from the original EKO overlay. We noticed that there is a conspicuous negatively charged cavity in PSCK9 comprising Ser383, Asp367, and Ser381 proximal to the C-terminus of A, but not interacting with it. Consequently, we made that cavity a priority in CombiGlide simulations. Those calculations indicated that the negative pocket was filled by addition of His, Lys, or, optimally, Arg at the A C-terminus; structures 1 were conceived in this way. Figure 2b illustrates how Arg in the small molecule H-bonds to Asp367.
Figure 2.

Docking of LDL-Adln (gold) and LDLL-1dlnr (silver) onto PCSK9. (a) Compounds LDL-Adln and LDLL-1dlnr overlay on the LDLR (shown in magenta wire), in which six Cα−Cβ atoms of chemotypes and LDLR side chains (green arrows) are compared; RMSD = 1.75 and 1.95 Å for LDL-Adln and LDLL-1dlnr, respectively. (b) The improving binding affinity of LDLL-1dlnr is likely due to the H-bonds of the Arg residue to the Asp367 of PCSK9 at a negative “cliff face” region, as indicated by green lines.
A solid-phase synthesis of chemotypes 1 on TentaGel-NH2 resin was developed (Scheme 1). Microwave-accelerated amino acid couplings installed the R4-bearing residue and then the protected amino acid fragment 10 that carries R3. Nosyl removal as indicated45 and then two more coupling–deprotection cycles using standard N-Fmoc-protected amino acids assembled the fragments bearing the R2 and R1 side chains. The Fmoc was removed, and the N-terminal dipeptide was converted to a hydantoin via a two-step process.46 Finally, cyanogen bromide was used to cleave the protected chemotypes 1 from the resin, giving the C-terminal lactone appendix in these structures.
Scheme 1.

Solid-Phase Syntheses of Chemotypes 1
Compounds in Scheme 1 and throughout this paper are numbered according to the scaffold (or scaffold intermediate). Lower case one-letter codes are used to delineate the amino acid side chains R1–R4 and relate them to the closest amino acid; primed letters indicate protected side chains [e.g., d′ for the –CH2CO2tBu of Asp and k′ for the –(CH2)4NHBoc of Lys].
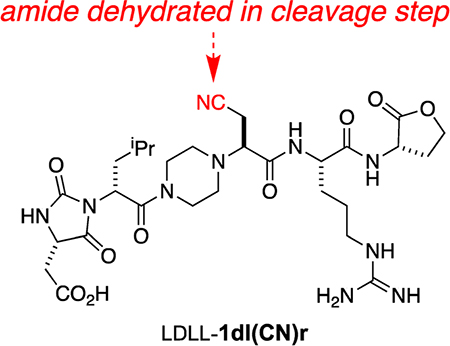
Implementation of Scheme 1 gave 15 compounds for screening, but one, LLLD-1ldnq, was surprisingly vulnerable to degradation in the air and was not considered further. One of the compounds that was considered, LDLL-1dl(CN)r, is a byproduct formed via dehydration of the Asn side chain in the cyanogen bromide cleavage step.
Binding Assays.
Surface plasmon resonance (SPR) was used to screen the library of 14 compounds indicated in Figure 3a. Pep2–8 is a 13-residue peptide developed by GenenTech that was reported to bind PCSK9 (Kd = 0.66 ± 0.11 μM)21 and was used here as a positive control. Compound LLLL-1aaar is a “partial negative control” having the same core as the EKO-implicated compounds but only Ala side chains and an all-L stereochemistry that was not predicted via EKO; i.e., LLLL-1aaar controls for random stereochemistry and lack of functional side chains. In the event, three compounds and Pep2–8 were selected (on the basis of this initial SPR data and the cellular uptake assays described below) for more thorough SPR analyses: LDLL-1dlnr (Kd = 24.8 ± 9.1 μM; Figure 3b), DLDD-1nclk (41.2 ± 17.5 μM; Figure S7b, SI), LDLL-1dl(CN)r (35.8 ± 11.4 μM; Figure S7d, SI), and Pep2–8 (3.56 ± 0.16 μM). The Kd for Pep2–8 determined here is slightly higher than the value reported previously;21 this discrepancy might be because of different techniques used to study the dissociation constant (the literature procedure used was biolayer inferometry).
Figure 3.

(a) Initial SPR screening of 14 compounds at 50 μM and Pep2–8 at 5 μM over PCSK9 supported on gold via amine coupling chemistry. (b) Sensorgram of LDLL-1dlnr within the 1–54 μM concentration range showing that the estimated Kd was 24.8 ± 9.1 μM [kon = (4.04 ± 2.20) × 103 M−1 s−1, koff = (8.74 ± 3.40) × 10−2 s−1]. (c) Selected compounds and Pep2−8 were screened at 50 μM for an inhibitory effect against 50 ng/mL of PCSK9 using a PCSK9-LDLR in vitro binding assay kit (MBLI Co.) following the manufacture instructions. Results are the averages ± SD of three independent experiments.
Seven compounds in the library (again, selected on the basis of the SPR data and the cellular assays described below) were subjected to an ELISA assay to obtain additional evidence that the small molecules bind PCSK9 (Figure 3c). Error limits in this assay are higher than in the SPR experiments. All the compounds tested showed increased inhibition of PCSK9 to the EGF-AB domain of LDLR relative to a blank control. Inhibition by the compounds in this assay was 1–2 orders of magnitude less than that of the positive control Pep2–8.
Photoaffinity Labeling.
Two derivatives of LDLL-1dlnr, compounds 2 and 3, were prepared to explore if binding of these to PCSK9 could be detected via photoaffinity labeling. Thus, a protected fragment of LDLL-1dlnr was prepared on chlorotrityl resin, cleaved with the protecting groups in place, and coupled to a photoaffinity fragment designed in these laboratories48 (see the SI).
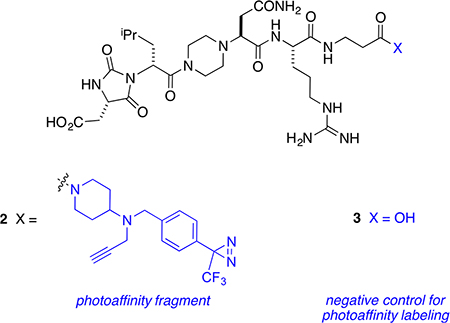
Preincubation of PCSK9 with 2 and (optionally) with a large excess of the blocking ligand 3, irradiation of some wells at 365 nm, copper-mediated click reaction with Alexa-488-azide, and then SDS–PAGE gave the data shown in Figure 4. A fluorescent band corresponding to the molecular mass of labeled PCSK9 (~60 kDa) was observed only in the wells that were irradiated in the absence of the blocking ligand 3.
Figure 4.

Photoaffinity labeling of human PCSK9 protein with 2. PCSK9 was incubated with PAL ligand 2 and optionally pretreated with a 50-fold excess of the competing ligand 3. UV (365 nm) irradiation and then Cu-mediated click with Alexa-488-azide gave the samples for analysis. These samples were diluted in SDS sample buffer and subjected to SDS–PAGE. Fluorescent proteins were detected by in-gel fluorescence (Alexa 488) and all proteins were stained with CBB (Coomassie Brilliant Blue) G250.
Liver-Cell Uptake Assays.
The seven select compounds shown in Figure 3c were tested for cytotoxicity using liver cells (hepatocytes; HepG2). All seven of these compounds showed no toxicity up to 50 μM. The compound with the lowest Kd in SPR studies, LDLL-1dlnr, was also checked at 100 μM and showed no cytotoxicity, even at this higher concentration (Figure S2, SI). In another prelude to the key cellular assays, the water solubilities of the featured compounds were measured. The solubility concentration gradients for these materials were linear beyond 100 μM; i.e., they were soluble at the maximum concentration used in the uptake assays below (Figure S5, SI). Compounds A and 1 were evaluated their drug likeliness by QikProp calculations47,49 to determine absorption, distribution, metabolism, and excretion (ADME) properties, and the results are reported in Tables S3 and S4 (SI).
An established assay for PCSK9·LDLR inhibition features uptake of fluorescently labeled LDL nanoparticles (BODIPY-LDL, Invitrogen) by hepatocytes; the cells become fluorescent as the particles are absorbed.21 Uptake of the BODIPY-containing particles is maximized in the absence of PCSK9, and the cells become fluorescent; conversely, adding PCSK9 diminishes that signal. Addition of PCSK9 and a compound that interferes with the PCSK9·LDLR interaction would be expected to give cells that are more fluorescent than those to which only PCSK9 was added but less so than cells to which none of that protein was present.
Figure 5a shows maximal uptake (calibrated to 100%, black bar) in the absence of PCSK9, while all the other data points correspond to 15 μg/mL of that protein; the “negative” corresponds to only PCSK9 added (calibrated to 0%). Pep2–8 at 30 μM restored the LDL uptake (yellow bar) to within 80% of its maximal value (black). Several of the featured chemotypes 1 showed promise insofar as they, like Pep2–8, also restored fluorescence; the ones marked with a red dagger were selected for further assays on the basis of this data and the binding studies above. Recall that LLLL-1aaar is a “partial control” as described above (same chemotype, just methyl side chains, and random stereochemistry); it did not induce significant BODIPY-LDL uptake.
Figure 5.

Uptake of BODIPY-LDL by hepatocytes. (a) Initial screen at 50 μM concentrations of the featured compounds. (b) Data of select compounds at three different doses. (c) A more extensive dose– response curve of one select lead: LDLL-1dlnr. All results are represented as means ± SD of three independent experiments. Significant differences between compounds and the negative control are determined using Student’ t test (*p ≤ 0.05, **p ≤ 0.01).
Figure 5b shows data derived from repetition of these experiments under identical conditions, except that three different doses of the test compounds were used. Overall, all of the compounds show a dose response, except LLLD-1qndr, DLLL-1qndr, and, as expected, the partial control LLLL-1aaar. Figure 5c shows a more extensive dose–response curve for one of these compounds, LDLL-1dlnr (again, selected on the basis of the overall data); this data shows an encouraging correspondence.
It is curious that LDL uptake in the HepG2 cells was significantly enhanced when 100 μM of compound was used (Figure 5c). This is consistent with the SPR binding data in which LDLL-1dlnr showed a longer resident time (15–20 fold slower off-rate) compared with Pep2–8. More particularly, the longer half-life of the LDLL-1dlnr·PCSK9 complex became more obvious when a higher concentration (100 μM) of compound was injected onto the PCSK9-functionalized surface (data not shown). One explanation for these observations is that there could be a synergistic target site for LDLL-1dlnr that only becomes significant at higher compound concentrations.
Cell-Surface LDLR Assay.
Two assays were attempted to probe if increased uptake of LDL particles correlates with increased expression of LDLR. Our initial attempts to do this featured monitoring LDLR levels on treated and untreated hepatocytes using flow cytometry. The data obtained (Figure S3, SI) indicated an increase in LDLR levels upon treatment with the seven hit compounds (as shown in Figure 5b), but the errors in the measurements were such that the increases had borderline statistical significance. Consequently, we resorted to a semi-quantitative approach in which the LDLRs on live hepatocytes (treated and untreated) were visualized using an LDLR-selective mAb in combination with an Alexa Fluor-labeled secondary mAb. Figure 6a shows that expression of LDLR in the cells was suppressed when they were treated with PCSK9 alone. When cells were treated with the test compound LDLL-1dlnr, they stained more brightly (Figure 6b), though not as brilliantly as the positive control culture that lacked PCSK9 and any test compound (Figure 6c). Similar data including bright-field and merged images were collected for another three of the featured compounds (see Figure S4, SI).
Figure 6.

Cell-surface LDLRs of hepatocytes were determined by fluorescent imaging. HepG2 cells were treated (a) with PCSK9 alone (negative control), (b) with PCSK9 and LDLL-1dlnr, and (c) without PCSK9 (positive control).
CONCLUSION
This study was undertaken to find validated, small molecule leads that disrupt PCSK9·LDLR. Structural modifications to chemotypes 1 are now planned to discover derivatives that retain and improve on their encouraging binding and increased LDL uptake characteristics, while simultaneously engineering in features to endow more favorable ADME properties. Thus, the next steps in the process will probably involve substitution of amino acid side chains with bioisosteres to improve bioavailability50 and perhaps targeting the liver, e.g., via attaching ligands implicated in galactose uptake.51–56
The chemical design component of this study involved conception of possible chemotypes, implementation of the EKO approach, and one cycle of virtual pharmacophore screening. On the basis of the two binding and three cellular assays presented, over half of the 13 test compounds showed significant, measurable activities. Thus, the strategy brought a degree of rationality to this process that led to a hit rate (~50%) that would be highly unlikely via high-throughput screening of random compounds against the same target. Moreover, the limitation of EKO that requires that the chemotypes considered must bear three amino acid side chains is also a strength insofar as it forces practitioners to explore virgin patent diversity space.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. John Kulp of Small Molecule PPI Mimics LLC for useful discussions, Mr. Zhengyang Jiang for help with the flow cytometry experiments, and Dr. Jay Horton of UT Southwestern for the gift of the PCSK9 protein used in these studies. We thank DoD BCRP Breakthrough Award (BC141561), CPRIT (RP150559 and RP170144), and The Robert A. Welch Foundation (A-1121) for financial support. The NMR instrumentation at Texas A&M University was supported by a grant from the National Science Foundation (DBI-9970232) and the Texas A&M University System. The SPR studies were conducted in the Protein Production and Analysis Core at IBT.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b09360.
Details of the solid-phase syntheses, characterization of compounds A and 1–3, protocols for the biological assays (LDL uptake, binding via SPR/ELISA/TR-FRET, MTT cell viability, and determination of relative levels of LDLR expression), determination of water solubilities, photo-affinity labeling, computational procedures, and predicted physiochemical characteristics (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Costet P; Krempf M; Cariou B Trends Biochem. Sci 2008, 33, 426–434. [DOI] [PubMed] [Google Scholar]
- (2).Li J; Tumanut C; Gavigan J-A; Huang W-J; Hampton EN; Tumanut R; Suen KF; Trauger JW; Spraggon G; Lesley SA; Liau G; Yowe D; Harris JL Biochem. J 2007, 406, 203–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).McNutt MC; Lagace TA; Horton JD J. Biol. Chem 2007, 282, 20799–20803. [DOI] [PubMed] [Google Scholar]
- (4).Norata GD; Tibolla G; Catapano AL Annu. Rev. Pharmacol. Toxicol 2014, 54, 273–293. [DOI] [PubMed] [Google Scholar]
- (5).Tveten K; Holla OL; Cameron J; Strom TB; Berge KE; Laerdahl JK; Leren TP Hum. Mol. Genet 2012, 21, 1402–1409. [DOI] [PubMed] [Google Scholar]
- (6).Yamamoto T; Lu C; Ryan RO J. Biol. Chem 2011, 286, 5464–5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Holla OL; Cameron J; Tveten K; Stroem TB; Berge KE; Laerdahl JK; Leren TP J. Lipid Res 2011, 52, 1787–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zhang D-W; Lagace TA; Garuti R; Zhao Z; McDonald M; Horton JD; Cohen JC; Hobbs HH J. Biol. Chem 2007, 282, 18602–18612. [DOI] [PubMed] [Google Scholar]
- (9).Horton JD; Cohen JC; Hobbs HH J. Lipid Res 2009, 50, S172–S177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Seidah NG Expert Opin. Ther. Targets 2009, 13, 19–28. [DOI] [PubMed] [Google Scholar]
- (11).Cohen JC; Boerwinkle E; Mosley TH Jr.; Hobbs HH N. Engl. J. Med 2006, 354, 1264–1272. [DOI] [PubMed] [Google Scholar]
- (12).Abifadel M; Varret M; Rabes J-P; Allard D; Ouguerram K; Devillers M; Cruaud C; Benjannet S; Wickham L; Erlich D; Derre A; Villeger L; Farnier M; Beucler I; Bruckert E; Chambaz J; Chanu B; Lecerf J-M; Luc G; Moulin P; Weissenbach J; Prat A; Krempf M; Junien C; Seidah NG; Boileau C Nat. Genet 2003, 34, 154–156. [DOI] [PubMed] [Google Scholar]
- (13).Zhao Z; Tuakli-Wosornu Y; Lagace TA; Kinch L; Grishin NV; Horton JD; Cohen JC; Hobbs HH Am. J. Hum. Genet 2006, 79, 514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Steinberg D; Witztum JL Proc. Natl. Acad. Sci. U. S. A 2009, 106, 9546–9547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Stein EA; Swergold GD Curr. Atheroscler. Rep 2013, 15, 310. [DOI] [PubMed] [Google Scholar]
- (16).Seidah NG; Prat A Nat. Rev. Drug Discovery 2012, 11, 367–383. [DOI] [PubMed] [Google Scholar]
- (17).Chan JCY; Piper DE; Cao Q; Liu D; King C; Wang W; Tang J; Liu Q; Higbee J; Xia Z; Di Y; Shetterly S; Arimura Z; Salomonis H; Romanow WG; Thibault ST; Zhang R; Cao P; Yang X-P; Yu T; Lu M; Retter MW; Kwon G; Henne K; Pan O; Tsai M-M; Fuchslocher B; Yang E; Zhou L; Lee KJ; Daris M; Sheng J; Wang Y; Shen WD; Yeh W-C; Emery M; Walker NPC; Shan B; Schwarz M; Jackson SM Proc. Natl. Acad. Sci. U. S. A 2009, 106, 9820–9825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ni YG; Di Marco S; Condra JH; Peterson LB; Wang W; Wang F; Pandit S; Hammond HA; Rosa R; Cummings RT; Wood DD; Liu X; Bottomley MJ; Shen X; Cubbon RM; Wang S.-p.; Johns DG; Volpari C; Hamuro L; Chin J; Huang L; Zhao JZ; Vitelli S; Haytko P; Wisniewski D; Mitnaul LJ; Sparrow CP; Hubbard B; Carfi A; Sitlani A J. Lipid Res 2011, 52, 78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Raal FJ; Stein EA; Dufour R; Turner T; Civeira F; Burgess L; Langslet G; Scott R; Olsson AG; Sullivan D; Hovingh GK; Cariou B; Gouni-Berthold I; Somaratne R; Bridges I; Scott R; Wasserman SM; Gaudet D Lancet 2015, 385, 331–340. [DOI] [PubMed] [Google Scholar]
- (20).Raal FJ; Honarpour N; Blom DJ; Hovingh GK; Xu F; Scott R; Wasserman SM; Stein EA Lancet 2015, 385, 341–350. [DOI] [PubMed] [Google Scholar]
- (21).Zhang Y; Eigenbrot C; Zhou L; Shia S; Li W; Quan C; Tom J; Moran P; Di Lello P; Skelton NJ; Kong-Beltran M; Peterson A; Kirchhofer DJ Biol. Chem 2014, 289, 942–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Mayer G; Poirier S; Seidah NG J. Biol. Chem 2008, 283, 31791–31801. [DOI] [PubMed] [Google Scholar]
- (23).Seidah NG; Poirier S; Denis M; Parker R; Miao B; Mapelli C; Prat A; Wassef H; Davignon J; Hajjar KA; Mayer G PLoS One 2012, 7, e41865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Shan L; Pang L; Zhang R; Murgolo NJ; Lan H; Hedrick JA Biochem. Biophys. Res. Commun 2008, 375, 69–73. [DOI] [PubMed] [Google Scholar]
- (25).Du F; Hui Y; Zhang M; Linton MF; Fazio S; Fan DJ Biol. Chem. 2011, 286, 43054–43061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Palmer-Smith H; Basak A Curr. Med. Chem 2010, 17, 2168–2182. [DOI] [PubMed] [Google Scholar]
- (27).Lammi C; Zanoni C; Aiello G; Arnoldi A; Grazioso G Sci. Rep. 2016, 6, 29931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Guay D; Crane S; Lachance N; Chiasson J-F; Truong VL; Lacombe P; Skorey K; Seidah NG Patent WO 2014139008, 2014.
- (29).Barta TE; Bourne JW; Monroe KD; Muehlemann MM; Pandey A; Bowers S Patent WO 2017034990, 2017.
- (30).Min D-K; Lee H-S; Lee N; Lee CJ; Song HJ; Yang GE; Yoon D; Park SW Yonsei Med. J 2015, 56, 1251–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Park SW; Lee HS; Min DK; Lee NR; Lee CJ; Yang GE Patent WO 2016108572, 2016.
- (32).Stucchi M; Grazioso G; Lammi C; Manara S; Zanoni C; Arnoldi A; Lesmaa G; Silvania A Org. Biomol. Chem 2016, 14, 9736–9740. [DOI] [PubMed] [Google Scholar]
- (33).Ko E; Raghuraman A; Perez LM; Ioerger TR; Burgess K J. Am. Chem. Soc 2013, 135, 167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Xin D; Holzenburg A; Burgess K Chem. Sci 2014, 5, 4914–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).McNutt MC; Kwon HJ; Chen C; Chen JR; Horton JD; Lagace TA J. Biol. Chem 2009, 284, 10561–10570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kwon HJ; Lagace TA; McNutt MC; Horton JD; Deisenhofer J Proc. Natl. Acad. Sci. U. S. A 2008, 105, 1820–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kortemme T; Kim DE; Baker D Sci. Signaling 2004, 2004, [Google Scholar]
- (38).Koes DR; Camacho CJ Bioinformatics 2012, 28, 784–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Accordino SR; Morini MA; Sierra MB; Fris JAR; Appignanesi GA; Fernandez A Proteins: Struct., Funct., Bioinf 2012, 80, 1755–1765. [DOI] [PubMed] [Google Scholar]
- (40).London N; Raveh B; Schueler-Furman O Curr. Opin. Chem. Biol 2013, 17, 952–959. [DOI] [PubMed] [Google Scholar]
- (41).Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS J. Med. Chem 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- (42).Halgren TA; Murphy RB; Friesner RA; Beard HS; Frye LL; Pollard WT; Banks JL J. Med. Chem 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- (43).Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT J. Med. Chem 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- (44).Kaminski GA; Friesner RA; Tirado-Rives J; Jorgensen WL J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar]
- (45).Kan T; Fukuyama T Chem. Commun 2004, 353–359. [DOI] [PubMed] [Google Scholar]
- (46).Meusel M; Guetschow M Org. Prep. Proced. Int 2004, 36, 391–443. [Google Scholar]
- (47).Jorgensen WL; Duffy EM Bioorg. Med. Chem. Lett 2000, 10, 1155–1158. [DOI] [PubMed] [Google Scholar]
- (48).Zhao B; Burgess K ACS Med. Chem. Lett 2018, 9, 155–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Duffy EM; Jorgensen WL J. Am. Chem. Soc 2000, 122, 2878–2888. [Google Scholar]
- (50).Wipf P; Xiao J; Stephenson CRJ Chimia 2009, 63, 764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Lee MH; Han JH; Kwon P-S; Bhuniya S; Kim JY; Sessler JL; Kang C; Kim JS J. Am. Chem. Soc 2012, 134, 1316–1322. [DOI] [PubMed] [Google Scholar]
- (52).Thao LQ; Lee C; Kim B; Lee S; Kim TH; Kim JO; Lee ES; Oh KT; Choi H-G; Yoo SD; Youn YS Colloids Surf., B 2017, 152, 183–191. [DOI] [PubMed] [Google Scholar]
- (53).Lai C-H; Chang T-C; Chuang Y-J; Tzou D-L; Lin C-C Bioconjugate Chem. 2013, 24, 1698–1709. [DOI] [PubMed] [Google Scholar]
- (54).Margarida Cardosos M; Peca IN; Raposo CD; Petrova KT; Teresa Barros M; Gardner R; Bicho A J. Microencapsulation 2016, 33, 315–322. [DOI] [PubMed] [Google Scholar]
- (55).Gankhuyag N; Singh B; Maharjan S; Choi Y-J; Cho C-S; Cho M-H Macromol. Biosci 2015, 15, 777–787. [DOI] [PubMed] [Google Scholar]
- (56).Feng L; Yu H; Liu Y; Hu X; Li J; Xie A; Zhang J; Dong W Polym. Chem 2014, 5, 7121–7130. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.