

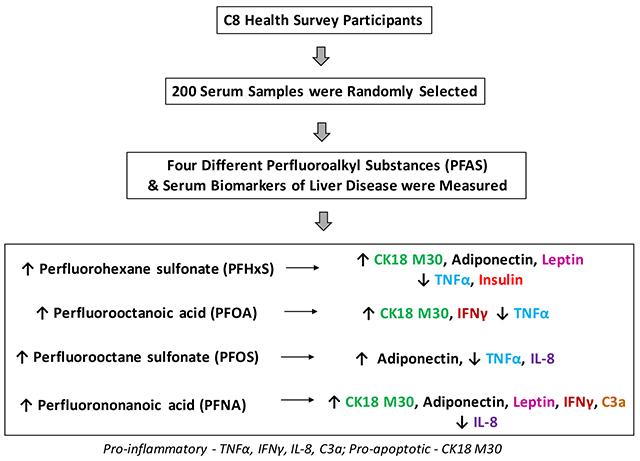
Abstract
Exposures to perfluoroalkyl substances (PFAS) including perfluoroalkyl acids (PFAAs) are associated with increased liver enzymes in cohort studies including the C8 Health Study. In animal models, PFAAs disrupt hepatic lipid metabolism and induce apoptosis to cause nonalcoholic fatty liver disease (NAFLD). PFAAs are immunotoxic and inhibit pro-inflammatory cytokine release from stimulated leukocytes in vitro. This cross-sectional study tests the hypothesis that environmental PFAAs are associated with increased hepatocyte apoptosis and decreased pro-inflammatory cytokines in serum. Biomarkers previously associated with PFAS exposures and/or NAFLD were evaluated as secondary endpoints. Two hundred adult C8 Health Study participants were included. Measured serum biomarkers included: perfluorohexane sulfonate (PFHxS); perfluorooctanoic acid (PFOA); perfluorooctane sulfonate (PFOS); perfluorononanoic acid (PFNA); cytokeratin 18 M30 (CK18 M30, hepatocyte apoptosis); adipocytokines; insulin; and cleaved complement 3 (C3a). Confounder-adjusted linear regression models determined associations between PFAS and disease biomarkers with cut-offs determined by classification and regression tree analysis. CK18 M30 was positively associated with PFHxS (β=0.889, p= 0.042); PFOA (β=2.1, p=0.005); and PFNA (β=0.567, p=0.03). Tumor necrosis factor α (TNFα) was inversely associated with PFHxS (β= −0.799, p=0.001); PFOA (β= − 1.242, p=0.001); and PFOS (β= −0.704, p<.001). Interleukin 8 was inversely associated with PFOS and PFNA. PFAAs were also associated with sexually dimorphic adipocytokine and C3a responses. Overall, PFAA exposures were associated with the novel combination of increased biomarkers of hepatocyte apoptosis and decreased serum TNFα. These data support previous findings from cohorts and experimental systems that PFAAs may cause liver injury while downregulated some aspects of the immune response. Further studies of PFAAs in NAFLD are warranted and should evaluate sex differences.
Keywords: Perfluoroalkyl acids (PFAAs), perfluorooctanoic acid (PFOA), non-alcoholic fatty liver disease (NAFLD), cytokeratin 18, tumor necrosis factor alpha (TNFα)
Graphical Abstract
Introduction
Perfluoroalkyl acids (PFAAs) are common manmade compounds within the larger group of per- and polyfluoroalkyl substances (PFAS). They have multiple useful consumer applications including lubrication as well as resistance to grease, stains, and water. Industrial applications include hydraulics, electronics, enhanced extraction of oil and natural gas, and firefighting. Recently, widespread groundwater contamination has been found at airports and especially military airbases where PFAAs were used for multiple purposes including firefighting foams to extinguish jet fuel fires (Milley et al., 2018; Sunderland et al., 2018). Unfortunately, PFAAs persist in the environment and bioaccumulate in humans. Human exposures occur primarily through ingestion of contaminated drinking water and food. PFAAs have been detected in the serum of almost all people tested in the developed world (Domingo and Nadal, 2017). Longer chain PFAAs have extremely long half-lives in humans (Worley et al., 2017) and accumulate in the liver and other organs (Perez et al., 2013).
PFAS are hepatotoxic, immunotoxic, and endocrine and metabolism disrupting chemicals (DeWitt et al., 2012; Heindel et al., 2017). Environmental cohort studies from North America, Europe, and Asia have consistently reported positive associations between PFAS exposures and liver enzymes (Darrow et al., 2016; Gallo et al., 2012; Gleason et al., 2015; Lin et al., 2010; Salihovic et al., 2018; Yamaguchi et al., 2013). The largest liver enzyme study (n=47,092) found positive associations between alanine aminotransferase (ALT) levels and exposures to perfluorooctanoic acid (PFOA, or C8) and perfluorooctane sulfonic acid (PFOS) (Gallo et al., 2012). The association between PFOA and ALT was subsequently confirmed in 30,723 C8 participants through different techniques; although no associations were found between exposure and self-reported (and subsequently medically validated) liver disease history (Darrow et al., 2016). However, because liver disease may be asymptomatic and subclinical early in its course, the present study applies mechanistic serologic biomarkers to further investigate liver toxicity in the C8 cohort.
Nonalcoholic fatty liver disease (NAFLD) is the most prevalent liver disease, affecting approximately 25% of the global population (Younossi et al., 2015). NAFLD occurs as a consequence of altered lipid metabolism, resulting in excess lipid accumulation (steatosis) within hepatocytes. PFAA exposures disrupt normal hepatic lipid metabolism to cause steatosis in animal models (Bijland et al., 2011; Das et al., 2017; Kim et al., 2011; Tan et al., 2013; Wan et al., 2012; Zhang et al., 2016). Mechanisms implicated in PFAA-induced steatosis include the upregulation of hepatic de novo lipogenesis and lipid uptake from blood (Das et al., 2017) as well as the downregulation of VLDL production/export, possibly due to a reduction of bioavailable choline (Bijland et al., 2011; Zhang et al., 2016).
Hyperlipidemia is the most common comorbid condition associated with human NAFLD (Younossi et al., 2015). PFAA exposures have reproducibly been positively associated with hyperlipidemia in adult and pediatric cohort studies including the C8 Health Study (Frisbee et al., 2010; Geiger et al., 2014; Nelson et al., 2010; Steenland et al., 2009). In contrast to the epidemiological data, PFAA exposures decreased blood lipids in rodents fed a standard diet. However, mice fed a hypercaloric ‘Western’ diet developed hypercholesterolemia with PFAS treatment (Rebholz et al., 2016). Possible receptor-based modes of action for PFAS-induced alterations in liver and blood lipids include: inhibition of hepatocyte nuclear factor 4 α (HNF4α); and activation of the xenobiotic receptors, pregnane X receptor (PXR) and constitutive androstane receptor (CAR); the lipid receptors, liver X receptor (LXR) and peroxisome proliferator activated receptors α and γ (PPARα and PPARγ); the bile acid receptor, farnesoid x receptor (FXR); and the sex hormone receptor, estrogen receptor α (ERα) (Bjork et al., 2011; Buhrke et al., 2015; Rosen et al., 2017). Importantly, all of these nuclear receptors have been implicated in NAFLD (Cave et al., 2016). ERα could potentially confer sex differences in PFAS-dysregulated hepatic lipid metabolism and NAFLD; and sex differences have been reported in rodent models (Kim et al., 2011; Rebholz et al., 2016).
Experimental systems demonstrate that PFAS exposures induced hepatocyte caspase 3-mediated apoptosis; increased oxidative stress while reducing NRF2-regulated hepatic antioxidant defenses; and elicited cytoskeletal remodeling (Kim et al., 2011; Wan et al., 2016; Zhang et al., 2016). However, PFAS are potent immunotoxic chemicals which suppress innate immune function. In experimental systems, PFAS reduced LPS-stimulated tumor necrosis factor α (TNFα) production from leukocytes ex vivo in whole blood from human volunteers and also in vitro, through a partially PPARα-dependent mechanism (DeWitt et al., 2012). This anti-inflammatory action is consistent with the significantly reduced lobular inflammation associated with PFAS exposures noted on liver biopsy in bariatric surgery patients with NAFLD (Rantakokko et al., 2015). Based on this literature, we postulate that PFAA exposures can cause human NAFLD - associated with hepatocyte apoptosis and paradoxically without increased and possibly with reduced, liver inflammation. This cross-sectional study measures mechanistic serological biomarkers in adult C8 Health Study participants, to test the hypothesis that PFAS exposures are associated with increased hepatocyte apoptosis and decreased pro-inflammatory cytokines. Secondary endpoints included a panel of other serum biomarkers previously implicated in NAFLD and also evaluation of potential sex differences. The significance of this study is that it could help to explain how environmental chemical exposures impact the genesis and progression of liver diseases including NAFLD.
Methods
Serum Specimens and Study Population
The C8 Health Study is a very large population enrollment study from six water districts that were contaminated with perfluorooctanoic acid (PFOA) in the mid-Ohio Valley of Ohio and West Virginia. Recruitment and data collection for the entire C8 Health Study have been described previously (Frisbee et al., 2009). Two hundred serum specimens were randomly selected from the banked specimens of the adult participant population age 40-70 (West Virginia University IRB protocol 190943735), with deliberate oversampling for African American participants and for BMI ≤25 in order to obtain a range of BMI. Individuals who reported that they had hepatitis of any origin (viral or otherwise) and from those reporting consumption of >3 alcoholic drinks/day were excluded from the sampling. Serum specimens were stored since 2006 at − 80°C and some had undergone freeze-thaw cycle before this investigation was performed in 2017. The sample size (N number) may differ slightly for some measurements due to missing values from the original data set. Missing values are assumed to be missing at random.
Serum Biomarkers of PFAA Exposure
Exposure biomarkers included serum PFOA, PFOS, perfluorohexane sulfonic acid (PFHxS), and perfluorononanoic acid (PFNA). Other PFAS in the data set featured many samples below the limit of detection and were unsuited for the purposes of this study. The analytical method for PFAS measurement was derived from Flaherty et al. (Flaherty et al., 2005) and has been previously reported (Frisbee et al., 2009; Steenland et al., 2010). Briefly, protein precipitation extraction was followed by reverse-phase high performance liquid chromatography-tandem mass spectrometry in selection mode for the individual PFAS species, compared to a 13C surrogate for quality assurance. Estimates of precision were in the range of ±10%.
Serum Disease Biomarkers
The selected serum disease biomarkers were similar to those measured in recent environmental or occupational liver disease cohort studies (Cave et al., 2011; Cave et al., 2010; Clair et al., 2018; Hsieh et al., 2017). Primary serologic outcome biomarkers include the caspase-cleaved cytokeratin 18 fragment (CK18 M30) and pro-inflammatory cytokines. CK18 is a cytoskeletal protein found in epithelial cells including hepatocytes. Dying hepatocytes release CK18 which can then be measured in serum. Levels of the whole protein, CK18 M65, reflect cell death from all causes, while the caspase 3-cleaved fragment (CK18 M30) reflects apoptotic cell death. CK18 M30 is a promising serologic NAFLD biomarker (Feldstein et al., 2009; He et al., 2017). CK18 M30 was positively associated with traffic-related air pollution in overweight and obese children with fatty liver (Hsieh et al., 2017). In contrast, the toxicant associated fatty liver disease (TAFLD) previously associated with VOC and PCB exposures was positively associated with CK18 M65, but not CK18 M30 (Cave et al., 2011; Cave et al., 2010; Clair et al., 2018). This difference is likely related to the predominant hepatocellular death mechanism: necrosis versus apoptosis. Because ALT was previously associated with PFAS exposures in the C8 Health Study (Darrow et al., 2016; Gallo et al., 2012), and animal exposure models demonstrated hepatic steatosis with cytoskeletal remodeling and caspase 3-mediated apoptosis (Kim et al., 2011; Wan et al., 2016); CK 18 M30 was selected as a primary outcome variable for this study. Serum CK18 M30 and M65 were measured by ELISA (Diapharma, West Chester, OH, kits #10011 and 10020) according to manufacturer’s instructions using an EPOCH II plate reader running Gen 5 software (Biotek Instruments Inc., Winooski, VT). Measurement of the serum pro-inflammatory cytokines, TNFα, interleukin 6 (IL-6), interleukin 8 (IL-8), and interferon γ (IFNγ), was performed using the Multispot Assay System for Human ProInflammatory Panel II (4-plex kit) per the accompanying manual. The data were acquired and calculated employing the Sector Imager 2400 running Discovery Workbench Software Version 4 (Mesoscale Discovery LLC, Rockville, MD).
Measurement of secondary outcome protein hormones adiponectin, insulin, and leptin was performed using the MSD single spot assay system specific for each analyte. Measurement of cleaved complement 3 (C3a) and plasminogen activator ihhibitor-1 (PAI-1) was performed using Human c3a Platinum Elisa Kit and Human PAI-1 ELISA kit (Affymetrix, ThermoFisher, Waltham, MA) according to kit manuals using the EPOCH II plate reader.
Statistical Analysis
SAS 9.4 and R software (version 3.4.4 R Foundation, Vienna, Austria) were used to examine the association between biomarker expressions and PFAS serum concentrations, adjusting for the potential confounders commonly considered in PFAS studies regarding hepatic effects such as age, alcohol consumption, BMI, sex, and the estimated glomerular filtration rate (eGFR, using the modification of diet in renal disease standard equation) (Levey et al., 2006). Alcohol consumption was categorized as zero or 1-3 drinks per day. Potential subjects with greater alcohol consumption were excluded. Exposure and disease biomarkers were natural log transformed to achieve approximate normality and variance stabilization during the data processing procedure. Exploratory analyses were used to summarize the data, including mean with standard deviation, summary tables and bar-plots. Additionally, a heat map was used to explore the biomarker data simultaneously, and to illustrate which the biomarkers were clustered using hierarchical algorithm with Euclidean distance.
Univariate analysis began with a preliminary test to establish a statistical cutoff level for each biomarker. Using the CART method to express the statistical cut point, a regression tree is built through a process known as binary recursive partitioning, which is an iterative process that splits the data into partitions or branches, and then continues splitting each partition into smaller groups as the method moves up each branch. As a result, the different subgroups are identified to predict the response variable. The cut point for each biomarker is individually assessed using the CART method. From these specified levels, the observed biomarker data are then dichotomized into either high, or low categories, depending on the cut point designated for each biomarker.
The goal of this method is to create a model that predicts biomarker values based on serum PFAS concentrations. The algorithm computes where to prune a tree based on the deviance and constructs a classification tree with the important conditions based on the biomarkers (high versus low). Decision tree analysis is increasingly used to support development of algorithms using biomarker screening candidates (Chen et al., 2011; Marshall, 2001). In order to identify a unique cutoff, a summed PFAS mixture concentration was computed based on the normalized z-scores of each PFAS variable; and log-transformed biomarker data were used. Using a t-test, or when necessary based on deviance from statistical assumptions, the Wilcoxon ranked Sum test are implemented to compare the dichotomized levels of each biomarker.
In the multivariate analysis, a linear regression model adjusting for potential confounding variables was used to assess PFAS concentrations by the categorized disease biomarkers. Cognizant of published sex differences in health outcomes associated with PFAS exposures, an interaction term of sex and biomarkers was included in the linear regression model. The results of the multivariate regression model are similar to that of a factorial ANOVA design, with comparable results. Statistical significance was set at both the 0.05 and 0.1 levels. The higher threshold p value (p=0.1) was included to reduce Type II errors, because this analysis could be considered a pilot study investigating only a small portion of the overall cohort. In all cases, the actual p value is provided.
Results
Demographic data are provided in Tables 1A–B. Mean participant age was 55.3±8.4, 54% were women, and 61% were currently non-drinkers. The oversampling procedures a distribution of BMI categories (normal weight 40.5%, overweight 27.5%, and obese 32%) as well as a significant number of African Americans (19% vs. 3% in the overall C8 Health Study). Mean serum levels of PFAS (ng/mL) were: PFOA= 94.6±183.6 (SD); PFOS = 26.9±16.7; PFHxS = 4.2±3.9; and PFNA = 1.6±0.7. Mean serum liver enzymes were: alanine aminotransferase (ALT) = 24.0±12.3 (U/L, SD) and aspartate aminotransferase (AST) = 22.8±7.9 (U/L). Serum exposure and aminotransferase biomarkers were similar between sexes (Table 1A).
Table 1A:
Descriptive statistics for continuous factors
| Variable | Overall | Male | Female | ||||
|---|---|---|---|---|---|---|---|
| Mean (SD) | N | Mean (SD) | N | Mean (SD) | N | ||
| Age (years) | 55.3 (8.4) | 200 | 55.4 (8.6) | 92 | 55.2 (8.3) | 108 | |
| Weight (lbs) | 181.1 (44.5) | 200 | 200.6 (39.7) | 92 | 164.4 (41.6) | 108 | |
| Height (inches) | 66.9 (4.2) | 200 | 70.6 (2.8) | 92 | 63.7 (2.3) | 108 | |
| BMI | 28.3 (6.0) | 200 | 28.2 (4.7) | 92 | 28.4 (7.0) | 108 | |
| ALT (U/L) | 24.0 (12.5) | 199 | 26.8 (14.5) | 92 | 21.7 (9.87) | 107 | |
| AST (U/L) | 22.8 (7.3) | 199 | 23.0 (6.5) | 92 | 22.50 (7.9) | 107 | |
| e-GFR (Calculated) | 71.6 (15.5) | 199 | 74.0 (15.9) | 92 | 69.6 (14.9) | 107 | |
| Analyte (ng/mL) | PFHxS | 4.2 (3.9) | 200 | 5.1 (4.6) | 92 | 3.4 (2.9) | 108 |
| PFOA | 94.6 (183.6) | 200 | 97.0 (199.9) | 92 | 92.6 (169.3) | 108 | |
| PFOS | 26.9 (16.7) | 200 | 29.1 (17.8) | 92 | 25.0 (15.5) | 108 | |
| PFNA | 1.6 (0.7) | 200 | 1.6 (0.8) | 92 | 1.6 (0.6) | 108 | |
All available data used for analysis
Table 1B:
Descriptive statistics for categorical factors
| Variable | N | Male 1 | Female 1 | |
|---|---|---|---|---|
| Frequency (%) | Frequency (%) | |||
| Current Alcohol User | Yes | 78 | 43 (55.1) | 35 (44.9) |
| No | 122 | 49 (40.2) | 73 (59.8) | |
| Average Alcohol Consumption | Not Classified | 57 | 31 (54.4) | 26 (45.6) |
| 1-3 Drinks per day | 12 | 10 (83.3) | 2 (16.7) | |
| A few drinks per week | 23 | 14 (60.9) | 9 (39.1) | |
| Less than one drink per month | 48 | 22 (45.8) | 26 (54.2) | |
| None | 60 | 15 (25.0) | 45 (75.0) | |
| African American | Yes | 38 | 22 (57.9) | 16 (42.1) |
| No | 162 | 70 (43.2) | 92 (56.8) | |
| BMI Classification | Normal BMI (19.0 - 24.9) | 81 | 34 (42.0) | 47 (58.0) |
| Overweight BMI (25.0 - 29.9) | 55 | 25 (45.5) | 30 (54.6) | |
| Obese BMI (≥ 30.0) | 64 | 33 (51.6) | 31 (48.4) | |
No missing values
A heat map of normalized disease biomarkers (Figure 1) demonstrates a visual picture of the correlations between high (red) and low (green) variables. The closest relationship was between the hepatocyte total cell death biomarker (CK18 M65) and the hepatocyte apoptosis biomarker (CK18 M30) as anticipated. Weaker correlations were observed between TNFα and IL-8; IFNγ and IL-6; as well as insulin and leptin. Disease biomarker data were grouped into ‘low’ or ‘high’ categories based on cutoffs determined by CART analysis. The cutoff values are provided in the Supplementary Table The univariate analysis (Supplementary Table) examined the relationships between mean PFAS levels in the ‘low’ vs. ‘high’ category for each disease biomarker. Means for univariate and multivariate analyses are compared using a t-test, and when necessary, the Wilcoxon Ranked Sum test is used when t-test assumptions, namely sample size requirements, falter. The Supplementary Figure depicts this information graphically. The multivariate models (Table 2) determined interaction coefficients (Interaction Model) investigating sexually dimorphic responses as well as adjusted beta coefficients (Final Model). Figure 2 provides the same information visually for each of the four evaluated PFAS. The univariate and multivariate results were found to be similar and this relationship is illustrated in Table 3. This table summarizes the results from univariate model between PFAS and biomarkers alone (univariate model) versus multivariable model with additional confounding variables/interaction term.
Figure 1. Disease Biomarker Heat Map.
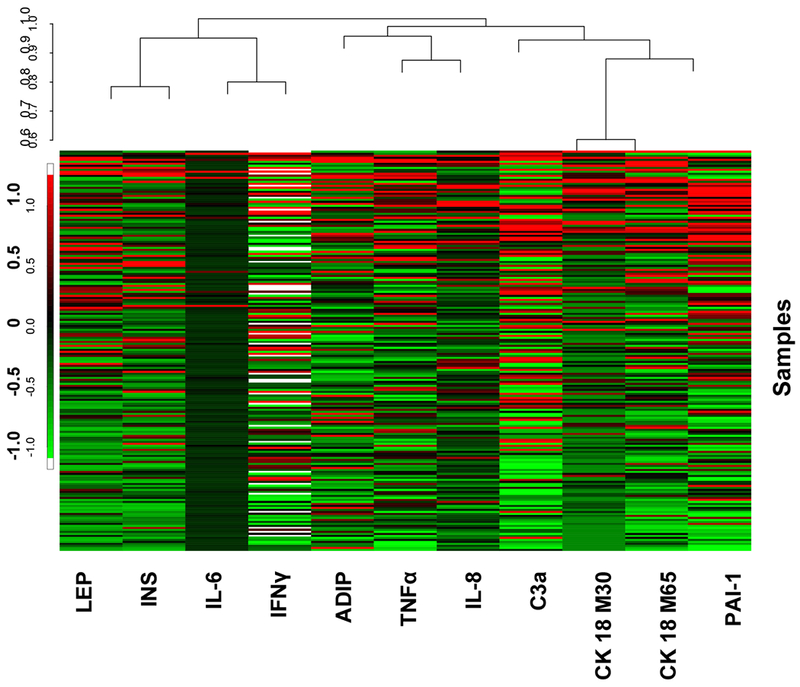
A heat map of the normalized logarithm of expression levels of biomarkers is demonstrated in colors that reflect expression levels (green represents low expression levels, red represents high expression levels). A color legend showing the normalized expression is on the left. The Venn diagram located in the upper part of the figure represents the exploratory hierarchical clustering analysis for associations among biomarkers based on their similarity, with the relative distance shown on the left axis (the smaller the closer).
Table 2:
Multivariate analysis, adjusting for e-GFR, alcohol consumption category, BMI, age and sex
| Analyte | Biomarker 1 | Interaction Model 2 | Final Model 3 | ||||
|---|---|---|---|---|---|---|---|
| Interaction Coefficient | Beta Coefficient (Analyte) | ||||||
| Estimate | SE | P Value | Estimate | SE | P Value | ||
| PFHxS | CK18 M30 | −0.081 | 0.882 | 0.927 | 0.889 | 0.434 | 0.042 |
| CK18 M65 | 0.411 | 0.838 | 0.624 | 0.966 | 0.418 | 0.022 | |
| TNFα | 0.166 | 0.430 | 0.700 | −0.799 | 0.214 | <0.001 | |
| IL-6 | −0.022 | 1.009 | 0.983 | 0.551 | 0.487 | 0.260 | |
| IL-8 | −1.027 | 0.524 | 0.051 | 0.371 | 0.350 | 0.291 | |
| Interferon γ | −0.261 | 0.383 | 0.498 | 0.032 | 0.191 | 0.868 | |
| C3a | 0.615 | 0.336 | 0.068 | −0.176 | 0.234 | 0.453 | |
| PAI-1 | 0.002 | 0.003 | 0.520 | <−0.001 | 0.002 | 0.826 | |
| Insulin | 0.503 | 0.478 | 0.293 | −0.442 | 0.231 | 0.057 | |
| Leptin | 0.172 | 0.338 | 0.611 | 0.471 | 0.168 | 0.006 | |
| Adiponectin | −0.675 | 0.348 | 0.054 | 0.562 | 0.259 | 0.031 | |
| PFOA | CK18 M30 | 0.118 | 1.500 | 0.937 | 2.100 | 0.739 | 0.005 |
| CK18 M65 | −0.736 | 1.445 | 0.611 | 1.460 | 0.720 | 0.044 | |
| TNFα | 0.479 | 0.743 | 0.520 | −1.242 | 0.370 | 0.001 | |
| IL-6 | −1.259 | 1.735 | 0.469 | 0.718 | 0.839 | 0.393 | |
| IL-8 | −1.172 | 0.902 | 0.195 | −0.593 | 0.441 | 0.180 | |
| Interferon γ | 0.162 | 0.651 | 0.804 | 0.580 | 0.325 | 0.076 | |
| C3a | 1.193 | 0.573 | 0.039 | −0.136 | 0.400 | 0.733 | |
| PAI-1 | −0.010 | 0.005 | 0.059 | 0.003 | 0.004 | 0.469 | |
| Insulin | 1.212 | 0.827 | 0.177 | −0.142 | 0.401 | 0.723 | |
| Leptin | −0.074 | 0.592 | 0.900 | 0.310 | 0.294 | 0.292 | |
| Adiponectin | 0.046 | 0.606 | 0.940 | 0.079 | 0.310 | 0.800 | |
| PFOS | CK18 M30 | 0.198 | 0.811 | 0.807 | 0.536 | 0.400 | 0.182 |
| CK18 M65 | −0.543 | 0.772 | 0.483 | 0.557 | 0.385 | 0.149 | |
| TNFα | −0.115 | 0.394 | 0.771 | −0.704 | 0.196 | <0.001 | |
| IL-6 | −0.065 | 0.925 | 0.944 | 0.322 | 0.446 | 0.471 | |
| IL-8 | −0.511 | 0.477 | 0.586 | −0.509 | 0.233 | 0.030 | |
| Interferon γ | −0.533 | 0.349 | 0.128 | 0.245 | 0.175 | 0.163 | |
| C3a | 0.505 | 0.306 | 0.100 | 0.018 | 0.213 | 0.933 | |
| PAI-1 | 0.002 | 0.003 | 0.390 | <−0.001 | 0.001 | 0.673 | |
| Insulin | 0.409 | 0.440 | 0.354 | −0.199 | 0.213 | 0.351 | |
| Leptin | 0.559 | 0.313 | 0.075 | −0.131 | 0.221 | 0.554 | |
| Adiponectin | −0.277 | 0.318 | 0.385 | 0.318 | 0.163 | 0.053 | |
| PFNA | CK18 M30 | −0.203 | 0.531 | 0.702 | 0.567 | 0.262 | 0.031 |
| CK18 M65 | −0.383 | 0.511 | 0.454 | 0.254 | 0.255 | 0.320 | |
| TNFα | 0.279 | 0.267 | 0.297 | −0.153 | 0.133 | 0.251 | |
| IL-6 | −0.166 | 0.611 | 0.786 | −0.046 | 0.295 | 0.876 | |
| IL-8 | −0.184 | 0.314 | 0.557 | −0.392 | 0.153 | 0.011 | |
| Interferon γ | −0.447 | 0.222 | 0.046 | 0.546 | 0.156 | 0.001 | |
| C3a | −0.074 | 0.200 | 0.710 | 0.282 | 0.101 | 0.006 | |
| PAI-1 | <0.001 | 0.002 | 0.861 | <−0.001 | 0.001 | 0.928 | |
| Insulin | 0.345 | 0.290 | 0.236 | −0.124 | 0.141 | 0.379 | |
| Leptin | 0.206 | 0.206 | 0.320 | 0.179 | 0.103 | 0.083 | |
| Adiponectin | −0.2700 | 0.209 | 0.198 | 0.231 | 0.107 | 0.033 | |
Units of biomarker: ADIP, LEP, INS, TNFα, IL-6, IL-8, IFNγ (pg/mL); M30, M65 (U/mL); C3a, PAI-1 (ng/mL)
Interaction Model: Evaluates the interaction between biomarker concentration and sex
Final Model: Interaction term only included if it was found to be significant from Interaction Model
Figure 2. Significance of Biomarker Concentration in the Final Model.
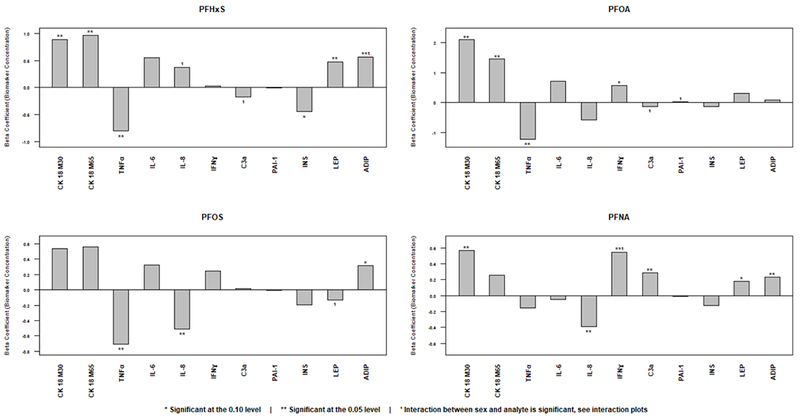
Adjusted beta coefficients are provided for each exposure and disease biomarker. These are graphical representations of the multivariate analysis results given in Table 2 (Final Model). Note that the range of the Y-axis varies between figures.
Table 3:
Summary of statistically significant results by model
| Exposure Variable | PFHxS | PFOA | PFOS | PFNA | ||||
|---|---|---|---|---|---|---|---|---|
| Statistical Model | Univariate1 | Multivariate | Univariate1 | Multivariate | Univariate1 | Multivariate | Univariate1 | Multivariate |
| Cytokeratin 18 M30 | ▴ | ▴ | ▴ | ▴ | ▴ | ▴ | ▴ | |
| Cytokeratin 18 M65 | ▴ | ▴ | ▴ | ▴ | ▴ | |||
| Tumor Necrosis Factor α | ▾ | ▾ | ▾ | ▾ | ▾ | ▾ | ||
| Interleukin 6 | ▴ | |||||||
| Interleukin 8 | * | ▾ | ▾ | ▾ | ▾ | |||
| Interferon γ | ▴ | ▴ | ▴ * | |||||
| Cleaved complement 3 | * | ▴ | * | ▴ | ▴ | ▴ | ||
| Plasminogen activator inhibitor 1 | * | ▾ | ||||||
| Insulin | ▾ | ▾ | ||||||
| Leptin | ▴ | * | ▴ | ▴ | ||||
| Adiponectin | ▴ * | ▴ | ▴ | ▴ | ▴ | |||
By Wilcoxon Ranked Sum Test
Denotes significant sex difference, statistical significance at the 0.10 level
▴▾ – Indicates positive or negative direction of associations
The primary disease biomarkers were CK18 and pro-inflammatory cytokines. In experimental systems, PFAS exposures were associated with caspase-3 dependent hepatocyte apoptosis such as CK18 (Kim et al., 2011). In the univariate analysis, participants with elevated caspase 3-cleaved CK18 (M30) had significantly higher mean levels of PFHxS, PFOA, PFOS, and PFNA (Supplementary Table). Likewise, those with elevated CK18 M65 had significantly higher mean levels of PFOA, PFOS, PFNA. In the multivariate model, M30 was positively associated with PFHxS (SE) (β=0.889±0.434, p=0.042); PFOA (β=2.1±0.739, p=0.005); and PFNA (β=0.567±0.262, p=0.03) with a trend for PFOS (p=0.182). M65 was positively associated with PFHxS (β=0.996±0.418, p=0.022); PFOA (β=1.460±0.720, p=0.044) with a trend for PFOS (p=0.149). Thus, M30 and M65 were positively associated with every PFAS tested in at least one statistical model. No sex differences were noted. These data suggest that there may be a class effect for PFAS in the promotion of liver disease and caspase-3 mediated hepatocellular apoptosis. This effect was independent of sex.
In our data, three PFAS were associated with significantly reduced serum TNFα in both the univariate analysis (Supplementary Table) and multivariate final model [PFHxS (β= −0.799±0.214, p<0.001); PFOA (β= −1.242±0.370, p=0.001); and PFOS (β= −0.704±0.196, p<0.001)]. Likewise, two PFAS were associated with significantly reduced IL-8 in both the univariate analysis and the multivariate final model [PFOS (β= −0.509±0.233, p<0.030) and PFNA (β= −0.392±0.153, p=0.011)]. The relationship between PFHxS and IL-8 varied with near statistical significance by sex (interaction coefficient= −1.027±0.524, p=0.051); and the association was inverse in males and positive in females (Figure 3). In contrast, IFNγ was positively associated with PFOA (multivariate final model, β= 0.580±0.325, p=0.076) and PFNA (univariate and multivariate final models, β=0.546±0.156, p<0.001). The PFNA association appears to have been driven primarily by effects in females (interaction coefficient= −0.447±0.222, p=0.046, Figure 3). In the univariate model, a significant positive association was seen between PFHxS and IL-6, while PFOS and PAI-1 were inversely associated. However, no significant associations were seen in the final multivariate model for any PFAS exposure and either IL-6 or PAI-1. A near-significant sex difference was noted for PFOA and PAI-1 (Table 2), but the interaction coefficient was small (−0.010±0.005, p=0.059). In summary, the final multivariate model demonstrated inverse relationships between three PFAS exposures with TNFα, and two PFAS exposures with IL-8. Exposures to two PFAS were positively associated with IFNγ. Sex differences were noted for IL-8, IFNγ, and PAI-1.
Figure 3. Significant Sex Differences in Biomarker Associations.
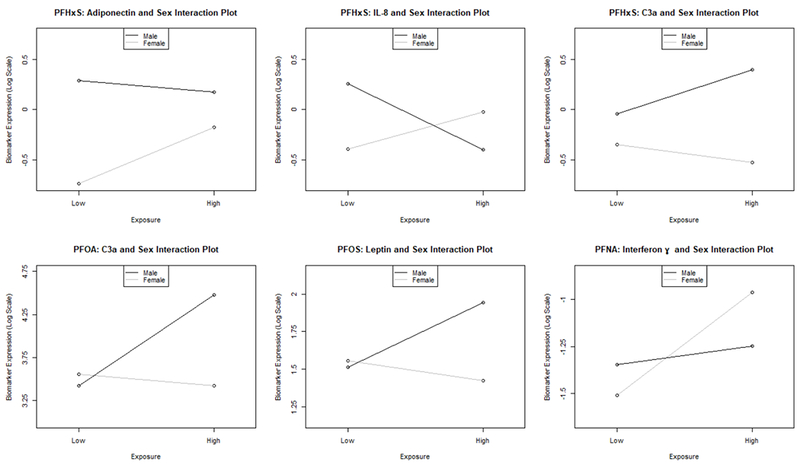
Significant relationships from the Interaction Model (Table 2) are depicted graphically. These figures are made using the coefficients from each respective linear model. The outcome variable (e.g., PFHxS) is continuous and log transformed, and the disease biomarker (e.g., adiponectin) is binary. One additional interaction was also statistically significant (PFOA and PAI-1) but is not displayed due to its small interaction coefficient estimate.
C3a was measured because a previous study showed that acute high-dose PFOA treatment induced steatosis and activated complement in mice (Botelho et al., 2015). On univariate analysis, C3a was positively associated with PFOA, PFOS, and PFNA (Supplementary Table). On multivariate analysis, the association with PFNA remained significant (β= 0.282±0.101, p=0.006). Sex differences were noted for C3a and PFHxS (interaction coefficient= 0.615±0.0336, p=0.068) and PFOA (interaction coefficient= 1.193±0.573, p=0.039). C3a increased with exposures to PFHxS and PFOA in men but decreased in women (Figure 3).
PFAS are endocrine disrupting hepatotoxic chemicals, and there are known bi-directional interactions between liver and the endocrine system (Heindel et al., 2017). Thus, the effects of PFAS on serum insulin, leptin and adiponectin were evaluated. In the multivariate final model, both leptin and adiponectin were significantly associated with increased PFHxS and PFNA exposures [PFHxS: leptin β= 0.471±0.168, p=.006; adiponectin β= 0.562±0.259, p=0.031; PFNA: leptin β= 0.179±0.103, p=.083; adiponectin β= 0.231±0.107, p=0.033]. The relationship between these two adipokines and PFNA was also significant in the univariate analysis (Supplementary Table). Adiponectin was also positively associated with PFOS in both univariate (Supplementary Table) and multivariate (β= 0.318±0.163, p=0.053) analyses. Sex differences were observed for PFHxS and adiponectin (interaction coefficient= −0.675±0.348, p=0.054) as well as for PFOS and leptin (interaction coefficient= 0.559±0.313, p=0.075). The significantly positive overall association between PFHxS and adiponectin was driven primarily by effects in women (Figure 3). Leptin was positively associated with PFOS in males only (Figure 3). PFHxS was significantly associated with decreased insulin in both the univariate (Supplementary Table) and multivariate (β= −0.442±0.231, p=0.057). However, the insulin data should be interpreted cautiously due to uncertainty relating to fasting status.
Discussion
This study demonstrated that PFAA serum concentrations were positively associated with increased serologic biomarkers of hepatocyte death/apoptosis as well as altered inflammatory and adipokine disease biomarkers in humans (Table 3). This combination of increased apoptosis and decreased inflammation is a novel finding in this study. Unlike these PFAAs, other hepatotoxic chemicals have often been associated with liver necrosis and increased inflammation (Cave et al., 2011; Clair et al., 2018). The CK18 data extend the previously published associations between PFAS exposures and liver enzymes in the C8 Health Study and other cohorts (Darrow et al., 2016; Gallo et al., 2012; Gleason et al., 2015; Lin et al., 2010; Salihovic et al., 2018; Yamaguchi et al., 2013) by demonstrating apoptosis as a hepatocyte cell death mechanism. This is the major finding of the current study, and it is consistent with results from an animal exposure model (Kim et al., 2011). While liver biopsies and imaging are not available in the C8 Health Study, data from in-vivo and in-vitro models implicate that the observed apoptosis in the study (CK18 M30) elevations could be due to PFAA-associated fatty liver disease (Bodin et al., 2016; Kim et al., 2011; Kleszczynski et al., 2009). Indeed, CK18 M30 has been proposed as fatty liver biomarker (Feldstein et al., 2009; He et al., 2017). Moreover, in cohorts, PFAS exposures were associated with increased blood lipids possibly reflective of altered hepatic lipid metabolism and NAFLD (Frisbee et al., 2010; Geiger et al., 2014; Nelson et al., 2010; Steenland et al., 2009). While PFOA exposures were not associated with self-reported (and subsequently physician-validated) liver disease history in the C8 Health Study, there was increase in both ALT and in clinically abnormal ALT (Darrow et al., 2016). It is well-known that liver diseases (including NAFLD) may be subclinical in the absence of decompensated cirrhosis. No sex-differences were noted for either CK18 biomarker suggesting that sex may not modulate the apoptotic hepatic response to PFAS exposures. PFAS could initiate NAFLD via disrupting hepatic lipid metabolism via its pleiotropic effects on nuclear receptors, but more data are required.
The second important finding of the present study is the consistent and novel finding of an inverse relationship between serum PFAA levels and serum TNFα in humans. Liver disease and hepatocyte death are typically associated with increased hepatic inflammation and serum pro-inflammatory cytokines. However, PFAS including PFAAs are already known to be immunotoxic and to downregulate some aspects of the immune response. TNFα is a cell-signaling cytokine involved in acute phase reactions and chronic systemic inflammation. Reduced TNFα production has been documented following PFAA exposure in experimental systems (Corsini et al., 2014). PFAA serum concentrations were also associated with decreased lobular inflammation on liver biopsy in NAFLD patients (Rantakokko et al., 2015). However, mice fed a high fat diet and exposed to PFOA experienced liver cell inflammation (Tan et al., 2013). The potentially anti-inflammatory action of PFAAs is also supported by the inverse association between PFAS and IL-8 in our data. While positive associations were seen for PFAS with IFNγ and C3a, the roles of these pro-inflammatory molecules are less well established than TNFα in fatty liver diseases. Additionally, sex differences were observed between several pro-inflammatory biomarkers and PFAS exposures. More data are required on the potentially sex-dependent effects of PFAS on histologic inflammation and liver disease progression. A third possible finding is the existence of sex differences for some markers, such as IL-8 and C3a and requires further evaluations in future, larger cohort studies.
NAFLD and other obesity-related diseases are typically associated with low adiponectin and high leptin. In our data, adiponectin was positively associated with PFHxS, PFOS, and PFNA exposures. While leptin usually moves discordantly to adiponectin, leptin was also positively associated with PFHxS and PFNA. Interestingly, adiponectin and leptin were also both increased in an animal PFOA exposure model (Du et al., 2018). The associations between PFAS and these adipokines have been investigated in several human developmental studies with conflicting results (Fleisch et al., 2017; Halldorsson et al., 2012; Minatoya et al., 2017). In the present study, sex differences were noted between the adipokines and PFAS exposures.
Only some patients with steatosis develop hepatic inflammation and nonalcoholic steatohepatitis (NASH), and only a subset of those progress to cirrhosis and hepatocellular carcinoma. Mechanisms implicated in the development of NASH include increased hepatocyte death, oxidative stress, and pro-inflammatory cytokines released by Kupffer cells, often in response to gut-derived lipopolysaccharide (LPS). Pollution exposures contributing to NASH include polychlorinated biphenyls (PCBs) or the volatile organic compound (VOC), vinyl chloride (VC); these appear be second hits mediating the progression of diet-induced steatosis to NASH (Lang et al., 2018; Wahlang et al., 2014). These pollutants have been associated with increased serum biomarkers of hepatocyte necrosis (PCBs, VC); pro-inflammatory cytokines (PCBs, VC); and reduced antioxidants (VC) in exposed cohorts with liver disease (Cave et al., 2010; Clair et al., 2018). The terms, toxicant associated fatty liver disease (TAFLD) and steatohepatitis (TASH), have been proposed to describe the spectrum of fatty liver disease associated with chemical exposures (Wahlang et al., 2013). The current data support the presence of apoptosis in the absence of additional inflammation with PFAA exposures. When considering data from other studies that used rodent, fish and human cell line models to demonstrate the actual presence of fatty infiltration of liver cells following PFAS exposure and mechanisms thereof, (Fai Tse et al., 2016; Peng et al., 2013; Quist et al., 2015; Wang et al., 2013), our data possibly support a role for PFAAs in the initiation stage of NAFLD, or steatosis.
Neither liver biopsy nor imaging were obtained in the C8 Health Study, and it is important to address weaknesses of the biomarker approaches. The serum samples are derived from a cross-sectional study, and therefore do not address the long term follow-up needed to definitively document the development and progression of liver disease in a population. This cross-sectional design is a limitation because it only address associations. The serum samples investigated are randomly selected from a much larger cohort, and some random variation is a possible explanation for findings.
Likewise, unmeasured confounding can affect any single study. Furthermore, because this is a pilot study, no multiple comparison analyses were performed which is another drawback. However, in future confirmatory studies, adjustments for false discovery will be performed. An important weakness of using frozen serum is the possible effects of long term storage and preexisting freeze-thaw cycles on results. In general, this concern would be most worrisome from a null bias perspective, but this study found hypothesized results for main outcomes (e.g., CK18 and TNFα) consistent with existing literature. Nevertheless, we wish to emphasize the scientific desirability of prospective studies on healthy humans with long term follow up and use of imaging and recently acquired sera for similar studies, and also note the strong ethical and logistical problems associated with that goal. Gene-environment interactions cannot be investigated using the C8 Health Study because only serum is available. Likewise, potential diet-PFAS interactions could not be evaluated in this study. In mice, short-term high fat diet feeding worsened PFAS-induced fatty liver disease and increased inflammation (Tan et al., 2013).
In summary, the most consistent findings of this study were associations between serum PFAA levels with increased CK18 M30, decreased TNFα, and increased adiponectin. Sex differences in the adipocytokine and C3a responses to PFAAs were observed. Based on the literature, the most plausible explanation for these findings is PFAA-induced toxicant associated fatty liver disease. We postulate that PFAAs disrupt hepatic lipid metabolism via nuclear receptor interactions to initiate steatosis with apoptosis while inhibiting Kupffer cell activation and upregulating adiponectin production. The latter two effects are expected to be protective. Thus, while PFAAs may initiate steatosis, they theoretically could also reduce the likelihood of subsequent progression of steatosis to steatohepatitis. However, based on the aforementioned limitations, this cross-sectional study must be considered preliminary with regards to liver disease progression. Nonetheless, these results demonstrate the need for more rigorously designed longitudinal studies of PFAS in the genesis and development of fatty liver disease. Because sex differences in disease mechanisms are also anticipated, sex as a biological variable must also be considered.
Supplementary Material
Highlights.
Serum levels of four perfluoroalkyl acids (PFAAs) and liver disease biomarkers were measured
CK18 M30, an indicator of hepatocyte death, was positively associated with exposures to PFAAs
Tumor necrosis factor α and interleukin 8 were inversely associated with exposures to PFAAs
PFAAs exposures led to hepatic apoptosis but decreased inflammation, which is a novel finding
Acknowledgments:
This work was supported, in part, by the National Institute of Environmental Health Sciences [R35ES028373, P42ES023716 and T32ES0011564], the National Institute of General Medical Sciences [P20GM113226], and the National Institute on Alcohol Abuse and Alcoholism [P50AA024337]. Alan Ducatman, Matt Cave, and John Barnett designed the study. John Bassler and Sijin Wen planned and executed the statistical approach to the data and its description. Elliott Meenal provided quality assurance and support for the laboratory work and its description. Alan Ducatman, Matt Cave, John Barnett, and Banrida Wahlang interpreted the results and prepared the manuscript. Drs. Ducatman, Cave, Barnett, and Wen are the guarantors of the several types of analytic work in the study.
Abbreviations:
- ADIP
adiponectin
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- C3a
cleaved complement 3
- CAR
constitutive androstane receptor
- CK18
cytokeratin 18
- ERα
estrogen receptor α
- eGFR
estimated glomerular filtration rate
- FXR
farnesoid x receptor
- HNF4α
hepatocyte nuclear factor 4 α
- IFNγ
interferon γ
- IL-6
interleukin 6
- IL-8
interleukin 8
- INS
insulin
- LEP
leptin
- LPS
lipopolysaccharide
- LXR
liver X receptor
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- PAI-1
plasminogen activator inhibitor 1
- PCBs
polychlorinated biphenyls
- PFAAs
perfluoroalkyl acids
- PFAS
perfluoroalkyl substances
- PFOA
perfluorooctanoic acid
- PFOS
perfluorooctane sulfonic acid
- PFHxS
perfluorohexane sulfonic acid
- PFNA
perfluorononanoic acid
- PPARα and PPARγ
peroxisome proliferator activated receptors α and γ
- PXR
pregnane X receptor
- TAFLD
toxicant associated fatty liver disease
- TASH
toxicant associated steatohepatitis
- TNFα
tumor necrosis factor α
- VC
vinyl chloride
- VOC
volatile organic compound
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: Dr. Cave reports financial research support from DiaPharma on an unrelated industry-sponsored clinical trial of the cytokeratin 18 liver disease biomarker. Alan Ducatman was supported to provide health communications for the C8 Health project and has provided support to a plaintiff class seeking medical monitoring following exposure to PFOA. All other authors declare they have no actual or potential competing financial interests.
References
- Bijland S, Rensen PC, Pieterman EJ, Maas AC, van der Hoorn JW, van Erk MJ, Havekes LM, van Dijk K. Willems, Chang SC, Ehresman DJ, Butenhoff JL, Princen HM, 2011. Perfluoroalkyl sulfonates cause alkyl chain length-dependent hepatic steatosis and hypolipidemia mainly by impairing lipoprotein production in APOE*3-Leiden CETP mice. Toxicol Sci 123, 290–303. [DOI] [PubMed] [Google Scholar]
- Bjork JA, Butenhoff JL, Wallace KB, 2011. Multiplicity of nuclear receptor activation by PFOA and PFOS in primary human and rodent hepatocytes. Toxicology 288, 8–17. [DOI] [PubMed] [Google Scholar]
- Bodin J, Groeng EC, Andreassen M, Dirven H, Nygaard UC, 2016. Exposure to perfluoroundecanoic acid (PFUnDA) accelerates insulitis development in a mouse model of type 1 diabetes. Toxicol Rep 3, 664–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botelho SC, Saghafian M, Pavlova S, Hassan M, DePierre JW, Abedi-Valugerdi M, 2015. Complement activation is involved in the hepatic injury caused by high-dose exposure of mice to perfluorooctanoic acid. Chemosphere 129, 225–231. [DOI] [PubMed] [Google Scholar]
- Buhrke T, Kruger E, Pevny S, Rossler M, Bitter K, Lampen A, 2015. Perfluorooctanoic acid (PFOA) affects distinct molecular signalling pathways in human primary hepatocytes. Toxicology 333, 53–62. [DOI] [PubMed] [Google Scholar]
- Cave M, Falkner KC, Henry L, Costello B, Gregory B, McClain CJ, 2011. Serum cytokeratin 18 and cytokine elevations suggest a high prevalence of occupational liver disease in highly exposed elastomer/polymer workers. J Occup Environ Med 53, 1128–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cave M, Falkner KC, Ray M, Joshi-Barve S, Brock G, Khan R, Homme M. Bon, McClain CJ, 2010. Toxicant-associated steatohepatitis in vinyl chloride workers. Hepatology 51, 474–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cave MC, Clair HB, Hardesty JE, Falkner KC, Feng W, Clark BJ, Sidey J, Shi H, Aqel BA, McClain CJ, Prough RA, 2016. Nuclear receptors and nonalcoholic fatty liver disease. Biochim Biophys Acta. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clair HB, Pinkston CM, Rai SN, Pavuk M, Dutton ND, Brock G, Prough RA, Falkner KC, McClain CJ, Cave MC, 2018. Liver Disease in a Residential Cohort with Elevated Polychlorinated Biphenyl Exposures. Toxicol Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsini E, Luebke RW, Germolec DR, DeWitt JC, 2014. Perfluorinated compounds: emerging POPs with potential immunotoxicity. Toxicol Lett 230, 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrow LA, Groth AC, Winquist A, Shin HM, Bartell SM, Steenland K, 2016. Modeled Perfluorooctanoic Acid (PFOA) Exposure and Liver Function in a Mid-Ohio Valley Community. Environ Health Perspect 124, 1227–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das KP, Wood CR, Lin MT, Starkov AA, Lau C, Wallace KB, Corton JC, Abbott BD, 2017. Perfluoroalkyl acids-induced liver steatosis: Effects on genes controlling lipid homeostasis. Toxicology 378, 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt JC, Peden-Adams MM, Keller JM, Germolec DR, 2012. Immunotoxicity of perfluorinated compounds: recent developments. Toxicol Pathol 40, 300–311. [DOI] [PubMed] [Google Scholar]
- Domingo JL, Nadal M, 2017. Per- and Polyfluoroalkyl Substances (PFASs) in Food and Human Dietary Intake: A Review of the Recent Scientific Literature. J Agric Food Chem 65, 533–543. [DOI] [PubMed] [Google Scholar]
- Du G, Sun J, Zhang Y, 2018. Perfluorooctanoic acid impaired glucose homeostasis through affecting adipose AKT pathway. Cytotechnology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse W.K. Fai, Li JW, Tse A.C. Kwan, Chan TF, Ho J.C. Hin, Wu R.S. Sun, Wong C.K. Chu, Lai KP, 2016. Fatty liver disease induced by perfluorooctane sulfonate: Novel insight from transcriptome analysis. Chemosphere 159, 166–177. [DOI] [PubMed] [Google Scholar]
- Feldstein AE, Wieckowska A, Lopez AR, Liu YC, Zein NN, McCullough AJ, 2009. Cytokeratin-18 fragment levels as noninvasive biomarkers for nonalcoholic steatohepatitis: a multicenter validation study. Hepatology 50, 1072–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty JM, Connolly PD, Decker ER, Kennedy SM, Ellefson ME, Reagen WK, Szostek B, 2005. Quantitative determination of perfluorooctanoic acid in serum and plasma by liquid chromatography tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 819, 329–338. [DOI] [PubMed] [Google Scholar]
- Fleisch AF, Rifas-Shiman SL, Mora AM, Calafat AM, Ye X, Luttmann-Gibson H, Gillman MW, Oken E, Sagiv SK, 2017. Early-Life Exposure to Perfluoroalkyl Substances and Childhood Metabolic Function. Environ Health Perspect 125, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisbee SJ, Brooks AP Jr., Maher A, Flensborg P, Arnold S, Fletcher T, Steenland K, Shankar A, Knox SS, Pollard C, Halverson JA, Vieira VM, Jin C, Leyden KM, Ducatman AM, 2009. The C8 health project: design, methods, and participants. Environ Health Perspect 117, 1873–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisbee SJ, Shankar A, Knox SS, Steenland K, Savitz DA, Fletcher T, Ducatman AM, 2010. Perfluorooctanoic acid, perfluorooctanesulfonate, and serum lipids in children and adolescents: results from the C8 Health Project. Arch Pediatr Adolesc Med 164, 860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo V, Leonardi G, Genser B, Lopez-Espinosa MJ, Frisbee SJ, Karlsson L, Ducatman AM, Fletcher T, 2012. Serum perfluorooctanoate (PFOA) and perfluorooctane sulfonate (PFOS) concentrations and liver function biomarkers in a population with elevated PFOA exposure. Environ Health Perspect 120, 655–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger SD, Xiao J, Ducatman A, Frisbee S, Innes K, Shankar A, 2014. The association between PFOA, PFOS and serum lipid levels in adolescents. Chemosphere 98, 78–83. [DOI] [PubMed] [Google Scholar]
- Gleason JA, Post GB, Fagliano JA, 2015. Associations of perfluorinated chemical serum concentrations and biomarkers of liver function and uric acid in the US population (NHANES), 2007-2010. Environ Res 136, 8–14. [DOI] [PubMed] [Google Scholar]
- Halldorsson TI, Rytter D, Haug LS, Bech BH, Danielsen I, Becher G, Henriksen TB, Olsen SF, 2012. Prenatal exposure to perfluorooctanoate and risk of overweight at 20 years of age: a prospective cohort study. Environ Health Perspect 120, 668–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Deng L, Zhang Q, Guo J, Zhou J, Song W, Yuan F, 2017. Diagnostic Value of CK-18, FGF-21, and Related Biomarker Panel in Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Biomed Res Int 2017, 9729107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heindel JJ, Blumberg B, Cave M, Machtinger R, Mantovani A, Mendez MA, Nadal A, Palanza P, Panzica G, Sargis R, Vandenberg LN, Vom Saal F, 2017. Metabolism disrupting chemicals and metabolic disorders. Reprod Toxicol 68, 3–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh S, Leaderer BP, Feldstein AE, Santoro N, McKay LA, Caprio S, McConnell R, 2017. Traffic-related air pollution associations with cytokeratin-18, a marker of hepatocellular apoptosis, in an overweight and obese paediatric population. Pediatr Obes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Kwack S. Jun, Han E. Sik, Kang T. Seok, Kim S. Hee, Han S. Young, 2011. Induction of apoptosis and CYP4A1 expression in Sprague-Dawley rats exposed to low doses of perfluorooctane sulfonate. J Toxicol Sci 36, 201–210. [DOI] [PubMed] [Google Scholar]
- Kleszczynski K, Stepnowski P, Skladanowski AC, 2009. Mechanism of cytotoxic action of perfluorinated acids II. Disruption of mitochondrial bioenergetics. Toxicol Appl Pharmacol 235, 182–190. [DOI] [PubMed] [Google Scholar]
- Lang AL, Chen L, Poff GD, Ding WX, Barnett RA, Arteel GE, Beier JI, 2018. Vinyl chloride dysregulates metabolic homeostasis and enhances diet-induced liver injury in mice. Hepatol Commun 2, 270–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levey AS, Coresh J, Greene T, Stevens LA, Zhang YL, Hendriksen S, Kusek JW, Van Lente F, 2006. Using standardized serum creatinine values in the modification of diet in renal disease study equation for estimating glomerular filtration rate. Ann Intern Med 145, 247–254. [DOI] [PubMed] [Google Scholar]
- Lin CY, Lin LY, Chiang CK, Wang WJ, Su YN, Hung KY, Chen PC, 2010. Investigation of the associations between low-dose serum perfluorinated chemicals and liver enzymes in US adults. Am J Gastroenterol 105, 1354–1363. [DOI] [PubMed] [Google Scholar]
- Milley SA, Koch I, Fortin P, Archer J, Reynolds D, Weber KP, 2018. Estimating the number of airports potentially contaminated with perfluoroalkyl and polyfluoroalkyl substances from aqueous film forming foam: A Canadian example. J Environ Manage 222, 122–131. [DOI] [PubMed] [Google Scholar]
- Minatoya M, Itoh S, Miyashita C, Araki A, Sasaki S, Miura R, Goudarzi H, Iwasaki Y, Kishi R, 2017. Association of prenatal exposure to perfluoroalkyl substances with cord blood adipokines and birth size: The Hokkaido Study on environment and children’s health. Environ Res 156, 175–182. [DOI] [PubMed] [Google Scholar]
- Nelson JW, Hatch EE, Webster TF, 2010. Exposure to polyfluoroalkyl chemicals and cholesterol, body weight, and insulin resistance in the general U.S. population. Environ Health Perspect 118, 197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng S, Yan L, Zhang J, Wang Z, Tian M, Shen H, 2013. An integrated metabonomics and transcriptomics approach to understanding metabolic pathway disturbance induced by perfluorooctanoic acid. J Pharm Biomed Anal 86, 56–64. [DOI] [PubMed] [Google Scholar]
- Perez F, Nadal M, Navarro-Ortega A, Fabrega F, Domingo JL, Barcelo D, Farre M, 2013. Accumulation of perfluoroalkyl substances in human tissues. Environ Int 59, 354–362. [DOI] [PubMed] [Google Scholar]
- Quist EM, Filgo AJ, Cummings CA, Kissling GE, Hoenerhoff MJ, Fenton SE, 2015. Hepatic Mitochondrial Alteration in CD-1 Mice Associated with Prenatal Exposures to Low Doses of Perfluorooctanoic Acid (PFOA). Toxicol Pathol 43, 546–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rantakokko P, Mannisto V, Airaksinen R, Koponen J, Viluksela M, Kiviranta H, Pihlajamaki J, 2015. Persistent organic pollutants and non-alcoholic fatty liver disease in morbidly obese patients: a cohort study. Environ Health 14, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebholz SL, Jones T, Herrick RL, Xie C, Calafat AM, Pinney SM, Woollett LA, 2016. Hypercholesterolemia with consumption of PFOA-laced Western diets is dependent on strain and sex of mice. Toxicol Rep 3, 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen MB, Das KP, Rooney J, Abbott B, Lau C, Corton JC, 2017. PPARalpha-independent transcriptional targets of perfluoroalkyl acids revealed by transcript profiling. Toxicology 387, 95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salihovic S, Stubleski J, Karrman A, Larsson A, Fall T, Lind L, Lind PM, 2018. Changes in markers of liver function in relation to changes in perfluoroalkyl substances - A longitudinal study. Environ Int 117, 196–203. [DOI] [PubMed] [Google Scholar]
- Steenland K, Tinker S, Frisbee S, Ducatman A, Vaccarino V, 2009. Association of perfluorooctanoic acid and perfluorooctane sulfonate with serum lipids among adults living near a chemical plant. Am J Epidemiol 170, 1268–1278. [DOI] [PubMed] [Google Scholar]
- Steenland K, Tinker S, Shankar A, Ducatman A, 2010. Association of perfluorooctanoic acid (PFOA) and perfluorooctane sulfonate (PFOS) with uric acid among adults with elevated community exposure to PFOA. Environ Health Perspect 118, 229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunderland EM, Hu XC, Dassuncao C, Tokranov AK, Wagner CC, Allen JG, 2018. A review of the pathways of human exposure to poly- and perfluoroalkyl substances (PFASs) and present understanding of health effects. J Expo Sci Environ Epidemiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X, Xie G, Sun X, Li Q, Zhong W, Qiao P, Sun X, Jia W, Zhou Z, 2013. High fat diet feeding exaggerates perfluorooctanoic acid-induced liver injury in mice via modulating multiple metabolic pathways. PLoS One 8, e61409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlang B, Beier JI, Clair HB, Bellis-Jones HJ, Falkner KC, McClain CJ, Cave MC, 2013. Toxicant-associated steatohepatitis. Toxicol Pathol 41, 343–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlang B, Song M, Beier JI, Cameron Falkner K, Al-Eryani L, Clair HB, Prough RA, Osborne TS, Malarkey DE, Christopher States J, Cave MC, 2014. Evaluation of Aroclor 1260 exposure in a mouse model of diet-induced obesity and nonalcoholic fatty liver disease. Toxicol Appl Pharmacol 279, 380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan C, Han R, Liu L, Zhang F, Li F, Xiang M, Ding W, 2016. Role of miR-155 in fluorooctane sulfonate-induced oxidative hepatic damage via the Nrf2-dependent pathway. Toxicol Appl Pharmacol 295, 85–93. [DOI] [PubMed] [Google Scholar]
- Wan HT, Zhao YG, Wei X, Hui KY, Giesy JP, Wong CK, 2012. PFOS-induced hepatic steatosis, the mechanistic actions on beta-oxidation and lipid transport. Biochim Biophys Acta 1820, 1092–1101. [DOI] [PubMed] [Google Scholar]
- Wang L, Wang Y, Liang Y, Li J, Liu Y, Zhang J, Zhang A, Fu J, Jiang G, 2013. Specific accumulation of lipid droplets in hepatocyte nuclei of PFOA-exposed BALB/c mice. Sci Rep 3, 2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worley RR, Moore SM, Tierney BC, Ye X, Calafat AM, Campbell S, Woudneh MB, Fisher J, 2017. Per- and polyfluoroalkyl substances in human serum and urine samples from a residentially exposed community. Environ Int 106, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Arisawa K, Uemura H, Katsuura-Kamano S, Takami H, Sawachika F, Nakamoto M, Juta T, Toda E, Mori K, Hasegawa M, Tanto M, Shima M, Sumiyoshi Y, Morinaga K, Kodama K, Suzuki T, Nagai M, Satoh H, 2013. Consumption of seafood, serum liver enzymes, and blood levels of PFOS and PFOA in the Japanese population. J Occup Health 55, 184–194. [DOI] [PubMed] [Google Scholar]
- Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M, 2015. Global Epidemiology of Non-Alcoholic Fatty Liver Disease-Meta-Analytic Assessment of Prevalence, Incidence and Outcomes. Hepatology. [DOI] [PubMed] [Google Scholar]
- Zhang L, Krishnan P, Ehresman DJ, Smith PB, Dutta M, Bagley BD, Chang SC, Butenhoff JL, Patterson AD, Peters JM, 2016. Editor’s Highlight: Perfluorooctane Sulfonate-Choline Ion Pair Formation: A Potential Mechanism Modulating Hepatic Steatosis and Oxidative Stress in Mice. Toxicol Sci 153, 186–197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.