

Abstract
The bacterial mechanosensitive channel of large conductance (MscL) normally functions as an emergency release valve discharging cytoplasmic solutes upon osmotic stress. Opening the large pore of MscL inappropriately is detrimental to the cell, and thus it has been speculated to be a potential antibiotic target. Although MscL is one of the best studied mechanosensitive channels, no chemical that influenced bacterial growth by modulating MscL is known. We therefore used a high-throughput screen to identify compounds that slowed growth in an MscL-dependent manner. We characterized 2 novel sulfonamide compounds identified in the screen. We demonstrated that, although both increase MscL gating, one of these compounds does not work through the folate pathway, as other antimicrobial sulfonamides; indeed, the sulfonamide portion of the compound is not needed for activity. The only mode of action appears to be MscL activation. The binding pocket is where an α-helix runs along the cytoplasmic membrane and interacts with a neighboring subunit; analogous motifs have been observed in several prokaryotic and eukaryotic channels. The data not only demonstrate that MscL is a viable antibiotic target, but also give insight into the gating mechanisms of MscL, and they may have implications for developing agonists for other channels.—Wray, R., Iscla, I., Kovacs, Z., Wang, J., Blount, P. Novel compounds that specifically bind and modulate MscL: insights into channel gating mechanisms.
Keywords: mechanosensitive, osmoregulation, antibiotic
Mechanosensitive (MS) channels in bacteria serve the physiologic role of biologic emergency-release valves to relieve high cell turgor caused by sudden extracellular decreases in osmolarity (1). When bacteria are exposed to high osmolarity they transport (K+, glutamate, betaine, and proline) and synthesize (glutamate, trehalose, proline, and betaine) solutes to balance the increase in external osmolarity to keep cell turgor high, a requisite for cell growth and division. When the osmotic environment acutely decreases, water rushes in, turgor increases, and cell integrity threatened. Bacterial cells prevent such a catastrophe by activation of membrane-tension–gated MS channels. Of these bacterial MS channels, mechanosensitive channel of large conductance (MscL), which has the largest gated pore of any known channel, about 30 Å in diameter (2), serves as the last-ditch effort for bacterial cell survival.
The MscL channel is a homopentamer with each subunit containing 2 transmembrane domains (TM1 and -2), an N-terminal α-helix along the membrane [surface domain (S)1], and a C-terminal region that forms a cytoplasmic bundle that is thought to be relatively intact upon gating (1, 3–6). One of the regions of the MscL channel that is thought to show dramatic conformational changes upon gating and has been suggested to be critical for channel opening is the constriction point of the pore, which is solely composed of the cytoplasmic half of TM1. Mutations and post-translational modifications that lead to more polar or charged residues often result in channels that gate inappropriately (7–10). These data lead to and supported the theory that the breaking of hydrophobic interactions and transient exposure of these residues to an aqueous solution may be an energy barrier for channel opening (11). Indeed, studies of dihydrostreptomycin (DHS), which uses MscL as one of the primary ways to enter the cell, also strongly support this notion (12, 13). A second region of the protein that appears to play a role in gating is the cytoplasmic region of TM2, close to a cluster of charges, where mutations and other studies have shown the region to enter and exit the lipid membrane in a piston-like manner (14).
Perhaps the most mysterious and least understood region of the MscL protein is the protein-protein and protein-lipid interacting area at the cytoplasmic-lipid interface where the S1 helix runs along the membrane, including how S1 interacts with the TM2 of a neighboring subunit. This area has been of intense interest because similar structures are found in many other channels including the bacterial inward rectifying K+ channel KirBac (15–17), transient receptor potential (TRP) channels (18–20), 2-pore domain K+ (K2P) channels (21), and even MscS, another bacterial mechanosensitive channel from an independent channel family (4, 22). For MscL, although an important role in gating was proposed for S1 over 10 yr ago (23) and subsequent studies have supported this notion by demonstrating that the region is very dynamic (24, 25), there are no data demonstrating that disrupting normal interactions in this region would increase the gating of the MscL channel.
We have used a high-throughput screen (HTS) to identify compounds that slowed bacterial growth in an MscL-dependent manner (12). The goal was to find compounds that gated the MscL channel inappropriately; such compounds could demonstrate that MscL is a viable antibiotic target, as well as be used to study channel gating. We characterize 2 compounds that were identified in the HTS that are in the sulfonamide family. Both compounds effected MscL-dependent slowed growth on Escherichia coli cells and both showed increased MscL channel activity in patch clamp. However, only one of the compounds, 011, did not work through the folate pathway, as do other sulfonamides. This is the first compound described that shows strong specificity, direct binding, and no other mode of action than increasing MscL activity. The binding site includes the S1 helix that runs along the membrane and the interface between S1 and TM2. Our findings thus have 3 implications: first, MscL may truly be a viable antibiotic target; second, interactions within the S1/TM2 region are important for MscL gating, and disrupting them pharmacologically makes channel gating more probable; third, because intersubunit regions analogous to the S1 helix are found in many other prokaryotic and eukaryotic channels, it seems possible that they are also viable targets for pharmacological manipulation.
MATERIALS AND METHODS
Strains and cell growth
E. coli Frag 1–derived cell lines MJF367 (ΔmscL::Cam), MJF451 (ΔmscS), MJF455 (ΔmscL::Cam, ΔmscS) (26), and MJF612 (ΔmscL::cm, ΔmscS, ΔmscK::kan, ΔybdG::aprΔ) (27) were used either alone for endogenous expression or to host the pb10 expression constructs. Cultures inoculated from a single colony were grown in either citrate-phosphate–defined medium (CphM; pH 7.0), consisting of (per liter): 8.57 g Na2HPO4, 0.87 g K2HPO4, 1.34 g citric acid, 1.0 g NH4SO4, 0.001 g thiamine, 0.1 g MgSO47H2O, and 0.002 g (NH4)2SO4.FeSO4.H2O, or K10 medium containing 46 mM Na2HPO4, 23 mM NaH2PO4, 8 mM (NH4)2SO4, 10 mM KCl, and 100 mM NaCl, supplemented with 0.4 mM MgSO4, 3 μM thiamine, 6 μM iron, and 0.04% glucose in a shaker incubator at 37°C, rotated at 250 cycles per minute. Ampicillin was added for strains carrying plasmid constructs (100 μg/ml).
For generation of the sulfonamide-resistant strain PB121, sulfonamide resistance in the MJF612 strain was produced as described by Skold (28). An overnight culture in CphM with 10 µg/ml chloramphenicol was diluted 1:100 in 10 new tubes, each containing either CphM with 5 µg/ml sulfamethoxazole (SMX) (MilliporeSigma, Burlington, MA USA) or CphM, 5 µg/ml SMX, and 10 µg/ml chloramphenicol. When growth was observed, each tube was then diluted into various concentrations of SMX from 5 to 30 µg/ml; from the concentration barely permitting growth, a 1:100 dilution was made into a slightly higher starting concentration series of SMX over a 10-d period. Final cultures were then plated on CphM plates with 20 µg/ml SMX and 10 µg/ml chloramphenicol, streak purified, and stocks made of the final 7 clones. Clones were then drug tested against 3 different sulfonamides (sulfamethoxazole, sulfamethizole, and sulfisoxazole) and kanamycin, chloramphenicol, in which the cell line is resistant. The folp gene, known to be mutated in sulfonamide-resistant strains, was sequenced for the top 3 clones, and 2 had the mutation P64L (29), one of which was used for this study.
Molecular techniques
Two new mutations were produced and were generated with a modified megaprimer technique, as previously described (30, 31). In brief, oligonucleotide primers 34 and 33 bases in length that incorporated the desired codon change in the center of the sequence were designed: B.sub R88K, 5′-CTCCATTTTTATCGTCATTAAGACTTTGAATGGG-3′; and Eco K97R, 5′-CCATCTTTATGGCGATTAGGCTAATCAACAAAC-3′. PCR products were then made in a 2-step process with oligos flanking the insert. The first product resulted in the mega primer using the flanking oligo 3′ long (5′-ATGAGCTTTACCGCAGGGTACCG-3′), which was used as a primer for the second reaction with the flanking oligo 5′ long (5′-GTTTTCACCGTCATCACCGAAACG-3′). The PCR products produced were then ligated into the pB10d vector (32, 33).
In vivo assays
To obtain minimal inhibitory concentration curves, overnight cultures of constructs in the MJF455 or MJF612 strain were diluted 1:50 in CphM and grown until an OD600 of 0.2 was reached. Expression was then induced by the addition of 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 30 min; 10-mM stocks of 120, 011, or 011A, solubilized in sterile DMSO (MilliporeSigma), were diluted to 2× their final concentration in prewarmed CphM with a final DMSO concentration of 0.9%; and 100 µl was added to wells of a prewarmed, sterile, 96-well flat-bottom plate (Greiner Bio-One, Monroe, NC, USA). Cultures were then diluted 1:200 in prewarmed CphM, 100 µg/ml ampicillin, and 2 mM IPTG, and 100 µl was added to wells with diluted or mock compound (DMSO only). For experiments with 011A as an adjuvant, the MJF455 stain was used to carry constructs and grown as previously described, or the MJF367, MJF451, and MJF455 strains were used for endogenous expression and were grown in the same medium without ampicillin until an OD600 of ∼0.35 was reached. Cultures were then diluted 1:200 into the same medium, with or without the antibiotic being tested at 2× their final concentrations. Final concentrations were DHS sesquisulfate at 2.25 µM (MilliporeSigma), kanamycin A at 1 µM (MilliporeSigma), tetracycline hydrochloride at 0.5 µM (Thermo Fisher Scientific, Waltham, MA, USA), and ampicillin sodium salt at 2 µM (Thermo Fisher Scientific). One hundred microliters of these mixtures was immediately added to 100 µl medium, with or without compound 011A (2× final concentration), diluted at various concentrations, and placed in a 96-well plate, as described above. Plates were then sealed with a sterile breathable film to prevent evaporation (Axygen, Union City, CA, USA), wrapped in aluminum foil, and placed in a 37°C shaker, rotated at 110 cycles per minute for 16–17 h, and OD620 was then taken with a Multiskan Ascent 354 Plate Reader (Thermo Fisher Scientific).
Cysteine library screen
Frozen stocks of the MJF455 strain carrying MscL single cysteine mutations (S2C-E107C) were placed in a 96-well plate format (omitting gain- and loss-of-function mutations), as previously described (13). The mutated channels were expressed by using pB10, an expression plasmid that induces only moderate expression levels (34). In brief, 10 µl of the overnight culture plate was diluted into 96-well plates containing 190 μl of prewarmed CphM, 1 mM IPTG or CphM, 1 mM IPTG, and final concentrations of compounds 011 or 011A at 40 µm and DMSO at 0.9%. Plates were sealed with breathable film, wrapped in foil, and placed in a 37°C shaker and rotated at 110 cycles per minute for 16–17 h. OD620 was then read with the Multiskan Ascent 354 (Thermo Fisher Scientific) plate reader, and the blank was subtracted. The difference in OD620 was calculated between the plate with and without compound added. The entire library was screened 3 times for 011 and 5 times for 011A. Hits were then selected as cultures showing at least 3 times more growth compared to WT MscL from E. coli (Eco-MscL).
Electrophysiology
Giant spheroplasts were generated from the E. coli strain MJF612 (27) and used in patch-clamp experiments, as described by Blount et al. (34). Excised inside-out patches were examined at room temperature under symmetrical conditions in a buffer comprising 200 mM KCl, 90 mM MgCl2, 10 mM CaCl2, and 5 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (pH 6–7; MilliporeSigma). Two different methods were used to present the compounds to the patch. The backfilled pipette method allows presenting the compound from the periplasmic side of the membrane; the tip of the recording pipette is filled with buffer without the compound, and the back of the pipette is filled with buffer with the compound. Control recordings are performed soon after forming the gigaohm seal, and the same patch is recorded later (∼45 min), after the compound has diffused to the tip. The compounds are also presented in the bath that corresponds to the cytoplasmic side of the membrane; in this case, the same patch is recorded before and after perfusion of the compound in the bath chamber. Recordings were performed at −20 mV (positive pipette). Data were acquired at a sampling rate of 20 kHz with a 5-kHz filter using an AxoPatch 200B amplifier in conjunction with Axoscope software (Molecular Devices, Sunnyvale, CA, USA). A piezoelectric pressure transducer (World Precision Instruments, Sarasota, FL, USA) was used to monitor the pressure throughout the experiments. Measurements were performed with ClampFit10 from pClamp10 (Molecular Devices).
Protein purification
Experiments were performed as previously described (13). In brief, frozen pellets were lysed in 50 mM KPi 7.5, 150 mM NaCl, 0.5 µg/ml DNAse, 1 mg/ml lysozyme, and protease inhibitor; 40 mM n-decyl β-d-maltopyranoside was then added. The lysate was then bound to, washed, and eluted from Talon resin (Takara Bio, Mountain View, CA, USA), per the manufacturer’s instructions, while the detergent concentration was reduced to 4 mM. Fractions were then concentrated, buffer exchanged, and protein assays were performed (DC Kit; Bio-Rad, Hercules, CA, USA).
MTS-Peg5000 competition experiments
Experiments were performed as we previously described (13). In brief, 5 µg of protein in buffer containing 50 mM KPi pH 7.5, 150 mM NaCl, 4 mM n-decyl β-d-maltopyranoside, was added to 0.2-ml PCR tubes. For each sample, the following conditions were set up: 1) mock-buffer and protein only, 2) PEG, buffer, and protein, and 3) PEG, buffer, protein, plus either compound 011 or 011A at 1.5, 3, and 5 mM. Samples were incubated for 15 min at 22°C. Methoxypoly (ethyleneglycol)-5000-amidopropionyl-methanethiosulfonate (MTS-Peg5000) at 50 µM was then added to all conditions, excluding mock, and incubated for 10 min. Reactions were then stopped by adding 20 μl nonreducing sample buffer with a final concentration of 5 mM iodoacetamide, run on a 4–20% criterion gel (Bio-Rad). Western blots were preformed, developed with Millipore horseradish peroxidase substrate, and exposed to film. Because of the size of the MTS-Peg5000 and the structure of the protein, not all sites are equally accessible, and thus are not PEGylated to the same extent. For example, a lessened extent of modification is seen with the I96C mutation. Indeed, we found 3 cysteine mutations, A89C, F90C and F93C, that could not be modified at all.
Growth inhibition at the stationary phase
A single colony was picked for each construct in the MJF455 strain and grown in degassed CphM with an argon gas overlay, capped, and sealed until an OD600 of 0.2 was reached. Cultures were then induced with the addition of 1 mM IPTG and grown overnight, after which argon was added and the tubes recapped and sealed. The next day 1-ml cultures were divided, compounds 011, 011A, or mock (DMSO only) were added at 80 µM with a final DMSO concentration of 0.9%, argon added and sealed in a 1.5-ml tube for 6 h in a 37°C shaker. Final OD600 was taken and cultures were then diluted 1:20 into prewarmed CphM and serially diluted from 103 to 106, and liquid drops of 5 µl for each dilution were placed on prewarmed Luria-Bertani plates and placed in a 37°C incubator. The next morning colony-forming units were calculated to determine cell viability, as previously described (23, 35).
Molecular docking
Molecular docking was conducted with a representative structure of a 150-ns molecular dynamics trajectory of Eco-MscL with a DHS molecule within the pocket formed by the periplasmic loops, as previously described (13). The binding pocket was identified by the SiteID module of the Sybyl-X2.11 software package (36). Then flexible-ligand docking was performed for 011A with the Glide module of the Schrodinger software package following the standard procedure (37). The best docking pose was manually examined and subject to further study.
Molecular dynamics simulations and binding free-energy calculations
Molecular dynamics simulation was performed for the Eco-MscL/011A complex from the docking study. The simulation box consists of 1 copy of MS channel protein, 1 copy of 011A, a lipid bilayer [230 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) molecules] with 0.1 M KCl and water. In total, there are 38,800 atoms. The basic molecular dynamic simulation protocol was as previously described (13). After the system was well equilibrized, 120 molecular dynamic (MD) snapshots were evenly selected from 120 ns for the subsequent free-energy calculations.
For each MD snapshot, the molecular mechanical (MM) energy (EMM) and the Poisson-Boltzmann surface area (PBSA) were calculated without further minimization. Unlike the regular MM-PBSA analysis for global proteins, 2 external dielectrics (80 for water and 4.0 for the lipid bilayer) were applied for this system. The membrane center offset parameter (mctrdz), which varies from snapshot to snapshot, was calculated with the coordinate centers of Eco-MscL and the POPC bilayer. The membrane thickness was set to 18 Å, as previously described. For 011A, the implicit membrane option was turned off and the external dielectric constant was set to 80. The nonpolar solvation energies were calculated by using solvent accessible surface (SAS) areas with the following equation: ΔGnonpolar = 0.0054 × SAS + 0.92. The entropic term was estimated by using a method described by Wang and Hou (38). All the MD simulations and the free-energy analyses were performed using the Amber 14 software package (39).
RESULTS
Compounds 011 and 120 show MscL-dependent decreased growth and increase MscL channel activity
As previously described, we performed an HTS to identify compounds that deceased bacterial growth in an MscL-dependent manner (12). Two compounds identified in this screen are in the sulfonamide class (see Supplemental Fig. S1 for structures). To determine the specificity of the compounds for the Eco-MscL channel, we assayed their ability to decrease growth of overnight cultures in strains expressing the Eco-MscL channel, the unrelated MscS channel, and only the expression vector. Compound 120 showed decreased growth, regardless of whether MscL or MscS from E. coli was expressed, with greater decreases in growth observed with MscS (Fig. 1A). These data are with the MS channels expressed in trans, but similar results were obtained using null strains expressing endogenous levels of the MscL proteins (Supplemental Fig. S2, 2 left panels). Given that both MscL and MscS are gated by membrane tension, the data are consistent with compound 120 intercalating in the membrane, adding tension, and slowing growth of cells expressing either E. coli MscS or MscL. Cells expressing MscS are presumably more sensitive because MscS, which also senses changes in the membrane pressure profile, is more sensitive to membrane tension (40). In contrast, compound 011 appeared to be highly specific: only cells expressing MscL, not MscS, showed decreased growth when this compound was added.
Figure 1.

Compound 011 specifically inhibits growth of MscL expressing bacteria. A) Minimal inhibitory concentration curves are shown for compound 120 (green) and 011 (red). Growth of the E. coli strain MJF612 (ΔmscL, ΔmscS, ΔmscK, and ΔybdG) carrying an empty plasmid (black) or expressing MscS (open squares) or Eco-MscL (solid squares) was measured as the OD600 of the cultures. Values are expressed as the percentage decrease in growth in the presence of compound 120 or 011, relative to the nontreated samples (n = 3–8). B) Representative traces showing the effects on MscL channel activity of compounds 120 (upper traces) and 011 (lower traces). The schemes on the right illustrate 2 different configurations where the patch is presented with the compound from the periplasmic side (pipette) of the cytoplasmic side (bath). Channel activity was recorded by patch clamp experiments of native membranes, in a single patch before (left traces) and after (right traces) treatment with the compounds, at the same indicated pressure.
To confirm that these compounds increased channel activity, we studied them at the single-channel level in patch clamp experiments. In brief, Eco-MscL channel activity was recorded by the patch clamp technique in native bacterial membranes with the giant-cell preparation (34). Representative traces taken from excised inside-out patches are shown in Fig. 1B, where Eco-MscL activity in the same patch was recorded before and after treatment with compounds 011 and 120. For each compound, we used 2 different configurations that allowed us to present the compound to the periplasmic (pipette) or cytoplasmic (bath) side of the membrane. Eco-MscL channel activity, measured as the open probability for MscL at the indicated pressure, increased from 0.18 ± 0.06 to 0.86 ± 0.2 (n = 7, P < 0.026, by paired Student’s t test), when 011 was presented from the periplasm, and from 0.14 ± 0.12 to 0.83 ± 1.17 (n = 6, P < 0.008, by paired Student t test), when presented from the cytoplasm. Consistent with the in vivo experiments, MscL channel activity also significantly increased when compound 120 was presented from the periplasm from 0.06 ± 0.05 to 0.53 ± 0.18 (n = 6; P < 0.02, by paired Student’s t test), but had no significant effect from the cytoplasm (0.04 ± 0.01 to 0.06 ± 0.01; n = 5, P > 0.2, paired Student’s t test). Note that no significant increase in MscL channel activity was observed for 1%-DMSO-only control experiments in either configuration. These results suggest that compounds 011 and 120 have different modes of action and that compound 011, but not 120, works on either side of the bacterial membrane.
Mechanisms of action: compound 011 leads to flux of K+ and glutamate and decreases viability of quiescent/stationary cells only through MscL activation
Sulfonamide antibiotics usually work by inhibiting folate synthesis. We used a previously published approach (28) to isolate sulfonamide-resistant MscL-null strains, which we characterized as having a mutation at proline 64 of the FolP protein (P64L). This mutant cell line was resistant to compound 120, but not to compound 011 (Fig. 2A). These data suggest that compound 120 opens MscL and uses it as a pathway to the cytoplasm, where it inhibits synthesis of folate; but compound 011 appears to inhibit growth solely by gating the MscL channel.
Figure 2.

The mode of action compound 011. A) Minimal inhibitory concentration curves are shown for compounds 120 or 011 in cultures of the sulfonamide resistant strain PB121 (open markers) and its parental MJF612 strain (closed markers). Bacterial cultures carrying an empty plasmid control (black) or expressing Eco-MscL, were treated with compound 120 (green) or compound 011 (red). Values represent the percentage decrease in growth (OD600) vs. nontreated. B) Minimal inhibitory concentration curves are shown for the MJF612 strain treated with compound 011 or 011A. Note that 011A is similar to 011 but lacking the sulfonamide group. Bacterial cultures carrying an empty plasmid (control) are shown in black, treated with 011 (closed squares) or 011A (open squares); cultures expressing Eco MscL treated with 011 (red) or 011A (blue) are shown. Values represent the percentage decreased in growth in the presence of compound 011 or 011A vs. nontreated. C) The reduction in viability of stationary cultures treated with 011 (red) or 011A (blue), expressed as the percent reduction of colony forming units (CFUs) vs. no treatment (n = 3–5). ***P < 0.0005, ****P < 0.00005, Eco WT vs. empty plasmid (2-tailed, 2-sample homoscedastic t test).
Given that compound 011 did not appear to work through the folate pathway, we decided to try a version of the compound that did not contain the sulfonamide moiety; we refer to this compound as 011A (see structure in Supplemental Fig. S1). Compound 011A had the same efficacy, but was close to 2 orders of magnitude more potent (Fig. 2B); this compound also worked at endogenous expression levels (Supplemental Fig. S2, right). Together, the data demonstrate that compounds 011 and 011A do not work as sulfonamides to inhibit the folate pathway, and the gating of MscL is its only apparent mechanism of action.
Potassium (K+) and glutamate (Glu) are natural osmoprotectants that accumulate at high osmolarity and fluxed out via MscL upon gating (9, 13, 41). As seen in Supplemental Table S1, when treated with 40 µM of 011A significant amounts of both K+ and Glu fluxed out of the cell; for 011, significant K+ fluxes were observed at this concentration. Compound 120 (4 µM) also led to K+ flux, consistent with its gating the MscL channel. No significant decrease in viability was observed in these cultures when controlled for OD600, suggesting that the compounds are largely static rather than cidal for growing cells, removing the possibility that the loss of K is related to cell lysis. In sum, these flux data are consistent with the notion that MscL is gated in vivo in response to treatment of these compounds.
The MscL channel does not require metabolic energy to be active and is upregulated at the stationary phase (42). We therefore speculated that, even though compounds 011 and 011A are shown to be largely static in growing cultures, the compounds may be cidal in quiescent or stationary cultures if enough metabolites are lost (i.e., the cells may not have the energy or resources to enter the logarithmic phase). We therefore grew cultures to the late stationary phase, and part of the culture was treated with 80 µM compound 011 or 011A and the viabilities determined. When Eco-MscL, but not Eco-MscS, was expressed, a significant decrease in viability was observed (Fig. 2C).
Compounds 011 and 011A bind between subunits at the cytoplasmic aqueous-membrane interface
As a first attempt to determine the binding site of compounds 011 and 011A, we screened a comprehensive library of Eco-MscL cysteine mutants containing 112 mutants in E. coli MscL (7, 23, 31, 43) (MscL is a cysteine-free protein). We have previously shown the viability of this approach using DHS as a model (13). Any single mutation that interferes with the compound 011 or 011A binding site is expected to partially suppress its normally observed growth inhibition. After screening the entire library 3 times for 011 and 5 times for 011A, we found 2 partial suppressors. These sites were consistent, not only among screens, but were also consistent between both the 011 and 011A compounds. Although separated in the secondary structure, both sites are along the MscL cytoplasmic membrane interface and have been shown to be in proximity (24); these sites were the E6C and I96C Eco-MscL (Fig. 3A).
Figure 3.

Compounds 011 and 011A block the binding of MTS-PEG5000 to specific cysteine mutants in a dose-dependent manner. A) The Eco MscL structure from molecular modeling, based on the Mycobacterium tuberculosis MscL crystal structure, is shown in a side view (left), with the approximate location of the bilayer indicated with horizontal bars; a single subunit is darkened for clarity. The location of the affected cysteine mutants is shown in a single subunit (middle) with residues E6 in green, V16 in blue, I96 in magenta, and K97 in red. A view from the cytoplasmic side (right) is also shown. B) Western blot analysis of MscL cysteine mutants after MTS-PEG5000 vs. compound 011 competition assay. The absence (−) or presence (+) of 50 μM MTS-PEG5000 (MTS-PEG), as well as the absence or different concentration of compound 011 used, are indicated in the table at the bottom of C. Top band: protein that has been PEGylated; this band disappears as the concentration of 011 is increased (1.5–5 mM). Both E6 and V16 dimmers can be seen slightly higher than the PEGylated protein band (PEG) (n = 3–7). Original magnification, ×2. C) Negative controls for compound 011: Eco-MscL with a mutation near the pore, I25C, shows a PEGylated band that is not inhibited or shifted by compound 011 (left), and WT Eco-MscL, which does not have any naturally occurring cysteine, shows no upper band with MTS-PEG5000 (right; n = 2). D) Western blot analysis after MTS-PEG5000 vs. compound 011A competition assay. The absence (−) or presence (+) of 50 μM MTS-PEG5000 (MTS-PEG) and the absence (−) or different concentration of compound 011A used, are indicated in the table at the bottom of E. Top band: protein that has been PEGylated; this band disappears as the concentration of 011A is increased (1.5–5 mM) (n = 3–5). E) Negative controls for compound 011A are shown: Eco-MscL I25C, shows a PEGylated band that is not inhibited or by compound 011A (left), and WT Eco-MscL, shows no top band with MTS-PEG5000 (right; n = 2). F) Docking site for 011A using Glide software. The position within the complex is shown on the left, and a close-up showing relative position to residues E6, F10, and K97 (K369 = K97, subunit 3).
We have developed a biochemical competition binding assay to determine the binding site of compounds to the MscL channel (13). In brief, the cysteine-mutated Eco-MscL protein is solubilized, purified, and treated with the thiol reagent Methoxypoly-(ethyleneglycol)-5000-amidopropionyl-meth-anethio-sulfonate (MTS-PEG5000), which binds to the mutated cysteine residue, decreasing its migration in SDS-PAGE. The compounds are assayed for their ability to block the MTS-PEG5000 reaction by competing for the binding site. We scanned several residues in this region (residues E6, F7, E9, F10, V16, I96, and K97). Addition of compound 011 inhibits MTS-PEG5000 modification of E6C, V16C, I96C, and K97C (Fig. 3B). This effect was site specific, given that wild-type Eco-MscL showed no shift in molecular weight when treated with MTS-PEG500, and compound 011 did not dramatically inhibit MTS-PEG5000 reaction with cysteine mutants in other locations, including I25C (Fig. 3C), previously identified by this assay for the DHS binding site (13). Similar experiments were performed for compound 011A, and consistent results were found for I96C and K97C (Fig. 3D, E). As with DHS (13), we also used computational docking for the 011A compound (Fig. 3F), which confirmed that E6, I96, and K97 are key residues of the binding pocket. The dynamics of 011A within the pocket is shown in Supplemental Fig. S3; this and the decreased size of 011A may explain why 011A binding is less affected by MTS-PEG5000 binding to specific cysteine mutants relative to 011 (e.g., E6C and V16C). We further studied the dynamics of Eco-MscL/011A binding using molecular dynamics (MD) simulations. As shown in Supplemental Fig. S4, the system is stable after the ∼20 ns equilibrium phase as measured by the root-mean-square deviation. The key residues for Eco-MscL/011A interaction revealed by MD simulations remain largely the same as those identified by docking simulation (Fig. 3F).
The free energy of Eco-MscL/011A binding was estimated by performing MM-PBSA analysis for 120 collected MD snapshots (Supplemental Table S2). For the Eco-MscL and Eco-MscL/011A, an implicit membrane was applied to accurately calculate the polar part of the solvation free energy (44). The total electrostatic energy (Eeel + 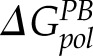 ) of −4.6 kcal/mol, is much smaller than the van der Waals energy, −30.59 kcal/mol, in agreement with the relatively hydrophobic binding pocket. The total MM-PBSA binding free energy, −10.32 kcal/mol, corresponds to 25 nM at 298K.
) of −4.6 kcal/mol, is much smaller than the van der Waals energy, −30.59 kcal/mol, in agreement with the relatively hydrophobic binding pocket. The total MM-PBSA binding free energy, −10.32 kcal/mol, corresponds to 25 nM at 298K.
Bacillus subtilis MscL was one of the few orthologs that had a substitution at a residue suggested to be critical for 011 and 011A binding: residue K97 was an R (Fig. 4A). We therefore assayed E. coli cells expressing the B. subtilis MscL (B.sub-MscL), and 3 other previously characterized (45) orthologs that do not have substitutions in this region: Clostridium perfringens, Haemophilus influenzae, and Staphylococcus aureus, for sensitivity to compounds 011 and 011A. B.sub-MscL was the only ortholog that showed no detectable sensitivity to 011A (Supplemental Fig. S5). However, as shown in Fig. 4B, mutating the B.sub-MscL to R88K (position 88 in B.sub-MscL is equivalent to position 97 in Eco-MscL) generated a channel that, when expressed, conferred sensitivity to compound 011A. On the other hand, mutating Eco-MscL to an R decreased the channel sensitivity to 011. These results were further confirmed by patch clamp Fig. 4C. The data suggest that lysine at position 97 in Eco-MscL is an important site for the binding of 011A, and that a single substitution at this position is enough to alter responsiveness to the compound.
Figure 4.

Mutational analysis provides additional evidence that residue K97 in Eco-MscL contributes to the binding site of compound 011A. A) Sequence alignment of 2 MscL homologs from Eco-MscL and B.sub-MscL showing the difference at amino acid 97 (boxed). B) Inhibition of growth (OD600) of E. coli strain MJF455 cultures, in the presence of compound 011A, expressed as the percentage of the nontreated. Note that changing the Eco-MscL K97 to R decreases 011A sensitivity, whereas changing B.sub-MscL R88 to K increases sensitivity (n = 3–6). ***P < 0.0005, ****P < 0.00005, mutations vs. WT for either Eco- or B.sub-MscL (2-tailed, 2-sample homoscedastic t test). C) Representative traces of the channel activity of Eco-MscL, B.sub-MscL, R88K B.sub-MscL, and K97R Eco-MscL, recorded from giant spheroplasts derived from the MJF612 strain. The effects of compound 011A on channel activity of MscL orthologs and mutants was recorded in the same patch before (left traces) and after (right traces) treatment with the 011A compound in the bath, at the indicated.
Thus, 4 independent approaches all support a single cytoplasmic/membrane-interface binding site for the 011 and 011A compounds: scanning a cysteine library for partial suppressors, a biochemical competition assay, computational docking energetics, and the study of orthologs and subsequent mutational analyses.
DISCUSSION
Several studies have found that when MscL gates at inappropriate times, it is detrimental to the cell (7–10, 31, 46), suggesting that MscL is a valid drug target. However, no specific agonists or modulators for the MscL channel were known. In this study, we identified the first compound that directly binds MscL and increases its activity.
Amphipaths that intercalate asymmetrically into the membrane are known to modulate MscL activity (47). However, these compounds simply change the pressures within the membrane and therefore are nonspecific, modulating many other MS channels including MscS (48). A study by Iscla et al. (49) of one compound, 1,3,5-Tris[(1E)-2′-(4″-benzoic acid)vinyl]benzene, now called Ramizol (Boulos and Cooper Pharmaceuticals, Balcatta, WA, Australia) (50), demonstrated that this potential antibiotic inhibits growth in an MscL-dependent manner, but also appears to have some influence on cells expressing MscS, strongly suggesting that Ramizol has at least some nonspecific influence on MS channels, presumably by intercalating into and adding tensions in the membrane. In addition, Ramizol still inhibits the growth of cells not expressing MscL or MscS, albeit at higher concentrations, thus demonstrating that modulating MS channels is not the only mode of action. Our HTS looking for MscL agonists surprisingly identified some known antibiotics (12). The influences on growth and viability of the aminoglycosides spectinomycin and DHS weres specific to MscL; MscS expression had no influence. Both drugs still inhibited growth of cells not expressing MscL, but higher concentrations were needed, consistent with the primary mode of action being the inhibition or disruption of protein synthesis. In a subsequent study, we demonstrated that DHS directly binds to, modulates, and passes through the MscL channel pore, appearing to be one of the major pathways for cytoplasm access (13). In sum, previous findings demonstrated that inappropriately gating MscL may have negative consequences for the cell and may even allow easier passage of antibiotics, but none of the studies found that modulating MscL activity is the primary mode of action of any compound or antibiotic drug that inhibits growth.
In this study, we have characterized 2 novel sulfonamide compounds that were identified in our HTS. Our data suggest that compound 120 works indirectly by increasing membrane tension and is a classic sulfonamide antibiotic that works through the folate pathway. In contrast, there is no indication that compound 011 works by adding tension in the membrane (no slowed growth was observed when only MscS was expressed), and no other mode of action than activating MscL is suggested. Notably, the compound even increases potency about 2 orders of magnitude when the sulfonamide portion is removed. Finally, it does not appear to be simply a conduit for cytoplasmic access, and has other cytoplasmic modes of action, because in patch clamp the compound increases MscL activity no matter what side of the membrane it is on, strongly suggesting it easily crosses the membrane.
The binding site of compound 011, and its smaller analog 011A, to MscL appears to be at the cytoplasmic interface, further supporting the notion that these compounds easily cross the membrane. We previously used a series of simple assays to determine the binding site for DHS (13); these assays were also used with the 011 compounds. Although the binding sites for DHS and the compounds 011 and 011A are quite different, all assays agreed in both cases, thus demonstrating the general usefulness of these approaches. The binding site for compounds 011 and 011A are at a subunit interface, reminiscent of a finding long ago that acetylcholine binds to the muscle nicotinic receptor (51); many channel agonists have since been shown to bind at subunit interfaces.
The significance of the binding site being at the S1 domain is amplified because many other channels have an analogous feature. For example, the bacterial inward rectifying K+ channel KirBac, in which the cytoplasmic α-helix running parallel to the membrane (slide-helix) (15, 16) was found to directly interact with the phospholipids headgroups to regulate channel gating (17). A similar structure has been found in 2-pore domain K+ (K2P) channels, where it has been proposed to play a role in mechanosensitive channel gating (21). In another study, the aromatic nature of a residue at the cytoplasmic end of the putative pore-forming TMD6 domain of the mechanosensitive yeast TRPY1 channel has been shown to be important in gating and has been referred to as a “gate anchor” (18); the region could form an amphipathic α-helix. The region also appears to be part of a latch for TRPV4 gating (19, 20). Finally, in crystallographic structures of MscS, another bacterial mechanosensitive channel from an independent channel family, there also is an α-helix along the cytoplasmic-membrane surface just adjacent to the pore (4, 22). Similar to MscL, current models of MscS gating predict a tilting of the pore domain (52), and a glycine is also present between these 2 domains (Gly113), probably serving as a hinge (4, 22). A previous study of this region of MscL suggested important interactions between the S1 region of the protein and TM2 of a neighboring subunit, including the E6/K97 interaction highlighted here, and that these regions “slide” relative to each other upon gating (24); this movement has since been confirmed by other approaches (25). Compounds 011 and 011A likely modify or disrupt protein-protein and protein-lipid interactions at this location that normally stabilize the closed state, encouraging this “sliding” and opening of the MscL channel. Capsaicin, a small-compound TRPV1 agonist, binds a structure that has clear mechanistic similarity—the S4-S5 linker helix, an α-helix running along the cytoplasmic membrane, contacting lipids as well as pore-lining helices near the gate (53). Given our data, this additional example for capsaicin binding to TRPV1 and the finding that α-helices are often found along the cytoplasmic membrane of channel structures, we propose that these analogous structures found in numerous channel proteins are targets for pharmacological manipulation.
Thus, the data demonstrate that MscL can be specifically activated by small compounds and, in doing so, can slow growth and even decrease viability of quiescent/stationary cells. Thus, MscL may indeed be a viable antibiotic target. The data also provide evidence that an α-helix that runs along the surface of the cytoplasmic membrane, S1, and its interactions with a transmembrane domain, TM2, of a neighboring subunit, are important for stabilizing the closed state; disrupting this with a small compound leads to inappropriate gating, and thus slowed bacterial growth. Finally, because the motif of a helix running along the cytoplasmic membrane that interacts with lipids and protein domains from neighboring subunits is a common feature among channels, the findings may have broader implications for pharmacologically manipulating other channels.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
ACKNOWLEDGMENTS
The authors thank Drs. Limin Yang (University of Texas, Southwestern Medical Center) and Andriy Anishkin (University of Maryland, College Park, MD, USA) for helpful suggestions and critical reading of the manuscript. This work was supported by U.S. National Institutes of Health (NIH) National Institute of General Medical Sciences (NIGMS) Grants R01 GM061028 and GM121780, and NIH National Institute of Biomedical Imaging and Bioengineering Grant P41-EB015908 (to Z.K.); Welch Foundation Grant I-1420 (to P.B.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or other funding organizations. The authors declare no conflicts of interest.
Glossary
- B.sub-MscL
MscL from Bacillus subtilis
- CphM
citrate-phosphate–defined medium
- DHS
dihydrostreptomycin
- Eco-MscL
MscL from Escherichia coli
- HTS
high-throughput screen
- ITPG
isopropyl-β-d-thiogalactopyranoside
- MD
molecular dynamics
- MM
molecular mechanical
- MS
mechanosensitive
- MscL
mechanosensitive channel of large conductance
- MTS-PEG5000
methoxypoly(ethyleneglycol)-5000-amidopropionyl-methanethio-sulfonate
- PBSA
Poisson-Boltzmann surface area
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- SMX
sulfamethoxazole
- TM
transmembrane domain
- TRP
transient receptor potential
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
R. Wray performed most of the whole-cell physiology assays including growth, viability, and in vivo flux studies; I. Iscla developed the HTS screen, performed the initial screening with the University of Texas Southwestern Medical Center core facility, performed all the electrophysiology, and helped to oversee the project; Z. Kovacs oversaw the chemical aspects of the project including suggestions for additional compounds for screens and chemical synthesis; J. Wang performed the computational analyses including docking and MD simulations; P. Blount oversaw and orchestrated the project; and all authors contributed to the writing of the manuscript.
REFERENCES
- 1.Booth I. R., Blount P. (2012) The MscS and MscL families of mechanosensitive channels act as microbial emergency release valves. J. Bacteriol. 194, 4802–4809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cruickshank C. C., Minchin R. F., Le Dain A. C., Martinac B. (1997) Estimation of the pore size of the large-conductance mechanosensitive ion channel of Escherichia coli. Biophys. J. 73, 1925–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iscla I., Blount P. (2012) Sensing and responding to membrane tension: the bacterial MscL channel as a model system. Biophys. J. 103, 169–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steinbacher S., Bass R., Strop P., Rees D. C. (2007) Structures of the prokaryotic mechanosensitive channels MscL and MscS: mechanosensitive ion channels. Curr Topics Membranes. 58, 1–20, [Google Scholar]
- 5.Cox C. D., Bavi N., Martinac B. (2018) Bacterial mechanosensors. Annu. Rev. Physiol. 80, 71–93 [DOI] [PubMed] [Google Scholar]
- 6.Blount P., Iscla I., Li Y. (2008) Mechanosensitive channels and sensing osmotic stimuli in bacteria Sensing with Ion Channels 11, 25–47 [Google Scholar]
- 7.Bartlett J. L., Levin G., Blount P. (2004) An in vivo assay identifies changes in residue accessibility on mechanosensitive channel gating. Proc. Natl. Acad. Sci. USA 101, 10161–10165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bartlett J. L., Li Y., Blount P. (2006) Mechanosensitive channel gating transitions resolved by functional changes upon pore modification. Biophys. J. 91, 3684–3691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ou X., Blount P., Hoffman R. J., Kung C. (1998) One face of a transmembrane helix is crucial in mechanosensitive channel gating. Proc. Natl. Acad. Sci. USA 95, 11471–11475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maurer J. A., Dougherty D. A. (2001) A high-throughput screen for MscL channel activity and mutational phenotyping. Biochim. Biophys. Acta 1514, 165–169 [DOI] [PubMed] [Google Scholar]
- 11.Blount P., Moe P. C. (1999) Bacterial mechanosensitive channels: integrating physiology, structure and function. Trends Microbiol. 7, 420–424 [DOI] [PubMed] [Google Scholar]
- 12.Iscla I., Wray R., Wei S., Posner B., Blount P. (2014) Streptomycin potency is dependent on MscL channel expression. Nat. Commun. 5, 4891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wray R., Iscla I., Gao Y., Li H., Wang J., Blount P. (2016) Dihydrostreptomycin directly binds to, modulates, and passes through the MscL channel pore. PLoS Biol. 14, e1002473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iscla I., Wray R., Blount P. (2011) An in vivo screen reveals protein-lipid interactions crucial for gating a mechanosensitive channel. FASEB J. 25, 694–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuo A., Domene C., Johnson L. N., Doyle D. A., Vénien-Bryan C. (2005) Two different conformational states of the KirBac3.1 potassium channel revealed by electron crystallography. Structure 13, 1463–1472 [DOI] [PubMed] [Google Scholar]
- 16.Kuo A., Gulbis J. M., Antcliff J. F., Rahman T., Lowe E. D., Zimmer J., Cuthbertson J., Ashcroft F. M., Ezaki T., Doyle D. A. (2003) Crystal structure of the potassium channel KirBac1.1 in the closed state. Science 300, 1922–1926 [DOI] [PubMed] [Google Scholar]
- 17.Enkvetchakul D., Jeliazkova I., Bhattacharyya J., Nichols C. G. (2007) Control of inward rectifier K channel activity by lipid tethering of cytoplasmic domains. J. Gen. Physiol. 130, 329–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou X., Su Z., Anishkin A., Haynes W. J., Friske E. M., Loukin S. H., Kung C., Saimi Y. (2007) Yeast screens show aromatic residues at the end of the sixth helix anchor transient receptor potential channel gate. Proc. Natl. Acad. Sci. USA 104, 15555–15559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teng J., Loukin S. H., Anishkin A., Kung C. (2016) A competing hydrophobic tug on L596 to the membrane core unlatches S4-S5 linker elbow from TRP helix and allows TRPV4 channel to open. Proc. Natl. Acad. Sci. USA 113, 11847–11852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teng J., Loukin S. H., Anishkin A., Kung C. (2015) L596-W733 bond between the start of the S4-S5 linker and the TRP box stabilizes the closed state of TRPV4 channel. Proc. Natl. Acad. Sci. USA 112, 3386–3391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brohawn S. G., del Mármol J., MacKinnon R. (2012) Crystal structure of the human K2P TRAAK, a lipid- and mechano-sensitive K+ ion channel. Science 335, 436–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bass R. B., Strop P., Barclay M., Rees D. C. (2002) Crystal structure of Escherichia coli MscS, a voltage-modulated and mechanosensitive channel. Science 298, 1582–1587 [DOI] [PubMed] [Google Scholar]
- 23.Iscla I., Wray R., Blount P. (2008) On the structure of the N-terminal domain of the MscL channel: helical bundle or membrane interface. Biophys. J. 95, 2283–2291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iscla I., Wray R., Blount P. (2012) The dynamics of protein-protein interactions between domains of MscL at the cytoplasmic-lipid interface. Channels (Austin) 6, 255–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bavi N., Cortes D. M., Cox C. D., Rohde P. R., Liu W., Deitmer J. W., Bavi O., Strop P., Hill A. P., Rees D., Corry B., Perozo E., Martinac B. (2016) The role of MscL amphipathic N terminus indicates a blueprint for bilayer-mediated gating of mechanosensitive channels. Nat. Commun. 7, 11984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levina N., Tötemeyer S., Stokes N. R., Louis P., Jones M. A., Booth I. R. (1999) Protection of Escherichia coli cells against extreme turgor by activation of MscS and MscL mechanosensitive channels: identification of genes required for MscS activity. EMBO J. 18, 1730–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schumann U., Edwards M. D., Rasmussen T., Bartlett W., van West P., Booth I. R. (2010) YbdG in Escherichia coli is a threshold-setting mechanosensitive channel with MscM activity. Proc. Natl. Acad. Sci. USA 107, 12664–12669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sköld O. (1976) R-factor-mediated resistance to sulfonamides by a plasmid-borne, drug-resistant dihydropteroate synthase. Antimicrob. Agents Chemother. 9, 49–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vedantam G., Guay G. G., Austria N. E., Doktor S. Z., Nichols B. P. (1998) Characterization of mutations contributing to sulfathiazole resistance in Escherichia coli. Antimicrob. Agents Chemother. 42, 88–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blount P., Krause J. E. (1993) Functional nonequivalence of structurally homologous domains of neurokinin-1 and neurokinin-2 type tachykinin receptors. J. Biol. Chem. 268, 16388–16395 [PubMed] [Google Scholar]
- 31.Levin G., Blount P. (2004) Cysteine scanning of MscL transmembrane domains reveals residues critical for mechanosensitive channel gating. Biophys. J. 86, 2862–2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blount P., Sukharev S. I., Schroeder M. J., Nagle S. K., Kung C. (1996) Single residue substitutions that change the gating properties of a mechanosensitive channel in Escherichia coli. Proc. Natl. Acad. Sci. USA 93, 11652–11657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blount P., Sukharev S. I., Moe P. C., Schroeder M. J., Guy H. R., Kung C. (1996) Membrane topology and multimeric structure of a mechanosensitive channel protein of Escherichia coli. EMBO J. 15, 4798–4805 [PMC free article] [PubMed] [Google Scholar]
- 34.Blount P., Sukharev S. I., Moe P. C., Martinac B., Kung C. (1999) Mechanosensitive channels of bacteria. Meth. Enzymol 294, 458–482 [DOI] [PubMed] [Google Scholar]
- 35.Batiza A. F., Kuo M. M., Yoshimura K., Kung C. (2002) Gating the bacterial mechanosensitive channel MscL invivo. Proc. Natl. Acad. Sci. USA 99, 5643–5648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sybyl-X (2013) Molecular modeling software package version 2.1.1, Tripos International, St. Louis [Google Scholar]
- 37.Glide 6.8 (2015) Schrodinger, Inc., New York [Google Scholar]
- 38.Wang J., Hou T. (2012) Develop and test a solvent accessible surface area-based model in conformational entropy calculations. J. Chem. Inf. Model. 52, 1199–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Case D. A., Berryman J. T., Betz R. M., Cerutti D. S., Cheatham I. T. E., Darden T. A., Duke R. E., Giese T. J., Gohlke H., Goetz A. W., Homeyer N., Izadi S., Janowski P., Kaus v., Kovalenko A., Lee T. S., LeGrand S., Li P., Luchko T., Luo R., Madej B. D., Merz K. M., Monard G., Needham P., Nguyen H., Nguyen H. T., Omelyan I., Onufriev A., Roe D. R., Roitberg A., Salomon-Ferrer R., Simmerling C. L., Smith W., Swails J., Walker R. C., Wang J., Wolf R. M., Wu X., York D. M., Kollman P. A. (2015) AMBER 14: tools for molecular simulations, University of California, San Francisco [Google Scholar]
- 40.Moe P., Blount P. (2005) Assessment of potential stimuli for mechano-dependent gating of MscL: effects of pressure, tension, and lipid headgroups. Biochemistry 44, 12239–12244 [DOI] [PubMed] [Google Scholar]
- 41.Blount P., Schroeder M. J., Kung C. (1997) Mutations in a bacterial mechanosensitive channel change the cellular response to osmotic stress. J. Biol. Chem. 272, 32150–32157 [DOI] [PubMed] [Google Scholar]
- 42.Stokes N. R., Murray H. D., Subramaniam C., Gourse R. L., Louis P., Bartlett W., Miller S., Booth I. R. (2003) A role for mechanosensitive channels in survival of stationary phase: regulation of channel expression by RpoS. Proc. Natl. Acad. Sci. USA 100, 15959–15964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iscla I., Levin G., Wray R., Blount P. (2007) Disulfide trapping the mechanosensitive channel MscL into a gating-transition state. Biophys. J. 92, 1224–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiao L., Diao J., Greene D., Wang J., Luo R. (2017) A continuum Poisson-Boltzmann model for membrane channel proteins. J. Chem. Theory Comput. 13, 3398–3412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moe P. C., Blount P., Kung C. (1998) Functional and structural conservation in the mechanosensitive channel MscL implicates elements crucial for mechanosensation. Mol. Microbiol. 28, 583–592 [DOI] [PubMed] [Google Scholar]
- 46.Iscla I., Wray R., Eaton C., Blount P. (2015) Scanning MscL channels with targeted post-translational modifications for functional alterations. PLoS One 10, e0137994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perozo E., Kloda A., Cortes D. M., Martinac B. (2002) Physical principles underlying the transduction of bilayer deformation forces during mechanosensitive channel gating. Nat. Struct. Biol. 9, 696–703 [DOI] [PubMed] [Google Scholar]
- 48.Blount P., Li Y., Moe P. C., Iscla I. (2008) Mechanosensitive channels gated by membrane tension: bacteria and beyond: mechanosensitive ion channels. Mechanosensitivity in Cells and Tissues 1, 71–101, Springer Press, New York [Google Scholar]
- 49.Iscla I., Wray R., Blount P., Larkins-Ford J., Conery A. L., Ausubel F. M., Ramu S., Kavanagh A., Huang J. X., Blaskovich M. A., Cooper M. A., Obregon-Henao A., Orme I., Tjandra E. S., Stroeher U. H., Brown M. H., Macardle C., van Holst N., Ling Tong C., Slattery A. D., Gibson C. T., Raston C. L., Boulos R. A. (2015) A new antibiotic with potent activity targets MscL. J. Antibiot. (Tokyo) 68, 453–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rao S., Prestidge C. A., Miesel L., Sweeney D., Shinabarger D. L., Boulos R. A. (2016) Preclinical development of Ramizol, an antibiotic belonging to a new class, for the treatment of Clostridium difficile colitis. J. Antibiot. (Tokyo) 69, 879–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blount P., Merlie J. P. (1989) Molecular basis of the two nonequivalent ligand binding sites of the muscle nicotinic acetylcholine receptor. Neuron 3, 349–357 [DOI] [PubMed] [Google Scholar]
- 52.Edwards M. D., Li Y., Kim S., Miller S., Bartlett W., Black S., Dennison S., Iscla I., Blount P., Bowie J. U., Booth I. R. (2005) Pivotal role of the glycine-rich TM3 helix in gating the MscS mechanosensitive channel. Nat. Struct. Mol. Biol. 12, 113–119 [DOI] [PubMed] [Google Scholar]
- 53.Yang F., Zheng J. (2017) Understand spiciness: mechanism of TRPV1 channel activation by capsaicin. Protein Cell 8, 169–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.